

Abstract
Type I interferon (IFN), as its name implies, ‘interferes’ with virus replication by activating numerous genes. Further, virus-induced type I IFN regulates the magnitude and functions of cells directing the host immune system. Importantly, recent exploration into how type I IFN operates following virus infection has advanced our understanding of its role with respect to modulation of host innate and adaptive immune responses. Such activities include the activation of antigen-presenting dendritic cells and the localization, expansion or differentiation of virus-specific T lymphocytes and antibody-producing B lymphocytes. However, type I IFN not only benefits the host but can also induce unnecessary or extremely pathogenic immune responses. This review focuses on such interactions and the manner in which type I IFN induces dynamic changes in the host immune network, particularly adaptive immune responses to viral invasion. Manipulating the type I IFN-mediated host immune response during virus infections could provide new immunotherapeutic interventions to remedy viral diseases and implement more effective and sustainable type I IFN therapy.
I. Introduction
Type I interferon (IFN) is renowned as the most powerful of antiviral molecules, because it effectively obstructs the replication of numerous viruses. Its inhibitory activity was initially uncovered by Isaacs and Lindenmann (1957), who coined the name (interfere + -on). Since then, its potency has been reaffirmed in multiple experimental systems in vitro and in vivo and occasionally even in virus-infected humans. The success of genetics technology for creating knockout (ko) mice deficient in type I IFN signaling highlighted the importance of type I IFN for protection of the host from virus-induced pathogenicity in vivo. Type I IFN is a cytokine family, which includes IFN-α (13 subtypes in mice), IFN-β, IFN-ɛ, IFN-κ, and IFN-ω; therefore, deleting the gene encoding the known receptor subunit (IFNAR1) for all these components blocked the activity of the entire system (Muller et al., 1994). Mice lacking the cognate receptor for type I IFN became highly susceptible to severe infections with multiple viruses such as vesicular stomatitis virus (VSV), Semliki Forest virus (Muller et al., 1994), and the A/WSN/33 strain of influenza virus (Garcia-Sastre et al., 1998). Similarly, other strains of influenza virus propagated much faster in mice deficient in a single IFN-β gene with higher virulence when compared to wild-type (wt) mice (Koerner et al., 2007). Although viruses appear to have devised multiple strategies to evade or counteract the type I IFN response, the results obtained from experiments with IFNAR ko mice confirmed that the type I IFN system is required for effective host protection from diverse viral diseases. Multiple research fields deal with type I IFN responses to virus infections. These specialties include investigations of (1) molecular mechanisms for type I IFN induction pathways, such as recognition of viral components by Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and nucleotide oligomerization domain (NOD)-like receptors (NLRs); (2) the JAK/STAT signaling pathway; (3) IFN-stimulated genes (ISGs) such as PKR; (4) viral evasion strategies of type I IFN induction, signaling, or function of ISGs; and (5) the effect of type I IFN on host adaptive immune responses. This review documents recent results from work on type I IFN's interaction with the host immune system, particularly the responses of dendritic cells (DCs) and T and B lymphocytes to virus infections.
II. Regulation of DC Responses by Type I IFN Released Following Virus Infections
A. Production of type I IFN from DCs
Identification of plasmacytoid DCs (PDCs) as major IFN-producing cells (IPCs) has advanced study of IFN generation and the specification of cell types that mediate its synthesis (Colonna et al., 2004). Although all nucleated cells can produce type I IFN, PDCs are especially abundant producers upon virus infection (Barchet et al., 2005; Fig. 4.1A). They synthesize up to 10 pg of type I IFN/cell and are 10–100-fold more efficient than other cell types. For example, the antiviral activity of type I IFN-producing PDCs was compared to that of myeloid DCs. The myeloid DCs were shown to be susceptible to H5N1 influenza virus infection, and that susceptibility was reversed by pretreatment with type I IFN, whereas type I IFN-producing PDCs were resistant to the viral infection (Thitithanyanont et al., 2007). Similarly, after infection with the coronavirus, mouse hepatitis virus (MHV), splenic PDCs, but not myeloid DCs, produced significant amount of type I IFN and rapidly contained MHV replication (Cervantes-Barragan et al., 2007). However, the expression of IFNAR on macrophages and conventional DCs was pivotal for the control of fatal cytopathic MHV infection (Cervantes-Barragan et al., 2009). These findings suggest that PDCs release type I IFN, which acts directly on conventional DCs and macrophages to protect the host from coronaviral pathogenicity. Depletion of PDCs by injecting mice with antibodies that react with a bone marrow (BM) stromal cell antigen 2 (BST-2), such as 120G8 or PDC antigen-1 (PDCA-1), substantially decreased the level of type I IFN following virus infection in vivo and increased the animals' susceptibility to this infection (Swiecki and Colonna, 2010). Further, the CpG-dependent OVA-specific CD8 T cell response in spleens was drastically impaired by depletion of PDCs from mice treated with 120G8 antibody (Honda et al., 2005), revealing the importance of PDCs for the induction of adaptive immunity. However, although BST-2 is exclusively expressed on PDCs and plasma cells, it was inducible on multiple cell types by type I or type II IFN or by virus infection (Blasius et al., 2006), prompting reevaluation of results from experiments with antibody-mediated PDC depletion (Swiecki and Colonna, 2010).
Figure 4.1.
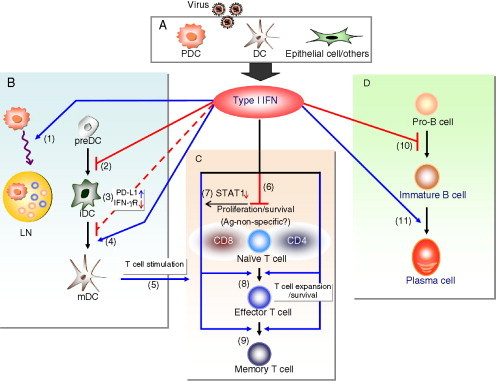
The effect of type I IFN on differentiation and function of DCs, T cells, and B cells. (A) Following virus infection, cells including PDCs, DCs, and epithelial cells produce type I IFN, which affects the differentiation and function of DCs (B), T cells (C), and B cells (D). B. Type I IFN promotes migration of PDCs (1) as well as transit of T and B cells into lymph nodes leading to transient lymphopenia in the bloodstream (not shown); inhibits development of committed immature DCs (iDC) from precursor DCs (preDC) via STAT2-specific signaling (2); could inhibit DC function by increasing PD-L1 expression or downregulating the receptor for IFN-γ (3); increases the maturation of DCs (4) and upgrades the capacity of mature DCs (mDC) to stimulate T cells (5). C. Type I IFN inhibits T cell proliferation and induces T cell apoptosis under certain conditions (6); virus-specific CD8 T cells, but not CD4 T cells, display low levels of STAT1 and evade type I IFN's antiproliferative activity (7); type I IFN is critical for viral antigen-specific T cell expansion and survival (8), and the formation of memory T cells (9). D. The effect of type I IFN on B cell responses: type I IFN inhibits the development of B cells (10) but enhances antibody-mediated B cell responses to viral infections and the differentiation of B cells into plasma cells (11).
PDCs express large amounts of IRF7; the expression and activation of IRF7 are essential for type I IFN production (Honda et al., 2005). These cells respond efficiently to stimulation with TLR7 or TLR9 ligands by producing type I IFN massively in murine systems. However, following infection with lymphocytic choriomeningitis virus (LCMV), PDC depletion failed to abrogate type I IFN production (Dalod et al., 2002). Indeed, myeloid DCs (CD11chighLy6C−B220−) isolated from spleens of mice infected with LCMV Clone 13 (Cl 13) released high levels of type I IFN (Diebold et al., 2003). Similarly, influenza virus lacking NS1 as well as synthetic dsRNA poly(I:C) induced substantial amounts of type I IFN from splenic DCs or BM-derived DCs in GM-CSF-supplemented culture, indicating that non-PDCs could become plentiful IFN producers under certain circumstances such as specific virus infections. Measles virus (MV) was reported to block signaling for type I IFN induction mediated by TLR7 and TLR9 on PDCs (Schlender et al., 2005), whereas type I IFN's synthesis was detected when GM-CSF-driven conventional DCs were infected with MV (Hahm, 2009). Thus, the nature of infecting viruses strongly affects the cell types synthesizing type I IFN and the amount produced.
Type I IFN is upregulated and detected in sera for several days upon LCMV Cl 13 infection in vivo (Zuniga et al., 2007). However, the cytokine's presence is nearly undetectable in sera during chronic infection, although the virus might continue to replicate in cells and, thus, persist in the host. This process is attributable to the negative regulatory circuits of type I IFN synthesis such as activation of suppressor of cytokine signaling-3 (SOCS-3) viral inhibition of type I IFN signaling, that is, suppression of the host's innate immune response (Martinez-Sobrido et al., 2009), persistence of the virus in a specific target tissue, and/or decreased virus amplification efficiency during the state of persistence. However, when DCs from spleens were isolated at 30 days postinfection with LCMV Cl 13, which preferentially infects DCs via α-dystroglycan receptor, type I IFN mRNA was detected in those DCs, suggesting the sustained local production of type I IFN by the infected cells during viral persistence (Hahm et al., 2005, Truong et al., 2009). Type I IFN was suggested to inhibit DC development, presumably explaining the decreased number of DCs observed during the LCMV Cl 13 persistence (Lee et al., 2009). However, elevated IFN-β contributes to the upregulation of MHC class I detected in the CNS and the peripheral tissues during LCMV persistence and seemed to continuously display antiviral activity, since the virus titer increased in IFNAR-deficient LCMV carrier mice (Truong et al., 2009). Thus, the precise role of locally produced type I IFN during chronic virus infections needs further investigation.
B. Dual opposite effects of type I IFN on DC development versus DC maturation
DCs are the most potent of all antigen-presenting cells and adept at priming naïve T cells (Steinman, 2007). Following the recognition of viral components, DCs mature, migrate to secondary lymphoid organs, and interact with antigen-specific T cells to stimulate them. The capacity of DCs to prime T cells is strongly influenced by the former's maturation status. DC maturation is determined mainly by the expression level of costimulatory molecules such as B7-1, B7-2 and CD40, and MHC molecules (MHC-I and MHC-II). Type I IFN was reported to enhance DC maturation, since the treatment of DCs with type I IFN increased the amounts of those proteins on the surfaces of DCs (Fig. 4.2 ) and rendered them more efficient in stimulating T cells. Exposure of human conventional DCs and PDCs to recombinant IFN-β before influenza virus infection enhanced the cells' expression of multiple cytokines and ISGs, indicating that type I IFN can prime DC activation to enhance the immune response to such infection (Phipps-Yonas et al., 2008). In support of the type I IFN's DC stimulatory activity, impaired DC maturation was observed when IFNAR-deficient mice were infected with Newcastle disease virus (NDV; Honda et al., 2003). However, migration of IFNAR-deficient DCs into secondary lymphoid tissues and the expression of CCR7 mRNA on the IFNAR-defective DCs were unaltered by NDV infection. These results suggest that type I IFN is critical for DC maturation but not for DC migration upon NDV infection.
Figure 4.2.
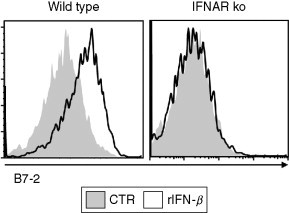
Type I IFN induces maturation of DCs. Wild-type or IFNAR-deficient BM-derived DCs were untreated (control, CTR) or treated with recombinant IFN-β (rIFN-β) (1000 U/mL). At day 2 after treatment, CD11c+ cells were analyzed for the expression of B7-2 by flow cytometry.
Experiments performed with human monocyte-derived DCs revealed two opposite functions of type I IFN. DCs derived from type I IFN-treated human monocytes were not competent in producing IL-12 and inefficient in stimulating T helper (Th) 1 type immune responses (Dauer et al., 2003). In contrast, type I IFN induced a rapid maturation of monocytes into potent DCs that effectively induced IL-15 and promoted a strong Th1 cell response (Santini et al., 2000). Further analysis of murine BM-derived DCs clarified the contrasting effect of type I IFN on DC precursors versus committed immature DCs (Hahm et al., 2005). Recombinant IFN-β markedly impaired the development and generation of myeloid DCs or PDCs from DC precursors, whereas it enhanced the maturation of committed immature DCs by elevating the level of MHC molecules and costimulatory molecules (Fig. 4.1B). This result indicates that the differentiation status of DCs (DC precursor vs. committed DC) is a decisive factor in the outcome of type I IFN treatment. Blockade of DC development was also demonstrated by using immune suppressive viruses such as MV and LCMV Cl 13 and was mediated via a STAT1-independent but STAT2-dependent novel IFN signaling pathway. Presumably, these viruses utilize the pathogenic effect of type I IFN via STAT2-specific signaling to suppress the host immune system. Interestingly, induction of a type I IFN-induced protein, adenosine deaminase acting on RNA (ADAR) 1a (p150) is dependent on STAT2 but not STAT1 (George et al., 2008). ADAR1a displays proviral activity by antagonizing PKR activation and enhancing VSV amplification (Li et al., 2010, Nie et al., 2007). Still unknown is whether ADAR1a is induced in DCs to affect the development or activation of DCs upon virus infections.
C. Type I IFN–DC interaction during chronic human virus infections
In the blood of human immunodeficiency virus (HIV)-1-infected donors, the absolute number of myeloid DCs and PDCs was shown to be decreased (Donaghy et al., 2001, Pacanowski et al., 2001). The loss of DC quantity inversely correlated with the viral load in the plasma of HIV patients. Whether the reduction of DC quantity is caused by the inhibition of DC development by type I IFN (Hahm et al., 2005) or by DC apoptosis or necrosis, as observed in vitro (Meyers et al., 2007), is uncertain. However, very similar findings were reported when patients persistently infected with hepatitis C virus (HCV) were examined for DC frequency (Kanto et al., 2004) and apoptosis (Siavoshian et al., 2005). An alternative possibility is that the DC quantity in the blood decreased because DCs accumulated, instead, in the secondary lymphoid tissues (Dillon et al., 2008). Although it is unclear whether DC accumulation in the lymph nodes is mediated by type I IFN induced by HIV or HCV infection, type I IFN can cause PDCs to gather in the lymph nodes (Gao et al., 2009).
Viral persistence can be achieved when the pathogen evades or suppresses its host's immune system, especially the DC-mediated adaptive T cell immune response. Eventually, multiple T cells specifically reactive with the infecting virus are deleted or exhausted even though they had expanded soon after infection (Yi et al., 2010). These exhausted T cells become unable to respond to the viral antigen so fail to proliferate, do not synthesize effector cytokines (IL-2, IFN-γ, or TNF-α), and do not perform cytotoxic activity. These exhausted T cells have been detected during chronic infections with HIV, hepatitis B virus (HBV), and HCV and found to express several inhibitory proteins such as programmed death-1 (PD-1), which was initially identified in mice infected with LCMV Cl 13 (Barber et al., 2006). The continual interaction of inhibitory receptor PD-1 on T cells with its ligand PD-L1 on DCs or other cells is critical to sustain T cell exhaustion. In fact, type I IFN treatment was shown to upregulate PD-L1 on DCs during HCV infection (Groschel et al., 2008, Muhlbauer et al., 2006, Urbani et al., 2008; Fig. 4.1B), but why and how type I IFN would induce PD-L1 on DCs remains elusive. Possibly type I IFN produced by the host is designed to induce apoptosis on certain cells via PD-L1 to block viral propagation. Currently, pegylated IFN-α2 and ribavirin are being used as treatment for HCV infection, although this regimen is effective in only approximately 50% of such patients. Tests to discern how PD-L1 blockade affects IFN treatment of patients acutely infected with HCV have provided promising results (Urbani et al., 2008), but further detailed assessment is necessary for patients with chronic infections. Nevertheless, the effectiveness of current antiviral type I IFN therapy could be enhanced by elevating the immune regulatory activity of type I IFN or blocking the adverse pathogenic properties of IFN.
III. Type I IFN Modulation of T Cell Responses to Virus Infections
A. Mobilization of T lymphocytes
Upon virus infections, a transient lymphopenia in the bloodstream is frequently observed. This temporary reduction in the quantity of lymphocytes in the blood comes from the swift migration of lymphocytes into secondary lymphoid organs where they take actions in response to pathogenic invasion. Interestingly, transient blood lymphopenia and the subsequent accumulation of T cells as well as B cells and PDCs in the lymph nodes were shown to be largely dependent on type I IFN signaling resulting from viral infection (Gao et al., 2009, Kamphuis et al., 2006). Highly pathogenic H5N1 virus caused depletion of circulating T cells, which correlated with massive induction of a type I IFN response (Baskin et al., 2009). Since sphingosine 1-phosphate (S1P) receptor signaling is involved in the egress of lymphocytes from lymph nodes (Rosen and Goetzl, 2005), possibly type I IFN acts on S1P signaling to induce lymphocyte retention in lymph nodes (Shiow et al., 2006). However, the lymphopenia in blood following virus infection was shown to be independent of G protein-coupled receptors and chemokines (Kamphuis et al., 2006), excluding the possibility for the involvement of S1P receptor signaling. Thus, further work is needed to reveal the molecular mechanisms of type I IFN-mediated lymphocyte mobilization and the role of lymphocytes' redistribution upon virus infection.
B. Shaping the virus-specific T cell response
Type I IFN was reported to regulate T cell responses positively or negatively yielding contrasting results (Fig. 4.1C). Type I IFN inhibited proliferation of naïve T cells and sensitized T cells for activation-induced cell death in vitro (Kaser et al., 1999, Petricoin et al., 1997), whereas this molecule enhanced the expansion of cytotoxic T cells and prolonged the survival of stimulated T cells (Aichele et al., 2006, Biron, 2001, Brinkmann et al., 1993, Marrack et al., 1999). Tough et al. showed that the direct injection of mice with poly(I:C) or recombinant IFN-β promoted the generation and survival of CD44hiCD8+ cells, indicating bystander memory CD8 T cell amplification and maintenance by virus-induced type I IFN. Recombinant IFN-α/β directly acted on activated CD4 and CD8 T cells to prevent them from undergoing activation-induced death, yet the IFN did not increase the viability of resting T cells (Marrack et al., 1999). In studies of human T cells, recombinant IFN-α drove the development of CCR7high/CXCR3low central memory CD8 T cells (Ramos et al., 2009).
Comparison between IFNAR-intact (wt), and IFNAR-deficient, antigen-specific T cells using LCMV GP33-41 epitope-specific P14 T cell receptor transgenic CD8 T cells extended our understanding of the impact type I IFN signaling has on T cells (Aichele et al., 2006, Kolumam et al., 2005). Upon recognition of LCMV in wt mice so-infected, adoptively transferred IFNAR-deficient P14 T cells retained their proliferative activity and IFN-γ/TNF-α-producing effector functions, but these virus-specific T cells failed to expand efficiently or form memory CD8 cells. Thus, the intactness of type I IFN signaling on CD8 T cells is critical for these cells to survive and expand at the T cell expansion phase but is not necessary for the cells' division per se. Although CD8 T cells lacking IFNAR could secrete IFN-γ/TNF-α, the diminished expression of granzyme B from the cells raises a question as to whether type I IFN signaling on T cells is important for complete functional competence of effector CD8 T cells (Kolumam et al., 2005). Further, the expansion of CD8 T cells deficient in IFNAR appears to be strongly influenced by the pathogenic context of a virus infection. That is, P14 CD8 T cell expansion in wt mice was less dependent on the intactness of type I IFN signaling on the T cells when recombinant vaccinia virus-expressing LCMV glycoprotein was used to infect mice instead of LCMV (Aichele et al., 2006). Similar to CD8 T cells, LCMV-specific CD4 T cells (SMARTA) depended on type I IFN signaling for clonal expansion and survival, but the loss of IFNAR on the CD4 T cells scarcely affected IFN-γ secretion or their capacity to proliferate (Havenar-Daughton et al., 2006). In contrast, upon infection with recombinant Listeria monocytogenes-bearing OVA, OVA-reactive CD4 T cells did not require type I IFN signaling for their expansion.
Type I IFN signaling often leads to the inhibition of cell division or induction of cellular apoptosis via transcriptional activation of ISGs. The effectiveness of type I IFN in the treatment of tumors or blockade of viral spread seems to be attributed to this property of type I IFN, since it contributes to removal of unwanted cells (tumor cells and virus-infected cells). This property also accounts for the inhibitory effect of type I IFN on T cells such as inhibition of T cell proliferation and enhanced T cell apoptosis observed under certain conditions. For instance, respiratory syncytial virus (RSV)-induced IFN-α and IFN-λ suppressed CD4 T cell proliferation, and neutralization of cognate receptors for IFN-α and IFN-λ reversed viral inhibition of CD4 T cell division (Chi et al., 2006). Intriguingly, when STAT1 or STAT2 is deficient in T cells, type I IFN does not inhibit mitogen-induced T cell proliferation, but rather facilitates T cells' proliferative activity and enhances T cell viability (Gimeno et al., 2005). Additionally, LCMV-specific CD8+ T cells, but not CD4+ T cells, display a significantly decreased level of STAT1 following LCMV infection, thereby evading type I IFN-mediated antiproliferative activity (Gil et al., 2006; Fig. 4.1C). Therefore, the function of type I IFN is thought to be influenced by multiple factors including the activation/expression of JAK/STAT type I IFN signaling components, the nature of a pathogen, the cytokine milieu, and the cell types in play.
C. CD4 T cell differentiation
Type I IFN also has the feature of affecting CD4 T cell polarization by inducing Th1 cells that produce IFN-γ (Brinkmann et al., 1993, Rogge et al., 1998; Fig. 4.1C). Additionally, type I IFN stimulates activated CD4 T cells to produce IL-10 (Aman et al., 1996). Recent study of human memory CD4 T cells demonstrated that IFN-α acted directly on the memory CD4 T cells in response to recalled antigens and modulated the responses differentially: (1) Upon challenge with tuberculin purified protein derivative, type I IFN enhanced proliferation of CD4 memory T cells with elevated IFN-γ production relative to IL-10 secretion. (2) Challenge with tetanus toxoid protein or influenza A hemagglutinin protein resulted in slightly inhibited cellular proliferation by type I IFN as well as a decreased ratio of IFN-γ/IL-10 (Gallagher et al., 2009).
IV. Type I IFN Interaction with B Cells
A. B cell development and migration
At an early stage of B cell development, type I IFN was reported to inhibit IL-7-induced growth of pre-B cells and induce apoptosis of the cells in vitro (Wang et al., 1995). Moreover, the development of CD19+ pro-B cells and their B lineage progeny was impaired when newborn mice were injected with IFN-α2/α1 hybrid molecule (Lin et al., 1998). To a similar but lesser extent, type I IFN also inhibited the development of pro-T cells. Other experiments employing oral administration of type I IFN into mice yielded a drastic reduction of B cell numbers and minor alteration of the T cell population in the spleens of these animals (Bosio et al., 2001). Thus, these previous observations suggest that type I IFN inhibits early B cell survival and development and may alter T cells as well (Fig. 4.1D).
Type I IFN can also affect the mobilization of B cells. A recent study showed that type I IFN was produced from PDCs in autoimmune BXD2 mice and contributed to follicular entry of marginal zone precursor B cells in secondary lymphoid organs, promoting antigen transport (Wang et al., 2010). Further, IFN-α enhanced the chemotaxis of human B cells to CCL20, CCL21, and CXCL12, by regulating chemokine receptor signaling and decreasing ligand-induced chemokine receptor internalization (Badr et al., 2005). On the contrary, IFN-β suppressed the migration of B cells purified from spleens of mice to CCL19, CCL21, and CXCL12 in a transwell experiment (Chang et al., 2007). Type I IFN signaling appeared important for B cell accumulation in the lymph nodes early after influenza virus infection by upregulating CD69 on B cells. Studies of the role of virus-induced type I IFN in the development and migration of B lymphocytes, and its impact on viral pathogenesis, however, remain incomplete.
B. B cell-mediated host immune responses
In contrast to the inhibitory effects of type I IFN on B cells at their early developmental stage, several lines of evidence indicate that type I IFN enhances the function of committed B cells in mediating immune responses to virus infections. Type I IFN enhanced CD69 and B7-2 on B cells in lymph nodes upon influenza virus infection, suggesting that type I IFN induces an early signal for B cell accumulation at these sites and mediates B7-2-induced local IgG secretion (Chang et al., 2007, Coro et al., 2006, Rau et al., 2009). Importantly, intact IFNAR signaling on B cells was shown to be critical for ensuring a sufficient quantity and quality of local antibody production in response to influenza (Coro et al., 2006). Selective deletion of the IFNAR on either T cells or B cells inhibited the type I IFN-mediated antibody response to a soluble protein antigen (Le Bon et al., 2006), denoting that type I IFN signaling on B and T cells contributes to the IFN-mediated stimulation of antibody responses. When BDCA4-coupled magnetic beads were used to deplete type I IFN-producing human PDCs from human peripheral blood mononuclear cells (PBMC), the cells' production of IgG specific for influenza virus was abrogated (Jego et al., 2003). Also, local B cell activation in the draining lymph nodes after West Nile virus infection was dependent on signals through the type I IFN receptor (Purtha et al., 2008). Following VSV infection, genetic deletion of IFNAR on VSV-specific B cells resulted in significant impairment of plasma cell formation and the antiviral IgM response (Fink et al., 2006). Enhancement of human plasma cell differentiation by type I IFN was also demonstrated by a direct treatment with IFN-α; neutralization of the IFN or IFNAR on the cells nullified type I IFN's stimulatory activity (Jego et al., 2003). Further, type I IFN was shown to promote isotype switching and stimulate long-term antibody production and immunological memory in response to chicken gamma globulin (Le Bon et al., 2001; Fig 4.3D). Collectively, these studies underscore the important role of type I IFN in the antibody response and, in particular, antiviral adaptive B cell immunity against virus infections.
Figure 4.3.
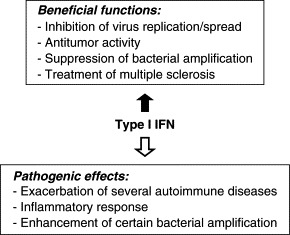
Diverse functions of type I IFN. Shown are diagrams depicting beneficial or pathogenic activities of type I IFN.
Type I IFN was reported to play a critical role in B cell survival. For instance, signaling by IFNAR was required for enhancement of B cell receptor-dependent B cell responses and increased cellular resistance to Fas-mediated apoptosis (Braun et al., 2002). Badr et al. (2010) showed that type I IFN protects both naïve and memory B cells from apoptosis via cellular signaling mechanisms including PI3Kδ/Akt, Rho-A, NFκB, and Bcl-2/BclXL.
These stimulatory effects of type I IFN on B cell responses could explain why type I IFN signaling is closely related to the development of autoimmune responses. In support of this notion, type I IFN was shown to promote the proliferation and development of peritoneal B-1 cells, which are an important producer of autoantibodies (Santiago-Raber et al., 2003). As also reported, type I IFN contributed to TLR-mediated naïve B cell activation and antibody production in the absence of B cell receptor engagement (Bekeredjian-Ding et al., 2005, Giordani et al., 2009). Indeed, type I IFN signaling is a factor in strengthening the pathogenesis of systemic lupus erythematosus (Baechler et al., 2003, Thibault et al., 2009).
V. Type I IFN in Bacterial Pathogenesis
Type I IFN was reported to downregulate the expression of a receptor for type II IFN (IFN-γ) on DCs and macrophages upon L. monocytogenes infection, demonstrating that this cytokine can magnify the pathogenicity of bacterial infections (Rayamajhi et al., 2010). That is, type I IFN signaling increased the susceptibility of lymphocytes to infection by L. monocytogenes, since the IFNAR ko mice were relatively resistant to that infection (Auerbuch et al., 2004, Carrero et al., 2004, O'Connell et al., 2004). Similarly, the IFNAR ko mice were more resistant to Francisella novicida infection than their wt counterparts (Henry et al., 2010). Additionally, influenza virus-induced type I IFN appeared to impair production of the neutrophil chemoattractants KC (Cxcl1) and Mip2 (Cxcl2), rendering the infected mice highly susceptible to bacterial superinfection (Shahangian et al., 2009). These findings were surprising because type I IFN previously proved to protect victims from bacterial as well as viral infections, as evidenced when type I IFN inhibited the replication of Legionella pneumophila (Opitz et al., 2006) and facilitated the clearance of Leishmania infection (Mattner et al., 2004). Possibly type I IFN incites harmful immune responses in pathogenic conditions and at certain locations (Vilcek, 2006; Fig. 4.3).
VI. Perspectives
Type I IFN has been used as clinical treatment for such diseases as hepatitis, hairy cell leukemia, condyloma acuminatum, multiple sclerosis, and Kaposi sarcoma (Pitha & Kunzi, 2007, Vilcek, 2006). However, type I IFN is occasionally associated with the induction or exacerbation of such autoimmune diseases as systemic lupus erythematosus and insulin-dependent diabetes mellitus, implying this treatment's pathogenic potential (Baccala et al., 2005, Kunzi & Pitha, 2003, Theofilopoulos et al., 2005). Direct administration of type I IFN has also induced flu-like symptoms in humans, indicative of a harmful inflammatory immune response (Vilcek, 1984, Vilcek, 2006). Interestingly, although several doses of IFN-αA (100, 1000, or 10,000 IU) inhibited virus replication to similar extents, only small amounts (100 IU), but not high doses (1000 or 10,000 IU), of IFN-αA protected mice from lethal influenza virus challenge (Beilharz et al., 2007). These results emphasize the importance of a balanced host immune response in conjunction with the antiviral effect of type I IFN for host protection. Further, if a factor (IFN-mediator) that induces local production of type I IFN is introduced near virus-infected cells, that mediator could block virus propagation locally without causing a systemic inflammatory response in vivo. Importantly, the H1N1 influenza virus responsible for 2009s pandemic was described as a weaker inducer of type I IFN from DCs and macrophages than seasonal influenza viruses but was highly sensitive to the IFN's antiviral activity (Osterlund et al., 2010). Thus, discovery of a small molecule(s) or cellular factor(s) that induces local type I IFN synthesis from virus-infected cells, but not from other ordinary cells could yield a product for curing multiple viral diseases.
Although type I IFN's antiviral activity is well established as an inhibitor of viral spread (Isaacs & Lindenmann, 1987, Vilcek, 2006), its regulation of individual immune components that influence innate and adaptive immune responses against invading viral infections requires extensive investigation. That research will not only provide a detailed understanding of host–virus interactions but can also lead to the development of therapeutic interventions to help those afflicted with pathogenic viral diseases.
Acknowledgments
We thank editors of Advances in Applied Microbiology for the invitation of this review. This work was supported by NIH/NIAID AI088363 (B. H.), University of Missouri (MU) Research Board (B. H.), Startup grant from the MU (B. H.), and Fellow/Mentor Research Grant from the Department of Surgery (Y. S. and B. H.).
References
- Aichele P. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- Aman M.J. Interferon-alpha stimulates production of interleukin-10 in activated CD4+ T cells and monocytes. Blood. 1996;87:4731–4736. [PubMed] [Google Scholar]
- Auerbuch V. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccala R. Interferons as pathogenic effectors in autoimmunity. Immunol. Rev. 2005;204:9–26. doi: 10.1111/j.0105-2896.2005.00252.x. [DOI] [PubMed] [Google Scholar]
- Badr G. IFN{alpha} enhances human B-cell chemotaxis by modulating ligand-induced chemokine receptor signaling and internalization. Int. Immunol. 2005;17:459–467. doi: 10.1093/intimm/dxh227. [DOI] [PubMed] [Google Scholar]
- Badr G. Type I interferon (IFN-alpha/beta) rescues B-lymphocytes from apoptosis via PI3Kdelta/Akt, Rho-A, NFkappaB and Bcl-2/Bcl(XL) Cell Immunol. 2010;263:31–40. doi: 10.1016/j.cellimm.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Baechler E.C. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber D.L. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Barchet W. Plasmacytoid dendritic cells—virus experts of innate immunity. Semin. Immunol. 2005;17:253–261. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Baskin C.R. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. USA. 2009;106:3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz M.W. Protection from lethal influenza virus challenge by oral type 1 interferon. Biochem. Biophys. Res. Commun. 2007;355:740–744. doi: 10.1016/j.bbrc.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Bekeredjian-Ding I.B. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J. Immunol. 2005;174:4043–4050. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- Biron C.A. Interferons alpha and beta as immune regulators—a new look. Immunity. 2001;14:661–664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- Blasius A.L. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- Bosio E. Low-dose orally administered type I interferon reduces splenic B cell numbers in mice. J. Interferon Cytokine Res. 2001;21:721–728. doi: 10.1089/107999001753124453. [DOI] [PubMed] [Google Scholar]
- Braun D. IFN-alpha/beta enhances BCR-dependent B cell responses. Int. Immunol. 2002;14:411–419. doi: 10.1093/intimm/14.4.411. [DOI] [PubMed] [Google Scholar]
- Brinkmann V. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J. Exp. Med. 1993;178:1655–1663. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero J.A. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragan L. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragan L. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J. Immunol. 2009;182:1099–1106. doi: 10.4049/jimmunol.182.2.1099. [DOI] [PubMed] [Google Scholar]
- Chang W.L. Influenza virus infection causes global respiratory tract B cell response modulation via innate immune signals. J. Immunol. 2007;178:1457–1467. doi: 10.4049/jimmunol.178.3.1457. [DOI] [PubMed] [Google Scholar]
- Chi B. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J. Virol. 2006;80:5032–5040. doi: 10.1128/JVI.80.10.5032-5040.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- Coro E.S. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J. Immunol. 2006;176:4343–4351. doi: 10.4049/jimmunol.176.7.4343. [DOI] [PubMed] [Google Scholar]
- Dalod M. Interferon alpha/beta and interleukin 12 responses to viral infections: Pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer M. Interferon-alpha disables dendritic cell precursors: Dendritic cells derived from interferon-alpha-treated monocytes are defective in maturation and T-cell stimulation. Immunology. 2003;110:38–47. doi: 10.1046/j.1365-2567.2003.01702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold S.S. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- Dillon S.M. Plasmacytoid and myeloid dendritic cells with a partial activation phenotype accumulate in lymphoid tissue during asymptomatic chronic HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2008;48:1–12. doi: 10.1097/QAI.0b013e3181664b60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy H. Loss of blood CD11c(+) myeloid and CD11c(−) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood. 2001;98:2574–2576. doi: 10.1182/blood.v98.8.2574. [DOI] [PubMed] [Google Scholar]
- Fink K. Early type I interferon-mediated signals on B cells specifically enhance antiviral humoral responses. Eur. J. Immunol. 2006;36:2094–2105. doi: 10.1002/eji.200635993. [DOI] [PubMed] [Google Scholar]
- Gallagher K.M. Type I interferon (IFN alpha) acts directly on human memory CD4+ T cells altering their response to antigen. J. Immunol. 2009;183:2915–2920. doi: 10.4049/jimmunol.0801607. [DOI] [PubMed] [Google Scholar]
- Gao Y. Dynamic accumulation of plasmacytoid dendritic cells in lymph nodes is regulated by interferon-beta. Blood. 2009;114:2623–2631. doi: 10.1182/blood-2008-10-183301. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A. The role of interferon in influenza virus tissue tropism. J. Virol. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George C.X. Organization of the mouse RNA-specific adenosine deaminase Adar1 gene 5′-region and demonstration of STAT1-independent, STAT2-dependent transcriptional activation by interferon. Virology. 2008;380:338–343. doi: 10.1016/j.virol.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil M.P. Modulation of STAT1 protein levels: A mechanism shaping CD8 T-cell responses in vivo. Blood. 2006;107:987–993. doi: 10.1182/blood-2005-07-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno R. Stat1 and Stat2 but not Stat3 arbitrate contradictory growth signals elicited by alpha/beta interferon in T lymphocytes. Mol. Cell. Biol. 2005;25:5456–5465. doi: 10.1128/MCB.25.13.5456-5465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordani L. IFN-alpha amplifies human naive B cell TLR-9-mediated activation and Ig production. J. Leukoc. Biol. 2009;86:261–271. doi: 10.1189/jlb.0908560. [DOI] [PubMed] [Google Scholar]
- Groschel S. TLR-mediated induction of negative regulatory ligands on dendritic cells. J. Mol. Med. 2008;86:443–455. doi: 10.1007/s00109-008-0310-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm B. Hostile communication of measles virus with host innate immunity and dendritic cells. Curr. Top. Microbiol. Immunol. 2009;330:271–287. doi: 10.1007/978-3-540-70617-5_13. [DOI] [PubMed] [Google Scholar]
- Hahm B. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Havenar-Daughton C. Cutting edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- Henry T. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J. Immunol. 2010;184:3755–3767. doi: 10.4049/jimmunol.0902065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA. 2003;100:10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Isaacs A., Lindenmann J. Virus interference. I. The interferon. J. Interferon Res. 1987;7:429–438. doi: 10.1089/jir.1987.7.429. By A. Isaacs and J. Lindenmann, 1957. [DOI] [PubMed] [Google Scholar]
- Jego G. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- Kamphuis E. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- Kanto T. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J. Infect. Dis. 2004;190:1919–1926. doi: 10.1086/425425. [DOI] [PubMed] [Google Scholar]
- Kaser A. Interferon alpha augments activation-induced T cell death by upregulation of Fas (CD95/APO-1) and Fas ligand expression. Cytokine. 1999;11:736–743. doi: 10.1006/cyto.1998.0484. [DOI] [PubMed] [Google Scholar]
- Koerner I. Protective role of beta interferon in host defense against influenza A virus. J. Virol. 2007;81:2025–2030. doi: 10.1128/JVI.01718-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam G.A. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzi M.S., Pitha P.M. Interferon targeted genes in host defense. Autoimmunity. 2003;36:457–461. doi: 10.1080/08916930310001605855. [DOI] [PubMed] [Google Scholar]
- Le Bon A. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–470. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- Le Bon A. Cutting edge: Enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 2006;176:2074–2078. doi: 10.4049/jimmunol.176.4.2074. [DOI] [PubMed] [Google Scholar]
- Lee L.N. Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J. Immunol. 2009;182:7178–7189. doi: 10.4049/jimmunol.0802526. [DOI] [PubMed] [Google Scholar]
- Li Z. RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology. 2010;396:316–322. doi: 10.1016/j.virol.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q. Impairment of T and B cell development by treatment with a type I interferon. J. Exp. Med. 1998;187:79–87. doi: 10.1084/jem.187.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack P. Type I interferons keep activated T cells alive. J. Exp. Med. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sobrido L. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2009;83:11330–11340. doi: 10.1128/JVI.00763-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattner J. Protection against progressive leishmaniasis by IFN-beta. J. Immunol. 2004;172:7574–7582. doi: 10.4049/jimmunol.172.12.7574. [DOI] [PubMed] [Google Scholar]
- Meyers J.H. Impact of HIV on cell survival and antiviral activity of plasmacytoid dendritic cells. PLoS ONE. 2007;2:e458. doi: 10.1371/journal.pone.0000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlbauer M. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J. Hepatol. 2006;45:520–528. doi: 10.1016/j.jhep.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Muller U. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nie Y. Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J. Virol. 2007;81:917–923. doi: 10.1128/JVI.01527-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell R.M. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz B. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 2006;281:36173–36179. doi: 10.1074/jbc.M604638200. [DOI] [PubMed] [Google Scholar]
- Osterlund P. Pandemic H1N1 2009 influenza A virus induces weak cytokine responses in human macrophages and dendritic cells and is highly sensitive to the antiviral actions of interferons. J. Virol. 2010;84:1414–1422. doi: 10.1128/JVI.01619-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacanowski J. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood. 2001;98:3016–3021. doi: 10.1182/blood.v98.10.3016. [DOI] [PubMed] [Google Scholar]
- Petricoin E.F., 3rd Antiproliferative action of interferon-alpha requires components of T-cell-receptor signalling. Nature. 1997;390:629–632. doi: 10.1038/37648. [DOI] [PubMed] [Google Scholar]
- Phipps-Yonas H. Interferon-beta pretreatment of conventional and plasmacytoid human dendritic cells enhances their activation by influenza virus. PLoS Pathog. 2008;4:e1000193. doi: 10.1371/journal.ppat.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitha P.M., Kunzi M.S. Type I interferon: The ever unfolding story. Curr. Top. Microbiol. Immunol. 2007;316:41–70. doi: 10.1007/978-3-540-71329-6_4. [DOI] [PubMed] [Google Scholar]
- Purtha W.E. Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J. Virol. 2008;82:10964–10974. doi: 10.1128/JVI.01646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos H.J. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood. 2009;113:5516–5525. doi: 10.1182/blood-2008-11-188458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F.C. B7-1/2 (CD80/CD86) direct signaling to B cells enhances IgG secretion. J. Immunol. 2009;183:7661–7671. doi: 10.4049/jimmunol.0803783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayamajhi M. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J. Exp. Med. 2010;207:327–337. doi: 10.1084/jem.20091746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge L. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J. Immunol. 1998;161:6567–6574. [PubMed] [Google Scholar]
- Rosen H., Goetzl E.J. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat. Rev. Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber M.L. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J. Exp. Med. 2003;197:777–788. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini S.M. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000;191:1777–1788. doi: 10.1084/jem.191.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlender J. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J. Virol. 2005;79:5507–5515. doi: 10.1128/JVI.79.9.5507-5515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahangian A. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 2009;119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow L.R. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- Siavoshian S. Hepatitis C virus core, NS3, NS5A, NS5B proteins induce apoptosis in mature dendritic cells. J. Med. Virol. 2005;75:402–411. doi: 10.1002/jmv.20283. [DOI] [PubMed] [Google Scholar]
- Steinman R.M. Lasker Basic Medical Research Award. Dendritic cells: Versatile controllers of the immune system. Nat. Med. 2007;13:1155–1159. doi: 10.1038/nm1643. [DOI] [PubMed] [Google Scholar]
- Swiecki M., Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 2010;234:142–162. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos A.N. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- Thibault D.L. Type I interferon receptor controls B-cell expression of nucleic acid-sensing Toll-like receptors and autoantibody production in a murine model of lupus. Arthritis Res. Ther. 2009;11:R112. doi: 10.1186/ar2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thitithanyanont A. High susceptibility of human dendritic cells to avian influenza H5N1 virus infection and protection by IFN-alpha and TLR ligands. J. Immunol. 2007;179:5220–5227. doi: 10.4049/jimmunol.179.8.5220. [DOI] [PubMed] [Google Scholar]
- Truong P. Persistent viral infection elevates central nervous system MHC class I through chronic production of interferons. J. Immunol. 2009;183:3895–3905. doi: 10.4049/jimmunol.0803085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani S. Restoration of HCV-specific T cell functions by PD-1/PD-L1 blockade in HCV infection: Effect of viremia levels and antiviral treatment. J. Hepatol. 2008;48:548–558. doi: 10.1016/j.jhep.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Vilcek J. Adverse effects of interferon in virus infections, autoimmune diseases and acquired immunodeficiency. Prog. Med. Virol. 1984;30:62–77. [PubMed] [Google Scholar]
- Vilcek J. Fifty years of interferon research: Aiming at a moving target. Immunity. 2006;25:343–348. doi: 10.1016/j.immuni.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Wang J. Resident bone marrow macrophages produce type 1 interferons that can selectively inhibit interleukin-7-driven growth of B lineage cells. Immunity. 1995;3:475–484. doi: 10.1016/1074-7613(95)90176-0. [DOI] [PubMed] [Google Scholar]
- Wang J.H. Marginal zone precursor B cells as cellular agents for type I IFN-promoted antigen transport in autoimmunity. J. Immunol. 2010;184:442–451. doi: 10.4049/jimmunol.0900870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J.S. T-cell exhaustion: Characteristics, causes and conversion. Immunology. 2010;129:474–481. doi: 10.1111/j.1365-2567.2010.03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga E.I. Type I interferon during viral infections: Multiple triggers for a multifunctional mediator. Curr. Top. Microbiol. Immunol. 2007;316:337–357. doi: 10.1007/978-3-540-71329-6_16. [DOI] [PubMed] [Google Scholar]