

Abstract
Transmissible gastroenteritis virus (TGEV) causes severe diarrhea and high mortality in newborn piglets. It is well established that porcine intestinal epithelium is the target of the TGEV infection, however the mechanism that TGEV invades the host epithelium remains largely unknown. Aminopeptidase N (APN) is a known receptor of TGEV. This study discovered that the extracellular receptor binding domain 1 pertaining to epidermal growth receptor (EGFR) interact with TGEV spike protein. APN and EGFR synergistically promote TGEV invasion. TGEV promotes APN and EGFR clustering early in infection. Furthermore APN and EGFR synergistically stimulate PI3K/AKT as well as MEK/ERK1/2 endocytosis signaling pathways. TGEV entry is via clathrin and caveolin mediated endocytosis in IPEC-J2 cells. TGEV binds with EGFR, and subsequently promotes EGFR internalization by a clathrin-mediated endocytosis pathway. These results show that EGFR is a co-factor of TGEV, and that it plays a synergistic role with APN early in TGEV infection.
Keywords: TGEV, IPEC-J2 cells, Aminopeptidase N, Epidermal growth receptor, Clathrin, Caveolin
1. Introduction
Procine transmissible gastroenteritis virus (TGEV) is a member of the enteropathogenic alpha-coronavirus (αCoV) family. TGEV infects intestinal epithelial cells resulting in severe and frequently fatal diarrhea in newborn pigs, with mortality rates reaching 100% (Doyle and Hutchings, 1946). TGEV is an enveloped CoV, with a large positive-sense single-stranded RNA genome, about 28.5 kb in length. It has a diameter ranging from 80 to 120 nm, including surface projections. Porcine intestinal columnar epithelial cells (IPEC-J2) offer a practical model for studying porcine enteric pathogens (Brosnahan and Brown, 2012). We will use this model to study the entrance mechanism of TGEV.
Aminopeptidase N (APN), also known as CD13, is a typeⅡtransmembrane glycoprotein, about 150 kDa, belonging to a membrane-bound metalloprotease family (Delmas et al., 1994). Most alpha coronavirus use APN as cellular receptors for virus entry, such as human coronavirus 229E (HCoV-229E), feline infectious peritonitis virus (FIPV), canine coronavirus (CCoV), porcine epidemic diarrhea virus (PEDV), and transmissible gastroenteritis virus (TGEV) (Delmas et al., 1992, Delmas et al., 1993, Kolb et al., 1996, Li et al., 2007, Tresnan et al., 1996).
The high degree of tropism of TGEV for the villous enterocytes of newborn pigs is well established and has been suggested as being a factor in age sensitivity of newborn pigs to the virus (Schwegmann-Wessels and Herrler, 2006). There has been some confusion around the question that if APN is the only receptor for TGEV entry, why older piglets are not susceptible to TGEV, especially since APN was found to be highly expressed in villous enterocytes of both newborn and older piglets. Research suggest a known protein, approximately 200-kDa in size, only expressed in the upper villi of newborn piglets, it has high affinity for TGEV (Weingartl and Derbyshire, 1994). It has also been demonstrated that APN is not essential for PEDV cell entry (Ji et al., 2018, Li et al., 2017). It is most likely that TGEV do have more than one receptor.
Many cell surface components have been identified as virus receptors, including: chemokine receptors (Feng et al., 1996), fibroblast growth factor receptors (Qing et al., 1999), the tumor necrosis factor receptor family (Terry-Allison et al., 1998), and integrin (Wang et al., 2005). pidermal growth factor receptor (EGFR), a member of the receptor tyrosine kinases (RTK) family, is widely expressed on many cells including epithelial and mesenchymal cells (Wells, 1999b). It has been demonstrated that many viruses interact with EGFR to facilitate viral entrance, including: influenza A virus (IAV), hepatitis C virus (HCV), herpes simplex virus 1 (HSV), and human cytomegalovirus (HCMV) (Chan et al., 2009, Eierhoff et al., 2010a, Lupberger et al., 2011, Zheng et al., 2014b). Ligand binding to EGFR induce receptor dimerization and cross phosphorylation, which in turn actives the intracellular signaling cascades critical for cellular protein synthesis, cytoskeleton re-organisation, apoptotic inhibition, transcriptional activation, and cell motility. The ligand binding to EGFR results in rearrangement of the cytoskeleton network through EGFR-mediated signaling, and subsequently ligand-EGFR complexes are internalize through clathrin-coated pits (Zheng et al., 2014a). Many researchers have shown that numerous viruses utilize EGFR endocytosis to mediate virus internalization (Mercer et al., 2010a). The interactions between viruses and their receptors are specific, but the affinity is low. Many multiple receptor binding sites exist on virus particles which are likely to cluster receptor proteins. It is known that multiple viruses use more than one type receptor to aid uptake into host cells (Marsh and Helenius, 2006). EGFR also has been identified as a co-receptor for many viruses, such as human cytomegalovirus (HCMV), hepatitis C virus (HCV), and adeno-associated virus serotype 6 (AAV6) (Lupberger et al., 2011, Wang et al., 2003, Weller et al., 2010). Previous studies that we have conducted demonstrated that TGEV spike protein interacts with EGFR, and stimulates phosphorylation of cofilin as well as stimulating polymerization of F-actin through the PI3K-Rac1/Cdc42-PAK-LIMK signaling pathway. This is required for efficient TGEV entry (Hu et al., 2016). We further demonstrate whether EGFR is another co-factor for TGEV. EGFR is a transmembrane protein with two dimer forms, it can be divided into extracellular, transmembranal, and intracellular regions. Its extracellular region contains two receptor-binding domains Receptor 1 (57-168 aa) and Receptor 2 (361-481 aa). Its intracellular protein associated with tyrosine kinase PI3K/Akt and Ras/Raf/ERK1/2 pathways are activated by phosphorylated tyrosine located in EGFR cytoplasmic tails ( Fig. 1A). The objective of our present study was also to study the relationship between APN and EGFR in the early stage of TGEV infection.
Fig. 1.

Interaction between TGEV S1 protein and EGFR extracellular receptor binding domain 1. (A) Structure of EGFR. (B) His-tagged EGFR extracellular receptor binding domain 1 or 2 expressed in E.coli BL21 and purified in Ni-NTA columns, the purified products were separated using SDS-PAGE and stained with Coomassie brilliant blue. (C) The purified EGFR extracellular receptor-binding domain 1 or 2 were verified by Western-blot. (D) TGEV (MOI = 2) was incubated in DMEM containing His-32a, His-EGFR Receptor 1 or His-EGFR Receptor 2 at 37 °C for 2 h, then incubated with IPEC-J2 cells and cultured for 1 h. The invasion of TGEV was detected by RT-PCR. (E) TGEV (MOI = 2) was incubated in DMEM containing His-32a, His-EGFR Receptor 1 and His-EGFR Receptor 2 at 37 °C for 2 h, then incubated with IPEC-J2 cells, and cultured for 1 h, the viral titers of intracellular TGEV were analyzed by tissue culture infectivity dose 50 TCID50. (F) IPEC-J2 cells were pretreated with His-EGFR Receptor 1 at different concentrations at 37 °C for 2 h, then incubated with IPEC-J2 cells, and cultured for 1 h. The invasion of TGEV was detected by RT-PCR. (G) Intracellular TGEV were analyzed by viral plaque morphology in ST cells. (H) The lysates of TGEV-infected IPEC-J2 cells were immunoprecipitated with rabbit anti-EGFR or normal Rabbit IgG. Immunoblotting was then performed to determine the presence of EGFR and TGEV in the EGFR immunoprecipitate. (I and J) 293T cells were co-transfected with a HA-tagged TGEV S1 expression plasmid together with GFP-tagged EGFR Receptor 1 or GFP-tagged EGFR Receptor 2 expression plasmid, cell lysates were immunoprecipitated with an anti-HA antibody or an anti-GFP antibody, the resulting precipitates were examined by immunoblotting using an anti-HA or an anti-GFP antibody to examine the interaction between HA-TGEV S1 and GFP-tagged EGFR. (** p < 0.01).
2. Materials and methods
2.1. Cells culture
IPEC-J2 cells are porcine intestinal columnar epithelial cells that are isolated from the middle jejunum of neonatal piglets. IPEC-J2 cells were purchased from DSMZ (Germany). HEK293T cells and swine testis (ST) cells were purchased from ATCC (United States). IPEC-J2, ST, and HEK293T cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with high glucose, HEPES containing with 10% fetal bovine serum (FBS, GIBCO), 1% penicillin-streptomycin (Invitrogen) at 37 °C in a 5% CO2 incubator (ThermoFisher Scientific).
2.2. Virus infection and assays
Transmissible gastroenteritis virus (strain SHXB) was isolated in Shanghai, China. The complete genome sequence for TGEV SHXB is available in GenBank (KP202848.1) (Weiwei et al., 2014). To analyze viral entry, cells were incubated with TGEV at a multiplicity of infection of 2 (MOI = 2) for 1 h at 4 °C. Subsequently, the cells were washed with phosphate-buffered saline (PBS), and maintained in a maintenance medium (DMEM supplemented with 2% FBS and 1% penicillin-streptomycin) for 1 h at 37 °C in a 5% CO2 incubator.
For viral labeling, viruses were filtered with 0.22 µm filter, and then clarified by centrifugation at 10,000g for 2.5 h, followed by ultra-centrifugation using 20%, 40%, and 60% sucrose gradient at 10,000g for 2.5 h. Viruses were labeled with the fluorescent probe DyLight 488, 633, and 594 NHS Ester (ThermoFisher Scientific, Waltham, USA), according to the manufacturer's instruction. Unincorporated dye was removed by using commercial fluorescent dye removal columns (ThermoFisher Scientific).
Flow cytometry analysis for the entry of TGEV was performed as follows: Fluorescent probe labeled “TGEV particles” were incubated with IPEC-J2 cells for 1 h at 4 °C. Subsequently, the cells were washed with PBS and maintained in DMEM for 1 h at 37 °C in a 5% CO2 incubator, the cells were harvested by 0.25% trypsin, and then washed with PBS three times. Cells acquisition was performed by FACS (Becton Dickinson), and the date was analyzed using Flowjo software.
2.3. Antibodies and western blotting
Antibodies used in the present study were obtained from commercial sources. These antibodies included: rabbit anti-human EGFR, rabbit anti-human phospho-EGFR (Tyr1068), rabbit anti-human AKT, rabbit anti-human phospho-AKT, rabbit anti-human phospho-ERK1/2, and rabbit anti-human ERK1/2 (Cell Signaling Technology, Danvers, USA). Mouse anti-procine APN antibody was donated by prof. Zhu Guoqiang in Yangzhou University. Mouse monoclonal antibodies to HA and GFP (CMCTAG, Milwaukee, USA). Goat anti-rabbit IgG (H + L) secondary Antibody, DyLight 594 conjugate, goat anti-mouse IgG (H + L) secondary Antibody, DyLight 488 conjugate (ThermoFisher Scientific). Anti-GAPDH monoclonal antibody, HRP-conjugated goat anti-rabbit IgG (H + L), and HRP-conjugated Goat Anti-Mouse IgG (H + L) (Vazyme, Nanjing, China).
IPEC-J2 cells were washed with PBS and lysed in an ice-cold cell lysis buffer, phosphatase inhibitor and protease inhibitor (ThermoFisher Scientific) were added in the cell lysis buffer according to the manufacturer's instructions. The supernatant of lysates were obtained by centrifugation at 12,000g for 10 min at 4 °C, and subsequently equal protein levels of the prepared lysates were fractionated by SDS-PAGE (10–12% gradient). The separated proteins were transferred to PVDF (Merck Millipore), and the membranes were blocked for 2 h in Tris-buffered saline (TBS), containing 5% nonfat dry milk. After which they were reacted with indicated primary antibodies at 4 °C overnight. Membranes were exposed to species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies, (dilution 1:5000) followed by enhanced chemiluminescence (ECL, Thermofisher Scientific) detection by use of autoradiography. Western blotting was quantified by Quantity One (Quantity One 1-D Analysis Software 170-9600, Bio-Rad). The intensity of the bands in terms of density was measured and normalized against GAPDH expression. All data were expressed as means ± SD of three independent experiments.
2.4. Plasmid construction and DNA transfection
The pLVX-DsRed-Monomer-N1 is an HIV-1-based, lentiviral expression vector that expresses the gene of interest fused to the DsRed-Monomer (Clontech, Palo Aito, CA). EGFR and APN sequences were inserted into the EcoRI/BamHI site. EGFR receptor1 and EGFR receptor2 sequences were cloned into a pAcGFP1-C vector (SalI/BamHI) (Clontech), and TGEV Spike1 sequence was cloned into pCMV-C-HA (BamHI/XbaI) (D2639, Beyotime, China). Table 2 showed the primers used for cloning. All constructs were verified by DNA sequencing. HEK 293 T cells were optimized for lentivirus production, we transfected APN or EGFR lentiviral overexpression vector and Lenti-X HTX Packaging Mix (VSV-G, plp1, plp2) into HEK 293T cells using the X-tremeGENE HP DNA Transfection Reagent (Roche, Switzerland), according to the manufacturer's instructions. IPEC-J2 cells were treated with APN, EGFR overexpressing lentiviral particles (MOI = 1), and after 24 h of incubation, infected cells were maintained with fresh DMEM, and continued for extra 12–24 h to allow the overexpressing lentiviral particles to achieve their maximum effect.
Table 2.
Primers used for cloning.
| Name | Primer sequence (5′-3′) | Vector |
|---|---|---|
| EGFR | F: CTCAAGCTTCgaattcATGCGACGCTCCTGGGCG | pLVX-DsRed |
| R: GTGGCGACCGGTggatccTCATGCCCCAGTAAGG | ||
| APN | F: CTCAAGCTTCgaattcATGGCCAAGGGATTCTAC | pLVX-DsRed |
| R: GTGGCGACCGGTggatccTTAGCTGTGCTCTAT | ||
| EGFR Receptor1 | F: CGACGACAAGGccatgAACTGCGAGGTGGTCCTTG | pET-32a-c |
| R: GGTGGTGGTGctcgagTTAGACAATGTCCCTCCAC | ||
| EGFR Receptor2 | F: CGACGACAAGGccatgAACTGCACCTCGATCAGC | pET-32a-c |
| R: GGTGGTGGTGctcgagTTATTAAAATAGTTTTTTCCAG | ||
| EGFR Receptor1 | F: TTAAGGCCTCTgctagcATGAACTGCGAGGTGGTC | pAcGFP1-C |
| R: TGGATCCGCCAgaattcTTAGACAATGTCCCTCCAC | ||
| TGEV-S1 | F: TCTAGCCCGGGCggatccTGTGCTAGTTATGTGGCT | pCMV-C-HA |
| R: ATCGTATGGGTAtctagaATTTGTATAATTATATATAGAG |
2.5. His-EGFR Receptor1 and His-EGFR Receptor2 expression and purification
His-EGFR Receptor1 and His-EGFR Receptor2 were cloned into and expressed in Escherichia coli BL-21, then purified using Ni-NTA resin. The purified proteins were eluted with elution buffer containing 8 M urea, which was removed using a dialysis bag against buffer (50 mM NaH2PO4, 300 mM NaCl) with gradual reduction in the concentration of urea (6 M, 3 M, buffer alone). The concentrations of the purified proteins were measured with the BCA assay. The two purified proteins were verified by SDS-PAGE and Western blot, respectively.
2.6. Lentivirus-mediated RNA interference depletion experiments
pLVX-shRNA2 is an HIV-1-based, lentiviral expression vector designed to express a small hairpin RNA (shRNA) used for RNA interference (RNAi) studies (Clontech) ( Table 1). The best silencing efficiency was observed with clone KF280271 (porcine APN), NM_214007 (porcine EGFR), NM_001146127.1 (procine clathrin), NM_214438 (procine caveolin). These shRNA oligonucleotides were inserted into the BamHI/EcoRI site. All constructs were verified by DNA sequencing. HEK 293T cells were optimized for interference lentivirus production. We transfected APN, EGFR, clathrin, or caveolin lentiviral interference vector and Lenti-X HTX Packaging Mix (VSV-G, plp1, plp2) into HEK 293T cells using the X-tremeGENE HP DNA Transfection Reagent (Roche, Switzerland), according to the manufacturer's instructions. IPEC-J2 cells were treated with APN, EGFR interference lentiviral particles (MOI = 1), after 24 h of incubation, infected cells were maintained with fresh DMEM and continued for 12–24 h to allow the interference lentiviral particles to achieve their maximum effect.
Table 1.
List of APN, clathrin and caveolin RNA interference sequences.
| Name | Interference sequence (5′-3′) |
|---|---|
| APN | shAPN1: GGAAATCCTTCCCATGCTTTG |
| shAPN2: GCGAGATGTTTGACTCCATCT | |
| shAPN3: GGACCTCATTCGGAAGCAAGA | |
| shAPN ctrl: TTCGGAAGCAAGAGGACCTCA | |
| Clathrin | shCla1:GCAGATAAGTGAGAAACATGA |
| shCla2: GCAGTTTGCTCAAATGTTAGT | |
| shCla3: GCAGAGAAAGCAACTGTTATG | |
| shCla ctrl: CTCAAATGTTAGTGCAGTTTG | |
| Caveolin | shCav1: GGAAATGAACGAGAAGCAAGT |
| shCav2: GCGGTTGTACCCTGCATTAAG | |
| shCav3: GCAATATCCGCATCAACATGC | |
| shCav ctrl: TACCCTGCATTAAGGCGGTTG |
2.7. Co-Immunoprecipitation assay
In regards to detecting the interaction between TGEV S1 and EGFR, cells were lysed in a lysis buffer after infection of TGEV (MOI = 100). Cell lysates were cleared by centrifugation at 12,000g for 10 min, and the supernatant was immune-precipitated with rabbit anti-human EGFR monoclonal antibody (4267S, Cell Signaling Technology, Danvers, USA) or normal rabbit IgG (A7016, Beyotime, China) at 4 °C for 8 h, and fresh protein A/G magnetic beads (B23201, Bimake, USA) were added and incubated with cells at 4 °C for another 3 h. Magnetic beads containing protein complexes were washed five times with PBS and incubated with concentrated TGEV at 4 °C for 5 h. Magnetic beads containing EGFR proteins and TGEV complexes were washed five times with PBS. Complexes were subsequently boiled for 10 min in 1 × protein loading buffer (TakaRa, Janpan), then analyzed by Western blotting with specific primary and secondary antibodies. The primary antibodies used to probe membranes were rabbit anti-EGFR Mab and rabbit anti-TGEV N pAb.
For detecting the binding domain of TGEV S1, HEK293T cells lysates were prepared by ice-cold NP-40 lysis buffer (P0013F, Beyotime, China), and protease inhibitor (ThermoFisher Scientific) was thereafter added according to the manufacturer's instructions. Cell lysates were cleared by centrifugation at 12,000g for 10 min, and supernatant was incubated with protein A/G magnetic beads (B23201, Bimake, USA) and normal IgG (from the same species as that of the immunoprecipitating antibody) at 4 °C for 1 h, to eliminate nonspecific binding to the Magnetic beads or IgG. After centrifugation at 1000g for 5 min, the supernatant was incubated with HA (AT0024, CMCTAG) or GFP (AT0028, CMCTAG) antibody at 4 °C for 8 h. Fresh protein G PLUS- Magnetic beads were added and incubated with cells at 4 °C for another 3 h. Magnetic beads, containing protein complexes, were washed five times with PBS, and complexes were boiled for 10 min in 1 × protein loading buffer (TakaRa, Janpan). At this point they were analyzed by Western blotting with specific primary and secondary antibodies.
2.8. Virus titration assay
TGEV-infected and mock-infected cells were seeded onto 6-well plates (5 × 105) and cultured for 24 h. TGEV was collected by freezing and thawing the plates three multiple times, and were determined by the tissue culture infectious dose 50 (TCID50) in ST cells.
2.9. Plaque assay
TGEV plaque assays were performed on monolayers of ST cells seeded in 12-well plates. Cells were inoculated with ten-fold dilutions of stock virus, and incubated for 1 h at 37 °C. These cells were overlaid with DMEM containing 2% FBS and 1% low melting point agarose (Sigma-Aldrich). After which they were incubated for about 72 h until plaques were visible. Plaques were stained with 1% crystal violet in methanol.
2.10. Indirect immunofluorescence staining and microscopy
IPEC-J2 cells were grown on coverslips in 24-well tissue culture plates, after TGEV infection, at room temperature, cells were fixed in 4% paraformaldehyde in PBS for 15 min. They were washed three times with PBS, and incubated with 0.1% Triton X-100 in PBS for 5 min, washed three times with PBS. At this point, cells were blocked in 5% bovine serum albumin (BSA) for 20 min. For studies analyzing the co-localization of APN and p-EGFR, cells were incubated with APN and p-EGFR primary antibodies (1:1000) at 4 °C, overnight. For studies analyzing the internalization of EGFR, cells were incubated with EGFR primary antibody (1:1000) at 4 °C, overnight, and were followed by three washes with PBS, then subsequently incubated with fluorochrome-conjugated secondary antibodies (1:500) at room temperature for 1 h. Cells were washed three times with PBS and the nucleus were stained using 1 μg/ml DAPI (4′,6′diamidino-2-phenylindole dihydrochloride)-PBS for 5 min at room temperature. Images were captured with a Zeiss fluorescence microscopy system and a 40× objective lens. The co-localization of the two channels was detected using ZEN 2012 (Zeiss) software.
2.11. Membrane protein extraction
Cells were incubated with TGEV at MOI = 2 for 1 h at 4 °C and washed with PBS (pH7.2 at 4 °C) three times to remove unbound virus, then maintained in maintenance medium (DMEM supplemented with 2% FBS and 1% penicillinstreptomycin) at 37 °C in a 5% CO2 incubator, after the indicated time (5 min, 15 min, 30 min, 60 min), cells were washed with acidic PBS (pH 3.0 at 4 °C) to remove the virus bound to the cell membrane (not enter the cell), then cell membrance protein was performed according to the manufactures’instructions. Ligand EGF was used as a positive control, EGF (100 ng/ml) was added to the cell culture medium for 1 h at 4 °C, and washed with PBS (pH7.2 at 4 °C) three times to remove unbound EGF, then maintained in maintenance medium (DMEM supplemented with 2% FBS and 1% penicillinstreptomycin) at 37 °C in a 5% CO2 incubator, after the indicated time (5 min, 15 min, 30 min, 60 min), then cell membrance protein was performed according to the manufactures’instructions (Mem-PER™ Plus Membrane Protein Extraction Kit, Catalog number: 89842,Thermo).
2.12. Internalization assays for TGEV with endocytic markers
Human transferrin-FITC labele was obtained from Nanocs (USA). Cholera toxin beta subunit (Ct-B)-FITC labele was purchased from Sigma-Aldrich (USA). IPEC-J2 cells were prepared on coverslips in 24-well tissue culture plates, and incubated with DyLight 594-TGEV with 50 μg/ml of FITC-transferrin or 10 μg/ml of FITC-CtxB for 30 min at 4 °C to synchronize entry. At this point they were then shifted to 37 °C for 30 min. Cells were fixed in 4% paraformaldehyde for 15 min, and nucleus were stained using 1 μg/ml DAPI for 5 min. Images were captured with a Zeiss LSM710 confocal laser-scanning microscopy system and a 40× objective lens. Top view images were prepared as compacted Z-Stack images of non-permeabilised cells. X-y plane and z-axis views of confocal images were prepared using ZEN 2012 LE software from Zeiss, Germany.
2.13. Statistical analysis
All results were presented as means ± standard deviation (SD) from three independent experiments and performed using GraphPad Prism 5 software (SanDiego, CA), P values for all data were determined using Student's t-test and one-way analysis of variance (ANOVA) (* 0.01 < p < 0.05, ** p < 0.01).
3. Results
3.1. Involvement of the binding domain in the direct interaction between TGEV S1 and EGFR
Purified Escherichia coli-expressed fusion protein His-tagged Receptor binding protein 1 and 2 were verified by SDS-PAGE and Western blot, respectively (Fig. 1B and C). TGEV was incubated with Receptor 1 (200 ng/ml) and Receptor 2 proteins (200 ng/ml) at 37 °C incubator for two hours. Receptor 1 protein was able to bind with TGEV and inhibit TGEV invasion (Fig. 1D and E). Receptor 1 protein was able to inhibit TGEV invasion in a dose dependent manner (Fig. 1F). Receptor 1 protein inhibition TGEV invasion was proven by plaque assays (Fig. 1G). To investigate the direct interaction between TGEV S1 and EGFR, co-immunoprecipitation was performed on lysates from TGEV-infected cells precipitated with antibody against EGFR. EGFR and TGEV N protein were detected in the precipitates of TGEV-infected cells, indicating that TGEV interacts simultaneously with EGFR in infected cells (Fig. 1H). The interaction between TGEV S1 and EGFR Receptor 1 / Receptor 2 were validated in precipitates from 293T cells co-transfected with plasmids expressing TGEV S1-HA and EGFR Receptor 1 / Receptor 2-GFP (Fig. 1I and J). This further confirmed that TGEV S1 directly interacts with EGFR Receptor 1 but not EGFR Receptor 2.
3.2. EGFR is a co-factor for TGEV entry
APN is a receptor of TGEV, we wanted to determine whether EGFR had any interaction with APN. Hence, the localization of APN and EGFR was assessed by fluorescent microscopy. In Mock-infected cells, it was noticed that APN and p-EGFR distributed evenly on the surface of the cell membrane. In TGEV-infected cells, EGFR was activated. The level of p-EGFR increased, APN and EGFR were co-localization significantly ( Fig. 2). To determine the role of EGFR and APN in mediating TGEV infection, lentivirus interference methods were used to reduce the expression of EGFR and APN. IPEC-J2 cells were transfected with lentivirus constructs that expressed APN or EGFR targeting shRNAs. APN or EGFR expression level reduced significantly ( Fig. 3A and B). The results of Real-time reverse transcription PCR (RT-PCR) for detection of TGEV invasion revealed that APN-targeting shRNA, EGFR-targeting shRNA, and APN + EGFR-targeting shRNAs, significantly inhibited TGEV entry. The inhibition of APN + EGFR-targeting shRNAs was seen to be more significant (Fig. 3E). These results were further verified by flow cytometry, TGEV particles were labeled with fluorescent probe DyLight 488, APN-targeting shRNA, and EGFR-targeting shRNA inhibited the invasion of TGEV with the similar results. APN+EGFR-targeting shRNAs showed a more significant inhibitory effect (Fig. 3D and E). TGEV infection was measured in the supernatant of infected IPEC-J2 cells by TCID50 (Fig. 3F). Plaque formation in ST cells by the intracellular of infected IPEC-J2 cells showed that APN + EGFR-targeting shRNAs inhibited TGEV entry more significantly (Fig. 3G). All of these results indicated to us that EGFR and APN synergistically promote TGEV invasion.
Fig. 2.

TGEV infection causes the co-localization of APN and EGFR. IPEC-J2 cells were infected with TGEV (MOI = 2) and cultured for 30 min, then stained for fluorescence microscopy using mouse anti-APN pAb, followed by DyLight 488-conjugated goat anti-mouse IgG and rabbit anti-p-EGFR mAb, followed by DyLight 594-conjugated goat anti-rabbit IgG. Mock-infected cells served as controls. The data shown are from two independent experiments.
Fig. 3.

APN and EGFR synergistically promote TGEV invasion. (A and B) APN and EGFR interference verification, shAPN1 and shEGFR3 were later used in the subsequent experiments. (C) IPEC-J2 cells were transfected with interference vector pLVX-shRNA-APN, pLVX-shRNA-APNCtrl, pLVX-shRNA-EGFR, pLVX-shRNA-EGFRCtrl, or pLVX-shRNA-APN + pLVX-shRNA-EGFR through lentiviral supernatant. Normal cells served as controls. Cells were infected with TGEV at an MOI of 2, and cultured for 1 h. The invasion of TGEV was detected by RT-PCR. (D and E) IPEC-J2 cells were infected with Dylight 488-TGEV, and cultured for 1 h The invasion of TGEV was detected by Flow cytometry. (F) The viral titers of intracellular TGEV were analyzed by TCID50. (G) Intracellular TGEV was analyzed by viral plaque morphology in ST cells. The data shown are the mean results ± SD from three independent experiments. (* 0.01 < p < 0.05, ** p < 0.01).
3.3. APN is required for EGFR activation during TGEV infection
The activation of phosphatidylinositol-3 kinase (PI3K) and the extracellular signal-regulated kinase 1/2 (ERK1/2) has been identified in many viruses as part of their entry mechanism. EGFR is an upstream mediator of PI3K/AKT signaling pathway and ERK1/2. To determine whether EGFR can induce the endocytosis signaling pathway to support TGEV entry, the activation of EGFR, PI3K/AKT and ERK1/2 were investigated. Cells were incubated with TGEV at 4 °C for 1 h, and unbound viruses were removed. Then cells were incubated at 37 °C for 30 min, and the phosphorylation level of EGFR was increased in the early infection stage of TGEV. In APN overexpressed, EGFR overexpressed, and APN+EGFR overexpressed cells, Overexpression of EGFR and APN were confirmed in Figs. 4A and S1, TGEV infection caused an a comparably increased phosphorylation level of EGFR, Akt, as well as ERK1/2 than that of the TGEV infection control group (Fig. 4A and C). In APN-targeting shRNA, EGFR-targeting shRNA, and APN+EGFR-targeting shRNAs cells, TGEV infection resulted in the decreased phosphorylation level of EGFR, Akt, and ERK1/2 significantly in comparison to the TGEV infection control group (Fig. 4F–H). APN-targeting shRNA inhibited TGEV-induced EGFR activation more significantly than EGFR-targeting shRNA (Fig. 4B and F). These data demonstrated that APN is required for TGEV-induced EGFR activation. Hence, TGEV binding to APN induces EGFR activation and is required for viral entry.
Fig. 4.

APN and EGFR synergistically activate PI3K/AKT and MEK/ERK1/2 signaling pathways. (A) IPEC-J2 cells were transfected with overexpression vector pLVX-DsRed, pLVX-APN-DsRed, pLVX-EGFR-DsRed, or pLVX-APN-DsRed + pLVX-EGFR-DsRed through lentiviral supernatant. Cells were infected with TGEV at an MOI of 2 and cultured for 30 min. Normal cells and TGEV infected-normal cells served as controls. The activation of downstream signaling pathways analyzed by Western-blot with anti-p-EGFR, anti-EGFR, anti-p-AKT, anti-AKT, anti-p-ERK1/2, anti-ERK1/2, and anti-GAPDH antibodies. (B) IPEC-J2 cells were transfected with interference vector pLVX-shRNA-APN, pLVX-shRNA-APNCtrl, pLVX-shRNA-EGFR, pLVX-shRNA-EGFRCtrl, or pLVX-shRNA-APN + pLVX-shRNA-EGFR through lentiviral supernatant. Cells were infected with TGEV at an MOI of 2, and cultured for 30 min Normal cells and TGEV infected-normal cells served as controls. The activation of downstream signaling pathways analyzed by Western-blot with anti-p-EGFR, anti-EGFR, anti-p-AKT, anti-AKT, anti-p-ERK1/2, anti-ERK1/2, and anti-GAPDH antibodies. (C–H) The ratio of p-EGFR, EGFR, p-AKT, AKT, p-ERK1/2 or ERK1/2 to GAPDH was normalized to control conditions. Data shown are the mean results ± SD from three independent experiments. (* 0.01 < p < 0.05, ** p < 0.01).
3.4. TGEV entry via clathrin and caveolin mediated endocytosis in IPEC-J2 cells
To explore whether the endocytosis pathway supports TGEV entry into IPEC-J2 cells, IPEC-J2 cells were transfected with lentivirus that expressed clathrin or caveolin targeting shRNAs. The clathrin or caveolin expression level was reduced significantly ( Fig. 5A and B). RT-PCR for detection TGEV invasion results showed that both clathrin and caveolin targeting shRNAs significantly inhibited TGEV entry (Fig. 5C and D). This data was reinforced by flow cytometry, TGEV particles were labeled with fluorescent probe DyLight 633, clathrin-targeting shRNA, and caveolin-targeting shRNA which inhibited the invasion of TGEV (Fig. 5E and F). The viral titers of intracellular TGEV were also analyzed by TCID50 (Fig. 5G). Our previous studies have found that nystatin, an cholesterol removing agent which also functions by inhibiting the lipid/caveolin pathway can in fact inhibit TGEV binding and entry. This result also show us that caveolin-mediated endocytosis is a method that can be utilized in TGEV internalization. To verify and confirm our results, clathrin and caveolin mediated endocytosis specific markers were used to provide direct evidence in an attempt to prove the TGEV endocytosis pathway. We found that both FITC-transferrin and FITC-Ct-B were co-localized with DyLight 594-TGEV (Fig. 5H and I). Taken together, we confirmed that both clathrin and caveolin mediated endocytosis are important for TGEV entry.
Fig. 5.

Clathrin and Caveolin mediate the endocytosis of TGEV. (A and B) Clathrin and Caveolin interference verification, shClai3 and shCav2 were later used in the subsequent experiments. (C and D) IPEC-J2 cells were transfected with interference vector pLVX-shRNA-Clathrin, pLVX-shRNA-ClathrinCtrl, pLVX-shRNA-Caveolin, or pLVX-shRNA-CaveolinCtrl through lentiviral supernatant. Normal cells served as controls. Cells were infected with TGEV at an MOI of 2 and cultured for 1 h. The invasion of TGEV was detected by RT-PCR. (E–G) IPEC-J2 cells were transfected with interference vector pLVX-shRNA-Clathrin, pLVX-shRNA-ClathrinCtrl, pLVX-shRNA-Caveolin, or pLVX-shRNA-CaveolinCtrl through lentiviral supernatant. Cells were infected with TGEV at an MOI of 2, and cultured for 1 h. The invasion of TGEV was detected by Flow cytometry (E and F). The viral titers of intracellular TGEV were analyzed by TCID50 (F). (H and I) The co-localization of transferrin and cholera toxin with TGEV, (scale bar = 20 µm), the immunofluorescence experiment was repeated two times, every time there are three groups of parallel samples. The data shown are the mean results ± SD from three independent experiments. (* 0.01 < p < 0.05, ** p < 0.01).
3.5. TGEV infection causes EGFR internalization through clathrin-mediated endocytosis
To explore the role of EGFR in TGEV internalization, the internalization of EGFR was investigated by confocal laser-scanning microscopy. In normal IPEC-J2 cells, most EGFR was located at the cell surface membrane, and EGFR was internalized upon EGF stimulation. TGEV infection also promotes EGFR internalization ( Fig. 6A and B). We also detected cell surface membrane EGFR expression levels (Fig. 6C and D). EGFR ligands (EGF and TGF-α) induced receptor dimerization, activation, and internalization (Wells, 1999a). IPEC-J2 cells were incubated with TGEV for different times at 4 °C for 1 h, and ligand EGF was used as a positive control. When EGF-stimulated cells were transferred at 37 °C, EGFR internalized rapidly. At 15 min post-infection (mpi), most cell membrane EGFR was internalized into the cytoplasm. At 60 mpi, EGFR was recruited to cell membrane. TGEV infection caused EGFR internalization in a time dependent manner. EGFR was internalized into the cytoplasm from 15 mpi to 60 mpi. In normal cells, most EGFR resided on the cell membrane. This data suggests that TGEV particles cause the internalization of EGFR early in TGEV infection.
Fig. 6.

TGEV infection induced EGFR internalization. (A) IPEC-J2 cells were infected with TGEV (MOI = 2), and cultured for 1 h. Then stained for fluorescence microscope using rabbit anti-EGFR pAb followed by DyLight 594-conjugated goat anti-rabbit IgG, Mock-infected cells served as controls. EGFR distribution was observed by confocal microscope. (B) Three-dimensional rendering of representative images obtained using Imaris 7.2 software. (C) IPEC-J2 cells were infected with TGEV (MOI = 2), and cultured for 1 h. The protein of the cell membrane was extracted. Cell membrane EGFR was analyzed by Westernblot using rabbit anti-EGFR pAb. (D) The ratio of EGFR to the mean of E-cadherin and GAPDH was normalized to control conditions. The data shown are the mean results ± SD, from three independent experiments. (scale bar = 20 µm).
To explore the role of caveolin and clathrin in EGFR internalization early in TGEV infection, we reduced clathrin or caveolin down in normal IPEC-J2 cells through targeting shRNAs, and investigated cell membrane EGFR expression levels during TGEV invasion. Clathrin targeting shRNA significantly inhibited EGFR internalization. In caveolin targeting shRNA cells, EGFR was internalized from 15 mpi to 30 mpi, and recruited to the cell membrane at 60 mpi. EGFR circulation was faster than that of normal cell groups ( Fig. 7A and B). These results indicate that TGEV causes EGFR internalization through the clathrin-mediated endocytosis pathway.
Fig. 7.

EGFR internalization through clathrin endocytosis pathway. (A) IPEC-J2 cells were transfected with interference vector pLVX-shRNA-Clathrin3 or pLVX-shRNA-Caveolin2 through lentiviral supernatant. Normal cells served as controls. Cells were infected with TGEV at an MOI of 2. The protein of the cell membrane was extracted. Cell membrane EGFR was analyzed by Westernblot using rabbit anti-EGFR pAb. (B) The ratio of EGFR to the mean of E-cadherin and GAPDH was normalized to control conditions. The data shown are the mean results ± SD from three independent experiments.
4. Discussion
In this study, we demonstrated that EGFR is an another co-factor of TGEV, and the TGEV spike protein can interact with EGFR extracellular receptor binding domain 1. TGEV infection causes the co-localization of APN and EGFR. Furthermore APN is required for TGEV-induced EGFR activation. PI3K/Akt and ERK1/2 signaling pathways are involved in TGEV internalization. In addition to this, TGEV particles and EGFR internalize through clathrin-mediated endocytosis. EGFR has also been demonstrated to participate in the invasion of other viruses, including: adeno-associated virus serotype 6, influenza A, hepatitis C virus, and human cytomegalovirus (Chan et al., 2009, Eierhoff et al., 2010a, Lupberger et al., 2011, Weller et al., 2010), suggesting that EGFR activation and internalization may be common mechanisms utilized in virus invasion.
The activation of PI3K and ERK1/2 has been found in many virus entry mechanisms, such as: human cytomegalovirus, influenza A, and herpes simplex virus (Eierhoff et al., 2010b, Johnson et al., 2001a, Johnson et al., 2001b, Zheng et al., 2014b). PI3K activation has been demonstrated to regulate vesicular uptake, and trafficking of Ebola virus (Saeed et al., 2008). Ligand binding to EGFR on the cell surface induces receptor dimerization and cross phosphorylation, which leads to the activation of downstream signaling cascades of which include: MAPK, PI3K, JAK/STAT, and PLCγ signaling pathways (Zheng et al., 2014a). In this study, we found that early in the TGEV infection process, APN and EGFR synergistically stimulate PI3K/AKT and MEK/ERK1/2 signaling pathways, and promoted TGEV entry.
Many pathogens enter the host cell by endocytosis which results in cell surface receptor, ligand, and membrane component internalization (Mosesson et al., 2008). Enveloped viruses are capable of entering directly through the cell membrane surface receptors, or are able to be internalizing via endocytosis via fusion taking place in the endosomal compartment (Belouzard et al., 2012). Our previous published research found that TGEV can be internalized into IPEC-J2 cells, however the mechanism of TGEV entry is still not known. It has been confirmed that PEDV invades via clathrin-mediated endocytosis independent of caveolin-mediated endocytosis (Park et al., 2014). Our results suggest that TGEV infection reduces in clathrin or caveolin targeting shRNAs cells. Furthermore, co-localization between endocytosed FITC-transferrin/FITC-CtxB and fluorochrome-labeled TGEV support the conclusion that both clathrin and caveolin mediated endocytic uptake are the pathways of TGEV entry. Typically for particle sizes internalized through caveolin ranging from 50 to 100 nm and through clathrin less than 200 nm (Aleksandrowicz et al., 2011). TGEV particles sizes are between 100 and 150 nm, some are smaller TGEV particles that are internalized through caveolin-mediated endocytosis.
The actin cytoskeleton has been thought of as participating in the formation of the clathrin-mediated endocytosis structure, as well as providing mechanical force to enable complete endocytosis (Kaksonen et al., 2006, Smythe and Ayscough, 2006). Our previous research has also found that TGEV infection induces actin cytoskeleton rearrangement through cofilin, and actin surround TGEV particles in the spatial part of the virus co-localize with the actin, which indicates that clathrin and actin are involved in the early invasion of TGEV. Receptor clustering is an actin-dependent process that uses RHO-family GTPase signaling, and the actin regulatory proteins filamin and cofilin (Jimenez-Baranda et al., 2007, Yoder et al., 2008). The co-localization of APN and EGFR is also found in the early infection of TGEV, suggesting that EGFR phosphorylation and PI3K-Rac1/Cdc42-LIMK-Cofilin signaling pathways participate in the regulation of actin cytoskeleton. TGEV particles movement towards entry sites, the co-localization of APN and EGFR are all mediated by actin cytoskeleton re-modeling.
In the resting state, most EGFR reside on the cell membrane surface. Upon ligand binding, EGFR is activated and internalized via clathrin-coated pits (Wells, 1999a, Zheng et al., 2014b). Numerous viruses utilize EGFR endocytosis to mediate virus internalization and trafficking to the site of replication during the infection of the host cell (Mercer et al., 2010b). Some previous studies have suggested clathrin-coated pits can form naturally at 0 °C in the presence of EGF, but are not internalized into the cytoplasm (Brown and Petersen, 1998, Jiang et al., 2003). Clathrin-coated pits connected to the cell surface and clathrin-coated vesicles are intracellular membranal structures. Lipid rafts function as platforms to active intracellular EGFR effector signals and EGFR internalization (Puri et al., 2005). In this study we found that when EGF was incubated with cells at 4 °C for 1 h, EGF formed clathrin-coated pits connected to the cell surface and EGFR became localized in the coated pits. When cells were transferred at 37 °C, EGFR was rapidly internalized into the cytoplasm through clathrin-coated vesicles. For TGEV infected cells, TGEV particles bound with cells at 4 °C. As soon as cells were transferred at 37 °C, TGEV stimulated clustering of lipid rafts and activated EGFR and effector signals. This process took comparatively more time, hence at 15 mpi, EGFR was found to begin internalization of TGEV particles. In this study, we found that TGEV infection caused EGFR internalization, and clathrin-targeting shRNA inhibited EGFR internalization. EGFR also was identified as an another receptor for TGEV. We can get to the conclusion that in the early infection stage of TGEV, TGEV particles bound with APN and EGFR, the virus-receptors complex are subsequently internalized by clathrin. Caveolin targeting shRNA has no effect on the internalization of EGFR. However further research is needed to specify the mechanism of TGEV particles internalized by caveolin.
All our data confirms that EGFR is a co-factor for TGEV invasion, and that TGEV S1 protein interacts with EGFR extracellular receptor binding domain 1. TGEV infection induces EGFR internalization and causes APN and EGFR clustering. Plus, APN and EGFR not only synergistically promote TGEV invasion, but they also active PI3K/AKT and MEK/ERK1/2 signaling pathways. Clathrin and caveolin mediate endocytosis of TGEV and EGFR internalization through the clathrin endocytosis pathway ( Fig. 8). These findings are conducive to enhancing our understanding of the entry mechanism of TGEV, and for providing a potential target for the development of new anti-TGEV therapies.
Fig. 8.

APN and EGFR synergistically promote TGEV invasion. EGFR is a co-factor for TGEV invasion. TGEV S1 protein interacts with EGFR extracellular receptor binding domain 1. TGEV infection induces EGFR internalization and causes APN and EGFR clustering. APN and EGFR synergistically promote TGEV invasion. APN and EGFR synergistically activate PI3K/AKT and MEK/ERK1/2 signaling pathways. Clathrin and caveolin mediate the endocytosis of TGEV and EGFR internalization through clathrin endocytosis pathway.
Acknowledgments
This work was supported by 31772777 from the National Science Grant of China and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Acknowledgments
Conflicts of interest
There is no conflict of interest.
Author contribution
WW.H. S.Z. and YM.S. performed the experiments, WW.H. wrote the paper, WW.H. and Q.Y. analyzed the date and designed the overall experimental strategy.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.virol.2018.05.009.
Appendix A. Supplementary material
Fig. S1.
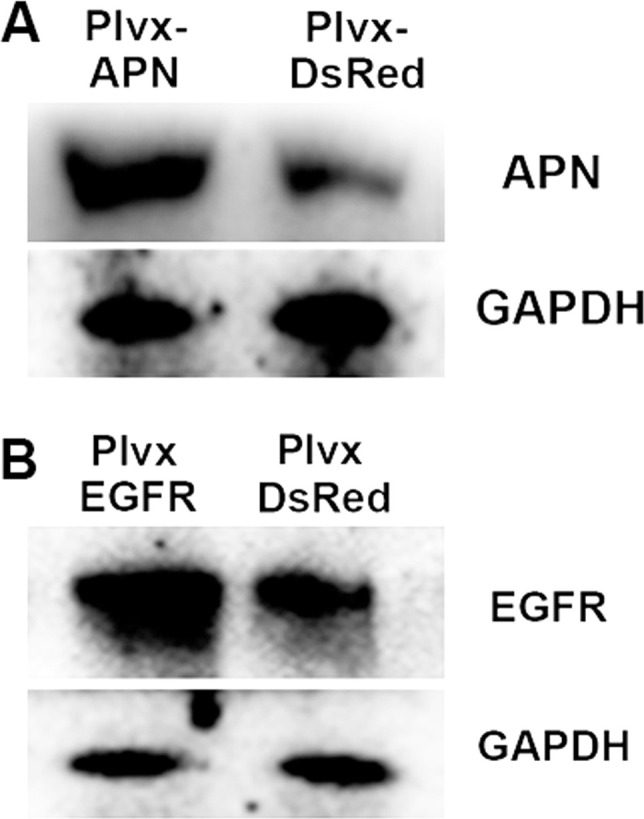
APN and EGFR overexpression verification.
References
- Aleksandrowicz P., Marzi A., Biedenkopf N., Beimforde N., Becker S., Hoenen T., Feldmann H., Schnittler H.J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011;204:S957–S967. doi: 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S., Millet J.K., Licitra B.N., Whittaker G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4:1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnahan A.J., Brown D.R. Porcine IPEC-J2 intestinal epithelial cells in microbiological investigations. Vet. Microbiol. 2012;156:229–237. doi: 10.1016/j.vetmic.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C.M., Petersen N.O. An image correlation analysis of the distribution of clathrin associated adaptor protein (AP-2) at the plasma membrane. J. Cell Sci. 1998;111:271–281. doi: 10.1242/jcs.111.2.271. [DOI] [PubMed] [Google Scholar]
- Chan G., Nogalski M.T., Yurochko A.D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. 2009;106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas B., Gelfi J., Kut E., Sjöström H., Norén O., Laude H. Determinants essential for the transmissible gastroenteritis virus-receptor interaction reside within a domain of aminopeptidase-N that is distinct from the enzymatic site. J. Virol. 1994;68:5216–5224. doi: 10.1128/jvi.68.8.5216-5224.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas, B., Gelfi, J., L'Haridon, R., Sjöström, H., Laude, H., 1992. Aminopeptidase N is a major receptor for the enteropathogenic coronavirus TGEV. [DOI] [PMC free article] [PubMed]
- Delmas B., Gelfi J., Sjöström H., Noren O., Laude H. Further characterization of aminopeptidase-N as a receptor for coronaviruses. Coronaviruses, Springer. 1993:293–298. doi: 10.1007/978-1-4615-2996-5_45. [DOI] [PubMed] [Google Scholar]
- Doyle L.P., Hutchings L.M. A transmissible gastroenteritis in pigs. J. Am. Vet. Med. Assoc. 1946;108:257–259. [PubMed] [Google Scholar]
- Eierhoff T., Hrincius E.R., Rescher U., Ludwig S., Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001099. (e1001099-e1001099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eierhoff T., Hrincius E.R., Rescher U., Ludwig S., Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Broder C.C., Kennedy P.E., Berger E.A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Hu W., Zhu L., Yang X., Lin J., Yang Q. The epidermal growth factor receptor regulates cofilin activity and promotes transmissible gastroenteritis virus entry into intestinal epithelial cells. Oncotarget. 2016;7:12206. doi: 10.18632/oncotarget.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.-M., Wang B., Zhou J., Huang Y.-W. Aminopeptidase-N-independent entry of porcine epidemic diarrhea virus into Vero or porcine small intestine epithelial cells. Virology. 2018 doi: 10.1016/j.virol.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.J., Huang F.T., Marusyk A., Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol. Biol. Cell. 2003;14:858–870. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Baranda S., Gomez-Mouton C., Rojas A., Martinez-Prats L., Mira E., Ana Lacalle R., Valencia A., Dimitrov D.S., Viola A., Delgado R., Martinez A.C., Manes S. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007;9:838–846. doi: 10.1038/ncb1610. [DOI] [PubMed] [Google Scholar]
- Johnson R.A., Ma X.L., Yurochko A.D., Huang E.S. The role of MKK1/2 kinase activity in human cytomegalovirus infection. J. Gen. Virol. 2001;82:493–497. doi: 10.1099/0022-1317-82-3-493. [DOI] [PubMed] [Google Scholar]
- Johnson R.A., Wang X., Ma X.L., Huong S.M., Huang E.S. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J. Virol. 2001;75:6022–6032. doi: 10.1128/JVI.75.13.6022-6032.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M., Toret C.P., Drubin D.G. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- Kolb A.F., Maile J., Heister A., Siddell S.G. Characterization of functional domains in the human coronavirus HCV 229E receptor. J. Gen. Virol. 1996;77:2515–2521. doi: 10.1099/0022-1317-77-10-2515. [DOI] [PubMed] [Google Scholar]
- Li B.X., Ge J.W., Li Y.J. Porcine aminopeptidase N is a functional receptor for the PEDV coronavirus. Virology. 2007;365:166–172. doi: 10.1016/j.virol.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Luo R., He Q., van Kuppeveld F.J., Rottier P.J., Bosch B.-J. Aminopeptidase N is not required for porcine epidemic diarrhea virus cell entry. Virus Res. 2017;235:6–13. doi: 10.1016/j.virusres.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J., Zeisel M.B., Xiao F., Thumann C., Fofana I., Zona L., Davis C., Mee C.J., Turek M., Gorke S. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M., Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J., Schelhaas M., Helenius A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- Mercer J., Schelhaas M., Helenius A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- Mosesson Y., Mills G.B., Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat. Rev. Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- Park J.-E., Cruz D.J.M., Shin H.-J. Clathrin-and serine proteases-dependent uptake of porcine epidemic diarrhea virus into Vero cells. Virus Res. 2014;191:21–29. doi: 10.1016/j.virusres.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri C., Tosoni D., Comai R., Rabellino A., Segat D., Caneva F., Luzzi P., Di Fiore P.P., Tacchetti C. Relationships between EGFR signaling-competent and endocytosis-competent membrane microdomains. Mol. Biol. Cell. 2005;16:2704–2718. doi: 10.1091/mbc.E04-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing K., Mah C., Hansen J., Zhou S., Dwarki V., Srivastava A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999;5:71–77. doi: 10.1038/4758. [DOI] [PubMed] [Google Scholar]
- Saeed M.F., Kolokoltsov A.A., Freiberg A.N., Holbrook M.R., Davey R.A. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008;4:e1000141. doi: 10.1371/journal.ppat.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwegmann-Wessels C., Herrler G. Sialic acids as receptor determinants for coronaviruses. Glycoconj. J. 2006;23:51–58. doi: 10.1007/s10719-006-5437-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe E., Ayscough K.R. Actin regulation in endocytosis. J. Cell Sci. 2006;119:4589–4598. doi: 10.1242/jcs.03247. [DOI] [PubMed] [Google Scholar]
- Terry-Allison T., Montgomery R.I., Whitbeck J.C., Xu R., Cohen G.H., Eisenberg R.J., Spear P.G. HveA (herpesvirus entry mediator A), a coreceptor for herpes simplex virus entry, also participates in virus-induced cell fusion. J. Virol. 1998;72:5802–5810. doi: 10.1128/jvi.72.7.5802-5810.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresnan D.B., Levis R., Holmes K.V. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J. Virol. 1996;70:8669–8674. doi: 10.1128/jvi.70.12.8669-8674.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Huang D.Y., Huong S.-M., Huang E.-S. Integrin αvβ3 is a coreceptor for human cytomegalovirus. Nat. Med. 2005;11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Huong S.-M., Chiu M.L., Raab-Traub N., Huang E.-S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature. 2003;424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- Weingartl H.M., Derbyshire J.B. Evidence for a putative second receptor for porcine transmissible gastroenteritis virus on the villous enterocytes of newborn pigs. J. Virol. 1994;68:7253–7259. doi: 10.1128/jvi.68.11.7253-7259.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwei H., Qinghua Y., Liqi Z., Haofei L., Shanshan Z., Qi G., Kongwang H., Qian Y. Complete genomic sequence of the coronavirus transmissible gastroenteritis virus SHXB isolated in China. Arch. Virol. 2014;159:2295–2302. doi: 10.1007/s00705-014-2080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M.L., Amornphimoltham P., Schmidt M., Wilson P.A., Gutkind J.S., Chiorini J.A. Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat. Med. 2010;16:662–664. doi: 10.1038/nm.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells A. EGF receptor. Int. J. Biochem. Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- Wells A. EGF receptor. Int. J. Biochem. Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- Yoder A., Yu D.Y., Dong L., Iyer S.R., Xu X.H., Kelly J., Liu J., Wang W.F., Vorster P.J., Agulto L., Stephany D.A., Cooper J.N., Marsh J.W., Wu Y.T. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K., Kitazato K., Wang Y. Viruses exploit the function of epidermal growth factor receptor. Rev. Med. Virol. 2014;24:274–286. doi: 10.1002/rmv.1796. [DOI] [PubMed] [Google Scholar]
- Zheng K., Xiang Y., Wang X., Wang Q., Zhong M., Wang S., Wang X., Fan J., Kitazato K., Wang Y. Epidermal growth factor receptor-PI3K signaling controls cofilin activity to facilitate herpes simplex virus 1 entry into neuronal cells. mBio. 2014;5:e00958–00913. doi: 10.1128/mBio.00958-13. [DOI] [PMC free article] [PubMed] [Google Scholar]