

Abstract
Sialic acids are cytoprotectors, mainly localized on the surface of cell membranes with multiple and outstanding cell biological functions. The history of their structural analysis, occurrence, and functions is fascinating and described in this review. Reports from different researchers on apparently similar substances from a variety of biological materials led to the identification of a 9-carbon monosaccharide, which in 1957 was designated “sialic acid.” The most frequently occurring member of the sialic acid family is N-acetylneuraminic acid, followed by N-glycolylneuraminic acid and O-acetylated derivatives, and up to now over about 80 neuraminic acid derivatives have been described. They appeared first in the animal kingdom, ranging from echinoderms up to higher animals, in many microorganisms, and are also expressed in insects, but are absent in higher plants. Sialic acids are masks and ligands and play as such dual roles in biology. Their involvement in immunology and tumor biology, as well as in hereditary diseases, cannot be underestimated. N-Glycolylneuraminic acid is very special, as this sugar cannot be expressed by humans, but is a xenoantigen with pathogenetic potential. Sialidases (neuraminidases), which liberate sialic acids from cellular compounds, had been known from very early on from studies with influenza viruses. Sialyltransferases, which are responsible for the sialylation of glycans and elongation of polysialic acids, are studied because of their significance in development and, for instance, in cancer. As more information about the functions in health and disease is acquired, the use of sialic acids in the treatment of diseases is also envisaged.
Keywords: Sialic acids, History, Sialochemistry, Sialobiochemistry, Sialobiology
Abbreviations
- Aci
acinetaminic acid (for abbreviations of Aci variants, see Table 4)
- ALL
acute lymphoblastic leukemia
- Asn
asparagine
- ATP
adenosine triphosphate
- BHK
baby hamster kidney
- CDP
cytidine diphosphate
- CE
capillary electrophoresis
- CIMS
chemical-ionization mass spectrometry
- CMAH
CMP-Neu5Ac hydroxylase
- CMP
cytidine monophosphate
- CTP
cytidine triphosphate
- DIFO
difluorinated cyclooctyne
- DMB
1,2-diamino-4,5-methylenedioxybenzene
- DTH
delayed-type hypersensitivity
- EIMS
electron-impact mass spectrometry
- EPR
electron paramagnetic resonance
- ESIMS
electrospray ionization mass spectrometry
- Fab
fragment antigen-binding (region on antibody)
- FABMS
fast-atom bombardment mass spectrometry
- Fc
fragment crystallizable (region on antibody)
- FLAG
Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys octapeptide
- Fuc
fucose
- Gal
galactose
- GalNAc
N-acetylgalactosamine
- Glc
glucose
- GLC
gas–liquid chromatography
- GlcNAc
N-acetylglucosamine
- GlcNGc
N-glycolylglucosamine
- GNE
UDP-GlcNAc 2-epimerase
- HD
Hanganutziu–Deicher
- HE
hemagglutinin-esterase
- HEF
hemagglutinin-esterase-fusion
- HEV
high endothelial venules
- HFB
heptafluorobutyryl
- HIBM
hereditary inclusion body myopathy
- HPAEC–PAD
high-performance anion-exchange chromatography with pulsed amperometric detection
- HPLC
high-performance liquid chromatography
- HSEA
hard-sphere exo-anomeric
- IR
infrared
- ISAV
Infectious Salmon Anemia Virus
- ISSD
Infantile Sialic Acid Storage Disease
- Kdn
ketodeoxynononic acid
- LAD II
leukocyte adhesion deficiency type II syndrome
- Leg
legionaminic acid (for abbreviations of derivatives and variants of Leg, see Table 4)
- LFA
Limax flavus agglutinin
- MAA
Maackia amurensis agglutinin
- MAG
myelin-associated glycoprotein
- MALDI-TOFMS
matrix-assisted laser-desorption ionization time-of-flight mass spectrometry
- Man
mannose
- ManNAc
N-acetylmannosamine
- ManNAz
N-azidoacetylmannosamine
- ManNGc
N-glycolylmannosamine
- MD
molecular dynamics
- MERS-CoV
Middle East Respiratory Syndrome Coronavirus
- MM
molecular mechanics
- MNK
ManNAc kinase
- MS
mass spectrometry
- NADH/NAD+
nicotinamide adenine dinucleotide (reduced and oxidized form)
- NADPH
nicotinamide adenine dinucleotide phosphate
- Neu
neuraminic acid [for abbreviations of derivatives (sialic acids), see Table 1]
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NulO
non-2-ulosonic acid
- OPD
o-phenylenediamine
- PAPS
peroxidase–antibody–peroxidase smear
- PAS
periodic acid–Schiff
- PPCA
protective protein cathepsin A
- Pse
pseudaminic acid (for abbreviations of derivatives of Pse, see Table 4)
- QM
quantum mechanical
- RDE
receptor-destroying enzyme (sialidase)
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- Ser
serine
- Sia
sialic acid
- SNA
Sambucus nigra agglutinin
- SOAT
sialate O-acetyltransferase
- Thr
threonine
- TLC
thin-layer chromatography
- TS
trans-sialidase
- UDP
uridine diphosphate
- UTP
uridine triphosphate
- UV
ultraviolet
- WGA
wheat germ agglutinin
- X-ray
Röntgen radiation
Table 4.
Survey of Reported Structures of Naturally Occurring 5,7-Diamino-3,5,7,9-tetradeoxynon-2-ulosonic Acid Derivatives, and Some Related Structures, Which Are Terminal or Internal Units of Bacterial Homo- and Hetero-polysaccharides and Glycoprotein Glycans595, 677, 679, 681
| Name | Abbreviation | References |
|---|---|---|
| *5,7-Diamino-3,5,7,9-tetradeoxy-l-glycero-l-manno-non-2-ulosonic acid/pseudaminic acid (Pse) | ||
| 5,7-Di-N-acetyl-pseudaminic acid | Pse5,7Ac2 | 688, 689 |
| 5,7-Di-N-acetyl-8-O-acetyl-pseudaminic acid | Pse5,7,8Ac3 | 689 |
| 5,7-Di-N-acetyl-8-O-glycyl-pseudaminic acid | Pse5,7Ac28Gly | 690 |
| 5,7-Di-N-glyceryl-pseudaminic acid | Pse5,7Gr2 | 689 |
| 5-N-Acetimidoyl-7-N-acetyl-pseudaminic acid | Pse7Ac5Am | 689, 691 |
| 5-N-Acetimidoyl-7-N-acetyl-8-O-acetyl-pseudaminic acid | Pse7,8Ac25Am | 692 |
| 5-N-Acetimidoyl-7-N-acetyl-8-O-(N-acetyl-glutaminyl)-pseudaminic acid | Pse7Ac5Am8GlnNAc | 690 |
| 5-N-Acetyl-7-N-formyl-pseudaminic acid | Pse5Ac7Fo | 682, 693 |
| 5-N-Acetyl-7-N-l-glyceryl-pseudaminic acid | Pse5Ac7Gr | 694 |
| 5-N-Acetyl-7-N-[(R)-3-hydroxybutyryl]-pseudaminic acid | Pse5Ac7(3RHb) | 682, 695, 696 |
| 5-N-Acetyl-7-N-[(R)-3-hydroxybutyryl]-4-O-acetyl-pseudaminic acid | Pse4,5Ac27(3RHb) | 697 |
| 5-N-Acetyl-7-N-[(S)-3-hydroxybutyryl]-pseudaminic acid | Pse5Ac7(3SHb) | 696 |
| 5-N-Acetyl-7-N-(4-hydroxybutyryl)-pseudaminic acid | Pse5Ac7(4Hb) | 698 |
| 5-N-Acetyl-7-N-(3,4-dihydroxybutyryl)-pseudaminic acid | Pse5Ac7(3,4Hb) | 602 |
| 7-N-Acetimidoyl-5-N-acetyl-pseudaminic acid | Pse5Ac7Am | 699 |
| 7-N-Acetimidoyl-5-N-(2,3-di-O-methyl-glyceryl)-pseudaminic acid | Pse7Am5Me2Gr | 700 |
| 7-N-Acetyl-5-N-(3-hydroxybutyryl)-pseudaminic acid | Pse7Ac5(3Hb) | 701 |
| 7-N-Acetyl-5-N-(2,3-di-O-methyl-glyceryl)-pseudaminic acid | Pse7Ac5Me2Gr | 700 |
| 7-N-Formyl-5-N-[(R)-3-hydroxybutyryl]-pseudaminic acid | Pse7Fo5(3RHb) | 682, 702, 703 |
| *5,7-Diamino-3,5,7,9-tetradeoxy-d-glycero-d-galacto-non-2-ulosonic acid/legionaminic acid (Leg) | ||
| 5,7-Di-N-acetyl-legionaminic acida | Leg5,7Ac2 | 684, 704, 705 |
| 5-N-Acetimidoyl-7-N-acetyl-legionaminic acida | Leg7Ac5Am | 684, 705 |
| 5-N-Acetimidoyl-7-N-acetyl-8-O-acetyl-legionaminic acidb | Leg7,8Ac25Am | 684, 706 |
| 5-N-Acetimidoyl-7-N-acetyl-5-N-methyl-legionaminic acid | Leg7Ac5Am5Me | 707 |
| 5-N-(N-Methyl-acetimidoyl)-7-N-acetyl-legionaminic acid | Leg7Ac5AmMe | 705, 707 |
| 5-N-(N,N-Dimethyl-acetimidoyl)-7-N-acetyl-legionaminic acid | Leg7Ac5AmMe2 | 707 |
| 5-N-Acetimidoyl-7-N-acetyl-8-O-acetyl-5-N-methyl-legionaminic acid | Leg7,8Ac25Am5Me | 706 |
| 5-N-(N,N-Dimethyl-acetimidoyl)-7-N-acetyl-8-O-acetyl-legionaminic acid | Leg7,8Ac25AmMe2 | 706 |
| 5-N-Acetyl-7-N-(N-acetyl-d-alanyl)-legionaminic acid | Leg5Ac7AlaNAc | 704 |
| 5-N-Acetyl-7-N-(d-alanyl)-legionaminic acid | Leg5Ac7Ala | 708 |
| 7-N-Acetyl-5-N-formyl-legionaminic acid | Leg7Ac5Fo | 709 |
| 7-N-Acetyl-5-N-[(S)-3-hydroxybutyryl]-legionaminic acidc | Leg7Ac5(3SHb) | 684 |
| 7-N-Acetyl-5-N-(N-methyl-5-glutamyl)-legionaminic acid | Leg7Ac5GluNMe | 710 |
| *5,7-Diamino-3,5,7,9-tetradeoxy-d-glycero-d-talo-non-2-ulosonic acid/4-epi-legionaminic acid (4eLeg) | ||
| 5,7-Di-N-acetyl-4-epi-legionaminic acid | 4eLeg5,7Ac2 | 711 |
| 5,7-Di-N-acetyl-8-O-acetyl-4-epi-legionaminic acidd | 4eLeg5,7,8Ac3 | 684 |
| 5-N-Acetimidoyl-7-N-acetyl-4-epi-legionaminic acid | 4eLeg7Ac5Am | 712 |
| 5-N-Acetimidoyl-7-N-acetyl-8-O-acetyl-4-epi-legionaminic acid | 4eLeg7,8Ac25Am | 712 |
| *5,7-Diamino-3,5,7,9-tetradeoxy-l-glycero-d-galacto-non-2-ulosonic acid/8-epi-legionaminic acid (8eLeg) | ||
| 5,7-Di-N-acetyl-8-epi-legionaminic acide | 8eLeg5,7Ac2 | 713, 714, 715 |
| 5,7-Di-N-acetyl-8-O-acetyl-8-epi-legionaminic acid | 8eLeg5,7,8Ac3 | 716 |
| 5-N-Acetimidoyl-7-N-acetyl-8-epi-legionaminic acid | 8eLeg7Ac5Am | 717 |
| 7-N-Acetimidoyl-5-N-acetyl-8-epi-legionaminic acid | 8eLeg5Ac7Am | 79 |
| 7-N-Acetimidoyl-5-N-acetyl-8-O-acetyl-8-epi-legionaminic acid | 8eLeg5,8Ac27Am | 79 |
| 7-N-Acetyl-5-N-[(R)-3-hydroxybutyryl]-8-epi-legionaminic acide | 8eLeg7Ac5(3RHb) | 684, 713, 714 |
| 7-N-Acetyl-5-N-(4-hydroxybutyryl)-8-epi-legionaminic acide | 8eLeg7Ac5(4Hb) | 684 |
| *5,7-Diamino-3,5,7,9-tetradeoxy-l-glycero-l-altro-non-2-ulosonic acid/acinetaminic acid (Aci) | ||
| 5,7-Di-N-acetyl-acinetaminic acid | Aci5,7Ac2 | 686 |
| *5,7-Diamino-3,5,7,9-tetradeoxy-d-glycero-l-altro-non-2-ulosonic acid/8-epi-acinetaminic acid (8eAci) | ||
| 5,7-Di-N-acetyl-8-epi-acinetaminic acid | 8eAci5,7Ac2 | 687 |
| *Some related 9-deoxynon-2-ulosonic acids | |
| 5- or 7-Acetamido-,7- or 5-(3-hydroxybutyramido)-5,7,9-trideoxynon-2-ulosonic acid | 718 |
| 5-Acetamido-7-[(S)-3-hydroxybutyramido]-8-amino-3,5,7,8,9-pentadeoxy-l-glycero-l-manno- or d-glycero-l-manno-non-2-ulosonic acid | 719 |
| 5-Acetamidino-3,5,9-trideoxy-l-glycero-l-gluco-non-2-ulosonic acid (tentatively assigned chirality; trivial name: fusaminic acid) | 720 |
| 5,7-Diacetamido-8-amino-3,5,7,8,9-pentadeoxy-d-glycero-d-galacto-non-2-ulosonic acid | 721 |
Included literature refers to first reports or revised reports.
Initially assigned with the d-glycero-l-galacto722 and later with the l-glycero-d-galacto713, 714, 723 configuration.
Initially assigned with the d-glycero-l-galacto,724, 725, 726 and later with the l-glycero-d-galacto713, 714 configuration.
Initially assigned with the l-glycero-d-galacto configuration.723
Initially assigned with the l-glycero-d-talo configuration.727
Table 1.
Survey of Reported Structures of Naturally Occurring Members of the Sialic Acid Familya
| Name | Abbreviation | References |
|---|---|---|
| Neuraminic acidb | Neu | 15, 16, 17, 18, 19, 20 |
| Neuraminic acid 1,5-lactamc | Neu1,5lactam | 20, 21 |
| 5-N-Acetyl-neuraminic acid | Neu5Ac | 6, 19, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 |
| 5-N-Acetyl-4-O-acetyl-neuraminic acid | Neu4,5Ac2 | 6, 19, 23, 24, 27, 29, 30, 31, 32, 33, 34 |
| 5-N-Acetyl-7-O-acetyl-neuraminic acid | Neu5,7Ac2 | 6, 19, 23, 24, 29, 30, 31, 32, 33, 35, 36, 37, 38, 38a |
| 5-N-Acetyl-8-O-acetyl-neuraminic acid | Neu5,8Ac2 | 6, 19, 30, 32, 33, 37 |
| 5-N-Acetyl-9-O-acetyl-neuraminic acid | Neu5,9Ac2 | 6, 19, 27, 29, 30, 31, 32, 33, 36, 37, 38a, 39 |
| 5-N-Acetyl-4,9-di-O-acetyl-neuraminic acid | Neu4,5,9Ac3 | 6, 19, 27, 30, 33 |
| 5-N-Acetyl-7,8-di-O-acetyl-neuraminic acidd | Neu5,7,8Ac3 | 35 |
| 5-N-Acetyl-7,9-di-O-acetyl-neuraminic acid | Neu5,7,9Ac3 | 6, 19, 27, 29, 30, 31, 32, 33, 37, 38a, 38, 40 |
| 5-N-Acetyl-8,9-di-O-acetyl-neuraminic acid | Neu5,8,9Ac3 | 6, 19, 29, 30, 31, 32, 33, 37, 38a, 38, 40 |
| 5-N-Acetyl-4,7,9-tri-O-acetyl-neuraminic acid | Neu4,5,7,9Ac4 | 19 |
| 5-N-Acetyl-7,8,9-tri-O-acetyl-neuraminic acid | Neu5,7,8,9Ac4 | 6, 30, 31, 32, 33, 37 |
| 5-N-Acetyl-4,7,8,9-tetra-O-acetyl-neuraminic acid | Neu4,5,7,8,9Ac5 | 19 |
| 5-N-Acetyl-4-O-glycolyl-neuraminic acid | Neu5Ac4Gc | 41 |
| 5-N-Acetyl-7-O-glycolyl-neuraminic acid | Neu5Ac7Gc | 41 |
| 5-N-Acetyl-9-O-lactyl-neuraminic acide | Neu5Ac9Lt | 6, 19, 30, 32, 33, 42, 43 |
| 5-N-Acetyl-4-O-acetyl-9-O-lactyl-neuraminic acid | Neu4,5Ac29Lt | 6, 19, 30, 33, 44 |
| 5-N-Acetyl-7-O-acetyl-9-O-lactyl-neuraminic acid | Neu5,7Ac29Lt | 19 |
| 5-N-Acetyl-8-O-acetyl-9-O-lactyl-neuraminic acid | Neu5,8Ac29Lt | 45 |
| 5-N-Acetyl-8-O-methyl-neuraminic acid | Neu5Ac8Me | 6, 19, 25, 33, 46 |
| 5-N-Acetyl-4-O-acetyl-8-O-methyl-neuraminic acid | Neu4,5Ac28Me | 19 |
| 5-N-Acetyl-9-O-acetyl-8-O-methyl-neuraminic acid | Neu5,9Ac28Me | 6, 19, 33, 46 |
| 5-N-Acetyl-9-O-methyl-neuraminic acid | Neu5Ac9Me | 47 |
| 5-N-Acetyl-4-O-sulfo-neuraminic acid | Neu5Ac4S | 48 |
| 5-N-Acetyl-8-O-sulfo-neuraminic acid | Neu5Ac8S | 19, 32, 49, 50 |
| 5-N-Acetyl-4-O-acetyl-8-O-sulfo-neuraminic acid | Neu4,5Ac28S | 19 |
| 5-N-Acetyl-9-O-phospho-neuraminic acidf, g | Neu5Ac9P | 51 |
| 5-N-Acetyl-2-deoxy-2,3-didehydro-neuraminic acidg, h | Neu2en5Ac | 6, 30, 33, 43, 52, 53, 54 |
| 5-N-Acetyl-9-O-acetyl-2-deoxy-2,3-didehydro-neuraminic acidg | Neu2en5,9Ac2 | 55, 56 |
| 5-N-Acetyl-2-deoxy-2,3-didehydro-9-O-lactyl-neuraminic acidg | Neu2en5Ac9Lt | 55, 56 |
| 5-N-Acetyl-2,7-anhydro-neuraminic acidg, i | Neu2,7an5Ac | 6, 33, 55, 57, 58 |
| 5-N-Acetyl-4,8-anhydro-neuraminic acidj | Neu4,8an5Ac | 31, 32, 59, 60 |
| 5-N-Acetyl-neuraminic acid 1,7-lactone | Neu5Ac1,7lactone | 19, 61, 62 |
| 5-N-Acetyl-9-O-acetyl-neuraminic acid 1,7-lactone | Neu5,9Ac21,7lactone | 19 |
| 5-N-Acetyl-4,9-di-O-acetyl-neuraminic acid 1,7-lactone | Neu4,5,9Ac31,7lactone | 19 |
| 1-Tauryl 5-N-acetyl-neuraminic amide | Neu5Ac1Tau | 63 |
| 5-N-Glycolyl-neuraminic acid | Neu5Gc | 6, 19, 23, 26, 27, 29, 30, 31, 32, 33, 64, 65, 66 |
| 4-O-Acetyl-5-N-glycolyl-neuraminic acid | Neu4Ac5Gc | 6, 27, 30, 31, 32, 33, 41 |
| 7-O-Acetyl-5-N-glycolyl-neuraminic acid | Neu7Ac5Gc | 6, 30, 32, 33, 37 |
| 8-O-Acetyl-5-N-glycolyl-neuraminic acid | Neu8Ac5Gc | 32, 55 |
| 9-O-Acetyl-5-N-glycolyl-neuraminic acid | Neu9Ac5Gc | 6, 19, 27, 29, 30, 31, 32, 33, 37 |
| 4,7-Di-O-acetyl-5-N-glycolyl-neuraminic acid | Neu4,7Ac25Gc | 19 |
| 4,9-Di-O-acetyl-5-N-glycolyl-neuraminic acid | Neu4,9Ac25Gc | 19 |
| 7,9-Di-O-acetyl-5-N-glycolyl-neuraminic acidk | Neu7,9Ac25Gc | 6, 19, 30, 31, 32, 33, 37 |
| 8,9-Di-O-acetyl-5-N-glycolyl-neuraminic acid | Neu8,9Ac25Gc | 6, 19, 30, 31, 32, 33, 37 |
| 4,7,9-Tri-O-acetyl-5-N-glycolyl-neuraminic acid | Neu4,7,9Ac35Gc | 19 |
| 7,8,9-Tri-O-acetyl-5-N-glycolyl-neuraminic acid | Neu7,8,9Ac35Gc | 6, 30, 31, 33, 37 |
| 4,7,8,9-Tetra-O-acetyl-5-N-glycolyl-neuraminic acid | Neu4,7,8,9Ac45Gc | 19 |
| 5-N-Glycolyl-9-O-lactyl-neuraminic acid | Neu5Gc9Lt | 19, 32, 55 |
| 4-O-Acetyl-5-N-glycolyl-9-O-lactyl-neuraminic acid | Neu4Ac5Gc9Lt | 19 |
| 7-O-Acetyl-5-N-glycolyl-9-O-lactyl-neuraminic acid | Neu7Ac5Gc9Lt | 19 |
| 8-O-Acetyl-5-N-glycolyl-9-O-lactyl-neuraminic acid | Neu8Ac5Gc9Lt | 19 |
| 4,7-Di-O-acetyl-5-N-glycolyl-9-O-lactyl-neuraminic acid | Neu4,7Ac25Gc9Lt | 19 |
| 7,8-Di-O-acetyl-5-N-glycolyl-9-O-lactyl-neuraminic acid | Neu7,8Ac25Gc9Lt | 19 |
| 5-N-Glycolyl-8-O-methyl-neuraminic acidl | Neu5Gc8Me | 6, 31, 32, 33, 46, 67, 68 |
| 4-O-Acetyl-5-N-glycolyl-8-O-methyl-neuraminic acid | Neu4Ac5Gc8Me | 69 |
| 7-O-Acetyl-5-N-glycolyl-8-O-methyl-neuraminic acid | Neu7Ac5Gc8Me | 32, 69 |
| 9-O-Acetyl-5-N-glycolyl-8-O-methyl-neuraminic acid | Neu9Ac5Gc8Me | 6, 31, 32, 33, 46, 55 |
| 4,7-Di-O-acetyl-5-N-glycolyl-8-O-methyl-neuraminic acid | Neu4,7Ac25Gc8Me | 69 |
| 7,9-Di-O-acetyl-5-N-glycolyl-8-O-methyl-neuraminic acid | Neu7,9Ac25Gc8Me | 31 |
| 5-N-Glycolyl-9-O-methyl-neuraminic acid | Neu5Gc9Me | 47, 70 |
| 5-N-Glycolyl-8-O-sulfo-neuraminic acid | Neu5Gc8S | 19, 32, 49, 71 |
| 5-N-Glycolyl-9-O-sulfo-neuraminic acid | Neu5Gc9S | 72 |
| 5-N-(O-Acetyl)glycolyl-neuraminic acid | Neu5GcAc | 6, 33, 73 |
| 5-N-(O-Methyl)glycolyl-neuraminic acid | Neu5GcMe | 74 |
| 2-Deoxy-2,3-didehydro-5-N-glycolyl-neuraminic acidg | Neu2en5Gc | 6, 33, 56, 75 |
| 9-O-Acetyl-2-deoxy-2,3-didehydro-5-N-glycolyl-neuraminic acidg | Neu2en9Ac5Gc | 56 |
| 2-Deoxy-2,3-didehydro-5-N-glycolyl-9-O-lactyl-neuraminic acidg | Neu2en5Gc9Lt | 55, 56 |
| 2-Deoxy-2,3-didehydro-5-N-glycolyl-8-O-methyl-neuraminic acidg | Neu2en5Gc8Me | 55 |
| 2,7-Anhydro-5-N-glycolyl-neuraminic acidg | Neu2,7an5Gc | 33, 55, 58 |
| 2,7-Anhydro-5-N-glycolyl-8-O-methyl-neuraminic acidg | Neu2,7an5Gc8Me | 55 |
| 4,8-Anhydro-5-N-glycolyl-neuraminic acidj | Neu4,8an5Gc | 32 |
| 5-N-Glycolyl-neuraminic acid 1,7-lactone | Neu5Gc1,7lactone | 19 |
| 9-O-Acetyl-5-N-glycolyl-neuraminic acid 1,7-lactone | Neu9Ac5Gc1,7lactone | 19 |
| 7-Acetamido-9-O-acetyl-7-deoxy-5-N-glycolyl-neuraminic acid | Neu9Ac5Gc7NAc | 19 |
| 7-Acetamido-8,9-di-O-acetyl-7-deoxy-5-N-glycolyl-neuraminic acidm | Neu8,9Ac25Gc7NAc | 19 |
| 2-Keto-3-deoxy-nononic acid | Kdn | 19, 32, 76, 77, 78 |
| 5-O-Acetyl-2-keto-3-deoxy-nononic acid | Kdn5Ac | 19, 32 |
| 7-O-Acetyl-2-keto-3-deoxy-nononic acid | Kdn7Ac | 19, 32 |
| 8-O-Acetyl-2-keto-3-deoxy-nononic acid | Kdn8Ac | 79 |
| 9-O-Acetyl-2-keto-3-deoxy-nononic acid | Kdn9Ac | 19, 32, 80, 81 |
| 4,5-Di-O-acetyl-2-keto-3-deoxy-nononic acid | Kdn4,5Ac2 | 19 |
| 4,7-Di-O-acetyl-2-keto-3-deoxy-nononic acid | Kdn4,7Ac2 | 19 |
| 5,9-Di-O-acetyl-2-keto-3-deoxy-nononic acid | Kdn5,9Ac2 | 32 |
| 7,9-Di-O-acetyl-2-keto-3-deoxy-nononic acid | Kdn7,9Ac2 | 19, 32 |
| 8,9-Di-O-acetyl-2-keto-3-deoxy-nononic acid | Kdn8,9Ac2 | 19 |
| 2-Keto-3-deoxy-5-O-methyl-nononic acid | Kdn5Me | 82 |
| 2-Keto-3-deoxy-9-O-phospho-nononic acidg, n | Kdn9P | 83 |
In the case of a structure proven by mass spectrometry and/or NMR spectroscopy, reference numbers refer in general to such studies.
Present only in bound form and considered to be derived from bound Neu5Ac in an enzymatic N-deacetylation/N-reacetylation cycle.18 In free open form, Neu is directly converted into its labile internal Schiff base 4-hydroxy-5-[1,2,3,4-tetrahydroxybutyl]-Δ1-pyrroline-2-carboxylic acid (pH-dependent equilibrium).15 However, Neu was picked up by GLC–EIMS of its heptafluorobutyrylated methyl ester derivative, and it was suggested that the heptafluorobutyric anhydride acylation is able to disrupt the Schiff base, followed by blocking the free amino group.19
Present only in bound form and considered to be derived from bound Neu in an enzymatic dehydration reaction, catalyzed by a so-called sialic acid cyclase. Neu1,5lactam was initially called “cyclic sialic acid.”21
Detected in bound form in (α2→9)-linked Neu5Ac polysaccharide.
Lactyl = l-Lactyl.
Biosynthetic intermediate to Neu5Ac.
Present only as free form.
Neu2en5Ac, originally known as a synthetic compound with a potent inhibitory effect on various sialidases,84 may be produced enzymatically from sialoglycoconjugates in tissues.85 Small amounts are formed by a water elimination side reaction from Neu5Ac during influenza-B-virus-sialidase-catalyzed desialylations of sialoglycoconjugates.86 Neu2en5Ac can be generated from CMP-Neu5Ac in a nonenzymatic elimination reaction, occurring under physiological and, much faster, under alkaline conditions.53, 56
Neu2,7an5Ac can be generated from Neu5Ac(α2→3)Gal(β1→-containing sialoglycoconjugates using a sialidase isolated from the leech Macrobdella decora.58, 87
Neu4,8an5Ac does not occur as such in nature. It is assumed to be formed under hydrolytic conditions from bound Neu4,5Ac2 in a multistep process, before cleavage of the glycosidic linkage.31, 59 Also alkaline treatment of free Neu4,5Ac2 yielded Neu4,8an5Ac.31 Alternatively, Neu4,8an5Ac is formed by thermal degradation of sodium N-acetylneuraminate.60 In addition, two other anhydro forms of Neu5Ac were suggested by ref. 32.
Three anhydro derivatives of Neu5Gc8Me were suggested by ref. 32.
Neu4,9Ac27Am5Gc in the text of ref. 19 is probably a typing error for Neu8,9Ac27Am5Gc.
Biosynthetic intermediate to Kdn.
1. Introduction
Alfred Gottschalk, discussing the state of the glycoprotein research at the “C.N.R.S. Colloque Internationale sur les Glycoconjugués” (later named the 2nd International Symposium on Glycoconjugates), organized by Jean Montreuil in 1973 at the Université des Sciences et Technologies de Lille (Villeneuve d’Ascq, France), stated: “We are not at the end of all progress, but at the beginning; we have but reached the shores of a great unexplored continent.”1 And this certainly held for his favorite topic: sialic acid research. Gottschalk encouraged students interested in research to work in the field of carbohydrate (bio)chemistry/glycobiology, which rapidly expanded in the 1970s. Since then, as this review will show, many questions regarding the chemistry, analysis, biochemistry, and biology of sialic acids could be answered, and the important influences of this family of sugar molecules on and in our lives have been documented in a myriad of publications.
Sialic acids are frequently found in Nature in many molecular forms, and detailed reviews have been available since the 1960s.2, 3, 4, 5, 6, 7 These compounds are regularly present in higher animals like Echinoderma, Hemichorda, Cephalocorda, and Vertebrata and could also sporadically be identified in some members of other groups of animals, such as the Platyhelminthes (Polychoerus carmelensis), Cephalopoda, and Crustaceae (Homarus americanus). It is, however, not clear whether the sialic acids are produced by the animals themselves or originate from nutrition or microorganisms. Sialic acid concentrations have been measured in subcellular membrane fractions of various cell types, showing highest values in the plasma membrane (65%–70% of the total sialic acid quantity of the cell) and in much lower amounts in the smooth endoplasmic reticulum.3 Generally, most of these sugars are bound to glycoproteins with the exception of nerve and brain cells, where they prevail on glycolipids. The overall sialic acid level in the various organelles was assumed to be influenced by endogenous sialidase, but no experimental evidence existed. Interestingly, also insects were shown to contain sialic acids and to express corresponding enzymes.8, 9, 10, 11 Sialic acids were found in some viruses, various bacteria, protozoa, and pathogenic fungi. They are absent in plants and in most animals in the evolutionary tree lower than the echinoderms. The various studies show that sialic acids appeared relatively late in evolution, which led to the assumption that acquisition of these monosaccharides accelerated differentiation of the higher animals.2, 4 Deaminated neuraminic acid is often seen in vertebrates and bacteria, but its abundant occurrence in animals is limited to the lower vertebrates.12 Further details are included in the various sections in this chapter, when applicable.
In naturally occurring sialic acid-containing structures, the sialic acid units play important roles in many physiological and pathological processes via carbohydrate–protein interactions, including cellular recognition and communication, cellular aggregation and development, controlling the lifetimes of glycoconjugates in organisms, mediating bacterial and viral infections, being involved in tumor growth and metastasis, playing a great role in immunology, in the biology of the microbiome, in cell signaling, in reproduction biology, and in neurobiology. Sialic acids, which occur mostly as the terminal part of many glycoconjugates (glycoproteins; glycolipids) and carbohydrate chains (oligosaccharides, capsular and tissue polysialic acids, bacterial lipooligo/polysaccharides) occur in various chemical forms, which can dramatically influence their biology. O-Acetyl and N-glycolyl groups, in particular, are most effective in determining sialic acid function. Many genes and enzymes control the anabolism and catabolism of sialic acids, which are of importance not only in health but also in disease situations. Increasing information about the regulation of sialic acid expression on the genetic level of various hereditary diseases can be gathered. Sialopharmacy is still in its infancy, but recently more publications show progress in this field. But it was not too long ago that groups of scientists worldwide were trying to elucidate the primary structures and the basic biochemical reactions of these unusual monosaccharides.
Nowadays, the basic structural features of the sialic acid (Sia) family, a subclass of the superfamily of naturally occurring non-2-ulosonic acids (NulOs), are well known (Fig. 1 ). The mother molecule of the family, neuraminic acid (Neu), which does not occur in free form in nature due to its immediate cyclization to form an internal Schiff base, is a nine-carbon-containing monosaccharide, chemically defined as a combination of a 2-keto-carboxylic acid, a deoxysugar, and an aminosugar. It is systematically named 5-amino-3,5-dideoxy-d-glycero-d-galacto-non-2-ulosonic acid (Neu; C9H17NO8) (C7 is the anomeric reference atom). The intramolecular (cyclic) hemiketal form of Neu (C2–O–C6) comprises a pyranose ring in a 2 C 5 chair conformation with an equatorially oriented glycerol side chain at C6. The amino group at C5 is generally N-acetylated [5-acetamido-3,5-dideoxy-d-glycero-d-galacto-non-2-ulopyranosonic acid (IUPAC/IUBMB Recommendations 1996/199713); N-acetylneuraminic acid, Neu5Ac; C11H19NO9] (for different views, see Fig. 2 ) or N-glycolylated [5-hydroxyacetamido-3,5-dideoxy-d-glycero-d-galacto-non-2-ulopyranosonic acid (IUPAC/IUBMB Recommendations 1996/199713); N-glycolylneuraminic acid, Neu5Gc; C11H19NO10]. Note that the full name for Neu5Ac, as presented in the IUPAC/IUB document 1969/1971,14 namely, 5-acetamido-3,5-dideoxy-d-glycero-d-galacto-2-nonulopyranosonic acid, is no longer used. The amino group at C5 can also be replaced by a hydroxyl function, yielding 3-deoxy-d-glycero-d-galacto-non-2-ulopyranosonic acid (ketodeoxynononic acid, Kdn; C9H16O9) (IUPAC/IUBMB Recommendations 1996/199713), usually called deaminated neuraminic acid (other names used: 2-keto-3-deoxy-nononic acid, 3-deoxynon-2-ulosonic acid, 2-keto-3-deoxy-d-glycero-d-galacto-nononic acid). To indicate the absolute configuration, up to the 1980s the trivial names of the mother molecule and the derived members were presented with the configurational prefix d, e.g., d-neuraminic acid, N-acetyl-d-neuraminic acid, N-glycolyl-d-neuraminic acid. According to the IUPAC/IUBMB Recommendations 1996/1997,13 this prefix should not be used, as the d-glycero-d-galacto configuration is implied in the trivial names.
Fig. 1.

The “mother” molecule of the family Sialic Acids (Neu), together with the three major “children” (Neu5Ac, Neu5Gc, Kdn), presented in the α-configuration, as occurring in sialic acid-containing carbohydrate chains.
Fig. 2.

Chemical structures of N-acetylneuraminic acid (5-acetamido-3,5-dideoxy-d-glycero-d-galacto-non-2-ulosonic acid, Neu5Ac) in different views according to IUPAC/IUBMB recommendations. (A) Neu5Ac in Fischer projection formula, open chain; (B) β-Neu5Ac/N-acetyl-β-neuraminic acid/5-acetamido-3,5-dideoxy-d-glycero-β-d-galacto-non-2-ulopyranosonic acid in Fischer projection formula, cyclic chain, pyranose ring; (C) β-Neu5Ac in Haworth representation, pyranose ring; and (D) β-Neu5Ac in 2C5 chair conformation.
The abbreviations used for sialic acids have a long and controversial history, i.e., for N-acetylneuraminic acid: NANA, N-AN, NAN, NANS, NeuNAc, AcNeu, and Ac-Neu; for N-glycolylneuraminic acid: NGNA, N-GN, NGN, NGNS, NeuNGl, NeuNGc, GlNeu, and Gc-Neu; for ketodeoxynononic acid: KDN. At present, the IUPAC/IUBMB recommendations are Neu5Ac (or NeuAc), Neu5Gc (or NeuGc), and Kdn. For neuraminic acid, the abbreviation is Neu, whereas the recommended abbreviation for sialic acid is Sia. A still frequently used old abbreviation for 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (Neu2en5Ac) is DANA.
Sialic acids are relatively strong acids, e.g., Neu5Ac has a pK a value found in the range of 2.0–3.0 in various studies with an average of 2.6. This strong acidity is responsible for processes such as autohydrolysis of sialic acid-containing glycan chains, and for repulsive effects, leading, for example, to the high viscosity of mucins, and for the binding and transport of cationic substances.
In Table 1 a survey of the over 80 known naturally occurring members of the sialic acid family, together with their abbreviations and relevant references,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38a, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87 is presented. In the case of Neu, besides N-acetyl or N-glycolyl groups at C5, the hydroxy groups at C4, C7, C8, and C9 may be free, esterified (acetylated, glycolylated, lactylated, sulfated, phosphorylated) or etherified (methylated). Most free sialic acids occur predominantly in the more thermodynamically stable β-anomeric form (>93%), whereas in nature bound sialic acids, with one exception, possess the α-anomeric configuration. The exception is the nucleotide-bound sialic acid, the activated CMP-donor, e.g., CMP-β-Neu5Ac (Fig. 3C). Crystalline Neu5Ac occurs specifically in the β-anomeric form. The mother molecule Neu and Neu1,5lactam (neuraminic acid 1,5-lactam) (Fig. 3A) only exist in bound form. A number of other members (can) only occur as free form, i.e., Neu5Ac9P (9-O-phospho-N-acetylneuraminic acid), (O-substituted) Neu2en5Ac (Fig. 3B), or Neu2en5Gc (2-deoxy-2,3-didehydro-N-glycolylneuraminic acid), (O-substituted) Neu2,7an5Ac (2,7-anhydro-N-acetylneuraminic acid) (Fig. 3D) or Neu2,7an5Gc (2,7-anhydro-N-glycolylneuraminic acid), and Neu4,8an5Ac (4,8-anhydro-N-acetylneuramic acid) (Fig. 3F) or Neu4,8an5Gc (4,8-anhydro-N-glycolylneuraminic acid). In the case of Kdn, the hydroxy groups at C4, C5, C7, C8, and C9 may be free, esterified (acetylated, phosphorylated) or etherified (methylated). In Neu5Gc, the hydroxy group at C7 can be replaced by an acetamido group, yielding in fact another variant of Neu, with the full name of 7-acetamido-5-hydroxyacetamido-3,5,7-trideoxy-d-glycero-d-galacto-non-2-ulosonic acid, so far only found with one or two O-acetyl groups.
Fig. 3.

Chemical structures of (A) Neu1,5lactam (5,2B conformation), (B) Neu2en5Ac (4H5 conformation), (C) CMP-β-Neu5Ac, (D) Neu2,7an5Ac (5C2 conformation), (E) Neu5Ac1,7lactone (5C2 conformation), and (F) Neu4,8an5Ac (7C4 conformation; two tautomers).
This chapter will review the fascinating story of the isolation and structural elucidation of the sialic acids and their importance as biological molecules in a historical perspective. It will describe how the founders of the sialic acid field since the mid-1930s, over a period of more than 20 years, were struggling with the chemical structure of these molecules in a period in which mass spectrometry and NMR spectroscopy did not exist. And it will show how sialic acid constituents of glycoproteins, glycolipids, oligosaccharides, and polysaccharides have grown to be considered biologically and medically highly essential factors of the daily life on earth.
Although not discussed in biological and medical detail in this chapter, also the expansion of the more recently well-established “sialic acid-like” subclasses of the superfamily of NulOs, namely, the 5,7-diamino-3,5,7,9-tetradeoxynon-2-ulosonic acids, with the mother molecules pseudaminic acid (Pse), legionaminic acid (Leg), 4-epi-legionaminic acid (4eLeg), 8-epi-legionaminic acid (8eLeg), acinetaminic acid (Aci), and 8-epi-acinetaminic acid (8eAci), so far exclusively found in bacterial polysaccharides and glycoproteins, will be subjects of a limited review.
Over the years several books88, 89, 90, 91, 92, 93, 94, 95 and book chapters (e.g., refs. 6 and 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114) dealing with the progress in the fields of sialic acid chemistry, biology, medicine, and food science have been published, as well as a number of reviews in scientific journals (e.g., refs. 7, 23, and 7, 23, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133a, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147) (additional review references are included in the specific sections), but no updated review in a historic perspective about this strongly growing field is available. It is therefore our intention to describe the historical development of the multifaceted sialochemistry/biology field and throw some light also on its present state and future.
2. Paving the Way With Ehrlich's Aldehyde and Bial's Orcinol Reagents
It was in 1877, more than 140 years ago, that the German scientist Hoppe-Seyler (1825–1895) described a substance released from epithelial mucins by mild acid treatment at elevated temperature as an acidic compound, being stronger than carbonic acid. When heated under alkaline conditions, the solution turned brown. The compound reduced cupric oxide or bismuth oxide or indigosulfuric acid, contained nitrogen, and was unfermentable by brewer's yeast. Its purification was a continual problem.148 Similar observations were made by Hoppe-Seyler,149 Green,150 Krukenberg,151 and Zeller,152 when investigating the mild acid-released material from nests of Asian swiftlets (genus Collocalia). For the construction of their nests, these birds use mucin secreted by their salivary glands. The bird's nests are edible and are used in several nutritional and medicinal applications.
Interestingly, Green claimed the formation of crystals of sugar from the released material, a claim that could not be confirmed by Zeller. Citation of Zeller: “Apparently, Green has inorganic material considered as sugar” (translated from the German).
Many years later, collocalia mucin would form an excellent source for the isolation of gram quantities of N-acetylneuraminic acid, simply by boiling pulverized edible bird's nest under reflux for 5 h.153, 154
In 1898, Ehrlich in Germany observed the formation of a purple color after heating mucins with p-dimethylaminobenzaldehyde in acid solution.155, 156 In the same year, Müller in Germany also described the formation of a splendid red color when mucus substances, made alkaline and heated slightly, were incubated with acidic p-dimethylaminobenzaldehyde at higher temperature.157 Although initially the mixture of p-dimethylaminobenzaldehyde in 50% (v/v) HCl, called Ehrlich's reagent, was frequently applied for the detection of indoles, pyrroles, and related nitrogen-containing compounds, this reaction was going to play an important role in the unraveling of the structure of sialic acids. It turned out that incubation of the newly discovered components, in bound and free form, with Ehrlich's reagent before (“direct” Ehrlich) and after (“indirect” Ehrlich) treatment with alkali yielded a red–violet color (see Section 7.1).
Later, in 1927, Walz in Germany and Levine and Landsteiner in the United States reported that on heating isolated lipid material of bovine spleen,158 or equine kidney/bovine kidney/bovine brain,159 respectively, with orcinol in hydrochloric acid containing ferric chloride158 or copper acetate,159 a red–violet color was formed. In fact, the orcinol/concd. HCl/FeCl3 mixture, introduced in 1902 by Bial160 and later called Bial's reagent, was used as a general test for the presence of carbohydrates. A pentose is dehydrated with concd. acid to form furfural, which condenses with orcinol to a product that gives in the presence of ferric ions a bluish-green color. With hexoses, whereby 5-hydroxymethylfurfural is formed, a yellow–brown color is seen. So, the observed red–violet/purple color was quite unusual (see Section 7.1).
2.1. The First Scoutings
The research groups who made the first steps in the discovery and isolation of sialic acids were located in different countries around the world: Gunnar Blix161 (Fig. 4A) and coworkers at the University of Uppsala (Sweden); Ernst Klenk162 (Fig. 4B) and coworkers at the University of Tübingen and the University of Cologne (Germany); and Alfred Gottschalk163, 164 (Fig. 4C) and coworkers at the Walter and Eliza Hall Institute in Melbourne (Australia) and the Max-Planck-Institute for Virology in Tübingen (Germany). Interestingly, materials from completely different biological sources played an important role in their journeys of exploration.
Fig. 4.

(A) Gunnar Blix (1894–1981), (B) Ernst Klenk (1896–1971), (C) Alfred Gottschalk (1894–1973).
Panel (A): Reprinted from Lundblad, A. Gunnar Blix and His Discovery of Sialic Acids—Fascinating Molecules in Glycobiology. Upsala J. Med. Sci.2015, 120, 104–112. With permission of the Board. Panel (B): Photo courtesy of Prof. Dr. Hans-Dieter Klenk. Panel (C): Photo courtesy of one of the authors (R.S.).
In 1936 Blix reported the isolation of a strongly acidic compound from a boiling water solution of bovine submaxillary gland mucin, that he called “Kohlenhydrat I.”165 The compound, generated via autohydrolysis and obtained in crystalline form, had reducing power. Elemental analyses yielded C14H24NO11 as the preferred chemical formula. Microacetyl determinations suggested the presence of two acetyl groups in one molecule, whereas a negative ninhydrin reaction supposed the presence of one N-acetyl group. The product had an mp of 140–150°C (dec.). UV spectroscopy showed the absence of ethylene bonds. The compound turned out to be very sensitive to heating with dilute mineral acid, giving rise to dark humin formation. When heated with Ehrlich's reagent, both with and without alkali pretreatment, an intense red–violet color was formed. Taking into account these and later observations, a disaccharide structure composed of an N-acetylated hexosamine part and a polyhydroxy acid part with six carbon atoms, being not a hexuronic acid, was suggested (note that hexosamine was not isolated from the substance).165, 166, 167 An initial suggestion that the hexosamine part should be chondrosamine (galactosamine)166 was withdrawn when chondrosamine was isolated, separated from “Kohlenhydrat I,” and it was stated that both compounds were “probably bound with an easily split linkage.”167 An additional earlier suggestion was that the polyhydroxy acid could be a deoxy-hexuronic acid.168 Because of the salivary origin of “Kohlenhydrat I” (the Greek name for saliva is “sialon”), Blix called the compound “sialic acid.”167
In further studies, Blix found that the so-called "protagon” lipid fraction of bovine brain tissue gave similar Ehrlich's and Bial's color reactions as found for the mucin-derived compound.168 A few years earlier, a red color with Bial's reagent had already been mentioned by Klenk for lipid material of the human brain,169 and the isolation of a nitrogen-containing organic acid in crystalline form, called “neuraminic acid” (related to the source: neurons), followed in 1941.170 Because of the observed lability in acidic medium, leading to dark humin formation, a methanolic-HCl release protocol was chosen. So, the lipid material was treated with 5% methanolic HCl (3 h, 105°C), and the released substance was converted, via its barium salt, into the free acid {elemental analysis: preferred chemical formula C11H21NO9; [α]20 D −54.9° (c 3.2, H2O)}. A positive ninhydrin reaction indicated a free amino group, whereas reducing power was not detected. The Bial and Ehrlich colorimetric assays were both strongly positive. Protocols directed to the detection of a hexosamine part in “neuraminic acid” were negative, and a suggestion for an aliphatic polyoxy-aminodicarboxylic acid structure was made. When Klenk realized that the isolated substance contained a methoxy group, introduced during the methanolic-HCl treatment (in the 1950s proved with 14C-labeled CH3OH171), he renamed “neuraminic acid” as “methoxyneuraminic acid” and kept the name “neuraminic acid” for the compound with chemical formula C10H19NO9 ( C11H21NO9 − CH2).172 Then, “neuraminic acid” might contain a reactive carbonyl function, which enabled the formation of a glycosidic linkage with other monosaccharides. In the latter study, the term “ganglioside” was introduced for brain glycolipids containing “neuraminic acid.”
Besides scientific interest from the mucin and brain glycolipid research areas for these new, but still unknown compounds, an interesting new angle of incidence came from the virology field. At the end of the 1940s, Burnet reported that the ability of various mucoproteins (i.e., serum mucoprotein, ovarian cyst mucoid, chorionic gonadotropin) to act as competitive inhibitors for hemagglutination by heated influenza B virus was lost after pretreatment of the mucoproteins with active virus.173 Then, Gottschalk and Lind found that incubation of the Melbourne strain of influenza A virus with ovomucin, a protein fraction containing a strong virus hemagglutinin inhibitor, yielded a water-soluble, nitrogen-containing compound with reducing power, that gave, after alkaline treatment, with Ehrlich's reagent, a purple color; furthermore, the Molisch carbohydrate test (concd. H2SO4, α-naphthol) was strongly positive.174 Acid treatment resulted in black insoluble humin formation. Incubation of ovomucin with a Vibrio cholerae filtrate yielded a substance with similar properties.175 In these years, the enzyme-like agent in the filtrates was called “receptor-destroying enzyme” (RDE) (see Section 11.7). Based on the results so far, the enzymatic liberation of a “carbohydrate–peptide complex” was suggested, and it was stated that the carbohydrate moiety should be “an oligosaccharide containing one or more N -acyl (acetyl) hexosamine residues linked by an alkali-labile glycosidic linkage to a non-amino-sugar.” Two years later, Gottschalk repeated the incubation experiments with human urinary mucoprotein176 and influenza B virus and suggested now that the released compound should be an “N-substituted iso glucosamine” (a fructosamine; an N-1-deoxy-1-ketose amino acid or peptide).177 Interestingly, the presence of a keto function was indicated by a positive Seliwanoff resorcinol/concd. HCl test, an assay whereby ketoses, in contrast to aldoses, rapidly give a red color (ketoses are more rapidly dehydrated than aldoses with concd. acid).
In 1975, Blix described in a letter to Klaus Störiko (Behringwerke AG, Marburg, Germany) about his first contact with Gottschalk: “… I recall how I at the first international biochemist congress in Cambridge 1949 talked with Stacey and Gottschalk, when the latter described the general properties of the carbohydrate component in mucins that inhibited virus haemagglutination and asked for advice from Stacey. It struck me immediately that some of the properties Gottschalk mentioned strongly reminded me about those of sialic acid. When I got home I mailed Gottschalk what we had written about sialic acid and told him that we intended to test the matter closer. Gottschalk obviously did not, at this time, know about sialic acid, which outside Cologne and Uppsala still was of low interest. I had on my part limited knowledge about Burnet's investigations. We agreed later on to study in each of our laboratories the carbohydrate component in the well defined Tamm–Horsfall urinary protein and to publish our results in 1952 simultaneously in Nature …” (from the archive of Blix's scientific correspondence, available at Uppsala University Library).161
2.2. The Growing Knowledge About the Substance
At the beginning of the 1950s, in their search for more structural details and to elucidate contrasting results, several overlapping studies were published by the Uppsala, Cologne, and Melbourne groups. Step by step the structural relationship among Blix's “sialic acid,” Klenk's “neuraminic acid”/“methoxyneuraminic acid,” and Gottschalk's “N-substituted isoglucosamine” was growing. Of course, it was also a period of close competition, reflected by several relatively short communications. But, although critical comments on each other's work can be picked up from the literature reports, the groups were also in good contact with each other, exchanging information when necessary.
To start with, Odin (Uppsala group)178 suggested in a paper published in parallel with Gottschalk's paper about the monosaccharide constituents of human urinary mucoprotein179 the close chemical relationship between Gottschalk's “N-substituted isoglucosamine” and Blix's “sialic acid.”
Earlier, the Uppsala group had stated that Klenk's “neuraminic acid” “is probably to be regarded as a degradation product of the native disaccharide compound” (the suggested structure by Blix, composed of an N-acetylated hexosamine part and a polyhydroxy acid part with six carbon atoms).166 However, this conclusion was contradicted by Klenk, saying that, based on further research, “there is no reason to doubt that “neuraminic acid” is a native residue of gangliosides” (translated from the German).180 So, it was not a hexosamine-containing cleavage product.
In further work, the Uppsala group reported the occurrence of “sialic acid” in many biological sources, i.e., egg white, ovomucin, allantoic fluid, ovarian cysts, bovine follicular fluid, cancer fluid, blood serum, plasma glycoproteins, human tears, bovine milk, epithelial mucins, human saliva, meconium, and gangliosides.178, 181, 182, 183, 184, 185, 186
In the same period, the Cologne group prepared “methoxyneuraminic acid” from bovine submaxillary gland mucin and human urinary mucoprotein,187 following their earlier applied protocol170 for gangliosides. In subsequent investigations, heating of an aqueous bovine submaxillary gland mucin preparation (2 h, 110°C) yielded, after isolation, a crystalline material with a chemical formula of C12H21NO10 and [α]20 D −31.7° (c 2.6, H2O) that they called “N -acetylneuraminic acid.”22 The various chemical reactions, carried out earlier on “sialic acid,” gave similar results for “N-acetylneuraminic acid” (Ehrlich and Bial reaction: red color; reducing power: positive; ninhydrin reaction: negative), except that “N-acetylneuraminic acid” contained only one acetyl group. Treatment of “N-acetylneuraminic acid” with 40% NaOH (2 h, 100°C) yielded pyrrole-2-carboxylic acid, a product identified 1 year earlier by Gottschalk188 in an alkaline hydrolysate of bovine submaxillary gland mucin (see Section 3). In additional studies, crystalline “N-acetylneuraminic acid” (C12H21NO10) could be obtained from enzymatic incubations of human urinary mucoprotein with influenza virus B-Lee,189 and of human urinary mucoprotein and bovine submandibular gland mucin with the RDE from V. cholerae.190 It was concluded that both the influenza virus and the RDE are acting as O-glycosidases. Following the methanolyis protocol,170 crystalline “methoxyneuraminic acid” (neuraminic acid methyl glycoside: C11H21NO9) could be isolated from porcine and equine submaxillary gland mucin and bovine colostrum mucin,191 from amyloidose liver,192 from fetuin,193 and from bovine stroma protein.194
The paper of Klenk and Uhlenbruck191 gives a clear insight into the competition between Klenk (“neuraminic acid”) and Blix (“sialic acid”) in the slowly developing field. At the end of a long criticism in a footnote (we write 1956), Klenk ends with “As long as the information provided by Blix can not be confirmed by other sources, it must be doubted that a chemically well-defined, uniform compound has been presented here” (translated from the German).
In these years, besides color reactions, elemental analyses, and optical rotations, IR spectroscopy and X-ray analysis were introduced as helpful techniques in comparing different isolates. As an illustration of the application of IR spectroscopy, in Fig. 5 the IR spectra of enzymatically and chemically released “N-acetylneuraminic acid” are depicted (indications for carboxyl and amide groups).190
Fig. 5.

IR spectra of “N-acetylneuraminic acid,” enzymatically released (V. cholerae) from (1) human urinary mucoprotein, (2) bovine submaxillary gland mucin (crystallization fraction I), and (3) bovine submaxillary gland mucin (crystallization fraction II), and chemically released from (4) bovine submaxillary gland mucin.
Reproduced from Faillard, H. Über die Abspaltung von N-Acetyl-neuraminsäure aus Mucinen durch das “Receptor-Destroying-Enzyme” aus Vibrio cholerae. Hoppe-Seyler’s Z. Physiol. Chem.1957, 307, 62–86/308, 187. Copyright De Gruyter Publishers.
Further studies of the Uppsala group focused on bovine, porcine, ovine, and equine submaxillary gland mucins.23, 195 Crystalline “sialic acids” were generated via hydrolytic protocols (1 h, 100°C; pH 3.5–4.0). An overview of some physical parameters (elemental analysis, optical rotation, decomposition range, type of N- and O-substituents, the number of hydroxyl functions) is presented in Table 2 . The chemical formulas for “methoxyneuraminic acid,” isolated from the mucins and purified “sialic acids” following the methanolic HCl protocol of Klenk, were found to be C10H19NO8 (preferred) or C11H21NO9, being the composition earlier reported by Klenk.170 Both the Blix and Klenk “methoxyneuraminic acid” samples showed the same X-ray powder patterns. Bial's and Ehrlich's reactions gave red–violet and red–purple colors for all four “sialic acids,” respectively. As the “sialic acids” can be reduced with sodium borohydride and subsequent hydrolysis did not produce a reducing substance, whereas the Bial and Ehrlich tests on the reduced compounds were negative, it was stated that, in contrast to earlier conclusions of the Uppsala group,165 this almost excludes the presence of a glycosidic linkage in “sialic acid.” Quantitative determination of hydroxy groups (via an O-acetylation/NaOH titration protocol) showed the presence of five hydroxy groups for the ovine preparation and six hydroxy groups for the porcine preparation. Amino-group determinations revealed the absence of significant amounts of amino nitrogen, so amino groups are substituted. Periodate studies on bovine “sialic acid” showed the consumption of 1 mol of periodate and no formic acid production (CH2OH–CHOH–CHOAc– element), whereas the periodate consumptions for the porcine and ovine “sialic acids” were 2 mol of periodate (CH2OH–CHOH–CHOH– element) and 1 mol of formic acid formation; in all cases 1 mol of formaldehyde was found. As shown in Table 2, the isolated porcine “sialic acid” contained an N-glycolyl group, a result confirmed one year later by the Cologne group. Here, “N -glycolylneuraminic acid” was chemically and enzymatically released from porcine submaxillary gland mucin, and a colorimetric method for the quantitative determination of glycolic acid was developed.64 See also ref. 196. For further discussions on the structure of the presented “sialic acids,” see Section 3. To give an impression of the use of X-ray powder diffraction analysis for comparison purposes, Fig. 6 illustrates the diagrams of various “sialic acids.”
Table 2.
Some Physical Parameters of Crystalline “Sialic Acids” Isolated From Bovine, Porcine, Ovine, and Equine Submaxillary Gland Mucins23
| Mucin Source | Elemental Analysisa | [α]20D (5%, H2O) | Decomposition Range (°C) | N,O-Acyl Groupsb | Hydroxy Groups |
|---|---|---|---|---|---|
| Bovinec | C13H21NO10 | + 8° ± 2° | 134–137 | NAc, OAc | n.a. |
| Ovine | C11H19NO9 | − 31° ± 2° | 185–187 | NAc | 5 |
| Porcine | C11H19NO10 | − 32° ± 2° | 185–187 | NGc | 6 |
| Equinec | C13H21NO10 | − 59° ± 2° | 183–187 | NAc, OAc | n.a. |
n.a., not available.
Note that in earlier reports it was not realized that during crystallization protocols, wherein methanol was used, partial methyl ester formation took place (all preparations contained one COOH function). The included chemical formulae from this study turned out to be in the end correct.
NAc = N-acetyl; OAc = O-acetyl; NGc = N-glycolyl. Acetyl determination as barium acetate after acid hydrolysis; glycolyl determination as crystalline calcium glycolate after alkaline treatment (see Section 7.1).
The bovine and equine “sialic acids” were suggested to be isomers.
Fig. 6.

X-ray powder diagrams of “sialic acid” isolated from submaxillary gland mucins [(A) bovine, (B) ovine, (C) porcine, (D) equine] and “methoxyneuraminic acid” (E). Guinier camera. Nickel-filtered copper K-radiation.
Reproduced from Blix, G.; Lindberg, E.; Odin, L.; Werner, I. Studies on Sialic Acids. Acta Soc. Med. Upsalien. 1956, 61, 1–25. With permission of the Board.
2.3. The Expanding Network of Researchers
Parallel to the research activities of the Uppsala, Cologne, and Melbourne groups, in the 1950s other reports with information about the existence of such Bial's and Ehrlich's reagent-positive products also appeared. In this context, among others, the groups of Tamio Yamakawa197 and coworkers at the University of Tokyo (Japan), Richard Kuhn198 and coworkers at the Max-Planck-Institute in Heidelberg (Germany), and Paul György199 and coworkers at the University of Pennsylvania in Philadelphia (USA) should be mentioned.
In 1951, Yamakawa and Suzuki reported the isolation of an unknown crystalline nitrogen-containing polyhydroxy-carboxylic acid (elemental analysis: C10H19NO8) from a methanolysate (8% methanolic HCl, 3 h, 100°C; work-up via a barium salt and treatment with diluted H2SO4) of an equine blood stroma glycolipid. The glycolipid was called “hematoside” and the released compound “hemataminic acid.”200 Both substances gave a red color with Bial's and Ehrlich's reagents. The ninhydrin test for “hemataminic acid” was positive only in the presence of a trace of alkali. After acid hydrolysis the reducing power changed from negative to positive. As “hemataminic acid” ([α]20 D −54.2°) was suspected to be a methyl glycoside (and suggested to correspond with Klenk's “methoxyneuraminic acid,” [α]20 D −54.9°,170 the hypothetical free compound should have the chemical formula C9H17NO8; it was called “prehemataminic acid,” described as a 2-amino-2-deoxy-nonuronic acid (corresponding with Klenk's “neuraminic acid”) (for a proposal of the structure, see Section 3).201
Here, a citation of Yamakawa and Suzuki201 illustrates the initial controversy with respect to “neuraminic acid” vs “hemataminic acid”: “According to a private communication from Dr. Klenk, which we received some days ago, he found chondrosamine in his preparation of ganglioside, and maintained that his ‘neuraminic acid’ gave, contrary to our ‘hemataminic acid’, no reducing hydrolysate after cleavage with hydrochloric acid. For this reason, he was doubtful of the identity of these two substances.” However, in the same year, after having analyzed the same material, Klenk withdrew his doubts by saying: “The “prehemataminic acid” occurring therein as a characteristic building block is identical with “neuraminic acid”” (translated from the German).202
From a hydrolysate of equine serum mucoprotein (pH 1–2, 1 h, 80°C), a similar crystalline compound could be isolated that was called “sero-lactaminic acid.”203
In another investigation by Kuhn et al., acid hydrolysis (pH 1, 40 min, 70–80°C) of a mucoprotein fraction of bovine colostrum afforded, after work-up (use of methanol), a low-molecular-mass ninhydrin-negative product, that they called the methoxy derivative of “lactaminic acid.”204 The crystalline material had similar Ehrlich and Bial colorimetric properties as reported for “sialic acid” and “methoxyneuraminic acid.” However, the paper chromatographic R f values of the methoxy derivative of “lactaminic acid” in two solvent systems were different from that of “methoxyneuraminic acid.” In a subsequent paper, the notation methoxy derivative of “lactaminic acid” was reformulated as “lactaminic acid methyl ester” (de-esterification with alkali).205 Furthermore, “lactaminic acid” (C11H19NO9; mp 183–185°C with decomposition; [α]21 D −31.0°, c 1, H2O)206 was established to be identical with Blix's ovine “sialic acid.”23 The same conclusion was drawn by Faillard for “lactaminic acid” in relation to N-acetylneuraminic acid.190 In further screening studies for low-molecular-mass compounds in bovine milk, human milk, and bovine, human, ovine, caprine, and porcine colostrum, thereby demonstrating the presence of Ehrlich- and Bial-positive materials, the Heidelberg group isolated an oligosaccharide from bovine colostrum, which turned out to be O-acetyl-lactaminic acid-lactose.205, 207
György and coworkers reported that nondialyzable preparations of human milk afforded red colors with Bial's and Ehrlich's reagents,208 and similarities with “sialic acid” and “neuraminic acid” were suggested. The methanolic-HCl protocol was followed to prepare crystalline “methoxyneuraminic acid” {[α]20 D −49.5° (c 3.3, H2O); elemental analysis: C11H21NO9}. In additional reports, a milk oligosaccharide fraction was hydrolyzed under mild conditions (pH 1, 2 h, 80°C), and subsequent work-up, including ion-exchange chromatography, yielded an anhydrous product in crystalline form (no use of methanol), that was called “gynaminic acid,” a name that referred to human milk.209, 210 The final substance210 was published with the following analytical parameters: elemental analysis, C11H19NO9; [α]25 D −32.1° (c 2, H2O); mp 176–177°C (dec.); positive with Bial's and Ehrlich's reagents; negative with ninhydrin reagent; presence of acetyl group; absence of methoxy group; consumption of 2 mol of periodate; four acetylizable hydroxy groups. In a similar way, “gynaminic acid” was isolated from human meconium. The X-ray powder diagrams of “gynaminic acid” were identical with that of “sialic acid,” isolated by the Uppsala group from ovine submaxillary gland mucin.
Other interesting studies from the 1950s by other authors focused on the detection of sialic acids in different biological materials. Those that should be mentioned are those on pooled and fractionated human serum proteins, transudates, urine, cerebrospinal fluid, milk, saliva, and a large variety of animal serum proteins,211, 212 and those on the isolation of neuramin-lactose from rat mammary glands.213, 214
3. The First Creation of Chemical Structures
The first creations of a chemical structure were directly influenced by the finding that the Ehrlich reaction should be based on the formation of pyrrole-2-carboxylic acid as reactant for p-dimethylaminobenzaldehyde to give the specific red–violet color.
Already in 1949, Hiyama published a first proposal.215 Bovine sublingual gland mucin was treated with 10% KOH for 10 h at 100°C. After neutralization and work-up, a product was obtained that gave a red color with p-dimethylaminobenzaldehyde (absorption max. near 550 nm), and was identified as pyrrole-2-carboxylic acid by comparison with its synthetic analogue. Subjecting Blix's “Kohlenhydrat I”165 (see Section 2.1), isolated from bovine submaxillary gland mucin, to the same alkaline treatment, also afforded pyrrole-2-carboxylic acid. Based on these results, and taking into account Blix's suggestion that “Kohlenhydrat I” should contain an acetylated hexosamine part (one N- and one O-acetyl group), a completely substituted pyrrolidine ring with a d-glucosamine part incorporated in its structure (C13H21NO10) was tentatively proposed (Scheme 1 ). The reduction of hot alkaline copper solution by “Kohlenhydrat I” was ascribed to the presence of d-erythrose.
Scheme 1.

Proposed structure of “Kohlenhydrat I” with the formation of pyrrole-2-carboxylic acid (and d-erythrose), according to Hiyama, taking into account Blix's findings.165, 215
A drawing of a probable structure for the reagent-positive “hemataminic acid” of Ehrlich and Bial was reported in 1952 by Yamakawa and Suzuki (Fig. 7 ).201 As mentioned in Section 2.3, “hemataminic acid” was isolated from hematoside via the methanolysis protocol and suggested to be identical to Klenk's “methoxyneuraminic acid.” The assumed structure was based on a series of observations, e.g.: (i) the gasometric α-amino-carboxyl determination with ninhydrin, used to detect α-amino acids,216 did not liberate CO2; (ii) reduction of alkaline ferricyanide or Fehling's reagent after heating with HCl: presence of an aldehyde group; (iii) presence of one methoxy group and a free amino group adjacent to a hydroxy group; (iv) absence of an acetyl group; and (v) consumption of 3 mol of periodate with concomitant formation of 1 mol each of formic acid and glyoxylic acid (CHO–COOH) which was an indication for an α,β-dihydroxy-carboxylic acid.
Fig. 7.
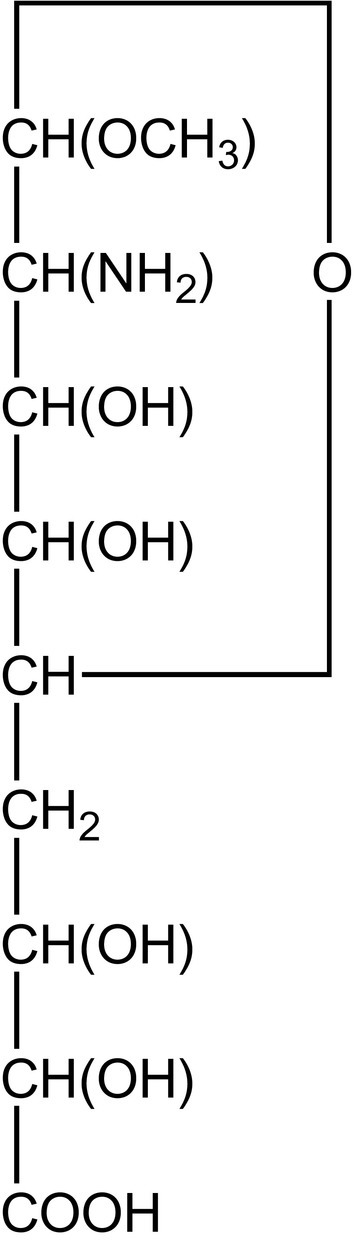
Structure proposal of Yamakawa and Suzuki for “hemataminic acid” (C10H19NO8), a methoxy derivative of a 2-amino-2-deoxy-nonuronic acid.201
In 1953, just like Hiyama, Gottschalk also reported that the alkaline hydrolysate of bovine submaxillary gland mucin was shown to contain pyrrole-2-carboxylic acid [comparison with synthetic pyrrole-2-carboxylic acid; formation of dl-proline (pyrrolidine-2-carboxylic acid) on reduction with PtO2/H2].188 Based on the different findings,174, 177, 188 it was hypothesized that the substance released with the influenza viral enzyme (Section 2.1) is built up from a pyrrole-2-carboxylic acid, whereby the pyrrole nitrogen is glycosidically linked to an undefined sugar residue (structure A) (Fig. 8 ).188, 217 In bound form, the N-glycosyl-pyrrole-2-carboxylic acid A was assumed to be connected via an amide linkage with the nitrogen of an adjacent hexosamine (structure B), probably d-chondrosamine (d-galactosamine). When incubated with the influenza viral/V. cholerae enzymes, and accepting these are amidases, the amide linkage is cleaved, thereby releasing structure A. Both nitrogen linkages of structure B were expected to be alkali labile, thereby explaining the formation of pyrrole-2-carboxylic acid from the mucoprotein in the Ehrlich's test with p-dimethylaminobenzaldehyde.
Fig. 8.

Gottschalk's N-glycosyl-pyrrole-2-carboxylic acid proposal (A, free form; B, bound form) for the structure of the enzymatically released substance from mucoprotein.188, 217
However, the latter hypothesis was withdrawn in a publication that followed.218 As UV absorption spectra of the untreated mucoprotein did not give indications for the presence of a pyrrole derivative (absence of a peak at 256 nm), it was suggested that pyrrole-2-carboxylic acid was formed by alkaline degradation (0.05 M Na2CO3, 20 min, 100°C) of the enzymatically or chemically released substance from the mucoprotein, in a way as visualized in Scheme 2A. The precursor of pyrrole-2-carboxylic acid was supposed to be pyrroline-4-hydroxy-2-carboxylic acid, stabilized by the engagement of its enolic hydroxy group in an O-glycosidic linkage apparently with a pentose, being the released substance. The latter compound is very sensitive to alkaline hydrolysis. The choice of position 4 for the hydroxy group was based on the biogenetic relationship to hydroxyproline. In a subsequent paper from 1955, however, an isomeric structure for the O-glycosyl-pyrrole-2-carboxylic was assumed (Scheme 2B).219 In aqueous solution, such a structure will undergo a reversible ring opening, yielding a product that would be unstable to alkali (or acid), and gives, under the expulsion of the sugar, pyrrole-2-carboxylic acid.
From a citation of Gottschalk218 discussing the earlier report of Hiyama215 about the formation of pyrrole-2-carboxylic acid (see Scheme 1): “It is not known whether pyrrolidines are transformed into pyrroles under the conditions applied, and Hiyama's results cannot therefore be interpreted.”
Scheme 2.

Gottschalk's O-glycosyl-pyrrole-2-carboxylic acid proposals for the structure of the (bio)chemically released substance from mucoprotein with the formation of pyrrole-2-carboxylic acid.218, 219
Then, inspired by the data of the Uppsala and Cologne groups, Gottschalk reinterpreted his earlier structural data218, 219 and formulated a new correlation among his finding of pyrrole-2-carboxylic acid, the bovine “sialic acid” data (Section 2), and the “methoxyneuraminic acid”/“neuraminic acid” data (Section 2). Taking into account the revised chemical formula C13H21NO10·H2O for bovine “sialic acid,”195 with one N- and one O-acetyl group, C9-sugar structures were constructed as depicted in Scheme 3 .220 In this proposal, “neuraminic acid” c is presented as an aldol type of condensation product of a 2-amino-2-deoxy-hexose with pyruvic acid. For the cyclization to pyrrole-2-carboxylic acid d, the α-keto function, the δ-amino function, and the γ-hydroxyl function of the open deacetylated chain (a → c) are in the right positions relative to the carboxyl group. Blix and coworkers stated that the supposed pyranose ring form is in accordance with their periodate experiments, carried out on bovine, porcine, ovine, and equine “sialic acids.”23 Gottschalk's proposal also included for terminal “N-acetylneuraminic acid” an enzymatic release from mucoproteins via a glycosidase instead of an amidase.97, 221 See also ref. 190. A first unraveling of the mechanism behind the enzymatic release had to wait until the 1980s.
The citation of Blix and coworkers (1956) in view of their finding that the O-acetyl group should be on C7 instead of C4 (Scheme 3): “The bovine ‘sialic acid’ consumes only 1 mole of oxidant per mole, and produces no formic acid. This indicates that the C7 group is acetylated in this substance. The position of the O-acetyl group in Gottschalk's formula is therefore probably wrong.”23 The structure was corrected in 1957.222
Scheme 3.

Reaction scheme to explain Gottschalk's proposal for the structures of “sialic acid” (a and b) (C13H21NO10), “neuraminic acid” (c), and “methoxyneuraminic acid” (f, C10H19NO8), including the formation of pyrrole-2-carboxylic acid (d) and the tetrose (e), and a possible intermediate g.220, 222
Experiments, whereby d-glucosamine and pyruvic acid were coupled in alkaline medium (pH 11) for 20 min at 100°C via an aldol condensation, and pyrrole-2-carboxylic acid was formed in trace amounts, formed a further support for this correlation.220, 222 Also a possible other degradation route a → g → d (g is an unstable 4-hydroxypyrroline derivative) has been included in Scheme 3.222 Although the positive aldol condensation with d-glucosamine also fixed in fact the stereochemistry around C5, C6, C7, and C8, these have been left open in Scheme 3 and will be further discussed in Section 4.1. Note that “sialic acid” (a), but not “methoxyneuraminic acid” (f), can be converted into pyrrole-2-carboxylic acid.
A different proposal for the structure of “methoxyneuraminic acid” appeared in 1956 by Klenk and coworkers.171 Based on the earlier investigations on “methoxyneuraminic acid,” now called “neuraminic acid methyl glycoside,” the following observations were taken together22, 170, 172, 201, 208 (see also Section 2): (i) presence of a carboxyl group; (ii) presence of a primary amino group, but not in an α-position to the carboxyl group; (iii) presence of a methoxyl group; (iv) absence of an acetyl group, an acetamido group, or a reducing group; (v) consumption of 3 mol of periodate, and formation of 1 mol of formic acid and 1 mol of formaldehyde in mild periodate oxidation experiments; (vi) IR support for a carboxyl group; (vii) for “N-acetylneuraminic acid” it holds that the acetamido group should be situated next to the glycosidic C-atom. Further, using a plethora of chemical reactions, the authors arrived finally at a structure for “methoxyneuraminic acid” (C11H21NO9), as formulated in Fig. 9 .
Fig. 9.
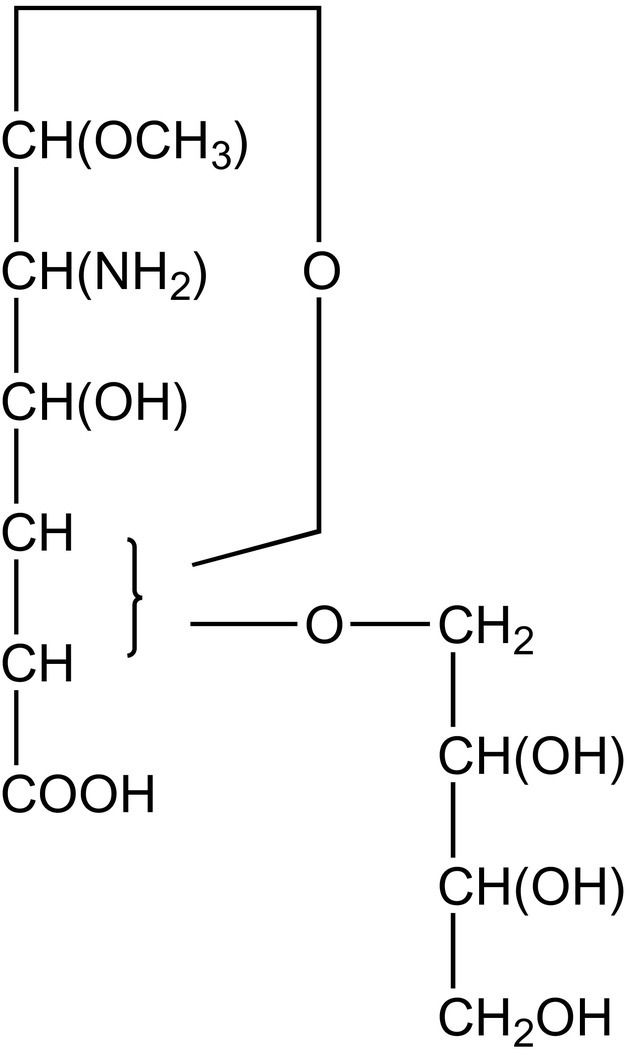
Structure of “methoxyneuraminic acid” according to Klenk and coworkers; C11H21NO9 (mw 311) was the best fit with the elemental analysis.171 Note that the ultimately correct chemical formula, C10H19NO8 (mw 281), was pushed aside. Interestingly, molecular weight determination, via cryoscopy in water, yielded 273 and 276 Da.
Taking together all structural data so far available, Gottschalk's proposal of a C9-sugar220, 222 (Fig. 10 , Starting Structure) turned out to be the right direction to continue the battle for the chiralities of the carbon atoms in the final structure. The problems with the great variations in elemental analyses (including separate nitrogen determinations) on different preparations of the isolated substances, published over a long period of time, certainly delayed the progress in the field. One of the causes for these variations was due to the frequent use of methanol in work-up procedures, leading to contaminating methyl-esterification of the relatively strong acids. Another complication was mentioned to be the hygroscopicity of the substances. But also the possible presence of closely related family members in specific preparations should not be excluded.
Fig. 10.

Survey of the search for the stereochemistry around C4, C5, C6, C7, and C8 of N-acetylneuraminic acid in a historical perspective: a path of trial and error (see Section 4).
In view of all the foregoing, after more than 20 years of activity in many laboratories, Blix, Gottschalk, and Klenk finally arrived, in 1957, at a joint statement on the chemical relationship of the substances designated over the years by various names, such as “sialic acid,” “neuraminic acid,” “methoxyneuraminic acid,” “lactaminic acid,” “sero-lactaminic acid,” “gynaminic acid,” “hemataminic acid,” and “prehemataminic acid.” It was formulated as follows: “In order to avoid further confusion, we propose to call the basic, unsubstituted compound neuraminic acid. Sialic acid is suggested as group name for the acylated neuraminic acids (for example, N-acetylneuraminic acid, N-glycolylneuraminic acid, and diacetylneuraminic acids). For the enzyme, which splits the glycosidic linkage joining the terminal sialic acid to the residual oligo- or polysaccharide, the names neuraminidase and sialidase may be used synonymously. We have agreed to use this nomenclature in future.”223 Looking back, one can say that this joint statement was really a breakthrough after many years of experiments and competition.
Gottschalk wrote in a letter to Klenk (in 1957) about the final phase of the Nature publication: “… I have just returned the corrected proofs of our joint note to Nature, ordered and paid 250 reprints in all and asked the printers to send all reprints to you. I am sorry that I could not consult you with regard to the reprints; however, I had to deal with the proof by return mail and the reprints have to be ordered when returning the proof. I have informed Blix accordingly and asked him to claim reprints (up to 85) from you. I hope you agree with this arrangement …” (from the archive of Ernst Klenk, held by his son Hans-Dieter Klenk).
Fifteen years later, Scott, Yamashina, and Jeanloz proposed to change the terminology by replacing the name neuraminic acid by aminosialosonic acid (the reference sugar should be a hypothetical 3-deoxynonulose, called sialose and abbreviated Sia), which means that N-acetylneuraminic acid should be named acetylaminosialosonic acid.224 But the proposal was rejected by the IUPAC-IUB Commission on Biochemical Nomenclature.225 Only the abbreviation Sia survived, but it is now used as an abbreviation for sialic acids in general.
4. The Battle for the Stereochemistry
4.1. The C5, C6, C7, and C8 Chiralities
As mentioned in Section 3, the sialic acid formula (C5–C8 chiralities) of Gottschalk was partly based on an aldol condensation product of d-glucosamine and pyruvic acid, yielding pyrrole-2-carboxylic acid in alkaline medium (Fig. 10, Structure I).220, 222 Support for such a structure was supplied by studies of Kuhn and Brossmer.206 Heating (90 min, 100°C) of “lactaminic acid” (N-acetylneuraminic acid) with Ni(OAc)2·4H2O in pyridine yielded N-acetyl-d-glucosamine (d-GlcNAc), which afforded the chiralities for C5, C6, C7, and C8 (Fig. 10, Structure I), and carbon dioxide [–CH(OH)–CH2–CO–COOH → –CH(OH)–CH2–CHO + CO2]. Because of the anomeric C2 atom, “lactaminic acid” should occur in α- and β-forms; therefore, it was surprising that no mutarotation was observed (see Section 4.3). Also Zilliken and Glick demonstrated that incubation of “gynaminic acid” (N-acetylneuraminic acid) with 0.1 M NaOH (5–10 min at 90°C) afforded d-GlcNAc and pyruvic acid.226 In another study, incubation of bovine submaxillary gland mucin with a V. cholerae filtrate, containing the RDE, yielded besides sialic acid, also d-GlcNAc, leading to the suggestion that still another enzyme should be present in the extract.227 Additional incubation of sialic acid with the same extract gave equimolar amounts of d-GlcNAc and pyruvic acid, and an aldolase mechanism for this splitting was suggested (see Section 12). Finally, N-acetylneuraminic acid could be synthesized from d-GlcNAc and oxaloacetic acid at pH 11 in 1%–2% yield (Scheme 4 ).228
Scheme 4.

Synthesis of N-acetylneuraminic acid (Structure I; Fig. 10) from N-acetyl-d-glucosamine and oxaloacetic acid.228
In the same period, however, another player in the field, i.e., the group of Saul Roseman229, 230 (Fig. 11 ) and coworkers at the University of Michigan, Ann Arbor (USA) and later at the Johns Hopkins University, Baltimore (USA), reported enzymatic experiments that were not in agreement with the assigned chirality around C5, as discussed earlier.231, 232 Incubation of N-acetyl- and N-glycolylneuraminic acid with an enzyme purified from Clostridium perfringens (pH 7.2, 3 h, 37°C) yielded in an aldol-type cleavage an N-acyl-hexosamine and pyruvic acid. In the case of N-acetylneuraminic acid, the N-acyl-hexosamine was identified as N-acetyl-d-mannosamine (d-ManNAc), not d-GlcNAc (Fig. 10, Structure II). Note that the d-configuration was established by a chemical degradation reaction of mannosamine, yielding d-arabinose. Note also that d-GlcNAc and d-ManNAc could only be separated by paper chromatography, when using borate-treated paper. The reaction was reversible; thus incubation of d-ManNAc and pyruvate using this enzyme led to the formation of N-acetylneuraminic acid (Scheme 5 ). The enzyme was inactive with d-GlcNAc and N-acetyl-d-galactosamine (d-GalNAc). Phosphoenolpyruvate could not replace pyruvate. In view of its activity, the enzyme was called N-acetylneuraminic acid aldolase [later called sialate (acylneuraminate)-pyruvate lyase; see Section 12]. Incubation of the enzyme with N-glycolyl-d-mannosamine and pyruvic acid afforded N-glycolylneuraminic acid.
Fig. 11.

Saul Roseman (1921–2011).
Photo courtesy of Prof. Dr. Ronald Schnaar.
Scheme 5.

N-Acetylneuraminic acid aldolase-catalyzed cleavage and synthesis of N-acetylneuraminic acid (Structure II; Fig. 10) with N-acetyl-d-mannosamine and pyruvic acid as counter compounds.231, 232
Pushed by the results of Comb and Roseman, Kuhn and Brossmer repeated the degradation of N-acetylneuraminic acid by Ni(OAc)2·4H2O in hot pyridine and concluded that the primary formation of d-ManNAc was followed by a rapid epimerization to d-GlcNAc.233 Besides the unexpected epimerization in hot pyridine + Ni(OAc)2 (an epimerization was hardly seen in hot pyridine alone), of which the balance lies strongly on the side of d-GlcNAc, a paper chromatography system used earlier by these authors was not able to separate d-ManNAc and d-GlcNAc. However, using borate-treated paper in a time-course experiment, the initial formation of d-ManNAc, next to d-GlcNAc, was clearly seen. The d-ManNAc ↔ d-GlcNAc reversible epimerization (ratio 1:4) was also observed in aqueous alkaline solution (pH 10–11) at room temperature,233, 234, 235 thereby explaining the earlier results of Zilliken and Glick226 and of Cornforth and coworkers.228 Repetition of the synthesis of N-acetylneuraminic acid from oxaloacetic acid and either d-GlcNAc or d-ManNAc confirmed this view. In both cases N-acetylneuraminic acid was formed (Scheme 6 ; Fig. 10, Structure II).235, 236 Replacing oxaloacetic acid by its di-tert-butyl ester gave higher yields.237 In a repeated study, incubating N-acetylneuraminic acid with a V. cholerae filtrate (pH 6.1, 70 h, 37°C), d-ManNAc, but not d-GlcNAc, was found.238 The earlier observation of splitting into d-GlcNAc and pyruvate227 was also attributable to a paper chromatography protocol that could not separate d-GlcNAc and d-ManNAc.238 In another report, the system d-ManNAc/phosphoenolpyruvate/ATP/rat liver extract (not d-GlcNAc or d-GalNAc; not pyruvic acid) was described for the synthesis of N-acetylneuraminic acid.239 Finally, several years later a protocol was described to determine the absolute configuration of N-acetylneuraminic acid by converting it into d-arabinose by chemical degradation.240
Scheme 6.

Synthesis of N-acetylneuraminic acid (Structure II; Fig. 10) from d-GlcNAc or d-ManNAc and oxaloacetic acid.235, 236
4.2. The C4 Chirality
Also the estimation of the stereochemistry around C4 required a few years, whereby the Heidelberg group of Kuhn and Brossmer played a major role. Reaction of N-acetylneuraminic acid with C2H5SH/concd. HCl yielded the γ-lactone of N-acetylneuraminic acid diethyl dithioacetal. With reference to Hudson's lactone rules,241 the strong levorotary value of the γ-lactone {[α]23 D −83° (c 1, CH3OH)} was interpreted as belonging to a Fischer projection formula structure with the lactone ring drawn to the left. This meant for N-acetylneuraminic acid that the OH group at C4 should project to the left (Fig. 10, Structure III; Scheme 7 ).242, 243 Using Raney nickel, the diethyl dithioacetal could easily be converted into the corresponding levorotating desulfurized γ-lactone. Oxidation of the desulfurized γ-lactone with HNO3 at 120°C yielded succinic acid.243
Scheme 7.

Chemical reactions to search for the chirality of the C4 atom of N-acetylneuraminic acid: The OH group at C4 should project to the left in the Fischer projection formula (Structure III; Fig. 10).243
However, additional experiments along three chemical pathways by Kuhn and Brossmer led to a correction of the stereochemistry around C4 (Scheme 8 ).244 First of all, the desulfurized γ-lactone mentioned earlier could be converted into (−)-(R)-1,4,5-pentanetriol {[α]20 D −18° (pyridine)} (route 1). The same pentanetriol could also be synthesized from (+)-l-glutamic acid via (−)-l-α-hydroxyglutaric acid (route 2), and from the Wittig condensation product of (+)-2,3-O-isopropylidene-d-glyceraldehyde and bromoacetic acid ethyl ester (route 3). So, in the Fischer projection formula the OH group at C4 should project to the right instead of to the left (Fig. 10, Structure IV).
Scheme 8.

Three routes to prove the correct chirality around C4 of N-acetylneuraminic acid: The OH group at C4 should project to the right in the Fischer projection formula (Structure IV; Fig. 10).244
A fourth chemical route, described by Kuhn and Baschang237 also led to an OH group at C4 projected to the right (Scheme 9 ). Here, ozonolysis of the generated γ-lactone of N-acetylneuraminic acid245 and reductive work-up afforded oxalic acid and 3-acetamido-3-deoxy-d-glycero-d-galacto-heptose, and not 3-acetamido-3-deoxy-d-glycero-d-talo-heptose.
Scheme 9.

Synthesis of 3-acetamido-3-deoxy-d-glycero-d-galacto-heptose from the γ-lactone of N-acetylneuraminic acid (Structure IV; Fig. 10).245
4.3. The C2 Chirality and the Ring Form/Conformation
From the time that a C9 structure with a keto group on C2 and an acetamido group at C5 was generally accepted for sialic acid, a pyranose ring form between C2 and C6 was drawn, although still in a Fischer projection formula. A furanose ring form was not possible, and a seven-membered ring was not obvious. As mentioned in Section 3, results from periodate oxidation studies were in agreement with such a pyranose ring form.23 Further support for a C2–C6 hemiacetal was supplied by detailed inspection of the IR spectra of Blix's bovine, porcine, ovine, and equine sialic acids.246
Studies focused on the α,β-anomericity at C2 of sialic acids started with a more detailed characterization of lactaminic acid-lactose (N-acetylneuraminyl-lactose) (Section 2.3) by the Heidelberg group. Isolated crystalline N-acetylneuraminic acid showed an increasing mutarotation in (CH3)2SO as follows: [α]23 D −115° (t = 7 min) → −37° (t = 23 min) → −24° (final value) (c 0.2) (extrapolated to t = 0 min, [α]23 D should be −150° to −200°). This observation was correlated with the β-form for the crystalline product, which means that the OH group at C2 in the Fischer formula should project to the left.242 As in H2O no change in mutarotation was observed, it was concluded that in this solvent the mutarotation was too fast (strong acid) to be followed in the polarimeter. Note that in other studies no mutarotation could be observed in (CH3)2SO,247 whereas in H2O mutarotation was seen.248 Applying Hudson's isorotation rules,249 comparison of the optical rotations in (CH3)2SO of (N-acetylneuraminic acid)-(α,β-lactose) ([α]23 D +6°), α,β-lactose ([α]23 D +53°), and assumed β-N-acetylneuraminic acid ([α]23 D −150° to −200°; see above), it was hypothesized that the bound N-acetylneuraminic acid occurred in the α-form. In case of a β-glycosidic linkage, a negative optical rotation was expected for (N-acetylneuraminic acid)-(α,β-lactose).250, 251 But it was stated that for further support pairs of epimeric α- and β-ketosides should be synthesized. Note that the Fischer projection formulae used in these reports (chiralities around C4 and C5) were still not the final ones.
Indeed, in the following years large series of glycosides were synthesized to study the α,β-anomericity at C2 (for a review, see ref. 252), and two examples will be given. Meindl and Tuppy253 prepared a series of alkyl and aryl glycosides of N-acetylneuraminic acid, starting from N-acetylneuraminic acid via 2,4,7,8,9-penta-O-acetyl-N-acetylneuraminic acid and 4,7,8,9-tetra-O-acetyl-N-acetyl-2-chloro-2-deoxy-neuraminic acid as intermediates (Koenigs–Knorr protocol). It turned out that all glycosides could be cleaved with V. cholerae and influenza virus sialidases, and based on the earlier studies of Kuhn and Brossmer, the compounds were assigned as α-ketosides.253, 254 Kuhn and coworkers converted N-acetylneuraminic acid methyl ester via 2,4,7,8,9-penta-O-acetyl-N-acetylneuraminic acid methyl ester and 4,7,8,9-tetra-O-acetyl-N-acetyl-2-chloro-2-deoxy-neuraminic acid methyl ester as intermediates (Koenigs–Knorr protocol) into N-acetylneuraminic acid methyl ester methyl glycoside with [α]20 D −6° (CH3OH), and N-acetylneuraminic acid, using a CH3OH/Dowex-50 [H+] (24 h, boiling under reflux) protocol, into N-acetylneuraminic acid methyl ester methyl glycoside with [α]20 D −46° (CH3OH).255 As the first preparation was less levorotatory than the second preparation, the first one was hypothesized to be the α-anomer, and the second one the β-anomer (Hudson's isorotation rules249). The first preparation and its free acid form were susceptible to V. cholerae sialidase, while the second preparation and its free acid form were resistant.
The application of 100-MHz 1H NMR (nuclear magnetic resonance) spectroscopy (1968) gave conclusive evidence for the occurrence of pyranose ring forms in 2 C 5 (1C) chair conformations.256, 257 Taking the per-O-methylated methyl ester benzyl glycosides α-3 and β-3 as examples, 1H NMR analysis in CDCl3 showed axial positions for H3a, H4, H5, and H6 (J 3a,4, J 4,5, J 5,6 >10 Hz) and an equatorial position for H3e (J 3e,4 ~5 Hz), so equatorial positions for OR2, NHAc, and the glycerol side chain at C4, C5, and C6, respectively (Fig. 12 ). Although not discussed,257 the large J coupling constant between H7 and H8 (J 7,8 ~8 Hz) in the per-O-acetylated methyl ester benzyl glycoside α-2 is in agreement with a staggered anti orientation of the OR2 groups at C7 and C8; the J-value for the coupling between H6 and H7 is small (~2 Hz). Finally, in the 1H NMR spectrum of methyl ester 1, recorded in dimethyl sulfoxide-d 6:benzene-d 6 = 8:3, a long-range coupling between H3a and HO2 (J 2.1 Hz) indicated that HO2 occurs in the axial orientation, in agreement with a β-configuration (H3a–C–C–HO2 in a W conformation). For a further discussion on the conformation of the glycerol side chain, see Section 6.
Fig. 12.

Conclusive evidence for the 2C5 chair conformation of the pyranose ring, and the anomeric configuration assignment, of N-acetylneuraminic acid methyl ester (derivatives), based on 100-MHz 1H NMR analysis.257
With guidance from the observed optical rotation measurements, combined with Hudson's isorotation rules, several studies suggested the α-configuration for bound N-acetylneuraminic acid. However, there was a general feeling of caution, given the initially wrong configurational assignment of the hydroxy group at C4 using Hudson's lactone rule.
Conclusive data about the C2 α,β-anomericity were published in 1969 by Yu and Ledeen.258 For this study, use was made of the earlier synthesized255 methyl esters of the sialidase-sensitive and sialidase-resistant forms of N-acetylneuraminic acid methyl glycoside. Both compounds were subjected to mild periodate oxidation followed by sodium borohydride reduction, and the degradation products that formed were treated with N,N′-dicyclohexylcarbodiimide in pyridine (Scheme 10 ). The C7 product related to the sialidase-sensitive methyl glycoside did not give rise to lactone formation, whereas the C7 product related to the sialidase-resistant methyl glycoside gave rise to lactone formation. IR analysis of the isolated lactone fraction [preparative thin-layer chromatography (TLC)] revealed evidence for a δ (major) + γ (minor) lactone mixture. The trimethylsilyl derivative of the major lactone, isolated via gas–liquid chromatography (GLC), was analyzed by electron-impact mass spectrometry (EIMS), and shown to be a δ-lactone. Boat conformations of the lactones were supported by Dreiding modeling. All these findings were in agreement with an axial orientation of the carboxyl function (α-configuration) in the case of the sialidase-susceptible methyl glycoside and with an equatorial orientation of the carboxyl function (β-configuration) in the case of the sialidase-resistant methyl glycoside. So, sialidases (here V. cholerae sialidase) were proven to be α-sialidases (see Section 11.7). Note that also the glycerol-side-chain-trimmed α-anomer was susceptible to sialidase, but the β-analogue not susceptible. Note also that N-acetyl-α-neuraminic acid methyl ester methyl glycoside was totally inactive with sialidase, which is contrary to results published three years earlier.255
Scheme 10.

Elucidation of the stereochemistry around the anomeric carbon atom via a search for lactone formation, by subjecting the two anomeric N-acetylneuraminic acid methyl ester methyl glycosides to sequential periodate degradation, borohydride reduction, and treatment with N,N′-dicyclohexylcarbodiimide (DCC).258 The two starting compounds differ, after de-esterification, in their susceptibility to sialidase.
4.4. The Replacement of NH2-C5 by HO-C5
The history with respect to deaminated neuraminic acid (ketodeoxynononic acid, Kdn; Fig. 1) started in 1986.12 In that year, the groups of Sadako and Yasuo Inoue et al. at the Showa University and the University of Tokyo, Tokyo (Japan) reported an unknown sialic acid-like component (SiaX) in the monosaccharide GLC analysis of some glycopeptide fractions prepared from Salmo gairdneri (rainbow trout) egg polysialoglycoprotein.259 In subsequent studies,76 SiaX could be isolated as its methyl ester methyl glycoside. In comparison with permethylated N-glycolylneuraminic acid, the EIMS fragmentation pattern of permethylated SiaX supported a structure in which the N(CH3)COCH2OCH3 group at C5 was replaced by an OCH3 group (for EIMS data of sialic acids, see Section 5). Further support for a hydroxy group instead of an N-acyl group at C5 was supplied by the 1H NMR spectrum of the SiaX methyl ester methyl glycoside (for NMR data of sialic acids, see Section 5). The spectrum turned out to be identical with deaminated neuraminic acid methyl ester methyl glycoside, synthesized from N-glycolylneuraminic acid.260 Referring to the earlier work of Roseman et al. (Section 4.1), incubation of free SiaX with N-acetylneuraminic acid aldolase yielded d-mannose and pyruvate. Finally, SiaX showed the same absorption spectrum as N-acetyl- and N-glycolylneuraminic acid in the thiobarbituric acid assay. In conclusion, SiaX could be assigned as 3-deoxy-d-glycero-d-galacto-non-2-ulosonic acid. For information with respect to the metabolism of Kdn and the enzymatic release of this sialic acid from glycoconjugates, see Sections 11.5.3 and 11.7, respectively.
5. Elucidating the O-Substitution Patterns
From the beginning, there were indications for the presence of O-acetyl groups in addition to N-acetyl or N-glycolyl groups in sialic acids. As mentioned in Section 2.1, the first studies, reported in 1936, spoke of the presence of an O-acetyl group in the unknown substance, isolated from bovine submandibular gland mucin.165 It is to be noted that for the release of (O-acylated) sialic acids from sialoglycoconjugates/sialooligosaccharides, the chosen acid hydrolysis conditions must be such that the labile glycosidic linkage between sialic acid and a neighboring monosaccharide is efficiently cleaved, but also that the applied conditions do not cause too much de-esterification. Note that conditions that are too harsh lead to acid- and heat-mediated degradation to chromogens (humin formation). Over the years, the three acid hydrolysis systems most applied are formic acid pH 2.0 (1 h, 70°C) [in later years followed by HCl pH 1.0 (1 h, 80°C)],24, 104 2 M acetic acid (3 h, 80°C),261 and 2 M propionic acid (4 h, 80°C)262; however, other systems have been advocated. Although these conditions do not lead to significant N-deacylation, O-deacylation has been shown to occur to an extent of approximately 30%–50%. The use of aqueous H2SO4 is no longer recommended.6, 33, 154 Besides mild acid hydrolysis, in later years sialidases made their appearance.6, 154 As mentioned in Section 2.1, even the acidity of mucin suspensions (pH 3.5–4.0) was low enough to initiate release of sialic acids.23
As reported in Section 3, analysis of the sialic acids released from bovine submaxillary gland mucin gave Neu5,7Ac2, as deduced from periodate oxidation experiments.23 In additional investigations based on periodate oxidation, besides Neu5,7Ac2 (consumption of 1 mol of periodate), also Neu5Ac/Neu5Gc (consumption of 2 mol of periodate), and Neu5,7,8/9Ac3 (no consumption of periodate) were found.263 In the same report, equine submaxillary gland mucin was shown to contain Neu4,5Ac2 (consumption of 2 mol of periodate). Also the RDE of V. cholerae was used to release N,O-acylneuramic acids from bovine submaxillary gland mucin, yielding Neu5Ac, Neu5Gc, and di- and triacetylated Neu5Ac.264
For the isolation of sialic acid variants, at the end of the 1960s Faillard and Schauer at the Ruhr-Universität Bochum (Germany) introduced a combination of anion-exchange and cellulose column chromatography.24, 119, 154 Subsequently, the positions of the O-acetyl groups in the isolated sialic acids were determined by a combined approach as follows:
-
(i)
Polarographic determination of the periodic acid consumption during mild periodate oxidation (pH 3). It was assumed that Neu5Ac, Neu5Gc, and Neu4,5Ac2 consumed 2 mol of periodate/mol sialic acid, Neu5,7Ac2 and Neu5,9Ac2 1 mol of periodate/mol sialic acid, and Neu5,8Ac2, 0 mol of periodate/mol sialic acid.
-
(ii)
Colorimetric assay using periodic acid/thiobarbituric acid according to Aminoff.265 The periodate oxidation was carried out at pH 1–2 and 37°C, whereby it was assumed that Neu5Ac, Neu5Gc, Neu4,5Ac2, and Neu5,9Ac2, but not Neu5,7Ac2 and Neu5,8Ac2, were degraded to β-formylpyruvic acid, the chromogen that forms a red chromophore with thiobarbituric acid (see Section 7.1).265, 266, 267, 268, 269
-
(iii)
TLC on cellulose and silica-gel layers. Neu5Ac and Neu4,5Ac2 are discriminated on the basis of different R f values. Also Neu5,7Ac2 and Neu5,8Ac2 have different R f values (see Section 7.2).269
Following this combined protocol, a series of N,O-acylneuraminic acids, released from bovine and equine submaxillary gland mucin via mild formic acid hydrolysis (pH 2–2.5, 1 h, 70°C), were characterized (Fig. 13 ).24 In case of the bovine source, Neu5Ac, Neu5Gc, Neu5,7Ac2, and Neu5,8Ac2 were identified, and in the case of the equine source, Neu5Ac, Neu5Gc, and Neu4,5Ac2 were identified. Furthermore, it was suggested that the bovine material also contained Neu5,7,8Ac3, and the equine material Neu4,5,?Ac3. The consumption lines of Neu5Ac and Neu4,5Ac2 showed overconsumption after 40 min, due to opening of the pyranose ring. In theory, Neu5,8Ac2 should give no consumption of periodate; the slow consumption of periodate (0.25 mol/mol sialic acid in 40 min) combined with only a partial positive Aminoff test (extinction coefficient 47% of that obtained with free Neu5Ac) was explained as the result of a slow O-deacetylation. In fact, a possible presence of Neu5,9Ac2 was excluded, because the combination of “1 mol periodate consumption and a positive Aminoff test” was not found. The number of N-glycolyl groups was estimated using a microanalysis protocol for glycolic acid,270 and the number of O-acetyl groups was determined with the colorimetric Hestrin assay (alkaline hydroxylamine/Fe(ClO4)3; see Section 7.1).271 Note that all bovine sialic acids could also be released with the sialidases of V. cholerae and C. perfringens, although at different rates, but equine Neu4,5Ac2 and di-O-acetylated Neu5Ac (presumably one of the O-acetyl groups attached at C4) were resistant to these enzymes (see Section 11.5.2). In the same period, Neu5,8Ac2, assigned via polarographic periodate oxidation studies, was found to occur in fish brain gangliosides.272
Fig. 13.

(A) Polarographic measurement of the periodic acid consumption at 0°C of isomeric N-acetyl-mono-O-acetyl-neuraminic acids.24 Neu5Ac, cross; Neu4,5Ac2, open circle; Neu5,7Ac2, closed circle; Neu5,8Ac2, closed square. (B) Expected cleavages in the C7–C8–C9 glycerol side chains are indicated with ------.
Panel (A): Reproduced from Schauer, R. Chemistry and Biology of the Acylneuraminic Acids. Angew. Chem., Int. Ed. Engl.1973, 12, 127–138. Copyright Wiley-VCH Verlag GmbH & Co. KGaA.
A few years later (1975), a novel technology for the analysis of N,O-acylneuraminic acids, namely, GLC–EIMS, was introduced by Vliegenthart/Kamerling and coworkers at Utrecht University (the Netherlands).26, 27 To this end, the isolated sialic acids were esterified with diazomethane, followed by trimethylsilylation of the free hydroxy groups. In a later protocol, the sialic acids were pertrimethylsilylated.33 Analysis of the mass spectra suggested strongly that all N,O-acylneuraminic acids with an earlier identified 8-O-acetyl group by periodate oxidation, in fact, had to be assigned with an O-acetyl group at C9. This change in assignment was based on mass spectrometric fragmentation patterns that originated from the mass spectral analysis of partially methylated alditol acetates.273, 274, 275 For cleavage of the latter compounds, at the end of the 1960s it had been demonstrated that the charge is preferentially located on an ether oxygen instead of on an ester oxygen atom. Translated to the sialic acid situation, it meant that, in general, cleavage occurs between two trimethylsiloxylated carbon atoms, or between an acetoxylated and a trimethylsiloxylated carbon atom with location of the charge on the ether oxygen, rather than between an acetoxylated and a trimethylsiloxylated carbon atom with location of the charge on the ester oxygen, or between two acetoxylated carbon atoms (Fig. 14A). The mass spectra obtained for the different sialic acid derivatives gave rise to highly specific fragmentation patterns A–H, both for N,O-acylneuraminic acids and for N-acyl-O-alkyl-neuraminic acids (Fig. 14B).6, 27, 30, 33, 275 Fragments A and B indicate the molecular mass of the sialic acid derivatives, and thus the number and type of substituents. Fragments C–H contain the information concerning the position of the different substituents. Fragment C only has significant abundance if C7 bears an ether group; when an ester group is present at C7, this fragment is absent or hardly observable. The occurrence of fragment D is dependent on the presence of fragment C. Fragment E is not seen if an O-acyl group is attached to C4. Fragment F can be readily formed only if an ether group is attached at C8. As an illustration, the mass spectra of Neu5Ac and Neu5,9Ac2 are presented in Fig. 15 . Additional mass spectra of various members of the sialic acid family can be found in refs. 6, 30, 33, 104, as well as in the references included in Table 1. For tables of m/z data of the A–H fragments and GLC retention times of various sialic acid derivatives, see refs. 6, 26, 27, 30, 33, as well as the references included in Table 1.
Fig. 14.

(A) EI mass spectrometric cleavages in the C7–C8–C9 glycerol side chain of N,O-acylneuraminic acids. (B) Fragmentation scheme for the analysis of sialic acid derivatives.6, 27Ac, acetyl; Lt, lactyl; Me, methyl; TMS, trimethylsilyl.
Fig. 15.

(A) EI mass spectrum (70 eV) of the per-O-trimethylsilylated methyl ester of β-Neu5Ac. (B) EI mass spectrum (70 eV) of the per-O-trimethylsilylated methyl ester of β-Neu5,9Ac2.
Reproduced from Kamerling, J. P.; Vliegenthart, J. F. G. Gas-Liquid Chromatography and Mass Spectrometry of Sialic Acids. In Sialic Acids—Chemistry, Metabolism and Function; Schauer, R., Ed.; Cell Biology Monographs, Vol. 10; Springer-Verlag: Vienna, Austria, 1982; pp 95–125. Copyright Springer-Verlag.
To prove the interpretation of the mass spectra of the 9-O-acetylated sialic acid derivatives, Neu5,9Ac2 and Neu9Ac5Gc were incubated with acylneuraminate pyruvate lyase. The products formed were 6-O-acetyl-N-acetylmannosamine and 6-O-acetyl-N-glycolylmannosamine, respectively, as proven by GLC–EIMS.27 Furthermore, synthetic β-Neu5,9Ac21,2Me2 and β-Neu4,5,9Ac31,2Me2, of which the structures were confirmed by 1H and 13C NMR spectroscopy, were subjected to mass spectrometry and periodate oxidation studies. Indeed, both derivatives showed mass spectrometric fragmentation patterns similar to those found for β-Neu5,9Ac2 and β-Neu4,5,9Ac3, and consumed only ~0.15 mol of periodate/mol of sialic acid within 10 min (β-Neu5Ac1,2Me2 consumed 2 mol of periodate/mol sialic acid within 10 min).276 An explanation for the resistance of 9-O-acetylated sialic acids to periodate oxidation was stated to originate from the staggered anti orientation of the OH groups at C7 and C8, stabilized by some hydrogen-bonding network, that hampers the formation of a diol–periodate complex (see Section 6). It seems to take time to create the complex with both OH groups in a cis configuration, as over a period of 48 h a higher periodate consumption (85% of their theoretical value) was seen for both compounds, only partially due to O-deacetylation. Neu5Ac9Lt behaved in the same way as Neu5,9Ac2 (consumption of only ~0.10 mol of periodate/mol sialic acid within 10 min).42 Periodate oxidation of Neu5Ac9P yielded glycolaldehyde phosphate, indicating a splitting of the C7–C8 bond, and a positive thiobarbituric acid color reaction (extinction coefficient 78% of that obtained with free Neu5Ac).51 Also Neu5Gc9S turned out to be easily susceptible to periodate oxidation.72 In view of the literature protocols followed, it seems that in both cases the oxidation had been carried out for several hours. It should be noted that on treatment of β-Neu5Ac1,2Me2 with low amounts of periodate, the C8–C9 diol group is preferentially oxidized, as was shown by the high ratio of the aldehyde of the 8-carbon analogue to the aldehyde of the 7-carbon analogue obtained.277 The aldehyde of the 8-carbon analogue, being a 2-hydroxyaldehyde, is further oxidized if the amount of periodate is sufficient. So, the oxidation of the C7–C8 diol group will be hindered if the formation of the mentioned aldehyde is blocked by the presence of an O-acetyl group at C9. See also refs. 6, 30, 277, and 278 for periodate/borohydride degradation studies on the glycoprotein level, generating bound C8-α-Neu5Ac, C7-α-Neu5Ac, C8-α-Neu5Gc, and/or C7-α-Neu5Gc.
In later years, several other MS protocols, combined with GLC or high-performance liquid chromatography (HPLC), were explored. Underivatized N,O-acylneuraminic acids were successfully analyzed by combined HPLC–CIMS (chemical ionization mass spectrometry), although localization of O-substituents based on CIMS data alone was not possible.279 Although CIMS of pertrimethylsilylated sialic acid derivatives gave less fragmentation than EIMS, GLC–CIMS also furnished suitable information for identification purposes.104, 280 Furthermore, a methodology based on HPLC, coupled with electrospray-ionization mass spectrometry (ESIMS), has been developed.32 To this end fluorescent sialic acid derivatives were prepared by reaction with 1,2-diamino-4,5-methylenedioxybenzene (DMB) (see Section 7.2). A combination of HPLC retention times and ESI mass spectra allowed the assignment of the type and position of the various substituents.32, 33 A similar approach, but now using matrix-assisted laser-desorption ionization time-of-flight mass spectrometry (MALDI-TOFMS), has also been developed.281 Analogous to the GLC–EIMS analysis of trimethylsilylated sialic acids, a GLC–EIMS methodology for the analysis of heptafluorobutyrylated (HFB) sialic acid methyl ester derivatives has been worked out.19 Here, the EI mass spectra are rather complex; a survey of reporter fragment ions and GLC data of specific members of the sialic acid family can be found in refs. 19, 33. For EI mass spectra of acetylated derivatives, see ref. 25. For fast-atom bombardment mass spectrometry (FABMS) studies of sialic acid derivatives, see ref. 31.
Also 1H NMR spectroscopy has been shown to be an excellent tool in the characterization of N,O-acylneuramic acids.6, 29, 282 1H chemical shift values of sialic acids are pH dependent. However, the pH of the solution is also important for the stability of the sialic acids under investigation: autohydrolysis in case of bound sialic acids at low pH; O-deacylation at low and high pH; exchange of H3 in D2O or tritiated H2O at high pH283, 284 (see Section 7.3), migration of O-acyl groups at neutral pH38a, 38, 261 (see Section 11.5.2). With respect to anomeric α:β ratios, interestingly, the ratio of Neu5,7Ac2 and Neu5,7,9Ac3 (~23:77) differed strongly from the ratio of the other N(O)-acylneuraminic acids (~7:93); the 5Ac signal of both Neu5,7Ac2 anomers resonates at clearly different positions, when compared with the positions of Neu5Ac and Neu5,9Ac2.29
6. Different Views on the Conformation of the Glycerol Side Chain
The studies related to the conformational aspects of sialic acids are mainly attributed to the understanding of the hydrogen-bonding network within α- and β-sialic acids that comes to an expression in the interaction between the “flexible” staggered C7–C8–C9 glycerol side chain and the rigid C2–C3–C4–C5–C6–O6 2 C 5 chair conformation.
6.1. X-ray Diffraction Analysis
At the beginning of the 1970s, first crystallographic data appeared on β-methoxyneuraminic acid trihydrate (β-Neu2Me·3H2O),285 β-N-acetylneuraminic acid dihydrate (β-Neu5Ac·2H2O),286 β-N-acetylneuraminic acid methyl ester monohydrate (β-Neu5Ac1Me·H2O),287 and at a later stage α-N-acetylneuraminic acid methyl ester methyl glycoside (α-Neu5Ac1,2Me2).288 All four substances were found to occur in a 2 C 5 chair conformation with equatorial groups at C4 (OH), C5 (NH2 or NHAc), and C6 (glycerol side chain). The first three compounds had an axial OR (R = CH3 or OH) group at C2 (proof for β-anomer), while the fourth compound had an equatorial OCH3 group at C2 (proof for α-anomer). The equatorial glycerol side chain of all four compounds was found to occur in an extended, staggered conformation with the OH groups at C7 and C8 in anti orientation. In general, extensive three-dimensional systems of intermolecular hydrogen bonds, also involving H2O molecules, were seen. In the context of the conformation of the glycerol side chain, intramolecular hydrogen bonds are of interest. For β-Neu5Ac1Me·H2O, 1 intramolecular hydrogen bond between HO7 and the carbonyl O of NHCOCH3 was detected. In the case of α-Neu5Ac1,2Me2, intramolecular hydrogen bonds were detected between HO7 and the carbonyl O of the planar NHCOCH3 group (trans configuration of the peptide bond), and between HO8 and the carbonyl O of the COOCH3 group (Fig. 16 ). Here, the 2 C 5 chair was shown to be somewhat distorted toward a 6 H 5 half-chair conformation.
Fig. 16.

Molecular conformation of crystalline α-Neu5Ac methyl ester methyl glycoside.
Reproduced from Kooijman, H.; Kroon-Batenburg, L. M. J.; Kroon, J.; Breg, J. N.; de Boer, J. L. Structure of α-d-N-Acetyl-1-O-methylneuraminic Acid Methyl Ester. Acta Crystallogr. C1990, 46, 407–410. With permission of the International Union of Crystallography.
6.2. NMR Spectroscopy
The first 60-MHz 1H NMR spectra of Neu5Ac, recorded in D2O, were published in 1968,289, 290 but these low-resolution spectra did not contain much information (Fig. 17 ). In fact, the 100-MHz 1H NMR spectrum of β-Neu5Ac1Me, already discussed in Section 4.3, delivered the first useful information with respect to the conformation of sialic acids.257 In a first high-resolution study, the 270-MHz 1H NMR spectrum of Neu5Ac in D2O was completely assigned.28 The wrong assignment of the Neu5Ac sample as α-Neu5Ac was corrected in a later report.291 In subsequent papers full assignments of various sialic acids were published, as reviewed in refs. 6, 282. As a typical example, Fig. 18 presents the 500-MHz 1H NMR spectrum of Neu5Ac, dissolved in D2O, at pD 7, whereas Table 3 summarizes chemical shifts and coupling constants for α- and β-Neu5Ac (note the 3 J 7,8 value of ~9 Hz, reflecting the staggered anti orientation of H7 and H8, and thus of HO7 and HO8 at the glycerol side chain). The spectrum shows a major and a minor set of protons, related to the subspectra of the β- and α-anomer of Neu5Ac, respectively (β:α = 93:7). The positions of the H3e and H3a signals, being highly pH dependent and resonating outside the proton bulk signal, turned out to be of special interest in the structural analysis of sialoglycan chains (see Section 8.6).
Fig. 17.

60-MHz 1H NMR spectrum of Neu5Ac, dissolved in D2O, recorded at ambient temperature.
Reproduced from Kimura, A.; Tsurumi, K. Studies on Colominic Acid—IV. Conformation and Configuration of the N-Acetyl-neuraminic Acid Moiety of Colominic Acid. Fukushima J. Med. Sci.1968, 15, 55–60. With permission of the Board.
Fig. 18.

500-MHz 1H NMR spectrum of Neu5Ac (β:α = 93:7), dissolved in D2O, recorded at pD 7 and 27°C (Table 3), relative to internal acetone in D2O (δ 2.225 ppm). The H3e and H3a signals of α-Neu5Ac are enlarged 10-fold.
Reproduced from Schauer, R.; Kamerling, J. P. Chemistry, Biochemistry and Biology of Sialic Acids. In Glycoproteins II; Montreuil, J.; Vliegenthart, J. F. G.; Schachter, H., Eds.; New Comprehensive Biochemistry, Neuberger, A.; van Deenen, L. L. M., Series Eds., Vol. 29b; Elsevier Science B.V.: Amsterdam, The Netherlands, 1997; pp 243–402. Copyright Elsevier Science B.V.
Table 3.
360-MHz 1H Chemical Shifts and Coupling Constants of β- and α-Neu5Ac at Different pH Values, in ppm Relative to Internal Acetone in D2O (δ 2.225) at 25°C6, 29, 282
| Compound | pD | H3a | H3e | H4 | H5 | H6 | H7 | H8 | H9 | H9′ | NAc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| β-Neu5Ac | 1.4 | 1.880 | 2.313 | 4.067 | 3.931 | 4.056 | 3.556 | 3.750 | 3.841 | 3.619 | 2.053 |
| α-Neu5Ac | 1.4 | 1.705 | 2.718 | n.d. | 3.85 | 3.684 | 3.53 | 3.75 | 3.85 | 3.62 | 2.036 |
| β-Neu5Ac | 7.0 | 1.827 | 2.208 | 4.024 | 3.899 | 3.984 | 3.514 | 3.753 | 3.835 | 3.608 | 2.050 |
| α-Neu5Ac | 7.0 | 1.621 | 2.730 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 2.030 |
| Compound | pD | 2J3a,3e | 3J3a,4 | 3J3e,4 | 3J4,5 | 3J5,6 | 3J6,7 | 3J7,8 | 3J8,9 | 3J8,9′ | 2J9,9′ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| β-Neu5Ac | 1.4 | − 13.2 | 11.8 | 5.0 | 10.4 | 10.7 | 1.2 | 9.4 | 2.8 | 6.4 | − 12.4 |
| α-Neu5Ac | 1.4 | − 13.0 | 11.5 | 4.5 | n.d. | 10.5 | 1.5 | 9.0 | n.d. | 6.5 | − 12.5 |
n.d., value could not be determined.
The first 22.6- and 25.2-MHz 13C NMR spectra of Neu5Ac (derivatives), recorded in H2O or D2O, appeared in the 1970s.35, 291, 292, 293, 294 For a review of early 13C NMR studies, including chemical shift data, see ref. 282. As a typical example, in Fig. 19 the 25.2-MHz 13C NMR spectrum of Neu5Ac, dissolved in D2O, at pD 2, is depicted.
Fig. 19.

Proton-noise-decoupled 25.2-MHz 13C NMR spectrum of Neu5Ac, dissolved in D2O, recorded at pD ~2 and 25°C, relative to external TMS (δ 0 ppm). Only the signals stemming from the β-anomer are clearly observable. For a first report on chemical shifts of both α- and β-Neu5Ac, see ref. 291.
Reproduced from Vliegenthart, J. F. G.; Dorland, L.; van Halbeek, H.; Haverkamp, J. NMR Spectroscopy of Sialic Acids. In Sialic Acids—Chemistry, Metabolism and Function; Schauer, R., Ed.; Cell Biology Monographs, Vol. 10; Springer-Verlag: Vienna, Austria, 1982; pp 127–172. Copyright Springer-Verlag.
Besides the full interpretation of the 1H and 13C NMR spectra of sialic acids in different solvents, the conformation of the glycerol side chain (average over rotamer states) as part of the whole molecule in D2O was studied in more detail and compared with the conformation in crystal structures. In a first study,28 taking into account the values of the vicinal coupling constants J 6,7, J 7,8, J 8,9, and J 8,9′ (Table 3), the lowest energy conformation of the extended C7–C8–C9 side chain was believed to be as depicted in Fig. 20A. Here, H6 and H7 are placed in a staggered gauche, H7 and H8 in a staggered anti, H8 and H9 in a staggered gauche, and H8 and H9′ in a staggered anti orientation. In this spatial model the H of HO7 can form a hydrogen bond with the ring O, and the H of HO8 a hydrogen bond with the O of HO9 (two five-membered ring interactions). In another study, using hard-sphere, exo-anomeric (HSEA) calculations, the latter model (Fig. 20A) got support.295
Fig. 20.

NMR Molecular conformations in aqueous solution of (A) β-Neu5Ac according to ref. 28, (B) β-Neu5Ac according to refs. 294, 296, and (C) and (D) α-Neu5Ac according to refs. 297, 298. For both (A) and (B), as measured in D2O solution, the H atoms of the OH and NH groups would be replaced by D atoms.
However, the spatial model depicted in Fig. 20A was not supported by results obtained from a 13C spin–lattice relaxation time (T 1) study of the sodium salts of β-Neu5Ac, β-Neu5Ac1Me, β-Neu5Ac2Me, β-Neu5Fo1,2Me2, α-Neu5Ac2Me, β-Neu5Gc, and Neu2en5Ac in D2O.294, 296 In this case, a conformational model was postulated, consistent with the 1H NMR data and the motional requirements of the T 1 values (Fig. 20B). In this model, intramolecular hydrogen bonds occur between the O of HO7 and the acetamido NH and between the H of HO8 and the ring O, keeping HO7 and HO8 in a fixed staggered anti orientation (two six-membered chair-form ring interactions). Furthermore, the model made a third intramolecular hydrogen bond between the acetamido C O and the H of HO4 (seven-membered ring interaction) possible. Apparently, the anomeric center is not involved in any hydrogen bond, leading to the same conformation for α- and β-anomers.
Using a new approach to the NMR study of OH and NH protons in aqueous solution (85% H2O/15% (CD3)2CO), α-Neu5Ac as part of Neu5Ac-containing trisaccharides was investigated.297 Careful NOE experiments showed that the strongest NOE was visible between the NH proton and H6. Further NOE contacts were seen between the HO8 proton and H6. Based on a comparison with a study on GM1 ganglioside in (CD3)2SO,298 it was concluded that HO8 was involved in a strong intramolecular hydrogen bond with the carboxylic acid function or the ring O, or both, and explained as the main source of rigidity of the glycerol side chain (Fig. 20C and D) (note that in case of β-Neu5Ac only a hydrogen bond between HO8 and the ring O is possible). The investigation on GM1 ganglioside mentioned also a hydrogen bond between the H of HO7 and the acetamido C O. Both intramolecular hydrogen bonds are also proposed for crystalline α-Neu5Ac1,2Me2 (Fig. 16).288
For NMR complexation studies of Neu5Ac and Neu5Gc (derivatives) with Ca2+, Mn2+ or Gd3+, see refs. 66, 291, 292, 299, and 300.
6.3. Molecular Mechanics, Molecular Dynamics, and Ab Initio Calculations
First potential energy calculations of the conformation of β-Neu5Ac suggested that in solution the compound might exist preponderantly in two conformations, differing only in the orientation of the terminal CH2OH (C9) group of the glycerol side chain.301 One of these two energy conformations accorded with the crystal structure of β-Neu5Ac∙2H2O.286 Although the two generated conformations were consisted with the earlier reported 1H and 13C NMR data, there is disagreement with the derived NMR models in Fig. 20A and B. For α-Neu5Ac, two similar minimum energy conformations have been reported.302 See also ref. 303 for theoretical studies on the conformation of mono- and disialogangliosides.
In a more recent study, density functional theory calculations of the conformation of α-Neu5Ac in the gas phase have been carried out.304 Out of many possibilities for relatively stable hydrogen bond networks, the most stable structure had an intramolecular hydrogen bond network of O9–H9…O8–H8…O C1–OH/O7–H7…O C–NH–C5, which shows similarity with the earlier reported crystal structure of α-Neu5Ac1,2Me2 (Fig. 16). One of the lower stable structures shows similarity with the model presented in Fig. 20A (H9–O9…H8–O8/O7–H7…O6 (ring)/O4–H4…O C–NH–C5/O2–H2…O C1–OH). In addition, α-Neu5Ac–H2O complexes of the various gas phase conformations were optimized. Here, the following hydrogen bond networks for the optimum complex were seen: O9–H9…OH2…O8–H8, O8–H8…OH2(…O C1–OH)…OH2(…H2–O2)…O6 (ring), O7–H7…OH2…O C–NH–C5, and HO–C1 O…H2O…HO–C1 O (compare with Fig. 20C).
A first molecular dynamics (MD) simulation of α-Neu5Ac was performed both in vacuum and in water.305 It was shown that the average conformation of the glycerol side chain was not influenced by the presence of explicit solvent molecules, and the rotation around the C6–C7 and C7–C8 bonds is highly restricted. Hydrogen bond positions were suggested for either O8–H8…O C1–OH or O8–H8…O6 (ring), for either O7–H7…O C–NH–C5 or O7–H7…O6 (ring), and for O4–H4…O C–NH–C5. The C8–C9 bond was found to be more flexible.
Furthermore, also an MD simulation and quantum mechanical (QM) calculations of α-Neu5Ac directed to the whole molecule have been published.306 Here, the optimized structure was in good agreement with the crystal structure published for α-Neu5Ac1,2Me2.288
Finally, ab initio calculations (gas phase) were carried out on a series of neuraminic acid derivatives: α-Neu5Ac, α-Neu5Ac1,2Me2, β-Neu5Ac, β-Neu4,5Ac2, β-Neu5,9Ac2, β-Neu5Gc, β-Neu4Ac5Gc, and Neu2en5Ac.307 The calculations for α-Neu5Ac1,2Me2 were in close agreement with those of the crystal structure.288 The calculated structure for α-Neu5Ac is depicted in Fig. 21 .
Fig. 21.

Ab initio calculated structure of α-Neu5Ac.
Reproduced from van Lenthe, J. H.; den Boer, D. H. W.; Havenith, R. W. A.; Schauer, R.; Siebert, H.-C. Ab initio Calculations on Various Sialic Acids Provide Valuable Information About Sialic Acid-Specific Enzymes. J. Mol. Struct. (Theochem)2004, 677, 29–37. Copyright Elsevier B.V.
6.4. Final Remarks
First drawings of the conformation of sialic acids, including the glycerol side chain, stem from the end of the 1960s. These drawings, in later years included in the carbohydrate IUPAC/IUBMB Recommendations 1996/1997,13 and generally used by many researchers including us, have a glycerol side chain with the H6–C6–C7–C8 part of the chain in an eclipsed conformation. The HO7 and HO8 groups are placed in a staggered anti orientation, whereby the HO7 group points in the direction of the ring O (Fig. 2/Fig. 22A).
Fig. 22.

(A, B) Two different drawings of the structure of α-Neu5Ac.
Going through the different reports, discussed in 6.1, 6.2, 6.3, it is evident that, although a discussion about the average conformation of the glycerol side chain is highly complex, the drawing as depicted in Fig. 22A does not reflect the most favorable conformation. In contrast, especially models with intramolecular hydrogen bonds between the HO7 and the NHCOCH3 group, and between the HO8 and the ring O or the COOH function (in case of the α-configuration), leading to a strong hydrogen-bonding network between the glycerol side chain and the 2 C 5 pyranose ring conformation, pop up. Inspection of the literature shows a great variety in drawing the glycerol side chain. Perhaps, one of the better drawings is shown in Fig. 22B.
7. An Impression of Analytical Techniques as Used in Sialic Acid Analysis
7.1. Spectrophotometric Assays
In the early days of the sialic acid history, colorimetry played a major role in the unraveling of sialic acid structures. As demonstrated in Section 2, initially two reagents were applied routinely, namely, Bial's reagent (orcinol/concd. HCl/FeCl3 mixture) and Ehrlich's reagent (p-dimethylaminobenzaldehyde/50% HCl). By the 1950s several additional assays were developed; however, the majority of these early methods were found to be unsuitable because of their lack of specificity and/or sensitivity required for sialic acid analysis in biological materials or for enzymatic assays.6, 101, 269 In the following, a number of spectrophotometric assays, still in use or used for a long time, will be explained in more detail.
In the Bial reaction,181, 308, 309 the sialic acid sample is mixed with the orcinol/~30% HCl/FeCl3 reagent and heated for 15 min at 96°C, whereafter the formed purple to red–violet chromophore is extracted with isoamyl alcohol, and its absorbance measured at 572 nm (for more details, see refs. 101, 104, and 269). Because of the strong acidic conditions used, the method is suitable for the quantification of total amounts of free and glycosidically bound sialic acid. Note that O-acyl groups are split off. When applied during the fractionation of biological materials, the Bial assay is therefore typically a reaction to monitor the presence of sialic acids. As other carbohydrates (pentoses, hexoses, uronic acids) can strongly disturb the determination of small amounts of sialic acid, quantification usually requires purified samples. To overcome some of the problems, different variants of the Bial reaction have been explored. With a focus on contaminating 2-ketohexoses, a suitable variant reagent to measure total sialic acid, namely, resorcinol/concd. HCl/CuSO4, was worked out by Svennerholm. It proved to be 30%–50% more sensitive than the Bial assay.310, 311 Here, free or bound sialic acid samples are mixed with the reagent and heated for 15 min at 110°C, and the formed blue–violet chromophore is extracted with amyl alcohol followed by measurement of the absorbance at 580 nm, and also at 450 nm to correct for 2-ketohexoses (for further information, see refs. 269, 312, and 313). Note that Kdn gives no color in the orcinol and resorcinol reactions.314
In trying to formulate a reaction mechanism for the Bial and Svennerholm reaction, it was proposed that the strong acidic conditions lead to N-deacetylation, yielding an internal Schiff base formed by the condensation of the free amino group at C5 and the C2 carbonyl group, followed by decarboxylation and dehydration, to yield a pyrrole derivative. The latter chromogen then gives a condensation product with orcinol or resorcinol (Scheme 11 ).313, 315 When combined with a preceding periodate oxidation step, the sensitivity of the Svennerholm assay can be increased significantly (between three- and sixfold).312, 313
Scheme 11.

Possible pyrrole chromogen formation [N-deacetylation, condensation NH2(C5) and CO(C2), decarboxylation, dehydration] in the Bial (orcinol/concd. HCl/FeCl3) and Svennerholm (resorcinol/concd. HCl/CuSO4) reaction, taking into account the proposals in refs. 313 and 315.
In the “direct” Ehrlich reaction (without previous treatment with alkali),165, 181 the sialic acid sample is usually mixed with the p-dimethylaminobenzaldehyde/~18% HCl reagent and heated for 30 min at 100°C, whereafter the absorbance of the formed red–violet chromophore is measured at 565 nm (for more details, see refs. 88 and 181). In the case of the “indirect” Ehrlich reaction, the test material is first incubated with mild alkali, e.g., borate buffer at pH 8.5 (45 min, 100°C), followed by the addition of Ehrlich's reagent (20 min, 70°C), whereafter the extinction is determined at 558 nm.265 To illustrate the chemical background of the color formation in the Ehrlich's test, and taking into account the proposals by different authors, the “direct” and “indirect” Ehrlich pathways for Neu5Ac are presented in Scheme 12 . Just like in the Bial/Svennerholm assays, N-deacetylation, internal Schiff base formation, and dehydration play a role. For the formulation of the pathway leading to structure A, the earlier scheme of Tuppy and Gottschalk, combined with information of Treibs and Herrmann, is followed.100, 316 For the formulation of the pathway leading to structure B (including a decarboxylation), the earlier scheme of Wirtz-Peitz, combined with information from Alexander and Butler, is followed.315, 317 Note that the “direct” Ehrlich test does not distinguish free from bound sialic acid. It should be mentioned that the Ehrlich assay as a quantitative method has lost its importance.
Scheme 12.

Possible pyrrole derivatives arising from Neu5Ac, following the “direct” Ehrlich (E = p-dimethylaminobenzaldehyde/concd. HCl) and the “indirect” Ehrlich (alkali; E = p-dimethylaminobenzaldehyde/concd. HCl) reaction, taking into account the proposals in refs. 100 and 315, 316, 317.
The periodic acid/thiobarbituric acid assay, independently developed at about the same time (1959) by Warren and by Aminoff, grew out to the most widely applied quantitative colorimetric method for measuring free sialic acid.266, 267 The methods differ in two ways from each other: (i) the acidity is that of the initial periodate oxidation step (concd. H3PO4 266 vs 0.06 M H2SO4 265), and (ii) the use of cyclohexanone266 or acidic n-butanol265 for the extraction of the chromophore. In the periodate oxidation step (followed by an arsenite step), a chromogen is generated, predicted to be β-formylpyruvic acid, which gives a red chromophore with 2-thiobarbituric acid with λ max 549 nm. A proposed reaction mechanism of the assay is presented in Scheme 13 .100, 268 In contrast to Warren,266 it was stated by Paerels and Schut268 that a conversion of the N-acyl group into a free amino group under the acidic periodate oxidation conditions (room temperature; incubation for a relatively short time) is not obvious, because of the high stability of an amide bond. As sialic acid glycosides did not react in the Warren and Aminoff tests, sialic acid glycosidic linkages are not hydrolyzed under the assay conditions. Furthermore, it was presumed that β-formylpyruvic acid is not a direct product of the periodate oxidation, but the result of an acid-catalyzed C4–C5 aldol splitting of the true oxidation product (for Neu5Ac: formed after consumption of 3 mol of periodate; see the upper part of Scheme 13) during the incubation with the acid solution of 2-thiobarbituric acid at 100°C. The red pigment was earlier prepared in the crystalline form from a derivative of β-formylpyruvic acid, and proven to be a condensation product of 2 mol of 2-thiobarbituric acid and 1 mol of β-formylpyruvic acid.318 It may be clear that O-substitutions of the glycerol side chain will influence the prechromogen formation (see discussion in Section 5), and a test after O-deacetylation was advised. In case of Neu4,5Ac2, an O-deacetylation at C4 during the reaction with 2-thiobarbituric acid before the aldol splitting was suggested. See the comments on the analysis of Kdn, which directly yields β-formylpyruvic acid.314
Scheme 13.

Proposed reaction mechanisms of the periodic acid/thiobarbituric acid assay.100, 268, 319, 320
However, in the 1980s, a positive periodic acid/thiobarbituric acid test with Neu5Ac4Me initiated a new discussion, wherein the postulated prechromogen was suggested to react directly with 1 mol of 2-thiobarbituric acid, followed by an acid-catalyzed reaction (cleavage of the C4–C5 bond) to yield the β-formylpyruvic acid derivative.319 This derivative can then react with a second mol of 2-thiobarbituric acid to give the final chromophore (lower part Scheme 13). The latter pathway, stating that free β-formylpyruvic acid is not an intermediate, was suggested in the 1970s to explain the positive Warren reaction for 3-deoxy-5-O-methyloctulosonic acid.320 However, in view of the positive Warren reaction for Neu5Ac4Me, a chromophoric 1:1 condensation product should be expected.
So, applying the periodic acid/thiobarbituric acid methodology on biological samples before and after mild acid hydrolysis, and eventually saponification, the levels of free, bound, and total sialic acid can be determined (for experimental details, see refs. 101, 104, and 269).
In attempts to lower the detection limits of sialic acid, since the 1960s a number of fluorimetric assays have also been developed (for reviews, see refs. 6, 101, 102, 269, and 269). In the following, some typical examples will be presented. Reaction of sialic acid with 3,5-diaminobenzoic acid dihydrochloride in hot dilute HCl yields an intense green fluorescent condensation product (405 nm excitation, 510 nm emission) (free and bound sialic acids react equally).321 Reaction of sialic acid with pyridoxamine in the presence of zinc acetate and pyridine/methanol for 45 min at 70°C yields a highly fluorescent chelate (395 nm excitation, 470 nm emission; Scheme 14 ) (free sialic acids).322, 323 The fluorescence of the chromophoric product, obtained from sialic acid in the periodate/2-thiobarbituric acid assay (Scheme 13), is readily detected by excitation at 550 nm and analysis of the emission at 570 nm. Here, special attention has been paid to contamination with 2-deoxyribose.324 Finally, a different approach has been worked out, whereby free or glycosidically bound sialic acid was subjected to mild periodate oxidation. This method also allowed the discrimination between sialic acids with or without O-acyl groups at the glycerol side chain by O-deacetylation of a parallel sample (for a discussion of the mild periodate oxidation of sialic acids, see Section 5). The released formaldehyde, derived from C9 in the case of nonsubstituted HO9 and HO8 functions, is derivatized with acetylacetone in the presence of ammonium acetate, yielding the slightly yellow chromophore 3,5-diacetyl-1,4-dihydro-2,6-dimethylpyridine,325 of which the fluorescence can be detected by excitation at 410 nm and analysis of the emission at 510 nm (Scheme 15 ).326 Using this methodology, it could be demonstrated that rapid O-deacetylation of Neu5,9Ac2 occurred only at pH values below 2 and above 11. When incubated for 10 min at 58°C, no significant loss of O-acetyl groups were seen in the range of pH 2–11.327
Scheme 14.

Proposed reaction mechanism for the transamination of sialic acid with pyridoxamine in the presence of zinc acetate and pyridine/methanol, yielding the fluorescent Zn-chelate.323
Scheme 15.

Reaction of formaldehyde with acetylacetone in the presence of ammonium acetate, yielding the fluorigen 3,5-diacetyl-1,4-dihydro-2,6-dimethylpyridine.325, 326
Based on the finding of the acylneuraminate pyruvate lyase (or aldolase)-catalyzed cleavage and synthesis of Neu5Ac with ManNAc and pyruvic acid as counter compounds by Roseman and coworkers (Section 4.1; Scheme 5),231, 232 indirect quantification protocols for sialic acids, via estimation of ManNAc or pyruvate, were also developed.269, 328, 329 The most sensitive method turned out to be a pyruvate assay, based on the lactic acid dehydrogenase-catalyzed interconversion of pyruvate and lactic acid with the concomitant interconversion of NADH and NAD+. Coupling of the aldolase-catalyzed cleavage reaction of N-acylneuraminic acids to the lactate dehydrogenase/NADH-catalyzed reduction of pyruvate to lactate drives the lyase reaction, otherwise an equilibrium reaction, to completion. Initially, the amount of pyruvate (and thus the amount of sialic acid) was calculated from the amount of oxidized NADH, measured spectrophotometrically by the change in absorbance at 340 nm.329 In the course of time, various commercial colorimetric and fluorimetric assays have been developed for the estimation of oxidized NADH. Note that the reaction rate of the cleavage by aldolase is influenced by the sialic acid O-substitution pattern, e.g., it has been found that O-acyl groups at the sialic acid glycerol side chain lead to a reduction in the reaction rate of 30%–50%, while O-acetylation at C4 gives 90% reduction.41, 330, 331
Originally, acetyl determinations for sialic acids were described using a protocol that included hydrolysis with p-toluenesulfonic acid, subsequent distillation to collect acetic acid, conversion into barium acetate, and determination of the Ba2+ content.23, 332 In the case of glycolyl groups, sialic acids were treated with 1 M NaOH at 100°C, and the glycolic acid that formed was quantified as crystalline calcium glycolate.23 At a later stage, among the different colorimetric methods to quantify the O-acyl content of sialic acids, the Hestrin assay, as modified by Ludowieg and Dorfman, played an important role.271, 333 Incubation of N,O-acylneuraminic acids with alkaline hydroxylamine affords acylhydroxamates that form red chromophores with Fe(ClO4)3, the absorbance of which are measured at 520 nm.334 O-Glycolyl (but also N-glycolyl) groups can be quantified using a protocol that includes treatment with ethanolic p-toluenesulfonic acid (8 h, 95°C), affording ethyl glycolate, which is collected via distillation in vacuo. After saponification, the amount of sodium glycolate is estimated colorimetrically using the Eegriwe's reagent, 2,7-dihydroxynaphtalene/concd. H2SO4.64, 270, 334, 335, 336 The reddish chromophore that is formed (20 min, 100°C), represents the condensation product of 1 mol of formaldehyde (oxidative decarboxylation of glycolic acid) and 2 mol of 2,7-dihydroxynaphtalene, and yields an absorption at λ max 546 nm. Finally, the configuration and the amount of O-lactyl groups in N,O-acylneuraminic acids can be measured as lactic acid with l-lactate dehydrogenase and d-lactate dehydrogenase after release with NaOH (1 h, room temperature) and neutralization.42, 43 So far, only O-l-lactyl groups have been found.
7.2. Chromatographic Techniques
Just as with structural investigations of many other natural products, in the early days paper chromatography played an important role in the analysis of sialic acids.88, 263, 269
However, at the end of the 1950s, thin-layer chromatography (TLC) on cellulose and silica gel plates slowly surpassed the use of paper. The greater resolution, combined with improved reproducibility, made TLC for many years the technique of choice for screening and (tentative) assignments of sialic acids. Especially, Schauer and coworkers at the Ruhr-Universität Bochum, and later at the Christian-Albrechts-Universität zu Kiel, contributed greatly to the development of TLC technologies. Although several solvents were developed, the most popular flow systems turned out to be propan-1-ol/butan-1-ol/0.1 M HCl (2:1:1, v/v/v) for cellulose sheets (Fig. 23 ), and propan-1-ol/water (7:3, v/v) for silica gel sheets. For R f values of different sialic acids, see refs. 6 and 269. For the visualization of the sialic acid members, in most cases, the Bial reagent (orcinol/concd. HCl/FeCl3) was used, yielding typical purple–blue bands. Periodate combined with resorcinol/CuSO4 and periodate combined with arsenite and thiobarbituric acid sprays were also developed. For a further differentiation between N,O-acylneuraminic acids with similar R f values in one direction, a two-dimensional TLC method with intermediate saponification, carried out on cellulose sheets with the solvent system mentioned earlier, was developed. The intermediate exposure to an atmosphere of ammonia vapor over a 5 M NH4OH solution (2 h, room temperature) generates Neu5Ac and Neu5Gc from their O-acyl derivatives.41, 334 A two-dimensional technique on cellulose sheets was also developed for O-acyl group identifications. After a separation in the first dimension using propan-1-ol/butan-1-ol/0.1 M HCl (2:1:1, v/v/v), the lane of migrated sialic acids is sprayed with Hestrin's alkaline hydroxylamine reagent and left for 60 min. Then, the plate is developed in the second dimension using propan-1-ol/10% aq (NH4)2CO3/5 M NH4OH (6:2:1, v/v/v). Visualization of the acylhydroxamates is carried out with a 10% FeCl3 spray.41, 334
Fig. 23.

TLC patterns of sialic acids on a cellulose plate, developed with propan-1-ol/butan-1-ol/0.1 M HCl (2:1:1, v/v/v), and stained with Bial's reagent. Lane 1: sialic acid mixture, released from bovine submandibular gland mucin. Lane 2: standard Neu5Ac and Neu5Gc.
Around the 1970s, GLC (in combination with EIMS) of volatile sialic acid derivatives was introduced.6, 30, 33 Major protocols that were explored are based on trimethylsilylation,260, 337, 338, 339, 340, 341, 342 trifluoroacetylation,343 and methylation (in case of partial methylation, followed by trimethylsilylation or acetylation).344, 345, 346, 347, 348 For the application of GLC–EIMS in studying sialic acid O-substitution patterns, see Section 5. The partially methylated derivatives of sialic acids play a role in methylation/linkage analysis studies (GLC–EIMS) of glycan chains with internal sialic acid residues.
As already mentioned in Section 5, the introduction of serial anion-exchange and cellulose column chromatography at the end of the 1960s was a real step forward in the isolation of sialic acid family members on a preparative scale.24 However, at a later stage the introduction of HPLC using different column materials, elution protocols, and detection techniques replaced this original approach. So, rapid screening of sialic acids in biological materials, affording assignments based on retention times, whereby contaminating substances (e.g., other monosaccharides) did not interfere, could be developed. Also the monitoring of fast transitions between members of the sialic acid family due to migration of substituents, the introduction of substituents, the cleavage of substituents, or other (enzymatic) modifications became possible.
First reports on the application of HPLC for the separation and quantitative analysis of sialic acids stem from the mid-1970s onward. Initially, methods for Neu5Ac were developed using an anion-exchange resin combined with a periodate/arsenite/thiobarbituric acid colorimetric detection assay,349 a cation-exchange resin combined with UV detection at 206 nm,350 and an amino column combined with UV detection at 195 nm.351 Chromophores, obtained by treatment of Neu5Ac and Neu5Gc with periodate/arsenite/thiobarbituric acid, were analyzed on a reversed-phase C18 column, monitored at 549 nm.352 As it was realized that analysis of complex mixtures of sialic acids should require other approaches, the focus was switched to generally applicable methods (for a review, see ref. 6). A first report proposed the use of an anion-exchange resin in borate buffer (pH 8.55), whereby nonderivatized sialic acids were separated as sugar–borate complexes. For the detection, the effluent was mixed with 4,4′-dicarboxy-2,2′-biquinoline disodium salt/CuSO4 (5.4 min, 100°C), and the chromophores (Cu+/4,4′-dicarboxy-2,2′-biquinoline complexes) that formed were monitored at 570 nm.353, 354, 355 At the same time a different anion-exchange chromatography method was reported, making use of sodium sulfate as mobile phase and UV detection at 195 or 215 nm.356 Great progress in HPLC approaches was made by the combination of reversed-phase HPLC with fluorometric detection systems and, in a later stage, with additional coupling to MS. To this end fluorescent sialic acid derivatives were prepared by reaction with DMB (373 nm excitation; 448 nm emission) (Scheme 16 )357, 358, 359 or o-phenylenediamine (OPD) (232 nm excitation; 420 nm emission).360 In an evaluation of seven different HPLC methods, including high-performacnce anion-exchange chromatography with pulsed amperometric detection (HPAEC–PAD) (see ref. 361), it turned out that no single method is adequate to completely separate and quantitate complex mixtures of sialic acids.362 For a quantification protocol of Neu5Ac and Neu5Gc by a stable isotope dilution assay using HPLC coupled with ESIMS, see ref. 363. For a more detailed overview of HPLC procedures, see ref. 6.
Scheme 16.

Reaction of Neu5Ac with 1,2-diamino-4,5-methylenedioxybenzene (DMB) to yield the corresponding fluorescent derivative.357, 358
Around the turn of the century, also capillary electrophoresis (CE) was introduced for the analysis of derivatized and nonderivatized Neu5Ac and Neu5Gc.364, 365, 366, 367, 368, 369
7.3. Instrumental Techniques
As indicated at several other places in this chapter, since the 1970s, mass spectrometry and NMR spectroscopy have played important roles in the further development of the sialic acid world, and detailed reviews are available.6, 30, 282
In the context of this section, attention will be paid to some NMR studies of sialic acids. As shown in 5, 11.5.2, the value of 1H NMR spectroscopy for the characterization of N,O-acylneuraminic acids and for the migration of O-acetyl groups along the glycerol side chain was clearly demonstrated. NMR spectroscopy also proved to be of great value for unraveling of the mechanism of the release of bound sialic acid and for the assignment of the type of glycosidic linkage [e.g., (α2→3), (α2→6), (α2→8); see also Section 8.6]. Following the incubation of Neu5Ac(α2→3 or 6) conjugates with bacterial (C. perfringens, Arthrobacter ureafaciens, V. cholerae, Bifidobacterium) and viral (fowl plague virus) sialidases in an NMR tube, NMR spectroscopy showed in a fast process at first instance the formation of free α-Neu5Ac in high amounts, which then mutarotates in a slower process to the equilibrium α-Neu5Ac:β-Neu5Ac = 8:92. The anomerization could be followed easily via the intensities of the H3e, H3a, and N-acetyl signals of both anomers (see Section 6.2; Table 3).370, 371, 372 Fortunately, the initial high concentration of α-Neu5Ac made it now possible to follow the mutarotation in more detail than described in Section 4.3. But it should be said that the rapidity of the α-Neu5Ac → β-Neu5Ac mutarotation depends on the pH. At pD 5.4, being the pH for an optimal cleavage of 2-azido-α-Neu5Ac with C. perfringens sialidase, the process turned out to be rather slow, but at higher and lower pD values a more rapid establishment of the equilibrium of mutarotation was observed. At pD 1.3 and pD 11.7 the mutarotation was too fast to be measured (no indications for open-chain structures or lactones).373 For GLC–EIMS studies on the release of Neu5Ac from substrates by sialidases in the presence of H2[18O], see ref. 374. The fact that the cleavage by these sialidases passed with retention of anomeric configuration received support in a later mechanistic study with the sialidase from influenza virus.375 These results were in contradiction with an earlier report that Neu5Ac, liberated by the enzymatic cleavage of the α-linked Neu5Ac, leaves the catalytic site of V. cholerae sialidase as the β-anomer, which meant an inversion of anomeric configuration in the mechanism.247 However, such a mechanism was later proposed in case of Salmonella typhimurium sialidase.376, 377 With the ability to generate α-Neu5Ac in situ, the aldolase-catalyzed degradation of Neu5Ac to pyruvate and ManNAc (in fact α-ManNAc, followed by a fast mutarotation to α,β-ManNAc) could also be investigated in more detail using 1H NMR spectroscopy.378, 379
In alkaline D2O solutions of Neu5Ac at pD 9.0, H3a was completely replaced by D3a, which led to the disappearance of the H3a signal in the 1H NMR spectrum and simpler splitting patterns for the H3e and H4 signals.284 Further increase of the pD to 12.4 also caused the exchange of H3e for D3e.283 When using tritiated H2O, tritium-labeled sialic acids could be prepared. A 13C NMR study of aqueous solutions of Neu5Ac, labeled with 13C at C1, C2, and/or C3 at varying pHs, made the detection and quantification of its acyclic forms possible. Thus, at pH 2 anomerization of β-Neu5Ac leads to α-pyranose:β-pyranose:keto:enol:keto hydrate (gem-diol) = 5.8:91.2:0.7:0.5:1.9.380
The importance of different MS techniques in the structural analysis of sialic acids and sialoglycans is discussed in Sections 5 and 8.6, respectively.
7.4. Histological Methods
Histochemistry is a most important area of glycobiology research, enabling insight into the chemical architecture of a cell, and especially its surface, the glycocalyx. For this purpose, the cell or tissue remains intact and can be investigated mostly in sliced form by different forms of microscopy. Nowadays, histological experiments are also possible with viable cells. This means that cell biological studies, during which the function of a cell varies, can now be visualized.
Histochemistry has a long history, and the methods applied are manifold. In a short note from 1946 in Nature, McManus from the University Museum in Oxford, UK, described the staining of mucins by Schiff's reagent following the action of 0.5% periodic acid.381 Originally, Schiff's reagent was defined as “a solution of fuchsine decolorized by treatment with sulfur dioxide that gives a useful test for aldehydes because they restore the dye's color.” Examples of the staining comprised mucins of goblet cells of the human intestine and bronchus, and also various other tissues from rat and man. A few years later, in 1950 and 1952, Leblond and coworkers used the technology in combination with light microscopy.382, 383 After periodate oxidation, tissues of different animals were stained with fuchsine–sulfurous acid, a technique developed by Hotchkiss.384 At these times, the nature of the molecules in tissues, oxidized by periodic acid, was not yet known.
Interestingly, in 1957, in a report by Leblond and coworkers385 describing the staining of glycogen by the periodic acid–Schiff (PAS) technique, sialic acid also came into play as the probable molecule responsible for periodate staining (sialic acid was found in various tissues along with galactose, fucose, and hexosamine). In subsequent years, the PAS technique developed into a powerful technique of sialic acid staining, because of the sensitivity of its glycerol side chain to mild periodate oxidation, thereby generating aldehyde functions. Typical examples are the detection of vicinal hydroxy groups (mostly of sialic acids) in plasma membranes by electron microscopy.386, 387 In an early review,388 the periodate acid–Schiff technique, together with other methods for the detection of acidic groups/acidic glycoproteins of plasma membranes, e.g., using cationic compounds such as colloidal iron, thorium hydroxide, Alcian blue, and ruthenium red, used earlier for light or electron microscopy, is summarized. The studies confirmed that all cell surfaces seem to be covered by a carbohydrate-rich coat. In a thorough investigation on complex carbohydrates (glycoproteins) in the Golgi apparatus of rat cells, two electron microscopy visualization methods, based on (1) successive treatment of sections with periodic acid, chromic acid, and silver methenamine, and (2) brief treatment of sections with a mixture of chromic acid and phosphotungstic acid, were compared with the PAS technique using light microscopy, and the staining results obtained were shown to be identical/similar.389
Soon after the beginning of the use of periodate for histological sialic acid determinations, it was noticed that pretreatment with alkali (0.5% KOH) increased the periodic acid–Schiff (1% PAS) reactivity of, e.g., certain mucins,390 and the authors confirmed the hypothesis that this KOH–PAS effect was due to the presence of O-acetylated sialic acids.391, 392 It should be noted that ester groups, which prevent aldehyde formation by periodate, were assumed to be located at C8 of the glycerol side chain of sialic acid. Until the mid-1970s, 9-O-acetylated sialic acids were believed to be cleavable between C7 and C8 (see fig. 1 of ref. 391). However, as discussed in Section 5, in the same period (1974), this assumption turned out to be incorrect.276 So, PAS-resistant sialic acids can be substituted at C9 and not necessarily at C8. Neu4,5Ac2 is fully PAS positive (but sialidase resistant). The specificity of this sialic acid-staining technique can be increased by previous sialidase or acid treatment of tissue slices. The difference of staining intensity with and without alkali treatment enables a rough estimation of the degree of O-acetylation in the glycerol side chain.
In the following years, staining of sialic acid by periodic acid and Schiff or thionine reagents, respectively, was carried out in numerous studies. For an early review by Culling and Reid that describes many details of the histochemical methodologies available for the location and identification of members of the sialic acid family in tissue sections, see ref. 393. It should be mentioned that the Hungarian G. Romhány developed in the 1970s a polarization optical electron microscopic analysis of the spatial arrangement of sialylated cell membrane components by periodate oxidation with the aldehyde bisulfite toluidine blue reaction (“anisotropic ABT reaction”), summarized by Makovitzky and Richter.394 Also new reagents for staining, making use of periodate oxidation as a first step, were developed, as illustrated by the two following examples. First, a light microscopic histochemical localization of sialic acids (sialoglycoconjugates) in tissue sections of eccrine glands of porcine snout skin could be carried out by employing a method that includes a selective periodate oxidation–phenylhydrazine blockade and a thiocarbohydrazide–silver protein sequence, followed by a physical development procedure.395, 396 Second, for the in situ imaging by confocal laser microscopy and flow cytometric quantification of sialylated glycoproteins on living human gastric normal and cancer cells, the cell lines were treated by mild periodate oxidation (1 mM, 4°C, up to 20 min), and the aldehyde functions generated on the glycoproteins were ligated with the fluorescence tag, fluorescein-5-thiosemicarbazide.397 Note that the chosen periodate oxidation conditions only introduce aldehyde groups into the sialic acid glycerol side chains of the glycoproteins on the living cell surface.
In the 1990s, staining of sialic acids in tissues or cells also became possible with lectins and antibodies. The specificities of the methods, developed for sialic acid linkage analysis, were higher than those developed using the periodate reaction.398 Especially, the sialyl(α2→3)-specific Maackia amurensis agglutinin (MAA) and the sialyl(α2→6)-specific Sambucus nigra agglutinin (SNA) found wide application [and more rarely Limax flavus agglutinin (LFA)] in the lectin histochemical localization of glycoconjugates containing Neu5Ac in (α2→3) and (α2→6) linkage, respectively, to Gal and GalNAc. Wheat germ agglutinin (WGA) also interacts with Neu5Ac (not Neu5Gc), but according to a discussion with Jürgen Roth (University of Zurich, Switzerland), the lectin is not suited for sialic acid analysis. For the detection of sialic acid with these lectins, for example, a peroxidase–antibody–peroxidase complex (PAPS), which can be visualized, is in use. In the same period, the labeling of lectins and immunoglobulins with ferritin, and especially colloidal gold particles, used in both light and electron microscopy, got wide attention. Jürgen Roth (“Immunogold Master,” “Mr. Compartment”399) and collaborators developed, most elegantly and frequently applied, the lectin–gold technique for human and animal tissues, including the Golgi complex.400, 401, 402 Typical examples of staining of normal human colonic mucosa and colon carcinoma with MAA and SNA are shown in Fig. 24 . Before these studies, the same research group had developed cationic (poly-l-lysine-coated) colloidal gold particles, which allow the detection of anionic, sialidase-sensitive, sites on erythrocytes.403
Fig. 24.

Detection of sialic acid residues in human colon by silver-intensified lectin–gold labeling of sections from paraffin-embedded tissues. (A) Normal colonic surface (arrowhead) and crypt epithelium (asterisks) are reactive for (α2→3)-linked sialic acid residues, as detected with the Maackia amurensis lectin. (B) In contrast, (α2→6)-linked sialic acid residues are undetectable in the colonic surface (arrowhead) and crypt epithelium (asterisks) with the Sambucus nigra lectin. Note the positive labeling of capillary endothelia (arrows). In two adjacent serial sections of a colon carcinoma, both (α2→3)-linked sialic acid residues (C) and (α2→6)-linked sialic acid residues (D) can be detected. Scale bar: 20 μm (A, B), 50 μm (C, D).
Photos courtesy of Prof. Dr. Jürgen Roth.
In more recent years, virolectins have shown to be also powerful reagents for histochemical detection of sialic acids. For example, influenza C viruses bind to 9-O-acetylated sialic acid in tissue slices from rat liver or human colon and are immunologically detected by incubation with rabbit antiserum, followed by treatment with fluorescent anti-rabbit IgG antibody for visualization.404 Staining is also possible by the esterase activity of the bound viruses. In a similar way, such staining has also been done with a soluble form of influenza C virus hemagglutinin-esterase, wherein the C-terminal transmembrane and cytoplasmic domains are replaced by the Fc portion of human IgG. This complex (CHE-Fc) retains both its recognition and enzymatic function. It is detected on the cryosections with alkaline phosphatase-conjugated goat antihuman IgG.405 Great progress in the identification of 4-mono-, 9-mono, and 7,9-di-O-acetylated sialic acids (Neu5Ac and Neu5Gc) in tissue slices or cells of man and mouse was recently achieved with hemagglutinin-esterase (HE) envelope proteins of nidoviruses (toro- and coronaviruses) and influenza C virus (HEF).406 For this purpose, HE and HEF ectodomains were fused with the C-terminal domain of human IgG1 or of mouse or bovine IgG2a. The authors showed that O-acetylated sialoglycans occur in a species-, organ-, tissue-, and cell-type-specific way, and that their expression is regulated at the level of the individual cell. They also observed that in several key tissues, especially in the brain, mice and humans share similar 9-O-acetyl-sialic acid expression profiles, which suggests an evolutionary and functional conservation. In a similar way, this method with the recombinant soluble virolectins was applied to various tissues of many kinds of animals, including humans, and demonstrated an astonishing variability of the occurrence of the three O-acetylated sialic acids in the tissues studied. While Neu4,5Ac2, for example, was prevalent in horse and guinea pig respiratory tissue, Neu5,9Ac2 and Neu5,7,9Ac3 were found especially in the same tissue of man and mouse.407 In an early study using a monoclonal antibody bound to gold particles against the Meningococci group B capsular polysaccharide, [→ 8)Neu5Ac(α2 →]n, this polymer was detected in a human Wilms tumor (Fig. 25 ).408, 409, 410 Treatment with endo-neuraminidase N as control abolished staining. Furthermore, in an immunocytochemical study on ganglioside expression in human breast cancer cells, specific monoclonal antibodies and antimouse IgM or antimouse IgG labeled with Alexa 488 showed the accumulation of gangliosides on the tumor cell surfaces in confocal microscopy.411
Fig. 25.

Reexpression of (α2→8)-linked polysialic acid in human Wilms tumor. (A) Silver-intensified immunogold staining with mAb 735 of a section of paraffin-embedded tissue shows intense labeling in the Wilms tumor (WT). The adjacent normal kidney (NK) is unlabeled. (B) Immunolabeling in a consecutive serial section for (α2→8)-linked polysialic acid is abolished by section pretreatment with endoneuraminidase N (Endo N). (C) Immunogold labeling for (α2→8)-linked polysialic acid of an ultrathin frozen section from a Wilms tumor is very intense at cell-surface regions with a thick surface coat. (D) In contrast, immunogold labeling is sparse and patchy in regions of close contact of tumor cells. Scale bar: 300 μm (A, B), 300 nm (C), 150 nm (D).
Photos courtesy of Prof. Dr. Jürgen Roth.
Finally, at the end of this short overview of the history of histological methods in sialobiology, attention will be paid to some new fluorescence microscopy investigations that allow the manipulation of glycans within their native environment, i.e., to study living cells and the localization, mobility, and lifetime of glycans. Since some sialyltransferases tolerate such large groups as fluorescein at C5 or C9, it was possible to incorporate fluorescent probes via suitably modified CMP-sialic acid donors into cell-surface glycoconjugates412 (for a general review, see ref. 413) or even enabled the kinetic analyses of the rates of different sialyltransferases within cells.414 So, it was possible to localize sialyltransferases in the Golgi compartments in rat liver with the CMP-9-fluoresceinyl-Neu5Ac donor substrate.415 Furthermore, Bertozzi and coworkers introduced the Staudinger ligation for covalent tagging of azido sugars, incorporated biosynthetically into cell-surface glycans. In case of sialic acid, cells are treated with per-O-acetylated N-azidoacetyl-d-mannosamine (ManNAz). After entering the cells, the product is O-deacetylated, transformed into the corresponding N-azidoacetylneuraminic acid (Neu5Az), subsequently converted into the activated CMP sugar, and finally incorporated into cell-surface glycans on human or other cells.416 Interestingly, the various biosynthetic enzymes, normally involved in the conversion of ManNAc into bound Neu5Ac (see Section 11.1), tolerate the unnatural substrates. The bound Neu5Az can be visualized using a copper-free click chemistry reaction with, for instance, a difluorinated cyclooctyne (DIFO) reagent, conjugated to an imaging probe, e.g., Alexa Fluor 488. This staining procedure is not toxic and can be carried out in cultured cells and in live mice. Here, a typical example is the study of the sialome during zebrafish development, which showed that the biosynthesis of cell-surface sialosides starts as early as 8.5 h postfertilization.417 In another example, Neu5Az(α2→3)Lac or Neu5Az(α2→3)Gal was incubated with live trypomastigotes, resulting in Neu5Az(α2→3)Gal-glycoconjugates by means of the parasite trans-sialidase (see Section 11.7). For visualization use was made of, for instance, the phosphine–FLAG conjugate, identified by anti-FLAG antibodies. It was learned from this approach that the transfer of sialic acid is mainly to the mucins of the parasites.418
8. Sialic Acids as a Part of Glycoproteins, Glycolipids, Oligosaccharides, and Polysaccharides
Although some members of the sialic acid family only occur in free form (see Section 1; Table 1), most members are constituents of glycoproteins, glycolipids, capsular polysaccharides, lipopolysaccharides, and (lipo)oligosaccharides. But also glycosylphosphatidylinositol membrane anchors and keratan sulfates have been shown to contain sialic acid. In the following sections a summary of sialic acid-containing structural elements will be given, as found in first/early reports.
8.1. Glycoprotein N- and O-Glycans
In most eukaryotic glycoproteins the N- and O-glycans are terminated with Neu5Ac or Neu5Gc, or both. First indications stem from the late 1950s. Over the years, the two sialic acids have been found to be attached to d-galactose (Gal) via (α2→3)419, 420, 421, 422, 423 or (α2→6)423, 424, 425 linkages, to N-acetyl-d-galactosamine (GalNAc) via (α2→3)426, 427, 428 or (α2→6)421, 429, 430, 431, 432 linkages, and to N-acetyl-d-glucosamine (GlcNAc) via an (α2→6) linkage.433 At the end of the 1970s, mid-1980s, terminal N-acylneuraminic acids were also discovered to be (α2→8)-linked434, 435, 436 or (α2→9)-linked437, 438 with internal N-acylneuraminic acids, a phenomenon that was already seen in the 1960s for glycolipids (see Section 8.2). Incidentally, terminal Neu5Ac(α2→4)Gal(β1 →439, 440, 441 and terminal Neu5Ac(α2→4)GlcNAc(β1→442 sequences have been published, but discussion about these results does exist. Furthermore, an internal →)Fuc(1→4)Neu5Gc(2→ sequence is known.443
Although a long list of naturally occurring N,O-acylneuraminic acids is known (Table 1), only for a few glycoproteins, intact glycans with such sialic acids have been published. In short, the terminal sequences comprise Neu4,5Ac2(α2→3)Gal(β1→,444, 445 Neu4,5Ac2(α2→6)Gal(β1→,445, 446, 447, 448, 449 Neu5,9Ac2(α2→3)Gal(β1→,450, 451, 452, 453, 454 and Neu5,7,9Ac3(α2→3)Gal(β1 → .452, 453 But also (α2→3) linkages with Gal have been reported for Neu5,7Ac2, Neu5,8Ac2, Neu5,7,8Ac3, and Neu5,8,9Ac3.452 Furthermore, [Neu5Ac(α2→8)]n, [Neu5Gc(α2→8)]n, and [Neu5Ac/5Gc(α2→8)]n sequences with 4Ac, 7Ac, 9Ac, or 9Lt substituents,455 a Neu5Ac8S(α2→9)[Neu5Ac(α2→9)]n sequence,456 and the [Neu5Gc(α2→O5)]n sequence (linkage between Neu5Gc C2 and the N-glycolyl OH function of the next Neu5Gc residue; Fig. 26 ) should be mentioned.457 The latter sequence can also be terminated with Neu5Gc9S, i.e., Neu5Gc9S(α2→O5)[Neu5Gc(α2→O5)]n.72
Fig. 26.

A terminal Neu5Gc(α2→O5)Neu5Gc sequence.
The N- and O-glycans can also be terminated by Kdn, and first reports are from the late 1980s and early 1990s. The sialic acid can be (α2→3)-linked with Gal458, 459 and (α2→3)- or (α2→6)-linked with GalNAc.458, 460 Furthermore, Kdn occurs in polysialyl sequences: [Kdn(α2→8)]n (eventually with 4Ac, 7Ac, or 9Ac substituents),458, 461 and Kdn(α2→8)[Neu5Ac(α2→8)]n, Kdn(α2→8)[Neu5Gc(α2→8)]n, Kdn(α2→8)[Neu5Ac/5Gc(α2→8)]n, and Kdn9Ac(α2→8)[Neu5Gc(α2→8)]n.76, 80, 461 Of interest are the disubstituted internal Kdn units in Fuc(α1→4)[Fuc(α1→5)]Kdn(α2→3)Gal(β1→462 and in Fuc(α1→4)[Fuc(α1→5)]Kdn(α2→6)GalNAc(α1→,462, 463 and the double Kdn-termination in Kdn(α2→3)[Kdn(α2→6)]GalNAc(α1→,464 all three occurring in O-glycans.
For extensive reviews of established sialic acid-containing glycoprotein glycan structures, see, e.g., refs. 6, 465, 466, 467, 468. For sialylation of glycosylphosphatidylinositol membrane anchors and keratan sulfates, see ref. 469 and ref. 470, respectively.
8.2. Glycolipids
As in glycoprotein glycans, in glycolipids/gangliosides (first reports from the early 1960s), terminal Neu5Ac and/or Neu5Gc can be coupled with Gal via (α2→3)471, 472, 473, 474 or (α2→6)475, 476 linkages, with GalNAc via (α2→3)477 or (α2→6)478 linkages, with GlcNAc via an (α2→6)479, 480 linkage, and with other N-acylneuraminic acids via (α2→8)473, 481, 482 or (α2→9)483 linkages. Also Neu5Gc(α2→4)Neu5Gc, Neu5Gc(α2→4)Neu5Ac, Neu5Ac(α2→6)Glc, and Neu5Gc(α2→6)Glc (Glc = d-glucose) elements have been reported.48, 484, 485, 486 Furthermore, terminal sequences with Neu itself have been identified: Neu(α2→3)Gal(β1→16, 17, 487, 488 and Neu(α2→8)Neu5Ac(α2→3)Gal(β1→.18 In the mid-1960s, it was predicted that gangliosides could occur in the lactonic form (inner esters),489, 490 which a few years later was supported by IR spectroscopy.491 Besides the inner ester formation between sialic acids (see Section 8.5), such lactone formation was also seen for the Neu5Ac(α2→3)Gal sequence.492 Here, Neu5Ac(α2→3,1→2)Gal and/or Neu5Ac(α2→3,1→4)Gal elements could be proven by NMR spectroscopy and shown not to be artifacts.493 More recently, both lactone forms were prepared from sialoglycopeptides (treatment with glacial acidic acid).494
In addition to the Neu5Ac/Neu5Gc-glycolipids, from the 1970s onward, intact glycan structures with O-acetylated and O-methylated N-acylneuraminic acid residues have been published. Established terminal sequences show
Neu5,9Ac2(α2→3)Gal(β1→,495
Neu9Ac5Gc(α2→3)Gal(β1→,498
Neu4,9Ac25Gc(α2→3)Gal(β1→,499
Neu5Gc8Me(α2→3)GalNAc(β1→,502
Neu5Ac9Me(α2→6)Glc(β1→,503
Fuc(α1→O5)Neu5Gc(α2→6)Glc(β1→,504
Neu4,5Ac2(α2→8)Neu5Ac(α2→3)Gal(β1→,34
Neu5,7Ac2(α2→8)Neu5Ac(α2→3)Gal(β1→,505
Neu5,7,9Ac3(α2→8)Neu5Ac(α2→3)Gal(β1→,505
Neu5Ac8Me(α2→O5)Neu5Gc(α2→3)Gal(β1→,508
Also sequences containing terminal sulfated sialic acid residues have been characterized:
Neu5Ac8S(α2→3)Gal(β1→ and Neu5Gc8S(α2→3)Gal(β1→,510
Neu5Ac4S(α2→6)Glc(β1→,48
Neu5Ac8S(α2→6)Glc(β1→,49
Neu5Ac8S/5Gc8S(α2→8)Neu5Ac/5Gc(α2→3)Gal(β1→,50
Neu5Ac8S(α2→8)Neu5Ac(α2→6)Glc(β1→.511
Furthermore, terminal and internal (branched) elements with sialic acid became known from the late 1970s onward:
Neu5Ac(α2→3)[Neu5Ac(α2→6)]Gal(β1→,512
Neu5Ac(α2→3)[Neu5Ac(α2→4)]Gal(β1→8)Neu5Ac(α2→3)GalNAc(β1→,513
Neu5Gc8Me(α2→3)[Neu5Gc8Me(α2→6)]GalNAc(β1→,483
→6)Gal(β1→4)[Gal(β1→8)]Neu5Gc(α2→3)Gal(β1→,514
→6)Gal(β1→4)Neu5Gc(α2→3)Gal(β1→,515
→6)Gal(β1→4)Neu5Gc8Me(α2→3)Gal(β1→,515
→3/4)Gal(α1→4)Neu5Ac(α2→3)Gal(β1→ [earlier reported with an (α2→4) linkage],516, 517, 518
→3)Gal(α1→4)Neu5Ac(α2→6)Galf(β1→3)Gal(α1→4)Neu5Ac(α2→3)Gal(β1→,519
→3)Gal(α1→4)Neu5Ac8Me(α2→3)Gal(β1→,501
→3)Gal(α1→9)Neu5Ac(α2→3)Gal(β1→,501
→6)Glc(β1→8)Neu5Gc(α2→6)Glc(β1→,520
Fuc(α1→8)Neu5Gc(α2→4)Neu5Ac(α2→,48
Fuc(α1→4)Neu5Ac(α2→O5)Neu5Gc(α2→4)Neu5Ac(α2→,521
Fuc(α1→O5)Neu5Gc(α2→4)Neu5Ac(α2→ .522
Finally, the [Neu5Gc8Me(α2→O5)]n 46, 68 and the Neu5Gc8Me(α2 → O5)[Neu5Gc(α2 → O5)]n 523 sequences, and the Kdn-containing sequences Kdn(α2 → 3)Gal(β1 →,524 Kdn9Ac(α2 → 3)Gal(β1→,525 and Kdn(α2 → 6)GalNAc(β1→526 have been established.
In fact, the large variety in sialylated structural elements found so far among glycolipid glycans is certainly influenced by the interest of researchers in the glycolipids of lower animals, such as starfish, sea urchins, sea cucumbers, brittle stars, and feather stars.
For extensive reviews of established sialic acid-containing glycolipid structures, see, e.g., refs. 6, 527, 528, 529.
8.3. Milk Oligosaccharides
Since the late 1950s, neutral and acidic milk oligosaccharides of a large variety of animals have been studied. The ensembles of oligosaccharides differ from species to species, and in the case of human milk over 200 different compounds are expected to be present, of which at the moment 163 have been characterized (up to tridecasaccharides), including 54 sialyloligosaccharides.530, 531, 532, 533, 534, 535
The established terminal sequences with sialic acid are similar to those found in glycoprotein and glycolipid glycans:
Neu5Ac(α2 → 6)GalNAc(β1 →,538
Neu5Ac(α2 → 3)GlcNAc(β1 →,539
Neu5Ac(α2 → 6)GlcNAc(β1 →,536
Neu5Ac(α2 → 6)Glc,542
Neu5Gc(α2 → 3)Gal(β1 →,536
Neu5Gc(α2 → 6)Gal(β1 →,543
Neu4,5,xAc3(α2 → 3)Gal(β1 → with x = 7, 8, or 9,546
Neu4,5Ac2(α2 → 6)Gal(β1 →,547 and
For Neu5,9Ac2 data in human and cow milk carbohydrates, see also ref. 548. Interestingly, besides Neu5Ac and Neu5Gc, porcine milk oligosaccharides (and also glycoproteins and gangliosides) contain very small amounts of Kdn as the building block.549
8.4. Urinary Oligosaccharides and Glycopeptides
In normal human urine of different biological origin only small ensembles of sialylated oligosaccharides have been demonstrated to occur, e.g.:
Neu5Ac(α2 → 3)Gal(β1 → 4)Glc/GlcNAc/GlcNAc(α1 → P/GalNAc(α1 → P,550, 551, 552, 553
Neu5Ac(α2 → 6)Gal(β1 → 4)Glc/GlcNAc/GlcNAc(α1 → P,550, 552, 553
Neu5Ac(α2 → 3)Gal(β1 → O)inositols,553
Neu5Ac(α2 → 3)Gal(β1 → 3)[Neu5Ac(α2 → 6)]GalNAc,552 and
Neu5Ac(α2 → 3)Gal(β1 → 3)[Neu5Ac(α2 → 6)]GalNAc(α1 → O)Ser.552
A disialylated hexasaccharide was found in human pregnancy urine, Neu5Ac(α2 → 3)Gal(β1 → 3)[Neu5Ac(α2 → 6)]GlcNAc(β1 → 3)Gal(β1 → 4)Glc.554
Studies on rat urine revealed the presence of Neu5Ac/Neu5Gc/Neu5,9Ac2(α2 → 3)Gal(β1 → 4)Glc.450, 551, 555
Several carbohydrate-related inborn errors of metabolism can lead to the accumulation of oligosaccharides in the body (see Sections 13.3 and 13.4). In case of sialidosis/galactosialidosis, (α2 → 3)- and (α2 → 6)-sialylated (Neu5Ac) glycoprotein N-glycans (nearly only the expected endo-β-N-acetylglucosaminidase-cleaved oligosaccharides) and O-glycans (glycopeptides) are accumulated in the patient's urine, blood serum, amniotic fluid, and ascitic fluid, stored in the placenta, and shown to be present in fibroblasts.556, 557, 558, 559, 560, 561, 562, 563, 564, 565, 566, 567, 568, 569 In a more recent study also relatively small Neu5Ac- and Neu5Ac–Neu5Ac-containing oligosaccharides with a reducing-end hexose were identified. It was suggested that they were derived from glycosphingolipids, whereby the existence of a hitherto unknown endoglycosylceramidase involved in an alternative glycosphingolipid catabolic pathway was assumed. Unexpectedly, also related sialylated C1-oxidized (aldohexonic acid) versions were detected.569
Interestingly, Neu5Ac(α2 → 6)Man(β1 → 4)GlcNAc was isolated from the urine of a patient with β-mannosidosis.570
Furthermore, in the urine of patients with aspartylglucosaminuria, a series of neutral and acidic glycoasparagines, including Neu5Ac(α2 → 3/6)Gal(β1 → 4)GlcNAc(β1 → N)Asn, Neu5Ac(α2 → 3/6)Gal(β1 → 4)GlcNAc(β1 → 3)Gal(β1 → 4)GlcNAc(β1 → N)Asn, and Neu5Ac(α2 → 8)Neu5Ac(α2 → 3)Gal(β1 → 4)GlcNAc(β1 → N)Asn, are accumulated.568 Note that the history of elucidating aspartylglucosaminuria carbohydrates showed, besides GlcNAc(β1 → N)Asn, initially several proposals for structures, including compounds with terminal Neu5Ac(α2 → 4)Gal(β1 →, Neu5Ac(α2 → 7)Neu5Ac(α2 → 3)Gal(β1 →, and Neu5Ac(α2 → 9)Neu5Ac(α2 → 3)Gal(β1 → elements, that were found to be incorrect in later studies.571, 572, 573, 574, 575
Patients with Schindler disease type I, type II (Kanzaki), and type III, having an α-N-acetylgalactosaminidase deficiency, excrete relatively high concentrations of mono- and disialylated O-glycopeptides and related oligosaccharides in their urine.568, 576, 577, 578, 579
In this context, also pathological abnormalities of (i) lysosomal sialic acid transport (Salla disease) and (ii) a biosynthetic defect in the sialic acid metabolism (CMP-Neu5Ac overproduction; sialuria), described for the first time in 1979 and 1967, respectively, should be mentioned. Depending on the defect, the urinary excretions of Neu5Ac vary from (i) 5- to 10-fold (Salla disease580, 581) and 200-fold (infantile free sialic acid storage disease582) to (ii) 10,000-fold (sialuria; hypersialylation of glycoprotein O-glycans583, 584, 585, 586, 587) over the normal values, and increased levels in tissues and cells (for further information, see Sections 13.1 and 13.2).
8.5. Capsular Polysaccharides, Lipopolysaccharides, and Lipooligosaccharides
With the first characterized polysialic acid, called colominic acid, in the late 1950s, over the years a continuous flow of structures of sialic acid-containing microbial polysaccharides has appeared. Colominic acid is an (α2 → 8)-linked homopolymer of Neu5Ac, [→ 8)Neu5Ac(α2 → 8)Neu5Ac(α2 →]n, isolated from Escherichia coli K235.117, 588, 589, 590 Such a homopolymer, isolated from E. coli K1, can also be partially acetylated at Neu5Ac O7 and/or O9.36 A polysialic acid from Neisseria meningitidis Group C was identified as [→ 9)Neu5Ac(α2 → 9)Neu5Ac(α2 →]n (partly Neu5,7,8Ac3 and Neu5,7Ac2 or Neu5,8Ac2).35, 591 Note that in the late 1950s this homopolymer was suggested to be built up from hexosamine and sialic acid residues.592 A [→ 8)Neu5Ac(α2 → 9)Neu5Ac(α2 →]n polysialic acid was reported to be present in the E. coli K92 strain.593 In several heteropolysaccharides and core regions of lipopolysaccharides (lipooligosaccharides), the (O-acetylated) Neu5Ac(α2 → 3/6) unit was found in the terminal (side-chain) position.6, 594, 595 Interestingly, a microbial decasaccharide revealed the presence of a terminal Neu5,9Ac2(α2 → 6)Gal(β1 → sequence.596 Also heteropolysaccharide structures with internal sialic acid, e.g., → 4)Neu5Ac(α2 →; → 4)Neu5,7/9Ac2(α2 →; → 4)Neu5,9Ac2(α2 →; → 4)Neu5,7/8,9Ac3(α2 →; and → 7)Neu5Ac(α2 → units, were established.6, 595, 597 Typical examples are [→ 4)Neu5Ac(α2 → 6)Gal(α1 →]n,598 [→ 3)GalNAc(β1 → 4)Gal(α1 → 4)Neu5,9Ac2(α2 → 3)Gal(β1 →]n,39, 599 and {→ 3)GalNAc(β1 → 7)Neu5Ac(α2 → 3)[Glc(α1 → 2)]Glc(β1 →}n,600 and, as core region oligosaccharide of a lipopolysaccharide, Gal(α1 → 6)Glc(β1 → 7)Neu5Ac.601 For Kdn-containing capsular polysaccharides, terminal [Kdn(α2 →] and internal [→ 9)Kdn5Me(α2 →] units have been shown. In case of teichulosonic acids, containing Kdn residues, (β2 → 4)-linked Kdn backbones, decorated with single monosaccharide side-chain units, have been determined, e.g., {→ 4)[Glc(β1 → 8)]Kdn(β2 →}n, {→ 4)[Gal(β1 → 9)]Kdn(β2 →}n, and {→ 4)[Glc(β1 → 8)][Glc(β1 → 9)]Kdn(β2 →}n, as well as tetrasaccharides with two Kdn residues, which are (α2 → 4)-interlinked, e.g., Gal(β1 → 9)Kdn(α2 → 4)[Gal(β1 → 9)]Kdn.595, 602 The Kdn residues in the tetrasaccharide can be acetylated at O7 and O8.603
In polysialyl sequences intramolecular lactone formation between sialic acids has been observed.6 In the case of a Neu5Ac(α2 → 8)Neu5Ac sequence, lactonization yields a Neu5Ac(α2 → 8,1 → 9)Neu5Ac element, whereby the COOH group of one residue reacts with HO9 of an adjacent residue to give a six-membered ring. In a similar way, Neu5Ac(α2 → 9)Neu5Ac can be converted into Neu5Ac(α2 → 9,1 → 8)Neu5Ac.
8.6. General Remarks About the Structural Analysis of Glycans
In the late 1960s and early 1970s, the protocols for the structural analysis of carbohydrates underwent a great change. At first instance, GLC combined with EIMS, developed for the linkage analysis on the monomer level (methylation analysis), caused a revolution in polysaccharide and glycoconjugate characterization.273, 275 And with the introduction of high-resolution 1H and 13C NMR spectroscopy in the mid-1970s, the analysis of glycoprotein glycans, glycolipids, and polysaccharide-derived oligosaccharides came within reach.275, 465, 466, 468 Concerning sialoglycans, the chemical shifts of H3e and H3a of sialic acids (and other non-2-ulosonic acids) turned out to be highly indicative for the anomeric configuration, the type of linkage with neighboring residues, and the further microenvironment of these acids. Note that the pH of the solution can influence these values (Table 3). For illustration, a few examples at pH 7 will be given. α-Neu5Ac: H3e, δ 2.730/H3a, δ 1.621; β-Neu5Ac: H3e, δ 2.208/H3a, δ 1.827; Neu5Ac(α2 → 6)Gal(β1 → 4): H3e, δ 2.67/H3a, δ 1.71–1.72; Neu5Ac(α2 → 3)Gal(β1 → 4): H3e, δ 2.75–2.76/H3a, δ 1.80; Neu5Gc(α2 → 6)Gal(β1 → 4): H3e, δ 2.69–2.70/H3a, δ 1.73–1.74; Neu5Gc(α2 → 3)Gal(β1 → 4): H3e, δ 2.77–2.78/H3a, δ 1.81–1.82; Neu4,5Ac2(α2 → 6)Gal(β1 → 4): H3e, δ 2.67–2.68/H3a, δ 1.85; Neu4,5Ac2(α2 → 3)Gal(β1 → 4): H3e, δ 2.77/H3a, δ 1.93; Neu5Ac(α2 → 6)[R → 3]GalNAc-ol: H3e, δ 2.72–2.74/H3a, δ 1.69–1.71. For complete tables of H3e/H3a structure–reporter-group NMR data of Neu5Ac/Neu5Gc/Neu4,5Ac2/Neu5,9Ac2-containing structural elements, see refs. 466, 468.
Fortunately, the discovery of softer ionization techniques than electron-impact (EIMS) also made mass spectrometry suitable for the analysis of glycoprotein glycans, glycolipids, and (polysaccharide-derived) oligosaccharides. This revolution started with the introduction of fast-atom-bombardment mass spectrometry (FABMS) in the 1980s. However, today, MALDI-TOFMS and ESIMS play the main roles in carbohydrate analysis (for recent reviews, see refs. 275 and 604, 605, 606, 607, 608, 609, 610). Even intact glycoproteins can be unraveled now by ESIMS.611 For a series of papers, specifically focused on mass spectrometry of intact sialoglycans, including information about the stabilization of sialic acid, see refs. 612, 613, 614, 615, 616, 617, 618. For the differentiation between (α2 → 3) and (α2 → 6) linkages several elegant protocols have been developed. For instance, treatment of Neu5Ac(α2 → x)Gal(β1 → 4)-glycans (x = 3 or 6) with 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (carboxylic acid activator) in methanol yielded methyl ester derivatives for (α2 → 6)-linked Neu5Ac and lactone derivatives for (α2 → 3)-linked Neu5Ac [Neu5Ac(α2 → 3,1 → 2)Gal(β1 → and/or Neu5Ac(α2 → 3,1 → 4)Gal(β1 →], providing a difference of 32 Da in mass between both glycosidic linkage types.617 A very recent example comprises the treatment of Neu5Ac(α2 → x)Gal(β1 → 4)-glycans (x = 3 or 6) with dimethylamine, 1-ethyl-3-(3-dimethylamino)propyl)carbodiimide (activator)/1-hydroxybenzotriazole (catalyst) in dimethyl sulfoxide, affording dimethylamidate derivatives for (α2 → 6)-linked Neu5Ac and lactone derivatives for (α2 → 3)-linked Neu5Ac (difference in mass: 45 Da).619 Subsequent treatment with ammonium hydroxide had only effect on the lactone ring of the (α2 → 3)-linked Neu5Ac, resulting in the formation of an amidate derivative (difference in mass: 28 Da).620
9. Growing Interest From the Organic and Enzymatic Chemistry
As has been described in Section 4.1, first reports on the organic and enzymatic synthesis of Neu5Ac date from the 1960s, in a period that questions around the different chiralities were still not completely solved. The protocols comprised chemical coupling of oxaloacetic acid (or its di-tert-butyl ester) and either d-GlcNAc or d-ManNAc, followed by decarboxylation (Scheme 6),228, 235, 236, 237 and enzymatic coupling of pyruvic acid and d-ManNAc in the presence of Neu5Ac aldolase (N-acylneuraminate pyruvate lyase) (Scheme 5).231, 232 For the preparation of Neu5Gc, d-ManNAc was replaced by d-ManNGc. Next routes to prepare Neu5Ac enzymatically were the condensation of d-ManNAc with phosphoenolpyruvic acid catalyzed by Neu5Ac synthase (rat liver extract) in the presence of ATP, or by Neu5Ac synthase using nonphosphorylated ManNAc (meningococcal extract).239, 621, 622 In the following years, different routes and improvements to obtain higher yields (even at the multikilogram level) were reported.6, 100, 623, 624, 625, 626 In this context, d-ManNAc was frequently generated from the cheaper d-GlcNAc in an alkaline epimerization process. Note that an excellent biological source for the isolation of large amounts of Neu5Ac was initially the urine of sialuria patients (see Sections 8.4 and 13.1),584 and later edible bird's nest mucin153, 548 and hen's egg.627 For the isolation of large amounts of Neu5Gc from holothuroidea gumi, Cucumaria echinata, see ref. 628. With the characterization of a long series of N,O-acylneuraminic acids in the late 1970s and early 1980s (Section 5), synthetic activities in this direction were also started. Via organic synthesis, several free O-acetylated sialic acids became available, e.g., Neu4,5Ac2, Neu5,9Ac2, Neu9Ac5Gc, Neu4,5,9Ac3, and Neu5,7,8,9Ac4.629, 630, 631, 632 Additionally, a large series of N,O-acetylneuraminic acid alkyl/aryl ester α-alkyl/α-aryl glycosides were synthesized: Neu4,5Ac2, Neu5,7Ac2, Neu5,8Ac2, Neu5,9Ac2, Neu5,7,8Ac3, Neu5,7,9Ac3, Neu5,8,9Ac3, Neu4,5,9Ac3, Neu4,5,8,9Ac4, and Neu5,7,8,9Ac4.633, 634, 635 Enzymatic protocols using Neu5Ac aldolase were developed for the synthesis of Neu5,9Ac2 and Neu5Ac9Lt.636, 637 A number of other naturally occurring sialic acids have also been synthesized along organic chemical or aldolase-catalyzed routes. Examples are as follows: Neu2en5Ac,638 Neu2en5Gc,75, 638 Neu2en5,9Ac2,56 Neu2en5,7,9Ac3,56 Neu2en9Ac5Gc,56 Neu2en5GcAc,56 Kdn,639, 640, 641 Kdn2en,642 Neu2,7an5Ac,643, 644 Neu5Ac8Me,645 Neu5Ac9P,51 Neu5Ac1,7lactone,646, 647, 648 Neu5Gc1,7lactone,647 and Kdn1,7lactone.647 See also refs. 6 and 252. More recently, a highly efficient, one-pot, three-enzyme system with flexible substrate specificity was developed, whereby sialic acid family members are generated as intermediates from corresponding N,O-acyl-d-mannosamines and O-acyl/O-alkyl-d-mannoses (catalyst: a sialic acid aldolase) in a protocol that leads via CMP-sialic acids (catalyst: a CMP-sialic acid synthetase) to sialic acid-containing glycosides or sialyl(α2 → 3 or α2 → 6)galacto-glycans (catalyst: a bacterial sialyltransferase).649, 650, 651, 652 Reported examples of intermediates comprise Neu5Ac, Neu5,9Ac2, Neu5Ac9Lt, Neu5Gc, Neu9Ac5Gc, Neu5GcAc, Neu5GcMe, Kdn, Kdn5Ac, Kdn5Me, Kdn9Ac, and Kdn5,9Ac2. For the study of biochemical pathways and to understand interactions at the molecular level, isotopically labeled and (stereochemically) modified sialic acids (at C1 and C3 to C9) were also prepared; for reviews, see refs. 6, 106, 108, 252, 626, and 626, 653, 654, 655, 656, 657, 658, 659, 660.
The scientific interest in the synthesis of sialo-oligosaccharides dates from the end of the 1960s. The first synthetic compounds comprised two disaccharides with Neu5Ac in a reducing position, i.e., Glc(β1 → 9)Neu5Ac1Me and Gal(β1 → 9)Neu5Ac1Me.661 Furthermore, a series of reducing disaccharides with Neu5Ac in a nonreducing position were synthesized, although in low yields: Neu5Ac(α2 → 3)Glc, Neu5Ac(α2 → 3)GlcNAc, Neu5Ac(α2 → 6)Glc, Neu5Ac(α2 → 6)Gal, and Neu5Ac(α2 → 6)GlcNAc.662 Other interesting disaccharides were nonreducing Neu5Ac(α2↔2α)Neu5Ac and Neu5Ac(α2↔2β)Neu5Ac.663 Higher yields for Neu5Ac(α2 → 6)Gal were obtained in the early 1980s,664 but efforts to synthesize Neu5Ac(α2 → 6)GlcNAc in larger amounts failed, as mainly Neu5Ac(β2 → 6)GlcNAc was formed.665
It was soon realized that stereoselective chemical sialylation leading to α-glycosidic linkages is one of the most difficult glycosylation reactions in carbohydrate chemistry. First, a neighboring participating group is missing to direct the stereochemical outcome of the coupling (C3 is a deoxy group). Second, the coupling reaction takes place at a sterically hindered, tertiary oxocarbenium-ion intermediate. Third, the deoxy moiety in combination with the electron-withdrawing carboxylic acid group (C1) at the anomeric center (C2) makes sialic acid donors prone to glycal formation. Because it was realized that the availability of labeled sialic acids, sialic acid derivatives, sialic acid O-, N-, C-, and S-glycosides, sialo-analogues, and sialo-oligosaccharides was of the utmost importance for unraveling the secrets of the sialobiology and sialomedicine, the interest in sialochemistry grew enormously. In this context, the creation of a great variety of suitable sialic acid donors and catalysts for organic chemical approaches was developed.6, 107, 137, 660, 666, 667, 668, 669, 670, 671, 672 Furthermore, the progress in the biotechnology of carbohydrate enzymes led also to the generation of suitable amounts of stereoselective sialyltransferases/CMP-Neu5Acyl and trans-sialidases, which made enzymatic approaches attractive.107, 126, 137, 139, 142, 660, 669, 673, 674, 675, 676
In this way, growing activities focused on a further exploration of the properties of modified sialic acids, the preparation of substrates and inhibitors for sialidases, sialyltransferases, trans-sialidases, or sialic acid-converting enzymes, the synthesis of sialo-glycans, and to make compounds accessible for analytical purposes or for studies related to metabolism or biological functions of sialic acids. Over the years, several detailed reviews, describing different aspects of sialic acid organic and enzymatic synthesis, were published (see references in the foregoing paragraphs of this section).
10. Structural Variants of the Sialic Acid Backbone
A series of naturally occurring nonulosonic acids (NulOs), which do not belong to the classical sialic acid (Sia) family, contain a terminal methyl group as C9 and aminodeoxy functions at C5 and C7, and compared with neuraminic acid (Neu), may exhibit differences in the configuration of the chiral centers at C4, C5, C7, and C8 (Fig. 27 ). According to IUPAC recommendations, these structural variants of the sialic acid backbone are systematically named 5,7-diamino-3,5,7,9-tetradeoxynon-2-ulosonic acids. In the early days of their discovery, the mid-1980s, they have sometimes been called “bacterial sialic acids.” They were found specifically in bacterial lipopolysaccharides and capsular polysaccharides, and since the turn of the century, also in bacterial glycoproteins. Over the years, a number of reviews about these nonulosonic acids have appeared.114, 595, 677, 678, 679, 680, 681
Fig. 27.

2C5 Chair conformations of α-Pse, β-Leg, β-4eLeg, β-8eLeg, α-Aci, and α-8eAci. Note the difference in orientation of the COOH and OH groups at the anomeric C2 atom.
The first data about the occurrence of these “sialic acid-like” monosaccharides as components of lipopolysaccharides are from 1984 and reported by Knirel and coworkers at the N.D. Zelinsky Institute of Organic Chemistry, Moscow (Russia).682 In view of the date of their first discovery, it may not be surprising that, in contrast to all the approaches used in the development of the sialic acid field, the analytical techniques had completely changed. Now, from the beginning onward, 1H and 13C NMR spectroscopy and mass spectrometry played major roles in their characterization. Since 2017, six subclasses (six isomers) have been identified, namely,
-
(i)
5,7-diamino-3,5,7,9-tetradeoxy-l-glycero-l-manno-non-2-ulosonic acid, called pseudaminic acid (Pse);
-
(ii)
5,7-diamino-3,5,7,9-tetradeoxy-d-glycero-d-galacto-non-2-ulosonic acid, called legionaminic acid (Leg);
-
(iii)
5,7-diamino-3,5,7,9-tetradeoxy-d-glycero-d-talo-non-2-ulosonic acid, called 4-epi-legionaminic acid (4eLeg);
-
(iv)
5,7-diamino-3,5,7,9-tetradeoxy-l-glycero-d-galacto-non-2-ulosonic acid, called 8-epi-legionaminic acid (8eLeg);
-
(v)
5,7-diamino-3,5,7,9-tetradeoxy-l-glycero-l-altro-non-2-ulosonic acid, called acinetaminic acid (Aci); and
-
(vi)
5,7-diamino-3,5,7,9-tetradeoxy-d-glycero-l-altro-non-2-ulosonic acid, called 8-epi-acinetaminic acid (8eAci).
It should be noted that during the starting period of this research field, some tentative structures were published with misassignment of the chiral configurations. Therefore, to solve these problems, at the end of the 1990s 5,7-di-N-acetyl derivatives with the d-glycero-d-galacto, l-glycero-d-galacto, d-glycero-d-talo, l-glycero-d-talo, d-glycero-l-manno, l-glycero-l-manno, d-glycero-l-altro-, l-glycero-l-altro, and l-glycero-l-gluco configuration were synthesized by condensation of suitable 2,4-diacetamido-2,4,6-trideoxyhexoses with oxalacetic acid under basic conditions.683, 684, 685 In contrast to reports from the time of their discovery, presently, the earlier chosen trivial names Pse, Leg, 4eLeg, and 8-Leg have been directly related to the nonsubstituted mother molecules with free amino functions,595 and the same system was followed for the recently established Aci and 8eAci,686, 687 in a similar way as usual for the Sia family (Neu). As in the case of neuraminic acid, in the full names C7 is the anomeric reference atom. The free sugars exist in the 2 C 5 pyranose form as mixtures of α- and β-anomers with a predominance of the thermodynamically more stable anomer having an equatorial carboxyl group. Substitutions of the amino groups at C5 and C7 comprise acetyl (Ac), formyl (Fo), (R)-3-hydroxybutyryl (3RHb), (S)-3-hydroxybutyryl (3SHb), 4-hydroxybutyryl (4Hb), 3,4-dihydroxybutyryl (3,4Hb), acetimidoyl (Am), N-methyl-acetimidoyl (AmMe), N,N-dimethyl-acetimidoyl (AmMe2), methyl (Me), d-alanyl (Ala), N-acetyl-d-alanyl (AlaNAc), N-methyl-5-glutamyl (GluNMe), l-glyceryl (Gr), and/or 2,3-di-O-methyl-glyceryl (Me2Gr) groups. Note that only N-acylated derivatives are known; there are no derivatives with free NH25 or free NH27 groups. Furthermore, acetyl, N-acetyl-glutaminyl (GlnNAc), and glycyl (Gly) groups have been detected at O8, and acetyl groups at O4.
A survey of over 45 known structures79, 602, 682, 684, 686, 687, 688, 689, 690, 691, 692, 693, 694, 695, 696, 697, 698, 699, 700, 701, 702, 703, 704, 705, 706, 707, 708, 709, 710, 711, 712, 713, 714, 715, 716, 717, 718, 719, 720, 721, 722, 723, 724, 725, 726, 727, 728, 729, 730 of naturally occurring 5,7-diamino-3,5,7,9-tetradeoxynon-2-ulosonic acid derivatives, together with some related 9-deoxynon-2-ulosonic acids, is presented in Table 4 . The symbolic abbreviations for the various derivatives of the 5,7-diamino-3,5,7,9-tetradeoxynon-2-ulosonic acids have been chosen in accordance with the Sia recommendations. The constituents can occur in homo- and heteropolysaccharides, as terminal and internal units, with (α2 → or (β2 → glycosidic linkages. In case of internal residues, the HO4 or HO8 functions can be substituted by another monosaccharide. The same holds for the hydroxy groups of the 3- or 4-hydroxybutyramido substituents at C7. In flagellins, the nonulosonic acids are directly connected to Ser or Thr, e.g., Pse5,7Ac2(α2→O)Ser.689 For pilins, an O-linked trisaccharide with Pse7Fo5(3Hb) as the terminal unit has been demonstrated to occur,703 whereas Leg7Ac5Fo was found to be a terminal unit of an N-linked pentasaccharide of a haloarchaeal virus glycoprotein.709 For further biochemical information, see Section 11.5.3.
11. Unraveling the Metabolism of Sialic Acids
First observations on the metabolism of sialic acids were made at the beginning of the 1940s when sialic acid (at that time an unknown low-molecular-weight substance) was released from cells by a viral or bacterial enzyme, termed the RDE (see Section 2.1). This catabolic enzyme was later called neuraminidase or sialidase. Another catabolic enzyme, the acylneuraminate–pyruvate lyase (N-acetylneuraminic acid aldolase) from bacteria, was shown to split N-acetylneuraminic acid into pyruvate and N-acetylmannosamine (see Section 4.1). This serendipitous discovery by the group of Saul Roseman231, 232 at the end of the 1950s turned out to be a milestone in (sialo)glycobiology, leading to Roseman being named “Dr. Saul Serendipity Roseman.”230 The finding of N-acetylmannosamine instead of N-acetylglucosamine also played a major role in the elucidation of the final structure of N-acetylneuraminic acid.
In a detailed personal letter handed over to Yuan C. Lee, and recently published by Schnaar and Lee,731 Roseman describes the full GlcNAc vs ManNAc story, wherein Don Comb played such a great role. With respect to the quantification of N-acetylhexosamine using the Morgan–Elson color reaction, he memorized that this assay was used for the acylneuraminate–pyruvate lyase degradation of N-acetylneuraminic acid. Although the amounts of N-acetylneuraminic acid (consumed) and pyruvate (formed) were equal, the amount of formed N-acetylhexosamine was only half of the expected amount (reaction time, 3 min). Citing Roseman: “There are two ways to run the Morgan–Elson reaction; one involves heating for 3 min, while the other requires 12 min. There was no way that Don would waste time and heat for 12 min when 3 min was sufficient. But the incredible thing, however, is that both of these sugars [ManNAc and GlcNAc (R.S.)] give the same color when they are heated for 12 min. In other words, the stoichiometry results would have been perfect if he had heated for 12 min and not 3 min. I have often wondered where we would be if Don had heated for 12 min. Would we still think that the Gottschalk structure is correct?” Note that up to this time, N-acetylneuraminic acid was thought to be the condensation product of pyruvate and GlcNAc (see Section 4.1).
For the biosynthesis of sialic acid, it was believed in the beginning that the process is reversibly catalyzed by an enzyme, first observed in extracts of V. cholerae, which degrades N-acetylneuraminic acid to an N-acetylhexosamine and pyruvate.227 The enzyme also occurred in mammalian tissues and was purified 1700-fold from hog kidney cortex.328 As said (see Section 4.1), Comb and Roseman found the same enzyme in C. perfringens and identified the N-acetylhexosamine product as N-acetylmannosamine.231, 732 Three arguments, however, were not in favor of this acylneuraminate–pyruvate lyase to be responsible for sialic acid synthesis: (i) in cells, the reaction equilibrium of this enzyme is largely in favor of sialic acid degradation; (ii) the enzyme does not exist in tissues producing large quantities of sialylated mucins328; and (iii) the bacteria V. cholerae and C. perfringens do not contain sialic acid, but express the sialic acid aldolase. Thus, this aldolase is a degradative enzyme, although the reaction is reversible. For an earlier proposed reaction mechanism of the catalysis by this N-acetylneuraminic acid lyase, as well as a recent proposal based on crystallography, QM/MM simulation, and mutagenesis, see ref. 6 and ref. 733, respectively.
At this time, at the end of the 1950s, the “golden” and laborious time of the study of the metabolism of sialic acids began.
11.1. The Biosynthesis of N-Acetylneuraminic Acid
The biosynthesis of sialic acids, both in animals and in microorganisms, is linked to glycolysis. This central metabolic pathway provides both fructose-6-phosphate and phosphoenol pyruvate as basic substances for the biosynthesis of sialic acids. The chemical steps involved were primarily elucidated in the laboratories of Roseman734 and Warren.735 It had been shown in the 1950s by several researchers (see, e.g., refs. 736, 737) that radioactive d-glucose is converted into d-glucosamine. Moreover, the precursor of d-glucosamine-6-phosphate was investigated in microorganisms738, 739 and in mammals (rat liver).739, 740 Here, the biosynthetic route comprised an amination of d-fructose-6-phosphate (not d-glucose-6-phosphate) with the amido group of l-glutamine catalyzed by l-glutamine-d-fructose-6-phosphate aminotransferase (glucosamine-6-phosphate synthetase), to give d-glucosamine-6-phosphate and l-glutamate.739 The product d-glucosamine-6-phosphate is then acetylated by an N-acetyltransferase, which occurs in many animals and microorganisms, and was first isolated 254-fold from Neurospora crassa.741
The next step is the transformation of N-acetylglucosamine-6-phosphate into N-acetylglucosamine-1-phosphate, catalyzed by a phosphoacetylglucosamine mutase, isolated from different sources.742, 743, 744 The sialic acid biosynthesis continues with the formation of UDP-N-acetylglucosamine from N-acetylglucosamine-1-phosphate and UTP, and the liberation of pyrophosphate by a pyrophosphorylase.745 UDP-N-acetylglucosamine (UDP-GlcNAc) was first detected in rat liver extracts and in yeast.746 This sugar nucleotide could then, in 1955, be synthesized in extracts of yeast and Rous sarcoma tumors.747
The epimerization of N-acetylglucosamine (GlcNAc) to N-acetylmannosamine (ManNAc) is a key step in sialic acid biosynthesis. In this reaction, the acetylamino group at C2 is epimerized, and the linkage between UDP and the amino sugar is hydrolyzed (UDP-ManNAc could not be detected) (Fig. 28A). It should be noted that this reaction was discovered in rat liver in 1957, but erroneously the reaction product was described as GalNAc.748 The rectification to ManNAc followed one year later732; the responsible enzyme was isolated from rat liver in 1966.749 The reaction was shown to be irreversible, and the enzyme occurred widely in vertebrate tissues; however, it was not found in bacteria.
Fig. 28.

Metabolism of non-2-ulosonic acids having the d-glycero-d-galacto configuration. (A) N-Acetylneuraminic acid (Neu5Ac) in vertebrates114; (B) N-acetylneuraminic acid (Neu5Ac) in bacteria660; (C) ketodeoxynononic acid (Kdn) in vertebrates12; and (D) 5,7-di-N-acetyllegionaminic acid (Leg5,7Ac2) in bacteria.595d-6dManNAc4NAc, 2,4-diacetamido-2,4,6-trideoxy-d-mannose; GDP-α-d-6dGlcNAc4NAc, GDP-2,4-diacetamido-2,4,6-trideoxy-α-d-glucose; Kdn9P, ketodeoxynononic acid 9-phosphate; d-ManNAc6P, N-acetyl-d-mannosamine 6-phosphate; d-Man6P, d-mannose 6-phosphate; Neu5Ac9P, N-acetylneuraminic acid 9-phosphate; PEP, phosphoenolpyruvate; Pi, phosphate; PPi, diphosphate. In case of CMP-β-Leg5,7Ac2, also another route, going from UDP-α-d-GlcNAc, via UDP-α-d-6dGlcNAc4NAc to d-6dManNAc4NAc, has been formulated.595
For the biosynthesis of sialic acid in vertebrates, ManNAc had to be converted into N-acetylmannosamine-6-phosphate (ManNAc6P), and a corresponding kinase was detected in mammalian liver and submandibular glands (see, e.g., ref. 750), as summarized in ref. 751. It can use both ManNAc and N-glycolylmannosamine (ManNGc). Many years later (1990s), it was shown by the group of Werner Reutter at the Freie Universität Berlin (Germany) that the conversion of UDP-GlcNAc into ManNAc6P is catalyzed by one bifunctional enzyme, the UDP-N-acetylglucosamine 2-epimerase (GNE)/N-acetylmannosamine kinase (MNK), a crucial step often discussed between Reutter and one of the authors (R.S.). This enzyme, purified from rat liver, both epimerizes UDP-GlcNAc under release of UDP and phosphorylates the resulting ManNAc by its kinase activity.752 In the same study, also feedback inhibition of the UDP-N-acetylglucosamine 2-epimerase part of the enzyme by CMP-Neu5Ac was demonstrated. In a parallel report, the molecular cloning and functional expression of the GNE/MNK enzyme were published.753 A selective loss of either the epimerase or the kinase activity was established, via site-directed mutagenesis based on sequence alignments. This technique also enabled the localization of the 2-epimerase activity in the N-terminus and the kinase activity in the C-terminus of the protein.754 GNE, the root of which is in the Bilateria, was found to have a complex evolutionary origin.755 It is encoded in the genomes of deuterostomes, acoelomorphs, and Xenoturbella, but is absent in protostomes and nonbilaterians.
In order to study the inhibition of the sialic acid biosynthesis, a search for inhibitors of human N-acetylmannosamine kinase by high-throughput screening of drug-like small molecules was carried out,756 and a few promising compounds binding to the ATP-binding pocket of the enzyme were found. 3-O-Methyl-N-acetylmannosamine inhibited the same enzyme and correspondingly affected the sialic acid concentration in cells.757
In vertebrate tissues, the formation of sialic acid as its 9-phosphate ester (Neu5Ac9P) (Roseman–Warren pathway, Fig. 28A) occurs by condensation of ManNAc6P and phosphoenolpyruvate in an irreversible manner. A synthesizing, anabolic “sialic acid-9-phosphate synthetase” responsible for this reaction was discovered in the early 1960s by the Warren and Roseman groups.51, 621, 735 The first sources were rat liver and bovine submandibular gland. The enzyme reaction favors the production of sialic acid.
Before the activation of Neu5Ac by CMP, the 9-phosphate group must be removed. This is achieved by a specific Neu5Ac-9-phosphate phosphatase,735 first seen in rat liver, and later isolated 800-fold from human erythrocytes (Fig. 28A).758 This enzyme, completing the Neu5Ac biosynthesis, only occurs in animals. It does not exist in bacteria, where Neu5Ac is formed from nonphosphorylated ManNAc and phosphoenolpyruvate under release of phosphate (Fig. 28B).759, 760
In summary, at the end of these challenging studies in the 1950s and 1960s, three pathways were described as leading to Neu5Ac:
-
(i)
The Neu5Ac aldolase (lyase) route, which condenses ManNAc and pyruvate and occurs in both animals and bacteria;
-
(ii)
The sialic acid-9-phosphate synthetase route, which condenses ManNAc6P with phosphoenolpyruvate and exists only in animal tissues;
-
(iii)
The bacterial sialic acid synthetase route, condensing ManNAc with phosphoenolpyruvate.
11.2. The Activation of Sialic Acids
The sialylation of glycan chains (see Section 8 for structural elements) requires in general an activated Neu5Ac donor substrate, which is obtained by reaction of Neu5Ac with CTP, resulting in the nucleotide sugar CMP-Neu5Ac and pyrophosphate (Fig. 28A) (for trans-sialidases, see Section 11.7). The glycosidic linkage between Neu5Ac and CMP has the β-configuration.761, 762, 763 The responsible enzyme, CMP-sialic acid synthetase, a pyrophosphorylase (EC 2.7.7.43), was first detected in bacteria and purified from N. meningitidis.764 At the same time, it was also found in animal tissues765, 766 and was first purified from pig submandibular glands,767 as reviewed in ref. 768. The enzyme from different sources was found to react with Neu5Ac, Neu5Gc, and O-acetylated sialic acids, and some of them also with Kdn.6 Remarkably, in contrast to the other sialic acid-synthesizing enzymes, CMP-Neu5Ac synthetase is located in cell nuclei, first observed by Ed Kean769 at Case Western Reserve University (Cleveland, USA). From the nucleus, CMP-Neu5Ac is transported into the Golgi, using a carrier-mediated transport system to penetrate liver microsomal vesicles (mice studies).770 For the cloning and expression of this CMP-sialic acid transporter, see ref. 771. Kean, who devoted most of his scientific life to the investigation of sialic acid activation, discovered also a CMP-Neu5Ac hydrolase in the plasma membranes of rat liver. Its activity was low in other tissues.772 The role of this hydrolase is still a subject of speculation. Especially in liver, with its most active sialic acid metabolism, the membrane-bound CMP-Neu5Ac hydrolase may regulate the extent of attachment of sialic acid residues to cell membrane glycoconjugates by membrane sialyltransferases.
In the Golgi, CMP-Neu5Ac is used as the substrate for sialyltransferases, which are responsible for sialic acid polymerization (polySia) and sialylation of oligo/polysaccharides, glycoproteins, and glycolipids (see Section 11.3). It additionally serves as a substrate for the N-acetyl-hydroxylation reaction leading to Neu5Gc (see Section 11.5.1) and for the O-acetylation of sialic acids (see Section 11.5.2). These modified CMP-sialic acids are also transferred to glycoconjugates in the Golgi. For the synthetic production of CMP-Neu5Ac, see, e.g., refs. 763, 773, and 774.
11.3. Sialic Acid Transfer to Glycans
Studies of the group of sialyltransferases comprise one of the largest and most important fields in sialobiology, and a wealth of papers during the last decades deals with their occurrence, properties, structure, and function. The enzymes are mostly Golgi-membrane-bound. Since several excellent reviews cover this subject, here only some of the milestones will be reported, and the reader is referred to refs. 6, 126, 138, 775, and 776.
In 1963, the first sialyltransferase activities were observed in studies on the biosynthesis of colominic acid589 and the sialylation of lactose.777 In the latter case in which details were worked out in Roseman's laboratory, a particulate preparation from the rat mammary gland was incubated with lactose and CMP-[1-14C]Neu5Ac to yield radiolabeled Neu5Ac(α2 → 3)lactose. In the field of glycolipids, the biosynthesis of GM3 ganglioside was shown from an incubation of lactosyl-ceramide, CMP-[1-14C]Neu5Ac, and a particulate preparation from a 9-day-old embryonic chicken brain.778 In Roseman's laboratory the period between 1963 and 1970 was most fruitful, and the detection of four different sialyltransferases for the biosynthesis of glycoproteins and four different sialyltransferases for the biosynthesis of gangliosides was reported by various coworkers (summarized in refs. 230, 779, and 780). These studies suggested that the different sialyltransferases were responsible for the formation of a variety of sialyl glycosidic linkages. The first purification of a sialyltransferase from bovine colostrum by affinity chromatography on CDP-hexanolamine agarose was achieved by the group of Robert Hill at Duke University Medical Center (Durham, NC, USA).780 The specific activity increased 440,000 times when compared with whole colostrum. The enzyme was pure and consisted of two bands of 43 and 56 kDa, respectively. The enzyme turned out to be a β-d-galactoside α2,6-sialyltransferase.781 The enzyme was cloned782 and is cleaved in a whole or post-Golgi compartment.783 The expression of this ST6Gal-I, regulated by multiple transcriptional promotors, is tissue specific.
Meanwhile, 20 distinct sialyltransferases, which are Golgi type II transmembrane glycosyltransferases, have been identified in murine and human genomes. This superfamily comprises four main sialyltransferase families: (i) an ST3 Gal family with six members (ST3Gal I–VI), whereby sialic acid is (α2 → 3)-linked to galactose (Gal); (ii) an ST6 family with two members (ST6Gal I–II), whereby sialic acid is (α2 → 6)-linked to Gal; (iii) an ST6GalNAc family with six members (ST6GalNAc I–VI), whereby sialic acid is (α2 → 6)-linked to N-acetylgalactosamine (GalNAc); and (iv) an ST8Sia family with six members (ST8Sia I–VI), which mediates the transfer of Neu5Ac residues to other Neu5Ac residues of glycoproteins and glycolipids via (α2 → 8) linkages.775 In the latter reference, a phylogenetic approach to animal sialyltransferases and the related genes was undertaken. In this excellent analysis the architecture of these enzymes, which is similar in all vertebrate sialyltransferases, is discussed. Also the precursor genes for animal sialyltransferases are suggested, e.g., for ST6Gal from insects (Drosophila melanogaster).10 There are also over 25 subfamilies in lower vertebrates, orthologs of the 20 mammalian subfamilies, as well as additional subfamilies in fish genomes.138 For the interesting evolutionary history of the ST8Sia gene family since the early deuterostomes, see ref. 784. Recently, it was shown that the cellular availability of sialic acids regulates the expression of various sialyltransferases at the level of transcription.785
Since sialyltransferases are the final enzymes of sialic acid anabolism, they are responsible for the physiologically correct sialylation of glycans in cells, and their activity should be decreased by inhibitors of cellular development or of aberrant oversialylation, such as that which may occur in cancer (see Section 12.3). A natural inhibitor was first purified in 1988 from the 100,000 × g supernatant of a rat-brain homogenate.786 It is a small, heat-stable protein composed of two components of 14.8 and 22.4 kDa, which inhibits brain α2,6-sialyltransferase noncompetitively. Inhibitory activity was also found in brains from other mammals and in nonbrain tissues. A similar, potent inhibitor was isolated from calf brain, which inhibited (95% by 4 μg of inhibitor) the α2,6-sialyltransferase from ovine submandibular glands.787 Experiments to chemically modify CMP-Neu5Ac in either its nucleotide or sialic acid moiety in order to gain effective sialyltransferase inhibitors were not successful. Instead, cytidine nucleotides inhibited rat liver α2,6-sialyltransferase (α2,6-ST) and porcine submandibular gland α2,3-sialyltransferase (α2,3-ST) well and at similar rates. The K i-values of 5′-CMP, 5′-CDP, and 5′-CTP varied between 0.13 and 0.05 mM for α2,6-ST.788 Probably the strongest sialyltransferase inhibitor presently known is 3-fluoroax-N-acetylneuraminic acid (3Fax-Neu5Ac), which is supplied to cells or tissues in per-O-acetylated methyl ester form.789 The hydrophobic molecules penetrate into cells, where they are O-deacetylated. The liberated free 3Fax-Neu5Ac more efficiently inhibits α2,3-sialyltransferases and, consequently, hypersialylation of cancer cells. In vivo, delayed growth of inhibitor-treated melanoma cells was observed.
11.4. The Regulation of Sialic Acid Biosynthesis
Such an important biochemical pathway as that of sialic acid synthesis, which results in a great variety of bioactive compounds, must be regulated, since the amount of CMP-sialic acids in a cell may be critical, and influences the level of the sialylation grade in glycoconjugates. The first hint came from studies by Kean.769 The fact that CMP-sialic acid synthetase is located in cell nuclei of various prominent mammalian tissues suggested that this location is an important control mechanism. Furthermore, two feedback inhibition mechanisms of key enzymes of sialic acid synthesis were detected, i.e., (i) UDP-GlcNAc efficiently inhibits the l-glutamine-d-fructose-6-phosphate aminotransferases, and (ii) CMP-Neu5Ac inhibits UDP-N-acetylglucosamine 2-epimerase (GNE).790 Newer views on the latter enzyme (GNE/MNK) have been discussed in Section 11.1. An example of the lack of the feedback inhibition by CMP-Neu5Ac, required for a physiologically controlled sialic acid metabolism, came from studies of the metabolism of a sialuria patient583 (see Section 13.1). This young patient excreted gram quantities of free Neu5Ac in urine, as well as Neu2en5Ac, 2-acetamidoglucal, ManNAc, and GlcNAc.52, 585 2-Acetamidoglucal represents the putative transition state between UDP-GlcNAc and ManNAc.
11.5. Modified Sialic Acids Different From N-Acetylneuraminic Acid
The most frequent natural sialic acid in Nature is Neu5Ac, as is understandable from its biosynthetic pathway. However, from the survey in Table 1 it is also evident that there exist a large number of other sialic acids, variously substituted at the amino and/or hydroxy groups as well as dehydro and anhydro forms. In the following subsections, various derivatives of Neu5Ac will be discussed.
11.5.1. N-Glycolylneuraminic Acid
Leonard Warren, then at the National Institutes of Health, Bethesda, USA, described in 1963 the occurrence of Neu5Gc in different animals.2 Neu5Gc belongs to the group of the most frequently encountered sialic acids, and it is widely distributed in Nature. Although it has never been found in microorganisms (with the exception of Trypanosoma cruzi cultivated in Neu5Gc-containing medium791), it is often expressed in species ranging from echinoderms to mammals.5, 6, 7 However, exceptions are known, such as sauropsides (birds and reptiles), and the monotremes, such as platypus and echidna,792 as well as ferrets, New World monkeys, and man. Very recently, a study appeared about the phylogenetic distribution of the CMP-Neu5Ac hydroxylase (CMAH) gene in 322 deuterostome genomes. It was found that the gene has been lost or inactivated at least 31 times during deuterostome evolution.793
Since humans frequently consume Neu5Gc (mainly with red meat), they incorporate a small portion of this sialic acid into their tissues.794 Neu5Gc is a xeno-antigen and may cause the production of anti-Neu5Gc antibodies. Evidence exists that this can lead to inflammations and tumors, e.g., in the colon.795 African Great Apes regularly synthesize Neu5Gc in contrast to man.796 Not much is known about the function of Neu5Gc.6 Some aberrations in behavior and physiology like retarded wound healing and hearing loss, similar to human pathologies, were observed in Neu5Gc-knockout mice.797 Neu5Gc may even be toxic. Remarkably, in mammalian brains Neu5Gc exists in only very small amounts. This may be of physiological significance, since experiments leading to a higher expression of Neu5Gc in mouse brain resulted in some neuronal disturbancies.798 On the other hand, as far as it has been investigated, fetal tissues express more Neu5Gc than adult tissues, as shown in calf and pig.799, 800
Neu5Gc was first isolated and structurally analyzed by Blix and coworkers,23 and later it was chemically synthesized by Faillard and Blohm.801 Elucidation of the biosynthesis of Neu5Gc was a tedious procedure; first attempts go back to 1967.270, 802 Theoretically, the origin of the N-glycolyl group is a glycolic acid residue, e.g., from coenzyme A, or it is generated by a direct oxidation of the N-acetyl group of the sialic acid precursors GlcNAc and ManNAc, or Neu5Ac itself. Evidence for the formation of glycolyl-CoA from hydroxypyruvate was demonstrated in porcine liver.803 However, glycolyl-CoA seemed to be a less likely N-glycolyl precursor [only traces of O-glycolylated sialic acids have been detected in bovine and equine submandibular glands (Table 1)]. Furthermore, radioactively labeled 3-hydroxypyruvate and serine were not precursors of (radioactive) Neu5Gc.804 Thus, a transfer of glycolyl groups during Neu5Gc biosynthesis was largely excluded. The following two studies were in agreement with a direct oxidation of the N-acetyl group, as had been (theoretically) proposed 10 years earlier805: (i) direct hydroxylation of N-acetyl anilids to N-glycolyl anilids in liver microsomes806; (ii) oxidation of UDP-N-acetylmuramic acid to UDP-N-glycolylmuramic acid before incorporation into the peptide glycan of the bacterial cell wall.807
With respect to the suggestion of a direct oxidation of the N-acetyl group in the case of Neu5Gc, first experiments with homogenates of porcine submandibular glands did not show the formation of N-glycolylglucosamine (GlcNGc) from the sialic acid precursor GlcNAc.270, 802 This pointed to a sialic acid modification at a higher biosynthetic step. Incubation of surviving slices of porcine submandibular glands with N-[1-14C]acetyl-glucosamine led to labeling of sialic acids, including Neu5Gc having a radioactive glycolyl group (for analysis of glycolyl groups, see Section 7.1). The direct incorporation of 18O from 18O2 gas, resulting in N-[18O]glycolyl groups in Neu5Gc, was also shown.808 Further incubations with [1-14C]acetate, N-[1-14C]acetyl-mannosamine, and N-[1-14C]acetyl-neuraminic acid led in each case to the identification of N-[1-14C]glycolyl-neuraminic acid.809, 810, 811 All these results led to the claim of the existence of an N-acetylhydroxylase, involved in Neu5Gc biosynthesis. Most of the newly formed, radioactive Neu5Gc was within the tissue still free and not yet incorporated into glycoconjugates. As shown by the group of one of the authors (R.S.), CMP-Neu5Gc (and CMP-Neu5Ac) is formed in porcine submandibular glands and can act as the donor for the transfer of Neu5Gc to nascent glycoproteins by sialyltransferases.812 This led to the assumption that Neu5Gc was formed before the transfer to glycoconjugates in the Golgi.
In the same period, using particle-free homogenates from porcine submandibular glands, it was found that the hydroxylation of the N-acetyl group of Neu5Ac required, besides of oxygen, the addition of NADPH or ascorbic acid as cofactors.811 The responsible enzyme was denominated N-acetyl-neuraminate:O2-oxidoreductase, and later Neu5Ac monooxygenase (EC 1.14.18.2). Although activities were found both in the cytosol and in microsomes, they were generally low, whereas glycan-bound Neu5Ac as a substrate was inactive. A later reevaluation of experiments, however, made clear that the positive tissue fractions were contaminated with CMP-Neu5Ac.
Then, in 1988, CMP-Neu5Ac was found to be the real and only substrate for the monooxygenase in porcine submandibular glands, exhibiting highest activity in the soluble protein fraction.813 The same activity was also detected in the high-speed supernatant fraction of mouse liver.814 The renamed enzyme, CMP-N-acetylneuraminate hydroxylase, was purified from porcine submandibular glands815 and mouse liver.816 The molecular weight of the monomeric enzyme from both sources was determined to be 65 kDa. In the same years, the group of Akemi Suzuki at Tokyo Metropolitan Institute of Medical Science (Japan) elucidated the complex mechanism of electron transport required for hydroxylase activity, in which NADH, cytochrome b5,817 and cytochrome b5 reductase818 in mouse liver cytosol were involved. This cytochrome b5-dependent electron transport chain is soluble or membrane-bound.819 The enzyme activity can be stimulated by the addition of iron and is inhibited by iron-binding agents, which suggested the existence of an iron-containing prosthetic group.820 Electron paramagnetic resonance (EPR) spectroscopy of the primary structure of the hydroxylase revealed that this enzyme is an iron–sulfur protein of the Rieske type (2Fe–2S). Interestingly, it was the first example of such an enzyme found in the cytosol in Eukarya.821 The Rieske center is located close to the N-terminus of the enzyme protein. For a first molecular cloning of the CMP-Neu5Ac hydroxylase (CMAH) gene of mouse liver, see ref. 822. As the CMAH gene is defective in humans, it became clear why Neu5Gc cannot be synthesized in man.823 Finally, the group of Ajit Varki at the University of California (San Diego, USA) discovered that the Rieske sulfur–iron center is missing, which is due to an Alu-mediated inactivation of the human CMP-Neu5Ac hydroxylase gene.824, 824a
11.5.2. O-Acetylated Sialic Acids
The existence of O-acetylated sialic acids was assumed very early, when Blix, in 1936, isolated and crystallized sialic acids from bovine submandibular gland mucin under mild acid conditions and found more than one acetyl group per sialic acid molecule (“Kohlenhydrat I”)165 (see Section 2.1). And in a report from 1956, more information about “7-O- and 7,8(9)-O-acetylated N-acetylneuraminic acids” after periodate oxidation studies was provided23, 263 (see Section 5). In the same period, O-acetylated sialic acids were also isolated from bovine mucin by incubation with sialidase (V. cholerae receptor-destroying enzyme)264 (see Section 5). Larger quantities of various O-acetylated sialic acids from bovine submandibular gland were obtained by a combination of anion-exchange and cellulose chromatography.24 The challenges and problems of analyzing O-acetylated sialic acids (Table 1) have already been worked out in Section 5. A turning point in the history of assigning O-acyl substitution patterns was a report from the laboratories of both authors (J.P.K. and R.S.) in 1975, describing the determination of “8-O-acetyl-N-acetylneuraminic acid” as 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2). An unexpected misinterpretation of periodate oxidation data24 was rectified by EIMS27 (see Section 5). And over the years it became clear that Neu5,9Ac2 is the most frequent O-acetylated sialic acid occurring in Nature.
The newer analytical techniques made also the study of O-acyl migration in sialic acids possible, and Neu4,5Ac2, Neu5,7Ac2, Neu5,9Ac2, Neu5,7,9Ac3, and Neu5,8,9Ac3 were subjected to such analyses.38, 38a, 101, 261 TLC, HPLC, and GLC–EIMS experiments showed that Neu5,7Ac2, dissolved in pH 7.5 buffer, was almost completely converted into Neu5,9Ac2 after 30 h at 37°C (only 10% O-deacetylation). At pH 5.0 the substance was relatively stable. This migration phenomenon was studied in more detail by 360-MHz 1H NMR spectroscopy.38,38a Using the peak intensities of the N-acetyl signals of α,β-Neu5,7Ac2 and α,β-Neu5,9Ac2, Fig. 29A illustrates the decrease of Neu5,7Ac2 and the increase of Neu5,9Ac2 as a function of the time of incubation in 0.1 M phosphate buffer/D2O (pD 7.2) at 37°C. In the migration process Neu5,8Ac2 could not be traced as an intermediate. This could mean that a direct intramolecular transfer Ac7 (secondary ester group) → Ac9 (primary ester group) occurs. But note that so far, an Ac7 → Ac8 → Ac9 mechanism has not been excluded. In Fig. 29B a similar plot is depicted for the conversion of Neu5,7,9Ac3 to Neu5,8,9Ac3 (0.1 M phosphate buffer/D2O, pD 7.5), yielding a molar equilibrium of approximately 1:1. A transient distortion of the anti orientation of the functional groups at C7 and C8 seems to be necessary to explain the migration of the O-acetyl group between the two secondary OH groups (see Section 6). For 4-O-acetyl-N-acetylneuraminic acid no migration of the O-acetyl group was observed.
Fig. 29.

Time plots for the migration of O-acetyl groups in (A) Neu5,7Ac2 (α:β = 23:77) → Neu5,9Ac2 (α:β = 9:91) and (B) Neu5,7,9Ac3 (α:β = 22:78) → Neu5,8,9Ac3 (α:β = 3:97) in 0.1 M phosphate, pD 7.2–7.5, at 37°C, as monitored by 360-MHz 1H NMR spectroscopy. As a result of the migration, an anomerization process will also occur.
Reproduced from Kamerling, J. P.; Schauer, R.; Shukla, A. K.; Stoll, S.; van Halbeek, H.; Vliegenthart, J. F. G. Migration of O-Acetyl Groups in N,O-Acetylneuraminic Acids. Eur. J. Biochem.1987, 162, 601–607. Copyright Wiley.
Application of HPLC and MS techniques revealed that O-acylated sialic acids are most widely distributed, much more than Neu5Gc. They occur in various bacterial species and in vertebrates and invertebrates, from echinoderms to mammals, including those species lacking Neu5Gc, such as monotremes, sauropsides, and humans. For extensive reviews, see refs. 5, 6, 7, 792, 825, and 826. In these references, the great variety of biological functions of O-acetylated sialic acids is also described.
The O-acetyl groups render the respective sialic acid in most cases completely resistant, as in Neu4,5Ac2, or partially resistant, as in Neu5,7Ac2 or Neu5,9Ac2, toward the action of sialidases or sialate lyases. They act as differentiation and tumor antigens, suppress apoptosis, and strongly influence receptor interactions, which also leads to modulation of the immune system. Note that 9-O-acetylation of Neu5Ac and Neu5Gc interferes with the binding of sialoadhesin.827, 827a, 828 Increase of virulence by O-acetylation was early observed in E. coli K1.36 Sialic acids, O-acetylated at C4 or at the side chain, are also ligands for some viruses,826 first studied with influenza C virus. It took some time to demonstrate that the RDE of this virus is a sialate-O-acetylesterase, which O-deacetylates Neu5,9Ac2.829 In contrast, O-acetylation of the sialic acids of mouse erythrocytes inhibits binding of the malaria parasite Plasmodium falciparum and infection of the red blood cells.830
For a long time, O-acetylated sialic acids appeared as enigmatic molecules in glycobiology, and they were treated as Cinderella molecules, because of their lability, the difficulties in purification, and characterization of the underlying O-acetylating enzyme proteins, as well as the elucidation of their molecular biology. So, it was difficult in the beginning to convince researchers that it may be rewarding to invest time and effort in the study of sialate O-acetyltransferases (SOATs), which were rather reluctant to reveal their secrets.
The experiments to gain first insight into the biosynthesis of the O-acetylation of sialic acids were methodologically similar and carried out at about the same time as the study on the Neu5Gc biosynthesis. When surviving slices of submandibular glands from cow and horse were incubated with [1-14C]acetate at various times at 37°C in an atmosphere of oxygen, the radioactivity was almost exclusively incorporated into the O-acetyl groups (and N-acetyl and N-glycolyl groups) of sialic acids within the time of incubation of maximum 4 h.831 Similar to the Neu5Gc synthesis, the highest specific radioactivity of O-acetylated sialic acids was found in the fraction of free sialic acids, followed by that in the microsomal fractions, and the cytosolic sialic acid of the mature glycoproteins, which were least labeled. These findings were interpreted as O-acetylation also occurs before the sialic acid transfer, either at the hexosamine level (which would not be possible for the horse 4-O-acetylated sialic acid) or at the “free sialic acid” level. In the latter case, free Neu5Ac, Neu5Ac-9-phosphate, or CMP-Neu5Ac were candidates of O-acetyltransferase substrates. Indeed, incubation of homogenates of bovine submandibular gland with N-[1-14C]acetyl-neuraminic acid and acetyl-CoA led to N-[1-14C]acetyl-7- and -8-O-acetylneuraminic acid, as shown by radio-TLC analysis and based on our knowledge of sialic acid derivatives at that time (at the end of the 1960s). Hints for the existence of O-acetylated N-acetylhexosamines were never obtained, and O-acetylation of mature mucin-bound cytosolic sialic acids did not occur.832 Therefore, a systematic name for the enzyme was proposed: “acetyl-CoA:N-acetylneuraminate-7(8)-O-acetyltransferase.” As mentioned earlier, the 8-O-acetyl position was later corrected to be a 9-O-acetyl position, and correspondingly the enzyme was denoted as a 7(9)-O-acetyltransferase (Fig. 30 ). In equine submandibular gland and guinea pig liver, a corresponding acetyl-CoA:sialate-4-O-acetyltransferase (EC 2.3.1.44) was detected. Kinetic and other properties of the enzyme were studied in microsomes, prepared from equine submandibular glands, and in an enzyme solubilized from guinea pig liver, respectively.833, 834 In the years that followed, 7(9)-O-acetyltransferase activity was seen in some bacteria and various tissues and cells, ranging from starfish to man.826
Fig. 30.

Metabolism of sialic acid O-acetylation (here Neu5Ac), including a survey of enzymes involved in the transfer and removal of O-acetyl groups. The flowers indicate the hydroxy groups found to be O-acetylated. The hands symbolize the position specificity of the O-acetyltransferase activities discovered: sialate-4-O-acetyltransferase, sialate-7-O-acetyltransferase, and sialate-9-O-acetyltransferase. The scissors represent the sialate-4-O-acetylesterase and sialylate-9-O-acetylesterase, involved in the hydrolysis of the ester groups. The O-acetyl group at the sialic acid glycerol side chain can migrate between C7 and C9 (via C8?), whereas the 4-O-acetyl group seems to be immobile.
If sialic acid is modified before the transfer to nascent glycoconjugates, the enzymatic coupling of various sialic acids to CMP should be possible. Indeed, this was shown to be the case for the CMP-sialate synthetase from bovine submandibular glands, which activates Neu5Ac, Neu5Gc, Neu5,7Ac2, and Neu5,8Ac2 (in fact Neu5,9Ac2; see above).835 Sialyltransferases were also shown to be not absolutely strict toward the kind (structure) of activated sialic acid.836 Only in 2007, it was established that CMP-Neu5Ac was the correct substrate for O-acetyltransferase, and CMP-Neu5,7Ac2 was identified as the primary reaction product. Accordingly, the enzyme's name was then modified into AcCoA-CMP-Neu5Ac 7-O-acetyltransferase (EC 2.3.1.45).837 Interestingly, ab initio calculations gave information concerning possible roles of the different functional groups occurring on sialic acids.307 Here, the relatively negative charge on the sialic acid side chain O7 supports the hypothesis that in O-acetyltransferase reactions this position is the primary insertion site of the acetyl group, and not the less negative oxygen at C9 (Fig. 30). Afterward, the O-acetyl group migrates to O9, and the participation of a “migrase” was discussed.838 Before the 2007 Kiel report,837 much effort had been undertaken by the group of Ajit Varki to elucidate the mechanism of sialic acid O-acetylation in rat liver.839, 840 A transmembrane transfer of acetyl groups was observed for which histidine and lysine residues were essential. And it was hypothesized that the O-acetyltransferase activity might be associated within a membrane-bound complex composed of the acetyl-CoA transporter, the O-acetyltransferase, and sialyltransferase activities, and possibly an acetylated intermediate.841 The same group solubilized acetyl-CoA:polysialic acid O-acetyltransferase from K1-positive E. coli and studied its basic properties.842 Unfortunately, it was not possible, in spite of a lot of effort in various laboratories, to isolate and characterize the transferase from this subcellular location, and to get molecular genetic insight on the basis of the amino acid sequence of the enzyme.
However, between 2005 and 2009 several bacterial sialic acid-O-acetyltransferase genes from E. coli and Neisseria meningitides were identified.843, 844 These genes and their encoded proteins do not exhibit significant similarities to animal genes and gene products. Campylobacter jejuni also expresses a sialate-O-acetyltransferase, which transfers the acetyl groups directly to the O9 position of terminal (α2→8)-linked sialic acids, as shown by NMR spectroscopy (Fig. 30).845
Great progress was achieved by using a rational approach to identify the mammalian sialate-O-acetyltransferase (EC 2.3.1.45).846 To this end, the human genome database was screened for genes that are predicted to possibly encode acetyltransferases and are potentially located in the Golgi membrane. The candidate gene turned out to be CasD1 (capsule structure1 domain containing 1), which is similar to a gene of the fungus Cryptococcus neoformans (this yeast contains O-acetylated mannose). Various experiments were described indicating a direct involvement of the human Cas1 including siRNA protein (Cas1p) in the O-acetylation of sialic acids. Expression of this protein and characterization as O-acetyltransferase were achieved more recently.847 CMP-Neu5Ac as substrate of the enzyme was confirmed.
11.5.3. Other Natural Sialic Acids
In addition to the N-glycolyl group and O-acetyl groups in various positions of the neuraminic acid molecule, other, partially rare substituents were also found, as well as dehydro, anhydro, and lactone derivatives. Since the various functions are often combined, nowadays, over 80 sialic acid derivatives have been identified in Nature (see Table 1; Fig. 31 ).
Fig. 31.

Survey of naturally occurring substituents of the sialic acid backbone. For a full survey of structures, see Table 1.
A first example is the 9-O-phosphorylated form of Neu5Ac (Neu5Ac9P). This sialic acid only occurs in free form in very low concentrations with high turnover rate as intermediate of the Neu5Ac biosynthesis (see Section 11.1; Fig. 28A). The 9-O-l-lactylation of sialic acids was first seen in 1976, by the identification of Neu5Ac9Lt in bovine submandibular gland mucin42 and in human serum and saliva.43 Strikingly, in the latter sources Neu5Ac9Lt was quite often the main N,O-acylneuraminic acid. 9-O-Lactylation of sialic acid was also found in horse and trout (see Table 1). The function of this sialic acid is not yet known, but it seems to be formed enzymatically, as was studied with a particulate fraction from horse liver.848
So far, O-methylated sialic acids in significant quantities have been found only in echinoderms, in various starfish species. The first sialic acid of that type, 8-O-methylated Neu5Gc (Neu5Gc8Me), was seen in the starfish Asterias forbesi.67 The Neu5Ac analogue was later traced.25 For O-acetylated derivatives of both Neu5Ac8Me and Neu5Gc8Me, see Table 1. S-Adenosyl-l-methionine:N-acetylneuraminate 8-O-methyltransferase (EC 2.1.1.78), involved in the sialic acid methylation, was detected in Asterias rubens 46 and purified 22,000-fold from this source.849 This enzyme methylates both free and bound sialic acids. More recently, a 9-O-methylated variant of Neu5Ac, Neu5Ac9Me, was also found.47
In the period between 1976 and 1990 much attention was paid to 8-O-sulfated sialic acids (see Table 1) being present in glycoproteins (see Section 8.1) and glycolipids (see Section 8.2). Here, the group of Kochetkov and coworkers at the Academy of Sciences in Moscow (Russia), working on sialoglycolipids of sea urchin species (Neu5Ac8S, Neu5Gc8S; e.g., ref. 49), was quite active. Neu5Ac8S was also described for a sialoglycosphingolipid from bovine gastric mucosa.50 With newly developed chemical and immunological techniques, the distribution of Neu5Ac8S and Neu5Gc8S in various cellular sites of sea urchin sperm and eggs was recently shown.850 Note also the occurrence of Neu5Ac4S in a ganglioside from the sea cucumber Holothuria pervicax 48 (see Section 8.2) and Neu5Gc9S in a ganglioside from the sea urchin Hemicentrotus pulcherrimus 72 (see Section 8.2). The enzyme responsible for the sulfation of sialic acid is not yet known.
2-Keto-3-deoxynononic acid (Kdn) is a sialic acid in which the amino function at C5 of neuraminic acid (Neu) is substituted by a hydroxy group (Fig. 31). As mentioned in Section 4.4, this substance was discovered in 1986 in cortical alveolar polysialoglycoproteins of rainbow trout eggs,259 composed mainly of Neu5Gc as sialic acid residue.76 These sialoglycoproteins were identified 10 years earlier as a novel class of glycoproteins in the unfertilized eggs of rainbow trout.851 Meanwhile, Kdn is found in all types of glycoconjugates of bacteria and vertebrates, including mammals, in various linkages like Neu5Ac, and in free form in tissues, including cancer tissue. A recent study on the occurrence of free sialic acids in head and neck cancers of the throat revealed relatively high levels of Kdn (2 μg/g tissue) when compared with Neu5Ac and also Neu5Gc. Thus, Kdn was discussed to be of prognostic value, as a biomarker, for detecting some early-stage cancers at biopsy.852 The occurrence of Kdn in bacteria was first reported in 1989, namely, in the acidic capsular polysaccharide from Klebsiella ozaenae, which causes respiratory diseases in man.77 Kdn was also detected in trout gangliosides524 and in different organs of the pig, although in smaller amounts than Neu5Ac and Neu5Gc.800
For a review from 2006, describing 20 years of history of biological studies on deaminated neuraminic acid, see ref. 12. Focusing here on the metabolism (Fig. 28C), a Kdn-9-phosphate synthase, which condenses mannose-6-phosphate and phosphoenol pyruvate to yield Kdn-9-phosphate, was identified in rainbow trout ovaries and in testes. Thus, the biosynthesis of Kdn is similar to that of Neu5Ac, using Man as the hexose precursor instead of ManNAc. After dephosphorylation, the Kdn that is formed can be linked to CMP (CMP-Kdn synthetase853) and transferred to glycans. There exist some enzymes specific for Kdn, including a KDNase (see Section 11.7), but there are also enzymes of the Neu5Ac metabolism that can react with (CMP-)Kdn. For example, a sialyltransferase activity, which catalyzes the transfer of Kdn from CMP-Kdn to the nonreducing termini of the polysialyl chains of polysialoglycoproteins, was found in rainbow trout ovary854 (see Section 8.1). This capping with Kdn of the polysialyl chains prevents their further elongation.
Information about structural variants of the sialic acid backbone, i.e., the 5,7-diamino-3,5,7,9-tetradeoxynon-2-ulosonic acids pseudaminic acid (Pse), legionaminic acid (Leg), 4-epi-legionaminic acid (4eLeg), 8-epi-legionaminic acid (8eLeg), acinetaminic acid (Aci), and 8-epi-acinetaminic acid (8eAci), has been given in Section 10 and Table 4. For data about homologous biosynthetic pathways for sialic acids and 5,7-diamino-3,5,7,9-tetradeoxynon-2-ulosonic acid derivatives, see refs. 595, 679, 680, 681, 686, 699, 705, 708, 716, 855, and 856. As an example, the biosynthetic pathway of 5,7-di-N-acetyllegionaminic acid (Leg5,7Ac2),856 having also the d-glycero-d-galacto configuration, has been included in Fig. 28D. The biosynthetic pathway of 5,7-di-N-acetylpseudaminic acid (l-glycero-l-manno-configuration) has also been unraveled.857, 858 Here, UDP-α-d-GlcNAc is converted via UDP-β-l-6dAltNAc4NAc and l-6dAltNAc4NAc into Pse5,7Ac2, and then into CMP-α-Pse5,7Ac2 (l-6dAltNAc4NAc = 2,4-diacetamido-2,4,6-trideoxy-l-altrose). For reviews dealing with biological/immunological functions of non-2-ulosonic acids, other than sialic acids, see refs. 595, 679, 681, and 855.
11.5.4. Nonnatural Sialic Acids
The study of the metabolism and biological functions of sialic acids with hexosamine precursors substituted at the amino group with nonnatural functions, inaugurated by Werner Reutter in a visionary way,859 opened a new area in chemical sialobiology, referred to as metabolic glycoengineering. By the end of the 1960s, Reinhard Brossmer at the Max-Planck-Institut für medizinische Forschung (Heidelberg, Germany) had synthesized, among others, N-formyl- and N-succinyl-neuraminic acid and had studied the influence of these foreign substituents on sialidase activity860 (see Section 11.7). At the beginning of the 1980s, the group of Reutter reported that N-propionylglucosamine (GlcNProp) and N-propionylmannosamine (ManNProp) inhibited the Neu5Ac biosynthesis in a cell-free system of rat liver,861 and the formation of N-propionylneuraminic acid (Neu5Prop) was demonstrated. Following these studies, the uptake of N-propionylhexosamines was studied in more detail.862, 863 In rat liver, Neu5Prop biosynthesized from ManNProp was found back in the membrane and serum glycoproteins, as well as in gangliosides. Although some N-acetylmannosamine analogues exhibited a degree of toxicity to cells, with this glycoengineering, various modulations of cell functions became possible. For example, human peripheral blood mononuclear cells could be immunomodulated by the replacement of about 10% of their sialic acids with Neu5Prop, after incubation with ManNProp. The cells proliferated faster, produced more IL-2, upregulated their receptor CD25, and expressed the transferrin receptor.864 Modified sialic acids were found to stimulate the proliferation of astrocytes and microglia cells in the rat central nervous system865 and modulate virus–receptor interactions.866 Growth of diploid human fibroblasts with ManNProp, N-butyrylmannosamine (ManNBut), and N-valerylmannosamine (ManNVar) destroyed their sensitivity to contact inhibition of growth.867 N-Acyl modification of sialic acids seems to be promising in cancer therapy. Treatment of human neuroblastoma cells with ManNProp or ManNVar reduced their sialylation and metastatic potential and increased their sensitivity toward radiation and chemotherapeutics. Furthermore, ManNPent inhibited polysialylation.868 The formation of cell membrane N-azidoacetylneuraminic acid (Neu5Az) after the application of per-O-acetylated N-azidoacetylmannosamine (ManNAz) also offers many possibilities for cell biological, including histochemical (see Section 7.4), experiments.413 Recently, this rapidly growing area of the metabolic glycan labeling strategy was excellently and thoroughly reviewed.869, 870 It reports many more applications than mentioned here, and it summarizes that mannosamines with modified aliphatic N-acyl functions, like ManNProp, can be used for the investigation of Neu5Ac-dependent biological processes. In contrast, mannosamines with bio-orthogonal modified N-acyl groups, like ManNAz, are mainly used to visualize sialylation and sialic acid metabolism in vitro and in vivo.
11.6. The De-esterification of O-Acetylated Sialic Acids
The discovery of more than 20 different naturally occurring O-acetylated sialic acids (see Table 1) implies the necessity of O-acetylesterases for the catabolism of these substances, especially, since sialidases and sialate–pyruvate lyases, the next enzymes in the catabolic reaction sequence of bound sialic acids, are strongly influenced in their activity by O-acetyl groups. Although 4-O-acetylated sialic acids are resistant toward the action of the latter enzymes [an exception is the fowl plague virus sialidase (see Section 11.7)], no accumulation of sialoglycoconjugates in corresponding tissues, like in the horse, leading to storage diseases, was observed. A first hint for the existence of a “missing link” in the catabolism of O-acetylated sialic acids was obtained in the 1980s when the ganglioside Neu4Ac5Gc-GM3, isolated from horse erythrocytes, was incubated with partially purified equine liver sialidase.871 Unexpectedly, non-O-acetylated Neu5Gc was slowly released, which was later explained by the possible presence of a 4-O-acetylesterase activity in the sialidase preparation.872 More recently, two esterases could be isolated from the 100,000 × g supernatant of horse liver by isoelectric focusing, one O-deacetylating both Neu4,5Ac2 and Neu5,9Ac2, and the other only Neu5,9Ac2.873 Both enzymes were inactive with 7-O-acetylated sialic acids, as was also observed for other sialate esterases.
Sialate-O-acetylesterases (EC 3.1.1.53) were studied in bacteria,125 in mammalian tissues, for example, rat liver,841 bovine brain,874 and horse liver,873 in corona and toro viruses,875 and in influenza C viruses. For a summary of the enzyme tests available, see ref. 876. The sialate-O-acetylesterase in influenza C virus was one of the first esterases discovered. Although an enzyme destroying the virus receptor had been demonstrated to exist on influenza C viruses in 1950,877 the specificity of this enzyme and the nature of the cellular receptor for this virus have remained puzzling ever since. Then, in the 1980s, it was found that sialic acid is involved in the receptor function for the virus, because sialidase destroyed a potent hemagglutination inhibitor present in rat serum, i.e., α1-macroglobulin.878 The nature of this sialidase-susceptible sialic acid was found to be Neu5,9Ac2, which lost its O-acetyl group by incubation of rat α1-macroglobulin or bovine submandibular gland mucin with influenza C virus.829 This pointed to the presence of an esterase in the virus, which was thereafter isolated and denoted as sialate 9(4)-O-acetylesterase, because it hydrolyzed both Neu4,5Ac2 and Neu5,9Ac2.879 For a further characterization and molecular cloning, see ref. 880. In additional studies, the group of José Cabezas at the Universidad de Salamanca (Spain) found that the influenza C virus esterase had broad substrate specificity, hydrolyzing not only natural substrates containing Neu5,9Ac2 (e.g., bovine submandibular gland mucin) but also many nonsialic acid O-acetyl-containing compounds.881 This esterase turned out to be part of the hemagglutinin-esterase-fusion (HEF) protein complex of influenza C virus, the crystal structure of which has been elucidated in 1998.882 In the same period, the HEF protein complex from mouse hepatitis virus strain S was shown to contain a sialate 4-O-acetylesterase part.883 It should be noted that, due to steric hindrance, all these esterases never can act on sialic acid 7-O-acetyl groups. Therefore, for O-deacetylation, the 7-O-acetyl group must first migrate to position 9 (Fig. 30) in order to get access to the catalytic center of the enzyme.838 In further investigations, the human sialate-9-O-acetylesterase, the gene of which is located on chromosome 11, gained much interest, since it was shown to play a key role in the regulation of B-cell response and the level of O-acetylation in human colon mucosa,884 and to be involved in immune and autoimmune events.885, 886, 887 In a thorough study of the human sialic acid acetylesterase, including a genomic and phylogenetic analysis, the size of the enzyme and its subcellular localization, the composition of the active center, and other properties have been addressed.888 One of the most interesting target molecules of this 9-O-acetylesterase is the ganglioside 9-O-acetylated GD3, which influences a variety of biological functions, from apoptosis to the growth and metastasis of tumor cells.826 Its elevated concentration in a variety of tumors was described.
11.7. exo-, endo-, and trans-Sialidases
Sialidases are involved in the catabolism of sialic acids. As is clear from the foregoing, sialidases were discovered much earlier than the enzymes involved in the biosynthesis of these sugars (see Section 11.1). In 1942, George Hirst, in the Walter and Eliza Hall Institute of Research in Melbourne (Australia), directed by Frank Macfarlane Burnet (1899–1985), observed a factor eluting influenza viruses from erythrocytes, to which they were bound, resulting in agglutination of the red cells after a period of incubation.889 Some years later, in the same institute, it was discovered that culture filtrates of various Clostridia species made erythrocytes nonagglutinable by influenza virus.890 They had lost the ligand for virus binding by an enzyme-like activity. In the same year, similar reactions were described with V. cholerae filtrates.175 Additionally, the enzyme-like agent, in the meantime called RDE, was found in various viruses of the mumps influenza group,891 and later also in pneumococci.227 As discussed in Section 2.1, the released low-molecular-weight substances turned out to be sialic acids.
The RDE that catalyzes the release of terminal sialic acid residues was initially incorporated in the IUB Nomenclature Recommendations of 1961 with the name of “neuraminidase,” as proposed in 1957 by Gottschalk.892 This name was changed in the IUB Enzyme Nomenclature 1984 edition893 into “sialidase,” but the name “neuraminidase” was kept as an “other name,” e.g., viral “sialidases” are quite often called “neuraminidases” because of the use of “N” in their strain designation. In this context, it should be noted that bound neuraminic acid (the mother molecule) is not a substrate for the enzyme. For further enzyme nomenclature discussion, see ref. 894.
After these observations of viral and bacterial sialidases, this enzyme was also discovered in vertebrate tissues, first in both human and bovine plasma,895 and then in the allantoic membrane of the embryonic chicken,896 and in several organs of the rat.897 The following years revealed an explosion of sialidase discoveries in microorganisms and animals. The enzyme turned out to be common in metazoan animals of the deuterostomate lineage from echinoderms to mammals. Various fungi, protozoa, and many pathogenic as well as nonpathogenic bacteria express sialidase. Viral sialidases have been found mainly in orthomyxoviruses, like avian, porcine, equine, and influenza viruses, and in paramyxoviruses to which belong Sendai, Simian, mumps, and Newcastle disease viruses. Presently, influenza virus sialidases, which undergo antigenic drift and are of great pathophysiological significance in influenza, are probably the most studied sialidases. Bacterial sialidases are gaining interest since they are involved in many infectious diseases. For example, C. perfringens sialidase, which is involved in the dangerous gas gangrene of muscle tissue, was purified898 and can be detected by an immunoassay.899
The numerous sialidases known are all different regarding their molecular weight, pH dependency (mostly slightly acidic), substrate specificity, sialic acid structure specificity, kinetics, subcellular site, solubility, and susceptibility to inhibitors. They can release α-glycosidically bound sialic acids from N- and O-glycoprotein glycans, gangliosides, oligosaccharides, polysialic acids, and synthetic substrates at rather different rates (Fig. 32A). The isolated bacterial sialidases from A. ureafaciens,900 C. perfringens,898 V. cholerae,901 and Salmonella typhimurium,902 used in structural analysis studies, have similar broad substrate specificities for Neu5Ac. However, a linkage preference [Neu5Ac(α2 → 6)Gal, Neu5Ac(α2 → 3)Gal, Neu5Ac(α2 → 8)Neu5Ac] does exist. The sialidases from Newcastle disease virus903, 904 and Streptococcus pneumoniae 905 do not cleave the Neu5Ac(α2 → 6)Gal linkage. The Newcastle disease virus sialidase is also resistant to the Neu5Ac(α2 → 6)GalNAc linkage.904 For more information about the activity of the latter two sialidases on reduced or reductive aminated Neu5Ac(α2 → 6)GalNAc (open ring form of GalNAc), see refs. 905 and 906. endo-Sialidase, which hydrolyzes (α2 → 8) linkages in sialic acid polymers, first observed with colominic acid, was detected in the bacteriophage E infecting E. coli bacteria.907 The latter studies comprised rather tedious procedures because the source of the enzyme was feces from elephants kept in a zoo, and the smell during work-up was difficult to be tolerated by the members of the laboratories (Stephan Stirm, personal communication). Nowadays, the gene structures of many isolated sialidases have been analyzed. For reviews over the years, see refs. 6, 89, 113, 676, and 908, 909, 910, 911, 912, 913, 914, 915.
Fig. 32.

(A) exo- and endo-Sialidase activities. (B) trans-Sialidase activities.
Microbial sialidases seem to be inducible by free or bound sialic acids. This was first observed with V. cholerae 916, 917 and C. perfringens 918 sialidases. In the case of the bacterial enzyme, an induction of a permease was observed, enabling the uptake of Neu5Ac into the bacteria. The K M value of such a (hypothetical) permease reaction was 3.3 × 10− 4 M. Furthermore, Neu5Ac lyase activity was also induced in the Clostridia by free Neu5Ac. Today, many sialic acid transporters are known,919 e.g., a first characterized bacterial sialic acid transporter from E. coli.920
Sialidases are active, although at rather variable rates, with many naturally or synthetically modified α-linked sialic acids.909, 910, 921 Two examples from the beginning in the late 1950s are the demonstration that, using V. cholerae culture filtrates, O-acetylated sialic acids are released from bovine submandibular mucin at a slower rate than Neu5Ac,264 and that Neu5Gc is hydrolyzed from the stroma of bovine erythrocytes.196 All studies revealed that Neu5Ac is released at the highest rate, and Neu5Gc and sialic acids O-acetylated at the glycerol side chain (Neu5,7Ac2, Neu5,8Ac2, or Neu5,9Ac2) are released at slower rates, while Neu4,5Ac2 shows no activity with most sialidases.6, 24, 101 The exception is the sialidase from fowl plague virus, which can slowly hydrolyze the glycosidic bond of Neu4,5Ac2.922 It should be noted that the sialic acid lyase behaves toward these naturally substituted sialic acids in a similar way, i.e., it, for example, cannot cleave Neu4,5Ac2.330 Using chemically modified sialic acids, it was shown that a smaller function at the sialic acid amino group, like an N-formyl group, or a larger one, like an N-succinyl substituent, much reduces the V. cholerae sialidase activity, when compared with an N-acetyl group as in Neu5Ac. While N-succinylneuraminic acid is not released at all by the enzyme, the N-formyl derivative is slowly hydrolyzed.860 For more details about this topic, see refs. 6 and 910.
Mammalian sialidases have been investigated very intensively, because they are involved in the regulation of sialic acid degradation, and play a great pathophysiological role in genetic, immunological, and tumor diseases. Among many other researchers, the group of Taeko Miyagi at the Miyagi Cancer Center Research Institute (Miyagi, Japan) has worked very early and intensively on this group of sialidases. Four of them, NEU1–NEU4,113 are of great importance: (i) lysosomal sialidase NEU1 initiates the degradation of sialoglycoconjugates923; (ii) cytosolic sialidase NEU2 exhibits highest activity with gangliosides. It was cloned from rat skeletal muscle924; (iii) the plasma membrane-associated sialidase NEU3 is specific for gangliosides. It was cloned from bovine brain925; and (iv) sialidase NEU4, bound to the outer mitochondrial membranes via protein–protein interactions, has a wide substrate specificity from glycoproteins to gangliosides and oligosaccharides. It also occurs in the lysosomal lumen. For the cloning of NEU4, see ref. 926.
All these sialidases exert many cell biological functions from cell growth to apoptosis. Being involved in cancer, NEU3 is downregulated in some tumors like colon cancer and acute lymphoblastic leukemia (ALL).113, 927 Importantly, increase of sialidase activity in tumor cells by transfection reduces their aggressiveness. In a recent study with transcripts of the sialidases (NEU1, NEU3, and NEU4) of 11 types of cancer tissues from 170 patients, it was found that NEU1 and NEU3 were upregulated, while NEU4 was downregulated in most cancer types. NEU3 was expressed highest in colorectal and ovarian tumors, and NEU1 in ovarian cancer.928 The four human sialidase isoenzymes exhibited pronounced differences in their ability to hydrolyze the Neu5,9Ac2 α-glycosidic linkage. With the exception of NEU4, they hydrolyzed the O-acetylated species slower than Neu5Ac and Neu5Gc.929
The mammalian sialidases, including the bacterial and protozoal enzymes, contain a sequence motif of eight amino acids Ser/Thr-X-Asp-X-Gly-X-Thr-Trp/Phe, called the “Asp-box” and a Phe/Tyr-Arg-Ile-Pro motif near the sialidase N-terminus. These motifs, probably involved in the enzymatic cleavage reaction, were observed by comparison of the genes of a variety of mainly Clostridial exo-sialidases and the T. cruzi trans-sialidase.930
The eukaryotic trans-sialidases (TS) are best studied in the pathogenic trypanosomes T. cruzi, Trypanosoma brucei, and Trypanosoma congolense. However, trans-sialidase activity was also discovered, for example, in Corynebacterium diphtheriae, Pasteurella multocida, and C. jejuni.113, 139 Trypanosomal trans-sialidases are membrane-anchored enzymes that mediate the reversible (α2→3)-sialylation of β-galactose-terminated glycans. In this way, the parasites scavenge sialyl residues from host blood serum components or cells and transfer them to their cell-surface glycans terminated with galactose (Fig. 32B). trans-Sialidases can also act as mere sialidases. They are virulence factors in the South American Chagas Disease, the African Sleeping Sickness, and in some African cattle diseases. For a review about their pathomechanism, see ref. 139.
First indications for the existence of such trans-sialidases, probably involved in the biology of trypanosomes, stem from 1983, when the group of one of the authors (R.S.) detected Neu5Ac and Neu5Gc in cultivated T. cruzi.791, 931 These two sialic acid species probably were not synthesized by the parasites themselves, because their relationship corresponded to that of the culture medium. It was assumed that a sialidase, later localized on the cell membrane of T. cruzi,932 was involved. This assumption was confirmed after the isolation of the first TS from T. cruzi,933, 934 followed by TSs from the African species T. brucei and T. congolense.935, 936, 937 Information about the development of a suitable test system, study of donor and acceptor specificities, reaction mechanism, inhibitors, and biotechnological application of these rare and unique trans-sialidases is summarized in refs. 113, 139, and 938. The T. brucei group expresses TS during parasite growth in the insect stage, while in T. cruzi this enzyme functions mainly in the mammalian host. A wealth of literature, originating especially from South American laboratories, documents the developments during the last 20 years; see ref. 139 for an overview.
As an intramolecular trans-sialidase, a sialidase from the leech Macrobdella decora can be considered. It catalyzes the formation of 2,7-anhydro-Neu5Ac (Neu2,7an5Ac) from (α2→3)-bound sialic acid of sialyllactose and various glycoconjugates58, 87, 939 (Fig. 32A). Whether this sialidase is autochthonous to the leech or of bacterial origin is so far unknown. S. pneumoniae sialidase B has also been recognized as an intramolecular trans-sialidase.940 The detection of Neu2,7an5Ac in ear secretion suggests the presence of such an enzyme in human cerumen.57 Interestingly, gut microbiota are also among the producers of this Neu5Ac derivative.941
Although deaminated neuraminic acid (Kdn) is α-glycosidically linked to neighboring residues,76 the substitution of an N-acyl group by a hydroxyl function at C5 blocks nearly completely the action of bacterial exo- or endo-sialidases, known up to the 1990s.942 A few years later, however, a bacterial sialidase from Sphingobacterium multivorum was found to be capable of releasing Kdn (α2 → 3, α2 → 6, α2 → 8), but not N-acylneuraminic acids.943 Therefore, this sialidase was called KDNase. In the same period, a sialidase from the liver of the loach, Misgurnus fossilis, released both bound N-acetylneuraminic acid and deaminated neuraminic acid residues.944 The same held for sialidase preparations isolated from the kidney, spleen, and ovary of rainbow trout.945
Finally, a unique sialidase was isolated from the hepatopancreas of the oyster species Crassostrea virginica.946 This enzyme was capable of releasing Neu5Gc from the [→O5)Neu5Gc(α2→]n polysialic acid sequence in a more efficient way than from the [→8)Neu5Ac(α2→]n or [→8)Neu5Gc(α2→]n polysialic acid sequences. On the disaccharide level, Neu5Gc(α2→O5)Neu5Gc and also Kdn(α2→8)Kdn were effectively cleaved, whereas Neu5Ac(α2 →8)Neu5Ac and Neu5Gc(α2→8)Neu5Gc were poor substrates.
11.7.1. Sialidase Inhibitors
trans-Sialidases and many members of the bacterial and viral sialidases, when involved in diseases, require the availability of inhibitory substances for the study of their mechanism of action and especially for therapeutic use. The best examples are those from influenza viruses, which are absolutely dependent on their sialidase (neuraminidase, symbolized by N) for the infection of host cells. In order to escape the immune defense of the host, they steadily mutate, which, together with the hemagglutinin (H) that also mutates, continuously produce new viruses with variable infection potential. Such mutations are indicated by numbers added to the HN of the viruses, e.g., H1N1.
Early studies on an effective inhibitor of flu were successful in the laboratory of Hans Tuppy and Peter Meindl at the Universität Wien (Vienna, Austria).84, 638 They synthesized various 2-deoxy-2,3-didehydrosialic acids, such as 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (Neu2en5Ac) and the corresponding N-glycolyl, N-propionyl, N-butyryl, N-benzoyl, and N-carbobenzoxy derivatives. Mainly Neu2en5Ac was used for inhibition studies of the propagation of influenza viruses in cell culture. The compound inhibited most viral, bacterial, and mammalian sialidases with an inhibitor constant K i of about 5 μM, but not trypanosomal trans-sialidases. Substitution of the N-acetyl group in Neu2en5Ac by an N-trifluoroacetyl group led to a twofold better inhibitor.947 However, these inhibitors were not strong enough to inhibit virus growth in vivo. Neu2en5Ac was also found in human urine and saliva, in the latter source at rather variable quantities up to 2 × 10− 5 M, which, in principle, can inhibit influenza virus sialidase and may be of pathophysiological significance.43
Zbiral and coworkers at the Universität Wien (Vienna, Austria) synthesized a great variety of Neu2en5Ac variants, among others, 4-amino-Neu2en5Ac,948 which inhibited viral sialidase about 100-fold stronger than Neu2en5Ac.6, 949 It only weakly inhibited bacterial, e.g., V. cholerae, sialidase. A really potent virus sialidase inhibitor was developed when Mark von Itzstein at Monash University, Melbourne (now Griffith University, Brisbane), Australia, substituted this amino group by a guanidino function.108, 949, 950 4-Guanidino-Neu2en5Ac (Fig. 33A) exhibited a K i-value of 3 × 10− 11 M with influenza virus A sialidase. This strong, selective binding to viral sialidases is possible, because a pocket exists in the binding site of viral sialidases near the 4-hydroxy group of Neu5Ac. This pocket also allows slow release of bound Neu4,5Ac2, which is not possible with bacterial and mammalian sialidases.922 4-Guanidino-Neu2en5Ac inhibited the growth of a wide range of influenza A and B viruses in a variety of in vitro systems. The resulting drug Relenza® (zanamivir) as a spray is of prophylactic significance in preventing the infection of human bronchial epithelia with influenza viruses and is well suited for medical use.
Fig. 33.

However, Relenza® cannot enter cells in order to fight against the viruses already present in the tissues. Therefore, a more hydrophobic derivative with similar configuration was synthesized,951, 952 namely, Tamiflu® (oseltamivir, Fig. 33B), which can successfully reduce the symptoms of flu by inhibition of the virus sialidase inside the infected organism.953, 954 Because Tamiflu® also has some inhibitory effect on mammalian (human) sialidases, it should not be used for an extended period of time, nor should it be used prophylactically. Since resistance against Tamiflu® has been observed,955 searches for new and even better flu inhibitors are under way. In a recent publication, such a highly specific inhibitor against influenza viral N1–N9 sialidases, 9-cyclopropyl-carbonylamino-4-guanidino-Neu2en5Ac (cPro-GUN), has been described, which is also highly effective against a Tamiflu®-resistant strain. Furthermore, most importantly, it does not inhibit human sialidases.956 With such inhibitors, as well as vaccination, it is hoped that such severe and deadly influenza pandemics as had occurred just 100 years ago (“Spanish flu”), claiming many millions of casualties,957 can be prevented.
12. The Biology of Sialic Acids
In the previous sections, according to the chronological development of the sialic acid field, the history of the sialic acid chemistry and biochemistry was highlighted. Although in recent years the biological roles of sialic acid have been broadly studied in great detail, some important biological discoveries originate from the beginning of the 1940s. As worked out in 2.1, 11.7, for the progress in sialic acid chemistry, the discoveries in the Walter and Eliza Hall Institute of Research in Melbourne, i.e., the enzymatic release (using virus and microbial filtrates) of a low-molecular-weight substance (later identified as sialic acid) by an RDE (later identified as sialidase), were of great importance. Here, the biological site of this finding will be further cleared up. As mentioned earlier, George Hirst observed that at 4°C influenza viruses bound to chicken erythrocytes, leading to agglutination of the red cells.889 However, the viruses eluted from the erythrocytes after increasing the temperature to 37°C. Remarkably, the erythrocytes became inagglutinable, but the viruses remained functional. It was concluded that the viruses attach to a receptor substance at the red cell surface, which is inactivated by a viral enzyme. Then, Frank Burnet showed that such an enzyme also existed in the culture filtrate of V. cholerae.175 A mucoprotein of hen egg white (ovomucin) inhibited the reversible virus–erythrocyte binding, a property that was destroyed when the ovomucin was first incubated with influenza virus.97, 174 Thus, the main property of sialic acid was identified in these early days, namely, to act in biological recognition as a receptor, but in due respect to its structure, a ligand. Virologists even today consider sialic acids as receptors that accept viruses. However, sialic acid as a sugar should be called a ligand as it interacts per definition (see below) with a proteinaceous receptor. The first “receptor” function of sialic acids just described represents the binding to influenza virus hemagglutinin, the history and properties of which are described in detail in refs. 958, 959.
The cell biology, pathophysiology, and pharmacology of sialic acids are a huge and fast developing area, of which only a few highlights can be discussed here. An early review describing the role of glycans and sialic acids was published in 1972.99 When studying the biological events in which sialic acids are involved, a dual role of these sugars can be recognized, i.e., they can mask recognition sites or they are directly recognized by a lectin-like receptor, for which they represent a ligand. This all mainly happens on the surface of the cell, the so-called glycocalyx.
12.1. The “Sweet Coat of the Cell”
In the 1960s, a study on the migration of cells on electrophoresis led to the discovery of the electronegative charge of cell membranes.960, 961 In fact, this finding contributed to the discovery of the glycocalyx. The term “glycocalyx” was coined by Bennett in 1963 (as “sweet husk”) and describes the glycoproteins and glycolipids linked to the external lamina of cells.962 The carbohydrate composition of the glycocalyx is rather variable and depends on the cell type and function. It represents a sensory system that interacts with the environment. Sialic acids are predominant components at this outer surface, and mainly here they act as biological masks or receptors.
12.2. The Masking Role of Sialic Acids in Serum Glycoproteins and Blood Cells
Sialic acids keep serum glycoproteins and blood cells fit, which is one of the most dominant roles of these monosaccharides. In fact, they regulate the lifetimes of soluble serum glycoproteins and cells, have great significance in all aspects of cell biology, and are most prominent in immunology and in disease states such as cancer (for reviews, see refs. 6 and 963). The sialobiology area was greatly stimulated when Ashwell and Morell in 1974 described in a review that desialylated glycoproteins of rabbit blood serum were rapidly cleared from the circulation and taken up by hepatocytes.964 This process was mediated by a galactose-specific receptor on the hepatocytes, which recognized galactose residues of the glycoprotein glycans, demasked by the loss of terminal sialic acids (cleavage of Neu5Ac(α2→3/6)Gal linkage). The first report, describing the fate of desialylated serum glycoproteins, is from 1971.965 The properties, molecular biology, and biosynthesis of this Ca2+-dependent (galactose) receptor have been intensively investigated, e.g., see ref. 966.
In other studies, it was observed that sialidase treatment of erythrocytes drastically decreased their lifetime.967 In man it is reduced from about 120 days to a few hours.968, 969 A similar phenomenon was also observed for rabbit erythrocytes.970 The clearance of asialo-erythrocytes is similar to the sequestration of asialo-glycoproteins, although the erythrocytes are degraded in Kupffer cells. The adhesion of asialo-erythrocytes is mediated by lectin-like receptors on Kupffer cells, which recognize the unmasked galactosyl residues.969, 971, 972 Adhesion can be inhibited by glycans that possess galactose in a terminal position. This binding and phagocytosis of sialidase-treated (rat) erythrocytes occurs by a mechanism independent of opsonins.973 Using rat peritoneal macrophages and desialylated rat erythrocytes, the attachment of the erythrocytes to the macrophages and partial phagocytosis could be clearly demonstrated (Fig. 34A).974, 975 The galactose receptor of rat peritoneal macrophages was identified by photoaffinity labeling as a protein of 42 kDa.976 It adds to the rapidly increasing family of lectins recognizing galactose and other sugars, as reviewed in ref. 977.
Fig. 34.

Binding of sialidase-treated rat erythrocytes, lymphocytes, and thrombocytes to homologues rat peritoneal macrophages by the galactose-specific acceptor. (A) Scanning electron microscopy of erythrocyte–macrophage interaction. After prolonged incubation, the erythrocytes were ingested; (B) scanning electron microscopy of cultured, sialidase-treated lymphocytes bound as a rosette by a peritoneal macrophage; (C) scanning electron microscopy of the characteristic binding of a single sialidase-treated lymphocyte by a rat peritoneal macrophage. In contrast to sialidase-treated, bound erythrocytes, no deformation of phagocytosis is observed. (D) Scanning electron microscopy of sialidase-treated thrombocytes to a macrophage adherent to a Petri dish.
Panels (A) and (D): Reproduced from Schauer, R. Sialic Acids Regulate Cellular and Molecular Recognition. In Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry; Ogura, H.; Hasegawa, A.; Suami, T., Eds.; Kodansha Ltd.: Tokyo, Japan, 1992; pp 340–354. Copyright Wiley-VCH Verlag GmbH & Co. KGaA.
Lymphocytes also alter dramatically their “homing” behavior after sialidase treatment. It was found that enzyme-treated rat thoracic duct lymphocytes accumulated in liver instead of the spleen and lymph nodes.978, 979 However, this loss of orientation was reversible after about 1 day, because the cells recovered, probably by resynthesis of their sialic acid coat. Correspondingly, binding studies with sialidase-treated rat lymphocytes and homologous peritoneal macrophages also showed reversible binding to these nucleated cells (in contrast to the erythrocytes), but no phagocytosis (Fig. 34B and C).980, 981 The interaction was inhibited by galactosides, showing the involvement of the galactose receptor. Also in this in vitro experiment the lymphocytes continuously were released from the macrophages within 1 day. This reversible binding may be due to rapid resynthesis of membrane sialic acids, as observed with human lymphocytes982 and with rat liver plasma membranes.983 The turnover rate of sialic acid in a rat liver cell membrane glycoprotein was 33 h, whereas that of the corresponding protein part was 70–78 h.983
Sialidase-treated thrombocytes from rats were also rapidly cleared from the circulation.984 The interaction of desialylated rat thrombocytes with homologous peritoneal macrophages also occurs in a galactose-dependent manner (Fig. 34D).73, 975
The various experiments described in this section have shown that sialic acids are of vital importance in the regulation of the lifetimes of these blood components and to maintain homeostasis. The behavior of the fully sialylated glycoproteins and cells is called “self” when they are intact and tolerated by the organism. As soon as sialic acid is lost, the molecules and cells become “nonself” and no longer fit to the chemical architecture required for normal “behavior”; hence, they are recognized by the defense system of the organism and are eventually destroyed. This defense system is part of the immune system; therefore, the dual system “self/nonself” plays a vital role in immunology (see Section 12.4). It functions as an inhibitory “self-signal” to the vertebrate immune system.985 Also in cancer biology, the masking role of sialic acids is of great significance, as will be discussed in Section 12.3.
12.3. Sialic Acids in Tumors
This is one of the most important areas of sialoglycobiology, and the great variety in the phenomena of tumor growth in which sialic acids are involved has been described in several thousands of publications. The most important feature is the altered, mostly elevated, sialylation of the surface of tumor cells. This is caused by α2,3-, α2,6-, and α2,8-sialyltransferases, which are often more expressed in malignant cells compared to the corresponding normal cells. This high sialylation often leads to accelerated cancer progression including cell–cell repulsion, altered binding of cancer cells to the extracellular matrix, enhanced migration, docking to the vascular endothelium, and invasion that facilitates the formation of metastases and leads to poor prognosis of the disease. An elevated expression of the selectin ligands SLea and SLex in a variety of tumor types has been observed. Higher sialylation increases the resistance of tumor cells to apoptosis. Some tumors also exhibit more Neu5Gc and changes in sialic acid O-acetylation or Kdn concentration in comparison to the normal tissues.
Many reviews have appeared before, of which only a few will be listed here. For older reviews, see refs. 986, 987, 988, 989, 990, 991, 992, 993, 994, 995, 996. From more recent overviews, refs. 997, 998, 999 are herein selected. Here, only the very beginning—according to the aim of this chapter—of tumor sialoglycobiology will be detailed on the basis of publications available. These show the long and tedious way researchers had to go to reach our present profound knowledge on cancer biology. However, many questions still have to be answered, especially in the field of molecular biology and immunology, and, of course, for therapy.
The year 1956 may be considered as the beginning of tumor sialoglycobiology, when Ambrose and coworkers discovered that tumor cells from hamster kidneys have a much greater electrophoretic mobility than normal homologous kidney cells.1000 In addition, mouse sarcoma cells migrated faster on electrophoresis when they acquired more malignant properties.1001 At that time, the nature of the molecules responsible for the electrophoretic mobility was not yet known. However, treatment of erythrocytes, as a model, with trypsin released a “sialomucopeptide” which contained terminally linked sialic acids. This was the first demonstration of a sialoglycopeptide at the periphery of a cell.960 The existence of sialylated carbohydrates on the surface of normal and malignant mammalian cells was nicely demonstrated by using a Hale staining technique and comparison with sialidase treatment.1002 Most interesting experiments were carried out by the groups of Leonard Warren and Mary C. Glick in the beginning of the 1970s, when they demonstrated a marked difference in cell-surface glycosylation/sialylation of normal and virus-transformed baby hamster kidney (BHK) cells by a double-label technique with [14C]- or [3H]-l-fucose. The radioactive pronase-released glycoproteins from the two cell types were mixed and fractionated on Sephadex G-50. The eluting radioactive glycopeptides of both biological sources showed different profiles.1003, 1004 In a publicaton1004 by Warren and coworkers, an elevated (up to 11 times) sialyltransferase activity in the transformed BHK cells, when compared to the normal cells, was reported; the sialic acid content was much higher in the transformed cells. Also invasive tumors of human breast and colon expressed elevated levels of glycoprotein sialyltransferases.1005 Since then, by using various techniques, increased sialylation and altered glycosylation have been observed in many tumors, mostly derived from ectodermal tissues, for example, in the most prevalent breast and colon cancers. A wealth of information about tumor biology was obtained in the early days of this research from the study of blood serum from tumor patients; an increase of sialic acids during tumor growth in humans and mice was identified.1006, 1007, 1008 The increase of serum sialic acid was later recognized to be due to high levels of sialylation of both glycoproteins and glycolipids.1009 In the case of β-myeloma proteins, three to six times more sialic acid was found in blood serum.1010
Another milestone in our understanding of tumor biology was the discovery that the four types of known human sialidases behave in different manners during carcinogenesis. Probably the first observation of higher lysosomal sialidase and lower cytosolic sialidase levels was made in rat hepatomas, when compared to that of normal liver.1011 More recently, human sialidase was established to be a cancer marker, which is downregulated in lysosomes, promoting the growth and metastatic ability of tumor cells. The enzyme is upregulated in the plasma membrane, which suppresses apoptosis of cancer cells.1012 As described in Section 11.7, the human sialidases NEU1–NEU4 are expressed differently in various tumor tissues.928 In general, transformed cell lines express higher activities of membrane-bound sialidase that preferably desialylates gangliosides.1013 Furthermore, the SLex epitope, which is often more expressed on tumor cells and interacts with selectins, facilitates metastasis (see below). Its biosynthesis has been studied in human lung carcinoma cells.1014
Tumor cells often express modified Neu5Ac, such as Neu5Gc, O-acetylated sialic acid, or Kdn, at concentrations different from the corresponding normal cells. A well-studied example are human colon cells, the mucin of which is highly O-acetylated in the healthy state. On tumor development, in the adenoma–carcinoma sequence, mucin O-acetylation gradually decreased with the lowest value in adenocarcinoma. This leads to a higher concentration of SLex in mucins, which had been masked before by O-acetylation in the metastatic cells, thus facilitating metastasis.1015 The O-acetyltransferase activity declined, also.1016 Another example are melanoma and basalioma tissues in which gangliosides with O-acetylated sialic acids, mostly 9-O-Ac-GD3, are much more expressed than in normal skin.1017 For example, in gangliosides from basalioma, up to 56 times higher concentration of Neu5,9Ac2 was determined. O-Acetylated gangliosides, which also occur in other tumors of neuroectodermal origin, represent markers for neoplastic development. Interestingly, they are also universal markers for rapidly growing normal cells and tissues. In spinalioma (squamous cell carcinoma) and in skin nevi, sialic acid O-acetylation was not increased (J. Gierthmühlen and R. Schauer, unpublished results). A ganglioside containing sialylated lacto-N-fucopentaose II turned out to be an antigen associated with human gastrointestinal cancer cells.1018 During the last 30 years, more knowledge about gangliosides has been gathered, especially about the signaling mechanisms, particularly in cancer cells, regulated by these sialoglycolipids. Antitumor therapy addressing these molecules has been gradually put into practice.1019
N-Glycolylneuraminic acid is also a type of sialic acid, which deserves attention in tumor biology.794 While Neu5Gc is almost completely absent from normal human tissues (only about 0.01% Neu5Gc of total sialic acid) (see Section 11.5.1), this value is often higher in tumor tissues, especially in their gangliosides (for example, 0.5% of total lipid-bound sialic acid). Elevated levels of Neu5Gc were described as chemical or tumor-associated markers in glycolipids of human breast carcinoma and other tumors and in avian lymphoma cell lines.1020, 1021, 1022 They are called Hanganutziu–Deicher (HD) antigens (see Section 12.4). HD antigens occur in blood serum in rather variable amounts from normal individuals to cancer patients, where the values are often increased. With regard to the origin of Neu5Gc, a dietary source is assumed, as was first studied with rats.1023 Tumor cells may be able to take up more exogenous Neu5Gc than normal cells. There is also some evidence, which, however, needs more solidification, that small amounts of Neu5Gc can also be produced in an endogeneous metabolic pathway (H.-G. Hanisch, personal communication). Hydroxylase activity was, however, never observed. Finally, an important aspect of the xenoantigen Neu5Gc is its immunological and inflammatory role. There is evidence that these processes may lead to malignancies.1024
12.4. The Role of Sialic Acids in Immunology
In this section some historical aspects of sialic acids playing a role in immunology will be summarized. The beginning of the classical immunology goes back to 1900, when Landsteiner discovered the ABO blood group system.1025 For a review, see ref. 1026. The system was originally defined in terms of the antigenic substances occurring at the surface of red blood cells. At that time the carbohydrate nature of the antigens was not yet known. In the following years, more than 100 red cell antigens were analyzed including the Lewis system and the M and N antigens.1026, 1027, 1028 The largest group of these glycans are human erythrocyte autoantigens that react with the corresponding antibodies. They can also occur on cells of human tissues and in animals. The antibodies are monoclonal in patients with chronic lymphoproliferation. The cold agglutinins mainly react with erythrocytes, best at 0°C. This can lead via complement activation to intravasal erythrocyte degradation, which leads to anemia. The antigens corresponding with the monoclonal anti-Gd, -Fl, -Li, -Lud, -Pr, -Sa, -Ss, and -Vo are sialylated and can be inactivated by sialidase. As an example, the abbreviation Gd means “ganglioside-dependent.” Gangliosides with Neu5Gc are Gd-inactive. Sialic acids, linked (α2 → 3) or (α2 → 6) at the glycophorin glycans, can also influence the Pr antigen specificity.1029 For comprehensive reviews about this complex topic, see refs. 1030, 1031, 1032, 1033, 1034.
The analysis of the sugar components of the human blood group substances was started in the late 1940s.1035, 1036 Highly purified A, B, H, and Lea samples from human ovarian cyst fluids were shown to contain l-fucose, d-galactose, N-acetyl-d-glucosamine, N-acetyl-d-galactosamine, and N-acetylneuraminic acid.1026 Neu5Ac was isolated from Lea blood group substance and crystallized.1037 The structural analysis of these macromolecular substances, which occur in blood, but also other human fluids, took many years. This and the early studies of the biosynthesis of blood group substances are thoroughly described in ref. 1026. Regarding the glycosylation of immunoglobulins, as early as 1934 covalently linked carbohydrates were reported to occur on serum proteins with high antitoxin titer after immunizing horses against diphtheria toxin.1038 For a review, see ref. 1039. But it was until 1967 that sialic acid was described to be present in human IgG.1040 The variable sialic acid content altered the electrophoretic mobility of this immunoglobulin. In one of the first attempts to elucidate the three-dimensional structure of human γG1, four possible ways of joining the Fab region to a nearby Fc region were shown.1041
A later milestone in the glycobiology of immunoglobulins was the discovery of changing sialylation and its influence on function. About 30 different glycan structures, with and without sialic acids, linked to the Fc region of the immunoglobulin are known,1042 and voluminous reviews on the impact of glycosylation on the biological function and structure of human immunoglobulins have been written.1043, 1044 Immunoglobulin G mediates pro- and antiinflammatory responses through the engagement of its Fc fragment. This difference depends on the sialylation of the Fc core oligosaccharide, and it explains why therapeutic intravenous γ-globulins, if they are sialylated, have antiinflammatory effects in autoimmune diseases.1045 In a remarkable experiment, it was seen that engineered sialylation of pathogenic antibodies in vivo, at the site of autoimmune inflammation, led to an attenuation of the autoimmune disease.1046
The pronounced significance of Neu5Gc, although its nature was not yet known, in immune reactions was observed as early as at the beginning of the last century. M. Hanganutziu in 1924 and H. Deicher in 1926 observed after the therapeutic injection of antisera from horse blood into humans the formation of antibodies, now called “HD antibodies.”1047, 1048 About 50 years later, the epitope, recognized by HD antibodies, turned out to be the Neu5Gc residue of gangliosides.1049, 1050 The significance of these antibodies in tumor biology and in nutrition has been discussed in Section 11.5.1. Additionally, monoclonal antibodies have been described that distinguish between Neu5Ac- and Neu5Gc-containing ganglioside GD3.1051
Monoclonal antibodies against O-acetylated sialic acids in human colon mucins were obtained by immunization of BALB/c mice with mucosal scrapings from normal human large intestine. Antibody PR.3A51052 and antibody 6G41053 bound to O-acetylated mucin and were useful in studying the histochemistry of the normal colon, colon cancer, and ulcerative colitis. Immunization of mice with highly purified human colonic mucin afforded the monoclonal antibody MMM-17, which strongly binds to the mucin of goblet cells of the normal colon, but not to those of colon cancer tissue.1054 Saponification of the O-acetyl groups in the mucin or sialidase treatment of the mucin abolished the binding. O-Acetylated gangliosides are also immunogenic, as was studied with GD3 and GD3 derivatives. Here, monoclonal antibodies were obtained that distinguish between GD3 (CD 60a), 9-O-acetyl-GD3 (CD 60b), and 7-O-acetyl-GD3 (CD 60c).1055 It was found in many investigations that the disialoganglioside GD3 is the most frequent carrier of terminal O-acetylated sialic acid. The occurrence and the roles known of O-acetylated GD3 have been reviewed826 (see also Section 11.5.2). Among the most striking and influential effects of O-acetylation are the inhibition of apoptosis of tumor cells,1056 the inhibition of the interaction of the siglec sialoadhesin with 9-O-acetylated sialic acid ligands, and the inhibition of the binding of human complement factor H, a negative regulator of the alternative complement pathway, to sialic acids.1057, 1058 Thus, a decrease of O-acetylation may help tumor cells to escape from complement-mediated lysis. A classical example of the protective effect of (unsubstituted) sialic acids from activation of the lytical alternative complement pathway is the channel catfish Ictalurus punctatus. Very little bactericidal activity is exerted by the alternative complement pathway against the fish pathogens containing sialic acids, in contrast to a strong response against the nonpathogenic bacteria that lack sialic acid. This phenomenon is sialidase sensitive.1059
The assumption of the existence of non-N-acetylated neuraminic acid (Neu) in the gangliosides of some tumors was based on the detection with monoclonal antibodies and pulse-chase experiments.1060 Note that the first chemical existence for natural Neu was obtained after N-propionylation.488 N-Deacetylation of capsular polysialic acids provided antibodies protective against N. meningitidis groups B and C.1061 Earlier, antibodies against the virulence factors (α2 → 8)- and (α2 → 9)-polysialic acids were produced for vaccination against meningococci and E. coli K1 after conjugation to tetanus toxoid.1062 An effective vaccine, applied to small-cell lung cancer patients by the application of N-propionylated group B/(α2 → 8) polysialic acid, conjugated to keyhole limpet hemocyanin, is known.1063 Natural polysialic acids are very weak antigens in humans.1064
The previous examples have shown that sialic acids are involved in antigenic structures. To these also belong the so-called differentiation and oncodevelopmental antigens of glycoproteins and glycolipids, which are expressed during ontogenesis and also in cancer.987 However, sialic acids often mask antigenic sites on glycoproteins, glycolipids, and carbohydrate oligo- and polymers and thus represent “antiantigens.” Thus, these monosaccharides have a remarkable regulatory role in immunology (see, e.g., refs. 963 and 1065), in a similar way as already discussed for the regulation of the lifetime of blood components and cells, namely, via a dual “self/nonself” system (see Section 12.2). So, the masking effect of sialic acids renders molecules and cells as “self.” After the loss of the sialic acid residues, the structures become “nonself” and may be recognized by the immune system, which may lead to autoimmune diseases. A masking role of sialic acids has early been detected for porcine submandibular gland glycoproteins. Sialidase treatment of these mucins revealed H blood group activity, because Neu5Gc had stereospecifically masked the H-determinant Fuc(α1 → 2).1066 A classical example is also the masking of the T-antigen Gal(β1 → 3)GalNAc(α1 → O)Ser/Thr and the Tn-antigen GalNAc(α1 → O)Ser/Thr of blood cells and some tumors by Neu5Ac in humans.1032 The Gal(β1 → 4)[Fuc(α1 → 3)]GlcNAc antigen was also found to be cryptic in different human tissues.1067 It is masked to a variable extent by Neu5Ac at the terminal Gal residue of the antigen {Neu5Ac(α2 → 3)Gal(β1 → 4)[Fuc(α1 → 3)]GlcNAc} or at adjacent carbohydrate chains (obscuring the antigen by steric hindrance) and can be made visible by histochemical means by using sialidase. One of the most striking and important examples for the masking of antigenic sites is the immunobarrier between mother and fetus due to the expression of highly sialylated glycoconjugates on syncytiotrophoblast cells.1068, 1069 These molecules prevent the formation of antibodies by the maternal organism against fetal cells. Loss of sialic acids in kidney, e.g., by viral or bacterial infection, may lead to immunological injuries of the glomeruli leading to chronic glomerulonephritis.1070 Note that tissues rich in sialic acids tend to develop autoimmune diseases.963 The pronounced masking of tumor antigens by sialic acids has already been discussed in Section 12.3. These observations, together with an old report1071 about the effect of sialidase to increase the immunogenicity of the Landschütz ascites tumor cells, stimulated attempts to cure cancer by sialidase treatment of tumor cells, reinjection of the cells, and thus vaccination of the patient with these cells.963 Injection of the cells into various places of the skin (“chessboard vaccination”) had been recommended. This method was successful in the treatment of dog mammary tumors,1072 while in humans the duration of the remission was extended in myelocytic or lymphatic leukemia.1073 Although some positive results were obtained, this method was not followed further.
Sialic acids on the surface of T cells mediating “delayed-type hypersensitivity” (DTH), which have developed an immune response before, control the entry of the lymphocytes into sites of antigen deposition, where an inflammatory reaction is induced. This could be shown by sialidase treatment of the sensitized T cells that caused a transient reduction of DTH reactions in the periphery. The cells were trapped in the liver, but were released after 24 h, and their biological activity returned.1074 The involvement of terminal sialic acids on the surface of T cells, in most aspects of T cell fate and function, from cell maturation, differentiation, and migration to cell survival and cell death, was reviewed 27 years later.1075
In the context of this section, attention should also be paid to the function of (sialylated) milk oligosaccharides. As mentioned (see Section 8.3 for an updated series of reviews), the extraordinary diversity of these glycans is great, varying from mammal to mammal, and comprising more than 200 different structures in man. They regulate the bacterial colonization of the intestine, prevent the growth of pathogenic bacteria, and feed the microorganisms. This protects especially children from bacterial and viral infections. The variability of the microorganisms, nowadays called “microbiome,” also seems to influence the human immune system, although the exact mechanism and the role of sialic acids wait for complete elucidation. The enhancement of the biosynthesis of glycoproteins and glycolipids in the developing brain by milk oligosaccharide feeding and the corresponding stimulation of the intellectual capacities are also under study.1076, 1077
A striking example of innate, humoral immunity is limulin, a sialic acid-binding lectin from the American horseshoe crab Limulus polyphemus. It was isolated and characterized from the hemolymph of this animal, has a molecular weight of 335 kDa, and consists of subunits of each 19 kDa.1078, 1079 Limulin binds to glycosidically linked sialic acids of glycoproteins and gangliosides, but preferably to Neu5Gc-containing gangliosides (i.e., Neu5Gc-GM3), and thus agglutinates erythrocytes and other cells.1080 A similar lectin was isolated by affinity chromatography from the hemolymph of the Asian horseshoe crab Tachypleus tridentatus.1081 SDS-PAGE showed three protein bands between 27 and 32 kDa. This lectin also agglutinates erythrocytes and preferably binds to sialylated O-glycans of mucins. These lectins of the phylogenetically very ancient horseshoe crabs are considered as potent innate defense systems against parasites with sialic acids or perhaps similar nonulosonic acids.
A most important chapter of modern immunology are the mammalian sialic acid-recognizing lectins, called siglecs, which, in most cases, occur on the surface of immune-competent cells, e.g., B-lymphocytes, and are involved in many immune reactions including autoimmunity.985, 1082, 1083 For further details, see Section 12.5.3.
12.5. Proteins Recognizing Sialic Acids
In this section the history of the studies of the interaction of sialic acids as ligands with proteins as receptors (lectins) will be described. Special groups are selectins and siglecs, which will be treated separately. For an overview written by the pioneers of the research on lectins, Nathan Sharon and Halina Lis from The Weizmann Institute of Science (Israel), see ref. 1084.
12.5.1. Plant and Other Lectins, and Virus Hemagglutinins
Lectins were first found almost exclusively in plants.398, 1084, 1085, 1086 Since they were detected by the agglutination of erythrocytes, they were named “hemagglutinins” or “phytohemagglutinins.” As plant lectins are able to distinguish between erythrocytes of different blood types, as early as 1954 Boyd and Shapleigh coined the name “lectin,” from the Latin legere, “to select.”1087 This definition was later used for all carbohydrate-binding proteins, irrespective of their occurrence.
To go back in history, before 1860, S. Weir Mitchell in Pennsylvania had observed agglutination of pigeon blood by the venom of a rattlesnake.1088 The first lectin, called “ricin,” was discovered in 1888 in the beans of the castor tree (Ricinus communis) by Hermann Stillmark at the University of Dorpat (now Tartu) in Estonia.1089 In fact, it was a mixture of weakly agglutinating protein toxins, which were later found to bind to galactose and N-acetylgalactosamine.1084, 1090 Furthermore, Simon Flexner and Hideyo Noguchi,1091 from the Rockefeller Institute (New York, USA), described in 1902 snake venom agglutinins in some detail in the horseshoe crab (L. polyphemus)1078 and the American lobster Homarus americanus. In the first decade of the 20th century also the first bacterial agglutinins were found, which were later identified as lectins. Investigations on blood type-specific lectins stem from the early 1950s, when it was demonstrated for the first time that lectins can bind monosaccharides.1092
The already described studies of Hirst in the 1940s on the agglutination of human erythrocytes by influenza virus (see Sections 2.1 and 11.7) can be considered as the first example of the involvement of a lectin/hemagglutinin, a sialic acid-binding agent. Influenza virus hemagglutination was found to be inhibited by a brain lipid fraction, known to contain gangliosides.1093 In the following decades the structure and pathophysiological significance of the viral hemagglutinin (designated “H” antigen) was most intensively studied, until today, by the group of Hans-Dieter Klenk at Philipps-Universität Marburg (Germany).958, 959, 1094 Influenza viruses demonstrate pronounced receptor binding specificity. Human influenza viruses preferentially bind to Neu5Ac(α2 → 6)Gal sequences, whereas avian influenza viruses prefer Neu5Ac(α2 → 3)Gal as ligand (or receptor in the terminology of virologists).1095 Competitive inhibitors of the binding of avian or human influenza viruses to sialylated glycans are soluble glycoconjugates carrying (α2 → 3)- or (α2 → 6)-linked sialic acids, respectively. The earlier discussed mucin from the nests of the Chinese swiftlet, genus Aerodramus collocalia (collocalia mucoid, edible bird's nest substance),1096 rich in sialic acids, is a potent inhibitor of the agglutination of myxoviruses.1097 This may have been the reason that it has been used in Chinese medicine for hundreds of years in order to prevent and cure influenza and its following bronchial diseases. The substance, which is a good substrate for sialidase, has additional health benefits and is therefore highly esteemed in Asia.1098
The best known sialic acid-recognizing plant lectins are WGA, accepting Neu5Ac and (β1 → 4)-linked GlcNAc residues, S. nigra agglutinin (SNA) from the bark of the elderberry bush, recognizing (α2 → 6)-linked sialic acid to galactose, and M. amurensis lectin (MAA), specific for (α2 → 3)-linked sialic acid to galactose.1084 The fractions of these plant lectins are believed to protect the plants from pathogenic microorganisms and fungi, as well as from herbivorous animals. It was found that the bark lectin SNA provoked toxic effects in animals and created a reaction of avoidance (Fig. 35 ).1099 One of the authors (R.S.) observed that during a very cold winter with much snow, mammals, mainly rabbits, fed on the bark of various bushes, but not on that of the elderberry bush. Both SNA and MAA are used regularly in structural analysis protocols, i.e., lectin affinity chromatography, histochemistry, or lectin microarrays.275
Fig. 35.

Lectins, for example, the sialic acid-binding Sambucus nigra agglutinin (SNA), protect plants against herbivorous and chewing animals.
Reproduced from Peumans, W. J.; Van Damme, E. J. M. Lectins as Plant Defense Proteins. Plant Physiol.1995, 109, 347–352. Copyright American Society of Plant Biologists.
Sialic acid-recognizing proteins are manifold in Nature. The first sialic acid-binding protein from vertebrates reported was Complement Factor H, an early response factor of the innate immune system.1100, 1101 In a thorough search about 230 sialic acid-specific lectins in viruses (being the largest group), bacteria, toxins, protozoa, fungi, plants, invertebrates, and vertebrates were described.1102 In the intervening time, more sialic acid receptors have been detected, especially in the field of virus research, e.g., of the human Noro virus,1103 the Middle East Respiratory Syndrome Coronavirus (MERS-CoV),1104 and the Infectious Salmon Anemia Virus (ISAV), the latter binding to Neu4,5Ac2.1105 Most of these sialic acid lectins bind to Neu5Ac, but a few only to Neu5Gc or to both sialic acid types. An example of Neu5Gc as virus receptor is the transmissible porcine gastroenteritis coronavirus.1106 The various lectins may discriminate between (α2 → 3) and (α2 → 6) linkages and rarely prefer (α2 → 8) linkages, like e.g., siglec-7 and -11. Some invertebrate and viral lectins bind to 4- or 9-O-acetylated sialic acids, the best known example being influenza C virus.826, 1102
Not much is known about how and where influenza C viruses enter the human organism, but the human respiratory tract is the most likely place. In the nasal secreted mucin of only one person studied so far, about 10% of the sialic acids were 9-O-acetylated. In a pool of nasal mucosa probes collected after surgery in 1994, a 9-O-acetylated sialoglycoprotein fraction of 100–130 kDa was detected by Western Blot analysis. This could be the influenza C virus receptor. Since also the ganglioside fraction was found to be O-acetylated, the influenza C virus receptor must be studied further.1107 Influenza C virus hemagglutinins, specific for (α2 → 3) and (α2 → 6) linkages, can differentiate between receptor determinants bearing N-acetyl-, N-glycolyl-, and O-acetylated sialic acids. This was shown by the agglutination of Neu5Ac, Neu5Gc, or Neu5,9Ac2 covering erythrocytes.1108
Very recently, probes from viral sialic acid-binding proteins were developed to detect 4-O-acetyl, 9-O-acetyl, and 7,9-di-O-acetyl sialic acids on cells and tissues of humans and a great variety of animals.407 It is astonishing how widely distributed these esterified sialic acids are. The HE proteins were expressed in insect cells and fused to the Fc region of human IgG1 and to a hexahistidine sequence for histochemical or microarray use (see also Section 7.4). Binding of influenza A virus and Siglec-2 (CD22) is inhibited by Neu5Ac O-acetylation.826
12.5.2. Selectins
The selectins mediate, as do other cell adhesion molecules, the adhesion of leukocytes from the blood stream to endothelial cells, followed by extravasation into the surrounding tissue. They are thus involved in the initiation of inflammation. Tumor cells can experience the same fate. The basis of this phenomenon is the recognition of terminal sialyl Lewis x {SLex; Neu5Ac(α2 → 3)Gal(β1 → 4)[Fuc(α1 → 3)]GlcNAc} and sialyl Lewis a {SLea; Neu5Ac(α2 → 3)Gal(β1 → 3)[Fuc(α1 → 4)]GlcNAc} glycan sequences on the mobile cells. The first observations of this complicated process were recorded in the 1880s by Julius Friedrich Cohnheim, a German pathologist, who also worked one year (1868) at Christian-Albrechts-Universität zu Kiel (Germany).1109, 1110 This scientist studied by real-time microscopy the microvessels in the frog and proposed that postcapillary venules at sites of inflammation underwent molecular alterations that mediated the attachment of flowing leukocytes on the vessel internal surface, and subsequent emigration at sites of inflammation. Heinrich (Karl Wilhelm) Schade from the same university also observed about 40 years later the extravasation of white blood cells into inflamed tissues. He assumed that substances penetrating into the blood make the leukocytes “sticky” resulting in adhesion of the cells to the wall of the blood vessels before they extravasate into the tissue guided by chemotaxis.1111 The molecular basis of these events could not be elucidated in those times. Heinrich Schade is considered the father of molecular pathology, a new area of medical research, in completion with the then existing cellular pathology.
It took a further 30 years before researchers began to study the extensive flux of lymphocytes between blood and lymph (“lymphocyte recirculation”).1110 Here, a “homing molecule” on the surface of lymphocytes was assumed, which directs their traffic to lymph nodes. Electron microscopic studies showed that microvascular structures exist in the lymph nodes, consisting of plump, cuboidal endothelial cells, known as high endothelial venules (HEV),1112 and lymphocyte adherence to HEV could be demonstrated.1113 Further studies on the leukocyte extravasation at sites of inflammation showed a dynamic process.1114 After 2 h of injury, postcapillary venules displayed adhesive properties that supported leukocyte binding, followed by endothelial transmigration. At this state of the research, it was not known that carbohydrates were involved in leukocyte extravasation, but soon after these reports the participation of carbohydrates in lymphocyte trafficking was recognized.978 In many further studies, the HEV receptor of the lymphocytes was shown to be a lectin, whereby sialylated glycans on HEV serve as ligand for this lectin.1115, 1116 Furthermore, a family of sulfated sialofucosylated glycoproteins (sulfated at position 6 of GlcNAc or Gal of SLex) was found which serve as L-selectin ligands on HEV, known as “peripheral lymph node addressins.”1117 While L-selectin is expressed on lymphocytes, and functions in the migration of these cells to lymphoid organs, E- and P-selectins are expressed on vascular endothelial cells and mediate the recruitment of neutrophils to sites of inflammation in tissues (Fig. 36 ). P-Selectin also occurs on lymphocytes. E-Selectin is constitutively expressed on bone marrow microvessels, and human hematopoietic stem cells express high amounts of an E-/L-selectin ligand.1110 The involvement of selectins in cancer metastasis has drawn many researchers to this field.1118
Fig. 36.

The selectins play a key role in the control of leukocyte traffic in the body. (A) L-Selectin functions in the migration of lymphocytes to lymphoid organs. (B) E- and P-Selectins mediate the recruitment of neutrophils to sites of inflammation.
Courtesy of Ohad Bairey, and reproduced from Sharon, N.; Lis, H. Lectins, 2nd ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003, p 353. Copyright Kluwer Academic Publishers.
In 1989, the lymph node homing receptor, as well as E- and P-selectins, were cloned.1119 This protein family was then named “selectins,” belonging to the “C-type” lectins. At the same time, studies revealed that, in addition to homing receptors, cell migration is orchestrated by chemoattractants (e.g., chemokines), which may be present within endothelial beds or inducibly expressed at sites of inflammation, and inaugurate intracellular signaling followed by endothelial transmigration.1110 According to this “multistep paradigm” the function of a homing receptor is not purely to direct cellular trafficking in a tissue-specific manner, but to function as a molecular brake in initiating binding to endothelial cells under hemodynamic shear stress.
The contribution of selectins to the well-being of mammals was shown in genetically engineered mice, lacking selectins, which were prone to severe bacterial infections, and demonstrated the role of these lectins in acute inflammatory responses.1120 In the rare leukocyte adhesion deficiency type II syndrome (LAD II),1121 the patients suffer from recurrent bacterial infections as well as from growth and mental retardation. Their neutrophils are deficient in SLex and therefore cannot bind E- and P-selectin. The reason of this defect is a decrease of the metabolism of fucose required for the synthesis of Lex. Correspondingly, supplementation of the patients with fucose efficiently reduced the pathological symptoms.
12.5.3. Siglecs
Siglecs are a group of the “latest” sialic acid-recognizing lectins discovered. In contrast to the selectins, they are involved in the regulation of inflammation. Due to this pathophysiological function and the manifold members of the siglec family, with a great variety of functions, the number of publications has exploded in the last 20 years. This area is one of the most rapidly evolving in glycobiology research, not only because of the impressive immunological and clinical potential. Therefore, only a short historical introduction to this field is presented, and a number of excellent reviews, which deal with the numerous aspects of siglec occurrence, structure, and biology, will be cited.
The investigations in this area started in 1986, when P. R. Crocker and S. Gordon found a lectin-like hemagglutinin expressed on murine tissue macrophages.1124 The lectin was called “sialoadhesin (Sn),” a sialic acid-binding receptor that was purified from murine macrophages.1125 It was shown to be a transmembrane protein with 17 extracellular Ig-like domains, 16 C2-set domains, and 1 unusual V-set domain that contains an intrasheet disulfide bridge.1126 Sialoadhesin recognizes the sequences Neu5Ac(α2 → 3)Gal(β1 → 3)GalNAc and Neu5Ac(α2 → 3)Gal(β1 → 3/4)GlcNAc on glycoproteins and glycolipids.827, 827a, 828 The macromolecule is restricted to macrophages and is highest expressed in resident macrophages of hemopoietic and secondary lymphoid tissues. The adhesion mediates interactions with developing myeloid cells in the bone marrow1125 and lymphocytes in spleen and peripheral lymph nodes.1127 Recent research has shown that adhesion is also involved both in clearance of sialylated human pathogens and in antigen presentation.1123
In the 1990s, a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily, namely, sialoadhesin, CD22, and the myelin-associated glycoprotein (MAG) was defined.827, 827a The number of extracellular repeats differs much in these three lectins, that of MAG being the shortest (five repeats). After the discovery of Sn, CD22, MAG, and CD33, the group of Ajit Varki proposed the term “siglec,” derived from “sialic acid,” “immunoglobulin,” and “lectin” for each member of the group.133, 133a Siglecs are now considered a subset of I-type lectins, and they are numbered as follows: Sn (siglec-1), CD22 (siglec-2), CD33 (siglec-3), and MAG (siglec-4). The list of siglecs today1123 continues up to siglec-16 on macrophages and microglia. Mice express fewer, but partially similar siglecs.
CD22 is a B-cell-specific molecule which binds to the Neu5Ac(α2 → 6)Gal(β1 → 4)GlcNAc element on glycoproteins.1128 It is the best studied member of the siglec family. By cis and trans interactions, CD22 plays a role in B-cell signal transduction,129 and it regulates immune tolerance.1123 It carries immune inhibitory domains (ITIM and ITIM-like), as do also the other eight of the human siglecs, numbers 3 and 5–11, which have been shown to suppress immune responses or are assumed to do so.1129
The MAG is produced on myelinating oligodendrocytes at Schwann cells.1130 MAG plays roles in myelination, axonal growth regulation, and signal transduction.129 It binds to Neu5Ac(α2 → 3)Gal(β1 → 3)GalNAc elements. Studies of the binding specificities of MAG and sialoadhesin with synthetic sialylated oligosaccharides demonstrated differences between these two siglecs.1131 Remarkably, MAG bound Kdn better than Neu5Ac, and sialoadhesin revealed the opposite.
A cDNA encoding CD33, being a differentiation antigen of myeloid progenitor cells, was isolated in 1988.1132 The antigen belongs to a related subgroup composed of siglec-3 and siglecs-5–13 in primates and similar ones in rodents.133a, 133 This subgroup appears to evolve rapidly. In contrast, siglecs-1, -2, and -4 represent an evolutionarily conserved subgroup.
The roles of siglecs in immunology and human–pathogen interactions are many and varied. Special attention was recently drawn to siglec-8, which seems to function in resolving allergic inflammation, while siglec-9 may dampen neutrophil-mediated inflammation.1123
Focusing on the influence of various sialic acids on the binding of siglecs,133, 133a it was observed that the frequently occurring 9-O-acetyl group has a strong negative effect on recognition by human CD221133 and mouse sialoadhesin.828 The masking of the ligand fraction of sialic acid has been observed also with influenza A/B virus hemagglutinin and with selectins. Aberrant O-acetylation of sialic acids, caused by the enzyme pair O-acetyltransferase and -esterase, is thought to be involved in autoimmune diseases like asthma.885 Furthermore, mouse and human Sn prefer Neu5Ac over Neu5Gc. However, murine CD22 strongly prefers Neu5Gc over Neu5Ac, but human CD22, as well as CD33, siglec-5 and siglec-6 accept both types of sialic acids. It can be assumed that these human-specific changes are due to the dramatic alteration in the human “sialome” after the CMAH mutation and Neu5Gc loss.133, 133a
Many of these binding studies with differently linked sialic acids and natural or chemically modified sialic acid derivatives, e.g., Neu5Gc and Neu5,9Ac2, which much helped to understand the biology of siglecs, were, in the beginning of siglec research, based on a close cooperation between Paul Crocker, Jim Paulson, and Sørge Kelm. Paul had the cells available, Jim had purified several sialyltransferases, and Sørge had developed a procedure to transfer various sialic acids to desialylated erythrocytes in order to obtain homogeneous ligands regarding sialic acid nature and linkage.
For a detailed overview of all aspects of “siglectology,” see refs. 129, 133a, 134, 776, 1123, 133, and 1134, 1135, 1136, 1137, 1138, 1139.
13. Hereditary Sialic Acid Diseases
The last section of this chapter will deal with some diseases with an origin in genetic errors of sialic acid metabolism. Although rare, they clearly show that sialic acids have a decisive impact on health. In the following subsections, the history of the elucidation of sialuria, Salla disease, sialidosis, galactosialidosis, and GNE myopathy, the main diseases known with disturbed sialic acid metabolism, will be described. See also Section 8.4 for more chemical information.
13.1. Sialuria
Sialuria was first reported in 1968, affecting a then 2½-year-old French boy who excreted a 10,000-fold higher amount of sialic acid (Neu5Ac) than normal children.568, 583, 584, 1140, 1141, 1142 The Neu5Ac concentration in the urine was so high (excretion of up to 7.2 g Neu5Ac per day) that this monosaccharide could easily be isolated in large quantities from the patient's urine. This helped research on this topic, since Neu5Ac was very expensive at that time, and scientists were interested in a commercial source of the compound. As said in Section 11.4, the underlying metabolic defect in sialuria is the loss of feedback inhibition of the rate-limiting enzyme of sialic acid biosynthesis, i.e., UDP-GlcNAc 2-epimerase (GNE), by CMP-Neu5Ac. This leads to enhanced enzyme activity and accumulation of Neu5Ac in sialuria fibroblasts.586, 1143 Also the mutations in the epimerase gene have been analyzed.1142 Sialuria is a very rare disease. The French patient, suffering from sialuria, remained unique for a long time, but other cases, also without lysosomal involvement, but with lower excretion of sialic acids, have been described later.568 Sialuria patients show mild mental retardation, seizures, and slightly dysmorphic features.
13.2. Salla Disease and Infantile Sialic Acid Storage Disease
Salla disease is an autosomal recessive lysosomal storage disorder, first described in 1978 in four patients580, 1144 presenting severe psychomotor retardation and increased urinary excretion of Neu5Ac, combined with enlarged cellular free Neu5Ac levels. So far, about 150 cases have been reported.1141, 1145 The name “Salla” is derived from a Northern area in Finland from where most of the patients originated. However, two patients with Salla disease were also detected in Turkey.1146 A similar, but more severe disease than Salla disease, with a much earlier onset, often occurring at birth, was described in 1982 in an infant of German–Swedish origin, and it was called Infantile Sialic Acid Storage Disease (ISSD).1147 Such patients are, rarely, found in several continents. The pathomechanism of these diseases is an impaired transport of free Neu5Ac across the lysosomal membrane.1148 The reason for the Neu5Ac accumulation in the lysosomes of various tissues is a defective H+-driven transport of sialic acid (and d-glucuronic acid) through the lysosomal membrane by a mutation of the integral lysosomal protein Sialin.1142, 1149
13.3. Sialidosis
Sialidosis belongs to the so-called group of “oligosaccharidoses,” which are metabolic disorders characterized by an excessive accumulation of glycoprotein-derived oligosaccharides.568, 1150, 1151 Initially, a severe deficiency of neuraminidase in the cultured fibroblasts of a patient, classified as “mucolipidosis I,” was seen.1152, 1153 The patient had a neurodegenerative disorder with myoclonus, skeletal changes like in Hurler disease, and cherry-red spots in the macula of the eyes. As reviewed in Section 8.4, excessive quantities of sialyloligosaccharides (sialic acid = Neu5Ac), derived from glycoprotein N- and O-glycans, were found in body fluids and urine. Fibroblasts of the patients exhibit a sialidase defect and accumulate abnormal amounts of sialic acid-containing substances. Besides this group of dysmorphic children with early beginning of the disease, there is a milder type of sialidosis with normal physical appearance of the patients, absent or mild mental retardation, and onset only in adolescence. These patients also have impaired sialidase activity and excrete unusual amounts of sialyloligosaccharides.1154 These two groups are categorized as type I and type II sialidosis, respectively. In summary, sialidosis is an autosomal recessive lysosomal storage disease, affecting the degradation of glycoproteins. It is caused by a genetic defect of the lysosomal sialidase (Neu 1) gene, first cloned in 1996.1155 Several mutations are known, which lead to different expressions of the phenotype of this disease.1156, 1157
13.4. Galactosialidosis
Galactosialidosis is a disease with a combined deficiency of sialidase and β-galactosidase activity, due to a defect of a 32 kDa protein [lysosomal serine carboxypeptidase, protective protein cathepsin A (PPCA)] that shields sialidase and β-galactosidase against lysosomal proteases.1158 See also refs. 568, 1142, and 1159, 1160, 1161. Since the result of this disturbed enzyme complex is a reduced sialidase activity, like in sialidosis, there is no significant difference in the pattern of excreted glycoprotein-derived urinary sialooligosaccharides between sialidosis and galactosialidosis.1162 This suggests that sialidase deficiency, not β-galactosidase deficiency, determines the phenotype of the disease.
13.5. Hereditary Inclusion Body Myopathy (GNE Myopathy)
Hereditary inclusion body myopathy (HIBM) encompasses several syndromes with autosomal recessive or dominant inheritance, and the patients suffer from a progressive course of muscle weakness leading to severe disability. In 2011 about 500 patients with HIBM were known worldwide.1163 The most common form is the quadriceps muscle-sparing autosomal recessive myopathy,1164 which was originally described in patients of Persian–Jewish heritage.1165 The muscle problems usually start in the second to third decade of life with weakness and atrophy of distal lower limb muscles that eventually spreads proximally. The reason for this disease is a reduced activity of the key enzyme UDP-N-acetyl-glucosamine-2-epimerase/N-acetylmannosamine kinase (GNE/MNK) (see Section 11.1), which leads to impaired Neu5Ac biosynthesis, and to rimmed vacuoles and Congo-Red-positive depositions in fibers in the muscles. So far, 154 mutations have been identified in the gene of GNE that are likely leading to reduced GNE enzyme activities.1166 It was shown that mutation of methionine 743 to threonine in GNE leads to 30% reduction of enzyme activity and to an aggressive form of GNE myopathy. The introduction of threonine offers an additional phosphorylation/O-GlcNAcylation site. Indeed, the O-GlcNAcylation of the M743T GNE variant was increased. Removal of that residue resulted in almost normal GNE activity. Hereby, the balance of phosphorylation and O-GlcNAcylation is decisively involved in regulation and efficiency of GNE. Evidence is accumulating, especially from experiments with mice, that hyposialylation is underlying the molecular mechanism of this muscle weakness.1167, 1168 Also in humans, a considerable lack of free sialic acids in muscle biopsies of patients, when compared to healthy individuals, has been described.1169 It was stated that the lack of sialic acids in GNE myopathy contributes to ROS production/oxidative stress followed by muscle weakness and atrophy.1168 Correspondingly, application of antioxidants had a curative effect.1168, 1170 Furthermore, oral administration to mice of Neu5Ac, sialyllactose, ManNAc, and especially per-O-acetylated ManNAc also decreased the disease symptoms.1170 In man, intravenous application of high doses of highly sialylated immunoglobulin G (IVIG) also improved the disease symptoms.1171
14. Concluding Remarks
In this chapter, we have presented the historical development of the different areas of sialic acid research. Most impressive was to follow the efforts of those early pioneers to identify the correct structure of the N-acylneuraminic acid molecule. To achieve this goal the often cooperative work of dedicated scientists during several decades was necessary, and they had to cope with erroneous conclusions. After the isolation of sialic acids with still unknown structure from bovine submandibular gland mucin, it was by pure chance that a similar substance was discovered by studies with influenza viruses, and that years later also the analysis of the product of a lyase reaction played an important role in solving the stereochemistry of the sialic acid structure. Another difficult and lengthy effort was the development of reliable techniques for the identification of sialic acids and their many derivatives in natural sources, and chemists, synthesizing, for example, sialic acid glycosides, encountered challenges by these compounds that exceeded those experienced from working with “ordinary” sugars.
Sialic acids are among the most striking molecules in higher animals and many microorganisms, and it seems that these monosaccharides play a role in all branches of cell biology. They are involved in probably most diseases, although this is a field where more research should be focused. Sialic acids are prominent molecules on the cell surface, on the glycocalyx, not only in position but also in function, and they can be considered as cytoprotectors. However, in this position, they are often attacked and conquered by pathogens, especially viruses, and they can also be misused by malignant cells. We highlighted the beginnings and discussed the following inquiries into the pathobiochemical functions of sialic acids. The increasing number of publications on this topic shows that in laboratories all over the world efforts are continuing to gather more information and to employ this knowledge, for example, to develop effective therapies for various diseases.
When citing relevant, mostly historical papers in each section, we had to select due to space restrictions. Therefore, many important publications could not be included. And since we could not cover all aspects of sialic acid research in detail, this chapter is accompanied by special reviews on the chemistry, evolution, neurobiology, tumor biology, and virology of these monosaccharides, written by other authors.
With appreciation of the support from numerous colleagues, we are closing this chapter by citing an aphorism from the 1970s by Gerhard Uhlenbruck from the University of Cologne (Germany), who is a devoted glycoimmunologist: “Neuraminic acid (sialic acid) is a lady whose fascination has not faded away. Again and again stimulating, exciting and good for all kinds of surprises. A lovable molecule, which is enthusiastically courted by many, although the advertising budget never was opulent.” (Translated from the original German version.)
Acknowledgments
More than 50 years of sialic acid research brought us in contact with many scientists from all over the world, and—although we could not mention everyone in this review—we acknowledge their contributions and their support. We also thank our numerous associates for their dedication and contributions to the sialoglycoscience field. A special word of thanks goes to Ms Elfriede Schauer, for her continuous assistance in the preparation of this chapter.
References
- 1.Montreuil J., Vliegenthart J.F.G., Schachter H. In: Glycoproteins II. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 29b. Elsevier Science B.V; Amsterdam, The Netherlands: 1997. Preface; pp. v–vi. (New Comprehensive Biochemistry). [Google Scholar]
- 2.Warren L. The Distribution of Sialic Acids in Nature. Comp. Biochem. Physiol. 1963;10:153–171. doi: 10.1016/0010-406x(63)90238-x. [DOI] [PubMed] [Google Scholar]
- 3.Warren L. In: Biological Roles of Sialic Acid. Rosenberg A., Schengrund C.-L., editors. Plenum Press; New York, NY, USA: 1976. The Distribution of Sialic Acids Within the Eukaryotic Cell; pp. 103–121. [Google Scholar]
- 4.Ng S.-S., Dain J.A. In: Biological Roles of Sialic Acid. Rosenberg A., Schengrund C.-L., editors. Plenum Press; New York, NY, USA: 1976. The Natural Occurrence of Sialic Acids; pp. 59–102. [Google Scholar]
- 5.Corfield A.P., Schauer R. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Occurrence of Sialic Acids; pp. 5–50. (Cell Biology Monographs). [Google Scholar]
- 6.Schauer R., Kamerling J.P. In: Glycoproteins II, New Comprehensive Biochemistry. Montreuil J., Vliegenthart J.F.G., Schachter H., Neuberger A., van Deenen L.L.M., editors. Vol. 29b. Elsevier Science B.V; Amsterdam, The Netherlands: 1997. Chemistry, Biochemistry and Biology of Sialic Acids; pp. 243–402. Series Eds. [Google Scholar]
- 7.Angata T., Varki A. Chemical Diversity in the Sialic Acids and Related α-Keto Acids: An Evolutionary Perspective. Chem. Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 8.Roth J., Kempf A., Reuter G., Schauer R., Gehring W.J. Occurrence of Sialic Acids in Drosophila melanogaster. Science. 1992;256:673–675. doi: 10.1126/science.1585182. [DOI] [PubMed] [Google Scholar]
- 9.Malykh Y.N., Krisch B., Gerardy-Schahn R., Lapina E.B., Shaw L., Schauer R. The Presence of N-Acetylneuraminic Acid in Malpighian Tubules of Larvae of the Cicada Philaenus spumarius. Glycoconjugate J. 1999;16:731–739. doi: 10.1023/a:1007115627708. [DOI] [PubMed] [Google Scholar]
- 10.Repnikova E., Koles K., Nakamura M., Pitts J., Li H., Ambavane A., Zoran M.J., Panin V.M. Sialyltransferase Regulates Nervous System Function in Drosophila. J. Neurosci. 2010;30:6466–6476. doi: 10.1523/JNEUROSCI.5253-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di W., Fujita A., Hamaguchi K., Delannoy P., Sato C., Kitajima K. Diverse Subcellular Localizations of the Insect CMP-Sialic Acid Synthetases. Glycobiology. 2017;27:329–341. doi: 10.1093/glycob/cww128. [DOI] [PubMed] [Google Scholar]
- 12.Inoue S., Kitajima K. KDN (Deaminated Neuraminic Acid): Dreamful Past and Exciting Future of the Newest Member of the Sialic Acid Family. Glycoconjugate J. 2006;23:277–290. doi: 10.1007/s10719-006-6484-y. [DOI] [PubMed] [Google Scholar]
- 13.McNaught, A.D. IUPAC/IUBMB Joint Commission on Biochemical Nomenclature. Nomenclature of Carbohydrates, Recommendations 1996. Carbohydr. Res.1997, 297, 1–92. [DOI] [PubMed]
- 14.IUPAC Commission on the Nomenclature of Organic Chemistry and IUPAC/IUB Commission on Biochemical Nomenclature Tentative Rules for Carbohydrate Nomenclature—Part 1, 1969. Eur. J. Biochem. 1971;21:455–477. [PubMed] [Google Scholar]
- 15.Gielen W. Beitrag zur Chemie der Neuraminsäure. Hoppe Seyler's Z. Physiol. Chem. 1967;348:329–333. [PubMed] [Google Scholar]
- 16.Hanai N., Dohi T., Nores G.A., Hakomori S.-I. A Novel Ganglioside, De-N-acetyl-GM3 (II3NeuNH2LacCer), Acting as a Strong Promoter for Epidermal Growth Factor Receptor Kinase and as a Stimulator for Cell Growth. J. Biol. Chem. 1988;263:6296–6301. [PubMed] [Google Scholar]
- 17.Nores G.A., Hanai N., Levery S.B., Eaton H.L., Salyan M.E.K., Hakomori S.-I. Synthesis and Characterization of Lyso-GM3 (II3Neu5Ac Lactosyl Sphingosine), De-N-acetyl-GM3 (II3NeuNH2 Lactosyl Cer), and Related Compounds. Carbohydr. Res. 1988;179:393–410. doi: 10.1016/0008-6215(88)84135-1. [DOI] [PubMed] [Google Scholar]
- 18.Sjoberg E.R., Chammas R., Ozawa H., Kawashima I., Khoo K.-H., Morris H.R., Dell A., Tai T., Varki A. Expression of De-N-acetyl-gangliosides in Human Melanoma Cells Is Induced by Genistein or Nocodazole. J. Biol. Chem. 1995;270:2921–2930. doi: 10.1074/jbc.270.7.2921. [DOI] [PubMed] [Google Scholar]
- 19.Zanetta J.-P., Pons A., Iwersen M., Mariller C., Leroy Y., Timmerman P., Schauer R. Diversity of Sialic Acids Revealed Using Gas Chromatography/Mass Spectrometry of Heptafluorobutyrate Derivatives. Glycobiology. 2001;11:663–676. doi: 10.1093/glycob/11.8.663. [DOI] [PubMed] [Google Scholar]
- 20.Hamada T., Hirota H., Yokoyama S., Otsubo N., Ishida H., Kiso M., Kanamori A., Kannagi R. NMR Analysis of Novel Ganglioside GM4 Analogues Containing De-N-acetyl and Lactamized Sialic Acid: Probes for Searching New Ligand Structures for Human L-Selectin. Magn. Reson. Chem. 2002;40:517–523. [Google Scholar]
- 21.Mitsuoka C., Ohmori K., Kimura N., Kanamori A., Komba S., Ishida H., Kiso M., Kannagi R. Regulation of Selectin Binding Activity by Cyclization of Sialic Acid Moiety of Carbohydrate Ligands on Human Leukocytes. Proc. Natl. Acad. Sci. U. S. A. 1999;96:1597–1602. doi: 10.1073/pnas.96.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klenk E., Faillard H. Zur Kenntnis der Kohlenhydratgruppen der Mucoproteide. Hoppe-Seyler’s Z. Physiol. Chem. 1954;298:230–238. [PubMed] [Google Scholar]
- 23.Blix G., Lindberg E., Odin L., Werner I. Studies on Sialic Acids. Acta Soc. Med. Upsalien. 1956;61:1–25. [PubMed] [Google Scholar]
- 24.Schauer R., Faillard H. Zur Wirkungsspezifität der Neuraminidase––Das Verhalten isomerer N,O-Diacetyl-neuraminsäureglykoside im Submaxillarismucin von Pferd und Rind bei Einwirkung bakterieller Neuraminidase. Hoppe-Seyler’s Z. Physiol. Chem. 1968;349:961–968. [PubMed] [Google Scholar]
- 25.Kochetkov N.K., Chizhov O.S., Kadentsev V.I., Smirnova G.P., Zhukova I.G. Mass Spectra of Acetylated Derivatives of Sialic Acids. Carbohydr. Res. 1973;27:5–10. [Google Scholar]
- 26.Kamerling J.P., Vliegenthart J.F.G., Vink J. Mass Spectrometry of Pertrimethylsilyl Neuraminic Acid Derivatives. Carbohydr. Res. 1974;33:297–306. [Google Scholar]
- 27.Kamerling J.P., Vliegenthart J.F.G., Versluis C., Schauer R. Identification of O-Acetylated N-Acylneuraminic Acids by Mass Spectrometry. Carbohydr. Res. 1975;41:7–17. doi: 10.1016/s0008-6215(00)87002-0. [DOI] [PubMed] [Google Scholar]
- 28.Brown E.B., Brey W.S., Jr., Weltner W., Jr. Cell-Surface Carbohydrates and Their Interactions—I. NMR of N-Acetyl Neuraminic Acid. Biochim. Biophys. Acta. 1975;399:124–130. doi: 10.1016/0304-4165(75)90218-4. [DOI] [PubMed] [Google Scholar]
- 29.Haverkamp J., van Halbeek H., Dorland L., Vliegenthart J.F.G., Pfeil R., Schauer R. High-Resolution 1H-NMR Spectroscopy of Free and Glycosidically Linked O-Acetylated Sialic Acids. Eur. J. Biochem. 1982;122:305–311. doi: 10.1111/j.1432-1033.1982.tb05881.x. [DOI] [PubMed] [Google Scholar]
- 30.Kamerling J.P., Vliegenthart J.F.G. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Gas-Liquid Chromatography and Mass Spectrometry of Sialic Acids; pp. 95–125. (Cell Biology Monographs). [Google Scholar]
- 31.Manzi A.E., Dell A., Azadi P., Varki A. Studies of Naturally Occurring Modifications of Sialic Acids by Fast-Atom Bombardment-Mass Spectrometry—Analysis of Positional Isomers by Periodate Cleavage. J. Biol. Chem. 1990;265:8094–8107. [PubMed] [Google Scholar]
- 32.Klein A., Diaz S., Ferreira I., Lamblin G., Roussel P., Manzi A.E. New Sialic Acids From Biological Sources Identified by a Comprehensive and Sensitive Approach: Liquid Chromatography–Electrospray Ionization-Mass Spectrometry (LC-ESI-MS) of SIA Quinoxalinones. Glycobiology. 1997;7:421–432. doi: 10.1093/glycob/7.3.421. [DOI] [PubMed] [Google Scholar]
- 33.Kamerling J.P., Gerwig G.J. In: Glycobiology Protocols. Brockhausen I., editor. Vol. 347. Humana Press Inc.; Totowa, NJ, USA: 2006. Structural Analysis of Naturally Occurring Sialic Acids; pp. 69–91. (Methods Mol. Biol.). [DOI] [PubMed] [Google Scholar]
- 34.Niimura Y., Ishizuka I. Unique Disialosyl Gangliosides From Salmon Kidney: Characterization of V3αFuc,IV3βGalNAc,II3(αNeuAc)2-Gg4Cer and Its Analogue With 4-O-Acetyl-N-acetylneuraminic Acid. Glycoconjugate J. 2006;23:489–499. doi: 10.1007/s10719-006-6562-1. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharjee A.K., Jennings H.J., Kenny C.P., Martin A., Smith I.C.P. Structural Determination of the Sialic Acid Polysaccharide Antigens of Neisseria meningitidis Serogroups B and C With Carbon 13 Nuclear Magnetic Resonance. J. Biol. Chem. 1975;250:1926–1932. [PubMed] [Google Scholar]
- 36.Ørskov F., Ørskov I., Sutton A., Schneerson R., Lin W., Egan W., Hoff G.E., Robbins J.B. Form Variation in Escherichia coli K1: Determined by O-Acetylation of the Capsular Polysaccharide. J. Exp. Med. 1979;149:669–685. doi: 10.1084/jem.149.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reuter G., Pfeil R., Stoll S., Schauer R., Kamerling J.P., Versluis C., Vliegenthart J.F.G. Identification of New Sialic Acids Derived From Glycoprotein of Bovine Submandibular Gland. Eur. J. Biochem. 1983;134:139–143. doi: 10.1111/j.1432-1033.1983.tb07542.x. [DOI] [PubMed] [Google Scholar]
- 38.Kamerling J.P., van Halbeek H., Vliegenthart J.F.G., Pfeil R., Shukla A.K., Schauer R. Migration of O-Acetyl Groups in Sialic Acids. Glycoconjugates; Proc. 7th Int. Symp., Lund-Ronneby, Sweden, July 17–23; 1983. pp. 160–161. [Google Scholar]
- Kamerling J.P., Schauer R., Shukla A.K., Stoll S., van Halbeek H., Vliegenthart J.F.G. Migration of O-Acetyl Groups in N,O-Acetylneuraminic Acids. Eur. J. Biochem. 1987;162:601–607. doi: 10.1111/j.1432-1033.1987.tb10681.x. [DOI] [PubMed] [Google Scholar]
- 39.Kogan G., Jann B., Jann K. Structure of the Escherichia coli O104 Polysaccharide and Its Identity With the Capsular K9 Polysaccharide. FEMS Microbiol. Lett. 1992;91:135–140. doi: 10.1016/0378-1097(92)90673-c. [DOI] [PubMed] [Google Scholar]
- 40.Gamian A., Romanowska E., Ulrich J., Defaye J. The Structure of the Sialic Acid-Containing Escherichia coli O104 O-Specific Polysaccharide and Its Linkage to the Core Region in Lipopolysaccharide. Carbohydr. Res. 1992;236:195–208. doi: 10.1016/0008-6215(92)85016-s. [DOI] [PubMed] [Google Scholar]
- 41.Buscher H.-P., Casals-Stenzel J., Schauer R. New Sialic Acids—Identification of N-Glycoloyl-O-acetylneuraminic Acids and N-Acetyl-O-glycoloylneuraminic Acids by Improved Methods for Detection of N-Acyl and O-Acyl Groups and by Gas–Liquid Chromatography. Eur. J. Biochem. 1974;50:71–82. doi: 10.1111/j.1432-1033.1974.tb03873.x. [DOI] [PubMed] [Google Scholar]
- 42.Schauer R., Haverkamp J., Wember M., Vliegenthart J.F.G., Kamerling J.P. N-Acetyl-9-O-l-lactylneuraminic Acid, a New Acylneuraminic Acid From Bovine Submandibular Gland. Eur. J. Biochem. 1976;62:237–242. doi: 10.1111/j.1432-1033.1976.tb10153.x. [DOI] [PubMed] [Google Scholar]
- 43.Haverkamp J., Schauer R., Wember M., Farriaux J.-P., Kamerling J.P., Versluis C., Vliegenthart J.F.G. Neuraminic Acid Derivatives Newly Discovered in Humans: N-Acetyl-9-O-l-lactoylneuraminic Acid, N,9-O-Diacetylneuraminic Acid and N-Acetyl-2,3-dehydro-2-deoxyneuraminic Acid. Hoppe-Seyler’s Z. Physiol. Chem. 1976;357:1699–1705. doi: 10.1515/bchm2.1976.357.2.1699. [DOI] [PubMed] [Google Scholar]
- 44.Reuter G., Pfeil R., Kamerling J.P., Vliegenthart J.F.G., Schauer R. Identification by Gas-Liquid Chromatography-Mass Spectrometry of 4-O-Acetyl-9-O-lactyl-N-acetyl-neuraminic Acid, a New Sialic Acid From Horse Submandibular Gland. Biochim. Biophys. Acta. 1980;630:306–310. doi: 10.1016/0304-4165(80)90435-3. [DOI] [PubMed] [Google Scholar]
- 45.Bulai T., Bratosin D., Pons A., Montreuil J., Zanetta J.-P. Diversity of the Human Erythrocyte Membrane Sialic Acids in Relation With Blood Groups. FEBS Lett. 2003;534:185–189. doi: 10.1016/s0014-5793(02)03838-3. [DOI] [PubMed] [Google Scholar]
- 46.Bergwerff A.A., Hulleman S.H.D., Kamerling J.P., Vliegenthart J.F.G., Shaw L., Reuter G., Schauer R. Nature and Biosynthesis of Sialic Acids in the Starfish Asterias rubens—Identification of Sialo-oligomers and Detection of S-Adenosyl-l-methionine:N-acylneuraminate 8-O-Methyltransferase and CPM-N-Acetylneuraminate Monooxygenase Activities. Biochimie. 1992;74:25–38. doi: 10.1016/0300-9084(92)90181-d. [DOI] [PubMed] [Google Scholar]
- 47.Inagaki M., Shiizaki M., Hiwatashi T., Miyamoto T., Higuchi R. Constituents of Crinoidea—5. Isolation and Structure of a New Glycosyl Inositolphosphoceramide-Type Ganglioside From the Feather Star Comanthina schlegeli. Chem. Pharm. Bull. 2007;55:1649–1651. doi: 10.1248/cpb.55.1649. [DOI] [PubMed] [Google Scholar]
- 48.Yamada K., Harada Y., Nagaregawa Y., Miyamoto T., Isobe R., Higuchi R. Constituents of Holothuroidea, 7.—Isolation and Structure of Biologically Active Gangliosides From the Sea Cucumber Holothuria pervicax. Eur. J. Org. Chem. 1998:2519–2525. [Google Scholar]
- 49.Kochetkov N.K., Smirnova G.P., Chekareva N.V. Isolation and Structural Studies of a Sulfated Sialosphingolipid From the Sea Urchin Echinocardium cordatum. Biochim. Biophys. Acta. 1976;424:274–283. doi: 10.1016/0005-2760(76)90195-8. [DOI] [PubMed] [Google Scholar]
- 50.Slomiany A., Kojima K., Banas-Gruszka Z., Slomiany B.L. Structure of a Novel Sulfated Sialoglycosphingolipid From Bovine Gastric Mucosa. Biochem. Biophys. Res. Commun. 1981;100:778–784. doi: 10.1016/s0006-291x(81)80242-2. [DOI] [PubMed] [Google Scholar]
- 51.Roseman S., Jourdian G.W., Watson D., Rood R. Enzymatic Synthesis of Sialic Acid 9-Phosphates. Proc. Natl. Acad. Sci. U. S. A. 1961;47:958–961. doi: 10.1073/pnas.47.7.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamerling J.P., Vliegenthart J.F.G., Schauer R., Strecker G., Montreuil J. Isolation and Identification of 2-Deoxy-2,3-dehydro-N-acetylneuraminic Acid From the Urine of a Patient With Sialuria. Eur. J. Biochem. 1975;56:253–258. doi: 10.1111/j.1432-1033.1975.tb02228.x. [DOI] [PubMed] [Google Scholar]
- 53.Beau J.-M., Schauer R., Haverkamp J., Kamerling J.P., Dorland L., Vliegenthart J.F.G. Chemical Behaviour of Cytidine 5′-Monophospho-N-acetyl-β-d-neuraminic Acid Under Neutral and Alkaline Conditions. Eur. J. Biochem. 1984;140:203–208. doi: 10.1111/j.1432-1033.1984.tb08087.x. [DOI] [PubMed] [Google Scholar]
- 54.Furuhata K., Sato S., Goto M., Takayanagi H., Ogura H. Studies on Sialic Acids—VII. The Crystal and Molecular Structure of N-Acetyl-2,3-dehydro-2-deoxyneuraminic Acid. Chem. Pharm. Bull. 1988;36:1872–1876. [Google Scholar]
- 55.Schauer R., Schröder C., Shukla A.K. New Techniques for the Investigation of Structure and Metabolism of Sialic Acids. Adv. Exp. Med. Biol. 1984;174:75–86. doi: 10.1007/978-1-4684-1200-0_7. [DOI] [PubMed] [Google Scholar]
- 56.Shukla A.K., Schröder C., Nöhle U., Schauer R. Natural Occurrence and Preparation of O-Acylated 2,3-Unsaturated Sialic Acids. Carbohydr. Res. 1987;168:199–209. doi: 10.1016/0008-6215(87)80026-5. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki M., Suzuki A., Yamakawa T., Matsunaga E. Characterization of 2,7-Anhydro-N-acetylneuraminic Acid in Human Wet Cerumen. J. Biochem. 1985;97:509–515. doi: 10.1093/oxfordjournals.jbchem.a135085. [DOI] [PubMed] [Google Scholar]
- 58.Li Y.-T., Nakagawa H., Ross S.A., Hansson G.C., Li S.-C. A Novel Sialidase Which Releases 2,7-Anhydro-α-N-acetylneuraminic Acid From Sialoglycoconjugates. J. Biol. Chem. 1990;265:21629–21633. [PubMed] [Google Scholar]
- 59.Pozsgay V., Jennings H., Kasper D.L. 4,8-Anhydro-N-acetylneuraminic Acid—Isolation From Edible Bird's Nest and Structure Determination. Eur. J. Biochem. 1987;162:445–450. doi: 10.1111/j.1432-1033.1987.tb10622.x. [DOI] [PubMed] [Google Scholar]
- 60.Saito K.-I., Sugai K., Fujikura K., Yamada N., Goto M., Ban C., Hayasaka E., Sugiyama N., Tomita K. Thermal Degradation of Sodium N-Acetylneuraminate. Carbohydr. Res. 1989;185:307–314. [Google Scholar]
- 61.Cebo C., Dambrouck T., Maes E., Laden C., Strecker G., Michalski J.-C., Zanetta J.-P. Recombinant Human Interleukins IL-1α, IL-1β, IL-4, IL-6, and IL-7 Show Different and Specific Calcium-Independent Carbohydrate-Binding Properties. J. Biol. Chem. 2001;276:5685–5691. doi: 10.1074/jbc.M008662200. [DOI] [PubMed] [Google Scholar]
- 62.Bratosin D., Palii C., Moicean A.D., Zanetta J.-P., Montreuil J. Reduced Diversity of the Human Erythrocyte Membrane Sialic Acids in Polycythemia Vera. Absence of N-Glycolylneuraminic Acid and Characterisation of N-Acetylneuraminic Acid 1,7-Lactone. Biochimie. 2007;89:355–359. doi: 10.1016/j.biochi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 63.Li Y.-T., Maskos K., Chou C.-W., Cole R.B., Li S.-C. Presence of an Unusual GM2 Derivative, Taurine-Conjugated GM2, in Tay–Sachs Brain. J. Biol. Chem. 2003;278:35286–35291. doi: 10.1074/jbc.M306126200. [DOI] [PubMed] [Google Scholar]
- 64.Klenk E., Uhlenbruck G. Über die Abspaltung von N-Glykolyl-neuraminsäure (P-Sialinsäure) aus dem Schweine-Submaxillarismucin durch das “Receptor Destroying Enzyme.”. Hoppe-Seyler’s Z. Physiol. Chem. 1957;307:266–271. [PubMed] [Google Scholar]
- 65.Handa S., Yamakawa T. The Chemistry of Lipids of Posthemolytic Residue or Stroma of Erythrocytes. XII. Chemical Structure and Chromatographic Behaviour of Hematosides Obtained From Equine and Dog Erythrocytes. Jpn. J. Exp. Med. 1964;34:293–304. [PubMed] [Google Scholar]
- 66.Jaques L.W., Riesco B.F., Weltner W., Jr. N.M.R. Spectroscopy and Calcium Binding of Sialic Acids: N-Glycolylneuraminic Acid and Periodate-Oxidized N-Acetylneuraminic Acid. Carbohydr. Res. 1980;83:21–32. [Google Scholar]
- 67.Warren L. N-Glycolyl-8-O-methylneuraminic Acid—A New Form of Sialic Acid in the Starfish Asterias forbesi. Biochim. Biophys. Acta. 1964;83:129–132. doi: 10.1016/0926-6526(64)90062-x. [DOI] [PubMed] [Google Scholar]
- 68.Smirnova G.P., Kochetkov N.K., Sadovskaya V.L. Gangliosides of the Starfish Aphelasterias japonica—Evidence for a New Linkage Between Two N-Glycolylneuraminic Acid Residues Through the Hydroxy Group of the Glycolic Acid Residue. Biochim. Biophys. Acta. 1987;920:47–55. doi: 10.1016/0005-2760(87)90309-2. [DOI] [PubMed] [Google Scholar]
- 69.Zanetta J.-P., Srinivasan V., Schauer R. Analysis of Monosaccharides, Fatty Constituents and Rare O-Acetylated Sialic Acids From Gonads of the Starfish Asterias rubens. Biochimie. 2006;88:171–178. doi: 10.1016/j.biochi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 70.Arao K., Inagaki M., Higuchi R. Constituents of Crinoidea, 2—Isolation and Structure of the Novel Type Gangliosides From the Feather Star Comanthus japonica. Chem. Pharm. Bull. 2001;49:695–698. doi: 10.1248/cpb.49.695. [DOI] [PubMed] [Google Scholar]
- 71.Kubo H., Irie A., Inagaki F., Hoshi M. Gangliosides From the Eggs of the Sea Urchin, Anthocidaris crassispina. J. Biochem. 1990;108:185–192. doi: 10.1093/oxfordjournals.jbchem.a123179. [DOI] [PubMed] [Google Scholar]
- 72.Kitazume S., Kitajima K., Inoue S., Haslam S.M., Morris H.R., Dell A., Lennarz W.J., Inoue Y. The Occurrence of Novel 9-O-Sulfated N-Glycolylneuraminic Acid-Capped α2 → 5-O-Glycolyl-Linked Oligo/PolyNeu5Gc Chains in Sea Urchin Egg Cell Surface Glycoprotein—Identification of a New Chain Termination Signal for Polysialyltransferase. J. Biol. Chem. 1996;271:6694–6701. doi: 10.1074/jbc.271.12.6694. [DOI] [PubMed] [Google Scholar]
- 73.Kluge A., Reuter G., Lee H., Ruch-Heeger B., Schauer R. Interaction of Rat Peritoneal Macrophages With Homologous Sialidase-Treated Thrombocytes In Vitro: Biochemical and Morphological Studies—Detection of N-(O-Acetyl)glycoloylneuraminic Acid. Eur. J. Cell Biol. 1992;59:12–20. [PubMed] [Google Scholar]
- 74.Higuchi R., Inukai K., Jhou J.X., Honda M., Komori T., Tsuji S., Nagai Y. Biologically Active Glycosides From Asteroidea—XXXI. Glycosphingolipids From the Starfish Asterias amurensis versicolor Sladen, 2—Structure and Biological Activity of Ganglioside Molecular Species. Liebigs Ann. Chem. 1993:359–366. [Google Scholar]
- 75.Nöhle U., Shukla A.K., Schröder C., Reuter G., Schauer R., Kamerling J.P., Vliegenthart J.F.G. Structural Parameters and Natural Occurrence of 2-Deoxy-2,3-didehydro-N-glycoloylneuraminic Acid. Eur. J. Biochem. 1985;152:459–463. doi: 10.1111/j.1432-1033.1985.tb09219.x. [DOI] [PubMed] [Google Scholar]
- 76.Nadano D., Iwasaki M., Endo S., Kitajima K., Inoue S., Inoue Y. A Naturally Occurring Deaminated Neuraminic Acid, 3-Deoxy-d-glycero-d-galacto-nonulosonic Acid (KDN)—Its Unique Occurrence at the Nonreducing Ends of Oligosialyl Chains in Polysialoglycoprotein of Rainbow Trout Eggs. J. Biol. Chem. 1986;261:11550–11557. [PubMed] [Google Scholar]
- 77.Knirel Y.A., Kocharova N.A., Shashkov A.S., Kochetkov N.K., Mamontova V.A., Solov’eva T.F. Structure of the Capsular Polysaccharide of Klebsiella ozaenae Serotype K4 Containing 3-Deoxy-d-glycero-d-galacto-nonulosonic Acid. Carbohydr. Res. 1989;188:145–155. doi: 10.1016/0008-6215(89)84067-4. [DOI] [PubMed] [Google Scholar]
- 78.Strecker G., Wieruszeski J.-M., Michalski J.-C., Alonso C., Boilly B., Montreuil J. Characterization of Lex, Ley and Ley Antigen Determinants in KDN-Containing O-Linked Glycan Chains From Pleurodeles waltlii Jelly Coat Eggs. FEBS Lett. 1992;298:39–43. doi: 10.1016/0014-5793(92)80018-c. [DOI] [PubMed] [Google Scholar]
- 79.Shashkov A.S., Torgov V.I., Nazarenko E.L., Zubkov V.A., Gorshkova N.M., Gorshkova R.P., Widmalm G. Structure of the Phenol-Soluble Polysaccharide From Shewanella putrefaciens Strain A6. Carbohydr. Res. 2002;337:1119–1127. doi: 10.1016/s0008-6215(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 80.Iwasaki M., Inoue S., Troy F.A. A New Sialic Acid Analogue, 9-O-Acetyl-Deaminated Neuraminic Acid, and α-2,8-Linked O-Acetylated Poly(N-glycolylneuraminyl) Chains in a Novel Polysialoglycoprotein From Salmon Eggs. J. Biol. Chem. 1990;265:2596–2602. [PubMed] [Google Scholar]
- 81.Plancke Y., Wieruszeski J.-M., Boilly B., Strecker G. Primary Structure of Seven New Acidic Oligosaccharide-Alditols From the Egg Jelly Coats of Axolotl mexicanum and Pleurodeles waltl. Ci. Cult. J. Braz. Assoc. Adv. Sci. 1994;46:273–279. [Google Scholar]
- 82.Gil-Serrano A.M., Rodríguez-Carvajal M.A., Tejero-Mateo P., Espartero J.L., Thomas-Oates J., Ruiz-Sainz J.E., Buendía-Clavería A.M. Structural Determination of a 5-O-Methyl-Deaminated Neuraminic Acid (Kdn)-Containing Polysaccharide Isolated From Sinorhizobium fredii. Biochem. J. 1998;334:585–594. doi: 10.1042/bj3340585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Angata T., Nakata D., Matsuda T., Kitajima K., Troy F.A., II Biosynthesis of KDN (2-Keto-3-deoxy-d-glycero-d-galacto-nononic Acid)—Identification and Characterization of a KDN-9-Phosphate Synthetase Activity From Trout Testis. J. Biol. Chem. 1999;274:22949–22956. doi: 10.1074/jbc.274.33.22949. [DOI] [PubMed] [Google Scholar]
- 84.Meindl P., Tuppy H. Über 2-Desoxy-2,3-dehydro-sialinsäuren—II. Kompetitive Hemmung der Vibrio-cholerae-Neuraminidase durch 2-Desoxy-2,3-dehydro-N-acyl-neuraminsäuren. Hoppe-Seyler’s Z. Physiol. Chem. 1969;350:1088–1092. [PubMed] [Google Scholar]
- 85.Saito M., Rosenberg A. Identification and Characterization of N-Acetyl-2,3-didehydro-2-deoxyneuraminic Acid as a Metabolite in Mammalian Brain. Biochemistry. 1984;23:3784–3788. doi: 10.1021/bi00311a034. [DOI] [PubMed] [Google Scholar]
- 86.Burmeister W.P., Henrissat B., Bosso C., Cusack S., Ruigrok R.W.H. Influenza B Virus Neuraminidase Can Synthesize Its Own Inhibitor. Structure. 1993;1:19–26. doi: 10.1016/0969-2126(93)90005-2. [DOI] [PubMed] [Google Scholar]
- 87.Chou M.-Y., Li S.-C., Kiso M., Hasegawa A., Li Y.-T. Purification and Characterization of Sialidase L, a NeuAcα2 → 3Gal-Specific Sialidase. J. Biol. Chem. 1994;269:18821–18826. [PubMed] [Google Scholar]
- 88.Gottschalk A. Cambridge University Press; London, UK: 1960. The Chemistry and Biology of Sialic Acids and Related Substances. [Google Scholar]
- 89.Rosenberg A., Schengrund C.-L. Plenum Press; New York, NY, USA: 1976. Biological Roles of Sialic Acid. [Google Scholar]
- 90.Schauer, R., Ed. Cell Biology Monographs, Vol. 10; Sialic Acids—Chemistry, Metabolism and Function; Springer-Verlag: Vienna, Austria, 1982.
- 91.Wiegandt, H., Ed. New Comprehensive Biochemistry, Vol. 10; Glycolipids; Elsevier Science Publishers: Amsterdam, The Netherlands, 1985.
- 92.Rosenberg A. Plenum Press; New York, NY, USA: 1995. Biology of the Sialic Acids. [Google Scholar]
- 93.Tiralongo J., Martinez-Duncker I., editors. Sialobiology: Structure, Biosynthesis and Function—Sialic Acid Glycoconjugates in Health and Disease. Bentham Science Publishers; Oak Park, IL, USA: 2013. [Google Scholar]
- 94.Gerardy-Schahn R., Delannoy P., von Itzstein M., editors. SialoGlyco Chemistry and Biology I—Biosynthesis, Structural Diversity and Sialoglycopathologies. Vol. 366. Springer; Berlin, Germany: 2015. (Topics in Current Chemistry). [Google Scholar]
- 95.Gerardy-Schahn R., Delannoy P., von Itzstein M., editors. SialoGlyco Chemistry and Biology II—Tools and Techniques to Identify and Capture Sialoglycans. Vol. 367. Springer; Berlin, Germany: 2015. (Topics in Current Chemistry). [Google Scholar]
- 96.Zilliken F., Whitehouse M.W. The Nonulosaminic Acids—Neuraminic Acids and Related Compounds (Sialic Acids) Adv. Carbohydr. Chem. 1958;13:237–263. [PubMed] [Google Scholar]
- 97.Gottschalk A. Neuraminidase: Its Substrate and Mode of Action. Adv. Enzymol. 1958;20:135–146. doi: 10.1002/9780470122655.ch5. [DOI] [PubMed] [Google Scholar]
- 98.Blix G., Jeanloz R.W. In: The Amino Sugars. Jeanloz R.W., editor. Vol. IA. Academic Press; New York, USA: 1969. Sialic Acids and Muramic Acid; pp. 213–265. [Google Scholar]
- 99.Faillard H., Schauer R. In: Glycoproteins—Their Composition, Structure and Function. 2nd ed. Gottschalk A., editor. Vol. 5, Part B. B.B.A. Library, Elsevier Publishing Company; Amsterdam, The Netherlands: 1972. Glycoproteins as Lubricants, Protective Agents, Carriers, Structural Proteins and as Participants in Other Functions; pp. 1246–1267. [Google Scholar]
- 100.Tuppy H., Gottschalk A. In: Glycoproteins—Their Composition, Structure and Function. 2nd ed. Gottschalk A., editor. Vol. 5, Part A. B.B.A. Library, Elsevier Publishing Company; Amsterdam, The Netherlands: 1972. The Structure of Sialic Acids and Their Quantitation; pp. 403–449. [Google Scholar]
- 101.Schauer R. Chemistry, Metabolism, and Biological Functions of Sialic Acids. Adv. Carbohydr. Chem. Biochem. 1982;40:131–234. doi: 10.1016/s0065-2318(08)60109-2. [DOI] [PubMed] [Google Scholar]
- 102.Schauer R. Analysis of Sialic Acids. Methods Enzymol. 1987;138:132–161. doi: 10.1016/0076-6879(87)38012-7. [DOI] [PubMed] [Google Scholar]
- 103.Schauer R. Sialic Acids: Metabolism of O-Acetyl Groups. Methods Enzymol. 1987;138:611–626. doi: 10.1016/0076-6879(87)38055-3. [DOI] [PubMed] [Google Scholar]
- 104.Reuter G., Schauer R. Determination of Sialic Acids. Methods Enzymol. 1994;230:168–199. doi: 10.1016/0076-6879(94)30012-7. [DOI] [PubMed] [Google Scholar]
- 105.Reuter G., Schauer R. Enzymic Methods of Sialic Acid Analysis. Methods Carbohydr. Chem. 1994;10:29–39. [Google Scholar]
- 106.Von Itzstein, M.; Thomson, R. J. The Synthesis of Novel Sialic Acids as Biological Probes. In: Glycoscience—Synthesis of Oligosaccharides and Glycoconjugates, Driguez, H.; Thiem, J., Eds.; Topics in Current Chemistry, Springer–Verlag: Berlin, Germany; Vol. 186, 1997; pp 119–170.
- 107.Boons G.-J., Demchenko A.V. In: Carbohydrate-Based Drug Discovery. Wong C.-H., editor. Vol. 1. Wiley–VCH; Weinheim, Germany: 2003. The Chemistry of Sialic Acid; pp. 55–102. [Google Scholar]
- 108.Thomson R., von Itzstein M. In: Carbohydrate-Based Drug Discovery. Wong C.-H., editor. Vol. 2. Wiley–VCH; Weinheim, Germany: 2003. N-Acetylneuraminic Acid Derivatives and Mimetics as Anti-influenza Agents; pp. 831–861. [Google Scholar]
- 109.Kohla G., Schauer R. In: Neuroglycobiology (Molecular and Cellular Neurobiology) Fukuda M., Rutishauser U., Schnaar R.L., editors. Oxford University Press; Oxford, UK: 2005. Sialic Acids in Gangliosides: Origin and Function; pp. 133–155. [Google Scholar]
- 110.Schauer R. In: Glycobiology. Sansom C., Markman O., editors. Scion Publishing Ltd; Bloxham, UK: 2007. The Diversity of Sialic Acids and Their Interplay With Lectins; pp. 136–149. [Google Scholar]
- 111.Miyagi T., Yamaguchi K. In: Comprehensive Glycoscience—From Chemistry to Systems Biology. Kamerling J.P., Boons G.-J., Lee Y.C., Suzuki A., Taniguchi N., Voragen A.G.J., editors. Vol. 3. Elsevier Ltd.; Oxford, UK: 2007. Sialic Acids; pp. 297–323. [Google Scholar]
- 112.Varki A., Schauer R. In: Essentials of Glycobiology. 2nd ed. Varki A., Cummings R.D., Esko J.D., Freeze H.H., Stanley P., Bertozzi C.R., Hart G.W., Etzler M.E., editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, USA: 2009. Sialic Acids; pp. 199–217. [PubMed] [Google Scholar]
- 113.Monti E., Bonten E., D’Azzo A., Bresciani R., Venerando B., Borsani G., Schauer R., Tettamanti G. Sialidases in Vertebrates: A Family of Enzymes Tailored for Several Cell Functions. Adv. Carbohydr. Chem. Biochem. 2010;64:403–479. doi: 10.1016/S0065-2318(10)64007-3. [DOI] [PubMed] [Google Scholar]
- 114.Varki A., Schnaar R.L., Schauer R. In: Essentials of Glycobiology. 3rd ed. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Darvill A., Kinoshita T., Packer N.H., Prestegard J.J., Schnaar R.L., Seeberger P.H., editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, USA: 2017. Sialic Acids and Other Nonulosonic Acids; pp. 179–195. [Google Scholar]
- 115.Klenk E. Chemie und Biochemie der Neuraminsäure. Angew. Chem. 1956;68:349–352. [Google Scholar]
- 116.Blix G. Sialic Acids. Proceedings of IVth International Congress of Biochemistry, Vienna, Austria; 1958. pp. 94–106. [Google Scholar]
- 117.Barry G.T. Colominic Acid, a Polymer of N-Acetylneuraminic Acid. J. Exp. Med. 1958;107:507–521. doi: 10.1084/jem.107.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wiegandt H. Struktur und Funktion der Ganglioside. Angew. Chem. 1968;80:89–98. [Google Scholar]
- 119.Schauer R. Chemistry and Biology of the Acylneuraminic Acids. Angew. Chem., Int. Ed. Engl. 1973;12:127–138. doi: 10.1002/anie.197301271. [DOI] [PubMed] [Google Scholar]
- 120.Yamakawa T. Thus Started Ganglioside Research. Trends Biochem. Sci. 1988;13:452–454. doi: 10.1016/0968-0004(88)90221-6. [DOI] [PubMed] [Google Scholar]
- 121.Faillard H. The Early History of Sialic Acids. Trends Biochem. Sci. 1989;14:237–241. doi: 10.1016/0968-0004(89)90034-0. [DOI] [PubMed] [Google Scholar]
- 122.Schauer R. Biosynthesis and Function of N- and O-Substituted Sialic Acids. Glycobiology. 1991;1:449–452. doi: 10.1093/glycob/1.5.449. [DOI] [PubMed] [Google Scholar]
- 123.Troy F.A., II. Polysialylation: From Bacteria to Brains. Glycobiology. 1992;2:5–23. doi: 10.1093/glycob/2.1.5. [DOI] [PubMed] [Google Scholar]
- 124.Varki A. Diversity in the Sialic Acids. Glycobiology. 1992;2:25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Corfield T. Bacterial Sialidases: Roles in Pathogenicity and Nutrition. Glycobiology. 1992;2:509–521. doi: 10.1093/glycob/2.6.509. [DOI] [PubMed] [Google Scholar]
- 126.Harduin-Lepers A., Recchi M.-A., Delannoy P. 1994, the Year of Sialyltransferases. Glycobiology. 1995;5:741–758. doi: 10.1093/glycob/5.8.741. [DOI] [PubMed] [Google Scholar]
- 127.Taylor G. Sialidases: Structures, Biological Significance and Therapeutic Potential. Curr. Opin. Struct. Biol. 1996;6:830–837. doi: 10.1016/s0959-440x(96)80014-5. [DOI] [PubMed] [Google Scholar]
- 128.Schauer R. Origin and Biological Role of the Great Chemical Diversity of Natural Sialic Acids. Trends Glycosci. Glycotechnol. 1997;9:315–330. [Google Scholar]
- 129.Kelm S., Schauer R. Sialic Acids in Molecular and Cellular Interactions. Int. Rev. Cytol. 1997;175:137–240. doi: 10.1016/S0074-7696(08)62127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Traving C., Schauer R. Structure, Function and Metabolism of Sialic Acids. Cell. Mol. Life Sci. 1998;54:1330–1349. doi: 10.1007/s000180050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schauer R. Achievements and Challenges of Sialic Acid Research. Glycoconjugate J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schauer R. Sialic Acids: Fascinating Sugars in Higher Animals and Man. Zoology. 2004;107:49–64. doi: 10.1016/j.zool.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 133.Crocker P.R., Clark E.A., Filbin M., Gordon S., Jones Y., Kehrl J.H., Kelm S., Le Douarin N., Powell L., Roder J., Schnaar R.L., Sgroi D.C., Stamenkovic I., Schauer R., Schachner M., van den Berg T.K., van der Merwe P.A., Watt S.M., Varki A. Siglecs: A Family of Sialic Acid-Binding Lectins. Glycobiology. 1998;8:v. doi: 10.1093/oxfordjournals.glycob.a018832. [DOI] [PubMed] [Google Scholar]
- Varki A., Angata T. Siglecs—The Major Subfamily of I-Type Lectins. Glycobiology. 2006;16:1R–27R. doi: 10.1093/glycob/cwj008. [DOI] [PubMed] [Google Scholar]
- 134.Crocker P.R., Paulson J.C., Varki A. Siglecs and Their Roles in the Immune System. Nat. Rev. Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 135.Schauer R. Sialic Acids as Regulators of Molecular and Cellular Interactions. Curr. Opin. Struct. Biol. 2009;19:1–8. doi: 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang B. Sialic Acid Is an Essential Nutrient for Brain Development and Cognition. Annu. Rev. Nutr. 2009;29:177–222. doi: 10.1146/annurev.nutr.28.061807.155515. [DOI] [PubMed] [Google Scholar]
- 137.Chen X., Varki A. Advances in the Biology and Chemistry of Sialic Acids. ACS Chem. Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Harduin-Lepers A. Comprehensive Analysis of Sialyltransferases in Vertebrate Genomes. Glycobiol. Insights. 2010;2:29–61. [Google Scholar]
- 139.Schauer R., Kamerling J.P. The Chemistry and Biology of Trypanosomal trans-Sialidases: Virulence Factors in Chagas Disease and Sleeping Sickness. ChemBioChem. 2011;12:2246–2264. doi: 10.1002/cbic.201100421. [DOI] [PubMed] [Google Scholar]
- 140.Ogura H. Development of Miracle Medicines From Sialic Acids. Proc. Jpn. Acad., Ser. B. 2011;87:328–361. doi: 10.2183/pjab.87.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Varki A., Gagneux P. Multifarious Roles of Sialic Acids in Immunity. Ann. N. Y. Acad. Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li Y., Chen X. Sialic Acid Metabolism and Sialyltransferases: Natural Functions and Applications. Appl. Microbiol. Biotechnol. 2012;94:887–905. doi: 10.1007/s00253-012-4040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vimr E.R. Unified Theory of Bacterial Sialometabolism: How and Why Bacteria Metabolize Host Sialic Acids. ISRN Microbiol. 2013;2013 doi: 10.1155/2013/816713. 26 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schnaar R.L., Gerardy-Schahn R., Hildebrandt H. Sialic Acids in the Brain: Gangliosides and Polysialic Acid in Nervous System Development, Stability, Disease, and Regeneration. Physiol. Rev. 2014;94:461–518. doi: 10.1152/physrev.00033.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang T., She Z., Huang Z., Li J., Luo X., Deng Y. Application of Sialic Acid/Polysialic Acid in the Drug Delivery Systems. Asian J. Pharm. Sci. 2014;9:75–81. [Google Scholar]
- 146.Stencel-Baerenwald J.E., Reiss K., Reiter D.M., Stehle T., Dermody T.S. The Sweet Spot: Defining Virus-Sialic Acid Interactions. Nat. Rev. Microbiol. 2014;12:739–749. doi: 10.1038/nrmicro3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bauer J., Osborn H.M.I. Sialic Acids in Biological and Therapeutic Processes—Opportunities and Challenges. Future Med. Chem. 2015;7:2285–2299. doi: 10.4155/fmc.15.135. [DOI] [PubMed] [Google Scholar]
- 148.Hoppe-Seyler F. Physiologische Chemie. I. Theil. Verlag von August Hirschwald; Berlin, Germany: 1877. Allgemeine Biologie—Schleimgewebe; pp. 94–95. [Google Scholar]
- 149.Hoppe-Seyler F. Verlag von August Hirschwald; Berlin, Germany: 1878. Die Verdauung und Resorption der Nährstoffe—Die Sublingualdrüse und ihr Secret. Physiologische Chemie. II. Theil; pp. 197–198. [Google Scholar]
- 150.Green J.R. The Edible Bird’s-Nest, or Nest of the Java Swift (Collocalia nidifica) J. Physiol. 1885;6:40–45. doi: 10.1113/jphysiol.1885.sp000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Krukenberg, C. Fr. W. Weitere Mitteilungen über die Hyalogene. Z. Biol.1886, 4, 261–271.
- 152.Zeller H. Untersuchungen über die essbaren indischen Schwalbennester. Hoppe-Seyler’s Z. Physiol. Chem. 1913;86:85–106. [Google Scholar]
- 153.Martin J.E., Tanenbaum S.W., Flashner M. A Facile Procedure for the Isolation of N-Acetylneuraminic Acid From Edible Bird’s-Nest. Carbohydr. Res. 1977;56:423–425. doi: 10.1016/s0008-6215(00)83368-6. [DOI] [PubMed] [Google Scholar]
- 154.Corfield A.P. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Isolation and Purification of Sialic Acids; pp. 51–57. (Cell Biology Monographs). [Google Scholar]
- 155.Ehrlich P. Über die Dimethylamidobenzaldehydreaction. Med. Woche balneol. Centralz. 1901:151–153. [Google Scholar]
- 156.Pröscher F. Zur Kenntniss der Ehrlich’schen Dimethylamidobenzaldehydreaction. Hoppe-Seyler’s Z. Physiol. Chem. 1900–1901;31:520–526. [Google Scholar]
- 157.Müller F. Beiträge zur Kenntnis des Mucins und einiger damit verwandter Eiweissstoffe. Z. Biol. 1901;42:468–564. [Google Scholar]
- 158.Walz E. Über das Vorkommen von Kerasin in der normalen Rindermilz. Hoppe-Seyler’s Z. Physiol. Chem. 1927;166:210–222. [Google Scholar]
- 159.Levene P.A., Landsteiner K. On Some New Lipoids. J. Biol. Chem. 1927;75:607–612. [Google Scholar]
- 160.Bial M. Die Diagnose der Pentosurie. Deutsche Med. Wochenschr. 1902;28:253–254. [Google Scholar]
- 161.Lundblad A. Gunnar Blix and His Discovery of Sialic Acids—Fascinating Molecules in Glycobiology. Upsala J. Med. Sci. 2015;120:104–112. doi: 10.3109/03009734.2015.1027429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Faillard H. Professor Ernst Klenk—Scientific Achievements. J. Am. Oil Chem. Soc. 1966;43 48A/50A/78A. [Google Scholar]
- 163.Neuberger A. Alfred Gottschalk (1894–1973) Adv. Carbohydr. Chem. Biochem. 1976;33:1–10. doi: 10.1016/s0065-2318(08)60279-6. [DOI] [PubMed] [Google Scholar]
- 164.Carubelli R. Alfred Gottschalk, His Contributions to the Biochemistry of Sialic Acids and Glycoproteins. Trends Glycosci. Glycotechnol. 1992;4:559–566. [Google Scholar]
- 165.Blix G. Über die Kohlenhydratgruppen des Submaxillarismucins. Hoppe-Seyler’s Z. Physiol. Chem. 1936;240:43–54. [Google Scholar]
- 166.Blix G., Svennerholm L., Werner I. Chondrosamine as a Component of Gangliosides and of Submaxillary Mucin. Acta Chem. Scand. 1950;4:717. [Google Scholar]
- 167.Blix G., Svennerholm L., Werner I. The Isolation of Chondrosamine From Gangliosides and From Submaxillary Mucin. Acta Chem. Scand. 1952;6:358–362. [Google Scholar]
- 168.Blix G. Einige Beobachtungen über eine hexosaminhaltige Substanz in der Protagonfraktion des Gehirns. Skand. Arch. Physiol. 1938;80:46–51. [Google Scholar]
- 169.Klenk E. Über die Natur der Phosphatide und anderer Lipoide des Gehirns und der Leber bei der Niemann-Pickschen Krankheit. Hoppe-Seyler’s Z. Physiol. Chem. 1935;235:24–36. [Google Scholar]
- 170.Klenk E. Neuraminsäure, das Spaltprodukt eines neuen Gehirnlipoids. Hoppe-Seyler’s Z. Physiol. Chem. 1941;268:50–58. [Google Scholar]
- 171.Klenk E., Faillard H., Weygand F., Schöne H.H. Untersuchungen über die Konstitution der Neuraminsäure. Hoppe-Seyler’s Z. Physiol. Chem. 1956;304:35–52. [PubMed] [Google Scholar]
- 172.Klenk E. Über die Ganglioside, eine neue Gruppe von zuckerhaltigen Gehirnlipoiden. Hoppe-Seyler’s Z. Physiol. Chem. 1942;273:76–86. [Google Scholar]
- 173.Burnet F.M. Mucins and Mucoids in Relation to Influenza Virus Action—III. Inhibition of Virus Haemagglutination by Glandular Mucins. Aust. J. Exp. Biol. Med. Sci. 1948;26:371–380. doi: 10.1038/icb.1948.38. [DOI] [PubMed] [Google Scholar]
- 174.Gottschalk A., Lind P.E. Product of Interaction Between Influenza Virus Enzyme and Ovomucin. Nature. 1949;164:232–233. doi: 10.1038/164232a0. [DOI] [PubMed] [Google Scholar]
- 175.Burnet F.M., Stone J.D. The Receptor-Destroying Enzyme of Vibrio cholerae. Aust. J. Exp. Biol. Med. Sci. 1947;25:227–233. doi: 10.1038/icb.1947.33. [DOI] [PubMed] [Google Scholar]
- 176.Tamm I., Horsfall F.L., Jr. Characterization and Separation of an Inhibitor of Viral Hemagglutination Present in Urine. Proc. Soc. Exp. Biol. Med. 1950;74:108–114. [PubMed] [Google Scholar]
- 177.Gottschalk A. N-Substituted Isoglucosamine Released From Mucoproteins by the Influenza Virus Enzyme. Nature. 1951;167:845–847. doi: 10.1038/167845a0. [DOI] [PubMed] [Google Scholar]
- 178.Odin L. Carbohydrate Residue of a Urine Mucoprotein Inhibiting Influenza Virus Haemagglutination. Nature. 1952;170:663–664. doi: 10.1038/170663a0. [DOI] [PubMed] [Google Scholar]
- 179.Gottschalk A. Carbohydrate Residue of a Urine Mucoprotein Inhibiting Influenza Virus Haemagglutination. Nature. 1952;170:662–663. doi: 10.1038/170662a0. [DOI] [PubMed] [Google Scholar]
- 180.Klenk E. Zur Kenntnis der Ganglioside. Hoppe-Seyler’s Z. Physiol. Chem. 1951;288:216–220. [PubMed] [Google Scholar]
- 181.Werner I., Odin L. On the Presence of Sialic Acid in Certain Glycoproteins and in Gangliosides. Acta Soc. Med. Upsalien. 1952;57:230–241. [PubMed] [Google Scholar]
- 182.Odin L. Sialic Acid in Pseudomyxomatous Gels. Acta Chem. Scand. 1955;9:714–715. [Google Scholar]
- 183.Odin L. Sialic Acid in Human Serum Protein and in Meconium. Acta Chem. Scand. 1955;9:862–864. [Google Scholar]
- 184.Odin L. Sialic Acid in Human Cervical Mucus, in Hog Seminal Gel, and in Ovomucin. Acta Chem. Scand. 1955;9:1235–1237. [Google Scholar]
- 185.Blix G., Odin L. Isolation of Sialic Acid From Gangliosides. Acta Chem. Scand. 1955;9:1541–1543. [Google Scholar]
- 186.Svennerholm L. Isolation of Sialic Acid From Brain Gangliosides. Acta Chem. Scand. 1955;9:1033. [Google Scholar]
- 187.Klenk E., Lauenstein K. Zur Kenntnis der Kohlenhydratgruppen des Submaxillarismucins und Harnmucoproteids—Die Isolierung von Neuraminsäure als Spaltprodukt. Hoppe-Seyler’s Z. Physiol. Chem. 1952;291:147–152. [PubMed] [Google Scholar]
- 188.Gottschalk A. Presence of 2-Carboxy-pyrrole in Mucoproteins and Its Relation to the Viral Enzyme. Nature. 1953;172:808. doi: 10.1038/172808a0. [DOI] [PubMed] [Google Scholar]
- 189.Klenk E., Faillard H., Lempfrid H. Über die enzymatische Wirkung von Influenzavirus. Hoppe-Seyler’s Z. Physiol. Chem. 1955;301:235–246. [PubMed] [Google Scholar]
- 190.Faillard H. Über die Abspaltung von N-Acetyl-neuraminsäure aus Mucinen durch das “Receptor-Destroying-Enzyme” aus Vibrio cholerae. Hoppe-Seyler’s Z. Physiol. Chem. 1957;307 62–86/308, 187. [PubMed] [Google Scholar]
- 191.Klenk E., Uhlenbruck G. Über das Vorkommen von Neuraminsäure in den Mucinen von Schweine- und Pferde-Submaxillaris, sowie von Kuh-Colostrum. Hoppe-Seyler’s Z. Physiol. Chem. 1956;305:224–231. [PubMed] [Google Scholar]
- 192.Klenk E., Faillard H. Über das Vorkommen von Neuraminsäure im Lebereiweiss bei amyloider Degeneration. Hoppe-Seyler’s Z. Physiol. Chem. 1955;299:191–192. [PubMed] [Google Scholar]
- 193.Klenk E., Stoffel W. Über neuraminsäurehaltige Glykoproteide des Blutserums. Hoppe-Seyler’s Z. Physiol. Chem. 1955;302:286–288. [PubMed] [Google Scholar]
- 194.Klenk E., Stoffel W. Zur Kenntnis des Zellreceptoren für das Influenzavirus—Über das Vorkommen von Neuraminsäure im Eiweiss des Erythrocytenstromas. Hoppe-Seyler’s Z. Physiol. Chem. 1956;303:78–80. [PubMed] [Google Scholar]
- 195.Blix G., Lindberg E., Odin L., Werner I. Sialic Acids. Nature. 1955;175:340–341. doi: 10.1038/175340a0. [DOI] [PubMed] [Google Scholar]
- 196.Klenk E., Uhlenbruck G. Über ein neuraminsäurehaltiges Mucoproteid aus Rindererythrocytenstroma. Hoppe-Seyler’s Z. Physiol. Chem. 1958;311:227–233. [PubMed] [Google Scholar]
- 197.Suzuki A. Tamio Yamakawa: Dawn of Glycobiology. J. Biochem. 2009;146:149–156. doi: 10.1093/jb/mvp103. [DOI] [PubMed] [Google Scholar]
- 198.Baer H.H. Obituary—Richard Kuhn (1900–1967) Adv. Carbohydr. Chem. Biochem. 1969;24:1–12. [PubMed] [Google Scholar]
- 199.Barness L.A., Tomarelli R.M. Paul György (1893–1976): A Bibliographical Sketch. J. Nutr. 1979;109:17–23. [PubMed] [Google Scholar]
- 200.Yamakawa T., Suzuki S. The Chemistry of the Lipids of Posthemolytic Residue or Stroma of Erythrocytes—I. Concerning the Ether-Insoluble Lipids of Lyophilized Horse Blood Stroma. J. Biochem. 1951;38:199–212. [Google Scholar]
- 201.Yamakawa T., Suzuki S. The Chemistry of the Lipids of Posthemolytic Residue or Stroma of Erythrocytes—II. On the Structure of Hemataminic Acid. J. Biochem. 1952;39:175–183. [Google Scholar]
- 202.Klenk E., Wolter H. Über die zuckerhaltigen Lipoide des Erythrocytenstromas vom Pferde. Hoppe-Seyler’s Z. Physiol. Chem. 1952;291:259–265. [PubMed] [Google Scholar]
- 203.Yamakawa T., Suzuki S. Presence of Sero-Lactaminic Acid and Glucosamine as Constituents of Serum Mucoprotein. J. Biochem. 1955;42:727–736. [Google Scholar]
- 204.Kuhn R., Brossmer R., Schulz W. Über die prosthetische Gruppe der Mucoproteine des Kuh-Colostrums. Chem. Ber. 1954;87:123–127. [Google Scholar]
- 205.Kuhn R., Brossmer R. Isolierung einer nieder-molekularen, durch Influenza-Virus spaltbaren Substanz aus Milch. Angew. Chem. 1956;68:211–212. [Google Scholar]
- 206.Kuhn R., Brossmer R. Abbau der Lactaminsäure zu N-Acetyl-d-glucosamin. Chem. Ber. 1956;89:2471–2475. [Google Scholar]
- 207.Kuhn R., Brossmer R. Über O-Acetyl-lactaminsäure-lactose aus Kuh-Colostrum und ihre Spaltbarkeit durch Influenza-Virus. Chem. Ber. 1956;89:2013–2025. [Google Scholar]
- 208.Hoover J.R.E., Braun G.A., György P. Neuraminic Acid in Mucopolysaccharides of Human Milk. Arch. Biochem. Biophys. 1953;47:216–217. doi: 10.1016/0003-9861(53)90451-2. [DOI] [PubMed] [Google Scholar]
- 209.Zilliken F., Braun G.A., György P. Gynaminic Acid—A Naturally Occurring Form of Neuraminic Acid in Human Milk. Arch. Biochem. Biophys. 1955;54:564–566. doi: 10.1016/0003-9861(55)90073-4. [DOI] [PubMed] [Google Scholar]
- 210.Zilliken F., Braun G.A., György P. “Gynaminic Acid” and Other Naturally Occurring Forms of N-Acetylneuraminic Acid. Arch. Biochem. Biophys. 1956;63:394–402. doi: 10.1016/0003-9861(56)90054-6. [DOI] [PubMed] [Google Scholar]
- 211.Böhm P., Baumeister L. Über die Isolierung der Methoxy-neuraminsäure als Spaltprodukt des Serumeiweisses. Hoppe-Seyler’s Z. Physiol. Chem. 1955;300:153–156. [PubMed] [Google Scholar]
- 212.Böhm P., Baumeister L. Über das Vorkommen neuraminsäurehaltiger Glykoproteide in Körperflüssigkeiten. Hoppe-Seyler’s Z. Physiol. Chem. 1956;305:42–50. [PubMed] [Google Scholar]
- 213.Trucco R.E., Caputto R. Neuramin-Lactose, a New Compound Isolated From the Mammary Gland of Rats. J. Biol. Chem. 1954;206:901–909. [PubMed] [Google Scholar]
- 214.Heyworth R., Bacon J.S.D. A Nitrogenous Derivative of Lactose From Lactating Rat Mammary Gland. Biochem. J. 1957;66:41–47. [PMC free article] [PubMed] [Google Scholar]
- 215.Hiyama N. Biochemical Studies on Carbohydrates. CXXI. On the Direct-Ehrlich Reaction-Giving Structure in Glycidamins. Tohoku J. Exp. Med. 1949;51:319–326. [Google Scholar]
- 216.Van Slyke D.D., Dillon R.T., MacFadyen D.A., Hamilton P. Gasometric Determination of Carboxyl Groups in Free Amino Acids. J. Biol. Chem. 1941;141:627–669. [Google Scholar]
- 217.Gottschalk A. The Influenza Virus Enzyme and Its Mucoprotein Substrate. Yale J. Biol. Med. 1954;26:352–364. [PMC free article] [PubMed] [Google Scholar]
- 218.Gottschalk A. The Precursor of 2-Carboxy-pyrrole in Mucoproteins. Nature. 1954;174:652–653. doi: 10.1038/174652b0. [DOI] [PubMed] [Google Scholar]
- 219.Gottschalk A. 2-Carboxypyrrole: Its Preparation From and Its Precursor in Mucoproteins. Biochem. J. 1955;61:298–307. doi: 10.1042/bj0610298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gottschalk A. Structural Relationship Between Sialic Acid, Neuraminic Acid and 2-Carboxy-Pyrrole. Nature. 1955;176:881–882. [Google Scholar]
- 221.Gottschalk A. The Linkage of Sialic Acid in Mucoprotein. Biochim. Biophys. Acta. 1956;20:560–561. doi: 10.1016/0006-3002(56)90356-0. [DOI] [PubMed] [Google Scholar]
- 222.Gottschalk A. The Synthesis of 2-Carboxypyrrole From d-Glucosamine and Pyruvic Acid: Its Bearing on the Structure of Neuraminic Acid. Arch. Biochem. Biophys. 1957;59:37–44. doi: 10.1016/0003-9861(57)90470-8. [DOI] [PubMed] [Google Scholar]
- 223.Blix F.G., Gottschalk A., Klenk E. Proposed Nomenclature in the Field of Neuraminic and Sialic Acids. Nature. 1957;179:1088. doi: 10.1038/1791088b0. [DOI] [PubMed] [Google Scholar]
- 224.Scott J.E., Yamashina I., Jeanloz R.W. A Proposal for a Terminology of ‘Sialic Acid’ Derivatives. Biochem. J. 1982;207:367–368. doi: 10.1042/bj2070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Dixon H.B.F., Cornish-Bowden A. Terminology for Sialic Acids. Biochem. J. 1983;215:711. doi: 10.1042/bj2150711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zilliken F., Glick M.C. Alkalischer Abbau von Gynaminsäure zu Brenztraubensäure und N-Acetyl-d-glucosamin. Naturwissenschaften. 1956;43:536–537. [Google Scholar]
- 227.Heimer R., Meyer K. Studies on Sialic Acid of Submaxillary Mucoid. Proc. Natl. Acad. Sci. U. S. A. 1956;42:728–734. doi: 10.1073/pnas.42.10.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Cornforth J.W., Firth M.E., Gottschalk A. The Synthesis of N-Acetylneuraminic Acid. Biochem. J. 1958;68:57–61. doi: 10.1042/bj0680057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Schnaar R.L., Jourdian G.W. Obituary Saul Roseman (1921–2011) Glycobiology. 2011;21:1393–1394. doi: 10.1093/glycob/cwr132. [DOI] [PubMed] [Google Scholar]
- 230.Basu S.C. Saul Roseman (1921–2011) Adv. Carbohydr. Chem. Biochem. 2013;69:38–54. [Google Scholar]
- 231.Comb D.G., Roseman S. Composition and Enzymatic Synthesis of N-Acetylneuraminic Acid (Sialic Acid) J. Am. Chem. Soc. 1958;80:497–499. [Google Scholar]
- 232.Comb D.G., Roseman S. The Sialic Acids—I. The Structure and Enzymatic Synthesis of N-Acetylneuraminic Acid. J. Biol. Chem. 1960;235:2529–2537. [PubMed] [Google Scholar]
- 233.Kuhn R., Brossmer R. Zur Konfiguration der Lactaminsäure—Epimerisierung von N-Acetyl-d-glucosamin und N-Acetyl-d-mannosamin. Hoppe-Seyler’s Z. Physiol. Chem. 1958;616:221–225. [Google Scholar]
- 234.Roseman S., Comb D.G. The Hexosamine Moiety of N-Acetylneuraminic Acid (Sialic Acid) J. Am. Chem. Soc. 1958;80:3166–3167. [Google Scholar]
- 235.Brug J., Paerels G.B. Configuration of N-Acetylneuraminic Acid. Nature. 1958;182:1159–1160. doi: 10.1038/1821159b0. [DOI] [PubMed] [Google Scholar]
- 236.Carroll P.M., Cornforth J.W. Preparation of N-Acetylneuraminic Acid From N-Acetyl-d-mannosamine. Biochim. Biophys. Acta. 1960;39:161–162. doi: 10.1016/0006-3002(60)90137-2. [DOI] [PubMed] [Google Scholar]
- 237.Kuhn R., Baschang G. Aminozucker-Synthesen—XXV. Synthese der Lactaminsäure. Liebigs Ann. Chem. 1962;659:156–163. [Google Scholar]
- 238.Brug J., Esser R.J.E., Paerels G.B. The Enzymic Cleavage of N-Acetylneuraminic Acid. Biochim. Biophys. Acta. 1959;33:241–242. doi: 10.1016/0006-3002(59)90521-9. [DOI] [PubMed] [Google Scholar]
- 239.Warren L., Felsenfeld H. The Biosynthesis of N-Acetylneuraminic Acid. Biochem. Biophys. Res. Commun. 1961;4:232–235. doi: 10.1016/0006-291x(61)90277-7. [DOI] [PubMed] [Google Scholar]
- 240.Kisa F., Yamada K., Miyamoto T., Inagaki M., Higuchi R. Determination of the Absolute Configuration of Sialic Acids in Gangliosides From the Sea Cucumber Cucumaria echinata. Chem. Pharm. Bull. 2007;55:1051–1052. doi: 10.1248/cpb.55.1051. [DOI] [PubMed] [Google Scholar]
- 241.Hudson C.S. A Relation Between the Chemical Constitution and the Optical Rotary Power of the Sugar Lactones. J. Am. Chem. Soc. 1910;32:338–346. [Google Scholar]
- 242.Kuhn R., Brossmer R. Die Konfiguration der Lactaminsäure. Angew. Chem. 1957;69:534. [Google Scholar]
- 243.Kuhn R., Brossmer R. Abbau der Lactaminsäure zu Bernsteinsäure. Justus Liebigs Ann. Chem. 1959;624:137–141. [Google Scholar]
- 244.Kuhn R., Brossmer R. Die Konfiguration der Sialinsäuren. Angew. Chem. 1962;74:252–253. [Google Scholar]
- 245.Kuhn R., Baschang G. Die Konfiguration der Sialinsäuren am C-atom 4. Chem. Ber. 1962;95:2384–2385. [Google Scholar]
- 246.Fischmeister I. Infrarotspektren von Sialinsäuren und Sialinsäuremethylestern. Ark. Kemi. 1958;13:247–258. [Google Scholar]
- 247.Holmquist L., Östman B. The Anomeric Configuration of N-Acetylneuraminic Acid Released by the Action of Vibrio cholerae Neuraminidase. FEBS Lett. 1975;60:327–330. doi: 10.1016/0014-5793(75)80741-1. [DOI] [PubMed] [Google Scholar]
- 248.Scheinthal B.M., Bettelheim F.A. Multiple Forms of Sialic Acids. Carbohydr. Res. 1968;6:257–265. [Google Scholar]
- 249.Hudson C.S. The Significance of Certain Numerical Relations in the Sugar Group. J. Am. Chem. Soc. 1909;31:66–86. [Google Scholar]
- 250.Kuhn R., Brossmer R. Die Konstitution der Lactaminsäure-lactose–α-Ketosidase-Wirkung von Viren der Influenza-Gruppe. Angew. Chem. 1958;70:25–26. [Google Scholar]
- 251.Kuhn R., Brossmer R. Über das durch Viren der Influenza-Gruppe spaltbare Trisaccharid der Milch. Chem. Ber. 1959;92:1667–1671. [Google Scholar]
- 252.Vliegenthart J.F.G., Kamerling J.P. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Synthesis of Sialic Acids and Sialic Acid Derivatives; pp. 59–76. (Cell Biology Monographs). [Google Scholar]
- 253.Meindl P., Tuppy H. Über synthetische Ketoside der N-Acetyl-d-neuraminsäure—1. Mitt.: Darstellung einer Reihe durch Neuraminidase spaltbarer Ketoside. Monatsh. Chem. 1965;96:802–815. [Google Scholar]
- 254.Meindl P., Tuppy H. Über synthetische Ketoside der N-Acetyl-d-neuraminsäure—2. Mitt.: Anomere n-Amyl- und n-Hexyl-ketoside der N-Acetyl-d-neuraminsäure. Monatsh. Chem. 1965;96:816–827. [Google Scholar]
- 255.Kuhn R., Lutz P., MacDonald D.L. Synthese anomerer Sialinsäure-methylketoside. Chem. Ber. 1966;99:611–617. doi: 10.1002/cber.19660990235. [DOI] [PubMed] [Google Scholar]
- 256.Jochims J.C., Taigel G., Seeliger A., Lutz P., Driesen H.E. Stereospecific Long-Range Couplings of Hydroxyl Protons of Pyranoses. Tetrahedron Lett. 1967:4363–4369. [Google Scholar]
- 257.Lutz P., Lochinger W., Taigel G. Zur Konformation der N-Acetyl-neuraminsaüre. Chem. Ber. 1968;101:1089–1094. doi: 10.1002/cber.19681010345. [DOI] [PubMed] [Google Scholar]
- 258.Yu R.K., Ledeen R. Configuration of the Ketosidic Bond of Sialic Acid. J. Biol. Chem. 1969;244:1306–1313. [PubMed] [Google Scholar]
- 259.Kitajima K., Inoue Y., Inoue S. Polysialoglycoproteins of Salmonidae Fish Eggs—Complete Structure of 200-kDa Polysialoglycoprotein From the Unfertilized Eggs of Rainbow Trout (Salmo gairdneri) J. Biol. Chem. 1986;261:5262–5269. [PubMed] [Google Scholar]
- 260.Mononen I. Quantitative Analysis, by Gas–Liquid Chromatography and Mass Fragmentography, of Monosaccharides After Methanolysis and Deamination. Carbohydr. Res. 1981;88:39–50. [Google Scholar]
- 261.Varki A., Diaz S. The Release and Purification of Sialic Acids From Glycoconjugates: Methods to Minimize the Loss and Migration of O-Acetyl Groups. Anal. Biochem. 1984;137:236–247. doi: 10.1016/0003-2697(84)90377-4. [DOI] [PubMed] [Google Scholar]
- 262.Mawhinney T.P., Chance D.L. Hydrolysis of Sialic Acids and O-Acetylated Sialic Acids With Propionic Acid. Anal. Biochem. 1994;223:164–167. doi: 10.1006/abio.1994.1564. [DOI] [PubMed] [Google Scholar]
- 263.Blix G., Lindberg E. The Sialic Acids of Bovine and Equine Submaxillary Mucins. Acta Chem. Scand. 1960;14:1809–1814. [Google Scholar]
- 264.Faillard H., Gebhard L. Die Wirkung der Neuraminidase auf verschiedene Formen des Rinder-Submaxillarismucins. Hoppe-Seyler’s Z. Physiol. Chem. 1959;317:257–268. doi: 10.1515/bchm2.1959.317.1.257. [DOI] [PubMed] [Google Scholar]
- 265.Aminoff D. Methods for the Quantitative Estimation of N-Acetylneuraminic Acid and Their Application to Hydrolysates of Sialomucoids. Biochem. J. 1961;81:384–392. doi: 10.1042/bj0810384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Warren L. The Thiobarbituric Acid Assay of Sialic Acids. J. Biol. Chem. 1959;234:1971–1975. [PubMed] [Google Scholar]
- 267.Aminoff D. The Determination of Free Sialic Acid in the Presence of the Bound Compound. Virology. 1959;7:355–357. doi: 10.1016/0042-6822(59)90207-7. [DOI] [PubMed] [Google Scholar]
- 268.Paerels G.B., Schut J. The Mechanism of the Periodate–Thiobarbituric Acid Reaction of Sialic Acids. Biochem. J. 1965;96:787–792. doi: 10.1042/bj0960787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Schauer R., Corfield A.P. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Colorimetry and Thin-Layer Chromatography of Sialic Acids; pp. 77–94. (Cell Biology Monographs). [Google Scholar]
- 270.Schoop H.J., Faillard H. Ein Verfahren zum spezifischen Nachweis kleinster Glykolylmengen in Glykoproteinen. Hoppe-Seyler’s Z. Physiol. Chem. 1967;348:1509–1517. [PubMed] [Google Scholar]
- 271.Hestrin S. The Reaction of Acetylcholine and Other Carboxylic Acid Derivatives With Hydroxylamine, and Its Analytical Application. J. Biol. Chem. 1949;180:249–261. [PubMed] [Google Scholar]
- 272.Ishizuka I., Kloppenburg M., Wiegandt H. Characterization of Gangliosides From Fish Brain. Biochim. Biophys. Acta. 1970;210:299–305. doi: 10.1016/0005-2760(70)90174-8. [DOI] [PubMed] [Google Scholar]
- 273.Lönngren J., Svensson S. Mass Spectrometry in Structural Analysis of Natural Carbohydrates. Adv. Carbohydr. Chem. Biochem. 1974;29:41–106. [Google Scholar]
- 274.Kamerling J.P., Vliegenthart J.F.G. In: Clinical Biochemistry—Principles, Methods, Applications—Mass Spectrometry. Lawson A.M., editor. Walter de Gruyter; Berlin, Germany: 1989. Carbohydrates; pp. 175–263. [Google Scholar]
- 275.Kamerling J.P., Gerwig G.J. In: Comprehensive Glycoscience—From Chemistry to Systems Biology. Kamerling J.P., Boons G.-J., Lee Y.C., Suzuki A., Taniguchi N., Voragen A.G.J., editors. Vol. 2. Elsevier Ltd.; Oxford, UK: 2007. Strategies for the Structural Analysis of Carbohydrates; pp. 1–68. [Google Scholar]
- 276.Haverkamp J., Schauer R., Wember M., Kamerling J.P., Vliegenthart J.F.G. Synthesis of 9-O-Acetyl- and 4,9-Di-O-acetyl Derivatives of the Methyl Ester of N-Acetyl-β-d-neuraminic Acid Methylglycoside—Their Use as Models in Periodate Oxidation Studies. Hoppe-Seyler’s Z. Physiol. Chem. 1975;356:1575–1583. doi: 10.1515/bchm2.1975.356.2.1575. [DOI] [PubMed] [Google Scholar]
- 277.McLean R.L., Suttajit M., Beidler J., Winzler R.J. N-Acetylneuraminic Acid Analogues—I. Preparation of the 8-Carbon and 7-Carbon Compounds. J. Biol. Chem. 1971;246:803–809. [PubMed] [Google Scholar]
- 278.Reuter G., Schauer R., Szeiki C., Kamerling J.P., Vliegenthart J.F.G. A Detailed Study of the Periodate Oxidation of Sialic Acids in Glycoproteins. Glycoconjugate J. 1989;6:35–44. doi: 10.1007/BF01047888. [DOI] [PubMed] [Google Scholar]
- 279.Shukla A.K., Schauer R., Schade U., Moll H., Rietschel E.T. Structural Analysis of Underivatized Sialic Acids by Combined High-Performance Liquid Chromatography–Mass Spectrometry. J. Chromatogr. 1985;337:231–238. doi: 10.1016/0378-4347(85)80036-0. [DOI] [PubMed] [Google Scholar]
- 280.Reuter G., Schauer R. Comparison of Electron and Chemical Ionization Mass Spectrometry of Sialic Acids. Anal. Biochem. 1986;157:39–46. doi: 10.1016/0003-2697(86)90193-4. [DOI] [PubMed] [Google Scholar]
- 281.Stehling P., Gohlke M., Fitzner R., Reutter W. Rapid Analysis of O-Acetylated Neuraminic Acids by Matrix Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Glycoconjugate J. 1998;15:339–344. doi: 10.1023/a:1006965600322. [DOI] [PubMed] [Google Scholar]
- 282.Vliegenthart J.F.G., Dorland L., van Halbeek H., Haverkamp J. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. NMR Spectroscopy of Sialic Acids; pp. 127–172. (Cell Biology Monographs). [Google Scholar]
- 283.Friebolin H., Schmidt H., Supp M. 1H-NMR-Untersuchungen an Sialinsäure-Markierung von N-Acetyl-d-neuraminsäure mit Deuterium. Tetrahedron Lett. 1981;22:5171–5174. [Google Scholar]
- 284.Dorland L., Haverkamp J., Schauer R., Veldink G.A., Vliegenthart J.F.G. The Preparation of Specifically Deuterium- or Tritium-Labeled N-Acetylneuraminic Acid and Cytidine-5′-monophospho-β-N-acetylneuraminic Acid as Precursors for Glycoconjugate Synthesis. Biochem. Biophys. Res. Commun. 1982;104:1114–1119. doi: 10.1016/0006-291x(82)91365-1. [DOI] [PubMed] [Google Scholar]
- 285.Biedl A. Die Kristallstruktur der Methoxy-neuraminsäure. Naturwissenschaften. 1971;58:95. [Google Scholar]
- 286.Flippen J.L. The Crystal Structure of β-d-N-Acetylneuraminic Acid Dihydrate (Sialic Acid), C11H19NO9.2H2O. Acta Crystallogr. B. 1973;29:1881–1886. [Google Scholar]
- 287.O’Connell A.M. The Crystal Structure of N-Acetylneuraminic Acid Methyl Ester Monohydrate. Acta Crystallogr. B. 1973;29:2320–2328. [Google Scholar]
- 288.Kooijman H., Kroon-Batenburg L.M.J., Kroon J., Breg J.N., de Boer J.L. Structure of α-d-N-Acetyl-1-O-methylneuraminic Acid Methyl Ester. Acta Crystallogr. C. 1990;46:407–410. [Google Scholar]
- 289.Kimura A., Tsurumi K. Studies on Colominic Acid—IV. Conformation and Configuration of the N-Acetyl-neuraminic Acid Moiety of Colominic Acid. Fukushima J. Med. Sci. 1968;15:55–60. [PubMed] [Google Scholar]
- 290.Chapman D., Kamat V.B., de Gier J., Penkett S.A. Nuclear Magnetic Resonance Studies of Erythrocyte Membranes. J. Mol. Biol. 1968;31:101–114. doi: 10.1016/0022-2836(68)90058-2. [DOI] [PubMed] [Google Scholar]
- 291.Jaques L.W., Brown E.B., Barrett J.M., Brey W.S., Jr., Weltner W., Jr. Sialic Acid—A Calcium-Binding Carbohydrate. J. Biol. Chem. 1977;252:4533–4538. [PubMed] [Google Scholar]
- 292.Behr J.-P., Lehn J.-M. The Binding of Divalent Cations by Purified Gangliosides. FEBS Lett. 1973;31:297–300. doi: 10.1016/0014-5793(73)80126-7. [DOI] [PubMed] [Google Scholar]
- 293.Eschenfelder V., Brossmer R., Friebolin H. 13C-Untersuchungen von anomeren Ketosiden der N-Acetyl-d-neuraminsäure. Tetrahedron Lett. 1975:3069–3072. [Google Scholar]
- 294.Czarniecki M.F., Thornton E.R. Carbon-13 Nuclear Magnetic Resonance Spin-Lattice Relaxation in the N-Acylneuraminic Acids—Probes for Internal Dynamics and Conformational Analysis. J. Am. Chem. Soc. 1977;99:8273–8279. [Google Scholar]
- 295.Sabesan S., Bock K., Lemieux R.U. The Conformational Properties of the Gangliosides GM2 and GM1 Based on 1H and 13C Nuclear Magnetic Resonance Studies. Can. J. Chem. 1984;62:1034–1045. [Google Scholar]
- 296.Czarniecki M.F., Thornton E.R. 13C Spin Lattice Relaxation in the Neuraminic Acids—Evidence for an Unusual Intramolecular Hydrogen-Bonding Network. J. Am. Chem. Soc. 1976;98:1023–1025. doi: 10.1021/ja00420a031. [DOI] [PubMed] [Google Scholar]
- 297.Poppe L., van Halbeek H. Nuclear Magnetic Resonance of Hydroxyl and Amido Protons of Oligosaccharides in Aqueous Solution: Evidence for a Strong Intramolecular Hydrogen Bond in Sialic Acid Residues. J. Am. Chem. Soc. 1991;113:363–365. [Google Scholar]
- 298.Acquotti D., Poppe L., Dabrowski J., von der Lieth C.-W., Sonnino S., Tettamanti G. Three-Dimensional Structure of the Oligosaccharide Chain of GM1 Ganglioside Revealed by a Distance-Mapping Procedure: A Rotating and Laboratory Frame Nuclear Overhauser Enhancement Investigation of Native Glycolipid in Dimethyl Sulfoxide and in Water–Dodecylphosphocholine Solutions. J. Am. Chem. Soc. 1990;112:7772–7778. [Google Scholar]
- 299.Czarniecki M.F., Thornton E.R. 13C NMR Chemical Shift Titration of Metal Ion-Carbohydrate Complexes—An Unexpected Dichotomy for Ca+ 2 Binding Between Anomeric Derivatives of N-Acetylneuraminic Acid. Biochem. Biophys. Res. Commun. 1977;74:553–558. doi: 10.1016/0006-291x(77)90339-4. [DOI] [PubMed] [Google Scholar]
- 300.Daman M.E., Dill K. 13C-N.M.R.-Spectral Study of the Mode of Binding of Mn2 + and Gd3 + to N-Acetyl-α-neuraminic Acid. Carbohydr. Res. 1982;102:47–57. [Google Scholar]
- 301.Veluraja K., Rao V.S.R. Theoretical Studies on the Conformation of β-d-N-Acetyl Neuraminic Acid (Sialic Acid) Biochim. Biophys. Acta. 1980;630:442–446. doi: 10.1016/0304-4165(80)90293-7. [DOI] [PubMed] [Google Scholar]
- 302.Rao V.S.R., Qasba P.K., Balaji P.V., Chandrasekaran R. Harwood Academic Publishers; Amsterdam, The Netherlands: 1998. Conformation of Carbohydrates; pp. 70–71. [Google Scholar]
- 303.Veluraja K., Rao V.S.R. Theoretical Studies on the Conformation of Monosialogangliosides and Disialogangliosides. Carbohydr. Polym. 1983;3:175–192. [Google Scholar]
- 304.Sawada T., Hashimoto T., Nakano H., Shigematsu M., Ishida H., Kiso M. Conformational Study of α-N-Acetyl-d-neuraminic Acid by Density Functional Theory. J. Carbohydr. Chem. 2006;25:387–405. [Google Scholar]
- 305.Mukhopadhyay C., Bush C.A. Molecular Dynamics Simulation of Oligosaccharides Containing N-Acetyl Neuraminic Acid. Biopolymers. 1994;34:11–20. doi: 10.1002/bip.360340103. [DOI] [PubMed] [Google Scholar]
- 306.Priyadarzini T.R.K., Subashini B., Selvin J.F.A., Veluraja K. Molecular Dynamics Simulation and Quantum Mechanical Calculations on α-d-N-Acetylneuraminic Acid. Carbohydr. Res. 2012;351:93–97. doi: 10.1016/j.carres.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 307.Van Lenthe J.H., den Boer D.H.W., Havenith R.W.A., Schauer R., Siebert H.-C. Ab Initio Calculations on Various Sialic Acids Provide Valuable Information About Sialic Acid-Specific Enzymes. J. Mol. Struct. (Theochem) 2004;677:29–37. [Google Scholar]
- 308.Klenk E., Langerbeins H., Schumann E. Über die Verteilung der Neuraminsäure im Gehirn (Mit einer Mikromethode zur quantitativen Bestimmung der Substanz im Nervengewebe) Hoppe Seyler's Z. Physiol. Chem. 1941;270:185–193. [Google Scholar]
- 309.Svennerholm L. Quantitative Estimation of Sialic Acids—I. A Colorimetric Method With Orcinol–Hydrochloric Acid (Bial) Reagent. Ark. Kemi. 1957;10:577–596. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- 310.Svennerholm L. Quantitative Estimation of Sialic Acids—II. A Colorimetric Resorcinol–Hydrochloric Acid Method. Biochim. Biophys. Acta. 1957;24:604–611. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- 311.Svennerholm L. Quantitative Estimation of Sialic Acids—III. An Anion-Exchange Resin Method. Acta Chem. Scand. 1958;12:547–554. [Google Scholar]
- 312.Jourdian G.W., Dean L., Roseman S. The Sialic Acids—XI. A Periodate–Resorcinol Method for the Quantitative Estimation of Free Sialic Acids and Their Glycosides. J. Biol. Chem. 1971;246:430–435. [PubMed] [Google Scholar]
- 313.Peters B.P., Aronson N.N., Jr. Reactivity of the Sialic Acid Derivative 5-Acetamido-3,5-dideoxy-l-arabino-heptulosonic Acid in the Resorcinol and Thiobarbituric Acid Assays. Carbohydr. Res. 1976;47:345–353. doi: 10.1016/s0008-6215(00)84204-4. [DOI] [PubMed] [Google Scholar]
- 314.Kitajima K., Inoue S., Kitazume S., Inoue Y. Analytical Methods for Identifying and Quantitating Deamidated Sialic Acid (2-Keto-3-deoxy-d-glycero-d-galactonononic Acid) and α2 → 8-Linked Poly(oligo)nonulosonate Residues in Glycoconjugates. Anal. Biochem. 1992;205:244–250. doi: 10.1016/0003-2697(92)90430-f. [DOI] [PubMed] [Google Scholar]
- 315.Wirtz-Peitz F. 1969. Chromogen-Bildung der Neuraminsäure und anderer 2-Keto-3-deoxy-aldonsäuren. Dissertation, Ruhr-Universität Bochum. [Google Scholar]
- 316.Treibs A., Herrmann E. Über die Ehrlichsche Reaktion der Pyrrole und Indole—IV. Mitteilung über Pyrrolfarbstoffe. Hoppe-Seyler’s Z. Physiol. Chem. 1955;299:168–185. [PubMed] [Google Scholar]
- 317.Alexander R.S., Butler A.R. Electrophilic Substitution in Pyrroles—Part 1. Reaction With 4-Dimethylaminobenzaldehyde (Ehrlich's Reagent) in Acid Solution. J. Chem. Soc., Perkin Trans. 2. 1976:696–701. [Google Scholar]
- 318.Kuhn R., Lutz P. Über Formyl-brenztraubensäure und den Farbstoff der Warren-Reaktion. Biochem. Z. 1963;338:554–560. [PubMed] [Google Scholar]
- 319.Beau J.-M., Schauer R. Metabolism of 4-O-Methyl-N-acetylneuraminic Acid—A Synthetic Sialic Acid. Eur. J. Biochem. 1980;106:531–540. doi: 10.1111/j.1432-1033.1980.tb04600.x. [DOI] [PubMed] [Google Scholar]
- 320.Charon D., Szabó L. The Synthesis of 3-Deoxy-5-O-methyloctulosonic Acid and Its Behaviour in the Warren Reaction. Eur. J. Biochem. 1972;29:184–187. doi: 10.1111/j.1432-1033.1972.tb01973.x. [DOI] [PubMed] [Google Scholar]
- 321.Hess H.H., Rolde E. Fluorometric Assay of Sialic Acid in Brain Gangliosides. J. Biol. Chem. 1964;239:3215–3220. [PubMed] [Google Scholar]
- 322.Murayama J.-I., Tomita M., Tsuji A., Hamada A. Fluorimetric Assay of Sialic Acids. Anal. Biochem. 1976;73:535–538. doi: 10.1016/0003-2697(76)90204-9. [DOI] [PubMed] [Google Scholar]
- 323.Matsushima Y., Martell A.E. Pyridoxal Analogs—X. Zinc(II)-Chelate Catalysis of Transamination in Methanol Solution. J. Am. Chem. Soc. 1967;89:1331–1335. doi: 10.1021/ja00982a008. [DOI] [PubMed] [Google Scholar]
- 324.Hammond K.S., Papermaster D.S. Fluorometric Assay of Sialic Acid in the Picomole Range: A Modification of the Thiobarbituric Acid Assay. Anal. Biochem. 1976;74:292–297. doi: 10.1016/0003-2697(76)90210-4. [DOI] [PubMed] [Google Scholar]
- 325.Nash T. The Colorimetric Estimation of Formaldehyde by Means of the Hantzsch Reaction. Biochem. J. 1953;55:416–421. doi: 10.1042/bj0550416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Shukla A.K., Schauer R. Determination of Unsubstituted and 9-O-Acetylated Sialic Acids in Erythrocyte Membranes of Different Animals Using a Fluorimetric Method. Hoppe Seyler's Z. Physiol. Chem. 1981;362:236–237. doi: 10.1515/bchm2.1982.363.1.255. [DOI] [PubMed] [Google Scholar]
- 327.Shukla A.K., Schauer R. Study of De-O-acetylation of N-Acetyl-9-O-acetylneuraminic Acid by a Fluorimetric Method. Fresenius’ Z. Anal. Chem. 1982;311:377. [Google Scholar]
- 328.Brunetti P., Jourdian G.W., Roseman S. The Sialic Acids—III. Distribution and Properties of Animal N-Acetylneuraminic Aldolase. J. Biol. Chem. 1962;237:2447–2453. [PubMed] [Google Scholar]
- 329.Brunetti P., Swanson A., Roseman S. Enzymatic Determination of Sialic Acids: N-Acylneuraminic Acid ⇌ N-Acyl-d-mannosamine + Pyruvate. Methods Enzymol. 1963;6:465–473. [Google Scholar]
- 330.Schauer R., Wember M., Wirtz-Peitz F., Ferreira do Amaral C. Studies on the Substrate Specificity of Acylneuraminate Pyruvate-Lyase. Hoppe-Seyler’s Z. Physiol. Chem. 1971;352:1073–1080. doi: 10.1515/bchm2.1971.352.2.1073. [DOI] [PubMed] [Google Scholar]
- 331.Corfield A.P., Ferreira do Amaral C., Wember M., Schauer R. The Metabolism of O-Acyl-N-acylneuraminic Acids—Biosynthesis of O-Acylated Sialic Acids in Bovine and Equine Submandibular Glands. Eur. J. Biochem. 1976;68:597–610. doi: 10.1111/j.1432-1033.1976.tb10848.x. [DOI] [PubMed] [Google Scholar]
- 332.Friedrich A., Rapoport S. Mikromethode für die Acetylbestimmung. Biochem. Z. 1932;251:432–446. [Google Scholar]
- 333.Ludowieg J., Dorfman A. A Micromethod for the Colorimetric Determination of N-Acetyl Groups in Acid Mucopolysaccharides. Biochim. Biophys. Acta. 1960;38:212–218. doi: 10.1016/0006-3002(60)91233-6. [DOI] [PubMed] [Google Scholar]
- 334.Schauer R. Characterization of Sialic Acids. Methods Enzymol. 1978;50:64–89. doi: 10.1016/0076-6879(78)50008-6. [DOI] [PubMed] [Google Scholar]
- 335.Eegriwe E. Reaktionen und Reagenzien zum Nachweis organischer Verbindungen I. Z. Anal. Chem. 1932;89:121–125. [Google Scholar]
- 336.Takahashi K. A Colorimetric Method for Quantitative Determination of Glycolic Acid With 2,7-Dihydroxynaphthalene. J. Biochem. 1972;71:563–565. [PubMed] [Google Scholar]
- 337.Sweeley C.C., Bentley R., Makita M., Wells W.W. Gas–Liquid Chromatography of Trimethylsilyl Derivatives of Sugars and Related Substances. J. Am. Chem. Soc. 1963;85:2497–2507. [Google Scholar]
- 338.Sweeley C.C., Walker B. Determination of Carbohydrates in Glycolipides and Gangliosides by Gas Chromatography. Anal. Chem. 1964;36:1461–1466. [Google Scholar]
- 339.Craven D.A., Gehrke C.W. Quantitative Determination of N-Acetylneuraminic Acid by Gas–Liquid Chromatography. J. Chromatogr. 1968;37:414–421. doi: 10.1016/s0021-9673(01)99137-x. [DOI] [PubMed] [Google Scholar]
- 340.Kamerling J.P., Gerwig G.J., Vliegenthart J.F.G., Clamp J.R. Characterization by Gas–Liquid Chromatography–Mass Spectrometry and Proton-Magnetic-Resonance Spectroscopy of Pertrimethylsilyl Methyl Glycosides Obtained in the Methanolysis of Glycoproteins and Glycopeptides. Biochem. J. 1975;151:491–495. doi: 10.1042/bj1510491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Casals-Stenzel J., Buscher H.-P., Schauer R. Gas–Liquid Chromatography of N- and O-Acylated Neuraminic Acids. Anal. Biochem. 1975;65:507–524. doi: 10.1016/0003-2697(75)90536-9. [DOI] [PubMed] [Google Scholar]
- 342.Sugawara Y., Iwamori M., Portoukalian J., Nagai Y. Determination of N-Acetylneuraminic Acid by Gas Chromatography–Mass Spectrometry With a Stable Isotope as Internal Standard. Anal. Biochem. 1983;132:147–151. doi: 10.1016/0003-2697(83)90438-4. [DOI] [PubMed] [Google Scholar]
- 343.Zanetta J.P., Breckenridge W.C., Vincendon G. Analysis of Monosaccharides by Gas–Liquid Chromatography of the O-Methyl Glycosides as Trifluoroacetate Derivatives—Application to Glycoproteins and Glycolipids. J. Chromatogr. 1972;69:291–304. doi: 10.1016/s0021-9673(00)92897-8. [DOI] [PubMed] [Google Scholar]
- 344.Bhattacharjee A.K., Jennings H.J. Determination of the Linkages in Some Methylated, Sialic Acid-Containing, Meningococcal Polysaccharides by Mass Spectrometry. Carbohydr. Res. 1976;51:253–261. doi: 10.1016/s0008-6215(00)83333-9. [DOI] [PubMed] [Google Scholar]
- 345.Rauvala H., Kärkkäinen J. Methylation Analysis of Neuraminic Acids by Gas Chromatography–Mass Spectrometry. Carbohydr. Res. 1977;56:1–9. doi: 10.1016/s0008-6215(00)84231-7. [DOI] [PubMed] [Google Scholar]
- 346.Van Halbeek H., Haverkamp J., Kamerling J.P., Vliegenthart J.F.G., Versluis C., Schauer R. Sialic Acids in Permethylation Analysis: Preparation and Identification of Partially O-Methylated Derivatives of Methyl N-Acetyl-N-methyl-β-d-neuraminate Methyl Glycoside. Carbohydr. Res. 1978;60:51–62. [Google Scholar]
- 347.Inoue S., Matsumura G. Identification of the N-Glycolylneuraminyl-(2 → 8)-N-glycolylneuraminyl Group in a Trout-Egg Glycoprotein by Methylation Analysis and Gas–Liquid Chromatography–Mass Spectrometry. Carbohydr. Res. 1979;74:361–368. [Google Scholar]
- 348.Bruvier C., Leroy Y., Montreuil J., Fournet B., Kamerling J.P. Gas–Liquid Chromatography and Mass Spectrometry of Methyl Ethers of Methyl N-Acetyl-N-methyl-β-d-neuraminate Methyl Glycoside. J. Chromatogr. 1981;210:487–504. [Google Scholar]
- 349.Krantz M.J., Lee Y.C. A Sensitive Autoanalytical Method for Sialic Acids. Anal. Biochem. 1975;63:464–469. doi: 10.1016/0003-2697(75)90370-x. [DOI] [PubMed] [Google Scholar]
- 350.Silver H.K.B., Karim K.A., Gray M.J., Salinas F.A. High-Performance Liquid Chromatography Quantitation of N-Acetylneuraminic Acid in Malignant Melanoma and Breast Carcinoma. J. Chromatogr. B: Biomed. Sci. Appl. 1981;224:381–388. [Google Scholar]
- 351.Bergh M.L.E., Koppen P., van den Eijnden D.H. High-Pressure Liquid Chromatography of Sialic Acid-Containing Oligosaccharides. Carbohydr. Res. 1981;94:225–229. doi: 10.1016/s0008-6215(00)80720-x. [DOI] [PubMed] [Google Scholar]
- 352.Powell L.D., Hart G.W. Quantitation of Picomole Levels of N-Acetyl- and N-Glycolylneuraminic Acids by a HPLC-Adaptation of the Thiobarbituric Acid Assay. Anal. Biochem. 1986;157:179–185. doi: 10.1016/0003-2697(86)90211-3. [DOI] [PubMed] [Google Scholar]
- 353.Mopper K., Gindler E.M. A New Noncorrosive Dye Reagent for Automatic Sugar Chromatography. Anal. Biochem. 1973;56:440–442. doi: 10.1016/0003-2697(73)90210-8. [DOI] [PubMed] [Google Scholar]
- 354.Sinner M., Puls J. Non-Corrosive Dye Reagent for Detection of Reducing Sugars in Borate Complex Ion-Exchange Chromatography. J. Chromatogr. 1978;156:197–204. [Google Scholar]
- 355.Shukla A.K., Scholz N., Reimerdes E.H., Schauer R. High-Performance Liquid Chromatography of N,O-Acylated Sialic Acids. Anal. Biochem. 1982;123:78–82. doi: 10.1016/0003-2697(82)90625-x. [DOI] [PubMed] [Google Scholar]
- 356.Shukla A.K., Schauer R. Analysis of N,O-Acylated Neuraminic Acids by High-Performance Liquid Anion-Exchange Chromatography. J. Chromatogr. 1982;244:81–89. [Google Scholar]
- 357.Hara S., Takemori Y., Yamaguchi M., Nakamura M., Ohkura Y. Fluorometric High-Performance Liquid Chromatography of N-Acetyl- and N-Glycolylneuraminic Acids and Its Application to Their Microdetermination in Human and Animal Sera, Glycoproteins, and Glycolipids. Anal. Biochem. 1987;164:138–145. doi: 10.1016/0003-2697(87)90377-0. [DOI] [PubMed] [Google Scholar]
- 358.Hara S., Yamaguchi M., Takemori Y., Furuhata K., Ogura H., Nakamura M. Determination of Mono-O-acetylated N-Acetylneuraminic Acids in Human and Rat Sera by Fluorometric High-Performance Liquid Chromatography. Anal. Biochem. 1989;179:162–166. doi: 10.1016/0003-2697(89)90218-2. [DOI] [PubMed] [Google Scholar]
- 359.Lin S.-L., Inoue S., Inoue Y. Acid-Base Properties of the Reaction Product of Sialic Acid With Fluorogenic Reagent, 1,2-Diamino-4,5-methylenedioxybenzene (DMB) Carbohydr. Res. 2000;329:447–451. doi: 10.1016/s0008-6215(00)00177-4. [DOI] [PubMed] [Google Scholar]
- 360.Anumula K.R. Rapid Quantitative Determination of Sialic Acids in Glycoproteins by High-Performance Liquid Chromatography With a Sensitive Fluorescence Detection. Anal. Biochem. 1995;230:24–30. doi: 10.1006/abio.1995.1432. [DOI] [PubMed] [Google Scholar]
- 361.Rohrer J.S. Analyzing Sialic Acids Using High-Performance Anion-Exchange Chromatography With Pulsed Amperometric Detection. Anal. Biochem. 2000;283:3–9. doi: 10.1006/abio.2000.4643. [DOI] [PubMed] [Google Scholar]
- 362.Manzi A.E., Diaz S., Varki A. High-Pressure Liquid Chromatography of Sialic Acids on a Pellicular Resin Anion-Exchange Column With Pulsed Amperometric Detection: A Comparison With Six Other Systems. Anal. Biochem. 1990;188:20–32. doi: 10.1016/0003-2697(90)90523-c. [DOI] [PubMed] [Google Scholar]
- 363.Allevi P., Femia E.A., Costa M.L., Cazzola R., Anastasia M. Quantification of N-Acetyl- and N-Glycolylneuraminic Acids by a Stable Isotope Dilution Assay Using High-Performance Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. A. 2008;1212:98–105. doi: 10.1016/j.chroma.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 364.Che F.-Y., Shao X.-X., Wang K.-Y., Xia Q.-C. Characterization of Derivatization of Sialic Acid With 2-Aminoacridone and Determination of Sialic Acid Content in Glycoproteins by Capillary Electrophoresis and High Performance Liquid Chromatography–Ion Trap Mass Spectrometry. Electrophoresis. 1999;20:2930–2937. doi: 10.1002/(SICI)1522-2683(19991001)20:14<2930::AID-ELPS2930>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 365.Dong X., Xu X., Han F., Ping X., Yuang X., Lin B. Determination of Sialic Acids in the Serum of Cancer Patients by Capillary Electrophoresis. Electrophoresis. 2001;22:2231–2235. doi: 10.1002/1522-2683(20017)22:11<2231::AID-ELPS2231>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 366.Taga A., Sugimura M., Suzuki S., Honda S. Estimation of Sialic Acid in a Sialoglycan and a Sialoglycoprotein by Capillary Electrophoresis With In-Capillary Sialidase Digestion. J. Chromatogr. A. 2002;954:259–266. doi: 10.1016/s0021-9673(02)00151-6. [DOI] [PubMed] [Google Scholar]
- 367.Strousopoulou K., Militsopoulou M., Stagiannis K., Lamari F.N., Karamanos N.K. A Capillary Zone Electrophoresis Method for Determining N-Acetylneuraminic Acid in Glycoproteins and Blood Sera. Biomed. Chromatogr. 2002;16:146–150. doi: 10.1002/bmc.143. [DOI] [PubMed] [Google Scholar]
- 368.Lamari F.N., Karamanos N.K. Separation Methods for Sialic Acids and Critical Evaluation of Their Biologic Relevance. J. Chromatogr. B. 2002;781:3–19. doi: 10.1016/s1570-0232(02)00432-4. [DOI] [PubMed] [Google Scholar]
- 369.Ortner K., Buchberger W. Determination of Sialic Acids Released From Glycoproteins Using Capillary Zone Electrophoresis/Electrospray Ionization Mass Spectrometry. Electrophoresis. 2008;29:2233–2237. doi: 10.1002/elps.200700801. [DOI] [PubMed] [Google Scholar]
- 370.Friebolin H., Brossmer R., Keilich G., Ziegler D., Supp M. 1H-NMR-Spektroskopischer Nachweis der N-Acetyl-α-d-neuraminsäure als primäres Spaltprodukt der Neuraminidasen. Hoppe-Seyler’s Z. Physiol. Chem. 1980;361:697–702. [PubMed] [Google Scholar]
- 371.Friebolin H., Baumann W., Brossmer R., Keilich G., Supp M., Ziegler D., von Nicolai H. 1H-NMR Spectroscopic Evidence for the Release of N-Acetyl-α-d-neuraminic Acid as the First Product of Neuraminidase Action. Biochem. Int. 1981;3:321–326. [Google Scholar]
- 372.Friebolin H., Baumann W., Keilich G., Ziegler D., Brossmer R., von Nicolai H. 1H-NMR-Spektroskopie—Eine aussagekräftige Methode zur Bestimmung der Substratspezifität von Sialidasen. Hoppe-Seyler’s Z. Physiol. Chem. 1981;362:1455–1463. [PubMed] [Google Scholar]
- 373.Friebolin H., Kunzelmann P., Supp M., Brossmer R., Keilich G., Ziegler D. 1H-NMR-Spektroskopische Untersuchungen zur Mutarotation der N-Acetyl-d-neuraminsäure—pH-Abhängigkeit der Mutarotationsgeschwindigkeit. Tetrahedron Lett. 1981;22:1383–1386. [Google Scholar]
- 374.Deijl C.M., Kamerling J.P., Vliegenthart J.F.G. Release of Sialic Acid From Substrates by Sialidase in the Presence of H2[18O] Carbohydr. Res. 1984;126:338–342. doi: 10.1016/0008-6215(84)85394-x. [DOI] [PubMed] [Google Scholar]
- 375.Chong A.K.J., Pegg M.S., Taylor N.R., von Itzstein M. Evidence for a Sialosyl Cation Transition-State Complex in the Reaction of Sialidase From Influenza Virus. Eur. J. Biochem. 1992;207:335–343. doi: 10.1111/j.1432-1033.1992.tb17055.x. [DOI] [PubMed] [Google Scholar]
- 376.Guo X., Sinnott M.L. Salmonella typhimurium Neuraminidase Acts With Inversion of Configuration. Biochem. J. 1993;296:291–292. doi: 10.1042/bj2960291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Guo X., Laver W.G., Vimr E., Sinnott M.L. Catalysis by Two Sialidases With the Same Protein Fold but Different Stereochemical Courses: A Mechanistic Comparison of the Enzymes From Influenza A Virus and Salmonella typhimurium. J. Am. Chem. Soc. 1994;116:5572–5578. [Google Scholar]
- 378.Deijl C.M., Vliegenthart J.F.G. Configuration of Substrate and Products of N-Acetylneuraminate Pyruvate-Lyase From Clostridium perfringens. Biochem. Biophys. Res. Commun. 1983;111:668–674. doi: 10.1016/0006-291x(83)90358-3. [DOI] [PubMed] [Google Scholar]
- 379.Baumann W., Freidenreich J., Weisshaar G., Brossmer R., Friebolin H. Spaltung und Synthese von Sialinsäuren mit Aldolase—1H-NMR-Untersuchungen zur Stereochemie, zur Kinetik und zum Mechanismus. Biol. Chem. Hoppe-Seyler. 1989;370:141–149. [PubMed] [Google Scholar]
- 380.Klepach T., Carmichael I., Serianni A.S. 13C-Labeled N-Acetyl-neuraminic Acid in Aqueous Solution: Detection and Quantification of Acyclic Keto, Keto Hydrate, and Enol Forms by 13C NMR Spectroscopy. J. Am. Chem. Soc. 2008;130:11892–11900. doi: 10.1021/ja077565g. [DOI] [PubMed] [Google Scholar]
- 381.McManus J.F.A. Histological Demonstration of Mucin After Periodic Acid. Nature. 1946;158:202. doi: 10.1038/158202a0. [DOI] [PubMed] [Google Scholar]
- 382.Leblond C.P. Distribution of Periodic Acid-Reactive Carbohydrates in the Adult Rat. Am. J. Anat. 1950;86:1–49. doi: 10.1002/aja.1000860102. [DOI] [PubMed] [Google Scholar]
- 383.Leblond, C. P.; Clermont, Y. Spermiogenesis of Rat, Mouse, Hamster and Guinea Pig as Revealed by the “Periodic Acid-Fuchsin Sulfurous Acid” Technique. Am. J. Anat.1952, 90, 167–215. [DOI] [PubMed]
- 384.Hotchkiss R.D. A Microchemical Reaction Resulting in the Staining of Polysaccharide Structures in Fixed Tissue Preparations. Arch. Biochem. 1948;16:131–141. [PubMed] [Google Scholar]
- 385.Leblond C.P., Glegg R.E., Eidinger D. Presence of Carbohydrates With Free 1,2-Glycol Groups in Sites Stained by the Periodic Acid-Schiff Technique. J. Histochem. Cytochem. 1957;5:445–458. doi: 10.1177/5.5.445. [DOI] [PubMed] [Google Scholar]
- 386.Gasic G., Gasic T. Removal of PAS Positive Surface Sugars in Tumor Cells by Glycosidases. Exp. Biol. Med. 1963;114:660–663. doi: 10.3181/00379727-114-28762. [DOI] [PubMed] [Google Scholar]
- 387.Rambourg A., Neutra M., Leblond C.P. Presence of a ‘Cell Coat’ Rich in Carbohydrate at the Surface of Cells in the Rat. Anat. Rec. 1966;154:41–71. doi: 10.1002/ar.1091540105. [DOI] [PubMed] [Google Scholar]
- 388.Winzler R.J. In: Glycoproteins—Their Composition, Structure and Function. 2nd ed. Gottschalk A., editor. Vol. 5, Part B. B.B.A. Library, Elsevier Publishing Company; Amsterdam, The Netherlands: 1972. Glycoproteins of Plasma Membranes—Chemistry and Function; pp. 1268–1293. [Google Scholar]
- 389.Rambourg A., Hernandez W., Leblond C.P. Detection of Complex Carbohydrates in the Golgi Apparatus of Rat Cells. J. Cell Biol. 1969;40:395–414. doi: 10.1083/jcb.40.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Culling C.F.A., Reid P.E., Dunn W.L. The Effect of Saponification Upon Certain Histochemical Reactions of the Epithelial Mucins of the Gastrointestinal Tract. J. Histochem. Cytochem. 1971;19:654–662. doi: 10.1177/19.11.654. [DOI] [PubMed] [Google Scholar]
- 391.Culling C.F.A., Reid P.E., Clay M.G., Dunn W.L. The Histochemical Demonstration of O-Acylated Sialic Acid in Gastrointestinal Mucins—Their Association With the Potassium Hydroxide–Periodic Acid–Schiff Effect. J. Histochem. Cytochem. 1974;22:826–831. doi: 10.1177/22.8.826. [DOI] [PubMed] [Google Scholar]
- 392.Culling C.F.A., Reid P.E., Dunn W.L. A New Histochemical Method for the Identification and Visualization of Both Side Chain Acylated and Nonacylated Sialic Acids. J. Histochem. Cytochem. 1976;24:1225–1230. doi: 10.1177/24.12.187689. [DOI] [PubMed] [Google Scholar]
- 393.Culling, C. F. A.; Reid, P. E. Histochemistry of Sialic Acids. In: Sialic Acids—Chemistry, Metabolism and Function, Schauer, R., Ed.; Cell Biology Monographs, Vol. 10; Springer-Verlag: Vienna, Austria. 1982; pp 173–193.
- 394.Makovitzky J., Richter S. The Relevance of the Aldehyde Bisulfite Toluidine Blue Reaction and Its Variants in the Submicroscopic Carbohydrate Research. Acta Histochem. 2009;111:273–291. doi: 10.1016/j.acthis.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Ueda T., Fujimori O., Yamada K. A New Histochemical Method for Detection of Sialic Acids Using a Physical Development Procedure. J. Histochem. Cytochem. 1995;43:1045–1051. doi: 10.1177/43.10.7560882. [DOI] [PubMed] [Google Scholar]
- 396.Fukui K., Yasui T., Gomi H., Sugiya H., Fujimori O., Meyer W., Tsukise A. Histochemical Localization of Sialic Acids and Antimicrobial Substances in Eccrine Glands of Porcine Snout Skin. Eur. J. Histochem. 2012;56 doi: 10.4081/ejh.2012.e6. (28–34) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Zhang Y., Yuan J., Song J., Wang Z., Huang L. An Efficient Method for Selectively Imaging and Quantifying In Situ the Expression of Sialylated Glycoproteins on Living Cells. Glycobiology. 2013;23:643–653. doi: 10.1093/glycob/cws148. [DOI] [PubMed] [Google Scholar]
- 398.Goldstein I.J., Winter H.C., Poretz R.D. In: Glycoproteins II. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 29b. Elsevier Science B.V; Amsterdam, The Netherlands: 1997. Plant Lectins: Tools for the Study of Complex Carbohydrates; pp. 403–474. (New Comprehensive Biochemistry). [Google Scholar]
- 399.Sedwick C. Jürgen Roth: Immunogold Master. J. Cell Biol. 2013;200:686–687. doi: 10.1083/jcb.2006pi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Taatjes D.J., Roth J., Peumans W., Goldstein I.J. Elderberry Bark Lectin–Gold Techniques for the Detection of Neu5Ac(α2,6)Gal/GalNAc Sequences: Applications and Limitations. Histochem. J. 1988;20:478–490. doi: 10.1007/BF01002646. [DOI] [PubMed] [Google Scholar]
- 401.Sata T., Lackie P.M., Taatjes D.J., Peumans W., Roth J. Detection of the Neu5Ac(α2,3)Gal(β1,4)GlcNAc Sequence With the Leukoagglutinin From Maackia amurensis: Light and Electron Microscopic Demonstration of Differential Tissue Expression of Terminal Sialic Acid in α2,3 and α2,6 Linkage. J. Histochem. Cytochem. 1989;37:1577–1588. doi: 10.1177/37.11.2478613. [DOI] [PubMed] [Google Scholar]
- 402.Sata T., Roth J., Zuber C., Stamm B., Heitz P.U. Expression of α2,6-Linked Sialic Acid Residues in Neoplastic but Not in Normal Human Colonic Mucosa—A Lectin–Gold Cytochemical Study With Sambucus nigra and Maackia amurensis Lectins. Am. J. Pathol. 1991;139:1435–1448. [PMC free article] [PubMed] [Google Scholar]
- 403.Skutelsky E., Roth J. Cationic Colloidal Gold—A New Probe for the Detection of Anionic Cell Surface Sites by Electron Microscopy. J. Histochem. Cytochem. 1986;34:693–696. doi: 10.1177/34.5.3701033. [DOI] [PubMed] [Google Scholar]
- 404.Harms G., Reuter G., Corfield A.P., Schauer R. Binding Specificity of Influenza C-Virus to Variably O-Acetylated Glycoconjugates and Its Use for Histochemical Detection of N-Acetyl-9-O-acetylneuraminic Acid in Mammalian Tissues. Glycoconjugate J. 1996;13:621–630. doi: 10.1007/BF00731450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Klein A., Krishna M., Varki N.M., Varki A. 9-O-Acetylated Sialic Acids Have Widespread but Selective Expression: Analysis Using a Chimeric Dual-Function Probe Derived From Influenza C Hemagglutinin-Esterase. Proc. Natl. Acad. Sci. U. S. A. 1994;91:7782–7786. doi: 10.1073/pnas.91.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Langereis M.A., Bakkers M.J.G., Deng L., Padler-Karavani V., Vervoort S.J., Hulswit R.J.G., van Vliet A.L.W., Gerwig G.J., de Poot S.A.H., Boot W., van Ederen A.M., Heesters B.A., van der Loos C.M., van Kuppeveld F.J.M., Yu H., Huizinga E.G., Chen X., Varki A., Kamerling J.P., de Groot R.J. Complexity and Diversity of the Mammalian Sialome Revealed by Nidovirus Virolectins. Cell Rep. 2015;11:1966–1978. doi: 10.1016/j.celrep.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Wasik B.R., Barnard K.N., Ossiboff R.J., Khedri Z., Feng K.H., Yu H., Chen X., Perez D.R., Varki A., Parrish C.R. Distribution of O-Acetylated Sialic Acids Among Target Host Tissues for Influenza Virus. mSphere. 2017;2 doi: 10.1128/mSphere.00379-16. e00379-16 (1–17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Roth J., Zuber C., Wagner P., Taatjes D.J., Weisgerber C., Heitz P.U., Goridis C., Bitter-Suermann D. Reexpression of Poly(sialic Acid) Units of the Neural Cell Adhesion Molecule in Wilms Tumor. Proc. Natl. Acad. Sci. U. S. A. 1988;85:2999–3003. doi: 10.1073/pnas.85.9.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Roth J., Zuber C. Immunoelectron Microscopic Investigation of Surface Coat of Wilms Tumor Cells—Dense Lamina Is Composed of Highly Sialylated Neural Cell Adhesion Molecule. Lab. Invest. 1990;62:55–60. [PubMed] [Google Scholar]
- 410.Zuber C., Roth J. The Relationship of Polysialic Acid and the Neural Cell Adhesion Molecule N-CAM in Wilms Tumor and Their Subcellular Distributions. Eur. J. Cell Biol. 1990;51:313–321. [PubMed] [Google Scholar]
- 411.Cazet A., Groux-Degroote S., Teylaert B., Kwon K.-M., Lehoux S., Slomianny C., Kim C.-H., Le Bourhis X., Delannoy P. GD3 Synthase Overexpression Enhances Proliferation and Migration of MDA-MB-231 Breast Cancer Cells. Biol. Chem. 2009;390:601–609. doi: 10.1515/BC.2009.054. [DOI] [PubMed] [Google Scholar]
- 412.Gross H.J., Brossmer R. Enzymatic Introduction of a Fluorescent Sialic Acid Into Oligosaccharide Chains of Glycoproteins. Eur. J. Biochem. 1988;177:583–589. doi: 10.1111/j.1432-1033.1988.tb14410.x. [DOI] [PubMed] [Google Scholar]
- 413.Goon S., Bertozzi C.R. Metabolic Substrate Engineering as a Tool for Glycobiology. J. Carbohydr. Chem. 2002;21:943–977. [Google Scholar]
- 414.Gross H.J., Brossmer R. Characterization of Human Plasma Sialyltransferase Using a Novel Fluorometric Assay. Clin. Chim. Acta. 1991;197:237–247. doi: 10.1016/0009-8981(91)90144-2. [DOI] [PubMed] [Google Scholar]
- 415.Chammas R., McCaffery J.M., Klein A., Ito Y., Saucan L., Palade G., Farquhar M.G., Varki A. Uptake and Incorporation of an Epitope-Tagged Sialic Acid Donor Into Intact Rat Liver Golgi Compartments. Functional Localization of Sialyltransferase Overlaps With β-Galactosyltransferase but Not With Sialic Acid O-Acetyltransferase. Mol. Biol. Cell. 1996;7:1691–1707. doi: 10.1091/mbc.7.11.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Saxon E., Bertozzi C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 417.Dehnert K.W., Baskin J.M., Laughlin S.T., Beahm B.J., Naidu N.N., Amacher S.L., Bertozzi C.R. Imaging the Sialome During Zebrafish Development With Copper-Free Click Chemistry. ChemBioChem. 2012;13:353–357. doi: 10.1002/cbic.201100649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Lantos A.B., Carlevaro G., Araoz B., Ruiz Diaz P., de los Milagros Camara M., Buscaglia C.A., Bossi M., Yu H., Chen X., Bertozzi C.R., Mucci J., Campetella O. Sialic Acid Glycobiology Unveils Trypanosoma cruzi Trypomastigote Membrane Physiology. PLoS Pathog. 2016;12(1–29) doi: 10.1371/journal.ppat.1005559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Popenoe E.A. The Linkage of Neuraminic Acid in Orosomucoid. Biochim. Biophys. Acta. 1959;32:584–585. doi: 10.1016/0006-3002(59)90651-1. [DOI] [PubMed] [Google Scholar]
- 420.Endo Y., Yamashita K., Tachibana Y., Tojo S., Kobata A. Structures of the Asparagine-Linked Sugar Chains of Human Chorionic Gonadotropin. J. Biochem. 1979;85:669–679. [PubMed] [Google Scholar]
- 421.Fournet B., Fiat A.-M., Alais C., Jollès P. Cow κ-Casein: Structure of the Carbohydrate Portion. Biochim. Biophys. Acta. 1979;576:339–346. doi: 10.1016/0005-2795(79)90409-4. [DOI] [PubMed] [Google Scholar]
- 422.Savage A.V., Koppen P.L., Schiphorst W.E.C.M., Trippelvitz L.A.W., van Halbeek H., Vliegenthart J.F.G., van den Eijnden D.H. Porcine Submaxillary Mucin Contains α2 → 3- and α2 → 6-Linked N-Acetyl- and N-Glycolyl-neuraminic Acid. Eur. J. Biochem. 1986;160:123–129. doi: 10.1111/j.1432-1033.1986.tb09948.x. [DOI] [PubMed] [Google Scholar]
- 423.Damm J.B.L., Bergwerff A.A., Hård K., Kamerling J.P., Vliegenthart J.F.G. Sialic Acid Patterns in N-Linked Carbohydrate Chains—Structural Analysis of the N-Acetyl-/N-Glycolyl-neuraminic-Acid-Containing N-Linked Carbohydrate Chains of Bovine Fibrinogen. Recl. Trav. Chim. Pays-Bas. 1989;108:351–359. [Google Scholar]
- 424.Jamieson G.A., Jett M., DeBernardo S.L. The Carbohydrate Sequence of the Glycopeptide Chains of Human Transferrin. J. Biol. Chem. 1971;246:3686–3693. [PubMed] [Google Scholar]
- 425.Breg J., van Halbeek H., Vliegenthart J.F.G., Lamblin G., Houvenaghel M.-C., Roussel P. Structure of Sialyl-Oligosaccharides Isolated From Bronchial Mucus Glycoproteins of Patients (Blood Group O) Suffering From Cystic Fibrosis. Eur. J. Biochem. 1987;168:57–68. doi: 10.1111/j.1432-1033.1987.tb13387.x. [DOI] [PubMed] [Google Scholar]
- 426.Inoue S., Iwasaki M., Matsumura G. Novel Carbohydrate Structures in Trout Egg Glycoprotein: Occurrence of a Neuraminidase-Resistant N-Glycolylneuraminosyl-(2 → 3)-N-acetylgalactosamine Linkage. Biochem. Biophys. Res. Commun. 1981;102:1295–1301. doi: 10.1016/s0006-291x(81)80152-0. [DOI] [PubMed] [Google Scholar]
- 427.Iwasaki M., Inoue S. Structures of the Carbohydrate Units of Polysialoglycoproteins Isolated From the Eggs of Four Species of Salmonid Fishes. Glycoconjugate J. 1985;2:209–228. [Google Scholar]
- 428.Pfeiffer G., Dabrowski U., Dabrowski J., Stirm S., Strube K.-H., Geyer R. Carbohydrate Structure of a Thrombin-Like Serine Protease From Agkistrodon rhodostoma—Structure Elucidation of Oligosaccharides by Methylation Analysis, Liquid Secondary-Ion Mass Spectrometry and Proton Magnetic Resonance. Eur. J. Biochem. 1992;205:961–978. doi: 10.1111/j.1432-1033.1992.tb16863.x. [DOI] [PubMed] [Google Scholar]
- 429.Gottschalk A., Graham E.R.B. 6-α-d-Sialyl-N-acetylgalactosamine: The Neuraminidase-Susceptible Prosthetic Group of Bovine Salivary Mucoprotein. Biochim. Biophys. Acta. 1959;34:380–391. doi: 10.1016/0006-3002(59)90290-2. [DOI] [PubMed] [Google Scholar]
- 430.Carlson D.M., Blackwell C. Structures and Immunochemical Properties of Oligosaccharides Isolated From Pig Submaxillary Mucins. J. Biol. Chem. 1968;243:616–626. [PubMed] [Google Scholar]
- 431.Van Halbeek H., Dorland L., Haverkamp J., Veldink G.A., Vliegenthart J.F.G., Fournet B., Ricart G., Montreuil J., Gathmann W.D., Aminoff D. Structure Determination of Oligosaccharides Isolated From A+, H+ and A− H− Hog-Submaxillary-Gland Mucin Glycoproteins, by 360-MHz 1H-NMR Spectroscopy, Permethylation Analysis and Mass Spectrometry. Eur. J. Biochem. 1981;118:487–495. doi: 10.1111/j.1432-1033.1981.tb05545.x. [DOI] [PubMed] [Google Scholar]
- 432.Spik G., Coddeville B., Montreuil J. Comparative Study of the Primary Structures of Sero-, Lacto- and Ovotransferrin Glycans From Different Species. Biochimie. 1988;70:1459–1469. doi: 10.1016/0300-9084(88)90283-0. [DOI] [PubMed] [Google Scholar]
- 433.Mizuochi T., Yamashita K., Fujikawa K., Kisiel W., Kobata A. The Carbohydrate of Bovine Prothrombin—Occurrence of Galβ1 → 3GlcNAc Grouping in Asparagine-Linked Sugar Chains. J. Biol. Chem. 1979;254:6419–6425. [PubMed] [Google Scholar]
- 434.Finne J., Krusius T., Rauvala H. Occurrence of Disialosyl Groups in Glycoproteins. Biochem. Biophys. Res. Commun. 1977;74:405–410. doi: 10.1016/0006-291x(77)90318-7. [DOI] [PubMed] [Google Scholar]
- 435.Iwasaki M., Inoue S., KItajima K., Nomoto H., Inoue Y. Novel Oligosaccharide Chains on Polysialoglycoproteins Isolated From Rainbow Trout Eggs—A Unique Carbohydrate Sequence With a Sialidase-Resistant Sialyl Group, dGalNAcβ1 → 4(NeuGc2 → 3)dGalNAc. Biochemistry. 1984;23:305–310. doi: 10.1021/bi00297a020. [DOI] [PubMed] [Google Scholar]
- 436.Finne J. Polysialic Acid—A Glycoprotein Carbohydrate Involved in Neural Adhesion and Bacterial Meningitis. Trends Biochem. Sci. 1985;10:129–132. [Google Scholar]
- 437.Fukuda M.N., Dell A., Oates J.E., Fukuda M. Embryonal Lactosaminoglycan—The Structure of Branched Lactosaminoglycans With Novel Disialosyl (Sialyl α2 → 9 Sialyl) Terminals Isolated From PA1 Human Embryonal Carcinoma Cells. J. Biol. Chem. 1985;260:6623–6631. [PubMed] [Google Scholar]
- 438.Inoue S., Poongodi G.L., Suresh N., Jennings H.J., Inoue Y. Discovery of an α2,9-PolyNeu5Ac Glycoprotein in C-1300 Murine Neuroblastoma (Clone NB41A3) J. Biol. Chem. 2003;278:8541–8546. doi: 10.1074/jbc.M212799200. [DOI] [PubMed] [Google Scholar]
- 439.Eylar E.H., Jeanloz R.W. Periodate Oxidation of the α1 Acid Glycoprotein of Human Plasma. J. Biol. Chem. 1962;237:1021–1025. [PubMed] [Google Scholar]
- 440.Wrann M. Methylation Analysis of the Carbohydrate Portion of Fibronectin Isolated From Human Plasma. Biochem. Biophys. Res. Commun. 1978;84:269–274. doi: 10.1016/0006-291x(78)90292-9. [DOI] [PubMed] [Google Scholar]
- 441.Takasaki S., Yamashita K., Suzuki K., Iwanaga S., Kobata A. The Sugar Chains of Cold-Insoluble Globulin—A Protein Related to Fibronectin. J. Biol. Chem. 1979;254:8548–8553. [PubMed] [Google Scholar]
- 442.Slomiany A., Slomiany B.L. Structures of the Acidic Oligosaccharides Isolated From Rat Sublingual Glycoprotein. J. Biol. Chem. 1978;253:7301–7306. [PubMed] [Google Scholar]
- 443.Van der Meer A., Kamerling J.P., Vliegenthart J.F.G., Schmid K., Schauer R. Fucopyranosyl-(1 → 4)-N-glycolylneuraminic Acid, a Constituent of Glycoproteins of the Cuvierian Tubules of the Sea Cucumber Holothuria forskali Della Chiaje. Biochim. Biophys. Acta. 1983;757:371–376. [Google Scholar]
- 444.Inoue S., Iwasaki M., Ishii K., Kitajima K., Inoue Y. Isolation and Structures of Glycoprotein-Derived Free Sialooligosaccharides From the Unfertilized Eggs of Tribolodon hakonensis, a Dace—Intracellular Accumulation of a Novel Class of Biantennary Disialooligosaccharides. J. Biol. Chem. 1989;264:18520–18526. [PubMed] [Google Scholar]
- 445.Van Kuik J.A., Hård K., Vliegenthart J.F.G. A 1H NMR Database Computer Program for the Analysis of the Primary Structure of Complex Carbohydrates. Carbohydr. Res. 1992;235:53–68. doi: 10.1016/0008-6215(92)80078-f. [DOI] [PubMed] [Google Scholar]
- 446.Damm J.B.L., Voshol H., Hård K., Kamerling J.P., Vliegenthart J.F.G. Analysis of N-Acetyl-4-O-acetylneuraminic-Acid-Containing N-Linked Carbohydrate Chains Released by Peptide-N4-(N-acetyl-β-glucosaminyl)asparagine Amidase F—Application to the Structure Determination of the Carbohydrate Chains of Equine Fibrinogen. Eur. J. Biochem. 1989;180:101–110. doi: 10.1111/j.1432-1033.1989.tb14620.x. [DOI] [PubMed] [Google Scholar]
- 447.Hanaoka K., Pritchett T.J., Takasaki S., Kochibe N., Sabesan S., Paulson J.C., Kobata A. 4-O-Acetyl-N-acetylneuraminic Acid in the N-Linked Carbohydrate Structures of Equine and Guinea Pig α2-Macroglobulins, Potent Inhibitors of Influenza Virus Infection. J. Biol. Chem. 1989;264:9842–9849. [PubMed] [Google Scholar]
- 448.Coddeville B., Stratil A., Wieruszeski J.-M., Strecker G., Montreuil J., Spik G. Primary Structure of Horse Serotransferrin Glycans—Demonstration That Heterogeneity Is Related to the Number of Glycans and to the Presence of N-Acetylneuraminic Acid and N-Acetyl-4-O-acetylneuraminic Acid. Eur. J. Biochem. 1989;186:583–590. doi: 10.1111/j.1432-1033.1989.tb15248.x. [DOI] [PubMed] [Google Scholar]
- 449.Damm J.B.L., Hård K., Kamerling J.P., van Dedem G.W.K., Vliegenthart J.F.G. Structure Determination of the Major N- and O-Linked Carbohydrate Chains of the β Subunit From Equine Chorionic Gonadotropin. Eur. J. Biochem. 1990;189:175–183. doi: 10.1111/j.1432-1033.1990.tb15474.x. [DOI] [PubMed] [Google Scholar]
- 450.Bergwerff A.A., van Oostrum J., Asselbergs F.A.M., Bürgi R., Hokke C.H., Kamerling J.P., Vliegenthart J.F.G. Primary Structure of N-Linked Carbohydrate Chains of a Human Chimeric Plasminogen Activator K2tu-PA Expressed in Chinese Hamster Ovary Cells. Eur. J. Biochem. 1993;212:639–656. doi: 10.1111/j.1432-1033.1993.tb17702.x. [DOI] [PubMed] [Google Scholar]
- 451.Hokke C.H., Bergwerff A.A., van Dedem G.W.K., Kamerling J.P., Vliegenthart J.F.G. Structural Analysis of the Sialylated N- and O-Linked Carbohydrate Chains of Recombinant Human Erythropoietin Expressed in Chinese Hamster Ovary Cells—Sialylation Patterns and Branch Location of Dimeric N-Acetyllactosamine Units. Eur. J. Biochem. 1995;228:981–1008. doi: 10.1111/j.1432-1033.1995.tb20350.x. [DOI] [PubMed] [Google Scholar]
- 452.Ylönen A., Kalkkinen N., Saarinen J., Bøgwald J., Helin J. Glycosylation Analysis of Two Cysteine Proteinase Inhibitors From Atlantic Salmon Skin: Di-O-acetylated Sialic Acids Are the Major Sialic Acid Species on N-Glycans. Glycobiology. 2001;11:523–531. doi: 10.1093/glycob/11.7.523. [DOI] [PubMed] [Google Scholar]
- 453.Llop E., Gutiérrez Gallego R., Belalcazar V., Gerwig G.J., Kamerling J.P., Segura J., Pascual J.A. Evaluation of Protein N-Glycosylation in 2-DE: Erythropoietin as a Study Case. Proteomics. 2007;7:4278–4291. doi: 10.1002/pmic.200700572. [DOI] [PubMed] [Google Scholar]
- 454.Yang Y., Liu F., Franc V., Halim L.A., Schellekens H., Heck A.J.R. Hybrid Mass Spectrometry Approaches in Glycoprotein Analysis and Their Usage in Scoring Biosimilarity. Nat. Commun. 2016;13397(1 − 10):7. doi: 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Sato C., Kitajima K., Tazawa I., Inoue Y., Inoue S., Troy F.A., II Structural Diversity in the α2 → 8-Linked Polysialic Acid Chains in Salmonid Fish Egg Glycoproteins—Occurrence of Poly(Neu5Ac), Poly(Neu5Gc), Poly(Neu5Ac, Neu5Gc), Poly(KDN), and Their Partially Acetylated Forms. J. Biol. Chem. 1993;268:23675–23684. [PubMed] [Google Scholar]
- 456.Miyata S., Sato C., Kitamura S., Toriyama M., Kitajima K. A Major Flagellum Sialoglycoprotein in Sea Urchin Sperm Contains a Novel Polysialic Acid, an α2,9-Linked Poly-N-acetylneuraminic Acid Chain, Capped by an 8-O-Sulfated Sialic Acid Residue. Glycobiology. 2004;14:827–840. doi: 10.1093/glycob/cwh100. [DOI] [PubMed] [Google Scholar]
- 457.Kitazume S., Kitajima K., Inoue S., Troy F.A., II., Cho J.-W., Lennarz W.J., Inoue Y. Identification of Polysialic Acid-Containing Glycoprotein in the Jelly Coat of Sea Urchin Eggs—Occurrence of a Novel Type of Polysialic Acid Structure. J. Biol. Chem. 1994;269:22712–22718. [PubMed] [Google Scholar]
- 458.Kanamori A., Inoue S., Iwasaki M., Kitajima K., Kawai G., Yokoyama S., Inoue Y. Deaminated Neuraminic Acid-Rich Glycoprotein of Rainbow Trout Egg Vitelline Envelope—Occurrence of a Novel α-2,8-Linked Oligo(Deaminated Neuraminic Acid) Structure in O-Linked Glycan Chains. J. Biol. Chem. 1990;265:21811–21819. [PubMed] [Google Scholar]
- 459.Tezuka T., Taguchi T., Kanamori A., Muto Y., Kitajima K., Inoue Y., Inoue S. Identification and Structural Determination of the KDN-Containing N-Linked Glycan Chains Consisting of Bi- and Triantennary Complex-Type Units of KDN-Glycoprotein Previously Isolated From Rainbow Trout Vitelline Envelopes. Biochemistry. 1994;33:6495–6502. doi: 10.1021/bi00187a016. [DOI] [PubMed] [Google Scholar]
- 460.Iwasaki M., Inoue S., Nadano D., Inoue Y. Isolation and Structure of a Novel Deaminated Neuraminic Acid Containing Oligosaccharide Chain Present in Rainbow Trout Egg Polysialoglycoprotein. Biochemistry. 1987;26:1452–1457. [Google Scholar]
- 461.Terada T., Kitazume S., Kitajima K., Inoue S., Ito F., Troy F.A., Inoue Y. Synthesis of CMP-Deaminoneuraminic Acid (CMP-KDN) Using the CTP:CMP-3-Deoxynonulosonate Cytidyltransferase From Rainbow Trout Testis—Identification and Characterization of a CMP-KDN Synthetase. J. Biol. Chem. 1993;268:2640–2648. [PubMed] [Google Scholar]
- 462.Maes E., Wieruszeski J.-M., Plancke Y., Strecker G. Structure of Three Kdn-Containing Oligosaccharide-Alditols Released From Oviducal Secretions of Ambystoma tigrinum: Characterization of the Carbohydrate Sequence Fuc(α1-5)[Fuc(α1-4)]Kdn(α2-3/6) FEBS Lett. 1995;358:205–210. doi: 10.1016/0014-5793(94)01425-z. [DOI] [PubMed] [Google Scholar]
- 463.Strecker G., Wieruszeski J.-M., Michalski J.-C., Alonso C., Leroy Y., Boilly B., Montreuil J. Primary Structure of Neutral and Acidic Oligosaccharide-Alditols Derived From the Jelly Coat of the Mexican Axolotl—Occurrence of Oligosaccharides With Fucosyl(α1-3)fucosyl(α1-4)-3-deoxy-d-glycero-d-galacto-nonulosonic Acid and Galactosyl(α1-4)[fucosyl(α1-2)]galactosyl(β1-4)-N-acetylglucosamine Sequences. Eur. J. Biochem. 1992;207:995–1002. doi: 10.1111/j.1432-1033.1992.tb17135.x. [DOI] [PubMed] [Google Scholar]
- 464.Kimura M., Hama Y., Sumi T., Asakawa M., Rao B.N.N., Horne A.P., Li S.-C., Li Y.-T., Nakagawa H. Characterization of a Deaminated Neuraminic Acid-Containing Glycoprotein From the Skin Mucus of the Loach, Misgurnus anguillicaudatus. J. Biol. Chem. 1994;269:32138–32143. [PubMed] [Google Scholar]
- 465.Vliegenthart J.F.G., Dorland L., van Halbeek H. High-Resolution, 1H-Nuclear Magnetic Resonance Spectroscopy as a Tool in the Structural Analysis of Carbohydrates Related to Glycoproteins. Adv. Carbohydr. Chem. Biochem. 1983;41:209–374. [Google Scholar]
- 466.Kamerling J.P., Vliegenthart J.F.G. High-Resolution 1H-Nuclear Magnetic Resonance Spectroscopy of Oligosaccharide-Alditols Released From Mucin-Type O-Glycoproteins. Biol. Magn. Reson. 1992;10:1–194. [Google Scholar]
- 467.Strecker G. In: Glycoproteins II. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 29b. Elsevier Science B.V; Amsterdam, The Netherlands: 1997. Amphibian Glycoproteins; pp. 163–172. (New Comprehensive Biochemistry). [Google Scholar]
- 468.Vliegenthart J.F.G. In: Comprehensive Glycoscience—From Chemistry to Systems Biology. Kamerling J.P., Kamerling J.P., Boons G.-J., Lee Y.C., Suzuki A., Taniguchi N., Voragen A.G.J., editors. Vol. 2. Elsevier Ltd.; Oxford, UK: 2007. 1H NMR Structural-Reporter-Group Concepts in Carbohydrate Analysis; pp. 133–191. [Google Scholar]
- 469.Stahl N., Baldwin M.A., Hecker R., Pan K.-M., Burlingame A.L., Prusiner S.B. Glycosylinositol Phospholipid Anchors of the Scrapic and Cellular Prion Proteins Contain Sialic Acid. Biochemistry. 1992;31:5043–5053. doi: 10.1021/bi00136a600. [DOI] [PubMed] [Google Scholar]
- 470.Huckerby T.N., Brown G.M., Nieduszynski I.A. 13C-NMR Spectroscopy of Keratan Sulphates—Assignments for Five Sialylated Pentasaccharides Derived From the Non-reducing Chain Termini of Bovine Articular Cartilage Keratan Sulphate by Keratanase II Digestion. Eur. J. Biochem. 1998;251:991–997. doi: 10.1046/j.1432-1327.1998.2510991.x. [DOI] [PubMed] [Google Scholar]
- 471.Klenk E., Heuer K. Über die Ganglioside der Hundeerythrozyten. Dtsch. Z. Verdau. Stoffwechselkr. 1960;20:180–183. [PubMed] [Google Scholar]
- 472.Kuhn R., Wiegandt H. Die Konstitution der Ganglio-N-tetraose und des Gangliosids GI. Chem. Ber. 1963;96:866–880. [Google Scholar]
- 473.Kuhn R., Wiegandt H. Die Konstitution der Ganglioside GII, GIII und GIV. Z. Naturforsch. B. 1963;18:541–543. [PubMed] [Google Scholar]
- 474.Wiegandt H., Baschang G. Die Gewinnung des Zuckeranteiles der Glykosphingolipide durch Ozonolyse und Fragmentierung. Z. Naturforschg. B. 1965;20:164–166. [PubMed] [Google Scholar]
- 475.Wiegandt H. Gangliosides of Extraneural Organs. Hoppe-Seyler’s Z. Physiol. Chem. 1973;354:1049–1056. doi: 10.1515/bchm2.1973.354.2.1049. [DOI] [PubMed] [Google Scholar]
- 476.Watanabe K., Powell M.E., Hakomori S.-I. Isolation and Characterization of Gangliosides With a New Sialosyl Linkage and Core Structures—II. Gangliosides of Human Erythrocyte Membranes. J. Biol. Chem. 1979;254:8223–8229. [PubMed] [Google Scholar]
- 477.Watanabe K., Hakomori S.-I. Gangliosides of Human Erythrocytes—A Novel Ganglioside With a Unique N-Acetylneuraminosyl-(2 → 3)-N-acetylgalactosamine Structure. Biochemistry. 1979;18:5502–5504. doi: 10.1021/bi00591a037. [DOI] [PubMed] [Google Scholar]
- 478.Taki T., Hirabayashi Y., Ishikawa H., Ando S., Kon K., Tanaka Y., Matsumoto M. A Ganglioside of Rat Ascites Hepatoma AH 7974F Cells—Occurrence of a Novel Disialoganglioside (GD1a) With a Unique N-Acetylneuraminosyl(α2-6)-N-acetylgalactosamine Structure. J. Biol. Chem. 1986;261:3075–3078. [PubMed] [Google Scholar]
- 479.Prieto P.A., Smith D.F. A New Ganglioside in Human Meconium Detected by Antiserum Against the Human Milk Sialyloligosaccharide, LS-Tetrasaccharide b. Arch. Biochem. Biophys. 1985;241:281–289. doi: 10.1016/0003-9861(85)90384-4. [DOI] [PubMed] [Google Scholar]
- 480.Fukushi Y., Nudelman E., Levery S.B., Higuchi T., Hakomori S.-I. A Novel Disialoganglioside (IV3NeuAcIII6NeuAcLc4) of Human Adenocarcinoma and the Monoclonal Antibody (FH9) Defining This Disialosyl Structure. Biochemistry. 1986;25:2859–2866. doi: 10.1021/bi00358a018. [DOI] [PubMed] [Google Scholar]
- 481.Handa N., Handa S. The Chemistry of Lipids of Posthemolytic Residue or Stroma of Erythrocytes. XIV. Chemical Structure of Glycolipid of Cat Erythrocyte Stroma. Jpn. J. Exp. Med. 1965;35:331–341. [PubMed] [Google Scholar]
- 482.Furukawa K., Chait B.T., Lloyd K.O. Identification of N-Glycolylneuraminic Acid-Containing Gangliosides of Cat and Sheep Erythrocytes—252Cf Fission Fragment Ionization Mass Spectrometry in the Analysis of Glycosphingolipids. J. Biol. Chem. 1988;263:14939–14947. [PubMed] [Google Scholar]
- 483.Kochetkov N.K., Smirnova G.P., Glukhoded I.S. Gangliosides With Sialic Acid Bound to N-Acetylgalactosamine From Hepatopancreas of the Starfish, Evasterias retifera and Asterias amurensis. Biochim. Biophys. Acta. 1982;712:650–658. [Google Scholar]
- 484.Kochetkov N.K., Zhukova I.G., Smirnova G.P., Glukhoded I.S. Isolation and Characterization of a Sialoglycolipid From the Sea Urchin Strongylocentrotus intermedius. Biochim. Biophys. Acta. 1973;326:74–83. doi: 10.1016/0005-2760(73)90029-5. [DOI] [PubMed] [Google Scholar]
- 485.Hoshi M., Nagai Y. Novel Sialosphingolipids From Spermatozoa of the Sea Urchin Anthocidaris crassispina. Biochim. Biophys. Acta. 1975;388:152–162. [PubMed] [Google Scholar]
- 486.Kochetkov N.K., Smirnova G.P., Glukhoded I.S. The Structure of Sialoglycolipids From Gonads of the Sea Urchin Strongylocentrotus nudus. Bioorg. Khim. 1978;4:1093–1099. [Google Scholar]
- 487.Hidari K.I.-P.J., Irie F., Suzuki M., Kon K., Ando S., Hirabayashi Y. A Novel Ganglioside With a Free Amino Group in Bovine Brain. Biochem. J. 1993;295:259–263. doi: 10.1042/bj2960259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Sonnenburg J.L., van Halbeek H., Varki A. Characterization of the Acid Stability of Glycosidically Linked Neuraminic Acid—Use in Detecting De-N-acetyl-gangliosides in Human Melanoma. J. Biol. Chem. 2002;277:17502–17510. doi: 10.1074/jbc.M110867200. [DOI] [PubMed] [Google Scholar]
- 489.Kuhn R., Müldner H. Über Glyko-lipo-sialo-proteide des Gehirns. Naturwissenschaften. 1964;51:635–636. [Google Scholar]
- 490.Wiegandt H. Gangliosides. Ergeb. Physiol. Biol. Chem. Exp. Pharmakol. 1966;57:190–222. [PubMed] [Google Scholar]
- 491.McCluer R.H., Evans J.E. Ganglioside Inner Esters. Adv. Exp. Med. Biol. 1972;19:95–102. [Google Scholar]
- 492.Gross S.K., Williams M.A., McCluer R.H. Alkali-Labile, Sodium Borohydride-Reducible Ganglioside Sialic Acid Residues in Brain. J. Neurochem. 1980;34:1351–1361. doi: 10.1111/j.1471-4159.1980.tb11215.x. [DOI] [PubMed] [Google Scholar]
- 493.Terabayashi T., Ogawa T., Kawanishi Y. Characterization of Ganglioside GM4 Lactones Isolated From the Whale Brain. J. Biochem. 1990;107:868–871. doi: 10.1093/oxfordjournals.jbchem.a123140. [DOI] [PubMed] [Google Scholar]
- 494.Pudelko M., Lindgren A., Tengel T., Reis C.A., Elofsson M., Kihlberg J. Formation of Lactones From Sialylated MUC1 Glycopeptides. Org. Biomol. Chem. 2006;4:713–720. doi: 10.1039/b514918e. [DOI] [PubMed] [Google Scholar]
- 495.Hirabayashi Y., Li Y.-T., Li S.-C. Occurrence of a New Hematoside in the Kidney of Guinea Pig. FEBS Lett. 1983;161:127–130. doi: 10.1016/0014-5793(83)80744-3. [DOI] [PubMed] [Google Scholar]
- 496.Hakomori S.-I., Saito T. Isolation and Characterization of a Glycosphingolipid Having a New Sialic Acid. Biochemistry. 1969;8:5082–5088. doi: 10.1021/bi00840a061. [DOI] [PubMed] [Google Scholar]
- 497.Gasa S., Makita A., Kinoshita Y. Further Study of the Chemical Structure of the Equine Erythrocyte Hematoside Containing O-Acetyl Ester. J. Biol. Chem. 1983;258:876–881. [PubMed] [Google Scholar]
- 498.Yachida Y., Tsuchihashi K., Gasa S. Characterization of Novel Mono-O-Acetylated GM3s Containing 9-O-Acetyl Sialic Acid and 6-O-Acetyl Galactose in Equine Erythrocytes. Glycoconjugate J. 1996;13:225–233. doi: 10.1007/BF00731497. [DOI] [PubMed] [Google Scholar]
- 499.Yachida Y., Tsuchihashi K., Gasa S. Novel Di-O-Acetylated GM3s From Equine Erythrocytes, One Containing 4,9-Di-O-acetyl-N-glycolylneuraminic Acid and Another Containing 4-O-Acetyl-N-glycolylneuraminic Acid and 6-O-Acetyl-d-galactose. Carbohydr. Res. 1997;298:201–212. doi: 10.1016/s0008-6215(96)00307-2. [DOI] [PubMed] [Google Scholar]
- 500.Kawatake S., Inagaki M., Isobe R., Miyamoto T., Higuchi R. Isolation and Structure of Monomethylated GM3-Type Ganglioside Molecular Species From the Starfish Luidia maculata. Chem. Pharm. Bull. 2002;50:1386–1389. doi: 10.1248/cpb.50.1386. [DOI] [PubMed] [Google Scholar]
- 501.Pan K., Tanaka C., Inagaki M., Higuchi R., Miyamoto T. Isolation and Structure Elucidation of GM4-Type Gangliosides From the Okinawan Starfish Protoreaster nodosus. Mar. Drugs. 2012;10:2467–2480. doi: 10.3390/md10112467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Muralikrishna G., Reuter G., Peter-Katalinic J., Egge H., Hanisch F.-G., Siebert H.-C., Schauer R. Identification of a New Ganglioside From the Starfish Asterias rubens. Carbohydr. Res. 1992;236:321–326. doi: 10.1016/0008-6215(92)85025-u. [DOI] [PubMed] [Google Scholar]
- 503.Yamada K., Tanabe K., Miyamoto T., Kusumoto T., Inagaki M., Higuchi R. Isolation and Structure of a Monomethylated Ganglioside Possessing Neuritogenic Activity From the Ovary of the Sea Urchin Diadema setosum. Chem. Pharm. Bull. 2008;56:734–737. doi: 10.1248/cpb.56.734. [DOI] [PubMed] [Google Scholar]
- 504.Yamada K., Hamada A., Kisa F., Miyamoto T., Higuchi R. Constituents of Holothuroidea, 13—Structure of Neuritogenic Active Ganglioside Molecular Species From the Sea Cucumber Stichopus chloronotus. Chem. Pharm. Bull. 2003;51:46–52. doi: 10.1248/cpb.51.46. [DOI] [PubMed] [Google Scholar]
- 505.Ren S., Scarsdale J.N., Ariga T., Zhang Y., Klein R.A., Hartmann R., Kushi Y., Egge H., Yu R.K. O-Acetylated Gangliosides in Bovine Buttermilk—Characterization of 7-O-Acetyl, 9-O-Acetyl, and 7,9-Di-O-acetyl GD3. J. Biol. Chem. 1992;267:12632–12638. [PubMed] [Google Scholar]
- 506.Ghidoni R., Sonnino S., Tettamanti G., Baumann N., Reuter G., Schauer R. Isolation and Characterization of a Trisialoganglioside From Mouse Brain, Containing 9-O-Acetyl-N-acetylneuraminic Acid. J. Biol. Chem. 1980;255:6990–6995. [PubMed] [Google Scholar]
- 507.Thurin J., Herlyn M., Hindsgaul O., Strömberg N., Karlsson K.-A., Elder D., Steplewski Z., Koprowski H. Proton NMR and Fast-Atom Bombardment Mass Spectrometry Analysis of the Melanoma-Associated Ganglioside 9-O-Acetyl-GD3. J. Biol. Chem. 1985;260:14556–14563. [PubMed] [Google Scholar]
- 508.Inagaki M., Isobe R., Higuchi R. Biologically Active Glycosides From Asteroidea, 39—Glycosphingolipids From the Starfish Linckia laevigata, 1—Isolation and Structure of a New Ganglioside Molecular Species. Eur. J. Org. Chem. 1999:771–774. doi: 10.1248/cpb.53.1551. [DOI] [PubMed] [Google Scholar]
- 509.Arao K., Inagaki M., Miyamoto T., Higuchi R. Constituents of Crinoidea, 3—Isolation and Structure of a Glycosyl Inositolphosphoceramide-Type Ganglioside With Neuritogenic Activity From the Feather Star Comanthus japonica. Chem. Pharm. Bull. 2004;52:1140–1142. doi: 10.1248/cpb.52.1140. [DOI] [PubMed] [Google Scholar]
- 510.Slomiany B.L., Kojima K., Banas-Gruszka Z., Murty V.L.N., Galicki N.I., Slomiany A. Characterization of the Sulfated Monosialosyltriglycosylceramide From Bovine Gastric Mucosa. Eur. J. Biochem. 1981;119:647–650. doi: 10.1111/j.1432-1033.1981.tb05656.x. [DOI] [PubMed] [Google Scholar]
- 511.Ijuin T., Kitajima K., Song Y., Kitazume S., Inoue S., Haslam S.M., Morris H.R., Dell A., Inoue Y. Isolation and Identification of Novel Sulfated and Nonsulfated Oligosialyl Glycosphingolipids From Sea Urchin Sperm. Glycoconjugate J. 1996;13:401–413. doi: 10.1007/BF00731473. [DOI] [PubMed] [Google Scholar]
- 512.Kundu S.K., Samuelsson B.E., Pascher I., Marcus D.M. New Gangliosides From Human Erythrocytes. J. Biol. Chem. 1983;258:13857–13866. [PubMed] [Google Scholar]
- 513.Kaneko M., Kisa F., Yamada K., Miyamoto T., Higuchi R. Structure of a New Neuritogenic-Active Ganglioside From the Sea Cucumber Stichopus japonicus. Eur. J. Org. Chem. 2003:1004–1008. [Google Scholar]
- 514.Sugita M. Studies on the Glycosphingolipids of the Starfish, Asterina pectinifera—II. Isolation and Characterization of a Novel Ganglioside With an Internal Sialic Acid Residue. J. Biochem. 1979;86:289–300. doi: 10.1093/oxfordjournals.jbchem.a132526. [DOI] [PubMed] [Google Scholar]
- 515.Sugita M. Studies on the Glycosphingolipids of the Starfish, Asterina pectinifera—III. Isolation and Structural Studies of Two Novel Gangliosides Containing Internal Sialic Acid Residues. J. Biochem. 1979;86:765–772. doi: 10.1093/oxfordjournals.jbchem.a132582. [DOI] [PubMed] [Google Scholar]
- 516.Kawano Y., Higuchi R., Komori T. Biologically Active Glycosides From Asteroidea, XIX. Glycosphingolipids From the Starfish Acanthaster planci—4. Isolation and Structure of Five New Gangliosides. Liebigs Ann. Chem. 1990:43–50. [Google Scholar]
- 517.Miyamoto T., Inagaki M., Isobe R., Tanaka Y., Higuchi R., Iha M., Teruya K. Biologically Active Glycosides From Asteroidea, 36—Re-examination of the Structure of Acanthaganglioside C, and the Identification of Three Minor Acanthagangliosides F, G and H. Liebigs Ann./Recl. 1997:931–936. [Google Scholar]
- 518.Miyamoto T., Yamamoto A., Wakabayashi M., Nagaregawa Y., Inagaki M., Higuchi R., Iha M., Teruya K. Biologically Active Glycosides From Asteroidea, 40—Two New Gangliosides, Acanthagangliosides I and J From the Starfish Acanthaster planci. Eur. J. Org. Chem. 2000:2295–2301. [Google Scholar]
- 519.Higuchi R., Inoue S., Inagaki K., Sakai M., Miyamoto T., Komori T., Inagaki M., Isobe R. Biologically Active Glycosides From Asteroidea, 42—Isolation and Structure of a New Biologically Active Ganglioside Molecular Species From the Starfish Asterina pectinifera. Chem. Pharm. Bull. 2006;54:287–291. doi: 10.1248/cpb.54.287. [DOI] [PubMed] [Google Scholar]
- 520.Prokazova N.V., Mikhailov A.T., Kocharov S.L., Malchenko L.A., Zvezdina N.D., Buznikov G., Bergelson L.D. Unusual Gangliosides of Eggs and Embryos of the Sea Urchin Strongylocentrotus intermedius—Structure and Density-Dependence of Surface Localization. Eur. J. Biochem. 1981;115:671–677. doi: 10.1111/j.1432-1033.1981.tb06255.x. [DOI] [PubMed] [Google Scholar]
- 521.Yamada K., Harada Y., Miyamoto T., Isobe R., Higuchi R. Constituents of Holothuroidea, 9—Isolation and Structure of a New Ganglioside Molecular Species From the Sea Cucumber Holothuria pervicax. Chem. Pharm. Bull. 2000;48:157–159. doi: 10.1248/cpb.48.157. [DOI] [PubMed] [Google Scholar]
- 522.Yamada K., Matsubara R., Kaneko M., Miyamoto T., Higuchi R. Constituents of Holothuroidea, 10—Isolation and Structure of a Biologically Active Ganglioside Molecular Species From the Sea Cucumber Holothuria leucospilota. Chem. Pharm. Bull. 2001;49:447–452. doi: 10.1248/cpb.49.447. [DOI] [PubMed] [Google Scholar]
- 523.Inagaki M., Miyamoto T., Isobe R., Higuchi R. Biologically Active Glycosides From Asteroidea, 43—Isolation and Structure of a New Neuritogenic-Active Ganglioside Molecular Species From the Starfish Linckia laevigata. Chem. Pharm. Bull. 2005;53:1551–1554. doi: 10.1248/cpb.53.1551. [DOI] [PubMed] [Google Scholar]
- 524.Song Y., Kitajima K., Inoue S., Inoue Y. Isolation and Structural Elucidation of a Novel Type of Ganglioside, Deaminated Neuraminic Acid (KDN)-Containing Glycosphingolipid, From Rainbow Trout Sperm—The First Example of the Natural Occurrence of KDN-Ganglioside, (KDN)GM3. J. Biol. Chem. 1991;266:21929–21935. [PubMed] [Google Scholar]
- 525.Song Y., Kitajima K., Inoue S., Muto Y., Kasama T., Handa S., Inoue Y. Structure of Novel Gangliosides, Deaminated Neuraminic Acid (KDN)-Containing Glycosphingolipids, Isolated From Rainbow Trout Ovarian Fluid. Biochemistry. 1993;32:9221–9229. doi: 10.1021/bi00086a030. [DOI] [PubMed] [Google Scholar]
- 526.Song Y., Kitajima K., Inoue S., Khoo K.-H., Morris H.R., Dell A., Inoue Y. Expression of New KDN-Gangliosides in Rainbow Trout Testis During Spermatogenesis and Their Structural Identification. Glycobiology. 1995;5:207–218. doi: 10.1093/glycob/5.2.207. [DOI] [PubMed] [Google Scholar]
- 527.Nagai Y., Iwamori M. In: Biology of the Sialic Acids. Rosenberg A., editor. Plenum Press; New York, USA: 1995. Cellular Biology of Gangliosides; pp. 197–241. [Google Scholar]
- 528.Higuchi R., Inagaki M., Yamada K., Miyamoto T. Biologically Active Gangliosides From Echinoderms. J. Nat. Med. 2007;61:367–370. [Google Scholar]
- 529.Yu R.K., Yanagisawa M., Ariga T. In: Comprehensive Glycoscience—From Chemistry to Systems Biology. Kamerling J.P., Boons G.-J., Lee Y.C., Suzuki A., Taniguchi N., Voragen A.G.J., editors. Vol. 1. Elsevier Ltd.; Oxford, UK: 2007. Glycosphingolipid Structures; pp. 73–122. [Google Scholar]
- 530.Urashima T., Asakuma S., Messer M. In: Comprehensive Glycoscience—From Chemistry to Systems Biology. Kamerling J.P., Boons G.-J., Lee Y.C., Suzuki A., Taniguchi N., Voragen A.G.J., editors. Vol. 4. Elsevier Ltd.; Oxford, UK: 2007. Milk Oligosaccharides; pp. 695–724. [Google Scholar]
- 531.Urashima T., Kitaoka M., Asakuma S., Messer M. In: Advanced Dairy Chemistry—Lactose, Water, Salts and Minor Constituents. McSweeney P.L.H., Fox P.F., editors. Vol. 3. Springer Science + Business Media; New York, NY, USA: 2009. Milk Oligosaccharides; pp. 295–349. [Google Scholar]
- 532.Kobata A. Structures and Application of Oligosaccharides in Human Milk. Proc. Jpn. Acad., Ser. B. 2010;86:731–747. doi: 10.2183/pjab.86.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Chen X. Human Milk Oligosaccharides (HMOS): Structure, Function, and Enzyme-Catalyzed Synthesis. Adv. Carbohydr. Chem. Biochem. 2015;72:113–190. doi: 10.1016/bs.accb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Kobata A. In: Prebiotics and Probiotics in Human Milk—Origins and Functions of Milk-Borne Oligosaccharides and Bacteria. McGuire M., McGuire M.A., Bode L., editors. Elsevier Inc.; Oxford, UK: 2017. Structures, Classification, and Biosynthesis of Human Milk Oligosaccharides; pp. 17–44. [Google Scholar]
- 535.Urashima T., Messer M., Oftedal O.T. In: Prebiotics and Probiotics in Human Milk—Origins and Functions of Milk-Borne Oligosaccharides and Bacteria. McGuire M., McGuire M.A., Bode L., editors. Elsevier Inc.; Oxford, UK: 2017. Oligosaccharides in the Milk of Other Mammals; pp. 45–139. [Google Scholar]
- 536.Kuhn R., Gauhe A. Bestimmung der Bindungsstelle von Sialinsäureresten in Oligosacchariden mit Hilfe von Perjodat. Chem. Ber. 1965;98:395–413. [Google Scholar]
- 537.Kuhn R. Biochemie der Rezeptoren und Resistenzfaktoren—Von der Widerstandsfähigkeit der Lebewesen gegen Einwirkungen der Umwelt. Naturwissenschaften. 1959;46:43–50. [Google Scholar]
- 538.Marino K., Lane J.A., Abrahams J.L., Struwe W.B., Harvey D.J., Marotta M., Hickey R.M., Rudd P.M. Method for Milk Oligosaccharide Profiling by 2-Aminobenzamide Labeling and Hydrophilic Interaction Chromatography. Glycobiology. 2011;21:1317–1330. doi: 10.1093/glycob/cwr067. [DOI] [PubMed] [Google Scholar]
- 539.Albrecht S., Lane J.A., Marino K., Al Busadah K.A., Carrington S.D., Hickey R.M., Rudd P.M. A Comparative Study of Free Oligosaccharides in the Milk of Domestic Animals. Br. J. Nutr. 2014;111:1313–1328. doi: 10.1017/S0007114513003772. [DOI] [PubMed] [Google Scholar]
- 540.Huang R.T.C. The Structures of Some Neuraminic Acid-Containing Oligosaccharides of Human Milk. Hoppe-Seyler’s Z. Physiol. Chem. 1971;352:1645–1652. doi: 10.1515/bchm2.1971.352.2.1645. [DOI] [PubMed] [Google Scholar]
- 541.Wiegandt H. Bemerkungen zur voranstehenden Arbeit “The Structures of Some Neuraminic Acid-Containing Oligosaccharides of Human Milk”. Hoppe-Seyler’s Z. Physiol. Chem. 1971;352:1653. doi: 10.1515/bchm2.1971.352.2.1653. [DOI] [PubMed] [Google Scholar]
- 542.Taufik E., Fukuda K., Senda A., Saito T., Williams C., Tilden C., Eisert R., Oftedal O., Urashima T. Structural Characterization of Neutral and Acidic Oligosaccharides in the Milks of Strepsirrhine Primates: Greater Galago, Aye-Aye, Coquerel's Sifaka and Mongoose Lemur. Glycoconjugate J. 2012;29:119–134. doi: 10.1007/s10719-012-9370-9. [DOI] [PubMed] [Google Scholar]
- 543.Veh R.W., Michalski J.-C., Corfield A.P., Sander-Wewer M., Gies D., Schauer R. New Chromatographic System for the Rapid Analysis and Preparation of Colostrum Sialyloligosaccharides. J. Chromatogr. 1981;212:313–322. doi: 10.1016/s0021-9673(01)84044-9. [DOI] [PubMed] [Google Scholar]
- 544.Messer M. Identification of N-Acetyl-4-O-acetylneuraminyl-lactose in Echidna Milk. Biochem. J. 1974;139:415–420. doi: 10.1042/bj1390415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Kamerling J.P., Dorland L., van Halbeek H., Vliegenthart J.F.G., Messer M., Schauer R. Structural Studies of 4-O-Acetyl-α-N-acetylneuraminyl-(2 → 3)-lactose, the Main Oligosaccharide in Echidna Milk. Carbohydr. Res. 1982;100:331–340. doi: 10.1016/s0008-6215(00)81046-0. [DOI] [PubMed] [Google Scholar]
- 546.Oftedal O.T., Nicol S.C., Davies N.W., Sekii N., Taufik E., Fukuda K., Saito T., Urashima T. Can an Ancestral Condition for Milk Oligosaccharides Be Determined? Evidence From the Tasmanian Echidna (Tachyglossus aculeatus setosus) Glycobiology. 2014;24:826–839. doi: 10.1093/glycob/cwu041. [DOI] [PubMed] [Google Scholar]
- 547.Urashima T., Inamori H., Fukuda K., Saito T., Messer M., Oftedal O.T. 4-O-Acetyl-sialic Acid (Neu4,5Ac2) in Acidic Milk Oligosaccharides of the Platypus (Ornithorhynchus anatinus) and Its Evolutionary Significance. Glycobiology. 2015;25:683–697. doi: 10.1093/glycob/cwv010. [DOI] [PubMed] [Google Scholar]
- 548.Schauer R. Sialic Acids as Link to Japanese Scientists. Proc. Jpn. Acad., Ser. B. 2016;92:109–120. doi: 10.2183/pjab.92.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Jahan M., Wynn P.C., Wang B. Molecular Characterization of the Level of Sialic Acids N-Acetylneuraminic Acid, N-Glycolylneuraminic Acid, and Ketodeoxynonulosonic Acid in Porcine Milk During Lactation. J. Dairy Sci. 2016;99:8431–8442. doi: 10.3168/jds.2016-11187. [DOI] [PubMed] [Google Scholar]
- 550.Huttunen J.K. Neuraminic Acid-Containing Oligosaccharides of Human Urine: Isolation and Identification of Di-N-acetylneuraminyl-3-galactosyl-N-acetylgalactosamine, 6-2-N-Acetylneuraminyl-lactose, 6-2-N-Acetylneuraminyl-N-acetyllactosamine and 3-2-N-Acetylneuraminyl-lactose. Ann. Med. Exp. Biol. Fenn. 1966;44(Suppl. 12):1–60. [PubMed] [Google Scholar]
- 551.Maury P. Identification of N,O-Diacetyl-, N-Acetyl-, and N-Glycolylneuraminyl-(2 → 3)-lactose in Rat Urine. Biochim. Biophys. Acta. 1971;252:472–480. doi: 10.1016/0304-4165(71)90150-4. [DOI] [PubMed] [Google Scholar]
- 552.Parkkinen J., Finne J. Isolation and Structural Characterization of Five Major Sialyloligosaccharides and a Sialylglycopeptide From Normal Human Urine. Eur. J. Biochem. 1983;136:355–361. doi: 10.1111/j.1432-1033.1983.tb07749.x. [DOI] [PubMed] [Google Scholar]
- 553.Parkkinen J., Finne J. Isolation of Sialyl Oligosaccharides and Sialyl Oligosaccharide Phosphates From Bovine Colostrum and Human Urine. Methods Enzymol. 1987;138:289–300. doi: 10.1016/0076-6879(87)38024-3. [DOI] [PubMed] [Google Scholar]
- 554.Lemonnier M., Bourrillon R. Characterization and Structure of a Sialic Acid-Containing Hexasaccharide Isolated From Human Pregnancy Urine. Carbohydr. Res. 1976;51:99–106. doi: 10.1016/s0008-6215(00)84039-2. [DOI] [PubMed] [Google Scholar]
- 555.Konradt T., Schlichting A., May B., Veh R.W. Histochemical Differentiation Between Tissue-Bound 9-O-Acetyl-sialic Acid and 7,9-Di-O-acetyl-sialic Acid: Periodate Oxidation Studies With Purified 9-O-Acetyl-sialyllactose From Rat Urine. Anat. Anz. 1987;163:159–160. [Google Scholar]
- 556.Strecker G., Hondi-Assah T., Fournet B., Spik G., Montreuil J., Maroteaux P., Durand P., Farriaux J.-P. Structure of the Three Major Sialyl-Oligosaccharides Excreted in the Urine of Five Patients With Three Distinct Inborn Diseases: “I Cell Disease” and Two New Types of Mucolipidosis. Biochim. Biophys. Acta. 1976;444:349–358. doi: 10.1016/0304-4165(76)90378-0. [DOI] [PubMed] [Google Scholar]
- 557.Michalski J.C., Strecker G., Fournet B., Cantz M., Spranger J. Structures of Sialyl-Oligosaccharides Excreted in the Urine of a Patient With Mucolipidosis I. FEBS Lett. 1977;79:101–104. doi: 10.1016/0014-5793(77)80359-1. [DOI] [PubMed] [Google Scholar]
- 558.Strecker G., Peers M.-C., Michalski J.-C., Hondi-Assah T., Fournet B., Spik G., Montreuil J., Farriaux J.-P., Maroteaux P., Durand P. Structure of Nine Sialyl-Oligosaccharides Accumulated in Urine of Eleven Patients With Three Different Types of Sialidosis—Mucolipidosis II and Two New Types of Mucolipidosis. Eur. J. Biochem. 1977;75:391–403. doi: 10.1111/j.1432-1033.1977.tb11540.x. [DOI] [PubMed] [Google Scholar]
- 559.Dorland L., Haverkamp J., Vliegenthart J.F.G., Strecker G., Michalski J.-C., Fournet B., Spik G., Montreuil J. 360-MHz 1H Nuclear-Magnetic-Resonance Spectroscopy of Sialyl-Oligosaccharides From Patients With Sialidosis (Mucolipidosis I and II) Eur. J. Biochem. 1978;87:323–329. doi: 10.1111/j.1432-1033.1978.tb12381.x. [DOI] [PubMed] [Google Scholar]
- 560.Kuriyama M., Ariga T., Ando S., Suzuki M., Yamada T., Miyatake T. Four Positional Isomers of Sialyloligosaccharides Isolated From the Urine of a Patient With Sialidosis. J. Biol. Chem. 1981;256:12316–12321. [PubMed] [Google Scholar]
- 561.Lecat D., Lemonnier M., Derappe C., Lhermitte M., van Halbeek H., Dorland L., Vliegenthart J.F.G. The Structure of Sialyl-Glycopeptides of the O-Glycosidic Type, Isolated From Sialidosis (Mucolipidosis I) Urine. Eur. J. Biochem. 1984;140:415–420. doi: 10.1111/j.1432-1033.1984.tb08118.x. [DOI] [PubMed] [Google Scholar]
- 562.Kuriyama M., Ariga T., Ando S., Suzuki M., Yamada T., Miyatake T., Igata A. Two Positional Isomers of Sialylheptasaccharides Isolated From the Urine of a Patient With Sialidosis. J. Biochem. 1985;98:1049–1954. doi: 10.1093/oxfordjournals.jbchem.a135351. [DOI] [PubMed] [Google Scholar]
- 563.Van Pelt J., Kamerling J.P., Vliegenthart J.F.G., Verheijen F.W., Galjaard H. Isolation and Structural Characterization of Sialic Acid-Containing Storage Material From Mucolipidosis I (Sialidosis) Fibroblasts. Biochim. Biophys. Acta. 1988;965:36–45. doi: 10.1016/0304-4165(88)90148-1. [DOI] [PubMed] [Google Scholar]
- 564.Van Pelt J., Kamerling J.P., Vliegenthart J.F.G., Hoogeveen A.T., Galjaard H. A Comparative Study of the Accumulated Sialic Acid-Containing Oligosaccharides From Cultured Human Galactosialidosis and Sialidosis Fibroblasts. Clin. Chim. Acta. 1988;174:325–336. doi: 10.1016/0009-8981(88)90059-9. [DOI] [PubMed] [Google Scholar]
- 565.Van Pelt J., van Kuik J.A., Kamerling J.P., Vliegenthart J.F.G., van Diggelen O.P., Galjaard H. Storage of Sialic Acid-Containing Carbohydrates in the Placenta of a Human Galactosialidosis Fetus—Isolation and Structural Characterization of 16 Sialyloligosaccharides. Eur. J. Biochem. 1988;177:327–338. doi: 10.1111/j.1432-1033.1988.tb14380.x. [DOI] [PubMed] [Google Scholar]
- 566.Van Pelt J., van Bilsen D.G.J.L., Kamerling J.P., Vliegenthart J.F.G. Structural Analysis of O-Glycosidic Type of Sialyloligosaccharide-Alditols Derived From Urinary Glycopeptides of a Sialidosis Patient. Eur. J. Biochem. 1988;174:183–187. doi: 10.1111/j.1432-1033.1988.tb14080.x. [DOI] [PubMed] [Google Scholar]
- 567.Van Pelt J., Hård K., Kamerling J.P., Vliegenthart J.F.G., Reuser A.J.J., Galjaard H. Isolation and Structural Characterization of Twenty-One Sialyloligosaccharides From Galactosialidosis Urine—An Intact N,N’-Diacetylchitobiose Unit at the Reducing End of a Diantennary Structure. Biol. Chem. Hoppe-Seyler. 1989;370:191–203. doi: 10.1515/bchm3.1989.370.1.191. [DOI] [PubMed] [Google Scholar]
- 568.Michalski J.-C. In: Glycoproteins and Disease. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 30. Elsevier Science B.V; Amsterdam, The Netherlands: 1996. Normal and Pathological Catabolism of Glycoproteins—Catabolic Pathways; pp. 55–97. (New Comprehensive Biochemistry). [Google Scholar]
- 569.Bruggink C., Poorthuis B.J.H.M., Piraud M., Froissart R., Deelder A.M., Wuhrer M. Glycan Profiling of Urine, Amniotic Fluid and Ascitic Fluid From Galactosialidosis Patients Reveals Novel Oligosaccharides With Reducing End Hexose and Aldohexonic Acid Residues. FEBS J. 2010;277:2970–2986. doi: 10.1111/j.1742-4658.2010.07707.x. [DOI] [PubMed] [Google Scholar]
- 570.Van Pelt J., Dorland L., Duran M., Hokke C.H., Kamerling J.P., Vliegenthart J.F.G. Sialyl-α2-6-mannosyl-β1-4-N-acetylglucosamine, a Novel Compound Occurring in Urine of Patients With β-Mannosidosis. J. Biol. Chem. 1990;265:19685–19689. [PubMed] [Google Scholar]
- 571.Pollitt R.J., Pretty K.M. The Glycoasparagines in Urine of a Patient With Aspartylglycosaminuria. Biochem. J. 1974;141:141–146. doi: 10.1042/bj1410141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Sugahara K., Funakoshi S., Funakoshi I., Aula P., Yamashina I. Characterization of One Neutral and Two Acidic Glycoasparagines Isolated From the Urine of Patients With Aspartylglycosylaminuria (AGU) J. Biochem. 1976;80:195–201. doi: 10.1093/oxfordjournals.jbchem.a131264. [DOI] [PubMed] [Google Scholar]
- 573.Sugahara K., Akasaki M., Funakoshi I., Aula P., Yamashina I. Structural Studies of Glycoasparagines From Urine of a Patient With Aspartylglycosylaminuria (AGU) FEBS Lett. 1977;78:284–286. doi: 10.1016/0014-5793(77)80324-4. [DOI] [PubMed] [Google Scholar]
- 574.Sugahara K., Akasaki M., Funakoshi I., Aula P., Yamashina I. Structure of Two Glycoasparagines Isolated From the Urine of Patients With Aspartylglycosylaminuria (AGU) J. Biochem. 1977;82:1499–1501. doi: 10.1093/oxfordjournals.jbchem.a131841. [DOI] [PubMed] [Google Scholar]
- 575.Irie F., Murakoshi H., Suzuki T., Suzuki Y., Kon K., Ando S., Yoshida K., Hirabayashi Y. Characterization of Four Monosialo and a Novel Disialo Asn N-Glycosides From the Urine of a Patient With Aspartylglycosaminuria. Glycoconjugate J. 1995;12:290–297. doi: 10.1007/BF00731332. [DOI] [PubMed] [Google Scholar]
- 576.Linden H.-U., Klein R.A., Egge H., Peter-Katalinic J., Dabrowski J., Schindler D. Isolation and Structural Characterization of Sialic Acid-Containing Glycopeptides of the O-Glycosidic Type From the Urine of Two Patients With an Hereditary Deficiency in α-N-Acetylgalactosaminidase Activity. Biol. Chem. Hoppe-Seyler. 1989;370:661–672. doi: 10.1515/bchm3.1989.370.2.661. [DOI] [PubMed] [Google Scholar]
- 577.Hirabayashi Y., Matsumoto Y., Matsumoto M., Toida T., Iida N., Matsubara T., Kanzaki T., Yokota M., Ishizuka I. Isolation and Characterization of Major Urinary Amino Acid O-Glycosides and a Dipeptide O-Glycoside From a New Lysosomal Storage Disorder (Kanzaki Disease)—Excessive Excretion of Serine- and Threonine-Linked Glycan in the Patient Urine. J. Biol. Chem. 1990;265:1693–1701. [PubMed] [Google Scholar]
- 578.Froesch M., Bindila L., Zamfir A., Peter-Katalinic J. Sialylation Analysis of O-Glycosylated Sialylated Peptides From Urine of Patients Suffering From Schindler's Disease by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Sustained Off-Resonance Irradiation Collision-Induced Dissociation. Rapid Commun. Mass Spectrom. 2003;17:2822–2832. doi: 10.1002/rcm.1273. [DOI] [PubMed] [Google Scholar]
- 579.Desnick R.J., Schindler D. In: The Molecular and Genetic Basis of Neurologic and Psychiatric Disease. 4th ed. Rosenberg R.N., DiMauro S., Paulson H.L., Ptacek L., Nestler E.J., editors. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2008. Schindler Disease: Deficient α-N-Acetylgalactosaminidase Activity; pp. 309–316. [Google Scholar]
- 580.Aula P., Autio S., Raivio K.O., Rapola J., Thodén C.-J., Koskela S.-L., Yamashina I. ‘Salla Disease’—A New Lysosomal Storage Disorder. Arch. Neurol. 1979;36:88–94. doi: 10.1001/archneur.1979.00500380058006. [DOI] [PubMed] [Google Scholar]
- 581.Renlund M., Chester M.A., Lundblad A., Aula P., Raivio K.O., Autio S., Koskela S.-L. Increased Urinary Excretion of Free N-Acetylneuraminic Acid in Thirteen Patients With Salla Disease. Eur. J. Biochem. 1979;101:245–250. doi: 10.1111/j.1432-1033.1979.tb04237.x. [DOI] [PubMed] [Google Scholar]
- 582.Baumkötter J., Cantz M., Mendla K., Baumann W., Friebolin H., Gehler J., Spranger J. N-Acetylneuraminic Acid Storage Disease. Hum. Genet. 1985;71:155–159. doi: 10.1007/BF00283373. [DOI] [PubMed] [Google Scholar]
- 583.Fontaine G., Gaudier B., Biserte G., Montreuil J., Dupont A., Farriaux J.-P., Strecker G., Spik G., Puvion E., Puvion-Dutilleul F., Sezille G. Elimination Urinaire Permanente d’Acide Sialique Libre chez un Enfant de Trois Ans Atteint de Troubles Cliniques Divers. Pediatrie. 1967;22:705–709. [PubMed] [Google Scholar]
- 584.Montreuil J., Biserte G., Strecker G., Spik G., Fontaine G., Farriaux J.-P. Description d’un Nouveau Type de Méliturie: La Sialurie. Clin. Chim. Acta. 1968;21:61–69. doi: 10.1016/0009-8981(68)90011-9. [DOI] [PubMed] [Google Scholar]
- 585.Kamerling J.P., Strecker G., Farriaux J.-P., Dorland L., Haverkamp J., Vliegenthart J.F.G. 2-Acetamidoglucal, a New Metabolite Isolated From the Urine of a Patient With Sialuria. Biochim. Biophys. Acta. 1979;583:403–408. doi: 10.1016/0304-4165(79)90465-3. [DOI] [PubMed] [Google Scholar]
- 586.Weiss P., Tietze F., Gahl W.A., Seppala R., Ashwell G. Identification of the Metabolic Defect in Sialuria. J. Biol. Chem. 1989;264:17635–17636. [PubMed] [Google Scholar]
- 587.Wopereis S., Abd Hamid U.M., Critchley A., Royle L., Dwek R.A., Morava E., Leroy J.G., Wilcken B., Lagerwerf A.J., Huijben K.M.L.C., Lefeber D.J., Rudd P.M., Wevers R.A. Abnormal Glycosylation With Hypersialylated O-Glycans in Patients With Sialuria. Biochim. Biophys. Acta. 2006;1762:598–607. doi: 10.1016/j.bbadis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 588.Barry G.T., Goebel W.F. Colominic Acid, a Substance of Bacterial Origin Related to Sialic Acid. Nature. 1957;179:206. doi: 10.1038/179206a0. [DOI] [PubMed] [Google Scholar]
- 589.Aminoff D., Dodyk F., Roseman S. Enzymatic Synthesis of Colominic Acid. J. Biol. Chem. 1963;238:PC1177–PC1178. [PubMed] [Google Scholar]
- 590.McGuire E.J., Binkley S.B. The Structure and Chemistry of Colominic Acid. Biochemistry. 1964;3:247–251. doi: 10.1021/bi00890a017. [DOI] [PubMed] [Google Scholar]
- 591.Liu T.-Y., Gotschlich E.C., Dunne F.T., Jonssen E.K. Studies on the Meningococcal Polysaccharides—II. Composition and Chemical Properties of the Group B and Group C Polysaccharide. J. Biol. Chem. 1971;246:4703–4712. [PubMed] [Google Scholar]
- 592.Watson R.G., Marinetti G.V., Scherp H.W. The Specific Hapten of Group C (Group II α) Meningococcus—II. Chemical Nature. J. Immunol. 1958;81:337–344. [PubMed] [Google Scholar]
- 593.Egan W., Liu T.-Y., Dorow D., Cohen J.S., Robbins J.D., Gotschlich E.C., Robbins J.B. Structural Studies on the Sialic Acid Polysaccharide Antigen of Escherichia coli Strain Bos-12. Biochemistry. 1977;16:3687–3692. doi: 10.1021/bi00635a028. [DOI] [PubMed] [Google Scholar]
- 594.Dzieciatkowska M., Brochu D., van Belkum A., Heikema A.P., Yuki N., Houliston R.S., Richards J.C., Gilbert M., Li J. Mass Spectrometric Analysis of Intact Lipooligosaccharide: Direct Evidence for O-Acetylated Sialic Acids and Discovery of O-Linked Glycine Expressed by Campylobacter jejuni. Biochemistry. 2007;46:14704–14714. doi: 10.1021/bi701229k. [DOI] [PubMed] [Google Scholar]
- 595.Knirel Y.A., Shevelev S.D., Perepelov A.V. Higher Aldulosonic Acids: Components of Bacterial Glycans. Mendeleev Commun. 2011;21:173–182. [Google Scholar]
- 596.Deutschmann R., Boncheff A.G., Daraban L., MacInnes J.I., Monteiro M.A. Common Sialylated Glycan in Actinobacillus suis. Glycobiology. 2010;20:1227–1232. doi: 10.1093/glycob/cwq079. [DOI] [PubMed] [Google Scholar]
- 597.Ali T., Weintraub A., Widmalm G. Structural Determination of the O-Antigenic Polysaccharide From the Shiga Toxin-Producing Escherichia coli O171. Carbohydr. Res. 2006;341:1878–1883. doi: 10.1016/j.carres.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 598.Bhattacharjee A.K., Jennings H.J., Kenny C.P., Martin A., Smith I.C.P. Structural Determination of the Polysaccharide Antigens of Neisseria meningitidis Serogroups Y, W-135, and BO. Can. J. Biochem. 1976;54:1–8. doi: 10.1139/o76-001. [DOI] [PubMed] [Google Scholar]
- 599.Dutton G.G.S., Parolis H., Parolis L.A.S. The Structure of the Neuraminic Acid-Containing Capsular Polysaccharide of Escherichia coli Serotype K9. Carbohydr. Res. 1987;170:193–206. doi: 10.1016/s0008-6215(00)90904-2. [DOI] [PubMed] [Google Scholar]
- 600.Gamian A., Kenne L., Mieszala M., Ulrich J., Defaye J. Structure of the Escherichia coli O24 and O56 O-Specific Sialic-Acid-Containing Polysaccharides and Linkage of These Structures to the Core Region in Lipopolysaccharides. Eur. J. Biochem. 1994;225:1211–1220. doi: 10.1111/j.1432-1033.1994.1211b.x. [DOI] [PubMed] [Google Scholar]
- 601.Krauss J.H., Himmelspach K., Reuter G., Schauer R., Mayer H. Structural Analysis of a Novel Sialic-Acid-Containing Trisaccharide From Rhodobacter capsulatus 37b4 Lipopolysaccharide. Eur. J. Biochem. 1992;204:217–223. doi: 10.1111/j.1432-1033.1992.tb16627.x. [DOI] [PubMed] [Google Scholar]
- 602.Tul’skaya E.M., Shashkov A.S., Streshinskaya G.M., Senchenkova S.N., Potekhina N.V., Kozlova Y.I., Evtushenko L.I. Teichuronic and Teichulosonic Acids of Actinomycetes. Biochemistry (Moscow) 2011;76:736–744. doi: 10.1134/S0006297911070030. [DOI] [PubMed] [Google Scholar]
- 603.Shashkov A.S., Tul’skaya E.M., Evtushenko L.I., Denisenko V.A., Ivanyuk V.G., Stomakhin A.A., Naumova I.B., Stackebrandt E. Cell Wall Anionic Polymers of Streptomyces sp. MB-8, the Causative Agent of Potato Scab. Carbohydr. Res. 2002;337:2255–2261. doi: 10.1016/s0008-6215(02)00188-x. [DOI] [PubMed] [Google Scholar]
- 604.Dell, A.; Chalabi, S.; Hitchen, P. G.; Jang-Lee, J.; Ledger, V.; North, S. J.; Pang, P.-C.; Parry, S.; Sutton-Smith, M.; Tissot, B.; Morris, H. R.; Panico, M.; Haslam, S. M. Mass Spectrometry of Glycoprotein Glycans: Glycomics and Glycoproteomics. In Comprehensive Glycoscience—From Chemistry to Systems Biology, Kamerling, J. P.; Boons, G.-J.; Lee, Y. C.; Suzuki, A.; Taniguchi, N.; Voragen, A. G. J., Eds.; Elsevier Ltd.: Oxford, UK, 2007; Vol. 2, pp 69–100.
- 605.Zauner G., Deelder A.M., Wuhrer M. Recent Advances in Hydrophilic Interaction Liquid Chromatography (HILIC) for Structural Glycomics. Electrophoresis. 2011;32:3456–3466. doi: 10.1002/elps.201100247. [DOI] [PubMed] [Google Scholar]
- 606.Leymarie N., Zaia J. Effective Use of Mass Spectrometry for Glycan and Glycopeptide Structural Analysis. Anal. Chem. 2012;84:3040–3048. doi: 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Wuhrer M. Glycomics Using Mass Spectrometry. Glycoconjugate J. 2013;30:11–22. doi: 10.1007/s10719-012-9376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 608.Mechref Y., Hu Y., Desantos-Garcia J.L., Hussein A., Tang H. Quantitative Glycomics Strategies. Mol. Cell. Proteomics. 2013;12:874–884. doi: 10.1074/mcp.R112.026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Kailemia M.J., Ruhaak L.R., Lebrilla C.B., Amster I.J. Oligosaccharide Analysis by Mass Spectrometry: A Review of Recent Developments. Anal. Chem. 2014;86:196–212. doi: 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Banazadeh A., Veillon L., Wooding K.M., Zabet-moghaddam M., Mechref Y. Recent Advances in Mass Spectrometric Analysis of Glycoproteins. Electrophoresis. 2017;38:162–189. doi: 10.1002/elps.201600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Yang Y., Franc V., Heck A.J.R. Glycoproteomics: A Balance Between High-Throughput and In-Depth Analysis. Trends Biotechnol. 2017;35:598–609. doi: 10.1016/j.tibtech.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 612.Powell A.K., Harvey D.J. Stabilization of Sialic Acids in N-Linked Oligosaccharides and Gangliosides for Analysis by Positive Ion Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 1996;10:1027–1032. doi: 10.1002/(SICI)1097-0231(19960715)10:9<1027::AID-RCM634>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 613.Yamagaki T., Nakanishi H. A New Technique Distinguishing α2-3 Sialyl Linkage From α2-6 Linkage in Sialyllactoses and Sialyl-N-acetyllactosamines by Post-Source Decay Fragmentation Method of MALDI-TOF Mass Spectrometry. Glycoconjugate J. 1999;16:385–389. doi: 10.1023/a:1007050909293. [DOI] [PubMed] [Google Scholar]
- 614.Sekiya S., Wada Y., Tanaka K. Derivatization for Stabilizing Sialic Acids in MALDI-MS. Anal. Chem. 2005;77:4962–4968. doi: 10.1021/ac050287o. [DOI] [PubMed] [Google Scholar]
- 615.Galuska S.P., Geyer R., Mühlenhoff M., Geyer H. Characterization of Oligo- and Polysialic Acids by MALDI-TOF-MS. Anal. Chem. 2007;79:7161–7169. doi: 10.1021/ac0712446. [DOI] [PubMed] [Google Scholar]
- 616.Miura Y., Shinohara Y., Furukawa J.-I., Nagahori N., Nishimura S.-I. Rapid and Simple Solid-Phase Esterification of Sialic Acid Residues for Quantitative Glycomics by Mass Spectrometry. Chem. Eur. J. 2007;13:4797–4804. doi: 10.1002/chem.200601872. [DOI] [PubMed] [Google Scholar]
- 617.Wheeler S.F., Domann P., Harvey D.J. Derivatization of Sialic Acids for Stabilization in Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry and Concomitant Differentiation of α(2 → 3)- and α(2 → 6)-Isomers. Rapid Commun. Mass Spectrom. 2009;23:303–312. doi: 10.1002/rcm.3867. [DOI] [PubMed] [Google Scholar]
- 618.Galuska S.P., Geyer H., Bleckmann C., Röhrich R.C., Maass K., Bergfeld A.K., Mühlenhoff M., Geyer R. Mass Spectrometric Fragmentation Analysis of Oligosialic and Polysialic Acids. Anal. Chem. 2010;82:2059–2066. doi: 10.1021/ac902809q. [DOI] [PubMed] [Google Scholar]
- 619.De Haan N., Reiding K.R., Haberger M., Reusch D., Falck D., Wuhrer M. Linkage-Specific Sialic Acid Derivatization for MALDI-TOF-MS Profiling of IgG Glycopeptides. Anal. Chem. 2015;87:8284–8291. doi: 10.1021/acs.analchem.5b02426. [DOI] [PubMed] [Google Scholar]
- 620.Holst S., Heijs B., de Haan N., van Zeijl R.J.M., Briaire-de Bruijn I.H., van Pelt G.W., Mehta A.S., Angel P.M., Mesker W.E., Tollenaar R.A., Drake R.R., Bovée J.V.M.G., McDonnell L.A., Wuhrer M. Linkage-Specific In Situ Sialic Acid Derivatization for N-Glycan Mass Spectrometry Imaging of Formalin-Fixed Paraffin-Embedded Tissues. Anal. Chem. 2016;88:5904–5913. doi: 10.1021/acs.analchem.6b00819. [DOI] [PubMed] [Google Scholar]
- 621.Warren L., Felsenfeld H. N-Acetylmannosamine-6-phosphate and N-Acetylneuraminic Acid-9-phosphate as Intermediates in Sialic Acid Biosynthesis. Biochem. Biophys. Res. Commun. 1961;5:185–190. doi: 10.1016/0006-291x(61)90107-3. [DOI] [PubMed] [Google Scholar]
- 622.Warren L., Blacklow R.S. Biosynthesis of N-Acetylneuraminic Acid and Cytidine-5′-monophospho-N-acetylneuraminic Acid in Neisseria meningitidis. Biochem. Biophys. Res. Commun. 1962;7:433–438. doi: 10.1016/0006-291x(62)90330-3. [DOI] [PubMed] [Google Scholar]
- 623.Kragl U., Kittelmann M., Ghisalba O., Wandrey C. N-Acetylneuraminic Acid—From a Rare Chemical From Natural Sources to a Multikilogram Enzymatic Synthesis for Industrial Application. Ann. N. Y. Acad. Sci. 1995;750:300–305. [Google Scholar]
- 624.Kuboki A., Okazaki H., Sugai T., Ohta H. An Expeditious Route to N-Glycolylneuraminic Acid Based on Enzyme-Catalyzed Reaction. Tetrahedron. 1997;53:2387–2400. [Google Scholar]
- 625.Maru I., Ohnishi J., Ohta Y., Tsukada Y. Simple and Large-Scale Production of N-Acetylneuraminic Acid From N-Acetyl-d-glucosamine and Pyruvate Using N-Acyl-d-glucosamine 2-Epimerase and N-Acetylneuraminate Lyase. Carbohydr. Res. 1998;306:575–578. doi: 10.1016/s0008-6215(97)10106-9. [DOI] [PubMed] [Google Scholar]
- 626.Hemeon I., Bennet A.J. Sialic Acid and Structural Analogues: Stereoselective Syntheses. Synthesis. 2007:1899–1926. [Google Scholar]
- 627.Juneja L.R., Koketsu M., Nishimoto K., Kim M., Yamamoto T., Itoh T. Large-Scale Preparation of Sialic Acid From Chalaza and Egg-Yolk Membrane. Carbohydr. Res. 1991;214:179–186. doi: 10.1016/s0008-6215(00)90540-8. [DOI] [PubMed] [Google Scholar]
- 628.Sumi T., Sallay I., Asakawa M., Park S.S., Miyazaki M., Ohba H. Purification and Identification of N-Glycolylneuraminic Acid (Neu5Gc) From the Holothuroidea Gumi, Cucumaria echinata. Prep. Biochem. Biotechnol. 2001;31:135–146. doi: 10.1081/PB-100103379. [DOI] [PubMed] [Google Scholar]
- 629.Ogura H., Furuhata K., Sato S., Anazawa K., Itoh M., Shitori Y. Synthesis of 9-O-Acyl- and 4-O-Acetyl-salic Acids. Carbohydr. Res. 1987;167:77–86. doi: 10.1016/0008-6215(87)80269-0. [DOI] [PubMed] [Google Scholar]
- 630.Forstner M., Freytag K., Paschke E. A Simple, One-Step Synthesis of N-Acetyl-9-O-acetylneuraminic Acid by Enzymic Transesterification Mediated by Porcine Pancreas Lipase in Pyridine. Carbohydr. Res. 1989;193:294–295. doi: 10.1016/0008-6215(89)85130-4. [DOI] [PubMed] [Google Scholar]
- 631.Hasegawa A., Murase T., Ogawa M., Ishida H., Kiso M. Synthetic Studies on Sialoglycoconjugates—17: Synthesis of 4-O-, 9-O-, and 4,9-Di-O-acetyl-N-acetylneuraminic Acids and their Derivatives. J. Carbohydr. Chem. 1990;9:415–428. [Google Scholar]
- 632.Hasegawa A., Murase T., Ogawa M., Ishida H., Kiso M. Synthetic Studies on Sialoglycoconjugates—18: Synthesis of 7-O-, 7,9-Di-O-, and 7,8,9-Tri-O-acetyl-N-acetyl-neuraminic Acid Derivatives. J. Carbohydr. Chem. 1990;9:429–439. [Google Scholar]
- 633.Anazawa K., Furuhata K., Ogura H. Synthesis of 7-O-Acetyl-N-acetylneuraminic Acid Derivative. Chem. Pharm. Bull. 1988;36:4976–4979. [Google Scholar]
- 634.Furuhata K., Ogura H. Studies on Sialic Acids—XIX. Syntheses of Partially O-Acetylated 4-Methylcoumarin-7-yl 5-Acetamido-3,5-dideoxy-α-d-glycero-d-galacto-2-nonulopyranosidonic Acids. Chem. Pharm. Bull. 1989;37:2037–2040. [Google Scholar]
- 635.Reinhard B., Faillard H. Regioselective Acetylations of Sialic Acid α-Ketosides. Liebigs Ann. Chem. 1994:193–203. [Google Scholar]
- 636.David S., Augé C. Immobilized Enzymes in Preparative Carbohydrate Chemistry. Pure Appl. Chem. 1987;59:1501–1508. [Google Scholar]
- 637.Kim M.-J., Hennen W.J., Sweers H.M., Wong C.-H. Enzymes in Carbohydrate Synthesis: N-Acetylneuraminic Acid Aldolase Catalyzed Reactions and Preparation of N-Acetyl-2-deoxy-d-neuraminic Acid Derivatives. J. Am. Chem. Soc. 1988;110:6481–6486. [Google Scholar]
- 638.Meindl P., Tuppy H. Über 2-Deoxy-2,3-dehydro-sialinsäuren—1. Mitt.: Synthese und Eigenschaften von 2-Deoxy-2,3-dehydro-N-acylneuraminsäuren und deren Methylestern. Monatsh. Chem. 1969;100:1295–1306. [Google Scholar]
- 639.Augé C., Gautheron C. The Use of an Immobilized Aldolase in the First Synthesis of a Natural Deaminated Neuraminic Acid. J. Chem. Soc., Chem. Commun. 1987:859–860. [Google Scholar]
- 640.Shirai R., Nakamura M., Hara S., Takayanagi H., Ogura H. Thermal Rearrangement of N-Acetyl-N-nitrosoneuraminic Acid Derivative: Synthesis of 3-Deoxy-d-nonulosonic Acid (KDN) Tetrahedron Lett. 1988;29:4449–4452. [Google Scholar]
- 641.Shirai R., Ogura H. Improved Syntheses of Two 3-Deoxyald-2-ulosonic Acids (KDN, KDO) by Condensation of Oxalacetic Acid With Aldoses Followed by Ni2 + Catalyzed Decarboxylation. Tetrahedron Lett. 1989;30:2263–2264. [Google Scholar]
- 642.Schreiner E., Zbiral E., Kleineidam R.G., Schauer R. 2,3-Didehydro-2-deoxysialic Acids Structurally Varied at C-5 and Their Behaviour Towards the Sialidase From Vibrio cholerae. Carbohydr. Res. 1991;216:61–66. doi: 10.1016/0008-6215(92)84150-q. [DOI] [PubMed] [Google Scholar]
- 643.Ogura H. In: Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry. Ogura H., Hasegawa A., Suami T., editors. Kodansha Ltd.; Tokyo, Japan: 1992. Sialic Acid Derivatives as Glycolipids; pp. 282–303. [Google Scholar]
- 644.Asressu K.H., Wang C.-C. Concise Synthesis of 2,7-Anhydrosialic Acid Derivatives and Its Application. Carbohydr. Res. 2017;453–454:44–53. doi: 10.1016/j.carres.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 645.Khorlin A.Y., Privalova I.M. Synthesis of N-Acetylneuraminic Acid 8-Methyl Ether. Carbohydr. Res. 1970;13:373–377. [Google Scholar]
- 646.Colombo R., Anastasia M., Rota P., Allevi P. The First Synthesis of N-Acetylneuraminic Acid 1,7-Lactone. Chem. Commun. (Cambridge, U. K.) 2008:5517–5519. doi: 10.1039/b810447f. [DOI] [PubMed] [Google Scholar]
- 647.Allevi P., Rota P., Scaringi R., Colombo R., Anastasia M. Chemoselective Synthesis of Sialic Acid 1,7-Lactones. J. Org. Chem. 2010;75:5542–5548. doi: 10.1021/jo100732j. [DOI] [PubMed] [Google Scholar]
- 648.Rota P., Allevi P., Anastasia L. The Sialic Acids Waltz: Novel Stereoselective Isomerization of the 1,7-Lactones of N-Acetylneuraminic Acids Into the Corresponding γ-Lactones and Back to the Free Sialic Acids. Asian J. Org. Chem. 2015;4:1315–1321. [Google Scholar]
- 649.Yu H., Chokhawala H., Karpel R., Yu H., Wu B., Zhang J., Zhang Y., Jia Q., Chen X. A Multifunctional Pasteurella multocida Sialyltransferase: A Powerful Tool for the Synthesis of Sialoside Libraries. J. Am. Chem. Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 650.Yu H., Huang S., Chokhawala H., Sun M., Zheng H., Chen X. Highly Efficient Chemoenzymatic Synthesis of Naturally Occurring and Non-Natural α-2,6-Linked Sialosides: A P. damsela α-2,6-Sialyltransferase With Extremely Flexible Donor–Substrate Specificity. Angew. Chem., Int. Ed. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Chokhawala H.A., Yu H., Chen X. High-Throughput Substrate Specificity Studies of Sialidases by Using Chemoenzymatically Synthesized Sialoside Libraries. ChemBioChem. 2007;8:194–201. doi: 10.1002/cbic.200600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Chokhawala H.A., Huang S., Lau K., Yu H., Cheng J., Thon V., Hurtado-Ziola N., Guerrero J.A., Varki A., Chen X. Combinatorial Chemoenzymatic Synthesis and High-Throughput Screening of Sialosides. ACS Chem. Biol. 2008;3:567–576. doi: 10.1021/cb800127n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Zbiral E. In: Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry. Ogura H., Hasegawa A., Suami T., editors. VCH Verlagsgesellschaft; Weinheim, Germany: 1992. Synthesis of Sialic Acid Analogs and Their Behavior Towards the Enzymes of Sialic Acid Metabolism and Hemagglutinin X-31 of Influenza A-Virus; pp. 304–339. [Google Scholar]
- 654.Brossmer R., Gross H.J. Sialic Acid Analogs and Application for Preparation of Neoglycoconjugates. Methods Enzymol. 1994;247:153–176. doi: 10.1016/s0076-6879(94)47013-8. [DOI] [PubMed] [Google Scholar]
- 655.Roy R. In: Carbohydrates in Drug Design. Witczak Z.J., Nieforth K.A., editors. Marcel Dekker; New York, NY, USA: 1997. Sialoside Mimetics as Anti-inflammatory Agents and Inhibitors of Flu Virus Infections; pp. 83–135. [Google Scholar]
- 656.Von Itzstein M., Kiefel M.J. In: Carbohydrates in Drug Design. Witczak Z.J., Nieforth K.A., editors. Marcel Dekker; New York, NY, USA: 1997. Sialic Acid Analogues as Potential Antimicrobial Agents; pp. 39–82. [Google Scholar]
- 657.Kiefel M.J., von Itzstein M. Recent Advances in the Synthesis of Sialic Acid Derivatives and Sialylmimetics as Biological Probes. Chem. Rev. 2002;102:471–490. doi: 10.1021/cr000414a. [DOI] [PubMed] [Google Scholar]
- 658.Wilson J.C., Kiefel M.J., von Itzstein M. In: Handbook of Carbohydrate Engineering. Yarema K.J., editor. Taylor & Francis CRC Press; Boca Raton, FL, USA: 2005. Sialic Acids and Sialylmimetics: Useful Chemical Probes of Sialic Acid-Recognizing Proteins; pp. 687–748. [Google Scholar]
- 659.De Meo C., Priyadarshani U. C-5 Modifications in N-Acetyl-neuraminic Acid: Scope and Limitations. Carbohydr. Res. 2008;343:1540–1552. doi: 10.1016/j.carres.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 660.Cao H., Chen X. In: Carbohydrate Microarrays: Methods and Protocols. Chevolot Y., editor. Vol. 808. Springer Science + Business Media; Berlin, Germany: 2012. General Consideration on Sialic Acid Chemistry; pp. 31–56. (Methods Mol. Biol). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Khorlin A.Y., Privalova I.M. Glycosidation of N-Acetylneuraminic Acid. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1968;17:217–219. [Google Scholar]
- 662.Khorlin A.Y., Privalova I.M., Bystrova I.B. The Synthesis of N-Acetylneuraminyl-(2 → 3)- and -(2 → 6)-hexoses. Carbohydr. Res. 1971;19:272–275. [Google Scholar]
- 663.Brossmer R., Friebolin H., Keilich G., Löser B., Supp M. Synthesis of Disaccharides Containing N-Acetyl-d-neuraminic Acid. Hoppe-Seyler’s Z. Physiol. Chem. 1978;359:1064. [Google Scholar]
- 664.Van der Vleugel D.J.M., Wassenburg F.R., Zwikker J.W., Vliegenthart J.F.G. Synthesis of 6-O-(5-Acetamido-3,5-dideoxy-α-d-glycero-d-galacto-2-nonulopyranosylonic Acid)-d-galactose [6-O-(N-Acetyl-α-d-neuraminyl)-d-galactose] Carbohydr. Res. 1982;104:221–233. [Google Scholar]
- 665.Van der Vleugel D.J.M., Zwikker J.W., Vliegenthart J.F.G., van Boeckel S.A.A., van Boom J.H. Synthesis of 2-Acetamido-6-O-(5-acetamido-3,5-dideoxy-β-d-glycero-d-galacto-2-nonulopyranosylonic acid)-2-deoxy-d-glucose [2-Acetamido-6-O-(N-acetyl-β-d-neuraminyl)-2-deoxy-d-glucose] Carbohydr. Res. 1982;105:19–31. [Google Scholar]
- 666.Okamoto K., Goto T. Glycosidation of Sialic Acid. Tetrahedron. 1990;46:5835–5857. [Google Scholar]
- 667.DeNinno M.P. The Synthesis and Glycosidation of N-Acetylneuraminic Acid. Synthesis. 1991:583–593. [Google Scholar]
- 668.Hasegawa A., Kiso M. In: Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry. Ogura H., Hasegawa A., Suami T., editors. Kodansha Ltd.; Tokyo, Japan: 1992. Systematic Synthesis of Gangliosides Toward the Elucidation and Biomedical Application of Their Biological Functions; pp. 243–266. [Google Scholar]
- 669.Boons G.-J., Demchenko A.V. Recent Advances in O-Sialylation. Chem. Rev. 2000;100:4539–4565. doi: 10.1021/cr990313g. [DOI] [PubMed] [Google Scholar]
- 670.Ress D.K., Linhardt R.J. Sialic Acid Donors: Chemical Synthesis and Glycosylation. Curr. Org. Synth. 2004;1:31–46. [Google Scholar]
- 671.De Meo C., Boons G.-J., Demchenko A.V. In: Comprehensive Glycoscience—From Chemistry to Systems Biology. Kamerling J.P., Boons G.-J., Lee Y.C., Suzuki A., Taniguchi N., Voragen A.G.J., editors. Vol. 1. Elsevier Ltd.; Oxford, UK: 2007. Synthesis of Glycosides of Sialic Acid; pp. 583–604. [Google Scholar]
- 672.Adak A.K., Yu C.-C., Liang C.-F., Lin C.-C. Synthesis of Sialic Acid-Containing Saccharides. Curr. Opin. Chem. Biol. 2013;17:1030–1038. doi: 10.1016/j.cbpa.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 673.Ichikawa Y., Look G.C., Wong C.-H. Enzyme-Catalyzed Oligosaccharide Synthesis. Anal. Biochem. 1992;202:215–238. doi: 10.1016/0003-2697(92)90099-s. [DOI] [PubMed] [Google Scholar]
- 674.Taniguchi N., Honke K., Fukuda M., editors. Handbook of Glycosyltransferases and Related Genes—Sialyltransferases (Multiauthors) Springer-Verlag; Tokyo, Japan: 2002. pp. 265–356. [Google Scholar]
- 675.Cheng J., Huang S., Yu H., Li Y., Lau K., Chen X. Trans-Sialidase Activity of Photobacterium damsela α2,6-Sialyltransferase and Its Application in the Synthesis of Sialosides. Glycobiology. 2010;20:260–268. doi: 10.1093/glycob/cwp172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Kim S., Oh D.-B., Kang H.A., Kwon O. Features and Applications of Bacterial Sialidases. Appl. Microbiol. Biotechnol. 2011;91:1–15. doi: 10.1007/s00253-011-3307-2. [DOI] [PubMed] [Google Scholar]
- 677.Knirel Y.A., Kochetkov N.K. 2,3-Diamino-2,3-dideoxyuronic and 5,7-Diamino-3,5,7,9-tetradeoxynonulosonic Acids: New Components of Bacterial Polysaccharides. FEMS Microbiol. Rev. 1987;46:381–385. [Google Scholar]
- 678.Knirel Y.A., Kochetkov N.K. The Structure of the Lipopolysaccharides of Gram-Negative Bacteria—III. The Structure of the O-Antigens. Biochemistry (Moscow) 1994;59:1325–1383. [Google Scholar]
- 679.Knirel Y.A., Shashkov A.S., Tsvetkov Y.E., Jansson P.-E., Zähringer U. 5,7-Diamino-3,5,7,9-tetradeoxynon-2-ulosonic Acids in Bacterial Glycopolymers: Chemistry and Biochemistry. Adv. Carbohydr. Chem. Biochem. 2003;58:371–417. doi: 10.1016/s0065-2318(03)58007-6. [DOI] [PubMed] [Google Scholar]
- 680.Kandiba L., Eichler J. Analysis of Putative Nonulosonic Acid Biosynthesis Pathways in Archaea Reveals a Complex Evolutionary History. FEMS Microbiol. Lett. 2013;345:110–120. doi: 10.1111/1574-6968.12193. [DOI] [PubMed] [Google Scholar]
- 681.Zunk M., Kiefel M.J. The Occurrence and Biological Significance of the α-Keto-Sugars Pseudaminic Acid and Legionaminic Acid Within Pathogenic Bacteria. RSC Adv. 2014;4:3413–3421. [Google Scholar]
- 682.Knirel Y.A., Vinogradov E.V., L’vov V.L., Kocharova N.A., Shashkov A.S., Dmitriev B.A., Kochetkov N.K. Sialic Acids of a New Type From the Lipopolysaccharides of Pseudomonas aeruginosa and Shigella boydii. Carbohydr. Res. 1984;133:C5–C8. doi: 10.1016/0008-6215(84)85213-1. [DOI] [PubMed] [Google Scholar]
- 683.Tsvetkov Y.E., Shashkov A.S., Knirel Y.A., Backinowsky L.V., Zähringer U. Synthesis of 5,7-Diacetamido-3,5,7,9-tetradeoxy-l-glycero-d-galacto- and -l-glycero-d-talo-nonulosonic Acids, Putative Components of Bacterial Lipopolysaccharides. Mendeleev Commun. 2000;10:90–91. [Google Scholar]
- 684.Tsvetkov Y.E., Shashkov A.S., Knirel Y.A., Zähringer U. Synthesis and Identification in Bacterial Lipopolysaccharides of 5,7-Diacetamido-3,5,7,9-tetradeoxy-d-glycero-d-galacto- and -d-glycero-d-talo-non-2-ulosonic Acids. Carbohydr. Res. 2001;331:233–237. doi: 10.1016/s0008-6215(01)00041-6. [DOI] [PubMed] [Google Scholar]
- 685.Tsvetkov Y.E., Shashkov A.S., Knirel Y.A., Zähringer U. Synthesis and NMR Spectroscopy of Nine Stereoisomeric 5,7-Diacetamido-3,5,7,9-tetradeoxynon-2-ulosonic Acids. Carbohydr. Res. 2001;335:221–243. doi: 10.1016/s0008-6215(01)00235-x. [DOI] [PubMed] [Google Scholar]
- 686.Kenyon J.J., Marzaioli A.M., De Castro C., Hall R.M. 5,7-Di-N-acetyl-Acinetaminic Acid: A Novel Non-2-ulosonic Acid Found in the Capsule of an Acinetobacter baumannii Isolate. Glycobiology. 2015;25:644–654. doi: 10.1093/glycob/cwv007. [DOI] [PubMed] [Google Scholar]
- 687.Kenyon J.J., Notaro A., Hsu L.Y., De Castro C., Hall R.M. 5,7-Di-N-Acetyl-8-epiacinetaminic Acid: A New Non-2-ulosonic Acid Found in the K73 Capsule Produced by an Acinetobacter baumannii Isolate From Singapore. Sci. Rep. 2017;7:11357. doi: 10.1038/s41598-017-11166-4. (1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Staaf M., Weintraub A., Widmalm G. Structure Determination of the O-Antigenic Polysaccharide From the Enteroinvasive Escherichia coli O136. Eur. J. Biochem. 1999;263:656–661. doi: 10.1046/j.1432-1327.1999.00531.x. [DOI] [PubMed] [Google Scholar]
- 689.Thibault P., Logan S.M., Kelly J.F., Brisson J.-R., Ewing C.P., Trust T.J., Guerry P. Identification of the Carbohydrate Moieties and Glycosylation Motifs in Campylobacter jejuni Flagellin. J. Biol. Chem. 2001;276:34862–34870. doi: 10.1074/jbc.M104529200. [DOI] [PubMed] [Google Scholar]
- 690.Schirm M., Schoenhofen I.C., Logan S.M., Waldron K.C., Thibault P. Identification of Unusual Bacterial Glycosylation by Tandem Mass Spectrometry Analyses of Intact Proteins. Anal Chem. 2005;77:7774–7782. doi: 10.1021/ac051316y. [DOI] [PubMed] [Google Scholar]
- 691.Kenne L., Lindberg B., Schweda E., Gustafsson B., Holme T. Structural Studies of the O-Antigen From Vibrio cholerae O:2. Carbohydr. Res. 1988;180:285–294. doi: 10.1016/0008-6215(86)80017-9. [DOI] [PubMed] [Google Scholar]
- 692.Hitchen P., Brzostek J., Panico M., Butler J.A., Moris H.R., Dell A., Linton D. Modification of the Campylobacter jejuni Flagellin Glycan by the Product of the Cj1295 Homopolymeric-Tract-Containing Gene. Microbiology. 2010;156:1953–1962. doi: 10.1099/mic.0.038091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Knirel Y.A., Kocharova N.A., Shashkov A.S., Kochetkov N.K. The Structure of the Pseudomonas aeruginosa Immunotype 6 O-Antigen: Isolation and Identification of 5-Acetamido-3,5,7,9-tetradeoxy-7-formamido-l-glycero-l-manno-nonulosonic Acid. Carbohydr. Res. 1986;145:C1–C4. doi: 10.1016/s0008-6215(00)90444-0. [DOI] [PubMed] [Google Scholar]
- 694.Vinogradov E., Wilde C., Anderson E.M., Nakhamchik A., Lam J.S., Rowe-Magnus D.A. Structure of the Lipopolysaccharide Core of Vibrio vulnificus Type Strain 27562. Carbohydr. Res. 2009;344:484–490. doi: 10.1016/j.carres.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 695.Knirel Y.A., Vinogradov E.V., Shashkov A.S., Kochetkov N.K., L’vov V., Dmitriev B.A. Identification of 5-Acetamido-3,5,7,9-tetradeoxy-7-[(R)-3-hydroxybutyramido]-l-glycero-l-manno-nonulosonic Acid as a Component of Bacterial Polysaccharides. Carbohydr. Res. 1985;141:C1–C3. doi: 10.1016/s0008-6215(00)90469-5. [DOI] [PubMed] [Google Scholar]
- 696.Gil-Serrano A.M., Rodríguez-Carvajal M.A., Tejero-Mateo P., Espartero J.L., Menendez M., Corzo J., Ruiz-Sainz J.E., Buendía-Clavería A.M. Structural Determination of a 5-Acetamido-3,5,7,9-tetradeoxy-7-(3-hydroxybutyramido)-l-glycero-l-manno-nonulosonic Acid-Containing Homopolysaccharide Isolated From Sinorhizobium fredii HH103. Biochem. J. 1999;342:527–535. [PMC free article] [PubMed] [Google Scholar]
- 697.Knirel Y.A., Vinogradov E.V., Shashkov A.S., Dmitriev B.A., Kochetkov N.K., Stanislavsky E.S., Mashilova G.M. Somatic Antigens of Pseudomonas aeruginosa—The Structure of O-Specific Polysaccharide Chains of P. aeruginosa O10 (Lányi) Lipopolysaccharides. Eur. J. Biochem. 1986;157:129–138. doi: 10.1111/j.1432-1033.1986.tb09648.x. [DOI] [PubMed] [Google Scholar]
- 698.Shashkov A.S., Tul’skaya E.M., Streshinskaya G.M., Senchenkova S.N., Avtukh A.N., Evtushenko L.I. New Cell Wall Glycopolymers of the Representatives of the Genus Kribbella. Carbohydr. Res. 2009;344:2255–2262. doi: 10.1016/j.carres.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 699.McNally D.J., Hui J.P.M., Aubry A.J., Mui K.K.K., Guerry P., Brisson J.-R., Logan S.M., Soo E.C. Functional Characterization of the Flagellar Glycosylation Locus in Campylobacter jejuni 81-176 Using a Focused Metabolomics Approach. J. Biol. Chem. 2006;281:18489–18498. doi: 10.1074/jbc.M603777200. [DOI] [PubMed] [Google Scholar]
- 700.Logan S.M., Hui J.P.M., Vinogradov E., Aubry A.J., Melanson J.E., Kelly J.F., Nothaft H., Soo E.C. Identification of Novel Carbohydrate Modifications on Campylobacter jejuni 11168 Flagellin Using Metabolomics-Based Approaches. FEBS J. 2009;276:1014–1023. doi: 10.1111/j.1742-4658.2008.06840.x. [DOI] [PubMed] [Google Scholar]
- 701.Reuhs B.L., Geller D.P., Kim J.S., Fox J.E., Kolli V.S.K., Pueppke S.G. Sinorhizobium fredii and Sinorhizobium meliloti Produce Structurally Conserved Lipopolysaccharides and Strain-Specific K Antigens. Appl. Environ. Microbiol. 1998;64:4930–4938. doi: 10.1128/aem.64.12.4930-4938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702.Knirel Y.A., Kocharova N.A., Shashkov A.S., Dmitriev B.A., Kochetkov N.K., Stanislavsky E.S., Mashilova G.M. Somatic Antigens of Pseudomonas aeruginosa—The Structure of O-Specific Polysaccharide Chains of the Lipopolysaccharides From P. aeruginosa O5 (Lányi) and Immunotype 6 (Fisher) Eur. J. Biochem. 1987;163:639–652. doi: 10.1111/j.1432-1033.1987.tb10913.x. [DOI] [PubMed] [Google Scholar]
- 703.Castric P., Cassels F.J., Carlson R.W. Structural Characterization of the Pseudomonas aeruginosa 1244 Pilin Glycan. J. Biol. Chem. 2001;276 doi: 10.1074/jbc.M102685200. 26479–26485/36058. [DOI] [PubMed] [Google Scholar]
- 704.Hashii N., Isshiki Y., Iguchi T., Hisatsune K., Kondo S. Structure and Serological Characterization of 5,7-Diamino-3,5,7,9-tetradeoxy-non-2-ulosonic Acid Isolated From Lipopolysaccharides of Vibrio parahaemolyticus O2 and O-Untypable Strain KX-V212. Carbohydr. Res. 2003;338:1055–1062. doi: 10.1016/s0008-6215(03)00077-6. [DOI] [PubMed] [Google Scholar]
- 705.McNally D.J., Aubry A.J., Hui J.P.M., Khieu N.H., Whitfield D., Ewing C.P., Guerry P., Brisson J.-R., Logan S.M., Soo E.C. Targeted Metabolomics Analysis of Campylobacter coli VC167 Reveals Legionaminic Acid Derivatives as Novel Flagellar Glycans. J. Biol. Chem. 2007;282:14463–14475. doi: 10.1074/jbc.M611027200. [DOI] [PubMed] [Google Scholar]
- 706.Kooistra O., Lüneberg E., Lindner B., Knirel Y.A., Frosch M., Zähringer U. Complex O-Acetylation in Legionella pneumophila Serogroup 1 Lipopolysaccharide— Evidence for Two Genes Involved in 8-O-Acetylation of Legionaminic Acid. Biochemistry. 2001;40:7630–7640. doi: 10.1021/bi002946r. [DOI] [PubMed] [Google Scholar]
- 707.Kooistra O., Lüneberg E., Knirel Y.A., Frosch M., Zähringer U. N-Methylation in Polylegionaminic Acid is Associated With the Phase-Variable Epitope of Legionella pneumophila Serogroup 1 Lipopolysaccharide—Identification of 5-(N,N-Dimethylacetimidoyl)amino- and 5-Acetimidoyl(N-methyl)amino-7-acetamido-3,5,7,9-tetradeoxynon-2-ulosonic Acid in the O-Chain Polysaccharide. Eur. J. Biochem. 2002;269:560–572. doi: 10.1046/j.0014-2956.2001.02683.x. [DOI] [PubMed] [Google Scholar]
- 708.Li X., Perepelov A.V., Wang Q., Senchenkova S.N., Liu B., Shevelev S.D., Guo X., Shashkov A.S., Chen W., Wang L., Knirel Y.A. Structural and Genetic Characterization of the O-Antigen of Escherichia coli O161 Containing a Derivative of a Higher Acidic Diamino Sugar, Legionaminic Acid. Carbohydr. Res. 2010;345:1581–1587. doi: 10.1016/j.carres.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 709.Kandiba L., Aitio O., Helin J., Guan Z., Permi P., Bamford D.H., Eichler J., Roine E. Diversity in Prokaryotic Glycosylation: An Archaeal-Derived N-Linked Glycan Contains Legionaminic Acid. Mol. Microbiol. 2012;84:578–593. doi: 10.1111/j.1365-2958.2012.08045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Twine S.M., Paul C.J., Vinogradov E., McNally D.J., Brisson J.-R., Mullen J.A., McMullin D.R., Jarrell H.C., Austin J.W., Kelly J.F., Logan S.M. Flagellar Glycosylation in Clostridium botulinum. FEBS J. 2008;275:4428–4444. doi: 10.1111/j.1742-4658.2008.06589.x. [DOI] [PubMed] [Google Scholar]
- 711.Nazarenko E.L., Perepelov A.V., Shevchenko L.S., Daeva E.D., Ivanova E.P., Shashkov A.S., Widmalm G. Structure of the O-Specific Polysaccharide From Shewanella japonica KMM 3601 Containing 5,7-Diacetamido-3,5,7,9-tetradeoxy-d-glycero-d-talo-non-2-ulosonic Acid. Biochemistry (Moscow) 2011;76:791–796. doi: 10.1134/S0006297911070091. [DOI] [PubMed] [Google Scholar]
- 712.Knirel Y.A., Senchenkova S.N., Kocharova N.A., Shashkov A.S., Helbig J.H., Zähringer U. Identification of a Homopolymer of 5-Acetamidino-7-acetamido-3,5,7,9-tetradeoxy-d-glycero-d-talo-nonulosonic Acid in the Lipopolysaccharides of Legionella pneumophila Non-1 Serogroups. Biochemistry (Moscow) 2001;66:1035–1041. doi: 10.1023/a:1012334012605. [DOI] [PubMed] [Google Scholar]
- 713.Knirel Y.A., Helbig J.H., Zähringer U. Structure of a Decasaccharide Isolated by Mild Acid Degradation and Dephosphorylation of the Lipopolysaccharide of Pseudomonas fluorescens Strain ATCC 49271. Carbohydr. Res. 1996;283:129–139. doi: 10.1016/0008-6215(95)00401-7. [DOI] [PubMed] [Google Scholar]
- 714.Edebrink P., Jansson P.-E., Bøgwald J., Hoffman J. Structural Studies of the Vibrio salmonicida Lipopolysaccharide. Carbohydr. Res. 1996;287:225–245. doi: 10.1016/0008-6215(96)00076-6. [DOI] [PubMed] [Google Scholar]
- 715.Knirel Y.A., Senchenkova S.N., Shashkov A.S., Shevelev S.D., Perepelov A.V., Liu B., Feng L., Wang L. First Isolation and Identification of a Di-N-acyl Derivative of 5,7-Diamino-3,5,7,9-tetradeoxy-l-glycero-d-galacto-non-2-ulosonic Acid (8-Epilegionaminic Acid) Adv. Sci. Lett. 2009;2:384–387. [Google Scholar]
- 716.Perepelov A.V., Wang Q., Liu B., Senchenkova S.N., Feng L., Shashkov A.S., Wang L., Knirel Y.A. Structure of the O-Polysaccharide of Escherichia coli O61, Another E. coli O-Antigen That Contains 5,7-Diacetamido-3,5,7,9-tetradeoxy-l-glycero-d-galacto-non-2-ulosonic (Di-N-acetyl-8-epilegionaminic) Acid. J. Carbohydr. Chem. 2009;28:463–472. [Google Scholar]
- 717.Kilcoyne M., Shashkov A.S., Senchenkova S.A., Knirel Y.A., Vinogradov E.V., Radziejewska-Lebrecht J., Galimska-Stypa R., Savage A.V. Structural Investigation of the O-Specific Polysaccharides of Morganella morganii Consisting of Two Higher Sugars. Carbohydr. Res. 2002;337:1697–1702. doi: 10.1016/s0008-6215(02)00181-7. [DOI] [PubMed] [Google Scholar]
- 718.Corsaro M.M., Evidente A., Lanzetta R., Lavermicocca P., Parrilli M., Ummarino S. 5,7-Diamino-5,7,9-trideoxynon-2-ulosonic Acid: A Novel Sugar From a Phytopathogenic Pseudomonas Lipopolysaccharide. Carbohydr. Res. 2002;337:955–959. doi: 10.1016/s0008-6215(02)00077-0. [DOI] [PubMed] [Google Scholar]
- 719.Vinogradov E., MacLean L.L., Crump E.M., Perry M.B., Kay W.W. Structure of the Polysaccharide Chain of the Lipopolysaccharide From Flexibacter maritimus. Eur. J. Biochem. 2003;270:1810–1815. doi: 10.1046/j.1432-1033.2003.03543.x. [DOI] [PubMed] [Google Scholar]
- 720.Vinogradov E., St. Michael F., Cox A.D. The Structure of the LPS O-Chain of Fusobacterium nucleatum Strain 25586 Containing Two Novel Monosaccharides, 2-Acetamido-2,6-dideoxy-l-altrose and a 5-Acetimidoylamino-3,5,9-trideoxy-d-gluco-non-2-ulosonic Acid. Carbohydr. Res. 2017;440–441:10–15. doi: 10.1016/j.carres.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 721.Mazumder K., Choudhury B.P., Nair G.B., Sen A.K. Identification of a Novel Sugar 5,7-Diacetamido-8-amino-3,5,7,8,9-pentadeoxy-d-glycero-d-galacto-non-2-ulosonic Acid Present in the Lipopolysaccharide of Vibrio parahaemolyticus O3:K6. Glycoconjugate J. 2008;25:345–354. doi: 10.1007/s10719-007-9080-x. [DOI] [PubMed] [Google Scholar]
- 722.Nazarenko E.L., Shashkov A.S., Knirel Y.A., Ivanova E.P., Ovodov Y.S. Unusual Acidic Monosaccharides Forming Components of the O-Specific Polysaccharides of Bacteria of the Genus Vibrio. Bioorg. Khim. 1990;16:1426–1429. [PubMed] [Google Scholar]
- 723.Haseley S.R., Wilkinson S.G. Structural Studies of the Putative O-Specific Polysaccharide of Acinetobacter baumannii O24 Containing 5,7-Diamino-3,5,7,9-tetradeoxy-l-glycero-d-galacto-nonulosonic Acid. Eur. J. Biochem. 1997;250:617–623. doi: 10.1111/j.1432-1033.1997.0617a.x. [DOI] [PubMed] [Google Scholar]
- 724.Knirel Y.A., Rietschel E.T., Marre R., Zähringer U. The Structure of the O-Specific Chain of Legionella pneumophila Serogroup 1 Lipopolysaccharide. Eur. J. Biochem. 1994;221:239–245. doi: 10.1111/j.1432-1033.1994.tb18734.x. [DOI] [PubMed] [Google Scholar]
- 725.Knirel Y.A., Grosskurth H., Helbig J.H., Zähringer U. Structures of Decasaccharide and Tridecasaccharide Tetraphosphates Isolated by Strong Alkaline Degradation of O-Deacylated Lipopolysaccharide of Pseudomonas fluorescens Strain ATCC 49271. Carbohydr. Res. 1995;279:215–226. doi: 10.1016/0008-6215(95)00274-x. [DOI] [PubMed] [Google Scholar]
- 726.Helbig J.H., Lück P.C., Knirel Y.A., Witzleb W., Zähringer U. Molecular Characterization of a Virulence-Associated Epitope on the Lipopolysaccharide of Legionella pneumophila Serogroup 1. Epidemiol. Infect. 1995;115:71–78. doi: 10.1017/s0950268800058131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Knirel Y.A., Moll H., Helbig J.H., Zähringer U. Chemical Characterization of a New 5,7-Diamino-3,5,7,9-tetradeoxynonulosonic Acid Released by Mild Acid Hydrolysis of the Legionella pneumophila Serogroup 1 Lipopolysaccharide. Carbohydr. Res. 1997;304:77–79. doi: 10.1016/s0008-6215(97)00211-5. [DOI] [PubMed] [Google Scholar]
- 728.Knirel Y.A., Vinogradov E.V., Shashkov A.S., Dmitriev B.A., Kochetkov N.K., Stanislavsky E.S., Mashilova G.M. Somatic Antigens of Pseudomonas aeruginosa—The Structure of the O-Specific Polysaccharide Chain of the Lipopolysaccharide From P. aeruginosa O13 (Lányi) Eur. J. Biochem. 1987;163:627–637. doi: 10.1111/j.1432-1033.1987.tb10912.x. [DOI] [PubMed] [Google Scholar]
- 729.Vinogradov E.V., Shashkov A.S., Knirel Y.A., Kochetkov N.K., Dabrowski J., Grosskurth H., Stanislavsky E.S., Kholodkova E.V. The Structure of the O-Specific Polysaccharide Chain of the Lipopolysaccharide of Salmonella arizonae O61. Carbohydr. Res. 1992;231:1–11. doi: 10.1016/0008-6215(92)84002-a. [DOI] [PubMed] [Google Scholar]
- 730.Beynon L.M., Richards J.C., Perry M.B. The Structure of the Lipopolysaccharide O Antigen From Yersinia ruckeri Serotype 01. Carbohydr. Res. 1994;256:303–317. doi: 10.1016/0008-6215(94)84215-9. [DOI] [PubMed] [Google Scholar]
- 731.Schnaar R.L., Lee Y.C. Discoveries of the Structures of Sialic Acid and CMP-Sialic Acid (1957-1960): A Letter From Saul Roseman. Glycobiology. 2017;27:513–517. doi: 10.1093/glycob/cwx015. [DOI] [PubMed] [Google Scholar]
- 732.Comb D.G., Roseman S. Enzymic Synthesis of N-Acetyl-d-mannosamine. Biochim. Biophys. Acta. 1958;29:653–654. doi: 10.1016/0006-3002(58)90031-3. [DOI] [PubMed] [Google Scholar]
- 733.Daniels A.D., Campeotto I., van der Kamp M.W., Bolt A.H., Trinh C.H., Phillips S.E.V., Pearson A.R., Nelson A., Mulholland A.J., Berry A. Reaction Mechanism of N-Acetylneuraminic Acid Lyase Revealed by a Combination of Crystallography, QM/MM Simulation, and Mutagenesis. ACS Chem. Biol. 2014;9:1025–1032. doi: 10.1021/cb500067z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Roseman S. Metabolism of Sialic Acids and d-Mannosamine. Fed. Proc. 1962;21:1075–1083. [PubMed] [Google Scholar]
- 735.Warren L., Felsenfeld H. The Biosynthesis of Sialic Acids. J. Biol. Chem. 1962;237:1421–1431. [PubMed] [Google Scholar]
- 736.Becker C.E., Day H.G. Utilization of Glucosone and the Synthesis of Glucosamine in the Rat. J. Biol. Chem. 1953;201:795–801. [PubMed] [Google Scholar]
- 737.Spiro R.G. Studies on the Biosynthesis of Glucosamine in the Intact Rat. J. Biol. Chem. 1959;234:742–748. [PubMed] [Google Scholar]
- 738.Lowther D.A., Rogers H.J. The Relation of Glutamine to the Synthesis of Hyaluronate or Hyaluronate-Like Substances by Streptococci. Biochem. J. 1953;53:XXXIX. [PubMed] [Google Scholar]
- 739.Ghosh S., Blumenthal H.J., Davidson E., Roseman S. Glucosamine Metabolism—V. Enzymatic Synthesis of Glucosamine 6-Phosphate. J. Biol. Chem. 1960;235:1265–1273. [PubMed] [Google Scholar]
- 740.Pogell B.M., Gryder R.M. Enzymatic Synthesis of Glucosamine 6-Phosphate in Rat Liver. J. Biol. Chem. 1957;228:701–712. [PubMed] [Google Scholar]
- 741.Davidson E.A., Blumenthal H.J., Roseman S. Glucosamine Metabolism—II. Studies on Glucosamine 6-Phosphate N-Acetylase. J. Biol. Chem. 1957;226:125–133. [PubMed] [Google Scholar]
- 742.Leloir L.F., Cardini C.E. Enzymes Acting on Glucosamine Phosphates. Biochim. Biophys. Acta. 1956;20:33–42. doi: 10.1016/0006-3002(56)90259-1. [DOI] [PubMed] [Google Scholar]
- 743.Reissig J.L. Phosphoacetylglucosamine Mutase of Neurospora. J. Biol. Chem. 1956;219:753–767. [PubMed] [Google Scholar]
- 744.Carlson D.M. Phosphoacetylglucosamine Mutase From Pig Submaxillary Gland. Methods Enzymol. 1966;8:179–182. [Google Scholar]
- 745.Smith E.E.B., Munch-Petersen A., Mills G.T. Uridyl Transferases and the Formation of Uridine Triphosphate: Pyrophosphorolysis of Uridine Diphosphoglucose and ‘UDPX’ by a Rat Liver Nuclear Fraction. Nature. 1953;172:1038–1039. doi: 10.1038/1721036a0. [DOI] [PubMed] [Google Scholar]
- 746.Cabib E., Leloir L.F., Cardini C.E. Uridine Diphosphate Acetylglucosamine. J. Biol. Chem. 1953;203:1055–1070. [PubMed] [Google Scholar]
- 747.Glaser L., Brown D.H. The Enzymatic Synthesis In Vitro of Hyaluronic Acid Chains. Proc. Natl. Acad. Sci. U. S. A. 1955;41:253–260. doi: 10.1073/pnas.41.5.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748.Cardini C.E., Leloir L.F. Enzymatic Formation of Acetylgalactosamine. J. Biol. Chem. 1957;225:317–324. [PubMed] [Google Scholar]
- 749.Spivak C.T., Roseman S. UDP-N-Acetyl-d-glucosamine 2′-Epimerase. Methods Enzymol. 1966;9:612–615. [Google Scholar]
- 750.Ghosh S., Roseman S. Enzymatic Phosphorylation of N-Acetyl-d-mannosamine. Proc. Natl. Acad. Sci. U. S. A. 1961;47:955–958. doi: 10.1073/pnas.47.7.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.McGuire E.J. In: Biological Roles of Sialic Acid. Rosenberg A., Schengrund C.-L., editors. Plenum Press; New York, NY, USA: 1976. Anabolic Reactions Involving Sialic Acids; pp. 123–158. [Google Scholar]
- 752.Hinderlich S., Stäsche R., Zeitler R., Reutter W. A Bifunctional Enzyme Catalyzes the First Two Steps in N-Acetylneuraminic Acid Biosynthesis of Rat Liver—Purification and Characterization of UDP-N-Acetylglucosamine 2-Epimerase/N-Acetylmannosamine Kinase. J. Biol. Chem. 1997;272:24313–24318. doi: 10.1074/jbc.272.39.24313. [DOI] [PubMed] [Google Scholar]
- 753.Stäsche R., Hinderlich S., Weise C., Effertz K., Lucka L., Moormann P., Reutter W. A Bifunctional Enzyme Catalyzes the First Two Steps in N-Acetylneuraminic Acid Biosynthesis of Rat Liver—Molecular Cloning and Functional Expression of UDP-N-acetyl-glucosamine 2-Epimerase/N-Acetylmannosamine Kinase. J. Biol. Chem. 1997;272:24319–24324. doi: 10.1074/jbc.272.39.24319. [DOI] [PubMed] [Google Scholar]
- 754.Effertz K., Hinderlich S., Reutter W. Selective Loss of Either the Epimerase or Kinase Activity of UDP-N-Acetylglucosamine 2-Epimerase/N-Acetylmannosamine Kinase Due to Site-Directed Mutagenesis Based on Sequence Alignments. J. Biol. Chem. 1999;274:28771–28778. doi: 10.1074/jbc.274.40.28771. [DOI] [PubMed] [Google Scholar]
- 755.De Mendoza A., Ruiz-Trillo I. The Mysterious Evolutionary Origin for the GNE Gene and the Root of Bilateria. Mol. Biol. Evol. 2011;28:2987–2991. doi: 10.1093/molbev/msr142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Hinderlich S., Neuenschwander M., Wratil P.R., Oder A., Lisurek M., Nguyen L.D., von Kries J.P., Hackenberger C.P.R. Small Molecules Targeting Human N-Acetylmannosamine Kinase. ChemBioChem. 2017;18:1279–1285. doi: 10.1002/cbic.201700066. [DOI] [PubMed] [Google Scholar]
- 757.Wratil P.R., Rigol S., Solecka B., Kohla G., Kannicht C., Reutter W., Giannis A., Nguyen L.D. A Novel Approach to Decrease Sialic Acid Expression in Cells by a C-3-Modified N-Acetylmannosamine. J. Biol. Chem. 2014;289:32056–32063. doi: 10.1074/jbc.M114.608398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758.Jourdian G.W., Swanson A.L., Watson D., Roseman S. Isolation of Sialic Acid 9-Phosphatase From Human Erythrocytes. J. Biol. Chem. 1964;239:PC2714–PC2716. [PubMed] [Google Scholar]
- 759.Blacklow R.S., Warren L. Biosynthesis of Sialic Acids by Neisseria meningitidis. J. Biol. Chem. 1962;237:3520–3526. [PubMed] [Google Scholar]
- 760.Vimr E.R., Kalivoda K.A., Deszo E.L., Steenbergen S.M. Diversity of Microbial Sialic Acid Metabolism. Microbiol. Mol. Biol. Rev. 2004;68:132–153. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Comb D.G., Watson D.R., Roseman S. The Sialic Acids—IX. Isolation of Cytidine 5′-Monophospho-N-acetylneuraminic Acid From Escherichia coli K-235. J. Biol. Chem. 1966;241:5637–5642. [PubMed] [Google Scholar]
- 762.Haverkamp J., Spoormaker T., Dorland L., Vliegenthart J.F.G., Schauer R. Determination of the β-Anomeric Configuration of Cytidine 5′-Monophospho-N-acetylneuraminic Acid by 13C NMR Spectroscopy. J. Am. Chem. Soc. 1979;101:4851–4853. [Google Scholar]
- 763.Ambrose M.G., Freese S.J., Reinhold M.S., Warner T.G., Vann W.F. 13C NMR Investigation of the Anomeric Specificity of CMP-N-Acetylneuraminic Acid Synthetase From Escherichia coli. Biochemistry. 1992;31:775–780. doi: 10.1021/bi00118a019. [DOI] [PubMed] [Google Scholar]
- 764.Warren L., Blacklow R.S. The Biosynthesis of Cytidine 5′-Monophospho-N-acetylneuraminic Acid by an Enzyme From Neisseria meningitidis. J. Biol. Chem. 1962;237:3527–3534. [PubMed] [Google Scholar]
- 765.Roseman S. Enzymatic Synthesis of Cytidine 5′-Monophospho-sialic Acids. Proc. Natl. Acad. Sci. U. S. A. 1962;48:437–441. doi: 10.1073/pnas.48.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Spiro M.J., Spiro R.G. Glycoprotein Biosynthesis: Studies on Thyroglobulin—Thyroid Sialyltransferase. J. Biol. Chem. 1968;243:6520–6528. [PubMed] [Google Scholar]
- 767.Kean E.L., Roseman S. The Sialic Acids—X. Purification and Properties of Cytidine 5′-Monophosphosialic Acid Synthetase. J. Biol. Chem. 1966;241:5643–5650. [PubMed] [Google Scholar]
- 768.Kean E.L., Münster-Kühnel A.K., Gerardy-Schahn R. CMP-Sialic Acid Synthetase of the Nucleus. Biochim. Biophys. Acta. 2004;1673:56–65. doi: 10.1016/j.bbagen.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 769.Kean E.L. Nuclear Cytidine 5′-Monophosphosialic Acid Synthetase. J. Biol. Chem. 1970;245:2301–2308. [PubMed] [Google Scholar]
- 770.Carey D.J., Sommers L.W., Hirschberg C.B. CMP-N-Acetylneuraminic Acid: Isolation From and Penetration Into Mouse Liver Microsomes. Cell. 1980;19:597–605. doi: 10.1016/s0092-8674(80)80036-5. [DOI] [PubMed] [Google Scholar]
- 771.Eckhardt M., Mühlenhoff M., Bethe A., Gerardy-Schahn R. Expression Cloning of the Golgi CMP-Sialic Acid Transporter. Proc. Natl. Acad. Sci. U. S. A. 1996;93:7572–7576. doi: 10.1073/pnas.93.15.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Kean E.L., Bighouse K.J. Cytidine 5′-Monophosphosialic Acid Hydrolase—Subcellular Location and Properties. J. Biol. Chem. 1974;249:7813–7823. [PubMed] [Google Scholar]
- 773.Liu J.L.-C., Shen G.-J., Ichikawa Y., Rutan J.F., Zapata G., Vann W.F., Wong C.-H. Overproduction of CMP-Sialic Acid Synthetase for Organic Synthesis. J. Am. Chem. Soc. 1992;114:3901–3910. [Google Scholar]
- 774.Hamamoto T., Takeda S., Noguchi T. Enzymatic Synthesis of Cytidine 5′-Monophospho-N-acetylneuraminic Acid. Biosci. Biotechnol. Biochem. 2005;69:1944–1950. doi: 10.1271/bbb.69.1944. [DOI] [PubMed] [Google Scholar]
- 775.Harduin-Lepers A., Mollicone R., Delannoy P., Oriol R. The Animal Sialyltransferases and Sialyltransferase-Related Genes: A Phylogenetic Approach. Glycobiology. 2005;15:805–817. doi: 10.1093/glycob/cwi063. [DOI] [PubMed] [Google Scholar]
- 776.Bhide G.P., Colley K.J. Sialylation of N-Glycans: Mechanism, Cellular Compartmentalization and Function. Histochem. Cell Biol. 2017;147:149–174. doi: 10.1007/s00418-016-1520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777.Jourdian G.W., Carlson D.M., Roseman S. The Enzymatic Synthesis of Sialyl-lactose. Biochem. Biophys. Res. Commun. 1963;10:352–358. doi: 10.1016/0006-291x(63)90537-0. [DOI] [PubMed] [Google Scholar]
- 778.Basu S., Kaufman B. Ganglioside Biosynthesis in Embryonic Chicken Brain. Fed. Proc. 1965;24:479. [Google Scholar]
- 779.Roseman S., Carlson D.M., Jourdian G.W., McGuire E.J., Kaufman B., Basu S., Bartholomew B. Animal Sialic Acid Transferases (Sialyl-transferases) Methods Enzymol. 1966;8:354–372. [Google Scholar]
- 780.Paulson J.C., Beranek W.E., Hill R.L. Purification of a Sialyltransferase From Bovine Colostrum by Affinity Chromatography on CDP-Agarose. J. Biol. Chem. 1977;252:2356–2362. [PubMed] [Google Scholar]
- 781.Paulson J.C., Rearick J.I., Hill R.L. Enzymatic Properties of β-d-Galactoside α2 → 6 Sialyltransferase From Bovine Colostrum. J. Biol. Chem. 1977;252:2363–2371. [PubMed] [Google Scholar]
- 782.Weinstein J., Lee E.U., McEntee K., Lai P.-H., Paulson J.C. Primary Structure of β-Galactoside α2,6-Sialyltransferase—Conversion of Membrane-Bound Enzyme to Soluble Forms by Cleavage of the NH2-Terminal Signal Anchor. J. Biol. Chem. 1987;262:17735–17743. [PubMed] [Google Scholar]
- 783.Ma J., Qian R., Rausa F.M., III, Colley K.J. Two Naturally Occurring α2,6-Sialyltransferase Forms With a Single Amino Acid Change in the Catalytic Domain Differ in Their Catalytic Activity and Proteolytic Processing. J. Biol. Chem. 1997;272:672–679. doi: 10.1074/jbc.272.1.672. [DOI] [PubMed] [Google Scholar]
- 784.Harduin-Lepers A., Petit D., Mollicone R., Delannoy P., Petit J.-M., Oriol R. Evolutionary History of the α2,8-Sialyltransferase (ST8Sia) Gene Family: Tandem Duplications in Early Deuterostomes Explain Most of the Diversity Found in the Vertebrate ST8Sia Genes. BMC Evol. Biol. 2008;8:258. doi: 10.1186/1471-2148-8-258. (1–18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 785.Bork K., Weidemann W., Berneck B., Kuchta M., Bennmann D., Thate A., Huber O., Gnanapragassam V.S., Horstkorte R. The Expression of Sialyltransferases Is Regulated by the Bioavailability and Biosynthesis of Sialic Acids. Gene Expression Patterns. 2017;23–24:52–58. doi: 10.1016/j.gep.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 786.Albarracin I., Lassaga F.E., Caputto R. Purification and Characterization of an Endogenous Inhibitor of the Sialyltransferase CMP-N-Acetylneuraminate:Lactosylceramide α2,6-N-Acetylneuraminyltransferase (EC 2.4.99.-) Biochem. J. 1988;254:559–565. doi: 10.1042/bj2540559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 787.Van den Eijnden D.H., Schiphorst W.E.C.M. Specificity and Inhibition of Sialyltransferases. In: Schauer R., Yamakawa T., editors. Sialic Acids; Proc. Japanese-German Symp. Sialic Acids; Kiel, Germany: Kieler Verlag Wissenschaft + Bildung; 1988. pp. 128–129. [Google Scholar]
- 788.Kleineidam R.G., Schmelter T., Schwarz R.T., Schauer R. Studies on the Inhibition of Sialyl- and Galactosyltransferase. Glycoconjugate J. 1997;14:57–66. doi: 10.1023/a:1018560931389. [DOI] [PubMed] [Google Scholar]
- 789.Büll C., Boltje T.J., Wassink M., de Graaf A.M.A., van Delft F.L., den Brok M.H., Adema G.J. Targeting Aberrant Sialylation in Cancer Cells Using a Fluorinated Sialic Acid Analog Impairs Adhesion, Migration, and In Vivo Tumor Growth. Mol. Cancer Ther. 2013;12:1935–1946. doi: 10.1158/1535-7163.MCT-13-0279. [DOI] [PubMed] [Google Scholar]
- 790.Kornfeld S., Kornfeld R., Neufeld E.F., O’Brien P.J. The Feedback Control of Sugar Nucleotide Biosynthesis in Liver. Proc. Natl. Acad. Sci. U. S. A. 1964;52:371–379. doi: 10.1073/pnas.52.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Schauer R., Reuter G., Mühlpfordt H., Andrade A.F.B., Pereira M.E.A. The Occurrence of N-Acetyl- and N-Glycoloylneuraminic Acid in Trypanosoma cruzi. Hoppe-Seyler’s Z. Physiol. Chem. 1983;364:1053–1057. doi: 10.1515/bchm2.1983.364.2.1053. [DOI] [PubMed] [Google Scholar]
- 792.Schauer R., Srinivasan G.V., Coddeville B., Zanetta J.-P., Guérardel Y. Low Incidence of N-Glycolylneuraminic Acid in Birds and Reptiles and Its Absence in the Platypus. Carbohydr. Res. 2009;344:1494–1500. doi: 10.1016/j.carres.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 793.Peri S., Kulkarni A., Feyertag F., Berninsone P.M., Alvarez-Ponce D. Phylogenetic Distribution of CMP-Neu5Ac Hydroxylase (CMAH), the Enzyme Synthetizing the Proinflammatory Human Xenoantigen Neu5Gc. Genome Biol. Evol. 2018;10:207–219. doi: 10.1093/gbe/evx251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794.Malykh Y.N., Schauer R., Shaw L. N-Glycolylneuraminic Acid in Human Tumours. Biochimie. 2001;83:623–634. doi: 10.1016/s0300-9084(01)01303-7. [DOI] [PubMed] [Google Scholar]
- 795.Hedlund M., Padler-Karavani V., Varki N.M., Varki A. Evidence for a Human-Specific Mechanism for Diet and Antibody-Mediated Inflammation in Carcinoma Progression. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18936–18941. doi: 10.1073/pnas.0803943105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 796.Uhlenbruck G., Schmitt J. Über das Vorkommen von N-Glykolyl-neuraminsäure bei Affen. Naturwissenschaften. 1965;52:163. doi: 10.1007/BF00609282. [DOI] [PubMed] [Google Scholar]
- 797.Hedlund M., Tangvoranuntakul P., Takematsu H., Long J.M., Housley G.D., Kozutsumi Y., Suzuki A., Wynshaw-Boris A., Ryan A.F., Gallo R.L., Varki N., Varki A. N-Glycolylneuraminic Acid Deficiency in Mice: Implications for Human Biology and Evolution. Mol. Cell. Biol. 2007;27:4340–4346. doi: 10.1128/MCB.00379-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798.Naito-Matsui Y., Davies L.R.L., Takematsu H., Chou H.-H., Tangvoranuntakul P., Carlin A.F., Verhagen A., Heyser C.J., Yoo S.-W., Choudhury B., Paton J.C., Paton A.W., Varki N.M., Schnaar R.L., Varki A. Physiological Exploration of the Long Term Evolutionary Selection Against Expression of N-Glycolylneuraminic Acid in the Brain. J. Biol. Chem. 2017;292:2557–2570. doi: 10.1074/jbc.M116.768531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 799.Schauer R., Stoll S., Reuter G. Differences in the Amount of N-Acetyl- and N-Glycoloyl-neuraminic Acid, as Well as O-Acylated Sialic Acids, of Fetal and Adult Bovine Tissues. Carbohydr. Res. 1991;213:353–359. doi: 10.1016/s0008-6215(00)90623-2. [DOI] [PubMed] [Google Scholar]
- 800.Ji S., Wang F., Chen Y., Yang C., Zhang P., Zhang X., Troy F.A., II., Wang B. Developmental Changes in the Level of Free and Conjugated Sialic Acids, Neu5Ac, Neu5Gc and KDN in Different Organs of Pig: A LC–MS/MS Quantitative Analyses. Glycoconjugate J. 2017;34:21–30. doi: 10.1007/s10719-016-9724-9. [DOI] [PubMed] [Google Scholar]
- 801.Faillard H., Blohm M. Synthese der N-Glykolyl-neuraminsäure. Hoppe-Seyler’s Z. Physiol. Chem. 1965;341:167–171. [PubMed] [Google Scholar]
- 802.Schoop H.J., Faillard H. Beitrag zur Biosynthese der Glykolyl-Gruppe der N-Glykolyl-neuraminsäure. Hoppe-Seyler’s Z. Physiol. Chem. 1967;348:1518–1524. [PubMed] [Google Scholar]
- 803.Da Fonseca-Wollheim F., Heesen H., Holzer H. The Oxidative Decarboxylation of Hydroxypyruvate to Glycolyl Coenzyme A. Biochem. Z. 1964;340:383–390. [PubMed] [Google Scholar]
- 804.Kent P.W., Draper P. Biosynthesis of Intestinal Mucins. Sialic Acids of Sheep Colonic Epithelial Mucin. Biochem. J. 1968;106:293–299. doi: 10.1042/bj1060293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Mårtensson E., Raal A., Svennerholm L. Sialic Acids in Blood Serum. Biochim. Biophys. Acta. 1958;30:124–129. doi: 10.1016/0006-3002(58)90248-8. [DOI] [PubMed] [Google Scholar]
- 806.Von Jagow R., Kiese M., Lenk W. Hydroxylation of Acetic Acid to Glycolic Acid in Rabbits. Biochim. Biophys. Acta. 1968;158:45–50. doi: 10.1016/0304-4165(68)90070-6. [DOI] [PubMed] [Google Scholar]
- 807.Petit J.-F., Adam A., Wietzerbin-Falszpan J. Isolation of UDP-N-glycolylmuramyl-(Ala, Glu, DAP) From Mycobacterium phlei. FEBS Lett. 1970;6:55–57. doi: 10.1016/0014-5793(70)80042-4. [DOI] [PubMed] [Google Scholar]
- 808.Schoop H.J., Schauer R. Über den Einbau von molekularem Sauerstoff bei der Biosynthese der N-Glykolylneuraminsäure durch eine Neuraminsäure-N-acetyl-Hydroxylase. Hoppe-Seyler’s Z. Physiol. Chem. 1969;350:13. [Google Scholar]
- 809.Schauer R., Schoop H.J., Faillard H. Zur Biosynthese der Glykolyl-Gruppe der N-Glykolyl-neuraminsäure—Die oxydative Umwandlung der N-Acetyl-Gruppe zur Glykolyl-Gruppe. Hoppe-Seyler’s Z. Physiol. Chem. 1968;349:645–652. [PubMed] [Google Scholar]
- 810.Schoop H.J., Schauer R., Faillard H. Zur Biosynthese der N-Glykolyl-neuraminsäure—Die oxydative Entstehung von N-Glykolyl-neuraminsäure aus N-Acetyl-neuraminsäure. Hoppe-Seyler’s Z. Physiol. Chem. 1969;350:155–162. [PubMed] [Google Scholar]
- 811.Schauer R. Biosynthese der N-Glykoloylneuraminsäure durch eine von Ascorbinsäure bzw. NADPH abhängige N-Acetyl-hydroxylierende “N-Acetylneuraminate:O2-Oxidoreduktase” in Homogenaten der Unterkieferspeicheldrüse vom Schwein. Hoppe-Seyler’s Z. Physiol. Chem. 1970;351:783–791. [PubMed] [Google Scholar]
- 812.Buscher H.-P., Casals-Stenzel J., Schauer R., Mestres-Ventura P. Biosynthesis of N-Glycolylneuraminic Acid in Porcine Submandibular Glands—Subcellular Site of Hydroxylation of N-Acetylneuraminic Acid in the Course of Glycoprotein Biosynthesis. Eur. J. Biochem. 1977;77:297–310. doi: 10.1111/j.1432-1033.1977.tb11668.x. [DOI] [PubMed] [Google Scholar]
- 813.Shaw L., Schauer R. The Biosynthesis of N-Glycoloylneuraminic Acid Occurs by Hydroxylation of the CMP-Glycoside of N-Acetylneuraminic Acid. Biol. Chem. Hoppe-Seyler. 1988;369:477–486. doi: 10.1515/bchm3.1988.369.1.477. [DOI] [PubMed] [Google Scholar]
- 814.Shaw L., Schauer R. Detection of CMP-N-Acetylneuraminic Acid Hydroxylase Activity in Fractionated Mouse Liver. Biochem. J. 1989;263:355–363. doi: 10.1042/bj2630355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Schlenzka W., Shaw L., Schneckenburger P., Schauer R. Purification and Characterization of CMP-N-Acetylneuraminic Acid Hydroxylase From Pig Submandibular Glands. Glycobiology. 1994;4:675–683. doi: 10.1093/glycob/4.5.675. [DOI] [PubMed] [Google Scholar]
- 816.Schneckenburger P., Shaw L., Schauer R. Purification, Characterization and Reconstitution of CMP-N-Acetylneuraminate Hydroxylase From Mouse Liver. Glycoconjugate J. 1994;11:194–203. doi: 10.1007/BF00731218. [DOI] [PubMed] [Google Scholar]
- 817.Kozutsumi Y., Kawano T., Yamakawa T., Suzuki A. Participation of Cytochrome b5 in CMP-N-Acetylneuraminic Acid Hydroxylation in Mouse Liver Cytosol. J. Biochem. 1990;108:704–706. doi: 10.1093/oxfordjournals.jbchem.a123268. [DOI] [PubMed] [Google Scholar]
- 818.Kawano T., Kozutsumi Y., Takematsu H., Kawasaki T., Suzuki A. Regulation of Biosynthesis of N-Glycolylneuraminic Acid-Containing Glycoconjugates: Characterization of Factors Required for NADH-Dependent Cytidine 5′Monophosphate-N-acetylneuraminic Acid Hydroxylation. Glycoconjugate J. 1993;10:109–115. doi: 10.1007/BF00731194. [DOI] [PubMed] [Google Scholar]
- 819.Shaw L., Schneckenburger P., Schlenzka W., Carlsen J., Christiansen K., Jürgensen D., Schauer R. CMP-N-Acetylneuraminic Acid Hydroxylase From Mouse Liver and Pig Submandibular Glands—Interaction With Membrane-Bound and Soluble Cytochrome b5-Dependent Electron Transport Chains. Eur. J. Biochem. 1994;219:1001–1011. doi: 10.1111/j.1432-1033.1994.tb18583.x. [DOI] [PubMed] [Google Scholar]
- 820.Shaw L., Schneckenburger P., Carlsen J., Christiansen K., Schauer R. Mouse Liver Cytidine-5′-Monosphosphate-N-acetylneuraminic Acid Hydroxylase—Catalytic Function and Regulation. Eur. J. Biochem. 1992;206:269–277. doi: 10.1111/j.1432-1033.1992.tb16925.x. [DOI] [PubMed] [Google Scholar]
- 821.Schlenzka W., Shaw L., Kelm S., Schmidt C.L., Bill E., Trautwein A.X., Lottspeich F., Schauer R. CMP-N-Acetylneuraminic Acid Hydroxylase: The First Cytosolic Rieske Iron-Sulphur Protein to Be Described in Eukarya. FEBS Lett. 1996;385:197–200. doi: 10.1016/0014-5793(96)00384-5. [DOI] [PubMed] [Google Scholar]
- 822.Kawano T., Koyama S., Takematsu H., Kozutsumi Y., Kawasaki H., Kawashima S., Kawasaki T., Suzuki A. Molecular Cloning of Cytidine Monophospho-N-acetylneuraminic Acid Hydroxylase—Regulation of Species- and Tissue-Specific Expression of N-Glycolylneuraminic Acid. J. Biol. Chem. 1995;270:16458–16463. doi: 10.1074/jbc.270.27.16458. [DOI] [PubMed] [Google Scholar]
- 823.Irie A., Koyama S., Kozutsumi Y., Kawasaki T., Suzuki A. The Molecular Basis for the Absence of N-Glycolylneuraminic Acid in Humans. J. Biol. Chem. 1998;273:15866–15871. doi: 10.1074/jbc.273.25.15866. [DOI] [PubMed] [Google Scholar]
- 824.Chou H.-H., Takematsu H., Diaz S., Iber J., Nickerson E., Wright K.L., Muchmore E.A., Nelson D.L., Warren S.T., Varki A. A Mutation in Human CMP-Sialic Acid Hydroxylase Occurred After the Homo-Pan Divergence. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T., Satta Y., Gagneux P., Varki A., Takahata N. Alu-Mediated Inactivation of the Human CMP-N-Acetylneuraminic Acid Hydroxylase Gene. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11399–11404. doi: 10.1073/pnas.191268198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Klein A., Roussel P. O-Acetylation of Sialic Acids. Biochimie. 1998;80:49–57. doi: 10.1016/s0300-9084(98)80056-4. [DOI] [PubMed] [Google Scholar]
- 826.Schauer R., Srinivasan G.V., Wipfler D., Kniep B., Schwartz-Albiez R. O-Acetylated Sialic Acids and Their Role in Immune Defense. Adv. Exp. Med. Biol. 2011;705:525–548. doi: 10.1007/978-1-4419-7877-6_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827.Sgroi D., Varki A., Braesch-Andersen S., Stamenkovic I. CD22, a B Cell-Specific Immunoglobulin Superfamily Member, Is a Sialic Acid-Binding Lectin. J. Biol. Chem. 1993;268:7011–7018. [PubMed] [Google Scholar]
- Kelm S., Pelz A., Schauer R., Filbin M.T., Tang S., de Bellard M.-E., Schnaar R.L., Mahoney J.A., Hartnell A., Bradfield P., Crocker P.R. Sialoadhesin, Myelin-Associated Glycoprotein and CD22 Define a New Family of Sialic Acid-Dependent Adhesion Molecules of the Immunoglobulin Superfamily. Curr. Biol. 1994;4:965–972. doi: 10.1016/s0960-9822(00)00220-7. [DOI] [PubMed] [Google Scholar]
- 828.Kelm S., Schauer R., Manuguerra J.-C., Gross H.-J., Crocker P.R. Modifications of Cell Surface Sialic Acids Modulate Cell Adhesion Mediated by Sialoadhesin and CD22. Glycoconjugate J. 1994;11:576–585. doi: 10.1007/BF00731309. [DOI] [PubMed] [Google Scholar]
- 829.Herrler G., Rott R., Klenk H.-D., Müller H.-P., Shukla A.K., Schauer R. The Receptor-Destroying Enzyme of Influenza C Virus Is Neuraminate-O-acetylesterase. EMBO J. 1985;4:1503–1506. doi: 10.1002/j.1460-2075.1985.tb03809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 830.Klotz F.W., Orlandi P.A., Reuter G., Cohen S.J., Haynes J.D., Schauer R., Howard R.J., Palese P., Miller L.H. Binding of Plasmodium falciparum 175-Kilodalton Erythrocyte Binding Antigen and Invasion of Murine Erythrocytes Requires N-Acetylneuraminic Acid but Not Its O-Acetylated Form. Mol. Biochem. Parasitol. 1992;51:49–54. doi: 10.1016/0166-6851(92)90199-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Schauer R. Biosynthese von N-Acetyl-O-acetylneuraminsäuren—I. Inkorporation von [14C]Acetat in Schnitte der Unterkieferspeicheldrüse von Rind und Pferd. Hoppe-Seyler’s Z. Physiol. Chem. 1970;351:595–602. [PubMed] [Google Scholar]
- 832.Schauer R. Biosynthese von N-Acetyl-O-acetylneuraminsäuren—II. Untersuchungen über Substrat und intracelluläre Lokalisation der Acetyl-Coenzyme A: N-Acetylneuraminat-7- und 8-O-Acetyltransferase vom Rind. Hoppe-Seyler’s Z. Physiol. Chem. 1970;351:749–758. [PubMed] [Google Scholar]
- 833.Tiralongo J., Schmid H., Thun R., Iwersen M., Schauer R. Characterisation of the Enzymatic 4-O-Acetylation of Sialic Acids in Microsomes From Equine Submandibular Glands. Glycoconjugate J. 2000;17:849–858. doi: 10.1023/a:1010965128335. [DOI] [PubMed] [Google Scholar]
- 834.Iwersen M., Dora H., Kohla G., Gasa S., Schauer R. Solubilisation and Properties of the Sialate-4-O-acetyltransferase From Guinea Pig Liver. Biol. Chem. 2003;384:1035–1047. doi: 10.1515/BC.2003.116. [DOI] [PubMed] [Google Scholar]
- 835.Schauer R., Wember M., Ferreira do Amaral C. Synthesis of CMP-Glycosides of Radioactive N-Acetyl-, N-Glycoloyl-, N-Acetyl-7-O-acetyl- and N-Acetyl-8-O-acetylneuraminic Acids by CMP-Sialate Synthase From Bovine Submaxillary Glands. Hoppe-Seyler’s Z. Physiol. Chem. 1972;353:883–886. doi: 10.1515/bchm2.1972.353.1.883. [DOI] [PubMed] [Google Scholar]
- 836.Schauer R., Wember M. Studies on the Substrate Specificity of Acylneuraminate Cytidylyltransferase and Sialyltransferase of Submandibular Glands From Cow, Pig and Horse. Hoppe-Seyler’s Z. Physiol. Chem. 1973;354:1405–1414. doi: 10.1515/bchm2.1973.354.2.1405. [DOI] [PubMed] [Google Scholar]
- 837.Lrhorfi L.A., Srinivasan G.V., Schauer R. Properties and Partial Purification of Sialate-O-acetyltransferase From Bovine Submandibular Glands. Biol. Chem. 2007;388:297–306. doi: 10.1515/BC.2007.033. [DOI] [PubMed] [Google Scholar]
- 838.Vandamme-Feldhaus V., Schauer R. Characterization of the Enzymatic 7-O-Acetylation of Sialic Acids and Evidence for Enzymatic O-Acetyl Migration From C-7 to C-9 in Bovine Submandibular Glands. J. Biochem. 1998;124:111–121. doi: 10.1093/oxfordjournals.jbchem.a022069. [DOI] [PubMed] [Google Scholar]
- 839.Diaz S., Higa H.H., Varki A. Glycoprotein Sialate 7(9)-O-Acetyltransferase From Rat Liver Golgi Vesicles. Methods Enzymol. 1989;179:416–422. doi: 10.1016/0076-6879(89)79141-2. [DOI] [PubMed] [Google Scholar]
- 840.Higa H.H., Butor C., Diaz S., Varki A. O-Acetylation and De-O-Acetylation of Sialic Acids—O-Acetylation of Sialic Acids in the Rat Liver Golgi Apparatus Involves an Acetyl Intermediate and Essential Histidine and Lysine Residues—A Transmembrane Reaction? J. Biol. Chem. 1989;264:19427–19434. [PubMed] [Google Scholar]
- 841.Diaz S., Higa H.H., Hayes B.K., Varki A. O-Acetylation and De-O-Acetylation of Sialic Acids—7- and 9-O-Acetylation of α2,6-Linked Sialic Acids on Endogenous N-Linked Glycans in Rat Liver Golgi Vesicles. J. Biol. Chem. 1989;264:19416–19426. [PubMed] [Google Scholar]
- 842.Higa H.H., Varki A. Acetyl-Coenzyme A:Polysialic Acid O-Acetyltransferase From K1-Positive Escherichia coli—The Enzyme Responsible for the O-Acetyl plus Phenotype and for O-Acetyl Form Variation. J. Biol. Chem. 1988;263:8872–8878. [PubMed] [Google Scholar]
- 843.Deszo E.L., Steenbergen S.M., Freedberg D.I., Vimr E.R. Escherichia coli K1 Polysialic Acid O-Acetyltransferase Gene, neuO, and the Mechanism of Capsule Form Variation Involving a Mobile Contingency Locus. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5564–5569. doi: 10.1073/pnas.0407428102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 844.Bergfeld A.K., Claus H., Lorenzen N.K., Spielmann F., Vogel U., Mühlenhoff M. The Polysialic Acid-Specific O-Acetyltransferase OatC From Neisseria meningitidis Serogroup C Evolved Apart From Other Bacterial Sialate O-Acetyltransferases. J. Biol. Chem. 2009;284:6–16. doi: 10.1074/jbc.M807518200. [DOI] [PubMed] [Google Scholar]
- 845.Houliston R.S., Endtz H.P., Yuki N., Li J., Jarrell H.C., Koga M., van Belkum A., Karwaski M.-F., Wakarchuk W.W., Gilbert M. Identification of a Sialate O-Acetyltransferase From Campylobacter jejuni—Demonstration of Direct Transfer to the C-9 Position of Terminal α-2,8-Linked Sialic Acid. J. Biol. Chem. 2006;281:11480–11486. doi: 10.1074/jbc.M512183200. [DOI] [PubMed] [Google Scholar]
- 846.Arming S., Wipfler D., Mayr J., Merling A., Vilas U., Schauer R., Schwartz-Albiez R., Vlasak R. The Human Cas1 Protein: A Sialic Acid-Specific O-Acetyltransferase? Glycobiology. 2011;21:553–564. doi: 10.1093/glycob/cwq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Baumann A.-M.T., Bakkers M.J.G., Buettner F.F.R., Hartmann M., Grove M., Langereis M.A., de Groot R.J., Mühlenhoff M. 9-O-Acetylation of Sialic Acids Is Catalysed by CASD1 via a Covalent Acetyl–Enzyme Intermediate. Nat. Commun. 2015;7673(1 − 12):6. doi: 10.1038/ncomms8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Kleineidam R.G., Hofmann O., Reuter G., Schauer R. Indications for the Enzymatic Synthesis of 9-O-Lactoyl-N-acetylneuraminic Acid in Equine Liver. Glycoconjugate J. 1993;10:116–119. doi: 10.1007/BF00731195. [DOI] [PubMed] [Google Scholar]
- 849.Kelm A., Shaw L., Schauer R., Reuter G. The Biosynthesis of 8-O-Methylated Sialic Acids in the Starfish Asterias rubens—Isolation and Characterisation of S-Adenosyl-l-methionine:Sialate-8-O-methyltransferase. Eur. J. Biochem. 1998;251:874–884. doi: 10.1046/j.1432-1327.1998.2510874.x. [DOI] [PubMed] [Google Scholar]
- 850.Yamakawa N., Sato C., Miyata S., Maehashi E., Toriyama M., Sato N., Furuhata K., Kitajima K. Development of Sensitive Chemical and Immunochemical Methods for Detecting Sulfated Sialic Acids and Their Application to Glycoconjugates From Sea Urchin Sperm and Eggs. Biochimie. 2007;89:1396–1408. doi: 10.1016/j.biochi.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 851.Inoue S., Iwasaki M. Isolation of a Novel Glycoprotein From the Eggs of Rainbow Trout: Occurrence of Disialosyl Groups on All Carbohydrate Chains. Biochem. Biophys. Res. Commun. 1978;83:1018–1023. doi: 10.1016/0006-291x(78)91497-3. [DOI] [PubMed] [Google Scholar]
- 852.Wang F., Xie B., Wang B., Troy F.A., II. LC-MS/MS Glycomic Analyses of Free and Conjugated Forms of the Sialic Acids, Neu5Ac, Neu5Gc and KDN in Human Throat Cancers. Glycobiology. 2015;25:1362–1374. doi: 10.1093/glycob/cwv051. [DOI] [PubMed] [Google Scholar]
- 853.Terada T., Kitajima K., Inoue S., Koppert K., Brossmer R., Inoue R. Substrate Specificity of Rainbow Trout Testis CMP-3-Deoxy-d-glycero-d-galacto-nonulosonic Acid (CMP-Kdn) Synthetase—Kinetic Studies of the Reaction of Natural and Synthetic Analogues of Nonulosonic Acid Catalyzed by CMP-Kdn Synthetase. Eur. J. Biochem. 1996;236:852–855. doi: 10.1111/j.1432-1033.1996.00852.x. [DOI] [PubMed] [Google Scholar]
- 854.Angata T., Kitazume S., Terada T., Kitajima K., Inoue S., Troy F.A., II., Inoue Y. Identification, Characterization, and Developmental Expression of a Novel α2 → 8-KDN-Transferase Which Terminates Elongation of α2 → 8-Linked Oligo-Polysialic Acid Chain Synthesis in Trout Egg Polysialoglycoproteins. Glycoconjugate J. 1994;11:493–499. doi: 10.1007/BF00731286. [DOI] [PubMed] [Google Scholar]
- 855.Lewis A.L., Desa N., Hansen E.E., Knirel Y.A., Gordon J.I., Gagneux P., Nizet V., Varki A. Innovations in Host and Microbial Sialic Acid Biosynthesis Revealed by Phylogenomic Prediction of Nonulosonic Acid Structure. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13552–13557. doi: 10.1073/pnas.0902431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 856.Schoenhofen I.C., Vinogradov E., Whitfield D.M., Brisson J.-R., Logan S.M. The CMP-Legionaminic Acid Pathway in Campylobacter: Biosynthesis Involving Novel GDP-Linked Precursors. Glycobiology. 2009;19:715–725. doi: 10.1093/glycob/cwp039. [DOI] [PubMed] [Google Scholar]
- 857.Schoenhofen I.C., McNally D.J., Brisson J.-R., Logan S.M. Elucidation of the CMP-Pseudaminic Acid Pathway in Helicobacter pylori: Synthesis From UDP-N-acetylglucosamine by a Single Enzymatic Reaction. Glycobiology. 2006;16:8C–14C. doi: 10.1093/glycob/cwl010. [DOI] [PubMed] [Google Scholar]
- 858.Liu F., Aubry A.J., Schoenhofen I.C., Logan S.M., Tanner M.E. The Engineering of Bacteria Bearing Azido-Pseudaminic Acid-Modified Flagella. ChemBioChem. 2009;10:1317–1320. doi: 10.1002/cbic.200900018. [DOI] [PubMed] [Google Scholar]
- 859.Hinderlich S., Tauber R., Bertozzi C.R., Hackenberger C.P.R. Werner Reutter: A Visionary Pioneer in Molecular Glycobiology. ChemBioChem. 2017;18:1141–1145. doi: 10.1002/cbic.201700277. [DOI] [PubMed] [Google Scholar]
- 860.Brossmer R., Nebelin E. Synthesis of N-Formyl- and N-Succinyl-d-neuraminic Acid on the Specificity of Neuraminidase. FEBS Lett. 1969;4:335–336. doi: 10.1016/0014-5793(69)80269-3. [DOI] [PubMed] [Google Scholar]
- 861.Grünholz H.-J., Harms E., Opetz M., Reutter W., Černȳ M. Inhibition of In Vitro Biosynthesis of N-Acetylneuraminic Acid by N-Acyl- and N-Alkyl-2-amino-2-deoxyhexoses. Carbohydr. Res. 1981;96:259–270. doi: 10.1016/s0008-6215(00)81876-5. [DOI] [PubMed] [Google Scholar]
- 862.Kayser H., Zeitler R., Kannicht C., Grunow D., Nuck R., Reutter W. Biosynthesis of a Nonphysiological Sialic Acid in Different Rat Organs, Using N-Propanoyl-d-hexosamines as Precursors. J. Biol. Chem. 1992;267:16934–16938. [PubMed] [Google Scholar]
- 863.Kayser H., Geilen C.C., Paul C., Zeitler R., Reutter W. Incorporation of N-Acyl-2-amino-2-deoxy-hexoses Into Glycosphingolipids of the Pheochromocytoma Cell Line PC12. FEBS Lett. 1992;301:137–140. doi: 10.1016/0014-5793(92)81233-c. [DOI] [PubMed] [Google Scholar]
- 864.Roehlecke C., Horstkorte R., Reutter W. Stimulation of Human Peripheral Blood Mononuclear Cells by the Sialic Acid Precursor N-Propanoylmannosamine. Glycoconjugate J. 2013;30:813–818. doi: 10.1007/s10719-013-9485-7. [DOI] [PubMed] [Google Scholar]
- 865.Schmidt C., Stehling P., Schnitzer J., Reutter W., Horstkorte R. Biochemical Engineering of Neural Cell Surfaces by the Synthetic N-Propanoyl-Substituted Neuraminic Acid Precursor. J. Biol. Chem. 1998;273:19146–19152. doi: 10.1074/jbc.273.30.19146. [DOI] [PubMed] [Google Scholar]
- 866.Keppler O.T., Stehling P., Herrmann M., Kayser H., Grunow D., Reutter W., Pawlita M. Biosynthetic Modulation of Sialic Acid-Dependent Virus-Receptor Interactions of Two Primate Polyoma Viruses. J. Biol. Chem. 1995;270:1308–1314. doi: 10.1074/jbc.270.3.1308. [DOI] [PubMed] [Google Scholar]
- 867.Wieser J.R., Heisner A., Stehling P., Oesch F., Reutter W. In Vivo Modulated N-Acyl Side Chain of N-Acetylneuraminic Acid Modulates the Cell Contact-Dependent Inhibition of Growth. FEBS Lett. 1996;395:170–173. doi: 10.1016/0014-5793(96)01029-0. [DOI] [PubMed] [Google Scholar]
- 868.Gnanapragassam V.S., Bork K., Galuska C.E., Galuska S.P., Glanz D., Nagasundaram M., Bache M., Vordermark D., Kohla G., Kannicht C., Schauer R., Horstkorte R. Sialic Acid Metabolic Engineering: A Potential Strategy for the Neuroblastoma Therapy. PLoS One. 2014;e105403(1–10):9. doi: 10.1371/journal.pone.0105403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Cheng B., Xie R., Dong L., Chen X. Metabolic Remodeling of Cell-Surface Sialic Acids: Principles, Applications, and Recent Advances. ChemBioChem. 2016;17:11–27. doi: 10.1002/cbic.201500344. [DOI] [PubMed] [Google Scholar]
- 870.Wratil P.R., Horstkorte R., Reutter W. Metabolic Glycoengineering With N-Acyl Side Chain Modified Mannosamines. Angew. Chem., Int. Ed. 2016;55:9482–9512. doi: 10.1002/anie.201601123. [DOI] [PubMed] [Google Scholar]
- 871.Schauer R., Veh R.W., Sander M., Corfield A.P., Wiegandt H. “Neuraminidase-Resistant” Sialic Acid Residues of Gangliosides. Adv. Exp. Med. Biol. 1980;125:283–294. [PubMed] [Google Scholar]
- 872.Schauer R., Reuter G., Stoll S. Sialate-O-acetylesterases: Key Enzymes in Sialic Acid Catabolism. Biochimie. 1988;70:1511–1519. doi: 10.1016/0300-9084(88)90288-x. [DOI] [PubMed] [Google Scholar]
- 873.Schauer R., Shukla A.K. Isolation and Properties of Two Sialate-O-Acetylesterases From Horse Liver With 4- and 9-O-Acetyl Specificities. Glycoconjugate J. 2008;25:625–632. doi: 10.1007/s10719-008-9109-9. [DOI] [PubMed] [Google Scholar]
- 874.Schauer R., Reuter G., Stoll S., Shukla A.K. Partial Purification and Characterization of Sialate O-Acetylesterase From Bovine Brain. J. Biochem. 1989;106:143–150. doi: 10.1093/oxfordjournals.jbchem.a122804. [DOI] [PubMed] [Google Scholar]
- 875.Smits S.L., Gerwig G.J., van Vliet A.L.W., Lissenberg A., Briza P., Kamerling J.P., Vlasak R., de Groot R.J. Nidovirus Sialate-O-acetylesterases—Evolution and Substrate Specificity of Coronaviral and Toroviral Receptor-Destroying Enzymes. J. Biol. Chem. 2005;280:6933–6941. doi: 10.1074/jbc.M409683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Srinivasan G.V., Schauer R. Assays of Sialate-O-acetyltransferases and Sialate-O-acetylesterases. Glycoconjugate J. 2009;26:935–944. doi: 10.1007/s10719-008-9131-y. [DOI] [PubMed] [Google Scholar]
- 877.Hirst G.K. The Relationship of the Receptors of a New Strain of Virus to Those of the Mumps-NDV-Influenza Group. J. Exp. Med. 1950;91:177–184. doi: 10.1084/jem.91.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 878.Herrler G., Rott R., Klenk H.-D. Neuraminic Acid Is Involved in the Binding of Influenza C Virus to Erythrocytes. Virology. 1985;141:144–147. doi: 10.1016/0042-6822(85)90190-4. [DOI] [PubMed] [Google Scholar]
- 879.Schauer R., Reuter G., Stoll S., Posadas del Rio F., Herrler G., Klenk H.-D. Isolation and Characterization of Sialate 9(4)-O-Acetylesterase From Influenza C Virus. Biol. Chem. Hoppe-Seyler. 1988;369:1121–1130. doi: 10.1515/bchm3.1988.369.2.1121. [DOI] [PubMed] [Google Scholar]
- 880.Herrler G., Klenk H.-D. Structure and Function of the Hef Glycoprotein of Influenza C Virus. Adv. Virus Res. 1991;40:213–234. doi: 10.1016/S0065-3527(08)60280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Garcia-Sastre A., Villar E., Manuguerra J.C., Hannoun C., Cabezas J.A. Activity of Influenza C Virus O-Acetylesterase With O-Acetyl-Containing Compounds. Biochem. J. 1991;273:435–441. doi: 10.1042/bj2730435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Rosenthal P.B., Zhang X., Formanowski F., Fitz W., Wong C.-H., Meier-Ewert H., Skehel J.J., Wiley D.C. Structure of the Haemagglutinin-Esterase-Fusion Glycoprotein of Influenza C Virus. Nature. 1998;396:92–96. doi: 10.1038/23974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Regl G., Kaser A., Iwersen M., Schmid H., Kohla G., Strobl B., Vilas U., Schauer R., Vlasak R. The Hemagglutinin-Esterase of Mouse Hepatitis Virus Strain S Is a Sialate-4-O-acetylesterase. J. Virol. 1999;73:4721–4727. doi: 10.1128/jvi.73.6.4721-4727.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Shen Y., Tiralongo J., Kohla G., Schauer R. Regulation of Sialic Acid O-Acetylation in Human Colon Mucosa. Biol. Chem. 2004;385:145–152. doi: 10.1515/BC.2004.033. [DOI] [PubMed] [Google Scholar]
- 885.Pillai S. Rethinking Mechanisms of Autoimmune Pathogenesis. J. Autoimmun. 2013;45:97–103. doi: 10.1016/j.jaut.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Surolia I., Pirnie S.P., Chellappa V., Taylor K.N., Cariappa A., Moya J., Liu H., Bell D.W., Driscoll D.R., Diederichs S., Haider K., Netravali I., Le S., Elia R., Dow E., Lee A., Freudenberg J., De Jager P.L., Chretien Y., Varki A., MacDonald M.E., Gillis T., Behrens T.W., Bloch D., Collier D., Korzenik J., Podolsky D.K., Hafler D., Murali M., Sands B., Stone J.H., Gregersen P.K., Pillai S. Functionally Defective Germline Variants of Sialic Acid Acetylesterase in Autoimmunity. Nature. 2010;466:243–247. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 887.Yamamoto M., Iguchi G., Bando H., Fukuoka H., Suda K., Takahashi M., Nishizawa H., Matsumoto R., Tojo K., Mokubo A., Ogata T., Takahashi Y.A. Missense Single-Nucleotide Polymorphism in the Sialic Acid Acetylesterase (SIAE) Gene Is Associated With Anti-PIT-1 Antibody Syndrome. Endocr. J. (Tokyo) 2014;61:641–644. doi: 10.1507/endocrj.ej13-0539. [DOI] [PubMed] [Google Scholar]
- 888.Orizio F., Damiati E., Giacopuzzi E., Benaglia G., Pianta S., Schauer R., Schwartz-Albiez R., Borsani G., Bresciani R., Monti E. Human Sialic Acid Acetyl Esterase: Towards a Better Understanding of a Puzzling Enzyme. Glycobiology. 2015;25:992–1006. doi: 10.1093/glycob/cwv034. [DOI] [PubMed] [Google Scholar]
- 889.Hirst G.K. Adsorption of Influenza Hemagglutinins and Virus by Red Blood Cells. J. Exp. Med. 1942;76:195–209. doi: 10.1084/jem.76.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890.McCrea J.F. Modification of Red-Cell Agglutinability by Cl. welchii Toxins. Aust. J. Exp. Biol. Med. Sci. 1947;25:127–136. [Google Scholar]
- 891.Burnet F.M., McCrea J.F., Stone J.D. Modification of Human Red Cells by Virus Action—I. The Receptor Gradient for Virus Action in Human Red Cells. Br. J. Exp. Pathol. 1946;27:228–236. [PMC free article] [PubMed] [Google Scholar]
- 892.Gottschalk A. Neuraminidase: The Specific Enzyme of Influenza Virus and Vibrio cholerae. Biochim. Biophys. Acta. 1957;23:645–646. doi: 10.1016/0006-3002(57)90389-x. [DOI] [PubMed] [Google Scholar]
- 893.IUB Enzyme Nomenclature 1984, Academic Press: New York, NY, USA, 1984.
- 894.Cabezas J.A. Some Questions and Suggestions on the Type References of the Official Nomenclature (IUB) for Sialidase(s) and Endosialidase. Biochem. J. 1991;278:311–312. doi: 10.1042/bj2780311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.Warren L., Spearing C.W. Mammalian Sialidase (Neuraminidase) Biochem. Biophys. Res. Commun. 1960;3:489–492. doi: 10.1016/0006-291x(60)90161-3. [DOI] [PubMed] [Google Scholar]
- 896.Ada G.L., Lind P.E. Neuraminidase in the Chorioallantois of the Chick Embryo. Nature. 1961;190:1169–1171. doi: 10.1038/1901169a0. [DOI] [PubMed] [Google Scholar]
- 897.Carubelli R., Trucco R.E., Caputto R. Neuraminidase Activity in Mammalian Organs. Biochim. Biophys. Acta. 1962;60:196–197. doi: 10.1016/0006-3002(62)90392-x. [DOI] [PubMed] [Google Scholar]
- 898.Cassidy J.T., Jourdian G.W., Roseman S. The Sialic Acids—VI. Purification and Properties of Sialidase From Clostridium perfringens. J. Biol. Chem. 1965;240:3501–3506. [PubMed] [Google Scholar]
- 899.Roggentin T., Kleineidam R.G., Majewski D.M., Tirpitz D., Roggentin P., Schauer R. An Immunoassay for the Rapid and Specific Detection of Three Sialidase-Producing Clostridia Causing Gas Gangrene. J. Immunol. Methods. 1993;157:125–133. doi: 10.1016/0022-1759(93)90078-l. [DOI] [PubMed] [Google Scholar]
- 900.Uchida Y., Tsukada Y., Sugimori T. Enzymatic Properties of Neuraminidases From Arthrobacter ureafaciens. J. Biochem. 1979;86:1573–1585. doi: 10.1093/oxfordjournals.jbchem.a132675. [DOI] [PubMed] [Google Scholar]
- 901.Ada G.L., French E.L., Lind P.E. Purification and Properties of Neuraminidase From Vibrio cholerae. J. Gen. Microbiol. 1961;24:409–421. doi: 10.1099/00221287-24-3-409. [DOI] [PubMed] [Google Scholar]
- 902.Hoyer L.L., Roggentin P., Schauer R., Vimr E.R. Purification and Properties of Cloned Salmonella typhimurium LT2 Sialidase With Virus-Typical Kinetic Preferences for Sialyl α2 → 3 Linkages. J. Biochem. 1991;110:462–467. doi: 10.1093/oxfordjournals.jbchem.a123603. [DOI] [PubMed] [Google Scholar]
- 903.Drzeniek R. Substrate Specificity of Neuraminidases. Histochem. J. 1973;5:271–290. doi: 10.1007/BF01004994. [DOI] [PubMed] [Google Scholar]
- 904.Paulson J.C., Weinstein J., Dorland L., van Halbeek H., Vliegenthart J.F.G. Newcastle Disease Virus Contains a Linkage-Specific Glycoprotein Sialidase—Application to the Localization of Sialic Acid Residues in N-Linked Oligosaccharides of α1-Acid Glycoprotein. J. Biol. Chem. 1982;257:12734–12738. [PubMed] [Google Scholar]
- 905.Royle L., Mattu T.S., Hart E., Langridge J.I., Merry A.H., Murphy N., Harvey D.J., Dwek R.A., Rudd P.M. An Analytical and Structural Database Provides a Strategy for Sequencing O-Glycans From Microgram Quantities of Glycoproteins. Anal. Biochem. 2002;304:70–90. doi: 10.1006/abio.2002.5619. [DOI] [PubMed] [Google Scholar]
- 906.Amano J., Nishimura R., Mochizuki M., Kobata A. Comparative Study of the Mucin-Type Sugar Chains of Human Chorionic Gonadotropin Present in the Urine of Patients With Trophoblastic Diseases and Healthy Pregnant Women. J. Biol. Chem. 1988;263:1157–1165. [PubMed] [Google Scholar]
- 907.Kwiatkowski B., Boschek B., Thiele H., Stirm S. Endo-N-Acetylneuraminidase Associated With Bacteriophage Particles. J. Virol. 1982;43:697–704. doi: 10.1128/jvi.43.2.697-704.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 908.Faillard H. In: Hoppe-Seyler/Thierfelder, Handbuch der physiologisch- und pathologisch-chemischen Analyse. 10. Aufl. Bd. VI/B. Lang K., editor. Springer; Berlin, Germany: 1966. Aminozuckerspaltende Enzyme; pp. 1234–1261. [Google Scholar]
- 909.Drzeniek R. Viral and Bacterial Neuraminidases. Curr. Top. Microbiol. Immunol. 1972;59:35–74. doi: 10.1007/978-3-642-65444-2_2. [DOI] [PubMed] [Google Scholar]
- 910.Corfield A.P., Michalski J.-C., Schauer R. In: Perspectives in Inherited Metabolic Diseases. Tettamanti G., Durand P., Di Donato S., editors. Vol. 4. Edi Ermes; Milan, Italy: 1981. The Substrate Specificity of Sialidases From Microorganisms and Mammals; pp. 3–70. [Google Scholar]
- 911.Saito M., Yu R.K. In: Biology of the Sialic Acids. Rosenberg A., editor. Plenum Press; New York, USA: 1995. Biochemistry and Function of Sialidases; pp. 261–313. [Google Scholar]
- 912.Achyuthan K.E., Achyuthan A.M. Comparative Enzymology, Biochemistry and Pathophysiology of Human exo-α-Sialidases (Neuraminidases) Comp. Biochem. Physiol. B: Biochem. Mol. Biol. 2001;129B:29–64. doi: 10.1016/s1096-4959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 913.Monti E., Preti A., Venerando B., Borsani G. Recent Development in Mammalian Sialidase Molecular Biology. Neurochem. Res. 2002;27:649–663. doi: 10.1023/a:1020276000901. [DOI] [PubMed] [Google Scholar]
- 914.Giacopuzzi E., Bresciani R., Schauer R., Monti E., Borsani G. New Insights on the Sialidase Protein Family Revealed by a Phylogenetic Analysis in Metazoa. PLoS One. 2012;e44193(1–17):7. doi: 10.1371/journal.pone.0044193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 915.Miyagi T., Yamaguchi K. Mammalian Sialidases: Physiological and Pathological Roles in Cellular Functions. Glycobiology. 2012;22:880–896. doi: 10.1093/glycob/cws057. [DOI] [PubMed] [Google Scholar]
- 916.Ada G.L., French E.L. Purification of Bacterial Neuraminidase (Receptor-Destroying Enzyme) Nature. 1959;183:1740–1741. doi: 10.1038/1831740b0. [DOI] [PubMed] [Google Scholar]
- 917.Kawamura K. Production and Purification of Receptor Destroying Enzyme (RDE) Tohoku J. Exp. Med. 1958;67:372. doi: 10.1620/tjem.67.372. [DOI] [PubMed] [Google Scholar]
- 918.Nees S., Schauer R. Induction of Neuraminidase From Clostridium perfringens and the Cooperation of This Enzyme With Acetylneuraminate Pyruvate-Lyase. Behring Inst. Mitt. 1974;55:68–78. [Google Scholar]
- 919.Thomas G.H. Sialic Acid Acquisition in Bacteria—One Substrate, Many Transporters. Biochem. Soc. Trans. 2016;44:760–765. doi: 10.1042/BST20160056. [DOI] [PubMed] [Google Scholar]
- 920.Vimr E.R., Troy F.A. Identification of an Inducible Catabolic System for Sialic Acids (nan) in Escherichia coli. J. Bacteriol. 1985;164:845–853. doi: 10.1128/jb.164.2.845-853.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 921.Corfield A.P., Schauer R. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Metabolism of Sialic Acids; pp. 195–261. (Cell Biology Monographs). [Google Scholar]
- 922.Kleineidam R.G., Furuhata K., Ogura H., Schauer R. 4-Methylumbelliferyl-α-Glycosides of Partially O-Acetylated N-Acetylneuraminic Acids as Substrates of Bacterial and Viral Sialidases. Biol. Chem. Hoppe Seyler. 1990;371:715–719. doi: 10.1515/bchm3.1990.371.2.715. [DOI] [PubMed] [Google Scholar]
- 923.Miyagi T., Tsuiki S. Rat-Liver Lysosomal Sialidase—Solubilization, Substrate Specificity and Comparison With the Cytosolic Sialidase. Eur. J. Biochem. 1984;141:75–81. doi: 10.1111/j.1432-1033.1984.tb08159.x. [DOI] [PubMed] [Google Scholar]
- 924.Miyagi T., Konno K., Emori Y., Kawasaki H., Suzuki K., Yasui A., Tsuiki S. Molecular Cloning and Expression of cDNA Encoding Rat Skeletal Muscle Cytosolic Sialidase. J. Biol. Chem. 1993;268:26435–26440. [PubMed] [Google Scholar]
- 925.Miyagi T., Wada T., Iwamatsu A., Hata K., Yoshikawa Y., Tokuyama S., Sawada M. Molecular Cloning and Characterization of a Plasma Membrane-Associated Sialidase Specific for Gangliosides. J. Biol. Chem. 1999;274:5004–5011. doi: 10.1074/jbc.274.8.5004. [DOI] [PubMed] [Google Scholar]
- 926.Monti E., Bassi M.T., Bresciani R., Civini S., Croci G.L., Papini N., Riboni M., Zanchetti G., Ballabio A., Preti A., Tettamanti G., Venerando B., Borsani G. Molecular Cloning and Characterization of NEU4, the Fourth Member of the Human Sialidase Gene Family. Genomics. 2004;83:445–453. doi: 10.1016/j.ygeno.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 927.Miyagi T., Takahashi K., Shiozaki K., Yamaguchi K., Hosono M. Plasma Membrane-Associated Sialidase Confers Cancer Initiation, Promotion and Progression. Adv. Exp. Med. Biol. 2015;842:139–145. doi: 10.1007/978-3-319-11280-0_9. [DOI] [PubMed] [Google Scholar]
- 928.Forcella M., Mozzi A., Stefanini F.M., Riva A., Epistolio S., Molinari F., Merlo E., Monti E., Fusi P., Frattini M. Deregulation of Sialidases in Human Normal and Tumor Tissues. Cancer Biomarkers. 2018;21:591–601. doi: 10.3233/CBM-170548. [DOI] [PubMed] [Google Scholar]
- 929.Hunter C.D., Khanna N., Richards M.R., Rezaei Darestani R., Zou C., Klassen J.S., Cairo C.W. Human Neuraminidase Isoenzymes Show Variable Activities for 9-O-Acetyl-sialoside Substrates. ACS Chem. Biol. 2018;13:922–932. doi: 10.1021/acschembio.7b00952. [DOI] [PubMed] [Google Scholar]
- 930.Roggentin P., Schauer R., Hoyer L.L., Vimr E.R. The Sialidase Superfamily and Its Spread by Horizontal Gene Transfer. Mol. Microbiol. 1993;9:915–921. doi: 10.1111/j.1365-2958.1993.tb01221.x. [DOI] [PubMed] [Google Scholar]
- 931.Pereira M.E. A Developmentally Regulated Neuraminidase Activity in Trypanosoma cruzi. Science. 1983;219:1444–1446. doi: 10.1126/science.6338592. [DOI] [PubMed] [Google Scholar]
- 932.Prioli R.P., Mejia J.S., Aji T., Aikawa M., Pereira M.E.A. Trypanosoma cruzi: Localization of Neuraminidase on the Surface of Trypomastigotes. Trop. Med. Parasitol. 1991;42:146–150. [PubMed] [Google Scholar]
- 933.Previato J.O., Andrade A.F.B., Pessolani M.C.V., Mendonça-Previato L. Incorporation of Sialic Acid Into Trypanosoma cruzi Macromolecules—A Proposal for a New Metabolic Route. Mol. Biochem. Parasitol. 1985;16:85–96. doi: 10.1016/0166-6851(85)90051-9. [DOI] [PubMed] [Google Scholar]
- 934.Schenkman S., Jiang M.-S., Hart G.W., Nussenzweig V. A Novel Cell Surface trans-Sialidase of Trypanosoma cruzi Generates a Stage-Specific Epitope Required for Invasion of Mammalian Cells. Cell. 1991;65:1117–1125. doi: 10.1016/0092-8674(91)90008-m. [DOI] [PubMed] [Google Scholar]
- 935.Engstler M., Reuter G., Schauer R. Purification and Characterization of a Novel Sialidase Found in Procyclic Culture Forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 1992;54:21–30. doi: 10.1016/0166-6851(92)90091-w. [DOI] [PubMed] [Google Scholar]
- 936.Engstler M., Schauer R., Brun R. Distribution of Developmentally Regulated trans-Sialidases in the Kinetoplastida and Characterization of a Shed trans-Sialidase Activity From Procyclic Trypanosoma congolense. Acta Trop. 1995;59:117–129. doi: 10.1016/0001-706x(95)00077-r. [DOI] [PubMed] [Google Scholar]
- 937.Tiralongo E., Schrader S., Lange H., Lemke H., Tiralongo J., Schauer R. Two Trans-Sialidase Forms With Different Sialic Acid Transfer and Sialidase Activities From Trypanosoma congolense. J. Biol. Chem. 2003;278:23301–23310. doi: 10.1074/jbc.M212909200. [DOI] [PubMed] [Google Scholar]
- 938.Wilbrink M.H., ten Kate G.A., Sanders P., Gerwig G.J., van Leeuwen S.S., Sallomons E., Klarenbeek B., Hage J.A., van Vuure C.A., Dijkhuizen L., Kamerling J.P. Enzymatic Decoration of Prebiotic Galacto-oligosaccharides (Vivinal GOS) With Sialic Acid Using Trypanosoma cruzi trans-Sialidase and Two Bovine Sialoglycoconjugates as Donor Substrates. J. Agric. Food Chem. 2015;63:5976–5984. doi: 10.1021/acs.jafc.5b01505. [DOI] [PubMed] [Google Scholar]
- 939.Luo Y., Li S.-C., Li Y.-T., Luo M. The 1.8 Å Structures of Leech Intramolecular trans-Sialidase Complexes: Evidence of Its Enzymatic Mechanism. J. Mol. Biol. 1999;285:323–332. doi: 10.1006/jmbi.1998.2345. [DOI] [PubMed] [Google Scholar]
- 940.Gut H., King S.J., Walsh M.A. Structural and Functional Studies of Streptococcus pneumoniae Neuraminidase B: An Intramolecular trans-Sialidase. FEBS Lett. 2008;582:3348–3352. doi: 10.1016/j.febslet.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 941.Juge N., Tailford L., Owen C.D. Sialidases From Gut Bacteria: A Mini-Review. Biochem. Soc. Trans. 2016;44:166–175. doi: 10.1042/BST20150226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Kitazume S., Kitajima K., Inoue S., Inoue Y. Detection, Isolation, and Characterization of Oligo/Poly(sialic Acid) and Oligo/Poly(deaminoneuraminic Acid) Units in Glycoconjugates. Anal. Biochem. 1992;202:25–34. doi: 10.1016/0003-2697(92)90200-q. [DOI] [PubMed] [Google Scholar]
- 943.Kitajima K., Kuroyanagi H., Inoue S., Ye J., Troy F.A., II., Inoue Y. Discovery of a New Type of Sialidase, “KDNase”, Which Specifically Hydrolyzes Deaminoneuraminyl (3-Deoxy-d-glycero-d-galacto-2-nonulosonic Acid) but Not N-Acylneuraminyl Linkages. J. Biol. Chem. 1994;269:21415–21419. [PubMed] [Google Scholar]
- 944.Li Y.-T., Yuziuk J.A., Li S.-C., Nematalla A., Hasegawa A., Kimura M., Nakagawa H. A Novel Sialidase Capable of Cleaving 3-Deoxy-d-glycero-d-galacto-2-nonulosonic Acid (KDN) Arch. Biochem. Biophys. 1994;310:243–246. doi: 10.1006/abbi.1994.1163. [DOI] [PubMed] [Google Scholar]
- 945.Angata T., Kitajima K., Inoue S., Chang J., Warner T.G., Troy F.A., II., Inoue Y. Identification, Developmental Expression and Tissue Distribution of Deaminoneuraminate Hydrolase (KDNase) Activity in Rainbow Trout. Glycobiology. 1994;4:517–523. doi: 10.1093/glycob/4.4.517. [DOI] [PubMed] [Google Scholar]
- 946.Inoue S., Lin S.-L., Inoue Y., Groves D.R., Thomson R.J., von Itzstein M., Pavlova N.V., Li S.-C., Li Y.-T. A Unique Sialidase That Cleaves the Neu5Gcα2→5-OglycolylNeu5Gc Linkage: Comparison of Its Specificity With That of Three Microbial Sialidases Toward Four Sialic Acid Dimers. Biochem. Biophys. Res. Commun. 2001;280:104–109. doi: 10.1006/bbrc.2000.4084. [DOI] [PubMed] [Google Scholar]
- 947.Meindl P., Bodo G., Palese P., Schulman J., Tuppy H. Inhibition of Neuraminidase Activity by Derivatives of 2-Deoxy-2,3-dehydro-N-acetylneuraminic Acid. Virology. 1974;58:457–463. doi: 10.1016/0042-6822(74)90080-4. [DOI] [PubMed] [Google Scholar]
- 948.Schreiner E., Zbiral E., Kleineidam R.G., Schauer R. Structural Variations on N-Acetylneuraminic Acid—20. Synthesis of Some 2,3-Didehydro-2-deoxysialic Acids Structurally Varied at C-4 and Their Behavior Towards Sialidase From Vibrio cholerae. Liebigs Ann. Chem. 1991:129–134. doi: 10.1016/0008-6215(92)84150-q. [DOI] [PubMed] [Google Scholar]
- 949.Von Itzstein M., Wu W.-Y., Kok G.B., Pegg M.S., Dyason J.C., Jin B., Van Phan T., Smythe M.L., White H.F., Oliver S.W., Colman P.M., Varghese J.N., Ryan D.M., Woods J.M., Bethell R.C., Hotham V.J., Cameron J.M., Penn C.R. Rational Design of Potent Sialidase-Based Inhibitors of Influenza Virus Replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 950.Von Itzstein M., Wu W.-Y., Jin B. The Synthesis of 2,3-Didehydro-2,4-dideoxy-4-guanidinyl-N-acetylneuraminic Acid: A Potent Influenza Virus Sialidase Inhibitor. Carbohydr. Res. 1994;259:301–305. doi: 10.1016/0008-6215(94)84065-2. [DOI] [PubMed] [Google Scholar]
- 951.Kim C.U., Lew W., Williams M.A., Liu H., Zhang L., Swaminathan S., Bischofberger N., Chen M.S., Mendel D.B., Tai C.Y., Laver W.G., Stevens R.C. Influenza Neuraminidase Inhibitors Possessing a Novel Hydrophobic Interaction in the Enzyme Active Site: Design, Synthesis, and Structural Analysis of Carbocyclic Sialic Acid Analogues With Potent Anti-influenza Activity. J. Am. Chem. Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- 952.Rohloff J.C., Kent K.M., Postich M.J., Becker M.W., Chapman H.H., Kelly D.E., Lew W., Louie M.S., McGee L.R., Prisbe E.J., Schultze L.M., Yu R.H., Zhang L. Practical Total Synthesis of the Anti-influenza Drug GS 4104. J. Org. Chem. 1998;63:4545–4550. [Google Scholar]
- 953.Bardsley-Elliot A., Noble S. Oseltamivir. Drugs. 1999;58:851–860. doi: 10.2165/00003495-199958050-00007. [DOI] [PubMed] [Google Scholar]
- 954.McNicholl I.R., McNicholl J.J. Neuraminidase Inhibitors: Zanamivir and Oseltamivir. Ann. Pharmacother. 2001;35:57–70. doi: 10.1345/aph.10118. [DOI] [PubMed] [Google Scholar]
- 955.Gubareva L.V., Webster R.G., Hayden F.G. Detection of Influenza Virus Resistance to Neuraminidase Inhibitors by an Enzyme Inhibition Assay. Antiviral Res. 2002;53:47–61. doi: 10.1016/s0166-3542(01)00192-9. [DOI] [PubMed] [Google Scholar]
- 956.Sriwilaijaroen N., Magesh S., Imamura A., Ando H., Ishida H., Sakai M., Ishitsubo E., Hori T., Moriya S., Ishikawa T., Kuwata K., Odagiri T., Tashiro M., Hiramatsu H., Tsukamoto K., Miyagi T., Tokiwa H., Kiso M., Suzuki Y. A Novel Potent and Highly Specific Inhibitor Against Influenza Viral N1-N9 Neuraminidases: Insight Into Neuraminidase–Inhibitor Interactions. J. Med. Chem. 2016;59:4563–4577. doi: 10.1021/acs.jmedchem.5b01863. [DOI] [PubMed] [Google Scholar]
- 957.Taubenberger J.K. The Virulence of the 1918 Pandemic Influenza Virus: Unraveling the Enigma. Arch. Virol. Suppl. 2005;19:101–115. doi: 10.1007/3-211-29981-5_9. [DOI] [PubMed] [Google Scholar]
- 958.Klenk H.-D., Nagai Y., Rott R., Nicolau C. The Structure and Function of Paramyxovirus Glycoproteins. Med. Microbiol. Immunol. 1977;164:35–47. doi: 10.1007/BF02121300. [DOI] [PubMed] [Google Scholar]
- 959.Herrler G., Hausmann J., Klenk H.-D. In: The Biology of Sialic Acids. Rosenberg A., editor. Plenum Press; New York, NY, USA: 1995. Sialic Acid as Receptor Determinant of Ortho- and Paramyxoviruses; pp. 315–336. [Google Scholar]
- 960.Cook G.M.W., Heard D.H., Seaman G.V.F. A Sialomucopeptide Liberated by Trypsin From the Human Erythrocyte. Nature. 1960;188:1011–1012. doi: 10.1038/1881011a0. [DOI] [PubMed] [Google Scholar]
- 961.Cook G.M.W. Glycobiology of the Cell Surface: Its Debt to Cell Electrophoresis 1940–65. Electrophoresis. 2016;37:1399–1406. doi: 10.1002/elps.201500476. [DOI] [PubMed] [Google Scholar]
- 962.Bennett H.S. Morphological Aspects of Extracellular Polysaccharides. J. Histochem. Cytochem. 1963;11:14–23. [Google Scholar]
- 963.Reutter W., Köttgen E., Bauer C., Gerok W. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Biological Significance of Sialic Acids; pp. 263–305. (Cell Biology Monographs). [Google Scholar]
- 964.Ashwell G., Morell A.G. The Role of Surface Carbohydrates in the Hepatic Recognition and Transport of Circulating Glycoproteins. Adv. Enzymol. Relat. Areas Mol. Biol. 1974;41:99–128. doi: 10.1002/9780470122860.ch3. [DOI] [PubMed] [Google Scholar]
- 965.Morell A.G., Gregoriadis G., Scheinberg I.H., Hickman J., Ashwell G. The Role of Sialic Acid in Determining the Survival of Glycoproteins in the Circulation. J. Biol. Chem. 1971;246:1461–1467. [PubMed] [Google Scholar]
- 966.Lodish H.F., Kong N., Wikström L. Calcium Is Required for Folding of Newly Made Subunits of the Asialoglycoprotein Receptor Within the Endoplasmic Reticulum. J. Biol. Chem. 1992;267:12753–12760. [PubMed] [Google Scholar]
- 967.Stewart W.B., Petenyi C.W., Rose H.M. The Survival Time of Canine Erythrocytes Modified by Influenza Virus. Blood. 1955;10:228–234. [PubMed] [Google Scholar]
- 968.Halbhuber K.-J., Helmke U., Geyer G. Zur Elimination digerierter Erythrozyten der Ratten. Folia Haematol. (Leipzig) 1972;97:196–203. [Google Scholar]
- 969.Jancik J.M., Schauer R., Andres K.H., von Düring M. Sequestration of Neuraminidase-Treated Erythrocytes—Studies on Its Topographic, Morphologic and Immunologic Aspects. Cell Tissue Res. 1978;186:209–226. doi: 10.1007/BF00225532. [DOI] [PubMed] [Google Scholar]
- 970.Jancik J., Schauer R. Sialic Acid—A Determinant of the Life-Time of Rabbit Erythrocytes. Hoppe-Seyler’s Z. Physiol. Chem. 1974;355:395–400. doi: 10.1515/bchm2.1974.355.1.395. [DOI] [PubMed] [Google Scholar]
- 971.Aminoff D., Vorder Bruegge W.F., Bell W.C., Sarpolis K., Williams R. Role of Sialic Acid in Survival of Erythrocytes in the Circulation: Interaction of Neuraminidase-Treated and Untreated Erythrocytes With Spleen and Liver at the Cellular Level. Proc. Natl. Acad. Sci. U. S. A. 1977;74:1521–1524. doi: 10.1073/pnas.74.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 972.Kolb H., Kolb-Bachofen V. A Lectin-Like Receptor on Mammalian Macrophages. Biochem. Biophys. Res. Commun. 1978;85:678–683. doi: 10.1016/0006-291x(78)91215-9. [DOI] [PubMed] [Google Scholar]
- 973.Müller E., Schröder C., Schauer R., Sharon N. Binding and Phagocytosis of Sialidase-Treated Rat Erythrocytes by a Mechanism Independent of Opsonins. Hoppe-Seyler’s Z. Physiol. Chem. 1983;364:1419–1429. doi: 10.1515/bchm2.1983.364.2.1419. [DOI] [PubMed] [Google Scholar]
- 974.Schauer R., Fischer C., Lee H., Ruch B., Kelm S. In: Lectins and Glycoconjugates in Oncology. Gabius H.-J., Nagel G.A., editors. Springer-Verlag; Berlin, Germany: 1988. Sialic Acids as Regulators of Molecular and Cellular Interactions; pp. 5–23. [Google Scholar]
- 975.Schauer R. In: Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry. Ogura H., Hasegawa A., Suami T., editors. Kodansha Ltd.; Tokyo, Japan: 1992. Sialic Acids Regulate Cellular and Molecular Recognition; pp. 340–354. [Google Scholar]
- 976.Kelm S., Schauer R. The Galactose-Recognizing System of Rat Peritoneal Macrophages; Identification and Characterization of the Receptor Molecule. Biol. Chem. Hoppe-Seyler. 1988;369:693–704. doi: 10.1515/bchm3.1988.369.2.693. [DOI] [PubMed] [Google Scholar]
- 977.Barondes S.H. In: The Lectins: Properties, Functions, and Applications in Biology and Medicine. Liener I.E., Sharon N., Goldstein I.J., editors. Academic Press, Inc.; Orlando, FL, USA: 1986. Vertebrate Lectins: Properties and Functions; pp. 438–467. [Google Scholar]
- 978.Gesner B.M., Ginsburg V. Effect of Glycosidases on the Fate of Transfused Lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 1964;52:750–755. doi: 10.1073/pnas.52.3.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 979.Woodruff J.J., Gesner B.M. The Effect of Neuraminidase on the Fate of Transfused Lymphocytes. J. Exp. Med. 1969;129:551–567. doi: 10.1084/jem.129.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 980.Fischer C., Kelm S., Ruch B., Schauer R. Reversible Binding of Sialidase-Treated Rat Lymphocytes by Homologous Peritoneal Macrophages. Carbohydr. Res. 1991;213:263–273. doi: 10.1016/s0008-6215(00)90613-x. [DOI] [PubMed] [Google Scholar]
- 981.Jibril S.E., von Gaudecker B., Kelm S., Schauer R. Interaction of Rat Peritoneal Macrophages With Homologous, Sialidase-Treated Lymphocytes In Vitro. Biol. Chem. Hoppe-Seyler. 1987;368:819–829. doi: 10.1515/bchm3.1987.368.2.819. [DOI] [PubMed] [Google Scholar]
- 982.Roffman E., Wilchek M. Selective Covalent Modification of Newly Synthesized Activation-Induced Sialoglycoconjugates on the T Lymphocyte Membrane. Biochem. Biophys. Res. Commun. 1986;135:473–479. doi: 10.1016/0006-291x(86)90018-5. [DOI] [PubMed] [Google Scholar]
- 983.Kreisel W., Volk B.A., Büchsel R., Reutter W. Different Half-Lives of the Carbohydrate and Protein Moieties of a 110,000-Dalton Glycoprotein Isolated From Plasma Membranes of Rat Liver. Proc. Natl. Acad. Sci. U. S. A. 1980;77:1828–1831. doi: 10.1073/pnas.77.4.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 984.Choi S.-I., Simone J.V., Journey L.J. Neuraminidase-Induced Thrombocytopenia in Rats. Br. J. Haematol. 1972;22:93–101. doi: 10.1111/j.1365-2141.1972.tb08790.x. [DOI] [PubMed] [Google Scholar]
- 985.Mahajan V.S., Pillai S. Sialic Acids and Autoimmune Disease. Immunol. Rev. 2016;269:145–161. doi: 10.1111/imr.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 986.Emmelot P. Biochemical Properties of Normal and Neoplastic Cell Surfaces. Eur. J. Cancer. 1973;9:319–333. doi: 10.1016/0014-2964(73)90047-9. [DOI] [PubMed] [Google Scholar]
- 987.Feizi T. Demonstration by Monoclonal Antibodies That Carbohydrate Structures of Glycoproteins and Glycolipids Are Onco-developmental Antigens. Nature. 1985;314:53–57. doi: 10.1038/314053a0. [DOI] [PubMed] [Google Scholar]
- 988.Alhadeff J.A. Malignant Cell Glycoproteins and Glycolipids. Crit. Rev. Oncol. Hematol. 1989;9:37–107. doi: 10.1016/s1040-8428(89)80014-9. [DOI] [PubMed] [Google Scholar]
- 989.Bhavanandan V.P., Furukawa K. In: Biology of the Sialic Acids. Rosenberg A., editor. Plenum Press; New York, NY, USA: 1995. Biochemistry and Oncology of Sialoglycoproteins; pp. 145–196. [Google Scholar]
- 990.Dabelsteen E. Cell Surface Carbohydrates as Prognostic Markers in Human Carcinomas. J. Pathol. 1996;179:358–369. doi: 10.1002/(SICI)1096-9896(199608)179:4<358::AID-PATH564>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 991.Dall’Olio F. Protein Glycosylation in Cancer Biology: An Overview. J. Clin. Pathol.: Mol. Pathol. 1996;49:M126–M135. doi: 10.1136/mp.49.3.m126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992.Nishimura R., Kobata A. In: Glycoproteins and Disease. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 30. Elsevier Science B.V; Amsterdam, The Netherlands: 1996. Cancer Cells and Metastasis—Choriocarcinoma and hCG; pp. 185–195. (New Comprehensive Biochemistry). [Google Scholar]
- 993.Yamashita K., Kobata A. In: Glycoproteins and Disease. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 30. Elsevier Science B.V; Amsterdam, The Netherlands: 1996. Cancer Cells and Metastasis—Hepatocellular Carcinoma; pp. 197–210. (New Comprehensive Biochemistry). [Google Scholar]
- 994.Yamashita K., Kobata A. In: Glycoproteins and Disease. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 30. Elsevier Science B.V; Amsterdam, The Netherlands: 1996. Cancer Cells and Metastasis—Carcinoembryonic Antigen and Related Normal Antigens; pp. 229–239. (New Comprehensive Biochemistry). [Google Scholar]
- 995.Kobata A. In: Glycoproteins and Disease. Montreuil J., Vliegenthart J.F.G., Schachter H., editors. Vol. 30. Elsevier Science B.V; Amsterdam, The Netherlands: 1996. Cancer Cells and Metastasis—The Warren-Glick Phenomenon—A Molecular Basis of Tumorigenesis and Metastasis; pp. 211–227. (New Comprehensive Biochemistry). [Google Scholar]
- 996.Hanisch F.-G., Baldus S.E. The Thomsen-Friedenreich (TF) Antigen: A Critical Review on the Structural, Biosynthetic and Histochemical Aspects of a Pancarcinoma-Associated Antigen. Histol. Histopathol. 1997;12:263–281. [PubMed] [Google Scholar]
- 997.Stephens A.S., Day C.J., Tiralongo J. In: Sialobiology: Structure, Biosynthesis and Function—Sialic Acid Glycoconjugates in Health and Disease. Tiralongo J., Martinez-Duncker I., editors. Bentham Science Publishers; Oak Park, IL, USA: 2013. Sialic Acids and Cancer; pp. 381–403. [Google Scholar]
- 998.Pearce O.M.T., Läubli H. Sialic Acids in Cancer Biology and Immunity. Glycobiology. 2016;26:111–128. doi: 10.1093/glycob/cwv097. [DOI] [PubMed] [Google Scholar]
- 999.Vajaria B.N., Patel P.S. Glycosylation: A Hallmark of Cancer? Glycoconjugate J. 2017;34:147–156. doi: 10.1007/s10719-016-9755-2. [DOI] [PubMed] [Google Scholar]
- 1000.Ambrose E.J., James A.M., Lowick J.H.B. Differences Between the Electrical Charge Carried by Normal and Homologous Tumour Cells. Nature. 1956;177:576–577. doi: 10.1038/177576a0. [DOI] [PubMed] [Google Scholar]
- 1001.Purdom L., Ambrose E.J., Klein G. A Correlation Between Electrical Surface Charge and Some Biological Characteristics During the Stepwise Progression of a Mouse Sarcoma. Nature. 1958;181:1586–1587. doi: 10.1038/1811586a0. [DOI] [PubMed] [Google Scholar]
- 1002.Gasic G., Gasic T. Removal of Sialic Acid From the Cell Coat in Tumor Cells and Vascular Endothellum, and Its Effects on Metastasis. Proc. Natl. Acad. Sci. U. S. A. 1962;48:1172–1177. doi: 10.1073/pnas.48.7.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1003.Buck C.A., Glick M.C., Warren L. Glycopeptides From the Surface of Control and Virus-Transformed Cells. Science. 1971;172:169–171. doi: 10.1126/science.172.3979.169. [DOI] [PubMed] [Google Scholar]
- 1004.Warren L., Fuhrer J.P., Buck C.A. Surface Glycoproteins of Normal and Transformed Cells: A Difference Determined by Sialic Acid and a Growth-Dependent Sialyl Transferase. Proc. Natl. Acad. Sci. U. S. A. 1972;69:1838–1842. doi: 10.1073/pnas.69.7.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1005.Bosmann H.B., Hall T.C. Enzyme Activity in Invasive Tumors of Human Breast and Colon. Proc. Natl. Acad. Sci. U. S. A. 1974;71:1833–1837. doi: 10.1073/pnas.71.5.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1006.Nisselbaum J.S., Bernfeld P. The Properties of Two Glycoproteins Isolated From the Plasma of Normal and Tumor Bearing Mice. J. Am. Chem. Soc. 1956;78:687–689. [Google Scholar]
- 1007.Turumi K.-I., Dawes S.M.L. Serum Sialic Acid Levels in Mice With Neoplasms. Cancer Res. 1958;18:575–577. [PubMed] [Google Scholar]
- 1008.Kessel D., Allen J. Elevated Plasma Sialyltransferase in the Cancer Patient. Cancer Res. 1975;35:670–672. [PubMed] [Google Scholar]
- 1009.Plucinsky M.C., Riley W.M., Prorok J.J., Alhadeff J.A. Total and Lipid-Associated Serum Sialic Acid Levels in Cancer Patients With Different Primary Sites and Differing Degrees of Metastatic Involvement. Cancer. 1986;58:2680–2685. doi: 10.1002/1097-0142(19861215)58:12<2680::aid-cncr2820581222>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 1010.Müller-Eberhard H.J., Kunkel H.G. The Carbohydrate of γ-Globulin and Myeloma Proteins. J. Exp. Med. 1956;104:253–269. doi: 10.1084/jem.104.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1011.Miyagi T., Goto T., Tsuiki S. Sialidase of Rat Hepatomas: Qualitative and Quantitative Comparison With Rat Liver Sialidase. Jpn. J. Cancer Res. 1984;75:1076–1082. [PubMed] [Google Scholar]
- 1012.Miyagi T., Wada T., Yamaguchi K., Shiozaki K., Sato I., Kakugawa Y., Yamanami H., Fujiya T. Human Sialidase as a Cancer Marker. Proteomics. 2008;8:3303–3311. doi: 10.1002/pmic.200800248. [DOI] [PubMed] [Google Scholar]
- 1013.Schengrund C.-L., Lausch R.N., Rosenberg A. Sialidase Activity in Transformed Cells. J. Biol. Chem. 1973;248:4424–4428. [PubMed] [Google Scholar]
- 1014.Holmes E.H., Ostrander G.K., Hakomori S.-I. Biosynthesis of the Sialyl-Lex Determinant Carried by Type 2 Chain Glycosphingolipids (IV3NeuAcIII3FucnLc4, VI3NeuAcV3FucnLc6, and VI3NeuAcIII3V3Fuc2nLc6) in Human Lung Carcinoma PC9 Cells. J. Biol. Chem. 1986;261:3737–3743. [PubMed] [Google Scholar]
- 1015.Mann B., Klussmann E., Vandamme-Feldhaus V., Iwersen M., Hanski M.-L., Riecken E.-O., Buhr H.J., Schauer R., Kim Y.S., Hanski C. Low O-Acetylation of Sialyl-Lex Contributes to Its Overexpression in Colon Carcinoma Metastases. Int. J. Cancer. 1997;72:258–264. doi: 10.1002/(sici)1097-0215(19970717)72:2<258::aid-ijc10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 1016.Corfield A.P., Myerscough N., Warren B.F., Durdey P., Paraskeva C., Schauer R. Reduction of Sialic Acid O-Acetylation in Human Colonic Mucins in the Adenoma-Carcinoma Sequence. Glycoconjugate J. 1999;16:307–317. doi: 10.1023/a:1007026314792. [DOI] [PubMed] [Google Scholar]
- 1017.Kohla G., Stockfleth E., Schauer R. Gangliosides With O-Acetylated Sialic Acids in Tumors of Neuroectodermal Origin. Neurochem. Res. 2002;27:583–592. doi: 10.1023/a:1020211714104. [DOI] [PubMed] [Google Scholar]
- 1018.Magnani J.L., Nilsson B., Brockhaus M., Zopf D., Steplewski Z., Koprowski H., Ginsburg V. A Monoclonal Antibody-Defined Antigen Associated With Gastrointestinal Cancer Is a Ganglioside Containing Sialylated Lacto-N-fucopentaose II. J. Biol. Chem. 1982;257:14365–14369. [PubMed] [Google Scholar]
- 1019.Groux-Degrootte S., Rodríguez-Walker M., Dewald J.H., Daniotti J.L., Delannoy P. Gangliosides in Cancer Cell Signaling. Prog. Mol. Biol. Transl. Sci. 2018;156:197–227. doi: 10.1016/bs.pmbts.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 1020.Higashi H., Ikuta K., Ueda S., Kato S., Hirabayashi Y., Matsumoto M., Naiki M. Characterization of N-Glycolylneuraminic Acid-Containing Glycosphingolipids From a Marek's Disease Lymphoma-Derived Chicken Cell Line, MSB1, as Tumor-Associated Heterophile Hanganutziu-Deicher Antigens. J. Biochem. 1984;95:785–794. doi: 10.1093/oxfordjournals.jbchem.a134670. [DOI] [PubMed] [Google Scholar]
- 1021.Higashi H., Sasabe T., Fukui Y., Maru M., Kato S. Detection of Gangliosides as N-Glycolylneuraminic Acid-Specific Tumor-Associated Hanganutziu-Deicher Antigen in Human Retinoblastoma Cells. Jpn. J. Cancer Res. 1988;79:952–956. doi: 10.1111/j.1349-7006.1988.tb00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1022.Kawai T., Kato A., Higashi H., Kato S., Naiki M. Quantitative Determination of N-Glycolylneuraminic Acid Expression in Human Cancerous Tissues and Avian Lymphoma Cell Lines as a Tumor-Associated Sialic Acid by Gas Chromatography-Mass Spectrometry. Cancer Res. 1991;51:1242–1246. [PubMed] [Google Scholar]
- 1023.Nöhle U., Beau J.-M., Schauer R. Uptake, Metabolism and Excretion of Orally and Intravenously Administered, Double-Labeled N-Glycoloylneuraminic Acid and Single-Labeled 2-Deoxy-2,3-dehydro-N-acetylneuraminic Acid in Mouse and Rat. Eur. J. Biochem. 1982;126:543–548. doi: 10.1111/j.1432-1033.1982.tb06815.x. [DOI] [PubMed] [Google Scholar]
- 1024.Okerblom J., Varki A. Biochemical, Cellular, Physiological, and Pathological Consequences of Human Loss of N-Glycolylneuraminic Acid. ChemBioChem. 2017;18:1155–1171. doi: 10.1002/cbic.201700077. [DOI] [PubMed] [Google Scholar]
- 1025.Landsteiner K. Zur Kenntnis der antifermentativen, lytischen und agglutinierenden Wirkung des Blutserums und der Lymphe. Zbl. Bakt. 1900;27:357–362. [Google Scholar]
- 1026.Watkins W.M. In: Glycoproteins—Their Composition, Structure and Function. 2nd ed. Gottschalk A., editor. Vol. 5, Part B. B.B.A. Library, Elsevier Publishing Company; Amsterdam, The Netherlands: 1972. Blood-Group Specific Substances; pp. 830–891. [Google Scholar]
- 1027.Race R.R., Sanger R. Blackwell Science; Oxford, UK: 1968. Blood Groups in Man. [Google Scholar]
- 1028.Lisowska E. Biochemistry of M and N Blood Group Specificities. Rev. Franç. Transf. Immuno-hématol. 1981;24:75–84. doi: 10.1016/s0338-4535(81)80028-1. [DOI] [PubMed] [Google Scholar]
- 1029.Kewitz S., Groß H.J., Kosa R., Roelcke D. Anti-Pr Cold Agglutinins Recognize Immunodominant α2,3- or α2,6-Sialyl Groups on Glycophorins. Glycoconjugate J. 1995;12:714–720. doi: 10.1007/BF00731269. [DOI] [PubMed] [Google Scholar]
- 1030.Roelcke D. Kälteagglutinine: Humane monoklonale Antikörper gegen Glykokonjugat-Antigene von Zelloberflächen. Funkt. Biol. Med. 1984;3:106–127. [Google Scholar]
- 1031.Kościelak J. A Hypothesis on the Biological Role of ABH, Lewis and P Blood Group Determinant Structures in Glycosphingolipids and Glycoproteins. Glycoconjugate J. 1986;3:95–108. [Google Scholar]
- 1032.Anstee D.J. The Blood Group MNSs-Active Sialoglycoproteins. Semin. Hematol. 1981;18:13–31. [PubMed] [Google Scholar]
- 1033.Dahr W., Kordowicz M., Moulds J., Gielen W., Lebeck L., Krüger J. Characterization of the Ss Sialoglycoprotein and Its Antigens in Rhnull Erythrocytes. Blut. 1987;54:13–24. doi: 10.1007/BF00326022. [DOI] [PubMed] [Google Scholar]
- 1034.Kannagi R., Roelcke D., Peterson K.A., Okada Y., Levery S.B., Hakomori S.-I. Characterization of an Epitope (Determinant) Structure in a Developmentally Regulated Glycolipid Antigen Defined by a Cold Agglutinin F1, Recognition of α-Sialosyl and α-l-Fucosyl Groups in a Branched Structure. Carbohydr. Res. 1983;120:143–157. doi: 10.1016/0008-6215(83)88013-6. [DOI] [PubMed] [Google Scholar]
- 1035.Aminoff D., Morgan W.T.J. Hexosamine Components of the Human Blood Group Substances. Nature. 1948;162:579–580. doi: 10.1038/162579a0. [DOI] [PubMed] [Google Scholar]
- 1036.Watkins W.M., Morgan W.T.J. Inhibition by Simple Sugars of Enzymes Which Decompose the Blood-Group Substances. Nature. 1955;175:676–677. doi: 10.1038/175676a0. [DOI] [PubMed] [Google Scholar]
- 1037.Pusztai A., Morgan W.T.J. Studies in Immunochemistry—18. The Isolation and Properties of a Sialomucopolysaccharide Possessing Blood-Group Lea Specificity and Virus–Receptor Activity. Biochem. J. 1961;78:135–146. doi: 10.1042/bj0780135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1038.Hewitt L.F. Chemistry of Antibodies and Serum-Proteins—I. Nitrogen Distribution and Amino-Acids. II. Protein Carbohydrate Groups. Biochem. J. 1934;28:2080–2087. doi: 10.1042/bj0282080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1039.Clamp J.R., Johnson I. In: Glycoproteins—Their Composition, Structure and Function. 2nd ed. Gottschalk A., editor. Vol. 5, Part A. B.B.A. Library, Elsevier Publishing Company; Amsterdam, The Netherlands: 1972. Immunoglobulins; pp. 612–652. [Google Scholar]
- 1040.Schultze H.E. Influence of Bound Sialic Acid on Electrophoretic Mobility of Human Serum Proteins. Arch. Biochem. Biophys. 1962;(Suppl. 1):290–294. [PubMed] [Google Scholar]
- 1041.Sarma V.R., Silverton E.W., Davies D.R., Terry W.D. The Three-Dimensional Structure at 6 Å Resolution of a Human γG1 Immunoglobulin Molecule. J. Biol. Chem. 1971;246:3753–3759. [PubMed] [Google Scholar]
- 1042.Burton D.R., Dwek R.A. Sugar Determines Antibody Activity. Science. 2006;313:627–628. doi: 10.1126/science.1131712. [DOI] [PubMed] [Google Scholar]
- 1043.Rademacher T.W., Parekh R.B., Dwek R.A. Glycobiology. Annu. Rev. Biochem. 1988;57:785–838. doi: 10.1146/annurev.bi.57.070188.004033. [DOI] [PubMed] [Google Scholar]
- 1044.Arnold J.N., Wormald M.R., Sim R.B., Rudd P.M., Dwek R.A. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu. Rev. Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 1045.Kaneko Y., Nimmerjahn F., Ravetch J.V. Anti-inflammatory Activity of Immunoglobulin G Resulting From Fc Sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 1046.Pagan J.D., Kitaoka M., Anthony R.M. Engineered Sialylation of Pathogenic Antibodies In Vivo Attenuates Autoimmune Disease. Cell. 2018;172:564–577. doi: 10.1016/j.cell.2017.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1047.Hanganutziu M. Hémagglutinines Hétérogénétiques après Injection de Sérum de Cheval. C. R. Séances Soc. Biol. 1924;91:1457–1459. [Google Scholar]
- 1048.Deicher H. Über die Erzeugung heterospezifischer Hämagglutinine durch Injektion artfremden Serums—I. Mitteilung. Z. Hyg. Infekt. Krankh.: Med. Mikrobiol. Immunol. Virol. 1926;106:561–579. [Google Scholar]
- 1049.Higashi H., Naiki M., Matuo S., Okouchi K. Antigen of “Serum Sickness” Type of Heterophile Antibodies in Human Sera: Identification as Gangliosides With N-Glycolylneuraminic Acid. Biochem. Biophys. Res. Commun. 1977;79:388–395. doi: 10.1016/0006-291x(77)90169-3. [DOI] [PubMed] [Google Scholar]
- 1050.Merrick J.M., Zadarlik K., Milgrom F. Characterization of the Hanganutziu-Deicher (Serum-Sickness) Antigen as Gangliosides Containing N-Glycolylneuraminic Acid. Int. Arch. Allergy Appl. Immun. 1978;57:477–480. doi: 10.1159/000232140. [DOI] [PubMed] [Google Scholar]
- 1051.Tai T., Kawashima I., Furukawa K., Lloyd K.O. Monoclonal Antibody R24 Distinguishes Between Different N-Acetyl- and N-Glycolylneuraminic Acid Derivatives of Ganglioside GD3. Arch. Biochem. Biophys. 1988;260:51–55. doi: 10.1016/0003-9861(88)90423-7. [DOI] [PubMed] [Google Scholar]
- 1052.Richman P.I., Bodmer W.F. Monoclonal Antibodies to Human Colorectal Epithelium: Markers for Differentiation and Tumour Characterization. Int. J. Cancer. 1987;39:317–328. doi: 10.1002/ijc.2910390309. [DOI] [PubMed] [Google Scholar]
- 1053.Smithson J.E., Campbell A., Andrews J.M., Milton J.D., Pigott R., Jewell D.P. Altered Expression of Mucins Throughout the Colon in Ulcerative Colitis. Gut. 1997;40:234–240. doi: 10.1136/gut.40.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1054.Milton J.D., Eccleston D., Parker N., Raouf A., Cubbin C., Hoffman J., Hart C.A., Rhodes J.M. Distribution of O-Acetylated Sialomucin in the Normal and Diseased Gastrointestinal Tract Shown by a New Monoclonal Antibody. J. Clin. Pathol. 1993;46:323–329. doi: 10.1136/jcp.46.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1055.Schwartz-Albiez R., Claus C., Kniep B. In: Leucocyte Typing VII. Mason D., editor. Oxford University Press; Oxford, UK: 2002. CD60 Workshop Report; pp. 185–188. [Google Scholar]
- 1056.Kniep B., Kniep E., Özkucur N., Barz S., Bachmann M., Malisan F., Testi R., Rieber E.P. 9-O-Acetyl GD3 Protects Tumor Cells From Apoptosis. Int. J. Cancer. 2006;119:67–73. doi: 10.1002/ijc.21788. [DOI] [PubMed] [Google Scholar]
- 1057.Varki A., Kornfeld S. An Autosomal Dominant Gene Regulates the Extent of 9-O-Acetylation of Murine Erythrocyte Sialic Acids—A Probable Explanation for the Variation in Capacity to Activate the Human Alternate Complement Pathway. J. Exp. Med. 1980;152:532–544. doi: 10.1084/jem.152.3.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1058.Shi W.-X., Chammas R., Varki N.M., Powell L., Varki A. Sialic Acid 9-O-Acetylation on Murine Erythroleukemia Cells Affects Complement Activation, Binding to I-Type Lectins, and Tissue Homing. J. Biol. Chem. 1996;271:31526–31532. doi: 10.1074/jbc.271.49.31526. [DOI] [PubMed] [Google Scholar]
- 1059.Ourth D.D., Bachinski L.M. Bacterial Sialic Acid Modulates Activation of the Alternative Complement Pathway of Channel Catfish (Ictalurus punctatus) Dev. Comp. Immunol. 1987;11:551–564. doi: 10.1016/0145-305x(87)90044-9. [DOI] [PubMed] [Google Scholar]
- 1060.Manzi A.E., Sjoberg E.R., Diaz S., Varki A. Biosynthesis and Turnover of O-Acetyl and N-Acetyl Groups in the Gangliosides of Human Melanoma Cells. J. Biol. Chem. 1990;265:13091–13103. [PubMed] [Google Scholar]
- 1061.Moe G.R., Bhandari T.S., Flitter B.A. Vaccines Containing De-N-acetyl Sialic Acid Elicit Antibodies Protective Against Neisseria meningitidis Groups B and C. J. Immunol. 2009;182:6610–6617. doi: 10.4049/jimmunol.0803677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1062.Devi S.J.N., Robbins J.B., Schneerson R. Antibodies to Poly[(2 → 8)-α-N-acetylneuraminic Acid] and Poly[(2 → 9)-α-N-acetylneuraminic Acid] Are Elicited by Immunization of Mice With Escherichia coli K92 Conjugates: Potential Vaccines for Groups B and C Meningococci and E. coli K1. Proc. Natl. Acad. Sci. U. S. A. 1991;88:7175–7179. doi: 10.1073/pnas.88.16.7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1063.Krug L.M., Ragupathi G., Ng K.K., Hood C., Jennings H.J., Guo Z., Kris M.G., Miller V., Pizzo B., Tyson L., Baez V., Livingston P.O. Vaccination of Small Cell Lung Cancer Patients With Polysialic Acid or N-Propionylated Polysialic Acid Conjugated to Keyhole Limpet Hemocyanin. Clin. Cancer Res. 2004;10:916–923. doi: 10.1158/1078-0432.ccr-03-0101. [DOI] [PubMed] [Google Scholar]
- 1064.Finne J., Bitter-Suermann D., Goridis C., Finne U. An IgG Monoclonal Antibody to Group B Meningococci Cross-Reacts With Developmentally Regulated Polysialic Acid Units of Glycoproteins in Neural and Extraneural Tissues. J. Immunol. 1987;138:4402–4407. [PubMed] [Google Scholar]
- 1065.Schauer R. Sialic Acids as Antigenic Determinants of Complex Carbohydrates. Adv. Exp. Med. Biol. 1988;228:47–72. doi: 10.1007/978-1-4613-1663-3_2. [DOI] [PubMed] [Google Scholar]
- 1066.Aminoff D., Morrow M.P. Effect of Sialidase on Blood Group Specificity of Hog Submaxillary Glycoproteins. FEBS Lett. 1970;8:353–358. doi: 10.1016/0014-5793(90)80012-8. [DOI] [PubMed] [Google Scholar]
- 1067.Howie A.J., Brown G. Effect of Neuraminidase on the Expression of the 3-Fucosyl-N-acetyllactosamine Antigen in Human Tissues. J. Clin. Pathol. 1985;38:409–416. doi: 10.1136/jcp.38.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1068.Kelley L.K., King B.F., Johnson L.W., Smith C.H. Protein Composition and Structure of Human Placental Microvillous Membrane. Exp. Cell Res. 1979;123:167–176. doi: 10.1016/0014-4827(79)90433-6. [DOI] [PubMed] [Google Scholar]
- 1069.Taylor P.V., Hancock K.W., Gowland G. Effect of Neuraminidase on Immunogenicity of Early Mouse Trophoblast. Transplantation. 1979;28:256–257. doi: 10.1097/00007890-197909000-00020. [DOI] [PubMed] [Google Scholar]
- 1070.Blau E.B., Haas J.E. Glomerular Sialic Acid and Proteinuria in Human Renal Disease. Lab. Invest. 1973;28:477–481. [PubMed] [Google Scholar]
- 1071.Currie G.A., Bagshawe K.D. The Effect of Neuraminidase on the Immunogenicity of the Landschütz Ascites Tumour: Site and Mode of Action. Br. J. Cancer. 1968;22:588–594. doi: 10.1038/bjc.1968.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1072.Sedlacek H.H., Weise M., Lemmer A., Seiler F.R. Immunotherapy of Spontaneous Mammary Tumors in Mongrel Dogs With Autologous Tumor Cells and Neuraminidase. Cancer Immunol. Immunother. 1979;6:47–58. [Google Scholar]
- 1073.Bekesi J.G., Holland J.F. Active Immunotherapy in Leukemia With Neuraminidase-Modified Leukemic Cells. Recent Results Cancer Res. 1977;62:78–89. doi: 10.1007/978-3-642-81174-6_12. [DOI] [PubMed] [Google Scholar]
- 1074.Kaufmann S.H.E., Schauer R., Hahn H. Carbohydrate Surface Constituents of T Cells Mediating Delayed-Type Hypersensitivity That Control Entry Into Sites of Antigen Deposition. Immunobiology. 1981;160:184–195. doi: 10.1016/S0171-2985(81)80046-0. [DOI] [PubMed] [Google Scholar]
- 1075.Bi S., Baum L.G. Sialic Acids in T Cell Development and Function. Biochim. Biophys. Acta. 2009;1790:1599–1610. doi: 10.1016/j.bbagen.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 1076.Wang B., Yu B., Karim M., Hu H., Sun Y., McGreevy P., Petocz P., Held S., Brand-Miller J. Dietary Sialic Acid Supplementation Improves Learning and Memory in Piglets. Am. J. Clin. Nutr. 2007;85:561–569. doi: 10.1093/ajcn/85.2.561. [DOI] [PubMed] [Google Scholar]
- 1077.Wang B. Molecular Mechanism Underlying Sialic Acid as an Essential Nutrient for Brain Development and Cognition. Adv. Nutr. 2012;3:465S–472S. doi: 10.3945/an.112.001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1078.Marchalonis J.J., Edelman G.M. Isolation and Characterization of a Hemagglutinin From Limulus polyphemus. J. Mol. Biol. 1968;32:453–465. doi: 10.1016/0022-2836(68)90022-3. [DOI] [PubMed] [Google Scholar]
- 1079.Roche A.-C., Monsigny M. Purification and Properties of Limulin: A Lectin (Agglutinin) From Hemolymph of Limulus polyphemus. Biochim. Biophys. Acta. 1974;371:242–254. doi: 10.1016/0005-2795(74)90174-3. [DOI] [PubMed] [Google Scholar]
- 1080.Maget-Dana R., Veh R.W., Sander M., Roche A.-C., Schauer R., Monsigny M. Specificities of Limulin and Wheat-Germ Agglutinin Towards Some Derivatives of GM3 Gangliosides. Eur. J. Biochem. 1981;114:11–16. doi: 10.1111/j.1432-1033.1981.tb06164.x. [DOI] [PubMed] [Google Scholar]
- 1081.Fischer E., Khang N.Q., Letendre G., Brossmer R. A Lectin From the Asian Horseshoe Crab Tachypleus tridentatus: Purification, Specificity and Interaction With Tumour Cells. Glycoconjugate J. 1994;11:51–58. doi: 10.1007/BF00732432. [DOI] [PubMed] [Google Scholar]
- 1082.Crocker P.R., Paulson J.C., Varki A. Siglecs and Their Roles in the Immune System. Nat. Rev. Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 1083.Varki A., Schnaar R.L., Crocker P.R. In: Essentials of Glycobiology. 3rd ed. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., Darvill A., Kinoshita T., Packer N.H., Prestegard J.J., Schnaar R.L., Seeberger P.H., editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, USA: 2017. I-Type Lectins; pp. 453–457. [Google Scholar]
- 1084.Sharon N., Lis H. 2nd ed. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2003. Lectins. [Google Scholar]
- 1085.Sharon N., Lis H. History of Lectins: From Hemagglutinins to Biological Recognition Molecules. Glycobiology. 2004;14:53R–62R. doi: 10.1093/glycob/cwh122. [DOI] [PubMed] [Google Scholar]
- 1086.Sharon N. Lectins: Carbohydrate-Specific Reagents and Biological Recognition Molecules. J. Biol. Chem. 2007;282:2753–2764. doi: 10.1074/jbc.X600004200. [DOI] [PubMed] [Google Scholar]
- 1087.Boyd W.C., Shapleigh E. Specific Precipitating Activity of Plant Agglutinins (Lectins) Science. 1954;119:419. doi: 10.1126/science.119.3091.419. [DOI] [PubMed] [Google Scholar]
- 1088.Kilpatrick D.C., Green C. Lectins as Blood Typing Reagents. Adv. Lectin Res. 1992;5:51–94. [Google Scholar]
- 1089.Stillmark H. 1888. Über Ricin, ein giftiges Ferment aus den Samen von Ricinus comm. L. und einigen anderen Euphorbiaceen. Dissertation University of Tartu (Dorpat), Estonia. [Google Scholar]
- 1090.Gabius H.-J., editor. The Sugar Code—Fundamentals of Glycosciences. Wiley–VCH Verlag GmbH; Weinheim, Germany: 2009. [Google Scholar]
- 1091.Flexner S., Noguchi H. Snake Venom in Relation to Haemolysis, Bacteriolysis, and Toxicity. J. Exp. Med. 1902;6:277–301. doi: 10.1084/jem.6.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1092.Morgan W.T.J., Watkins W.M. Unravelling the Biochemical Basis of Blood Group ABO and Lewis Antigenic Specificity. Glycoconjugate J. 2000;17:501–530. doi: 10.1023/a:1011014307683. [DOI] [PubMed] [Google Scholar]
- 1093.Rosenberg A., Howe C., Chargaff E. Inhibition of Influenza Virus Haemagglutination by a Brain Lipid Fraction. Nature. 1956;177:234–235. doi: 10.1038/177234a0. [DOI] [PubMed] [Google Scholar]
- 1094.Böttcher-Friebertshäuser E., Garten W., Matrosovich M., Klenk H.-D. The Hemagglutinin: A Determinant of Pathogenicity. Curr. Top. Microbiol. Immunol. 2014;385:3–34. doi: 10.1007/82_2014_384. [DOI] [PubMed] [Google Scholar]
- 1095.Matrosovich M., Herrler G., Klenk H.-D. In: SialoGlyco Chemistry and Biology II—Tools and Techniques to Identify and Capture Sialoglycans. Gerardy-Schahn R., Delannoy P., von Itzstein M., editors. Vol. 367. Springer; Berlin, Germany: 2015. Sialic Acid Receptors of Viruses; pp. 1–28. (Topics in Current Chemistry). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096.Howe C., Lee L.T., Rose H.M. Collocalia Mucoid: A Substrate for Myxovirus Neuraminidase. Arch. Biochem. Biophys. 1961;95:512–520. doi: 10.1016/0003-9861(61)90184-9. [DOI] [PubMed] [Google Scholar]
- 1097.Kathan R.H., Weeks D.I. Structure Studies of Collocalia Mucoid—I. Carbohydrate and Amino Acid Composition. Arch. Biochem. Biophys. 1969;134:572–576. doi: 10.1016/0003-9861(69)90319-1. [DOI] [PubMed] [Google Scholar]
- 1098.Ogura H., Shoji M. Yôkihi Publishers; Tokyo, Japan: 2004. Origin of Perennial Youth and Long Life From Tsubame no sȗ (Edible Bird's Nest)—For Your Beauty and Health—From Sialic Acids. [Google Scholar]
- 1099.Peumans W.J., Van Damme E.J.M. Lectins as Plant Defense Proteins. Plant Physiol. 1995;109:347–352. doi: 10.1104/pp.109.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1100.Ram S., Sharma A.K., Simpson S.D., Gulati S., McQuillen D.P., Pangburn M.K., Rice P.A. A Novel Sialic Acid Binding Site on Factor H Mediates Serum Resistance of Sialylated Neisseria gonorrhoeae. J. Exp. Med. 1998;187:743–752. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1101.Kazatchkine M.D., Fearon D.T., Austen K.F. Human Alternative Complement Pathway: Membrane-Associated Sialic Acid Regulates the Competition Between B and β1H for Cell-Bound C3b. J. Immunol. 1979;122:75–81. [PubMed] [Google Scholar]
- 1102.Lehmann F., Tiralongo E., Tiralongo J. Sialic Acid-Specific Lectins: Occurrence, Specificity and Function. Cell. Mol. Life Sci. 2006;63:1331–1354. doi: 10.1007/s00018-005-5589-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1103.Wegener H., Mallagaray A., Schöne T., Peters T., Lockhauserbäumer J., Yan H., Uetrecht C., Hansman G.S., Taube S. Human Norovirus GII.4 (MI 001) P Dimer Binds Fucosylated and Sialylated Carbohydrates. Glycobiology. 2017;27:1027–1037. doi: 10.1093/glycob/cwx078. [DOI] [PubMed] [Google Scholar]
- 1104.Li W., Hulswit R.J.G., Widjaja I., Raj V.S., McBride R., Peng W., Widagdo W., Tortorici M.A., van Dieren B., Lang Y., van Lent J.W.M., Paulson J.C., de Haan C.A.M., de Groot R.J., van Kuppeveld F.J.M., Haagmans B.L., Bosch B.-J. Identification of Sialic Acid-Binding Function for the Middle East Respiratory Syndrome Coronavirus Spike Glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2017;117:E8505–E8517. doi: 10.1073/pnas.1712592114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1105.Hellebø A., Vilas U., Falk K., Vlasak R. Infectious Salmon Anemia Virus Specifically Binds to and Hydrolyzes 4-O-Acetylated Sialic Acids. J. Virol. 2004;78:3055–3062. doi: 10.1128/JVI.78.6.3055-3062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1106.Schultze B., Krempl C., Ballesteros M.L., Shaw L., Schauer R., Enjuanes L., Herrler G. Transmissible Gastroenteritis Coronavirus, but Not the Related Porcine Respiratory Coronavirus, Has a Sialic Acid (N-Glycolylneuraminic Acid) Binding Activity. J. Virol. 1996;70:5634–5637. doi: 10.1128/jvi.70.8.5634-5637.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1107.Tack J. 1994. Nachweis 9-O-acetylierter Sialoglykoproteine in menschlicher Nasenschleimhaut. Diploma-Thesis Christian-Albrechts-Universität Kiel. [Google Scholar]
- 1108.Higa H.H., Rogers G.N., Paulson J.C. Influenza Virus Hemagglutinins Differentiate Between Receptor Determinants Bearing N-Acetyl-, N-Glycollyl-, and N,O-Diacetylneuraminic Acids. Virology. 1985;144:279–282. doi: 10.1016/0042-6822(85)90325-3. [DOI] [PubMed] [Google Scholar]
- 1109.Cohnheim J. The New Sydenham Society; London, UK: 1889. Lectures on General Pathology: A Handbook for Practitioners and Students. (Translated by McKee, A. B.) [Google Scholar]
- 1110.Sackstein R. Fulfilling Koch's Postulates in Glycoscience: HCELL, GPS and Translational Glycobiology. Glycobiology. 2016;26:560–570. doi: 10.1093/glycob/cww026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1111.Hadjamu J. Michael Triltsch Verlag; Düsseldorf, Germany: 1974. Professor Dr. med. Heinrich Schade—Begründer der Molekularpathologie 1876–1935—Leben und Werk. (Düsseldorfer Arbeiten zur Geschichte der Medizin, Heft 39) [Google Scholar]
- 1112.Gowans J.L. The Recirculation of Lymphocytes From Blood to Lymph in the Rat. J. Physiol. 1959;146:54–69. doi: 10.1113/jphysiol.1959.sp006177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1113.Marchesi V.T., Gowans J.L. The Migration of Lymphocytes Through the Endothelium of Venules in Lymph Nodes: An Electron Microscope Study. Proc. R. Soc. London, Ser. B. 1964;159:283–290. doi: 10.1098/rspb.1964.0002. [DOI] [PubMed] [Google Scholar]
- 1114.Marchesi V.T., Florey H.W. Electron Micrographic Observations on the Emigration of Leucocytes. Q. J. Exp. Physiol. Cogn. Med. Sci. 1960;45:343–348. doi: 10.1113/expphysiol.1960.sp001489. [DOI] [PubMed] [Google Scholar]
- 1115.Stoolman L.M., Rosen S.D. Possible Role for Cell-Surface Carbohydrate-Binding Molecules in Lymphocyte Recirculation. J. Cell Biol. 1983;96:722–729. doi: 10.1083/jcb.96.3.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1116.Rosen S.D., Singer M.S., Yednock T.A., Stoolman L.M. Involvement of Sialic Acid on Endothelial Cells in Organ-Specific Lymphocyte Recirculation. Science. 1985;228:1005–1007. doi: 10.1126/science.4001928. [DOI] [PubMed] [Google Scholar]
- 1117.Rosen S.D. Ligands for L-Selectin: Homing, Inflammation, and Beyond. Annu. Rev. Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 1118.Natoni A., Macauley M.S., O’Dwyer M.E. Targeting Selectins and Their Ligands in Cancer. Front. Oncol. 2016;6:93. doi: 10.3389/fonc.2016.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1119.Bevilacqua M.P., Stengelin S., Gimbrone M.A., Jr., Seed B. Endothelial Leukocyte Adhesion Molecule 1: An Inducible Receptor for Neutrophils Related to Complement Regulatory Proteins and Lectins. Science. 1989;243:1160–1165. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- 1120.Ley K. In: Mammalian Carbohydrate Recognition Systems. Crocker P., editor. Springer; Berlin, Germany: 2001. Functions of Selectins; pp. 177–200. [Google Scholar]
- 1121.Etzioni A., Frydman M., Pollack S., Avidor I., Phillips M.L., Paulson J.C., Gershoni-Baruch R. Brief Report: Recurrent Severe Infections Caused by a Novel Leukocyte Adhesion Deficiency. N. Engl. J. Med. 1992;327:1789–1792. doi: 10.1056/NEJM199212173272505. [DOI] [PubMed] [Google Scholar]
- 1122.Varki A. Selectins and Other Mammalian Sialic Acid-Binding Lectins. Curr. Opin. Cell Biol. 1992;4:257–266. doi: 10.1016/0955-0674(92)90041-a. [DOI] [PubMed] [Google Scholar]
- 1123.Schnaar R.L. Glycobiology Simplified: Diverse Roles of Glycan Recognition in Inflammation. J. Leukoc. Biol. 2016;99:825–838. doi: 10.1189/jlb.3RI0116-021R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1124.Crocker P.R., Gordon S. Properties and Distribution of a Lectin-Like Hemagglutinin Differentially Expressed by Murine Stromal Tissue Macrophages. J. Exp. Med. 1986;164:1862–1875. doi: 10.1084/jem.164.6.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1125.Crocker P.R., Kelm S., Dubois C., Martin B., McWilliam A.S., Shotton D.M., Paulson J.C., Gordon S. Purification and Properties of Sialoadhesin, a Sialic Acid-Binding Receptor of Murine Tissue Macrophages. EMBO J. 1991;10:1661–1669. doi: 10.1002/j.1460-2075.1991.tb07689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1126.Crocker P.R., Mucklow S., Bouckson V., McWilliam A., Willis A.C., Gordon S., Milon G., Kelm S., Bradfield P. Sialoadhesin, a Macrophage Sialic Acid Binding Receptor for Haemopoietic Cells With 17 Immunoglobulin-Like Domains. EMBO J. 1994;13:4490–4503. doi: 10.1002/j.1460-2075.1994.tb06771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1127.Van den Berg T.K., Brevé J.J.P., Damoiseaux J.G.M.C., Döpp E.A., Kelm S., Crocker P.R., Dijkstra C.D., Kraal G. Sialoadhesin on Macrophages: Its Identification as a Lymphocyte Adhesion Molecule. J. Exp. Med. 1992;176:647–655. doi: 10.1084/jem.176.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1128.Stamenkovic I., Seed B. The B-Cell Antigen CD22 Mediates Monocyte and Erythrocyte Adhesion. Nature. 1990;345:74–77. doi: 10.1038/345074a0. [DOI] [PubMed] [Google Scholar]
- 1129.Macauley M.S., Crocker P.R., Paulson J.C. Siglec-Mediated Regulation of Immune Cell Function in Disease. Nat. Rev. Immunol. 2014;14:653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1130.Trapp B.D. Myelin-Associated Glycoprotein. Location and Potential Functions. Ann. N. Y. Acad. Sci. 1990;605:29–43. doi: 10.1111/j.1749-6632.1990.tb42378.x. [DOI] [PubMed] [Google Scholar]
- 1131.Strenge K., Schauer R., Bovin N., Hasegawa A., Ishida H., Kiso M., Kelm S. Glycan Specificity of Myelin-Associated Glycoprotein and Sialoadhesin Deduced From Interactions With Synthetic Oligosaccharides. Eur. J. Biochem. 1998;258:677–685. doi: 10.1046/j.1432-1327.1998.2580677.x. [DOI] [PubMed] [Google Scholar]
- 1132.Simmons D., Seed B. Isolation of a cDNA Encoding CD33, a Differentiation Antigen of Myeloid Progenitor Cells. J. Immunol. 1988;141:2797–2800. [PubMed] [Google Scholar]
- 1133.Sjoberg E.R., Powell L.D., Klein A., Varki A. Natural Ligands of the B Cell Adhesion Molecule CD22β Can Be Masked by 9-O-Acetylation of Sialic Acids. J. Cell Biol. 1994;126:549–562. doi: 10.1083/jcb.126.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1134.Kelm S., Schauer R., Crocker P.R. The Sialoadhesins—A Family of Sialic Acid-Dependent Cellular Recognition Molecules Within the Immunoglobulin Superfamily. Glycoconjugate J. 1996;13:913–926. doi: 10.1007/BF01053186. [DOI] [PubMed] [Google Scholar]
- 1135.Cao H., Crocker P.R. Evolution of CD33-Related Siglecs: Regulating Host Immune Functions and Escaping Pathogen Exploitation? Immunology. 2010;132:18–26. doi: 10.1111/j.1365-2567.2010.03368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1136.Chang Y.-C., Nizet V. The Interplay Between Siglecs and Sialylated Pathogens. Glycobiology. 2014;24:818–825. doi: 10.1093/glycob/cwu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1137.Crocker P.R., Kannagi R. Introduction to Special Issue: 'Emerging Roles of Siglecs in Health and Disease'. Glycobiology. 2014;24:784. doi: 10.1093/glycob/cwu073. [DOI] [PubMed] [Google Scholar]
- 1138.Padler-Karavani V., Hurtado-Ziola N., Chang Y.-C., Sonnenburg J.L., Ronaghy A., Yu H., Verhagen A., Nizet V., Chen X., Varki N., Varki A., Angata T. Rapid Evolution of Binding Specificities and Expression Patterns of Inhibitory CD33-Related Siglecs in Primates. FASEB J. 2014;28:1280–1293. doi: 10.1096/fj.13-241497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1139.Bochner B.S. “Siglec”ting the Allergic Response for Therapeutic Targeting. Glycobiology. 2016;26:546–552. doi: 10.1093/glycob/cww024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1140.Fontaine G., Biserte G., Montreuil J., Dupont A., Farriaux J.-P., Strecker G., Spik G., Puvion E., Puvion-Dutilleul F., Sezille G., Picqué M.T. La Sialurie: Un Trouble Métabolique Original. Helv. Paediatr. Acta. 1968;23(Suppl. XVII):1–32. [PubMed] [Google Scholar]
- 1141.Strehle E.M. Sialic Acid Storage Disease and Related Disorders. Genet. Test. 2003;7:113–121. doi: 10.1089/109065703322146795. [DOI] [PubMed] [Google Scholar]
- 1142.Gopaul K.P., Crook M.A. The Inborn Errors of Sialic Acid Metabolism and Their Laboratory Investigation. Clin. Lab. 2006;52:155–169. [PubMed] [Google Scholar]
- 1143.Thomas G.H., Reynolds L.W., Miller C.S. Overproduction of N-Acetylneuraminic Acid (Sialic Acid) by Sialuria Fibroblasts. Pediatr. Res. 1985;19:451–455. doi: 10.1203/00006450-198505000-00009. [DOI] [PubMed] [Google Scholar]
- 1144.Aula P., Raivio K., Autio S., Thoden C.-E., Rapola J., Koskela S.-L., Yamashina I. Four Patients With a New Lysosomal Storage Disorder (Salla Disease) Monogr. Hum. Genet. 1978;10:16–22. doi: 10.1159/000401559. [DOI] [PubMed] [Google Scholar]
- 1145.Suzuki K. In: Biology of the Sialic Acids. Rosenberg A., editor. Plenum Press; New York, NY, USA: 1995. Sialic Acid in Biochemical Pathology; pp. 337–361. [Google Scholar]
- 1146.Ҫoker M., Kalkan-Uҫar S., Kitiş Ö., Uҫar H., Gökşen-Şimşek D., Darcan Ş., Gökben S. Salla Disease in Turkish Children: Severe and Conventional Type. Turk. J. Pediatr. 2009;51:605–609. [PubMed] [Google Scholar]
- 1147.Hancock L.W., Thaler M.M., Horwitz A.L., Dawson G. Generalized N-Acetylneuraminic Acid Storage Disease: Quantitation and Identification of the Monosaccharide Accumulating in Brain and Other Tissues. J. Neurochem. 1982;38:803–809. doi: 10.1111/j.1471-4159.1982.tb08701.x. [DOI] [PubMed] [Google Scholar]
- 1148.Tietze F., Seppala R., Renlund M., Hopwood J.J., Harper G.S., Thomas G.H., Gahl W.A. Defective Lysosomal Egress of Free Sialic Acid (N-Acetylneuraminic Acid) in Fibroblasts of Patients With Infantile Free Sialic Acid Storage Disease. J. Biol. Chem. 1989;264:15316–15322. [PubMed] [Google Scholar]
- 1149.Verheijen F.W., Verbeek E., Aula N., Beerens C.E.M.T., Havelaar A.C., Joosse M., Peltonen L., Aula P., Galjaard H., van der Spek P.J., Mancini G.M.S. A New Gene, Encoding an Anion Transporter, Is Mutated in Sialic Acid Storage Diseases. Nat. Genet. 1999;23:462–465. doi: 10.1038/70585. [DOI] [PubMed] [Google Scholar]
- 1150.Maroteaux P., Humbel R. Les Oligosaccharidoses—Un Nouveau Concept. Arch. Franç. Pédiat. 1976;33:641–643. [PubMed] [Google Scholar]
- 1151.Cantz M. In: Sialic Acids—Chemistry, Metabolism and Function. Schauer R., editor. Vol. 10. Springer-Verlag; Vienna, Austria: 1982. Sialidoses; pp. 307–320. (Cell Biology Monographs). [Google Scholar]
- 1152.Cantz M., Gehler J., Spranger J. Mucolipidosis I: Increased Sialic Acid Content and Deficiency of an α-N-Acetylneuraminidase in Cultured Fibroblasts. Biochem. Biophys. Res. Commun. 1977;74:732–738. doi: 10.1016/0006-291x(77)90363-1. [DOI] [PubMed] [Google Scholar]
- 1153.Spranger J., Gehler J., Cantz M. Mucolipidosis I—A Sialidosis. Am. J. Med. Genet. 1977;1:21–29. doi: 10.1002/ajmg.1320010104. [DOI] [PubMed] [Google Scholar]
- 1154.Durand P., Gatti R., Cavalieri S., Borrone C., Tondeur M., Michalski J.-C., Strecker G. Sialidosis (Mucolipidosis I) Helv. Paediatr. Acta. 1977;32:391–400. [PubMed] [Google Scholar]
- 1155.Bonten E., van der Spoel A., Fornerod M., Grosveld G., d’Azzo A. Characterization of Human Lysosomal Neuraminidase Defines the Molecular Basis of the Metabolic Storage Disorder Sialidosis. Genes Dev. 1996;10:3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- 1156.Itoh K., Naganawa Y., Matsuzawa F., Aikawa S., Doi H., Sasagasako N., Yamada T., Kira J.-I., Kobayashi T., Pshezhetsky A.V., Sakuraba H. Novel Missense Mutations in the Human Lysosomal Sialidase Gene in Sialidosis Patients and Prediction of Structural Alterations of Mutant Enzymes. J. Hum. Genet. 2002;47:29–37. doi: 10.1007/s10038-002-8652-7. [DOI] [PubMed] [Google Scholar]
- 1157.Seyrantepe V., Poupetova H., Froissart R., Zabot M.-T., Maire I., Pshezhetsky A.V. Molecular Pathology of NEU1 Gene in Sialidosis. Hum. Mutat. 2003;22:343–352. doi: 10.1002/humu.10268. [DOI] [PubMed] [Google Scholar]
- 1158.D’Azzo A., Hoogeveen A., Reuser A.J.J., Robinson D., Galjaard H. Molecular Defect in Combined β-Galactosidase and Neuraminidase Deficiency in Man. Proc. Natl. Acad. Sci. U. S. A. 1982;79:4535–4539. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Vinogradova M.V., Michaud L., Mezentsev A.V., Lukong K.E., El-Alfy M., Morales C.R., Potier M., Pshezhetsky A.V. Molecular Mechanism of Lysosomal Sialidase Deficiency in Galactosialidosis Involves Its Rapid Degradation. Biochem. J. 1998;330:641–650. doi: 10.1042/bj3300641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1160.Bonten E.J., Wang D., Toy J.N., Mann L., Mignardot A., Yogalingam G., d’Azzo A. Targeting Macrophages With Baculovirus-Produced Lysosomal Enzymes: Implications for Enzyme Replacement Therapy of the Glycoprotein Storage Disorder Galactosialidosis. FASEB J. 2004;18:971–973. doi: 10.1096/fj.03-0941fje. [DOI] [PubMed] [Google Scholar]
- 1161.D’Azzo A., Machado E., Annunziata I. Pathogenesis, Emerging Therapeutic Targets and Treatment in Sialidosis. Expert Opin. Orphan Drugs. 2015;3:491–504. doi: 10.1517/21678707.2015.1025746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1162.Van Pelt J., Kamerling J.P., Bakker H.D., Vliegenthart J.F.G. A Comparative Study of Sialyloligosaccharides Isolated From Sialidosis and Galactosialidosis Urine. J. Inherit. Metab. Dis. 1991;14:730–740. doi: 10.1007/BF01799942. [DOI] [PubMed] [Google Scholar]
- 1163.Yardeni T., Choekyi T., Jacobs K., Ciccone C., Patzel K., Anikster Y., Gahl W.A., Kurochkina N., Huizing M. Identification, Tissue Distribution, and Molecular Modeling of Novel Human Isoforms of the Key Enzyme in Sialic Acid Synthesis, UDP-GlcNAc 2-Epimerase/ManNAc Kinase. Biochemistry. 2011;50:8914–8925. doi: 10.1021/bi201050u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Broccolini A., Gidaro T., Morosetti R., Sancricca C., Mirabella M. Hereditary Inclusion-Body Myopathy With Sparing of the Quadriceps: The Many Tiles of an Incomplete Puzzle. Acta Myol. 2011;30:91–95. [PMC free article] [PubMed] [Google Scholar]
- 1165.Argov Z., Yarom R. “Rimmed Vacuole Myopathy” Sparing the Quadriceps—A Unique Disorder in Iranian Jews. J. Neurol. Sci. 1984;64:33–43. doi: 10.1016/0022-510x(84)90053-4. [DOI] [PubMed] [Google Scholar]
- 1166.Bennmann D., Weidemann W., Thate A., Kreuzmann D., Horstkorte R. Aberrant O-GlcNAcylation Disrupts GNE Enzyme Activity in GNE Myopathy. FEBS J. 2016;283:2285–2294. doi: 10.1111/febs.13729. [DOI] [PubMed] [Google Scholar]
- 1167.Malicdan M.C.V., Noguchi S., Nishino I. Autophagy in a Mouse Model of Distal Myopathy With Rimmed Vacuoles or Hereditary Inclusion Body Myopathy. Autophagy. 2007;3:396–398. doi: 10.4161/auto.4270. [DOI] [PubMed] [Google Scholar]
- 1168.Cho A., Malicdan M.C.V., Miyakawa M., Nonaka I., Nishino I., Noguchi S. Sialic Acid Deficiency Is Associated With Oxidative Stress Leading to Muscle Atrophy and Weakness in GNE Myopathy. Hum. Mol. Genet. 2017;26:3081–3093. doi: 10.1093/hmg/ddx192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1169.Chan Y.M., Lee P., Jungles S., Morris G., Cadaoas J., Skrinar A., Vellard M., Kakkis E. Substantial Deficiency of Free Sialic Acid in Muscles of Patients With GNE Myopathy and in a Mouse Model. PLoS One. 2017;12 doi: 10.1371/journal.pone.0173261. (1–17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1170.Malicdan M.C.V., Noguchi S., Tokutomi T., Goto Y.-I., Nonaka I., Hayashi Y.K., Nishino I. Peracetylated N-Acetylmannosamine, a Synthetic Sugar Molecule, Efficiently Rescues Muscle Phenotype and Biochemical Defects in Mouse Model of Sialic Acid-Deficient Myopathy. J. Biol. Chem. 2012;287:2689–2705. doi: 10.1074/jbc.M111.297051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1171.Huizing M., Krasnewich D.M. Hereditary Inclusion Body Myopathy: A Decade of Progress. Biochim. Biophys. Acta. 2009;1792:881–887. doi: 10.1016/j.bbadis.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]