

I. Introduction
This review covers the following groups of heterocycle-fused acridines: pyridoacridines, pyranoacridines, pyrroloacridines, thienoacridines, and furoacridines.
Heterocycle-fused acridines possess a variety of biological activities, including Ca2 + releasing, antiviral (e.g., anti-HIV), antimicrobial (e.g., antiamebic and antiplasmodium) and antitumor properties. They are also enzyme inhibitors (e.g., topoisomerase II inhibitors and protein tyrosine kinase inhibitors) and have DNA-intercalation and metal-chelating properties.
II. Pyridoacridines
Depending on the ring fusion, pyridoacridines can be classified into the following main classes: pyrido[a]acridines (benzo[j]phenanthrolines), pyrido[b]acridines, pyrido[c]acridines (benzo[b]phenanthrolines), and pyrido[kl] acridines (dibenzo[f,ij][2,7]naphthyridines).

A. PYRIDO[a]ACRIDINES (BENZO[j]PHENANTHROLINES)
The pyridine ring is fused at bond a of the acridine. Depending on the position of the nitrogen in the fused pyridine ring, four different types of pyrido[a]acridines are possible.
1. Pyrido[2,3-a]acridines (Benzo[j][1,7]phenanthrolines)
Dobson et al. (48JCS123) constructed this ring system by using the Ullmann-amine coupling reaction between 7-aminoquinoline 1 and potassium 2,4-dichlorobenzoate, followed by cyclization of the resultant diarylamine 2 with a mixture of POCl3 and PCl5 (Scheme 1 ). The pyridoacridine 3 was then converted to a potential antimalarial compound 4, but poor activity was observed.
SCHEME 1.

(a) Cu bronze, amyl alcohol, 150 °C, 6 h; (b) POCl3/PCl5, 150 °C, 6 h; (c) diethylaminopropylamine, phenol, 100 °C, 2 h.
Gordon et al. (90MI1) obtained a triquinobenzene 5 from a two-step reaction between 1,3,5-tribromobenzene and anthranilic acid. The reaction involved three Ullmann-amine couplings followed by intramolecular acylations.

Reisch et al. (93JHC1469) obtained quino[a]acridone 6 as the main product from the condensation of phloroglucinol and 6-methylanthranilic acid (Scheme 2 ). The synthesis of similar quino[a]acridones from phloroglucinol and anthranilic acids has been reported in patents (35FRP771486).
SCHEME 2.

The condensation product 7 of m-phenylenediamine and 2-formylcyclohexanone, on treatment with polyphosphoric acid (PPA), gave the octahydrobenzophenanthroline 8, which, on dehydrogenation, afforded quino[a]acridine 9 (Scheme 3 ) (74IJC1230). A rearrangement prior to cyclodehydration is involved. The synthesis of similar compounds from 1,3-diiodobenzene and 2-acylanilines, using the Ullmann-amine coupling reaction followed by cyclization, has been reported by Hellwinkel and Ittemann (85LA1501).
SCHEME 3.

(a) EtOH, rt, 65%; (b) PPA, 160–170 °C, 2 h, 40%; (c) Se, 300 °C, 6 h, 58%.
In an attempted reaction toward a pyrido[2,3,4-kl]acridine, Gellerman et al. (92TL5577) isolated a pyrido[2,3-a]acridine 11 from an acid-catalyzed reaction between 1-amino-4-methylacridin-9(10H)one 10 and acetylacetone (Scheme 4 ).
SCHEME 4.

(a) AmOH, H+, 130 °C, 1.5 h, 40%.
2. Pyrido[3,2-a]acridines (Benzo[j][4,7]phenanthrolines)
12-Chloropyrido[3,2-a]acridines 12 were prepared by the route described in Scheme 1, but with 6-aminoquinolines instead of 7-aminoquinoline 46JCS151, 47JCS678. The chloroderivatives 12 were then converted to potent antimalarial agents with dialkylaminoalkylamines or alkylaminoalkylamines in phenol at 100 °C.

The double Ullmann-amine coupling of p-phenylenediamine with 2-chlorobenzoic acid followed by two acid-catalyzed Friedel-Crafts acylations afforded quino[3,2-a]acridone 13 (52JCS1874). A similar regioselectivity was observed when 1,4-diiodobenzene was coupled with aminoacetophenone and the resultant diketone was treated with H2SO4 to give quino[3,2-a]acridine 14 (85LA1501). The quino[3,2-a]acridine 15 was obtained after dehydrogenation of the minor product from p-phenylenediamine and 2-formylcyclohexanone (see II, B, 1 on pyrido[2,3-b]acridines) (74IJC1324).
We have prepared pyrido[3,2-a]acridines, 20 and 21 (96TH1), by Lewis acid-catalyzed cyclization of enamines 18, formed by the condensation of 6-aminoquinoline 16 with 2-cyano- 17a (Z = CN) or 2-ethoxycarbonylcyclohexanone 17b (Z = CO2Et), to give the tetrahydropyrido[2,3-a]acridines 19. Oxidation of these tetrahydro derivatives, with palladium on charcoal, gave the fully aromatic systems 20 (Scheme 5 ). Oxidation of the amino derivative 19a also resulted in the formation of some of the deaminated product 21.
SCHEME 5.

a X = NH2,b X = OH. (a) PhCH3, reflux; (b) A1C13, 190 °C; (c) Pd-on-C, 270–300 °C.
The synthesis of some hexa- and heptacyclic molecules 24 and 25 containing a pyrido[3,2-a]acridine nucleus has been reported, the principal step being the ring closure of anthraquinone-derived precursors 22 and 23 (Scheme 6 ) (84KGS962).
SCHEME 6.

(a) PPA, 170 °C, 96%; (b) PPA, 180 °C, 79%.
3. Pyrido[3,4-a]acridines (Benzo[j][2,7]phenanthrolines)
No natural or synthetic compounds based on this ring system 26 have been reported.

4. Pyrido[4,3-a]acridines (Benzo[j][3,7]phenanthrolines)
No natural or synthetic compounds based on this ring system 27 have been reported.
B. PYRIDO[b]ACRIDINES
The auxiliary pyridine ring is fused at bond b of the acridine nucleus in this class of pyridoacridines and, depending on the position of nitrogen in the auxiliary pyridine, four different types of pyrido[b]acridines are possible.
1. Pyrido[2,3-b]acridines
This ring system can be seen in the pentacyclic alkaloids: ascididemin 28, 2-bromoleptoclinidone 29, 11-hydroxyascididemin 30, 8,9-dihydro-11-hydroxyascididemin 31, calliactine 32, the heptacyclic eilatin 33, and the octacyclic biemnadin 34. (See II, D, 1 on pyrido[2,3,4-kl]acridines.)

The organic pigments, the linear-trans-quinacridones, such as 36, also possess this ring system. These quinacridones can be used in printing ink and as a colorant for plastics (67CRV1). They also exhibit photovoltaic and photoconductive properties 84CL1305, 87CL609. A large number of quinacridones have been synthesized. The synthetic methods have been reviewed by Labana and Labana (67CRV1). The most useful method involves the dehydrogenation of dihydroquinones, such as 35, prepared from diethyl 2,5-dioxo-1,4-cyclohexanedicarboxylate and anilines (Scheme 7 ).
SCHEME 7.

(a) Heat; (b) e.g., chloranil.
Some soluble quinacridones 37 have been prepared by applying this strategy (92JHC167). The introduction of long alkylthio groups into the 4 and 11 positions weakened the intermolecular hydrogen bonding and increased the solubility in different solvents. Diethyl 2,5-dioxo-1,4-cyclohexanedicarboxylate, on acid-catalyzed condensation with 2-aminobenzophenone followed by dehydrogenation with chloranil, afforded quinacridine 38 (88JHC1063).

The synthesis of isoquino[3,4-b]acridine 40 from the interaction of p-phenylenediamine with 2-formylcyclohexanone, followed by ring closure (after in situ rearrangement) and then dehydrogenation of 39, has been reported by Berde et al. (72IJC332). Further examination of the cyclodehydration reaction also gave octahydroquino[3,2-a]acridine 41 as the minor product, which was then dehydrogenated to provide quino[3,2-a]acridine 15 (Scheme 8 ) (74IJC1324).
SCHEME 8.
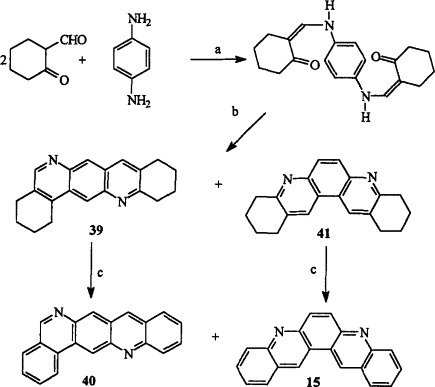
(a) EtOH, rt, 77%; (b) PPA, 180 °C, 3 h, 51% (39), 14% (41); (c) Se, 300–330 °C, 5–10 h, 25% (40), 38% (15).
2. Pyrido[3,2-b]acridines
Morton et al. (93H2757) have described a synthesis of pyrido[3,2-b]acridones 43 and 44 starting from 3-amino-4-methylacridin-9(10H)one 42 (Scheme 9 ). Both compounds were found to be inactive against cultured L1210 cells at concentrations up to 10 μM and showed negligible antibacterial activity.
SCHEME 9.

(a) NaBH4 AcOH, 10 h, 75%; (b) EtOCH = C(CO2Et)2, 155–160 °C, 1.5 h,; (c) PPE, 120–125 °C, 1.5 h, 72% (b,c); (d) NaOH, EtOH, 2 h, 67.5%.
The acid-catalyzed conversion of N,N′-bis(2′-carboxyphenyl)-1,3-diaminobenzene to quino[3,2-b]acridone 45 has been described in a patent (64USP3124581). Kumar and Jain [79IJC(B)623] have reported the synthesis of the octahydroquinolino[b]quinoline 46 from the base-catalyzed condensation of N-benzoyldecahydroquinolin-7-one with 2-aminobenzal-dehyde.

3. Pyrido[3,4-b]acridines
The synthesis of decahydropyrido[3,4-b]acridine 47, by a Friedländer reaction, and its opioid-antagonistic activity (IC50 = 54 nM) has been claimed [92JAP(K)92/275288].
4. Pyrido[4,3-b]acridines
Pyridoacidine alkaloids, eudistone A 48 and B 49, possess this ring as a part of their structures (see pyrido[2,3,4-kl]acridines).

C. PYRIDO[c]ACRIDINES (BENZO[b]PHENANTHROLINES)
The auxiliary pyridine is fused at bond c of the acridine nucleus. Depending upon the position of the nitrogen in the auxiliary pyridine ring, four different types of pyrido[c]acridine are possible.
1. Pyrido[2,3-c]acridines (Benzo[b][1,7]phenanthrolines)
Among the pyridoacridine alkaloids, meridine 50 and cystodamine 51 exhibit this ring skeleton (see II, D, 1 on pyrido[2,3,4-kl]acridines). This ring pattern can also be observed in quino[a]acridines 6 and 9 and triquinobenzene 5 (see II, A, 1 on pyrido[2,3-a]acridines).

In search of potential anti-malarial agents, Dobson et al. (48JCS123) prepared 7,10-dichlorobenzo[b][1,7]phenanthroline 52 by the route described in Scheme 1, but with 5-aminoquinoline 48 as a precursor of 7-diethylaminopropylamino-10-chlorobenzo[b][1,7]phenanthroline 53. No significant activity was observed when compound 53 was tested against Plasmodium gallinaceum (in vivo).

The coupling reaction between 5-aminoquinoline 54 and 2-chlorobenzoic acid gave a very poor yield (5%) of quinolylanthranilic acid 56. With diphenyliodonium-2-carboxylate 55, the yield increased to 80%. Quinolylanthranilic acid 56 was then converted to the sulfonamide 57 after cyclization with POCl3 (Scheme 10 ) (87MI1). The sulfonamide 57 showed negligible activity against L1210 leukemia cells in culture, against P388 leukemia in vivo, and against the Lewis lung solid tumor.
SCHEME 10.

(a) Cu2 + (CH3COO−)2, N-methylpyrrolidone, 95 °C, 12 h, 80%; (b) POCl3, Δ; (c) 4-amino-3-methoxymethanesulfonanilide, H+, MeOH.
Wardani and Lhomme (93TL6411) reported two routes for the synthesis of 10-aminobenzo[b][1,7]phenanthroline 64 from 3,6-diaminoacridine (proflavine) 58. In first route the proflavine 58 was monoacylated to give 59. Activation of the free amino group was achieved by tosylation. Alkylation of the monoacetyl monotosyl proflavine 60 with 3-bromopropionaldehyde ethylene acetal gave the intermediate 61. Acidic treatment afforded 10-aminobenzo[b][1,7]phenanthroline 64 in an 18% overall yield starting from proflavine 58 (Scheme 11 ). The second route involves the Skraup cyclization. The monoacylated proflavine 59 was reacted with acrolein diethyl acetal in refluxing acetic acid. The resultant mixture of 62 and 63 was treated with DDQ in acetic acid to afford 63 as the sole product. Deprotection with 4 M HCl produced 10-aminobenzo[b][1,7]phenanthroline 64 (Scheme 11). The Skraup synthesis of dipyridoacridine 65 from proflavine 58 has also been reported [67JCS(C)1415].
SCHEME 11.
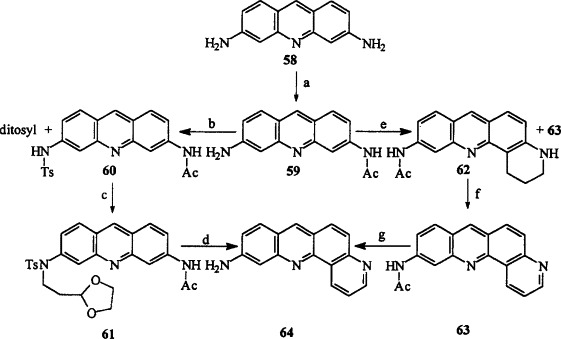
(a) Ac2O, EtCO2H, 20 °C, 10 h, 83%; (b) TsCl, pyridine, Et3N, 4 °C, 10 h, 65% (60); (c) DMF, K2CO3, BrCH2CH2CH(OCH2)2, 80 °C, 3 h, 72%; (d) H2SO4, 1 h, 40%; (e) AcOH, CH2 = CHCH2CH(OC2H5)2, reflux, 3 h; (f) DDQ, AcOH, 100 °C, 15 min.; (g) 4 M HCl, 80 °C, 1 h, 40% (e,f,g).
The synthesis of a number of benzo[b][1,7]phenanthroline anticancer agents from 10-aminobenzo[b][1,7]phenanothroline 64 has been claimed (91MIP1). Thus, 11-formyl-10-hydroxybenzo[b][1,7]phenanthroline 66 exhibited cytotoxicity, with an IC50 of 6.5 μM, against L1210 leukemia cells.

A large number of 1-hydroxy-2-ethoxycarbonylbenzo[b][1,7]phenanthrolines 68 have been synthesized from 3-aminoacridines 67 by using the route shown in Scheme 12 , and potent antimicrobial activities and low toxicities have been claimed (78USP4060527).
SCHEME 12.
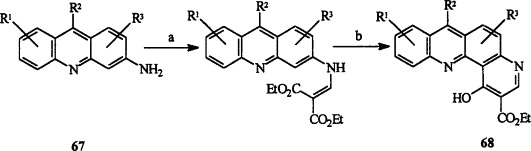
R1, R2, R3 = H, alkyl, aryl, alkylamino, alkylthio, nitro, cyano, alkylsulfonyl, etc. (a) EtOCH = C(CO2Et)2, Δ; (b) Ph2O, 260 °C.
Reisch et al. (93JHC981) described two different methods for the synthesis of 4-azaacronycine 71. One method involves the fusion of 1,3-dihydroxy-10-methyl-9(10H)-acridone 69 with 3-amino-3-methylbut-1-yne in the presence of CuCl2 in a closed ampule, followed by methylation (Scheme 13 ). The second method involves the N-alkylation of 3-amino-1-methoxy-10-methyl-9(10H)-acridone 70 with 3-chloro-3-methylbut-1-yne, followed by in situ cyclization (Scheme 13).
SCHEME 13.

(a) CuCl2, heat, closed ampule; (b) methylation, 10% (a,b); (c) DMF, K2CO3, KI, 120 °C, 8 h, N2, 20%.
We prepared a range of pyrido[2,3-c]acridines 74 by the base- or acid-catalyzed cyclization of the corresponding enamines 72, followed by oxidation of the dihydro derivatives 73 (Scheme 14 ) (96TH1). The aminopyrido[2,3-c]acridine 74a was tested for the inhibition of the spontaneous proliferation of a human gastric carcinoma cell line, MKN 45, and had an IC50 < 1 μmol dm− 3, but was noncytotoxic 95BRP9425409, 95M1P1.
SCHEME 14.

R = H or OMe, R1 = H or OH, R2 = H or OH. (a) NaNH2, DME, reflux, 3 h; (b) H3PO4, 150–160 °C; (c) MnO2, DMF, reflux.
Buu-Hoï [67JCS(C)213] used a quite different method for the construction of the benzo[b][1,7]phenanthroline ring system. Condensation of 2-naphthol with 5-aminoquinolines 75 and paraformaldehyde generated naphthaleno[b][1,7]-phenanthrolines 76 (Scheme 15 ).
SCHEME 15.

(a) Paraformaldehyde, 250 °C, 17% (R = H), 43% (R = Me).
2. Pyrido[3,2-c]acridines (Benzo[b][1,10]phenanthrolines)
Eilatin 33 and eudistone A 48 and B 49 are pyridoacridine alkaloids that possess this ring skeleton. This ring system is also found in a synthetic isomer, isoascidemin 77, of ascididemin 28 (see pyrido[b]acridines and pyrido[2,3,4-kl]acridines). Three other alkaloids, the plakinidines (A–C) 78–80 from Plakortis sponge, also share this ring system 90JA1, 90TL3271.

The synthesis of a number of 7-(mono and dialkylaminoalkylamino) derivatives 81 46JCS151, 47JA1543, 62JMC546, 72JMC739, and 7-anilino derivatives 82 87MI1, 93JPS262 of benzo[b][1,10]phenanthrolines by the route described in Scheme 1, but using 8-aminoquinolines instead of 7-aminoquinoline, and their biological evaluation has been reported. Benzo[b][1,10]phenanthrolin-7-ones 83 were also separated during these syntheses. Some of the alkylamino derivatives 81 were found to be highly effective against ascites tumors at low dosage (72JMC739). The anilino derivatives 82 were found to be active against L1210 murine leukemia (93JPS262), P388 leukemia cells (87MI1), and a Lewis lung solid tumor (87MI1). Using the same strategy, Wilkinson and Finar (48JCS288) have prepared some 7-aminobenzo[b][1,10]phenanthrolines 84 and related compounds. None of the amino derivatives showed significant antibacterial or trypanocidal activity. The same route was used to prepare 7-dimethylamino-propylthiobenzo[b][1,10]phenanthroline 85 as a potential platelet aggregation inhibitor (72JMC61). No significant activity was observed.

The synthesis and antileishmania activity of 8,9,10,11-tetrahydrobenzo[b][1,10]phenanthrolin-7(12H)-one 86 and 7-chloro-8,9,10,11-tetrahydrobenzo[b][1,10]phenanthroline 87 has been described by Satti et al. [93IJC(B)978].

A quite different approach to the construction of the benzo[b][1,10]-phenanthroline ring system, for example, 90, was used by Koft and Case (62JOC865). They used 4-aminoacridone 88 and 4-aminoacridine 89 as the starting materials (Scheme 16 ). The Skraup synthesis was also applied to the preparation of quino[8,7-b][1,10]phenanthroline 91 and its 7-oxo derivative 92 (62JOC865).

SCHEME 16.

(a) HOCH(CH2OH)2, H3AsO4, H2SO4, H2O, 130–140 °C, 2.5 h, 53%; (b) POCl3/PCl5, reflux, 5 h, 81%; (c) 10% Pd on charcoal, H2, EtOH, KOH, 3 h, 36%; (d) Na–Hg, EtOH, H2O, NaHCO3, 4–5 h, 47%; (e) H2C = CHCHO, H3AsO4, H3PO4, 100–110 °C, 1 h, 8.5%.
Condensation of 5,6-dihydroquinolin-8(7H)-ones, such as 93, with 2-aminoaromatic aldehydes, such as 94, afforded dihydrobenzo[b][1,10]phenanthrolines, such as 95 (Scheme 17 ), as precursors of a number of interesting compounds 88JA3673, 90AGE923, 93JOC1666. Condensation of 5,6-dihydroquinolin-8(7H)-one 93 with ternary iminium perchlorates, for example, 96, in the presence of ammonium acetate has been reported to give compounds such as 97 that contain the benzo[b][1,10]phenanthroline ring system (Scheme 18 ) (93TL1775).
SCHEME 17.

SCHEME 18.

Cyclocondensation of 2-aminobenzoylformic acid 98 and cyclohexane-1,2-dione dioxime 99, followed by decarboxylation with concomitant dehydrogenation of the diacid 100, gave quino[3,2-c]acridine 101 (Scheme 19 ) (70JPR1105). The same skeleton 102 was obtained from the Ullmann-amine coupling reaction of 2-aminobenzophenone and 1, 2-diiodobenzene, followed by ring closure (85LA1501).

SCHEME 19.

(a) H2O, reflux, 22%; (b) paraffin oil, N2, 320 °C, 93%.
In a search for dyestuffs, o-phenylenediamine was reacted with 1-nitroan-thraquinone and the resulting bisquinone 103 was cyclized with H2SO4 to give 104 (Scheme 20 ) (74MI1).
SCHEME 20.

(a) Na2CO3, Cu–bronze, PhNO2; (b) 70% H2SO4, 160 °C, 73% (a,b).
3. Pyrido[3,4-c]acridines (Benzo[b][1,8]phenanthrolines)
The pentacyclic pyridoacridine alkaloids (amphimedine 105, neoamphimedine 106, petrosamine 107, and debromopetrosamine 108) also contain this ring skeleton (see pyrido[2,3-4-kl]acridines).
Elslager and Tendick (62JMC546) prepared 7-chloro[b][1,8]phenanthrolines 109, via Ulllmann-amine coupling followed by cyclization, a route described in Scheme 1, but with 5-aminoisoquinoline, and converted them to potential amebicidal 7-(mono- and dialkylaminoalkylamino)benzo[b][1,-8]phenanthrolines 110.

7-Chlorobenzo[b][1,8]phenanthroline 109 (X = H) was later used by Creech et al. (72JMC739) to prepare nitrogen mustard derivatives 111 of benzo[b][1,8]phenanthroline as antitumor agents, and by Sánchez et al. (90H2003) to prepare a series of 7-anilino derivatives of benzo[b][1,8]phenanthroline 114. These anilino derivatives 114 were also prepared to cyclization of 2-(isoquinoline-5′-yl)benzanilides 113 with POCl3 (Scheme 21 ) (90H2003). Biological evaluation of these anilino derivatives 114 along with 7-chlorobenzo[b][1,8]phenanthroline 109 (X = H) and benzo[b][1,8]-phenanthrolin-7(12H)-one 112 showed a tight binding tendency with DNA. Significant inhibition of L1210 murine leukemia cells by benzo[b][1,8]phenanthrolin-7(12H)-one 112 demonstrated that the anilino side chain was unnecessary for activity. No change in the activity was observed on substitution of electron-withdrawing or electron-donating substituents onto the aniline ring. 7-Chlorobenzo[b][1,8]phenanthroline 109 was found to be inactive (93JPS262).
SCHEME 21.

R = F, OMe, NMe2, NHCOMe, SO2Me, SO2NH2, NHSO2Ph, NHSO2-p-C6H4Me. (a) POCl3, reflux, 39%; (b) H2SO4, heat, 60%; (c) PC15; (d) ArNH2, EtOH, MeSO3H; (e) ArNH, C6H6, moderate; (f) POCl3, reflux, 42–62%, (c,d,f); (g) POCl3, PC15; (h) H3O+.
Denny and Baguley (87MI1) used diphenyl iodonium-2-carboxylate 55 as an N-arylating agent in an Ullmann-amine coupling step, as described in Scheme 9, but with 5-aminoisoquinoline, and prepared some 7-anilino-benzo[b][1,8]phenanthrolines 115. None of the anilino derivatives showed significant activity against the P388 leukemia and Lewis lung solid tumor, although they displayed tight binding with DNA. Another derivative 116 of benzo[b][1,8]phenanthroline was tested as an antinociceptive agent but was found to be inactive (91MI1).

The Ullmann–Fetvadjian reaction has been applied to 5-aminoisoquinoline to prepare naphtho[2,1-b][1,8]phenanthrolines, such as 117 (Scheme 22 ) 67JCS(C)213, 80JCS(P1)1233.
SCHEME 22.

In the development of pyridone fused acridines, Kennedy et al. used a sulfoxide-based route to produce 3,7-dimethyl-5-methoxy-1-phenylthio-1,2,3,4-tetrahydro-2-oxobenzo[b][1,8]phenanthroline 118 (Scheme 23 ) [91JCS(P1)2499].
SCHEME 23.

(a) Na2CO3, Cu, PhNO2, reflux, 59%; (b) MCPBA, CH2C12, 0 °C to rt, 87%; (c) H2SO4, AcOH, 130 °C, 37%.
4. Pyrido[4,3-c]acridines (Benzo[b][1,9]phenanthrolines)
Kubo and Nakahara have reported the formation of an isomer 119 of amphimedine 105, which possesses this novel ring system (Scheme 24 ) (88H2095).
SCHEME 24.

(a) 10% Pd/C, Et3N, MeOH, rt 20 h, 18%.
D. PYRIDO[kl]ACRIDINES
In this class, the extra pyridine ring is fused at bonds k and l of the acridine. Three types of pyrido[kl]acridines 120 are possible, depending on the position of the nitrogen in the fused pyridine ring.

1. Pyrido[2,3,4-kl]acridines
a. Isolation and Biological Activity. The polycyclic aromatic alkaloids based on the pyrido[2,3,4-kl]acridine skeleton are members of a fast-growing class of marine sponge and ascidian (tunicate) metabolites. More than 50 alkaloids of this class have been isolated and characterized during the past 12 years.
Norsegoline 121 is the simplest member of this class isolated from Eudistoma sp., a tunicate 88TL3861, 89JOC5331. Other tetracyclic alkaloids include varamines A 122 and B 123, lissoclins A 124 and B 125, diplamine 126, cystodytins A–J 128–137, and pantherinine 138. Varamines A 122 and B 123, isolated from the ascidian Lissoclinum vareau, are brilliant red pigments that were found to be cytotoxic toward L1210 murine leukemia, with IC50 values of 0.03 and 0.05 μg/ml, respectively (89JOC4256). Lissoclins A 124 and B 125, isolated from Lissoclinum sp. collected from the Great Barrier Reef, Australia, did not show significant activity against the fungus Candida albicans (94JOC6600). Diplamine 126, another tetracyclic alkaloid isolated from the tunicate Diplosoma sp., showed cytotoxicity towards L1210 murine leukemia cells (IC50 = 0.02 μg/ml) (89TL4201) and human colon cancer cell lines (IC50 < 1.4 μM) (94JMC3819). DNA intercalation and topoisomerase II inhibition (IC90 = 9.2 μM) by diplamine 126 was also observed (94JMC3819). The isolation of another homolog of this series, “isobutyramide” 127, from an unidentified tunicate has been reported (93CRV1825).
The tunicate Cystodytes dellechiajei is a very rich source of pyrido[2,3,-4-kl]acridine alkaloids. Nine tetracyclic alkaloids, cystodytins A–I 128–136, have been isolated from this species 88JOC1800, 91JNP1634. Except for cystodytin C 130, all other cystodytins were isolated as inseparable isomeric pairs (3.5:1) of cystodytins, β,β-dimethylacrylate and tiglate amides [cystodytins A 128 and B 129, D 131 and E 132, F 133 and G 134, and H 135 and I 136], Cystodytin J 137, isolated from a Fijian Cystodytes sp., was found to be a good DNA intercalator, a potent inhibitor of topoisomerase II (IC90 = 8.0 μM), and a potent cytotoxin against the human colon tumor cell line HCT 116 (IC50 = 1.6 μM) (94JMC3819). Cystodytins A–C 128–130 showed powerful Ca2 + releasing activity in sarcoplasmic reticulum and cytotoxicity against L1210 murine leukemia cells (IC50 ~ 0.2 μg/ml) (88JOC1800). Cytotoxic activity (IC50s = 0.68–1.4 μg/ml) against both L1210 cells and epidermoid carcinoma KB cells was also observed for other cystodytins (91JNP1634). A bromo-substituted tetracyclic alkaloid pantherinine 138 has been isolated from the ascidiam Aplidium pantherinum, and a moderate cytotoxic activity (ED50 = 4.5 μg/ml) against P388 murine leukemia cells has been reported by Kim et al. (93JNP1813).

Pentacyclic alkaloids contain an additional fused heterocyclic ring, such as tetrahydropyridine, pyridine (pyridone), thiazine, or thiazole. Calliactine was shown to be a pyridoacridine alkaloid (87T4023) nearly half a century after its isolation from the Mediterranean anemone Calliactis parasitica in 1940 (40BSF608). Although the exact structure of the alkaloid is still unclear, the structural analysis shows that it contains an additional tetrahydropyridine ring (87T4023). The structure of neocalliactine acetate 139, derived from calliactine by heating with water (aromatization) followed by reaction with acetic anhydride, has been established by a total synthesis 92LA1205, 93H943. On the basis of spectral data and the establishment of the neocalliactine acetate 139 structure, structure 32 is the most favorable among the four proposed by Cimino et al. for calliactine (87T4023).

Amphimedine 105, the first pyridoacridine to be fully characterized, was isolated from the Guamanian sponge, Amphimedon sp. (83JA4835). Its regioisomer, neoamphimedine 106, was isolated from the Micronesian sponge Xestospongia cf. carbonaria, along with amphimedine 105 and debromopetrosamine 108 (93CRV1825). Neoamphimedine 106 was found to be a potent inhibitor of mammalian topoisomerase II (IC50 = 1.3 μM), but not of topoisomerase I. Intercalation of neoamphimedine 106 into DNA was observed with a K m of 2.8 × 105 M − 1 and a binding site size of 1.8 base pairs per molecule. Amphimedine 105, debromopetrosamine 108, and petrosamine 107 (from the sponge (Petrosia sp.) have little effect on topoisomerase I or II activity, despite comparable cytotoxicity (40BSF608).
Ascididemin 28, from Didenum sp. (88TL1177), and 2-bromoleptoclinidone 29, from Leptoclinides sp. 87JA6134, 89TL1069, were the first polycyclic aromatic metabolites to be isolated from ascidians. Both compounds show cytotoxcity toward leukemia cell lines with IC50s of 0.4 μg/ml, whereas ascididemin also inhibits topoisomerase II (IC50 = 75 μM) and causes release of calcium ions in the sarcoplasmic reticulum (91JOC804). Two regioisomers, meridine 50 and 11-hydroxyascididemin 30 were isolated from the ascidians Amphicarpa meridina and Leptoclinides sp., respectively (91JOC804). Both of these isomers, along with 8,9-dihydro-11-hydroxyascididemin 31, have also been isolated from the Okinawan marine sponge Biemna sp. (93T8337). Meridine 50 exhibits cytotoxicity against P388 murine leukemia cells (IC50 = 0.3–0.4 μg/ml) (91JOC804), and 8,9-dihydro-11-hydroxyascididemin 31 exhibits cytotoxicity against human epidermoid carcinoma KB (IC50 = 0.2 μg/ml) and murine lymphoma L1210 (IC50 = 0.7 μg/ml) cells in vitro (93T8337). A new pentacyclic alkaloid, cystodamine 51, has been isolated from the ascidian Cystodytes dellechiajei (94TL7023). The new alkaloid shows cytotoxicity against CEM human leukemic lymphoblasts (IC50 = 1.0 μg/ml).
Shermilamines A 140, B 141, and C 142 are thiazinone-containing pentacyclic alkaloids isolated from the purple tunicate Trididemum sp. (140 and 141) 88JOC4619, 89JOC4231 and a Fijian Cystodytes sp. (141 and 142) (94JMC3819). Shermilamine B 141 has been reported to exhibit cytotoxicity against KB cells (IC50 = 5.0 μg/ml) (91JOC804) and HCT cells (IC50 = 13.8 μM) (94JMC3819), and shermilamine C 142 exhibits cytotoxicity against HCT cells (IC50 = 16.3 μM). Shermaline B 141 and C 142 also inhibit topoisomerase II (IC90 = 118 μM and 138 μM, respectively) (94JMC3819).

A series of pentacyclic aromatic alkaloids that incorporate a thiazole ring were isolated from sponges, ascidians (tunicates), and the lamellariidae molusk Chelynotus semperi. Dercitin 143 88JA4356, 92JOC1523, a metabolite of deep-water sponge Dercitus sp., exhibits a remarkable biological activity. It inhibits a variety of cultured cell clones at nanomolar concentrations and exhibits antitumor activity (in mice) and antiviral activity (against herpes simplex and A-59 murine corona virus) at micromolar concentrations. Burres et al. (89MI2) observed the inhibition of both DNA and RNA syntheses by dercitin 143 by up to 83% at 400 nM and inhibition of protein synthesis to a lesser extent. Inhibition of DNA polymerase and DNase nick translation at 1.0 nM by dercitin 143 was also reported. Dercitin showed a potent intercalation into nucleic acids and little inhibition of topoisomerases (89MI2). New structural types of anti-HIV drugs based on dercitin have been proposed by Taraporewala et al. (92JMC2744).

Nordercitin 144, dercitamine 145, and dercitamide (kuanoniamine C) 148 were also isolated from Dercitus sp. and Stelleta sp., along with dercitin 143 89TL4359, 92JOC1523. Kuanoniamines A–D 146–149 and shermilamine B 141 were found in the mollusc Chelynotus semperi and its prey, an unidentified tunicate (90JOC4426). A new kuanoniamine, the dehydrokuanoniamine B 150, has been isolated along with kuanoniamine D 149 and other alkaloids from a Fijian Cystodytes sp. (94JMC3819). Kuanoniamines A 146, B 147, and D 149 exhibit cytotoxicities against KB cells, with IC50 values of 2.0, > 10, and 1.0 μg/ml, respectively (90JOC4426). Cytotoxicities against HCT cells (IC50 7.8 and 8.3 μM) for dehydrokuanoniamine B 150 and kuanoniamine D 149 were also reported (94JMC3819). Kuanoniamine D 149 can form complexes with Fe(II), Co(II), Cu(II), and Zn(II) ions (92JOC1523).
Two hexacyclic alkaloids, cyclodercitin 151 and stelletamine 152 from Stelleta sp. 89TL4359, 92JOC1523, and three optically active hexacyclic alkaloids, segoline A 153, segoline B 154, and isosegoline A 155 from the Red Sea tunicate, Eudistoma sp., have been isolated along with tetra- and pentacyclic alkaloids 88TL3861, 89JOC5331. Another interesting compound isolated from Eudistoma sp. was the symmetrical, heptacyclic eilatin 33 88TL6655, 94JMC3819. Cytotoxicity (IC50 = 5.3 μM) of eilatin 33 against HCT cell lines has been reported (94JMC3819). This compound was also found to regulate cell growth and to affect cAMP-mediated cellular processes (93MI1). Because of the presence of the 1,10-phenanthroline skeleton, eilatin 33 is capable of chelating metal ions such as Ni(II) (88TL6655).
Two octacyclic alkaloids, eudistones A 48 and B 49, along with ascididemin 28, have been isolated from another tunicate of the genus Eudistoma (from the Seychelles) (91JOC5369). These compounds are optically active, but their absolute configurations are still unknown. Another octacyclic alkaloid, biemnadin 34, isolated from the Okinawan marine sponge Biemna sp., has been shown to exhibit cytotoxicity against human epidermoid carcinoma KB and murine lymphoma L 1210 cells in vitro (93T8337).

b. Syntheses. The biological activity and the novel ring systems of these pyridoacridine alkaloids make them appealing targets for synthesis. A number of approaches have been developed for the synthesis of these compounds.
Imine formation. An example of this route is Echavarren and Stille’s use of a simple intramolecular imine formation between a quinone moiety and an amino group to complete the nucleus (Scheme 25 ) of amphimedine 105 (88JA4051). The quinone 159 was prepared by a palladiumcatalyzed cross-coupling of 5,8-dimethoxyquinolin-4-yl triflate 157 (from 5,8-dimethoxyquinolin-4-one 156) with 2-t-butoxycarbonyl-aminophenyltrimethyltin 158, followed by deprotection of the amino group, reprotection by the triflouroacetyl group, and then oxidation with eerie ammonium nitrate (CAN). The aza-Diels–Alder reaction of the resultant quinone 159 with Ghosez’s diene 160 afforded an intermediate 161. Deprotection of the amino group with aq. HC1 and in situ formation of the imine gave a precursor 162 of the amphimedine 105, which was obtained by methylation with dimethyl sulfate.
SCHEME 25.

(a) (Tf)2O, 2,6-Iutidine, CH2Cl2, 92–95%; (b) Pd(PPh3)4, LiCl, dioxane, 7 h, 100 °C; (c) TFA; (d) TFAA, (iPr)2NEt, 82–87% (b,c,d); (e) CAN, CH3CN/H2O, 23 °C, 15 min; (f) THF, 23 °C, 16 h; (g) pyridinium–HF, 48% (e,f,g); (h) 6 M HCl, THF aq., 70–80 °C, 86% (i) Me2SO4, K2CO3, DME, 96%.
Similar strategies have been employed by Kubo and Nakahara (88H2095) for the synthesis of amphimedine 105; by Szczepankiewicz and Heathcock (94JOC3512) for the synthesis of diplamine 126; by Nakahara et al. (93H1139) for the synthesis of eilatin 33; by Gómez-Bengoa and Echavarren (91JOC3497) for the synthesis of isoascididemin 77, a regioisomer of the naturally occuring ascididemin 28; and by Jolivet et al. for the synthesis of a series of quino- and pyranoquinoacridines, such as 163 and 164 [95JCS(P1)2333].

Nitrene insertion. A nitrene insertion reaction is central to many syntheses of pyridoacridine alkaloids and their analogues. For example, Labarca et al. [87JCS(P1)927] have reported a three-step synthesis of a pyridoacridine 169 starting from 2-methoxyacridine-9-carboxaldehyde 165 (Scheme 26 ). Cyclization of the vinyl azide 166 by thermolysis is believed to involve a nitrene insertion reaction, to give either 167 or 168.
SCHEME 26.

(a) MeO2CCH2N3, NaOMe, MeOH, –10 to 0 °C, 48%; (b) xylene, 140 °C, 78% (167), 19% (168); (c) MnO2/H2SO4, 75%.
Ciufolini and his co-workers have completed the total syntheses of pyridoacridine alkaloids such as cystodytins A 128 (91JA8016), B 129 (91JA8016), and J 137 (92JA10081), diplamine 126 (92JA10081), dercitin 143 92JA10081, 95JA12460, nordercitin 144 92JA10081, 95JA12460, kuanoniamine D 149 92JA10081, 95JA12460, and shermilamine B 141 (92JA10081) by using nitrene insertion methodology. Nitrene insertion is also involved in the Gellerman synthesis of the pyrido [2,3,4-kl] acridines (92TL5577), and a similar approach was used by McKillop and his coworkers for the synthesis of norsegoline 121 and other analogs 92JCS(CC)1453, 93JCS(P1)879.
Cyclodehydration. This route has been used by Bracher for the synthesis of ascididemin 28 (Scheme 27 ) (89H2093). Freshly prepared 2-aminoacetophenone was used for oxidative amination of p-quinolinoquinone 170 in the presence of air and cerium ions, to give intermediate 171, which cyclized to the linear pyridoacridine 172 on heating in a mixture of conc. sulfuric and acetic acids. Condensation of the side-chain methyl group of 172 with dimethylformamide diethyl acetal afforded an enamine 173, which cyclized to ascididemin 28.
SCHEME 27.

(a) O2, Ce(SO4)2, 78%; (b) AcOH/H2SO4, 94%; (c) Me2NCH(OEt)2, DMF; (d) NH4Cl, AcOH, 59% (c,d).
The same strategy has been applied to the preparation of a number of pyridoacridine alkaloids, which include 2-bromoleptoclinidone 29 (90LA205), 11-hydroxyascididemin 30 (93H943) and kuanoniamine A 146 (93H943), and also for the synthesis of neocalliactine acetate 139 92LA1205, 93H943 (a derivative of calliactine 32).
Biomimetic synthesis. Kashman and his co-workers have reported novel biomimetic syntheses of pyrido[2,3,4-kl]acridines by the reaction of kinuramine 174 or its derivatives, such as 177, and other analogs, such as 178, with a variety of diones (e.g., 175), quinones (e.g., 170), and hydroquinones (e.g., 179) (Scheme 28 ) 93TL1823, 93TL1827, 94S239, 94T12959. Using this strategy they have prepared a number of pyridoacridines, including the marine alkaloids, eilatin 33 (93TL1827), ascididemin 28 (94S239), their derivatives, and other analogs such as 176 and 180 93TL1823, 94T12959.
SCHEME 28.

Miscellaneous syntheses. Moody et al. 90TL4375, 92T3589 have described a synthesis of ascididemin 28 that involves the epoxidation of 1,10-phenanthroline 181, ring opening of the epoxide 182 with 2-iodoaniline to afford the amino alcohol 183, and oxidation followed by photocyclization of the o-iminoquinone 184 (Scheme 29 ).
SCHEME 29.

(a) NaCIO, aq.; (b) 2-iodoaniline, Et3Al, CH2Cl2, 79%; (c) Ba(MnO4)2, CH2Cl2, 83%; (d) hv, quartz, H2SO4, 32%.
A novel synthesis of amphimedine 105 has been reported by Prager et al. 89H847, 91AJC277. It involves an azido ring expansion of a pyridyla-zafluorenol 185, by the Schmidt reaction, to 5-(4-pyridyl)benzo[a]-[2,7]naphthyridin-4-one 186, and then refunctionalization to the α-cyano precursor 187, followed by cyclization in polyphosphoric acid (Scheme 30 ). Guillier et al. (95JOC292) have described a new synthesis of the intermediate 186.
SCHEME 30.

(a) Me3SiCl, Et3N, THF, 60 °C; (b) 4-pyridylithium, –40 to 20 °C, 2 h, 87% (a,b); (c) NaN3, PPA, 45 °C, 20 h, 69%; (d) PCl5, DMF (cat.) POCl3,180 °C, 20 h; (e) MeOS2OF, 20 °C, 40 min; (f) KOH, K3[Fe(CN)6], 20 °C, 10 h; (g) CuCN, DMSO, 150 °C, 4 h, 38% (d,e,f,g); (h) PPA, 90 °C, 5 h, 35%.
Taking advantage of the activity of the 9-position of acridines toward active methylenes, Gellerman et al. (92TL5577) have developed a method for the preparation of pyrido[2,3,4-kl]acridines starting from 1-aminoacridines. Thus, acid-catalyzed condensation of acetylacetone with 1-amino-4-methylacridine 188 gave a pyridoacridine 189 (Scheme 31 ).

SCHEME 31.

(a) AmOH, cat. H2SO4, 130 °C, 1.5 h.
2. Pyrido[3,4,5-kl]acridines
No natural or synthetic compounds based on this ring system 190 have been reported.
3. Pyrido[4,3,2-kl]acridines
Only a few examples of this ring system have been reported. Grout et al. [68JCS(C)2689] have cyclized N-acyl derivatives 191 of 9-aminobenzo[a]acridines to pyrido[4,3,2-kl]acridines 192 by heating with AlCl3/NaCl at 200–220 °C (Scheme 32 ).
SCHEME 32.
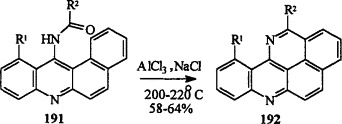
R1 = H, Me; R2 = H, Me.
The only natural product based on this ring system is necatorone 193. This alkaloid was isolated from a toadstool, Lactarius necator (84TL3575). This fungal metabolite showed a considerable mutagenic activity in the Ames test. A synthesis of necatorone 193 involving oxidative cyclization has been reported (Scheme 33 ) (85TL5975).
SCHEME 33.

(a) POCl3, CH3CN, 85–93%; (b) MnO2, C6H6, reflux, 24 h, 90–98%; (c) 48% HBr, 64%; (d) H2/Pd–C, 82–85%; (e) aq. NaOH (5%), O2, 67%.
III. Pyranoacridines
Only a few pyranoacridine ring systems have been reported in the literature.
A. PYRANO[2,3-a]ACRIDINES
The pentacyclic alkaloid bicyclo-N-methylatalaphylline 194, isolated from Atlantia monophylla Correa, possesses this ring system as a part of its structure (72JOC3035). Formation of this ring system (e.g., 196) from isoprene-containing acridone alkaloids or their derivatives (3-OH protected, e.g., 195) has been reported (Scheme 34 ) 70T2905, 82P1771, 83JCS(P1)1681.

SCHEME 34.

Bhavsar and his co-workers reported the formation of pyrano[2,3-a] acridin-2-ones, such as 199, from 8-aroyl-7-hydroxy-benzopyran-2-ones, such as 197, by using the Chapman–Mumm rearrangement of an imino ester 198 (Scheme 35 ) 87MI2, 87MI3.
SCHEME 35.
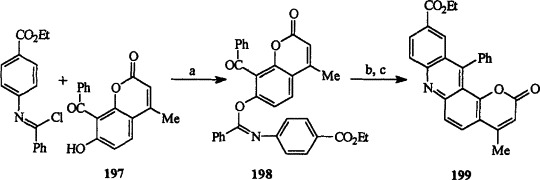
(a) Base, (b) heat, (c) H3O+.
We have described a novel method for the construction of this ring system that involves the condensation of 7-oxo-5,6,7,8-tetrahydroflavone 200 with ethyl anthranilate or anthranilonitrile, followed by base-catalyzed cyclization and then dehydrogenation of the dihydro derivatives, 201 and 203 (Scheme 36 ) [94JCS(P1)173], 12-Amino-2-phenylpyrano[2,3-a]acridin-4-one APPA 204 was found to be a very potent inhibitor (IC50 = 1.9 μmol dm− 3) of the EGF-dependent proliferation of DHER cells, and of the spontaneous proliferation of a human gastric carcinoma cell line, MKN 45, with an IC50 of 0.1 μmol dm− 3. In addition, APPA 204 was tested against 60 cancer cell lines as part of the NCI Developmental Therapeutics Program and was shown to have an activity profile similar to that of known topoisom-erase II inhibitors.
SCHEME 36.

(a) PTSA, toluene, reflux, 47%; (b) PTSA, toluene, reflux, 65%; (c) NaNH2, DME, reflux, 64% (201), 29% (203); (d) Hg(OAc)2, DMSO, 51%; (e) MnO2, toluene, reflux, 72%.
We have also developed an alternative synthesis [97JCS(P1)601] of this ring system that utilizes the von Strandtmann flavone annellation procedure. Thus, the esters 205 were treated with the dimsyl anion to give the β-ketosulfoxides 206, which were cyclized to the pyranoacridinones 207 upon treatment with an aromatic aldehyde in the presence of piperidine (Scheme 37 ).
SCHEME 37.

(a) −CH2SOCH3, DMSO; (b) ArCHO, piperidine, DMSO.
B. PYRANO[2,3-c]ACRIDINES
1. Isolation and Biological Activity
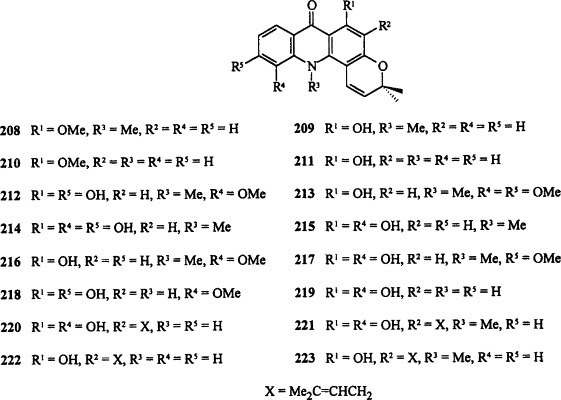
Most of the pyranoacridone alkaloids are based on this ring system. Acronycine (acronine) 208 was the first pyranoacridine alkaloid to be isolated, in 1948 by Hughes and co-workers from the bark of Baurella simplicifolia (Acronychia baueri, an Australian Rutaceae plant) [48NAT(L)223] and in 1949 by Lahey and Thomas from Vepris amphody (49MI1). The correct structure was established in 1966 by degradative studies supported by NMR studies 66AJC275, 66T3245 and finally by X-ray crystallographic studies [70AX(B)853]. Acronycine 208 has broad-spectrum antineoplastic activity 85MI1, 89MI1, 92MI1, although its poor solubility in aqueous media is a major hindrance to its development as a clinical agent. Efforts were continued to isolate more alkaloids based on this skeleton, from other plants of Rutaceae family and also through molecular variation, to improve the cytostatic activity of acronycine 208. Other alkaloids based on this system include noracronycine 209 66T3245, 78MI1, 83JCS(P1)1681, 84JNP285, de-N-methylacronycine 210 78MI1, 83JCS(P1)1681, de-N-methylnoracronycine 211 78MI1, 83JCS(P1)1681, citracridone-I 212 78MI1, 82H273, 83CPB901, 83JCS(P1)1681, 90JCS(P1)1593, citracridone-II 213 82H273, 83CPB895, 90JCS(P1)1593, citracridone-III 214 (91H1781), 11-hydroxynoracronycine 215 82H273, 83CPB895, 83JCS(P1)1681, 90JCS(P1)1593, 11-methoxynoracronycine (baiyamine A) 216 (86H1595), 11-hydroxy-20-methoxynoracronycine 217 (84JNP325), acrifoline 218 (95JNP1629), atalaphyllidine 219 75E1387, 82IJC16, atalaphyllinine 220 82IJC16, 82P1771, 83JCS(P1)1681, N-methylatalaphyllinine (11-hydroxy-N-methylseverifoline) 221 82IJC16, 82P1771, 83JCS(P1)1681, 96P235, severifoline 222 (82P1771), N-methylseverifoline 223 82P1771, 83JCS(P1)1681, acronycine epoxide 224 (88MI3), trans-1,2-dihydroxy-1,2-dihydrocitracridone-I 225 (95H187), (+)-1-hydroxy-1,2-dihydro-de-N-methylacronycine 226 (87H2057), (−)-cis-1,2-dihydroxy-1,2-dihydro-de-N-methylacronycine 227 (86JNP1091), 1-oxo-1,2-dihydro-de-N-methylacro-nycine 228 (87H2057), bicyclo-N-methylatalaphylline 194 (72JOC3035), acrignine 229 (93CPB406), neoacrimarines C 230 and D 232 (93CPB1757), ataline 231 [73JCS(CC)615], glycobisamines A–C 233, 234a, and 234b [93JCS(P1)471], and buntanbismine 235 (96P221). These alkaloids have been isolated from various species of Citrus, Glycosmis, Severinia, Sarcomelicope, and other plants (all Rutaceae family), and some of them have demonstrated significant biological activity. For example, 11-hydroxynoracronycine 215 showed significant effects on Epstein–Barr virus-EA activation (95MI1), whereas glycobisamine A 233 showed in vitro antimalarial activity comparable to that of chloroquine diphosphate (91AAC377).


2. Syntheses
Various approaches have been used to synthesize acronycine 208 and its derivatives. Lahey and Stick’s synthesis (Scheme 38 ) involves the condensation of 2,3-dimethylchroman-5,7-diol 236 with anthranilic acid, followed by N-methylation of the minor product 238 to produce 1,2-dihydronoracronycine 239 (73AJC2311).
SCHEME 38.

(a) ZnCl2, BuOH, reflux, 2 h; (b) Mel, K2CO3, acetone.
Beck et al. (68JA4706) have reported three interrelated syntheses of acronycine. One synthesis (Scheme 39 ) used 5,7-dimethoxy-1,2,3,4-tetrahydroquinolin-2(1H)-one 240, which was coupled with 2-iodobenzoic acid to give the acid 241. Ring closure with PPA, followed by refluxing with methanolic HCl, afforded methyl 1,3-dimethoxy-9-oxacridin-4-propionate 242. Ether cleavage at C-1, followed by reaction with an excess of MeLi, yielded the tertiary alcohol 243. Fusion with pyridinium chloride at high temperature caused O-demethylation at C-3 with concomitant ring closure. Treatment of the crude product with MeI under basic conditions produced dihydronoracronycine 239. Dehydrogenation with DDQ afforded noracronycine 209, which was converted to acronycine 208 by O-methylation with dimethyl sulfate.
SCHEME 39.

(a) 2-Iodobenzoic acid, CuI, nitrobenzene, K2CO3, 165–175 °C, 6.5 h, 40%; (b) PPA, 90 °C, 1.5 h; (c) MeOH, HCl, reflux, 2.5 h, 82% (b,c); (d) BCl3, CH2Cl2; (e) MeLi, ether/HCl, 64% (d,e); (f) pyridinium chloride, 190–200 °C, (g) MeI, K2CO3, acetone, 20% (f,g); (h) DDQ, toluene, 40–45%; (i) Me2SO4, K2CO3, acetone, 16 h, 60%.
Loughhead (90JOC2445) coupled 5-amino-2,2-dimethyl-7-methoxychromene 244 with 2-bromobenzoic acid, and the resultant product 245 was cyclized with TFAA. The de-N-methylacronycine 210 was then converted to acronycine 208 by methylation under phase-transfer conditions (Scheme 40 ). The same approach has been used by Elomri et al. to prepare 6-demethoxyacronycine 246 which was found to be more potent than acronycine 208 in some biological assays (92H799).

SCHEME 40.

(a) 2-Bromobenzoic Acid, Cu(AcO)2, KAcO, Et3N, tBuOH, 58%; (b) TFAA, CH2Cl2, rt, 3 days, 62%; (c) CH3I, PhCH2NEt3Cl, NaOH (aq.), 2-butanone, 96%.
Hlubucek et al. annelated dimethylpyran rings onto 3-hydroxyacridones 247 by a Claisen-type rearrangement of their α,α-dimethyl propargyl ethers 248 (Scheme 41 ) 69CI(L)1809, 70AJC1881. Similar strategies, with some modifications, were used by others to prepare acronycine 208 and its derivatives and analogs 72JMC266, 76JNP399, 82BSB33, 84LA31, 84T5181, 89AP31, 91AP67, 92JHC1293, 92M473, 93JHC1469.
SCHEME 41.

(a) HC ≡ C–C(CH3)2Cl, K2CO3, KI, DMF, 50–70 °C, N2,14–18 h; (b) DMF, 130 °C, 5 h; (c) Me2SO4, DMF, NaH, N2, 45 °C, 1 h, 85%; (d) Me2SO4, DMF, K2CO3, N2, 60 °C, 18 h, 86%.
Bandaranayaka et al. [74JCS(P1)998] devised an efficient synthesis of acronycine 208 that involves the condensation of 1,3-dihydroxyacridin-9(10H)-one 247 (R1 = R2 = H) with 1,1-dimethoxy-3-hydroxy-3-methylbutane in pyridine at 150 °C, followed by methylation of the angular pyranoacridine 211, which was isolated by repeated crystallization. Methylation of the unpurified condensation product also gave isoacronycine 249 and noracronycine 209, in addition to acronycine 208. Use of citral and farnesal in place of 1,1-dimethoxy-3-hydroxy-3-methylbutane provided mono- or diprenyl-substituted acridones and their cyclized product. The same approach was used by Ramesh and Kapil [86IJC(B)684] to prepare 11-hydroxynoracronycine 215 and atalaphyllidine 219.

Lewis and his co-workers have reported three interrelated syntheses of acronycine 208 (81T209). In one synthesis, 2,6-dimethoxy-4-hydroxy-2′-nitrobenzophenone 250, obtained as a minor product from Friedel–Crafts acylation of 3,5-dimethoxyphenol with 2-nitrobenzoylchloride, was treated with 3-chloro-3-methylbut-1f-yne under basic conditions. The resultant nitro compound 251 was reduced to the amine 252 with zinc. Upon reaction with sodium hydride in DMSO, this amine provided a mixture of de-N-methylisoacronycine 253 and de-N-methylacronycine 210. De-N-methylacronycine 210 was converted to acronycine 208 by methylation with methyl iodide (Scheme 42 ).
SCHEME 42.

(a) HC ≡ C–C(CH3)2Cl, DMF, K2CO3, KI, 65 °C, N2, 14 h, 90%; (b) Zn/EtOH, rt 5 days, 98%; (c) NaH, DMSO, rt, 6 days, 29% (210), 43% (253); (d) MeI, K2CO3, acetone, reflux, 11 h, 80%.
Coppola (84JHC913) condensed N-methylisatoic anhydride 254 with the lithium enolate of 2,6,7,8-tetrahydro-2,2-dimethylbenzopyran-5-one 255 to obtain 5,6-dihydro-6-demethoxyacronycine 256. Dehydrogenation with DDQ provided 6-demethoxyacronycine 246 (Scheme 43 ).
SCHEME 43.

(a) LDA, THF, N2, − 65 °C; 56%; (b) DDQ, 88%.
Watanabe et al. (84CPB1264) have reported a one-step synthesis of acronycine 208. Lithium methyl(2-methoxycarbonyl)phenyl amide 258 generated from methyl N-methylanthranilate 257 in situ in the presence of excess lithium cyclohexylamide (LCA), was reacted with 6-bromo-2, 2-dimethyl-7-methoxychromene 259 to produce acronycine 208. A benzyne intermediate 260 is believed to be involved in this reaction (Scheme 44 ).
SCHEME 44.

(a) LCA, THF, N2, − 78 °C; (b) LCA, THF, N2, − 10 °C, 41% (a,b).
A regiospecific synthesis of acronycine 208 from 3-acetyl-4-chloro-2-cyanomethylquinoline 261 has been described by Anand and Sinha (Scheme 45 ) 90H1733, 91JCS(P1)2339. This synthesis involves alkylation of the cyanomethylene 261 with 1-bromo-3-methylbut-2-ene, methanolysis of the resulting nitrile 262 to give the ester 263, and finally, ring closure and hydroxy-dechlorination to afford norglycocitrine II 264. Oxidative cyclization of this acridone 264 with DDQ gave de-N-methylnoracronycine 211, which was converted to acronycine 208 by methylation with methyl iodode. In another reaction, glycocitrine II 265 was converted to noracronycine 209 by oxidative cyclization with benzeneselenyl chloride followed by hydrogen peroxide [83JCS(P1)1681].
SCHEME 45.

(a) (CH3)2C = CHCH2Br, K2CO3, DMF, reflux, 30 h, 48%; (b) MeOH, HCl, 70%; (c) NaH, THF, 3 h; (d) PhOH, 100 °C, 3 h; (e) HCl, MeOH, 10 h, 47% (c,d,e); (f) DDQ, toluene, 52%; (g) NaH, DMF, MeI, 85%.
The syntheses of acronycine 208 and its derivatives, such as 266, 267 reported by Blechert et al., have novelty in that they involve the formation of ring “A” (Scheme 46 ) 78CB439, 80LA503.
SCHEME 46.

(a) TiCl4, CH2Cl2, rt 2 h, 60% (X = Br); (b) heat, 16%; (c) Ac2O; (d) tBuOK; (e) DMSO, pyridine, TFA, DCC, rt 20 h; (f) MeI, K2CO3, acetone, 68% (X = Br); (g) BuLi, ether, 0 °C, 4 h, 38%.
Microbial conversions of acronycine 208 to its hydroxy derivatives have been reported by two research groups 74JMC599, 74JMC653. Among many microbial agents, Aspergillus alleaceus, Cunninghamella echinulata, and Streptomyces spectablicus are found to be effective.
The reaction of organolithium compounds with noracronycine provided 7-substituted derivatives [95JCS(P1)511]. The reaction of acronycine with P4S10 produced the thio analog 268 79JPS36, 82S493, and oxidation of acronycine 208 resulted in one or more products that include acronycine epoxide 224, 1-hydroxy-2-oxo-1,2-dihydroacronycine 269, cis-1,2-dihydroxy-1,2-dihydroacronycine 270, and 5-hydroxyacronycine 271, depending on the nature of the oxidizing agent 86JNP1091, 90M709, 94LA317, 94M731. The synthesis of 1-hydroxy-1,2-dihydroacronycine 272 and 2-hydroxy-1,2-dihydroacronycine 273 from acronycine 208 has also been reported (88MI2).

Reisch and Gunaherath [89JCS(P1)1047] have reported the synthesis of 2,12-dimethyl-6-hydroxypyrano[2,3-c]acridine-3,7(12H)-dione 275 by two different routes starting from the alkylation of 1,3-dihydroxy-10-methylacridin-9(10H)-one 69 to give the α,β-unsaturated ester 274 (Scheme 47 ).
SCHEME 47.
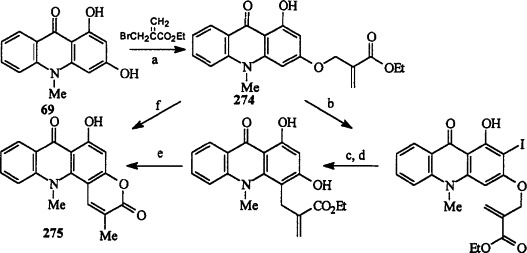
(a) K2CO3, acetone, reflux, 2 h, 73%; (b) I2, HIO4 (aq.), EtOH, rt, 2 h, 90%; (c) Ac2O, pyridine, 100 °C, 3 h, 32%; (d) K2CO3, MeOH, reflux, 15 min, 77%; (e) PEG, 200 °C, 30 min, 71%, (f) PEG, 220 °C, 15 min, 45%.
C. PYRANO[3,2-a]ACRIDINES
The coupling of 2,3-dihydroxy-10-methylacridin-9(10H)-one 276 with 3-chloro-3-methylbut-1-yne afforded a pyrano[3,2-a]acridine 277 (Scheme 48 ) (83MI1).
SCHEME 48.

6-Aminobenzopyran-2-ones 279 undergo the Conrad–Limpach reaction with 2-ethoxycarbonylcyclohexanone 278 to give anils 280, which, on heating in diphenyl ether at reflux, give cyclized products 281 (Scheme 49 ) (83JHC775).
SCHEME 49.

(a) Xylene, reflux, 29–30%; (b) Ph2O, heat, 52%.
We have prepared a series of pyrano[3,2-a]acridines (e.g., 282) by the route described in Scheme 34, but using 6-oxo-5,6,7,8-tetrahydroflavone instead of 7-oxo-5,6,7,8-tetrahydroflavone (84TH1).
D. PYRANO[3,2-b]ACRIDINES
Linear pyranoacridines are very rare in nature. Only a few acridone alkaloids based on the pyrano[3,2-b]acridine ring system have been isolated and characterized. These include pyranofoline 283 and glycofoline 284 from Glycosmis citrifolia (Willd.) Lindl. [83JCS(P1)1681], honyumine 285 from Citrus grandis (86H41) and Citrus funadoko [90JCS(P1)1593], junosidine 286 from Citrus junos Tanaka (87H2077), and yukocitrine 287 from Citrus yuko Hort plant (92H2123) and, with 4-(2′-hydroxy-3′-methylbut-3′-enyl)-yukocitrine 288, from Bosistoa transversa (96P235). Isoacronycine 249 or its de-N-methyl or de-O-methyl precursors have been separated as side products in most of the acronycine 208 syntheses (see pyrano[2,3-c] acridines).

Reisch et al. (91LA685) have reported a regioselective synthesis of isoacronycine 249 from 1,3-dihydroxy-10-methylacridin-9(10H)-one 69 that involves iodination at C-2, followed by palladiumcatalyzed Heck condensation with 3-hydroxy-3-methylbut-1-ene to give isonoracronycine 289. Methylation of isonoracronycine 289 with MeI gave isoacronycine 249 (Scheme 50 ).
SCHEME 50.

(a) I2, H5IO6, EtOH, rt, 92%; (b) Pd(OAc)2, (nBu)4 N+Br, CH2 = CH–C(OH)Me2; NaHCO3, DMSO/DMF, N2, 80 °C, 36 h, 48%; (c) MeI, NaH, THF, 12 h, 80%.
We have prepared a linear pyranoacridine 291 by the condensation of N-benzyl anthranilonitrile with 7-oxo-5,6,7,8-tetrahydroflavone 200, followed by base-catalyzed cyclization of the resultant enamine 290 (Scheme 51 ) (94TH1).
SCHEME 51.

(a) PTSA, toluene, reflux; (b) LDA, THF, N2, − 78 °C.
IV. Pyrroloacridines
Pyrroloacridines have been scarcely reported in the literature; only a few ring systems have been described.
A. PYRROLO[2,3-b]ACRIDINES
Bilgic and Young have reported the formation of the benzo[j]pyrrolo[2,3-b]acridine 294 in a reaction between 1-(N,N-dimethylaminomethyl)naphth-2-ol 292 and 5-aminoindole. A quinone methide 293 is believed to be involved as an intermediate (Scheme 52 ). The reaction of the naphthol 292 with 5-aminoindazole gave the angular pyrazoloacridine 295 [80JCS(P1)1233].
SCHEME 52.

(a) Ph2O, N2, reflux, 16 h, 67% 294, 61% 295.
B. PYRROLO[2,3-c]ACRIDINES
Takagi et al. have synthesized a number of 4,5-dihydropyrrolo[2,3-c] acridines 297 from 4-oxo-4,5,6,7-tetrahydroindoles 296 (Scheme 53 ) (73BSF2807). Syntheses of condensed heterocyclic compounds based on 4-oxotetrahydroindoles have also been reported in the Russian literature (75MI1).
SCHEME 53.

R1 = H, Me, Et, Ph, p An, β-Naph; R2 = Me, Ph.
Another strategy involves the fusion of a pyrrole ring onto the acridine nucleus. The Japp–Klingemann reaction of the diazonium salt 298 of 3-aminoacridine with ethyl 2-methylacetoacetate provided a hydrazone 299, which was cyclized to pyrrolo[2,3-c]acridine 300 in the presence of ZnCl2 (Scheme 54 ) 78KGS1277, 79KGS1092.
SCHEME 54.

(a) Ethyl 2-methylacetoacetate/base, 51%; (b) ZnCl2.
Wardani and Lhomme (93TL6411) used a different pathway for the construction of the pyrrole ring. Base-catalyzed alkylation of N-acetyl-N′-tosylproflavine 60 with bromoacetaldehyde diethyl acetal, followed by deprotection of the acetal function with concomitant ring closure and deacylation in acidic media yielded 9-amino-3-tosylpyrrolo[2,3-c]acridine 301. Detosylation was achieved by basic hydrolysis to give 9-aminopyrrolo[2,3-c]acridine 302 (Scheme 55 ).
SCHEME 55.

(a) DMF, K2CO3, BrCH2CH(OEt)2, 80 °C, 4 days; (b) CH3SO3H/CH2Cl2 (1:9) reflux, 24 h, 40% (a,b); (c) KOH; DMF–H2O, 78 °C., 5 h, 75%.
We have prepared pyrrolo[2,3-c]acridines 304 by our standard method, involving the base-catalyzed cyclization of the enamines 303, followed by oxidation (Scheme 56 ) (96TH1).
SCHEME 56.

(a) NaNH2, DME; (b) MnO2, DMF, reflux.
C. PYRROLO[2,3,4-kl]ACRIDINES
Three polycyclic alkaloids that contain this ring system as part of their structures, plakinidines A 78, B 79, and C 80, were isolated from the marine sponge Plakortis sp. (see II, C, 1 on pyrido[3,2-c]acridines).
Gellerman et al. (94T12959) have described the biomimetic synthesis of a pyrrolo[2,3,4-kl]acridine 305 (Scheme 57 ).
SCHEME 57.

V. Thienoacridines
Only a few thienoacridine ring systems are known, and all are synthetic.
A. THIENO[2,3-c]ACRIDINES
The Pfitzinger reaction of 6,7-dihydrobenzothiophen-4(5H)-ones 306 with isatins 307 produced 4,5-dihydrothieno[2,3-c]acridine-6-carboxylic acids 308 (Scheme 58 ) 50RTC1053, 55BSF1252, 55JCS21, 58JCS2418. Decarboxylation of the acid 308 (R = Me, X = H) upon heating above the melting point has been reported to give the dihydro derivative 309 50RTC1053, 55BSF1252, and Buu-Hoï has reported the decarboxylation with concomitant dehydrogenation of the acid 308 (R = Et, X = Br) to give 6-bromo-2-ethylthieno[2,3-c]acridine 310 (Scheme 58) (58JCS2418).
SCHEME 58.
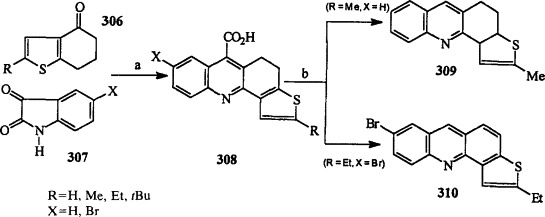
(a) KOH, EtOH, reflux, 10–24 h; (b) Heat > 300 °C.
Remers et al. (71JMC1127) have used a quite different approach, which involves the Vilsmeier–Haack formylation of 6,7-dihydrobenzothiophen-4(5H)-one 306 (R = H) followed by cyclocondensation with aniline to give 4,5-dihydrothieno[2,3-c]acridine 311 (Scheme 59 ).
SCHEME 59.

(a) POCl3–DMF, 100 °C, 1 h, 17%; (b) aniline, AcOH, reflux, 3 h, 67%.
Strekowski et al. (90JOC4777) condensed the 6,7-dihydrobenzothiophen-4(5H)-one 306 (R = H) with 2-trifluoromethylaniline and then cyclized the resultant imine 312 with lithium 4-methylpiperazide to afford this ring system 313 (Scheme 60 ).
SCHEME 60.

(a) Lithium 4-methylpiperazide, Et2O, − 10 °C, 30 min; (b) H3O+.
The Fetvadjian–Ullmann reaction between 4-hydroxy-7-(p-tolyl)benzothiophene 314, aniline, and paraformaldehyde provides another pathway for the construction of thieno[2,3-c]acridines, such as 315 (Scheme 61 ) (81JHC1519).
SCHEME 61.

Suresh et al. (93SUL7) have described a novel route for the preparation of thieno[2,3-c]acridines 316 that involves photocyclization (Scheme 62 ).
SCHEME 62.

We have used the strategy developed for the synthesis of pyrido[2,3-c]acridines to prepare thieno[2,3-c]acridines, such as 317 (96TH1).

B. THIENO[3,2-c]ACRIDINES
Buu-Hoï and Royer (46CR806) obtained a series of thieno[3,2-c]acridines 320 by using the Pfitzinger reaction between 4,5-dihydrobenzothiophene-7(6H)-one 318 and isatins 319, followed by decarboxylation at high temperature (Scheme 63 ). One of the decarboxylated products 320 (R1 = Me, R2 = H) was dehydrogenated with PbO at 310 °C to give the fully aromatic system 321.
SCHEME 63.

R1 = H, Me, Br; R2 = H, Me. (a) KOH, EtOH; (b) heat; (c) PbO, 310 °C.
C. THIENO[3,4-c]ACRIDINES
Isatins 307 on reaction with 1,3-dimethyl-6,7-dihydroisothiophene-4(5H)-one 322 gave 2,3-dimethyl-4,5-dihydrothieno[3,4-c]acridine-6-carboxylic acids 323 (Scheme 64 ) (50RTC1053).
SCHEME 64.

VI. Furoacridines
Only three furoacridine ring systems have been reported.
A. FURO[2,3-a]ACRIDINES
The reactions of copper(I) phenylacetylide 325a and copper(I) isopropenylacetylide 325b with 1-hydroxy-2-iodo-3-methoxy-10-methylacridin-9(10H)-one 324 gave furo[2,3-a]acridones 236a and 236b (Scheme 65 ) (84LA31).
SCHEME 65.

B. FURO[2,3-c]ACRIDINES
1. Isolation
This ring system is the basic skeleton of a number of acridone alkaloids isolated from intact plants or cell tissue cultures of various species from the Rutaceae family. Most of the dihydrofuroacridone alkaloids have been isolated from Ruta graveolens: rutacridone 327 67MI1, 81MI1, 87PHA67, 88MI1, 90PHA500, 91MI3, rutacridone epoxide 328 81MI1, 81ZN200, 82ZN132, 85MI2, 87PHA67, 88MI1, 90PHA500, 91MI2, 20-hydroxyrutacridone epoxide 329 82ZN132, 85MI2, 1-hydroxyrutacrodine epoxide 330 (85MI2), gravacridonol 331 81MI1, 85MI2, gravacridone chloride 332 72P2359, 87PHA67, 88MI1, isogravacridone chloride 333 77P151, 91MI1, gravacridondiol 334 72P2121, 76MI1, 76P240, gravacridondiol acetate 335 (91MI2), gravacridondiol monomethyl ether 336 (72P2121), gravacridontriol 337 76MI1, 76P240, gravacridonolchlorine 338 72P2121, 72P2359, and rutagravin 339 (85MI2). Rutacridone 327 and its epoxide 328 have also been detected in Boenninghausenia albiflora (78P169).


Some of these dihydrofuroacridones, 327, 328, 331, 333, 334, and 337, have been separated by Baumert et al. from the cell culture of Thamnosma montana (94MI1). They have also obtained the glucosides of gravacridonol, gravacridondiol, and gravacridontriol. The last two glucosides were also isolated from roots and tissue culture of Ruta graveolens 76MI1, 76P240.
Three bisacridone alkaloids, citbisamines A 341, B 342, and C 343, containing a C–C bond linkage between the dihydrofuroacridone and the acridone ring systems, have been isolated by Takemura et al. from the roots of Marsh grapefruit (Citrus paradisi) and Hirado-buntan (Citrus grandis) (95CPB1340). Previously, Fraser and Lewis reported the isolation of two dimeric alkaloids (containing O-linkages), atalanine 344 and ataline 231, from Atlantia ceylanica [73JCS(CC)615].

A fully dehydrogenated furoacridine (furacridone = furofoline-I) 345 was detected for the first time in Ruta graveolens by Reisch et al. (77P151) as a mixture with 1-hydroxy-3-methoxy-10-methylacridin-9(10H)-one. Wu et al. [83JCS(P1)1681] were able to isolate furofoline-I 345 and furofoline II 346 from Glycosmis citrifolia (willd.) Lindl.

Hallacridone 347 was isolated from Ruta graveolens by Baumert et al., along with the dihydrofuroacridones 327, 328, and 332 87PHA67, 88MI1. Its structure was revised by Reisch and Gunaherath [89JCS(P1)1047] on the basis of spectroscopic evidence and total synthesis. It was also isolated from tissue cultures of Ruta graveolens (90PHA500) and Thamnosma montana (94MI1). Isolation of two new alkaloids, thehaplosine 348 (93MI2) and furoparadine 349 (95H187), has been achieved from the aerial parts of Halophyllum thesioides and roots of Marsh grapefruit (Rutaceae), respectively.
2. Syntheses
Rutacridone 327 was synthesized for the first time by Mester et al. (81H77) by base-catalyzed alkylation, with concomitant cyclization, of 1,3-dihydroxy-10-methylacridin-9(10H)-one 69 with 1,4-dibromo-2-methylbut-2-ene. The linear isomer, isorutacridone 350, was also obtained as a byproduct (Scheme 66 ). A better yield of rutacridone 327 was obtained when A12O3 was used as the catalyst (90M829). Once again, isorutacridone 350 was obtained as a by-product (Scheme 66).
SCHEME 66.

(a) Na, MeOH, BrCH2CH = C(CH3)CH2Br, 15.5% (327), 5.2% (350); (b) A12O3, ClCH2CH = C(CH3)CH2Br.
Maier et al. have observed that microsomes (from Ruta graveolens cell cultures) catalyze the condensation of 1,3-dihydroxy-10-methylacridin-9(10H)-one 69 with isopentenyl pyrophosphate or dimethylallyl pyrophosphate, in the presence of NADPH and O2, to produce rutacridone 327, and also that the reaction involved glycocitrine-II 265 as an intermediate 90MI2, 93P691. A possible precursor 351 of rutacridone 327 has also been isolated from a reaction of glycocitrine-II 265 with m-chloroperbenzoic acid (MCPBA) (Scheme 67 ) (93CPB383).
SCHEME 67.

Selective hydroxylation of rutacridone 327 with SeO2 in the presence of tBuOOH provided gravacridonol 331 (91LA299), and oxidation with KMnO4 afforded rutagravin 339, isorutagravin 340, gravacridondiol 334, and dihydrohallacridone 353 [69CI(L)1809]. Dehydrogenation of dihydrohallacridone with DDQ produced hallacridone 347 [94JCR(S)157].

To confirm its structure, Reisch et al. have synthesized hallacridone 347 (Scheme 68 ) [89JCS(P1)1047]. Ullmann-amine coupling of 2-chlorobenzoic acid and 3,5-dimethoxyaniline gave an amine 354 that, on treatment with DMF-POCl3, provided 4-formyl-1,3-dimethoxyacridin-9(10H)-one 355. N-Methylation, O-demethylation, and subsequent condensation with 1-chloropropan-2-one in basic media gave hallacridone 347.
SCHEME 68.

(a) POCl3, DMF, rt, 1.5 h, 15%; (b) MeI, Ag2O, DMF, 16 h, 76%; (c) BCI3, CH2C12, rt 72 h, 64%; (d) CICH2COCH3, K2CO3, acetone, reflux, 2 h, 50%.
Takagi and Ueda have prepared a number of 4,5-dihydrofuro[2,3-c]acridines 358a–c from 4,5,6,7-tetrahydrobenzofuran-4-ones 356 by condensing with isatin, anthranilic acid, and 2-aminophenylcarbonyl hydrochlorides 357 using a range of conditions (Scheme 69 ) 71CPB1218, 72CPB380, 72CPB2051. Dehydrogenation of 4,5-dihydrofuro[2,3-c]acridines 358 has also been reported to give the aromatic systems 359.
SCHEME 69.
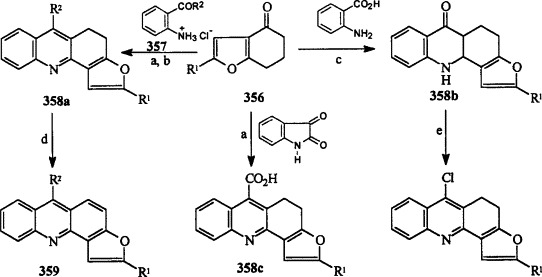
R1 = Me, Ph, pMeO-Ph, pBr-Ph; R2 = Me, Ph. (a) KOH, EtOH, reflux, 50–64 h, 18–31% (358c); (b) heat 110–140 °C, 1 h, 40–71%, (a,b); (c) 120–200 °C, 1 h, 16–38%; (d) Pd/C, 260–290 °C, 15 min, 40–54%; (e) POCl3, 135 °C, 2 h, 71%.
Coppola (84JHC1569) condensed N-methylisatoic anhydride 254 with the lithium enolate of 4,5,6,7-tetrahydrobenzofuran-4-one 356 (R = H) and obtained N-methylfuro[2,3-c]acridin-6-one 361 after dehydrogenation of the resultant 4,5-dihydrofuroacridone 360 (Scheme 70 ).
SCHEME 70.

(a) LDA, − 65 to − 40 °C, 2 h, 67%; (b) DDQ, toluene, 70 °C, 15 min., 100%.
The method of Jayabalan and Shanmugan is novel in that it involves the construction of a ring between a quinoline and furan moieties to complete this skeleton (Scheme 71 ) (91ZN558).
SCHEME 71.

Once again, we have used the strategy developed for the synthesis of pyrido[2,3-c]acridines to prepare furo[2,3-c]acridines 362 (96TH1).
C. FURO[3,2-b]ACRIDINES
Reisch and co-workers have isolated isorutacridone 350 as a by-product during their base-catalyzed (81H77) and A12O3-catalyzed (90M829) synthesis of rutacridone 327 (Scheme 66). They observed that the use of an ion-exchange resin as the catalyst favored the formation of isorutacridone 350 as the major product (81H77). The same group also reported the formation of another linear furoacridine, 4-hydroxy-3-methylene-2,2,10-trimethyl-2,3-dihydrofuro[3,2-b]acridin-5(10H)-one 363 (Scheme 72 ) (89JHC1849).
SCHEME 72.

ACKNOWLEDGMENTS
We thank the Government of Pakistan for the award of a C. O. T. Studentship (to M. A. M.).
REFERENCES
- I. G. Farbenindustrie, A.-G., Fr.Pat. 771,486 (1935) [CA29, 1435 (1935)].
- Lederer E., Tessier G., Huttrer C. Bull. Soc. Chim. Fr. 1940;7:608. [Google Scholar]
- Buu-Hoï Ng.Ph., Royer R. C. R. Hebd. Seances Acad. Sci. 1946;233:806. [PubMed] [Google Scholar]
- Dobson J., Kermack W.O. J. Chem. Soc. 1946;151 doi: 10.1039/jr9460000150. [DOI] [PubMed] [Google Scholar]
- Snyder H.R., Freier H.E. J. Am. Chem. Soc. 1947;69:1543. doi: 10.1021/ja01198a515. [DOI] [PubMed] [Google Scholar]
- Dobson J., Hutchison W.C., Kermack W.O. J. Chem. Soc. 1947;678 doi: 10.1039/jr9470000678. [DOI] [PubMed] [Google Scholar]
- Dobson J., Hutchison W.C., Kermack W.O. J. Chem. Soc. 1948;123 doi: 10.1039/jr9480000123. [DOI] [PubMed] [Google Scholar]
- Wilkinson J.W., Finar I.L. J. Chem. Soc. 1948;288 doi: 10.1039/jr9480000288. [DOI] [PubMed] [Google Scholar]
- Hughes G.K., Lahey F.N., Price J.R., Webb L.J. Nature (London) 1948;162:223. doi: 10.1038/162223a0. [DOI] [PubMed] [Google Scholar]
- Lahey F.N., Thomas W.C. Aust. J. Sci. Res., Ser. A. 1949;2:423. [Google Scholar]
- Buu-Hoï Ng.Ph., Hoán N., Khôi Ng.H. Recl Trav. Chim. Pays-Bas. 1950;69:1053. [Google Scholar]
- Badger G.M., Pettit R. J. Chem. Soc. 1952;1874 [Google Scholar]
- Cagniant P., Cagniant P. Bull. Soc. Chim. Fr. 1955;1252 [Google Scholar]
- Sy M., Buu-Hoï Ng.Ph., Xuong Ng.D. J. Chem. Soc. 1955;21 [Google Scholar]
- Buu-Hoï Ng.Ph. J. Chem. Soc. 1958;2418 [Google Scholar]
- Elslager E.F., Tendick F.H. J. Med. Pharm. Chem. 1962;546 doi: 10.1021/jm01238a015. [DOI] [PubMed] [Google Scholar]
- Koft E., Case F.H. J. Org. Chem. 1962;27:865. [Google Scholar]
- H. Bohler and F. Kehner, U.S. Pat. 3,124,581 (1964) [CA61, 13462 (1964)].
- Macdonald P.L., Robertson A.V. Aust. J. Chem. 1966;19:275. [Google Scholar]
- Govindachari T.R., Pai B.R., Subramanian P.S. Tetrahedron. 1966;22:3245. [Google Scholar]
- Labana S.S., Labana L.L. Chem. Rev. 1967;67:1. [Google Scholar]
- Buu-Hoï Ng.Ph. J. Chem. Soc. C. 1967;213 doi: 10.1039/j39670000213. [DOI] [PubMed] [Google Scholar]
- Dufour M., Buu-Höi Ng.Ph., Jacquignon P. J. Chem. Soc. C. 1967;1415 doi: 10.1039/j39670001415. [DOI] [PubMed] [Google Scholar]
- Reisch J., Szendrei K., Minker E., Novák I. Acta Pharm. Suec. 1967;4:265. [CA68, 39861k (1969)]. [PubMed] [Google Scholar]
- Beck J.R., Kowk R., Booher R.N., Brown A.C., Patterson L.E., Pranc P., Rockey B., Pohland A. J. Am. Chem. Soc. 1968;90:4706. doi: 10.1021/ja01019a036. [DOI] [PubMed] [Google Scholar]
- Grout R.T., Partrige M.W., Sparke J.M., Vipond H.J. J. Chem. Soc. C. 1968;2689 [Google Scholar]
- Hlubucek J., Ritchie E., Taylor W.C. Chem. Ind. (London) 1969;1809 [PubMed] [Google Scholar]
- Hlubucek J., Ritchie E., Taylor W.C. Aust. J. Chem. 1970;23:1881. [Google Scholar]
- Gougoutas J.Z., Kaski B.A. Acta Crystallogr., Sect. B. 1970;B26:853. [Google Scholar]
- Uhlemann E., Kurze P. J. Prakt. Chem. 1970;312:1105. [Google Scholar]
- Govindachari T.R., Viswanathan N., Pai B.R., Ramachandran V.N., Subramaniam P.S. Tetrahedron. 1970;26:2905. [Google Scholar]
- Takagi K., Ueda T. Chem. Pharm. Bull. 1971;19:1218. [Google Scholar]
- Remer W.A., Gibs G.J., Poletto J.F., Weiss M.J. J. Med. Chem. 1971;14:1127. doi: 10.1021/jm00293a033. [DOI] [PubMed] [Google Scholar]
- Takagi K., Ueda T. Chem. Pharm. Bull. 1972;20:380. [Google Scholar]
- Takagi K., Ueda T. Chem. Pharm. Bull. 1972;20:2051. [Google Scholar]
- Berde H.V., Gogte V.N., Tilak B.D. Indian J. Chem. 1972;10:332. [Google Scholar]
- Elslager E.F., Haley N.F., McLean J.R., Potoczak D., Veloso H., Wheelock R.H. J. Med. Chem. 1972;15:61. doi: 10.1021/jm00271a016. [DOI] [PubMed] [Google Scholar]
- Schneider J., Evans E.L., Grunberg E., Fryer R.I. J. Med. Chem. 1972;15:266. doi: 10.1021/jm00273a014. [DOI] [PubMed] [Google Scholar]
- Creech H.J., Preston R.K., Peck R.M., O’Connell A.P. J. Med. Chem. 1972;15:739. doi: 10.1021/jm00277a011. [DOI] [PubMed] [Google Scholar]
- Basu D., Basa S.C. J. Org. Chem. 1972;37:3035. [Google Scholar]
- Reisch J., Rózsa Zs., Szendrei K., Novák I., Minker E. Phytochemistry. 1972;11:2121. [Google Scholar]
- Reisch J., Szendrei K., Rózsa Zs., Novák I., Minker E. Phytochemistry. 1972;11:2359. [Google Scholar]
- Lahey F.N., Stick R.V. Aust. J. Chem. 1973;26:2311. [Google Scholar]
- Takagi K., Kobayashi N., Ueda T. Bull. Soc. Chim. Fr. 1973;9–10:2807. [Google Scholar]
- Fraser A.W., Lewis J.R. J. Chem. Soc., Chem. Commun. 1973;615 [Google Scholar]
- Dali H.M., Gogte V.N., Mullick G.B., Tilak B.D. Indian J. Chem. 1974;12:1230. [Google Scholar]
- Gogte V.N., Mullick G.B., Tilak B.D. Indian J. Chem. 1974;12:1324. [Google Scholar]
- Bandaranayake W.M., Begley M.J., Brown B.O., Clarke D.G., Crombie L., Whiting D.A. J. Chem. Soc., Perkin Trans. 1974;1:998. [Google Scholar]
- Betts R.E., Walters D.E., Rosazza J.P. J. Med. Chem. 1974;17:599. doi: 10.1021/jm00252a006. [DOI] [PubMed] [Google Scholar]
- Brannon D.R., Horton D.H., Svoboda G.H. J. Med. Chem. 1974;17:653. doi: 10.1021/jm00252a019. [DOI] [PubMed] [Google Scholar]
- Ragnekar D.W., Sunthankar S.V. Indian J. Technol. 1974;12:548. [Google Scholar]
- 75E1387.Basa S.C. Experientia. 1975;31:1387. [Google Scholar]
- Shvedov V.I., Altukhova L.B., Grinev A.N., Ermakov A.I., Sheinker Yu.N. v sb. Khim. Farmakol. Indol’n. Soedin. 1975;57 Zh. Khim. Abstr. No. 232ZH1213 [CA84, 121749u] [Google Scholar]
- Adams J.H., Bruce P.J., Lewis J.R. J. Nat. Prod. 1976;39:399. [Google Scholar]
- Rózsa Zs., Kuzovkina I.N., Reisch J., Novák I., Szendrei K., Minker E. Fitoterapia. 1976;47:147. [Google Scholar]
- Reisch J., Rózsa Zs., Szendrei K., Novák I., Minker E. Phytochemistry. 1976;15:240. [Google Scholar]
- Reisch J., Rózsa Zs., Szendrei K., Novák I., Minker E. Phytochemistry. 1977;16:151. [Google Scholar]
- Blechert S., Fichter K.-E., Winterfeldt E. Chem. Ber. 1978;111:439. [Google Scholar]
- Suvorov N.N., Alyab’eva T.M., Khoshtariya T.E. Khim. Geterotsikl. Soedin. 1978;1277 [Google Scholar]
- Fauvel M.T., Gleye J., Moulis C., Fouraste I. Planta Med. Phytother. 1978;12:207. doi: 10.1055/s-2006-957647. [DOI] [PubMed] [Google Scholar]
- Rózsa Zs., Szendrei K., Novák I., Reisch J., Minker E. Phytochemistry. 1978;17:169. [Google Scholar]
- H. Nakamoto, S.-I. Nakamoto, H. Amemiya, S. Miyamura, M. Shiba, and N. Nakamura, U.S. Pat. 4,060,527 (1977) [CA88, 121145b(1978)].
- Kumar Y., Jain P.C. Indian J. Chem., Sect. B. 1979;B17:623. [Google Scholar]
- Dimmock J.R., Repta A.J., Kaminski J. J. Pharm. Sci. 1979;68:36. doi: 10.1002/jps.2600680113. [DOI] [PubMed] [Google Scholar]
- Alyab’eva T.M., Khoshtariya T.E., Vasil’ A.M., ev Tret’yakova L.G., Efimova T.K., Suvorov N.N. Khim. Geterotsikl. Soedin. 1979;1092 [Google Scholar]
- Bilgic O., Young D.W. J. Chem. Soc., Perkin Trans. 1980;1:1233. [Google Scholar]
- Blechert S., Fichter K.-E., Lindner J., Winterfeldt E. Liebigs Ann. Chem. 1980;503 [Google Scholar]
- Mester I., Reisch J., Rózsa Zs., Szendrei K. Heterocycles. 1981;16:77. [Google Scholar]
- Moussa H.H., Abdel-Meguid S. J. Heterocycl. Chem. 1981;18:1519. [Google Scholar]
- Rózsa Zs., Reisch J., Szendrei K., Minker E. Fitoterapia. 1981;52:93. [Google Scholar]
- Adams J.H., Brown P.M., Gupta P., Khan M.S., Lewis J.R. Tetrahedron. 1981;37:209. [Google Scholar]
- Nahrstedt A., Eilert U., Wolters B., Wary V. Z. Naturforsch. C. 1981;36:200. [Google Scholar]
- Smolders R.R., Waefelaer A., Coomans R., Francart D., Hanuise J., Voglet N. Bull. Soc. Chim. Belg. 1982;91:33. [Google Scholar]
- Wu T.-S., Furukawa H., Kuoh C.-S. Heterocycles. 1982;19:273. [Google Scholar]
- Shah J.S., Sabata B.K. Indian J. Chem. B. 1982;21:16. [Google Scholar]
- Wu T.-S., Kuoh C.-S., Furukawa H. Phytochemistry. 1982;21:1771. [Google Scholar]
- Smolders R.R., Hanuise J., Coomans R., Proietto V., Voglet N., Waefelaer A. Synthesis. 1982;493 [Google Scholar]
- Eilert U., Wolters B., Nahrstedt A., Wary V. Z. Naturforsch. C. 1982;37:132. [Google Scholar]
- Wu T.-S., Kuoh C.-S., Furukawa H. Chem. Pharm. Bull. 1983;31:895. [Google Scholar]
- Wu T.-S., Furukawa H. Chem. Pharm. Bull. 1983;31:901. doi: 10.1248/cpb.31.1719. [DOI] [PubMed] [Google Scholar]
- Schmitz F.J., Agarwal S.K., Gunasekera S.P., Schmidt P.G., Shoolery J.N. J. Am. Chem. Soc. 1983;105:4835. [Google Scholar]
- Wu T.S., Furukawa H., Kuoh C.-S., Hsu K.-S. J. Chem. Soc., Perkin Trans. 1983;1:1681. [Google Scholar]
- Merchant J.R., Martyres G., Koshti N.M. J. Heterocycl. Chem. 1983;20:775. [Google Scholar]
- Smolders R.R., Blondiau T., Hanuise J., Voglet N. Ing. Chim. (Brussels. 1983;64:79. [Google Scholar]
- Tomita M., Kusabayashi S., Yokoyama M. Chem. Lett. 1984;1305 [Google Scholar]
- Watanabe M., Kurosaki A., Furukawa S. Chem. Pharm. Bull. 1984;32:1264. [Google Scholar]
- Coppola G.M. J. Heterocycl. Chem. 1984;21:913. [Google Scholar]
- Coppola G.M. J. Heterocycl. Chem. 1984;21:1569. [Google Scholar]
- Funayama S., Cordell G.A. J. Nat. Prod. 1984;47:285. doi: 10.1021/np50032a009. [DOI] [PubMed] [Google Scholar]
- Basa S.C., Tripathy R.N. J. Nat. Prod. 1984;47:325. [Google Scholar]
- Kazankov M.V., Bernadskii M.I., Mustafina M.Y. Khim. Geterosikl. Soedin. 1984;962 [Google Scholar]
- Reisch J., Mester I., El-Moghazy Aly S.M. Liebigs Ann. Chem. 1984;31 [Google Scholar]
- Smolders R.R., Hanuise J., Lepoint T., Voglet N., Tinant B., Declercq J.P., Van Meerssche M. Tetrahedron. 1984;40:5181. [Google Scholar]
- Fugmann B., Steffan B., Steglich W. Tetrahedron Lett. 1984;25:3575. [Google Scholar]
- Hellwinkel D., Ittemann P. Liebigs Ann. Chem. 1985;1501 [Google Scholar]
- Suffnes M., Cordell G.A. In: Brossi A., editor. Vol. 25. Academic Press; New York: 1985. (The Alkaloids). and references therein. [Google Scholar]
- Nahrstedt A., Wary V., Engel B., Reinhard E. Planta Med. 1985;51:517. doi: 10.1055/s-2007-969580. [DOI] [PubMed] [Google Scholar]
- Hilger C.S., Fugmann B., Steglich W. Tetrahedron Lett. 1985;26:5975. [Google Scholar]
- Wu T.-S., Huang S.-C., Joung T.-T., Lai J.-S., Furukawa H. Heterocycles. 1986;24:41. [Google Scholar]
- Ju-ichi M., Inoue M., Aoki K., Furukawa H. Heterocycles. 1986;24:1595. [Google Scholar]
- Ramesh K., Kapil R.S. Indian J. Chem., Sect. B. 1986;B25:684. [Google Scholar]
- Mitaku S., Skaltsounis A.-L., Tillequin F., Koch M., Pusset J., Chauvière G. J. Nat. Prod. 1986;49:1091. [Google Scholar]
- Manabe K., Kusabayashi S., Yokoyama M. Chem. Lett. 1987;609 [Google Scholar]
- Mitaku S., Skaltsounis A.-L., Tillequin F., Koch M. Heterocycles. 1987;26:2057. [Google Scholar]
- Ju-ichi M., Inoue M., Sakiyama K., Yoneda M., Furukawa H. Heterocycles. 1987;26:2077. [Google Scholar]
- Bloor S.J., Schmitz F.J. J. Am. Chem. Soc. 1987;109:6134. [Google Scholar]
- Labarca C.V., MacKenzie A.R., Moody C.J., Rees C.W., Vaquero J.J. J. Chem. Soc., Perkin Trans. 1987;1:927. [Google Scholar]
- Denny W.A., Baguley B.C. Anti-Cancer Drug Des. 1987;2:61. [PubMed] [Google Scholar]
- Bhavsar M.D., Lakhani J.D. Man-Made Text., India. 1987;30:107. 111, 113, 115, 117, 119, 120 [CA108, 96160z (1987)] [Google Scholar]
- Bhavsar M.D., Chavan U.G. Man-Made Text India. 1987;30:224. 225, 230 [CA109, 92715g (1987)] [Google Scholar]
- Baumert A., Gröger D., Schmidt J., Mügge C. Pharmazie. 1987;42:67. [Google Scholar]
- Cimino G., Crispino A., de Rosa S., de Stefano S., Gavagnin M., Sodano G. Tetrahedron. 1987;43:4023. [Google Scholar]
- Kubo A., Nakahara S. Heterocycles. 1988;27:2095. [Google Scholar]
- Bell T.W., Liu J. J. Am. Chem. Soc. 1988;110:3673. [Google Scholar]
- Echavarren A.M., Still J.K. J. Am. Chem. Soc. 1988;110:4051. [Google Scholar]
- Gunawardana G.P., Kohmoto S., Gunasekera S.P., McConnell O.J., Koehn F.E. J. Am. Chem. Soc. 1988;110:4356. [Google Scholar]
- Kitahara K., Nishi H. J. Heterocycl. Chem. 1988;25:1063. [Google Scholar]
- Kobayashi J., Cheng J., Walchli M.R., Nakamura H., Hirata Y., Sasaki T., Ohizumi Y. J. Org. Chem. 1988;53:1800. [Google Scholar]
- Cooray N.M., Scheuer P.J., Parkanyi L., Clardy J. J. Org. Chem. 1988;53:4619. [Google Scholar]
- Baumert A., Gröger D., Schmidt J., Kuzovkina I.N., Mügge C. Fitoterapia. 1988;59:83. [Google Scholar]
- Mitaku S., Skaltsounis A.-L., Tillequin F., Koch M. Planta Med. 1988;54:24. doi: 10.1055/s-2006-962323. [DOI] [PubMed] [Google Scholar]
- Brum-Bousquet M., Mitaku S., Skaltsounis A.-L., Tillequin F., Koch M. Planta Med. 1988;54:470. doi: 10.1055/s-2006-962511. [DOI] [PubMed] [Google Scholar]
- Kobayashi J., Cheng J., Nakamura H., Ohizumi Y., Hirata Y., Sasaki T., Ohta T., Nozoe S. Tetrahedron Lett. 1988;29:1177. [Google Scholar]
- Rudi A., Benayahu Y., Goldberg I., Kashman Y. Tetrahedron Lett. 1988;29:3861. [Google Scholar]
- Rudi A., Benayahu Y., Goldberg I., Kashman Y. Tetrahedron Lett. 1988;29:6655. [Google Scholar]
- Reisch J., Probst W. Arch. Pharm. (Weinheim, Ger.) 1989;322:31. doi: 10.1002/ardp.19893220108. [DOI] [PubMed] [Google Scholar]
- Prager R.H., Tsopelas C., Heisler T. Heterocycles. 1989;29:847. [Google Scholar]
- Bracher F. Heterocycles. 1989;29:2093. [Google Scholar]
- Reisch J., Gunaherath G.M.K.B. J. Chem. Soc., Perkin Trans. 1989;1:1047. [Google Scholar]
- Reisch J., Gunaherath G.M.K.B. J. Heterocycl. Chem. 1989;26:1849. [Google Scholar]
- Carroll A.R., Cooray N.M., Poiner A., Scheuer P.J. J. Org. Chem. 1989;54:4231. [Google Scholar]
- Molinski T.F., Ireland C.M. J. Org. Chem. 1989;54:4256. [Google Scholar]
- Rudi A., Kashman Y. J. Org. Chem. 1989;54:5331. [Google Scholar]
- Dorr R.T., Liddil J.D., Van Hoff D.D., Soble M., Osborne C.K. Cancer Res. 1989;49:340. [PubMed] [Google Scholar]
- Burres N.S., Sazesh S., Gunawardana G.P., Clement J.J. Cancer Res. 1989;49:5267. [PubMed] [Google Scholar]
- de Guzman F.S., Schmitz F.J. Tetrahedron Lett. 1989;30:1069. [Google Scholar]
- Charyulu A.G., McKee T.C., Ireland C.M. Tetrahedron Lett. 1989;30:4201. [Google Scholar]
- Gunawardana G.P., Kohmoto S., Burres N.P. Tetrahedron Lett. 1989;30:4359. [Google Scholar]
- Bell T.W., Liu J. Angew. Chem., Int. Ed. Engl. 1990;29:923. [Google Scholar]
- Anand R.C., Sinha A.K. Heterocycles. 1990;31:1733. [Google Scholar]
- Sánchez E., del Campo C., Avendaño C., Llama E. Heterocycles. 1990;31:2003. [Google Scholar]
- Inman W.D., O’Neill-Johnson M., Crews P. J. Am. Chem. Soc. 1990;112:1. [Google Scholar]
- Furukawa H., Ito C., Mizuno T., Ju-ichi M., Inoue M., Kajiura I., Omura M. J. Chem. Soc., Perkin Trans. 1990;1:1593. [Google Scholar]
- Loughhead D. J. Org. Chem. 1990;55:2445. [Google Scholar]
- Carroll A.R., Scheuer P.J. J. Org. Chem. 1990;55:4426. [Google Scholar]
- Strekowski L., Wydra R.L., Cegla M.T., Czarny A., Harden D.B., Patterson S.E., Battiste M.A., Coxon J.M. J. Org. Chem. 1990;55:4777. [Google Scholar]
- Bracher F. Liebigs Ann. Chem. 1990;205 [Google Scholar]
- Reisch J., Wickramasinghe A. Monatsh. Chem. 1990;121:709. [Google Scholar]
- Reisch J., Wickramasinghe A., Probst W. Monatsh. Chem. 1990;121:829. [Google Scholar]
- Gordon M.D., Jaffe E.E., Foris A. Dyes Pigm. 1990;12:301. [Google Scholar]
- Maier W., Schumann B., Gröger D. FEBS Lett. 1990;263:289. [Google Scholar]
- Probst W., Gröeger D. Pharmazie. 1990;45:500. [Google Scholar]
- West R.R., Mayne C.L., Ireland C.M., Brinen L.C., Clardy J. Tetrahedron Lett. 1990;31:3271. [Google Scholar]
- Moody C.J., Rees C.W., Thomas R. Tetrahedron Lett. 1990;31:4375. [Google Scholar]
- Queener S.F., Fujioka H., Nishiyama Y., Furukawa H., Bartlett M.S., Smith J.W. Antimicrob. Agents Chemother. 1991;35:377. doi: 10.1128/aac.35.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager R.H., Tsopelas C., Heisler T. Aust. J. Chem. 1991;44:277. [Google Scholar]
- Reisch J., Dziemba P. Arch. Pharm. (Weinheim, Ger.) 1991;324:67. doi: 10.1002/ardp.19913240202. [DOI] [PubMed] [Google Scholar]
- Ju-ichi M., Takemura Y., Ito C., Furukawa H. Heterocycles. 1991;32:1781. [Google Scholar]
- Ciufolini M.A., Byrne N.E. J. Am. Chem. Soc. 1991;113:8016. [Google Scholar]
- Anand R.C., Sinha A.K. J. Chem. Soc., Perkin Trans. 1991;1:2339. [Google Scholar]
- Kennedy M.J., Moody C.J., Rees C.W., Thomas R. J. Chem. Soc., Perkin Trans. 1991;1:2499. [Google Scholar]
- Kobayashi J., Tsuda M., Tanabe A., Ishibashi M., Cheng J.F., Yamamura S., Sasaki T. J. Nat. Prod. 1991;54:1634. doi: 10.1021/np50078a022. [DOI] [PubMed] [Google Scholar]
- Schmitz F.J., de Guzman F.S., Hussain M.B., van der Helm D. J. Org. Chem. 1991;56:804. [Google Scholar]
- Gómez-Bengoa E., Echavarren A.M. J. Org. Chem. 1991;56:3497. [Google Scholar]
- He H.Y., Faulkner D.J. J. Org. Chem. 1991;56:5369. [Google Scholar]
- Reisch J., Voerste A.A. Liebigs Ann. Chem. 1991;299 [Google Scholar]
- Reisch J., Hearth H.M.T.B., Kumar N.S. Liebigs Ann. Chem. 1991;685 [Google Scholar]
- Llama E., del Campo C., Capo M. J. Pharm. Pharmacol. 1991;43:68. doi: 10.1111/j.2042-7158.1991.tb05456.x. [DOI] [PubMed] [Google Scholar]
- Paulini H., Popp R., Schimmer O., Ratka O., Röder E. Planta Med. 1991;57:59. doi: 10.1055/s-2006-960019. [DOI] [PubMed] [Google Scholar]
- Paulini H., Waibel R., Kiefer J., Schimmer O. Planta Med. 1991;57:82. doi: 10.1055/s-2006-960027. [DOI] [PubMed] [Google Scholar]
- D. Bigg, J. Lhomme, H. Salez, and A. Wardani, PCT Int. Appl. WO 91/07,403 (1991) [CA115, 207973h(1991)].
- Jayabalan L., Shanmugan P. Z. Naturforsch. B. 1991;46:558. [Google Scholar]
- Elomri A., Michel S., Tillequin F., Koch M. Heterocycles. 1992;34:799. [Google Scholar]
- Takemura Y., Uchida H., Ju-ichi M., Ito C., Nakagawa K., Ono T., Furukawa H. Heterocycles. 1992;34:2123. [Google Scholar]
- Bishop M.J., Ciufolini M.A. J. Am. Chem. Soc. 1992;114:10081. [Google Scholar]
- H. Nagase, H. Wakita, K. Kawai, T. Endo, and O. Matsumoto, Jpn. Kokai Tokkyo Koho JP 92/275,288 (1992) [CA119, 49367a (1993)].
- Ali N.M., Chattopadhyay S.K., McKillop A., Perret-Gentil R.M., Ozturk T., Rebelo R.A. J. Chem. Soc., Chem. Commun. 1992;1453 [Google Scholar]
- Kitahara K., Yanagimoto H., Nakajima N., Nishi H. J Heterocycl. Chem. 1992;29:167. [Google Scholar]
- Reisch J., Dziemba P. J. Heterocycl. Chem. 1992;29:1293. [Google Scholar]
- Taraporewala I.B., Cessac J.W., Chanh T.C., Delgado A.V., Schinazi R.F. J. Med. Chem. 1992;35:2744. doi: 10.1021/jm00093a005. [DOI] [PubMed] [Google Scholar]
- Gunawardana G.P., Koehn F.E., Lee A.Y., Clardy J., He H.Y., Faulkner J.D. J. Org. Chem. 1992;57:1523. [Google Scholar]
- Bracher F. Liebigs Ann. Chem. 1992;1205 [Google Scholar]
- Reisch J., Voerste A.W., Top M., Dziemba P. Monatsh. Chem. 1992;123:473. [Google Scholar]
- Shieh H.L., Pezzuto J.M., Cordeil G.A. Chem.-Biol. Interact. 1992;81:35. doi: 10.1016/0009-2797(92)90025-g. [DOI] [PubMed] [Google Scholar]
- Moody C.J., Rees C.W., Thomas R. Tetrahedron. 1992;48:3589. [Google Scholar]
- Gellerman G., Rudi A., Kashman Y. Tetrahedron Lett. 1992;33:5577. [Google Scholar]
- Ito C., Ono T., Hatano K., Furukawa H. Chem. Pharm. Bull. 1993;41:383. [Google Scholar]
- Takemura Y., Abe M., Ju-ichi M., Ito C., Hatano K., Omura M., Furukawa H. Chem. Pharm. Bull. 1993;41:406. [Google Scholar]
- Takemura Y., Kurozumi T., Ju-ichi M., Okano M., Fukamiya N., Ito C., Ono T., Furukawa H. Chem. Pharm. Bull. 1993;41:1757. [Google Scholar]
- Molinski T.F. Chem. Rev. 1993;93:1825. [Google Scholar]
- Kitahara Y., Nakahara S., Yonezawa T., Nagatsu M., Kubo A. Heterocycles. 1993;36:943. [Google Scholar]
- Nakahara S., Tanaka Y., Kubo A. Heterocycles. 1993;36:1139. [Google Scholar]
- Morton J., Huel C., Bisagni E. Heterocycles. 1993;36:2757. [Google Scholar]
- Satti N.K., Suri K.A., Suri O.P., Kapil A. Indian J. Chem., Sect. B. 1993;B32:978. [Google Scholar]
- Furukawa H., Ito C., Ono T., Wu T.-S., Kuoh C.-S. J. Chem. Soc., Perkin Trans. 1993;1:471. [Google Scholar]
- Dunn S.H., McKillop A. J. Chem. Soc., Perkin Trans. 1993;1:879. [Google Scholar]
- Reisch J., Dziemba P., La Mura M., Rao A.R.R. J. Heterocycl. Chem. 1993;30:981. [Google Scholar]
- Reisch J., Dziema P., Adam T. J. Heterocycl. Chem. 1993;30:1469. [Google Scholar]
- Kim J., Pordesimo E.O., Toth S.I., Schmitz F.J., van Altena I. J. Nat. Prod. 1993;56:1813. doi: 10.1021/np50100a023. [DOI] [PubMed] [Google Scholar]
- Thummel R.P., Chirayil S., Hery C., Lim J.-L., Wang T.-L. J. Org. Chem. 1993;58:1666. [Google Scholar]
- Llama E., del Campo C., Capo M., Anadon M. J. Pharm. Sci. 1993;82:262. doi: 10.1002/jps.2600820309. [DOI] [PubMed] [Google Scholar]
- Shochet N.R., Rudi A., Kashman Y., Hod Y., El-Maghrabi M.R., Spector I. J. Cell. Physiol. 1993;157:481. doi: 10.1002/jcp.1041570307. [DOI] [PubMed] [Google Scholar]
- Ulubelen A., Mericli A.H., Mericli F., Sonmez U., Harsalan R. Nat. Prod. Lett. 1993;1:269. [Google Scholar]
- Maier W., Baumert A., Schumann B., Furukawa H., Gröger D. Pytochemistry. 1993;32:691. [Google Scholar]
- Suresh J.R., Jayabalan L., Shanmugam P. Sulfur Lett. 1993;17:7. [Google Scholar]
- Zeng C.-M., Ishibashi M., Matsumoto K., Nakaike S., Kobayashi J. Tetrahedron. 1993;49:8337. [Google Scholar]
- Westerwelle U., Risch N. Tetrahedron Lett. 1993;34:1775. [Google Scholar]
- Gellerman G., Rudi A., Kashman Y. Tetrahedron Lett. 1993;34:1823. [Google Scholar]
- Gellerman G., Barbad M., Kashman Y. Tetrahedron Lett. 1993;34:1827. [Google Scholar]
- Wardani A., Lhomme J. Tetrahedron Lett. 1993;34:6411. [Google Scholar]
- Reisch J., Dharmaratne H.R.W., Schiwek K., Henkel G. J. Chem. Res., Synop. 1994;157 [Google Scholar]
- Groundwater P.W., Solomons K.R.H. J. Chem. Soc., Perkin Trans. 1994;1:173. [Google Scholar]
- McDonald L.A., Eldredge G.S., Barrows L.R., Ireland C.M. J. Med. Chem. 1994;37:3819. doi: 10.1021/jm00048a017. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz B.G., Heathcock C.H. J. Org. Chem. 1994;59:3512. [Google Scholar]
- Searle P.A., Molinski T.F. J. Org. Chem. 1994;59:6600. [Google Scholar]
- Reisch J., Schiwek K. Liebigs Ann. Chem. 1994;317 [Google Scholar]
- Reisch J., Schiwek K. Monatsh. Chem. 1994;124:731. [Google Scholar]
- Baumert A., Maier W., Matern U., Schmidt J., Schumann B., Gröger D. Planta Med. 1994;60:143. doi: 10.1055/s-2006-959437. [DOI] [PubMed] [Google Scholar]
- Gellerman G., Rudi A., Kashman Y. Synthesis. 1994;239 [Google Scholar]
- Gellerman G., Rudi A., Kashman Y. Tetrahedron. 1994;50:12959. [Google Scholar]
- Solomons K.R.H. University of Wales; Cardiff: 1994. Ph.D. Thesis. [Google Scholar]
- Bontemps N., Bonnard I., Banaigs B., Combaut G., Francisco C. Tetrahedron Lett. 1994;35:7023. [Google Scholar]
- P. M. Evans, P. W. Groundwater, M. A. Munawar, and K. R. H. Solomons, Br. Pat. Appl. 9,425,409.1(1995).
- Takemura Y., Matsushita Y., Nagareya N., Abe M., Takaya J., Ju-Ichi M., Hashimoto T., Kan Y., Takaoka S., Asakawa Y., Omura M., Ito C., Furukawa H. Chem. Pharm. Bull. 1995;43:1340. [Google Scholar]
- Takemura Y., Matsushita Y., Onishi S., Atarashi T.A., Kunitomo Y., Ju-ichi M., Omura M., Ito C., Furukawa H. Heterocycles. 1995;41:187. [Google Scholar]
- Ciufolini M.A., Shen Y.-C., Bishop M.J. J. Am. Chem. Soc. 1995;117:12460. [Google Scholar]
- Jolivet C., Rivalle C., Bisagni E. J. Chem. Soc., Perkin Trans. 1995;1:511. [Google Scholar]
- Jolivet C., Rivalle C., Huel C., Bisagni E. J. Chem. Soc., Perkin Trans. 1995;1:2333. [Google Scholar]
- Ono T., Ito C., Furukawa H., Wu T.-S., Kuoh C.S., Hsu K.-S. J. Nat. Prod. 1995;58:1629. [Google Scholar]
- Guillier F., Nivoliers F., Godard A., Marsais F., Quéguiner G., Siddique M.A., Snieckus V. J. Org. Chem. 1995;60:292. [Google Scholar]
- Takemura Y., Ju-ichi M., Ito C., Furukawa H., Tokuda H. Planta Med. 1995;61:366. doi: 10.1055/s-2006-958104. [DOI] [PubMed] [Google Scholar]
- P. M. Evans, P. W. Groundwater, M. A. Munawar, and K. R. H. Solomons, Br. Pat Appl. 9,425,409.1; PCT Int. Appl. GB95/02948(1995).
- Wu T.-S., Huang S.-C., Wu P.-L. Phytochemistry. 1996;42:221. doi: 10.1016/0031-9422(96)00212-9. [DOI] [PubMed] [Google Scholar]
- Auzi A.A., Hartely T.G., Waigh R.D., Waterman P.G. Phytochemistry. 1996;42:235. [Google Scholar]
- Munawar M.A. University of Wales; Cardiff: 1996. Ph.D. Thesis. [Google Scholar]
- Drewe J.A., Groundwater P.W. J. Chem. Soc., Perkin Trans. 1997;1:601. [Google Scholar]