

Graphical abstract

Keywords: Imidazo[2,1-b]thiazole; Ketone hydrazone; Spirothiazolidinone; Synthesis; Biological activity
Highlights
-
•
Development of novel antimycobacterial drugs is an obligation.
-
•
Spirothiazolidinone moiety is potent for antitubercular activity.
-
•
Ring closure of acyl-hydrazone derivatives increases antitubercular activity.
Abstract
A series of novel acyl-hydrazone (4a-d) and spirothiazolidinone (5a-d, 6a-d) derivatives of imidazo[2,1-b]thiazole were synthesized and evaluated for their antiviral and antimycobacterial activity. The antituberculosis activity was evaluated by using the Microplate Alamar Blue Assay and the antiviral activity was evaluated against diverse viruses in mammalian cell cultures. According to the biological activity studies of the compounds, 5a-c displayed hope promising antitubercular activity, 6d was found as potent for Coxsackie B4 virus, 5d was found as effective against Feline corona and Feline herpes viruses. Consequently, the obtained results displayed that, 5a-d and 6d present a leading structure for future drug development due to its straightforward synthesis and relevant bioactivity.
1. Introduction
Lately, much interest has been focused on the chemistry and the biological activity of fused heterocyclic systems because of their broad spectrum of physiological activities [1], [2]. Among these heterocyclic compounds carrying nitrogen atom, imidazo[2,1-b]thiazole derivatives possess specific importance because of their diverse pharmacological activities. The imidazo[2,1-b]thiazole derivatives have been reported in the literature as antibacterial [3], [4], antitubercular [5], antifungal [6], antitumoral [7], antiviral [8], [9], antihelmintic [10], analgesic [11], anti-inflammatory [12], antihypertensive [13], cardiotonic [14], [15], diuretic [16], herbicide [17] and insecticide [18] agents.
Levamisole [(6S)-2,3,5,6-tetrahydro-6-phenylimidazo[2,1-b]thiazole] (Fig. 1 ), which is the levogyre isomer of antihelminthic Tetramisol and contains imidazo[2,1-b]thiazole moiety, is a drug that has significant immunomodulatory properties [19] in addition to its antihelminthic activity [20].
Fig. 1.
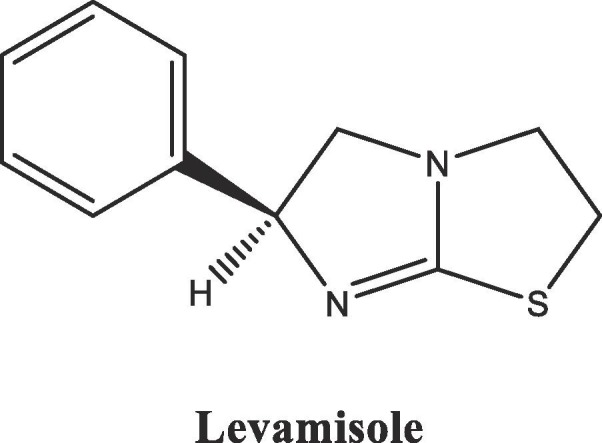
Chemical structure of the antihelminthic compound Levamisole.
Besides the wide biological activity spectrum of imidazo[2,1-b]thiazole derivatives, also the compounds bearing hydrazide, acyl-hydrazone and spirothiazolidinone moiety, have been reported in the literature with their various effects such as antibacterial [21], antifungal [22], antitubercular [23], antiviral [24], anticonvulsant [25] and antidepressant [26].
On the other hand, the two compounds BMY-27709 and BMS-199945, which are known in the literature for their antiviral activity and especially for their effects on influenza virus, have attracted the attention of researchers as compounds, that has a specific chemical character by an amide structure which connects an aliphatic cyclic system with an aromatic system [27], [28].
In this study, we further explored the scaffold containing the imidazo[2,1-b]thiazole ring as the aromatic moiety, that is linked by an amide to a spirothiazolidinone ring system as the aliphatic cyclic moiety and from this point forward, novel derivatives were synthesized (Table 1 ), and broadly evaluated for their antiviral and antimycobacterial activity (Fig. 2 ). Two hit compounds were found to inhibit Feline coronavirus or Coxsackie B4 virus. Besides, we observed some activity against Mycobacterium tuberculosis, suggesting the versatility and relevance of this compound class to achieved anti-infective agents.
Table 1.
Synthesis of the new compounds bearing imidazo[2,1-b]thiazole moiety (4a-d, 5a-d, and 6a-d).
| Compound | R1 | R2 | Yield (%) | M.p. (oC) |
|---|---|---|---|---|
| 4a | – | – | 92.7 | 227–229 |
| 4b | H | – | 95 | 245–248 |
| 4c | C6H5 | – | 93.7 | 225–227 |
| 4d | C6H4OH(4-) | – | 72.5 | 160–162 |
| 5a | – | – | 59.5 | 153–155 |
| 5b | H | – | 54.4 | 162–163 |
| 5c | C6H5 | – | 66.4 | 178–180 |
| 5d | C6H4OH(4-) | – | 87.8 | 285–286 |
| 6a | – | CH3 | 95 | 200–202 |
| 6b | H | CH3 | 90 | 242–243 |
| 6c | C6H5 | CH3 | 70.3 | 258–259 |
| 6d | C6H4OH(4-) | CH3 | 54 | 285–286 |
Fig. 2.

Chemical structure of the antiviral compounds BM27709, BMS-199945 and the novel compounds.
2. Materials and methods
2.1. Materials
IR spectra were recorded on KBr discs, using a Shimadzu IR Affinity-1 FT-IR instrument. 1H NMR (500 MHz), and 13C NMR (125 MHz) spectra were recorded on Varian UNITY INOVA spectrometer in DMSO-d 6 solution. Chemical shifts (δ) were reported in ppm; coupling constants (J) were recorded in hertz (Hz). Mass spectra were obtained on a Finnigan LCQ Advantage Max mass spectrometer. The reactions were monitored by TLC aluminum plates with silica gel Kieselgel 60 F254 thickness 0.25 mm (Merck), using UV light as a visualizing agent. All reagents and solvents were purchased from Merck, Fluka and Sigma-Aldrich and were used without further purification.
2.2. Chemical synthesis
The procedure for the synthesis of 2-amino-3-[(4-bromobenzoyl)methyl]-4-(ethoxycarbonylmethyl)thiazolium bromide (1)
Compound 1 was obtained according to the procedure described by Robert et al [29].
The procedure for the synthesis of ethyl [6-(4-bromophenyl)imidazo[2,1-b]thiazole-3-yl]acetate hydrobromide (2)
Compound 2 was obtained according to the procedure described by Robert et al [29].
The procedure for the synthesis of 2-[6-(4-bromophenyl)imidazo[2,1-b]thiazole-3-yl]acetohydrazide (3)
Compound 3 was obtained according to the procedure described by Harraga et al [30].
General procedure for the synthesis of 6-(4-bromophenyl)–N2-(substituted/non-substituted cycloalkylidene)imidazo[2,1-b]thiazole-3-acetohydrazides (4a-d)
0,005 mol of 3 was boiled in a water bath under reflux with 30 mL of ethanol until a clear solution was obtained. 0.01 mol of cyclic ketone was added and heated for 6 h. After cooling the mixture to room temperature, it was filtered and purified by crystallization with warm ethanol or washing.
2.2.1. 6-(4-Bromophenyl)-N2-(cyclopentylidene)imidazo[2,1-b]thiazole-3-acetohydrazide (4a)
Straw yellow solid, mp 227–229 °C, yield: 92.7%. Anal. Calcd. for C18H17BrN4OS: C, 51.80; H, 4.11; N, 13.43%. Found: C, 51.56; H, 3.78; N, 13.39. IR υmax (KBr, cm−1): 3190 (N—H stretching), 3101, 3041 (ar. C—H stretching), 2951, 2881, 2870 (al. C—H asymmetrical and symmetrical stretching), 1670 (amide I C O stretching), 1591 (hydrazone C N stretching), 1552, 1535, 1465 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1404, 1342 (al. C—H asymmetrical and symmetrical bending), 1222 (amide III N—H bending vibrations combined with C—N stretching), 1055 (ar. C—Br stretching), 829 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.69–2.34 (m, 8H, cyclopent.), 3.87 and 4.15 (2 s, 2H, CH2CO), 7.02 (s, 1H, imid.thia. C 2-H), 7.56 (t, 2H, J = 8.29 Hz, Br-Ph C3,5-H), 7.77 (d, 2H, J = 8.78 Hz, Br-Ph C2,6-H), 8.21 and 8.22 (2 s, 1H, imid.thia. C5-H), 10.23 and 10.28 (2 s, 1H, CONH). APCI (+) MS m/z (%): 419 ([M+H+2]+, 78), 417 ([M+H]+, 100). APCI (+) MS2 m/z (%): 417 ([M+H]+, 19), 335 (13), 334 (1 0 0), 319 (38), 293 (5), 255.
2.2.2. 6-(4-Bromophenyl)-N2-(cyclohexylidene)imidazo[2,1-b]thiazole-3-acetohydrazide (4b)
White solid, mp 245–248 °C, yield: 95%. Anal. Calcd. for C19H19BrN4OS: C, 52.90; H, 4.44; N, 12.99%. Found: C, 52.44; H, 4.02; N, 13.11%. IR υmax (KBr, cm−1): 3205, 3176 (N—H stretching), 3103, 3035 (ar. C—H stretching), 2924, 2858 (al. C—H asymmetrical and symmetrical stretching), 1668 (amide I C O stretching), 1591 (hydrazone C N), 1546, 1465 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1406, 1338 (al. C—H bending), 1209 (amide III N—H bending vibrations combined with C—N stretching), 1051 (ar. C-Br stretching), 829 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.56–2.40 (m, 10H, cyclohex.), 3.86 and 4.17 (2 s, 2H, CH2CO), 7.02 (s, 1H, imid.thia. C2-H), 7.56 (t, 2H, J = 8,29 Hz, Br-Ph C3,5-H), 7.76 and 7.78 (2d, 2H, J = 8,78; 8,79 Hz, Br-Ph C2,6-H), 8.22 (s, 1H, imid.thia. C5-H), 10.50 and 10.55 (2 s, 1H, CONH). 13C NMR (DEPT) (125 MHz) (DMSO-d 6/TMS) δ (ppm): 26.37 (cyclohex. C4), 26.95 (cyclohex. C2,6), 28.15 and 28.21 (cyclohex. C3,5), 36,45 (CH2), 110,19 and 110,54 (imid.thia. C5), 111.28 (imid.thia. C2), 127.85 (Br-Ph C2,6), 132.82 (Br-Ph C3,5). APCI (+) MS m/z (%): 433 ([M+H + 2]+, 100), 431 ([M+H]+, 77). APCI (+) MS2 m/z (%): 431 ([M+H]+, 43), 335 (10), 334 (1 0 0), 319 (17), 293 (4), 255 (29), 96 (7).
2.2.3. 6-(4-Bromophenyl)–N2-(4-phenylcyclohexylidene)imidazo[2,1-b]thiazole-3-acetohydrazide (4c)
White solid, mp 225–227 °C, yield: 93.7%. Anal. Calcd. for C25H23BrN4OS: C, 59.17; H, 4.57; N, 11.04%. Found: C, 58.80; H, 4.65; N, 10.69%. IR υmax (KBr, cm−1): 3205, 3172, 3142 (N—H stretching), 3030 (N—H and ar. C—H), 2956, 2912, 2854 (al. C—H asymmetric and symmetric stretching), 1666 (amide I C O stretching), 1589 (hydrazone C N), 1546, 1467 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1328, 1282 (al. C—H asymmetrical and symmetrical bending.), 1238 (amide III N—H bending vibrations combined with C—N stretching), 1072 (ar. C-Br stretching), 837 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.51–3.15 (m, 9H, cyclohex.), 3.88 and 4,20 (2 s, 2H, CH2CO), 7.03 and 7.04 (2 s, 1H, imid.thia. C2-H), 7.17–7.20 (m, 1H, Ph C4-H), 7.23–7.30 (m, 4H, cyclohex. 4-C6H5), 7.56 (t, 2H, J = 8.79 Hz, Br-Ph C3,5-H), 7.77 and 7.78 (2d, 2H, J = 8.79; 8,78 Hz, Br-Ph C2,6-H), 8,23 (s, 1H, imid.thia. C5-H), 10.58 and 10.63 (2 s, 1H, CONH). APCI (+) MS m/z (%):509 ([M+H + 2]+, 100), 507 ([M+H]+, 100). APCI (+) MS2 m/z (%): 507 ([M+H]+, 100), 336 (9), 335 (16), 334 (63), 319 (31), 293 (6), 253 (23), 174 (7).
2.2.4. 6-(4-Bromophenyl)–N2-(4-hydroxyphenylcyclohexylidene)imidazo[2,1-b]thiazole-3-acetohydrazide (4d)
White solid, mp 160–162 °C, yield: 72.5%. Anal. Calcd. for C25H23BrN4O2S: C, 57.36; H, 4.43; N, 10.70%. Found: C, 56.89; H, 4,74; N, 10.51%. IR υmax (KBr, cm−1): 3273 (Phenol O—H stretching), 3157 (N—H stretching), 3107, 3022 (ar. C—H), 2962, 2931, 2860 (al. C—H asymmetric and symmetric stretching), 1676 (amide I C O stretching), 1587 (hydrazone C N stretching), 1517, 1467 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1330 (al. C—H asymmetric and symmetric bending), 1265 (Phenol C—O stretching vibrations combined with O—H bending), 1234 (amide III N—H bending vibrations combined with C—N stretching), 1080 (ar. C-Br stretching), 825 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.44–3.13 (m, 9H, cyclohex.), 3.88 and 4.19 (2 s, 2H, CH2CO), 6.67 (d, 2H, J = 8,29 Hz, OH-Ph C3,5-H), 7.01–7.04 (m, 3H, imid.thia. C2-H and OH-Ph C2,6-H), 7.56 (t, 2H, J = 8.29 Hz, Br-Ph C3,5-H), 7.77 and 7.78 (2d, 2H, J = 8.29; 8,30 Hz, Br-Ph C2,6-H), 8.23 (s, 1H, imid.thia. C5-H), 9,13 (s, 1H, OH), 10,57 and 10,62 (2 s, 1H, CONH).
General procedure for the synthesis of 2-[6-(4-bromophenyl)imidazo[2,1-b]thiazole-3-yl]N-(2-nonsubstituted/methyl-6,7,8-nonsubstituted/alkyl/aryl-3-oxo-1-thia-4-azaspiro[4.4]non/[4.5]dec-4-yl]acetamides (5a-d, 6a-d)
To a suspension of 0.005 mol of 4a-d in 30 mL of anhydrous benzene is added 2 mL of mercaptoacetic acid / 2-mercaptopropionic acid. The reaction mixture is heated under a reflux condenser using a Dean-Stark trap in a water bath for 6 h. It is concentrated under reduced pressure and the excess acid is neutralized with NaHCO3 solution. The resulting product is kept in the refrigerator until solidified. The crude product is filtered, washed with water, dried and purified by crystallization or elution with ethanol.
2.2.5. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(3-oxo-1-thia-4-azaspiro[4.4]non-4- yl)acetamide (5a)
White solid, mp 153–155 °C, yield: 59.5%. Anal. Calcd. for C20H19BrN4O2S2·2H2O: C, 45.54; H, 4.40; N, 10.62%. Found: C, 44.88; H, 3.96; N, 11.06%. IR υmax (KBr, cm−1): 3441 (O—H stretching), 3365, 3174 (N—H stretching), 3091, 2997 (ar. C—H stretching), 2964, 2873, 2837 (al. C—H asymmetric and symmetric stretching), 1712 (s.thia. C O stretching), 1678 (amide I C O stretching), 1533, 1465 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1396, 1344 (al. C—H asymmetric and symmetric bending), 1247, 1215 (amide III N—H bending vibrations combined with C—N stretching), 1070 (ar. C-Br stretching), 823 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.60 (broad s, 4H, s.thia. C7-H, C8-H) 1.79–1.82 (m, 2H, s.thia. C6-H / C9-H), 2.05 (broad s, 2H, s.thia. C6-H / C9-H), 3.60 and 3.68 (2 s, 2H, S-CH2), 3.83 and 3.93 (2 s, 2H, CH2CO), 7.12 (s, 1H, imid.thia. C2-H), 7.55–7.59 (m, 2H, Br-Ph C3,5-H), 7.73–7.78 (m, 2H, Br-Ph C2,6-H), 8,27 (s, 1H, imid.thia. C5-H), 10.52 (s, 1H, CONH).
2.2.6. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(3-oxo-1-thia-4-azaspiro[4.5]dec-4-yl)acetamide (5b)
White solid, mp 162–163 °C, yield: 54.4%. Anal. Calcd. for C21H21BrN4O2S2·H2O: C, 48.18; H, 4.43; N, 10.70%. Found: C, 48.65; H, 4.44; N, 10.49%. IR υmax (KBr, cm−1): 3427 (O—H stretching), 3371, 3180, 3140 (N—H stretching), 3101 (ar. C—H stretching), 2989, 2931, 2856 (al. C—H asymmetric and symmetric stretching), 1718 (s.thia. C O stretching), 1680 (amide I C O
stretching), 1564, 1529, 1465, 1444 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1392 (al. C—H asymmetric and symmetric bending), 1242, 1205 (amide III N—H bending vibrations combined with C—N stretching), 1068 (ar. C-Br stretching), 819 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.03–1.75 (m, 10H, s.thia), 3.60 (s, 2H, S-CH2), 3.93 (s, 2H, CH2CO), 7.12 (s, 1H, imid.thia. C2-H), 7.58 (d, 2H, J = 8.29 Hz, Br-Ph C3,5-H), 7,75 (d, 2H, J = 8.78 Hz, Br-Ph C2,6-H), 8.29 (s, 1H, imid.thia. C5-H), 10.50 (s, 1H, CONH).
2.2.7. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(3-oxo-8-phenyl-1-thia-4-azaspiro[4.5]dec-4-yl)acetamide (5c)
White solid, mp, 178–180 °C, yield: 66.4%. Anal. Calcd. for C27H25BrN4O2S2: C, 55.76; H, 4.33; N, 9.63%. Found: C, 54.91; H, 4.05; N, 9.18%. IR υmax (KBr, cm−1): 3510, 3194 (N—H stretching), 3107, 3020 (ar. C—H stretching), 2929, 2873 (al. C—H asymmetric and symmetric stretching), 1722 (s.thia. C O stretching.), 1689 (amide I C O stretching), 1556, 1535, 1471 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1398, 1354 (al. C—H asymmetric and symmetric bending), 1257 (amide III N—H bending vibrations combined with C—N stretching), 1075 (ar. C-Br stretching), 827 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.60–2.37 (m, 9H, s.thia.), 3.65 (s, 2H, S-CH2), 3.95 (broad s, 2H, CH2CO), 7.07 (d, 2H, J = 7.32 Hz, 8-C6H5(C2,6-H)), 7.25 (t, 2H, J = 7.32 Hz 8-C6H4(C3,5-H)), 7.15–7.18 (m, 2H, 8-C6H5(C4-H) and imid.thia. C2-H), 7.58 (d, 2H, J = 8.78 Hz, Br-Ph C3,5-H), 7.78 (d, 2H, J = 8.78 Hz, Br-Ph C2,6-H), 8.33 (s, 1H, imid.thia. C5-H), 10,57 (s, 1H, CONH).
2.2.8. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(3-oxo-8-(4-hydroxyphenyl)-1-thia-4-azaspiro[4.5]dec-4-yl)acetamide (5d)
White solid, mp, 285–286 °C, yield: 87.8%. Anal. Calcd. for C27H25BrN4O3S2·H2O: C, 52.68; H, 4.42; N, 9.10%. Found: C, 52.11; H, 4.46; N, 9.35%. IR υmax (KBr, cm−1): 3446 (O—H stretching), 3228, 3130 (N—H stretching), 3111 (ar. C—H stretching), 2981, 2931 (al. C—H asymmetric and symmetric stretching), 1720 (s.thia. C O stretching), 1685 (amide I C O stretching), 1537, 1516, 1473 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1398, 1369 (al. C—H asymmetric and symmetric bending), 1263 (Phenol C—O stretching vibrations combined with O—H bending), 1220 (amide III N—H bending and C—N stretching), 1078 (ar. C-Br stretching), 827 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.54–2.23 (m, 9H, s.thia), 3.65 (s, 2H, S-CH2), 3.95 (s, 2H, CH2CO), 6.65 (d, 2H, J = 8.79 Hz, HO-Ph C3,5-H), 6.85 (d, 2H, J = 8,30 Hz, HO-Ph C2,6-H), 7.16 (s, 1H, imid.thia. C2-H), 7.58 (d, 2H, J = 8.78 Hz, Br-Ph C3,5-H), 7.77 (d, 2H, J = 8.79 Hz, Br-Ph C2,6-H), 8.33 (s, 1H, imid.thia. C5-H), 9.11 (s, 1H, OH), 10.55 (s, 1H, CONH).
2.2.9. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(2-methyl-3-oxo-1-thia-4-azaspiro[4.4]non-4-yl)acetamide (6a)
White solid, mp, 200–202 °C, yield: 95%. Anal. Calcd. for C21H21BrN4O2S2: C, 49.90; H, 4.19; N, 11.08%. Found: C, 50.23; H, 4.53; N, 10.12%.IR υmax (KBr, cm−1): 3143 (N—H stretching), 3111, 3049 (ar. C—H stretching), 2962, 2931, 2870 (al. C—H asymmetric and symmetric stretching), 1716 (s.thia. C O stretching), 1683 (amide I C O stretching), 1537, 1463 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1396, 1367, 1340 (al. C—H asymmetric and symmetric bending), 1253, 1228 (amide III N—H bending vibrations combined with C—N stretching), 1076 (ar. C-Br stretching),
829 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm): 1.42 (d, 3H, J = 6.83 Hz 2-CH3), 1.60 (broad s, 4H, s.thia. C7-H, C8-H), 1.63–2.12 (m, 4H, s.thia), 3.91 (q, 1H, J = 6.83 Hz, S-CH), 3.97 and 4.15 (2 s, 2H, CH2CO), 7.09 and 7.13 (2 s, 1H, imid.thia. C2-H), 7.47 and 7.55–7.60 (d, m, 2H, J = 8.30 Hz, Br-Ph C3,5-H), 7.67 and 7.74–7.78 (d, m, 2H, J = 8,79 Hz, Br-Ph C2,6-H), 8.27 and 8.30 (2 s, 1H, imid.thia. C5-H), 10.50 and 10.56 (2 s, 1H, CONH). 13C NMR (HSQC) (125 MHz) (DMSO-d 6/TMS) δ (ppm): 19.88 (2-CH3), 23.27 (s.thia. C7,8), 33.31 (CH2), 38.27, 38.36 (S-CH), 39.06 (s.thia. C6,9), 74.88 (s.thia. C5), 109.53, 109.76, 109.92 (imid.thia. C5), 111.23, 111.49, 111.55 (imid.thia. C2), 120.45, 120.55 (Br-Ph C4), 126.46, 126.79 (imid.thia. C3), 127.19, 127.69 (Br-Ph C2,6), 132.29, 132.34 (Br-Ph C3,5), 134.16, 134.21, 134.35 (Br-Ph C1), 145.51 (imid.thia. C6), 149.48, 149.55 (imid.thia. C7), 164.01, 167.16, 167.39 (CONH), 169.59, 171.20 (C O).
2.2.10. 2-[6-(4-Bromophenyl)imizado[2,1-b]thiazole-3-yl]-N-(2-methyl-3-oxo-1-thia-4-azaspiro[4], [5]dec-4-yl)acetamide (6b)
Straw yellow solid, mp, 242–243 °C, yield: 90%. Anal. Calcd. for C22H23BrN4O2S2: C, 50.87; H, 4.46; N, 10.79%. Found: C, 50.83; H, 4.23; N, 10.75%. IR υmax (KBr, cm−1): 3522, 3136 (N—H stretching), 3093 (ar. C—H stretching), 2974, 2935, 2854 (al. C—H asymmetric and symmetric stretching), 1707 (s.thia. C O stretching), 1678 (amide I C O stretching), 1537, 1473 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1392, 1348 (al. C—H asymmetric and symmetric bending), 1247, 1236 (amide III N—H bending and C—N stretching), 1076 (ar. C-Br stretching), 829 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6/TMS) δ (ppm):0.98–1.48 (m, 3H, s.thia), 1.40 (d, 3H, J = 6.83 Hz, 2-CH3), 1.51–1.54 (m, 1H, s.thia.), 1.71 (broad s, 6H, s.thia), 3.90 (q, 3H, J = 6.83 Hz, S-CH and CH2CO), 7.13 (s, 1H, imid.thia. C2-H), 7.59 (d, 2H, J = 8.78 Hz, Br-Ph C3,5-H), 7.76 (d, 2H, J = 8.78 Hz, Br-Ph C2,6-H), 8.30 (s, 1H, imid.thia.C5-H), 10.54 (s, 1H, CONH).
2.2.11. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(2-methyl-8-phenyl-3-oxo-1-thia-4-azaspiro[4.5]dec-4-yl)acetamide (6c)
White solid, mp, 258–259 °C, yield: 70.3. Anal. Calcd. for C28H27BrN4O2S2: C, 56.47; H, 4.57; N, 9.41%. Found: C, 56.29; H, 4.60; N, 9.54%. IR υmax (KBr, cm−1): 3170, 3122 (N—H stretching), 3057, 3024 (ar. C—H stretching), 2972, 2922, 2877 (al. C—H asymmetric and symmetric stretching), 1722 (s.thia. C O stretching), 1689 (amide I C O stretching), 1535, 1463 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1381, 1334 (al. C—H asymmetric and symmetric bending), 1246 (amide III N—H bending vibrations combined with C—N stretching), 1072 (ar. C-Br stretching), 829 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6 / TMS) δ (ppm): 1.43 (d, 3H, J = 6,84 Hz, 2-CH3), 1.45–2.38 (m, 9H, s.thia.), 3.95 (q, 3H, J = 6.34 Hz, CH2CO and S-CH), 7.08 (d, 2H, J = 6.84 Hz, 8-C6H4(H2,6)), 7.16–7.19 (m, 2H, 8-C6H4(H4) and imid.thia. C2-H), 7.26 (t, 2H, J = 7.32 Hz, 8-C6H4(H3,5)), 7.59 (d, 2H, J = 8,29 Hz, Br-Ph C3,5-H), 7.79 (d, 2H, J = 8.29 Hz, Br-Ph C2,6-H), 8.34 (s, 1H, imid.thia. C5-H), 10.61 (s, 1H, CONH). 13C NMR (DEPT) (125 MHz) (DMSO-d 6/TMS) δ (ppm):18.04 (2-CH3), 31.45 (s.thia. C7,9), 33.49 (CH2), 36.88 (s.thia. C6,10), 37.39 (S-CH), 42.31 (s.thia. C8), 109.53 (imid.thia. C5), 111.77 (imid.thia. C2), 126.83 (8-Ph C4′), 127.17 (Br-Ph C2,6 and 8-Ph C2′,C6′), 129.07 (8-Ph C3′,C5′), 132.38 (Br-Ph C3,5). ESI (+) MS m/z (%):597 ([M+H + 2]+, 100), 595 ([M+H]+, 75). ESI (+) MS2 m/z (%):327 (1 0 0).
2.2.12. 2-[6-(4-Bromophenyl)imidazo[2,1-b]thiazole-3-yl]-N-(2-methyl-8-(4-hydroxyphenyl)-3-oxo-1-thia-4-azaspiro[4.5]dec-4-yl)acetamide (6d)
Straw yellow solid, mp, 285–286 °C, yield: 54%. Anal. Calcd. for C28H27BrN4O3S2.2C2H5OH: C, 54.62; H, 5.59; N, 7.96. Found: C, 54.16; H, 5.75; N, 7.35%. IR υmax (KBr, cm−1): 3226, 3138 (O—H and N—H stretching), 3101 (ar. C—H stretching), 2970, 2929, 2868 (al. C—H asymmetric and symmetric stretching), 1724 (s.thia. C O stretching), 1670 (amide I C O stretching), 1533, 1517, 1465 (imid.thia. C N, C C, ar. C C stretching and amide II N—H bending vibrations combined with C—N stretching), 1400, 1323 (al. C—H asymmetric and symmetric bending), 1274 (phenol C—O stretching vibrations combined with O—H bending), 1249 (amide III N—H bending vibrations combined with C—N stretching), 1076 (ar. C-Br stretching), 831 (ar. 1,4-disubstitution). 1H NMR (500 MHz) (DMSO-d 6 / TMS) δ (ppm): 1,42 (d, 3H, J = 6,84 Hz, 2-CH3), 1.46–2.25 (m, 9H, s.thia.), 3.94 (q, 3H, J = 6,83 Hz, CH2CO and S-CH), 6.65 (d, 2H, J = 8.30 Hz, HO-Ph C3,5-H), 6.85 (d, 2H, J = 8.29 Hz, HO-Ph C2,6-H), 7.16 (s, 1H, imid.thia. C2-H), 7.59 (d, 2H, J = 8.29 Hz, Br-Ph C3,5-H), 7.79 (d, 2H, J = 8.30 Hz, Br—Ph C2,6-H), 8.33 (s, 1H, imid.thia.C5-H), 9.11 (s, 1H, OH), 10.59 (s, 1H, CONH).
2.3. Pharmacological evaluation
2.3.1. In vitro evaluation of antituberculosis activity
The antimycobacterial activity studies of the compounds were performed by the TAACF (Tuberculosis Antimicrobial Acquisition and Coordinating Facility by The National Institute of Health of the US government). Primary screening was conducted at 6.25 µg/mL against Mycobacterium tuberculosis H37Rv in BACTEC 12B medium using a broth microdilution assay the Microplate Alamar Blue Assay (MABA) [31]. Compounds exhibiting fluorescence were tested in the BACTEC 460 radiometric system. Compounds affecting less than 90% inhibition in the primary screen were not generally evaluated further. Compounds demonstrating at least 90% inhibition in the primary screen were re-tested at lower concentrations against M. tuberculosis H37Rv in order to determine the actual minimum inhibitory concentration (MIC) using MABA. Rifampin was utilized as the standard compound in the assays and each assay was replicated four times. The MIC was defined as the lowest concentration affecting a reduction in fluorescence of 90% relative to controls. Concurrently with the determination of MICs, compounds were tested for cytotoxicity (IC50) in VERO cells at concentrations ≤6.25 µg/mL or 10 times the MIC for M. tuberculosis H37Rv (solubility in media permitting). After 72 h exposure, viability was assessed on the basis of cellular conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) into a formazan product using the Promega CellTiter 96 Non-radioactive Cell Proliferation Assay. Compounds for which the selectivity index IC50:MIC ratio) SI > 10 were assumed to possess in vitro activity confirmed in the BACTEC 460 at 6.25 µg/mL.
2.3.1.1. Microplate alamar blue susceptibility assay (MABA)
Antimicrobial susceptibility testing was performed in black, clear-bottomed, 96-well microplates (black view plates; Packard Instrument, Meriden, Connecticut, USA) in order to minimize background fluorescence. Outer perimeter wells were filled with sterile water to prevent dehydration in experimental wells. Initial drug dilutions were prepared in either DMSO or distilled deionized water, and subsequent twofold dilutions were performed in 0.1 cm3 of 7H9GC (no Tween 80) in the microplates. BACTEC 12B-passaged inocula were initially diluted 1:2 7H9GC, and 0.1 cm3 was added to wells. Subsequent determination of bacterial titers yielded 1 × 106, 2.5 × 106 and 3.25 × 105 CFU cm−3 in plate wells for M. tuberculosis H37Rv. Frozen inocula were initially diluted 1:20 in BACTEC 12B medium followed by a 1:50 dilution in 7H9GC. Addition of 0.1 cm3 to wells resulted in final bacterial titers of 2.0x105 and 5x105 CFU cm−3 for H37Rv. Wells containing drugs only were used to detect autofluorescence of compounds. Additional control wells consisted of bacteria only (B) and medium only (M). Plates were incubated at 37 °C. Starting at day 4 of incubation, 20 mm3 of 10x Alamar Blue solution (Alamar Biosciences/Accumed, Westlake, Ohio, USA) and 12.5 mm3 of 20% Tween 80 were added to one B well and one M well, and plates were reincubated 37 °C. Wells were observed at 12 and 24 h for a color change from blue to pink and for a reading of ≥50,000 fluorescence units (FU). Fluorescence was measured in a Cytofluor II microplate fluorometer (PerSeptive Biosystems, Framingham, Massachusetts, USA) in bottom-reading mode with excitation at 530 nm and emission at 590 nm. If the B wells became pink by 24 h, the reagent was added to the entire plate. If the well remained blue or 50,000 ≤FU was measured, additional M and B wells were tested daily until a color change occurred, at which time reagents were added to all remaining wells. Plates were then incubated at 37 °C, and results were recorded at 24 h post-reagent addition. Visual MICs were defined as the lowest concentration of drug that had prevented a color change. For fluorometric MICs, background subtraction was performed on all wells with a mean of triplicate M wells. Percent inhibition was defined as 1-(test well FU/mean triplicate B wells) × 100. The lowest drug concentration affecting an inhibition of ≥90% was considered the MIC.
2.3.1.2. BACTEC radiometric method of susceptibility testing
A total of 0.1 cm3 of BACTEC 12B-passaged inoculum was delivered without prior dilution into 4 cm3 of the test medium. Subsequent determination of bacterial titers yielded average titers (three experiments) of 1 × 105, 2.5 × 105, and 3.25 × 104 CFU cm−3 of BACTEC 12B medium for M. tuberculosis H37Rv. Frozen inocula were initially diluted 1:20 in BACTEC 12B medium, and then 0.1 cm3 was delivered to test medium. This yielded 5.0 × 105 and 1.25 × 105 CFU per BACTEC vial for H37Rv. Twofold drug dilutions were prepared in either DMSO (Sigma) or distilled deionized water and delivered via a 0.5-cm3 insulin syringe in a 50-mm3 volume. Drug-free control vials consisted of solvent with bacterial inoculum and solvent with a 1:100 dilution of bacterial inoculum (1:100 controls). Vials were incubated at 37 °C, and the GI was determined in a BACTEC 460 instrument (Becton Dickinson) until the GI of the 1:100 controls reach at least 30. All vials were read the following day, and the GI and daily changes in GI (ΔGI) were recorded for each drug dilution. The MIC was defined as the lowest concentration for which the ΔGI was less than the ΔGI of the 1:100 control. If the GI of the test sample was greater than 100, the sample was scored as resistant even if the ΔGI was less than the ΔGI of the 1:100 control.
2.3.2. Antiviral procedures
The synthesized compounds (4a-d, 5a-d and 6a-d) were evaluated against diverse RNA and DNA viruses, using the following cell-based assays: (a) Crandell Rees feline kidney cells infected with Feline coronavirus or Feline herpes virus; (b) human embryonic lung (HEL) fibroblast cells infected with Herpes simplex virus-1 or −2, an acyclovir-resistant (ACVr) thymidine kinase-deficient Herpes simplex virus-1 strain, Vaccinia virus or Vesicular stomatitis virus; (c) Human cervix carcinoma HeLa cells infected with vesicular stomatitis virus, Coxsackie B4 virus or respiratory syncytial virus; (d) African green monkey kidney VERO cells infected with Parainfluenza-3 virus, Reovirus-1, Sindbis virus, Coxsackie B4 virus or Punta toro virus; (e) Mardin-Darby canine kidney (MDCK) cells infected with influenza A/H1N1, A/H3N2 or influenza B virus; and (f) human T-lymphoblast MT-4 cells infected with HIV-1 or HIV-2.
To perform the antiviral assays, the virus was added to subconfluent cell cultures in 96-well plates, and at the same time, the test compounds were added at serial dilutions. Appropriate reference compounds were included such as the virus entry inhibitors Urtica dioica agglutinin lectin and dextran sulfate; broad virus inhibitors Ribavirin and mycophenolic acid; antiherpetic drugs Ganciclovir and Cidofovir; and HIV inhibitor azidothymidine and nevirapine. After 3–6 days incubation at 37 °C (or 35 °C in the case of influenza virus), the cultures were examined by microscopy to score the compounds’ inhibitory effect on virus-induced cytopathic effect (CPE) or their cytotoxicity. For some viruses, antiviral and cytotoxic activities were confirmed by the colorimetric MTS cell viability assay [32].
3. Results and discussion
3.1. Chemistry
The target compounds 4a-d, 5a-d and 6a-d were synthesized from 2-(6-(4-bromophenyl)imidazo[2,1-b]thiazol-3-yl)acetohydrazide by a four and five-step synthesis through the pathways shown in Fig. 3 . 4-bromophenacyl bromide and ethyl 2-aminothiazole-4-acetate were dissolved in acetone and mixed in room temperature. After standing for 3 days, 2-amino-3-[(4-bromobenzoyl)methyl]-4-(ethoxycarbonylmethyl)thiazolium bromide (1) was obtained [29]. Thereafter, compound 1 was boiled under reflux in absolute ethanol and via the ring closure of compound 1, ethyl [6-(4-bromophenyl)imidazo[2,1-b]thiazole-3-yl]acetate hydrobromide (2) was obtained [29]. By heating the compound 2 and hydrazine hydrate in ethanol, 2-[6-(4-bromophenyl)imidazo[2,1-b]thiazole-3-yl]acetohydrazide (3) was obtained [30]. The compound 3 and cyclic ketones were heated under reflux in ethanol to yield 6-(4-bromophenyl)-N 2-(substituted/non-substituted cycloalkylidene)imidazo[2,1-b]thiazole-3-acetohydrazides (4a-d). The reaction yields of compounds 4a-d were between the interval of 95%-72.5%. 4a-d were then reacted with mercaptoacetic acid / 2-mercaptopropionic acid in anhydrous benzene under reflux by using a Dean-Stark water separator. The obtained reaction mixture was eventually neutralized with NaHCO3 solution and by that way 2-[6-(4-bromophenyl)imidazo[2,1-b]thiazol-3-yl]-N-(2-nonsubstituted/methyl-6,7,8-nonsubstituted/alkyl/aryl-3-oxo-1-thia-4-azaspiro[4.4]non/[4.5]dec-4-yl]acetamides (5a-d, 6a-d) were obtained. The reaction yields of the compounds 5a-d and 6a-d were between the interval of 87.8%-54.4% and 95%-54%, respectively. When the chemical structures of the synthesized compounds were examined, it was observed that the carbonyl group was not present in the compounds 4a-d whereas the same group was present in the spirothiazolidinone ring of the compounds 5a-d and 6a-d. Also according to the IR spectroscopy results, the stretching band belonging to the carbonyl group was not visible in the spectrum of the compounds 4a-d whereas the mentioned band was able to be observed in the range of 1724–1707 cm−1 for 5a-d and 6a-d. The IR values belonging to the carbonyl groups of spirothiazolidinone ring of 5a-d and 6a-d were in agreement with the literature [33], [34]. In the 1H NMR analysis, the NH2 protons of compound 3, which were observed as a broad singlet peak (2H) at 4.38 ppm, was disappeared in compounds 4a-d, whereas the peaks of aliphatic CH2 groups belonging to cyclic ketone arose as multiplet in the range of 3.15–1.44 ppm. When the results obtained from compounds 5a-d examined, it was determined that the peaks of S-CH2 protons were added to the spectrum in the range of 3.68–3.60 ppm as a distinctive indicator related to the formation of the mentioned compound [33], [35], [36]. Also from the 1H NMR analysis data obtained from compounds 6a-d, it was determined that the S-CH and CH-CH3 peaks of the spirothiazolidinone ring were in the range of 3.95–3.90 and 1.43–1.40 ppm, respectively and the values were in compliance with the literature [37], [38].
Fig. 3.

Synthesis of the title compounds (4a-d, 5a-d, and 6a-d).
The IR, 1H NMR, 13C NMR and MS spectra of the novel compounds are in agreement with the assigned structures. No unacceptable side reactions were observed, and products were obtained in moderate to good yields.
3.2. Biological studies
3.2.1. Antiviral activity
The broad antiviral evaluation demonstrated that compound 5d was quite effective against Feline coronavirus in CRFK cells (Table 2 ), with an antiviral EC50 value of 4.8 µg/mL and a superior selectivity index (ratio of cytotoxic to antiviral concentration) higher than 20. 5d was four-fold less active against Feline herpesvirus. Similarly, two compounds (4c and 4d) had weak anti-DNA virus activity in HEL cells, with antiviral EC50 values about 45 µg/mL (Table 3 ). The most effective compound against Feline coronavirus was 5d.
Table 2.
Antiviral activity and cytotoxicity in CRFKa feline kidney cells.
| Compound | Cytotoxicity CC50b (µg/mL) |
Antiviral EC50c (µg/mL) |
|
|---|---|---|---|
| Feline coronavirus | Feline herpesvirus | ||
| 4a | >100 | >100 | >100 |
| 4b | >100 | >100 | >100 |
| 4c | >100 | >100 | >100 |
| 4d | 68 | >100 | >100 |
| 5a | 20 | >100 | >100 |
| 5b | >100 | >100 | >100 |
| 5c | 9.8 | >100 | >100 |
| 5d | >100 | 4.8 | 21 |
| 6a | 20 | >100 | >100 |
| 6b | 9.2 | >100 | >100 |
| 6c | 9.6 | >100 | >100 |
| 6d | 54 | >100 | >100 |
| UDAd | 29 | 2.4 | 1.4 |
| Ganciclovir (µM) | >100 | >100 | 1.9 |
CRFK: Crandell Rees feline kidney cells.
CC50: 50% cytotoxic concentration in the MTS cell viability assay.
EC50: 50% effective concentration to produce 50% reduction in virus-induced cytopathicity, assessed by microscopy (mean values of three independent experiments).
UDA: Urtica dioica agglutinin; for this lectin compound, concentrations are expressed in µg per mL.
Table 3.
Antiviral activity and cytotoxicity in human embryonic lung (HEL) fibroblast cells.
| Compound | Cytotoxicity MCCa(µg/mL) |
Antiviral EC50b (µg/mL) |
||||
|---|---|---|---|---|---|---|
| HSV-1 | ACVr-HSV-1 | HSV-2 | Vaccinia virus | Vesicular stomatitis virus | ||
| 4a | >100 | >100 | >100 | >100 | >100 | >100 |
| 4b | 20 | >100 | >100 | >100 | >100 | >100 |
| 4c | ≥100 | 45 | ≥45 | ≥45 | 45 | >100 |
| 4d | >100 | 45 | 45 | 45 | 45 | >100 |
| 5a | 100 | >100 | >100 | >100 | >100 | >100 |
| 5b | >100 | >100 | >100 | >100 | >100 | >100 |
| 5c | 20 | >100 | >100 | >100 | >100 | >100 |
| 5d | 20 | >100 | >100 | >100 | >100 | >100 |
| 6a | ≥20 | >100 | >100 | >100 | >100 | >100 |
| 6b | 20 | >100 | >100 | >100 | >100 | >100 |
| 6c | 20 | >100 | >100 | >100 | >100 | >100 |
| 6d | ≥20 | >100 | >100 | >100 | >100 | >100 |
| Brivudine (µM) | >250 | 0.015 | 250 | 50 | 10 | >250 |
| Cidofovir (µM) | >250 | 0.80 | 2.0 | 0.85 | 10 | >250 |
| Acyclovir (µM) | >250 | 0.20 | ≥112 | 0.50 | >250 | >250 |
| Ganciclovir (µM) | >100 | 0.025 | 60 | 0.030 | >100 | >100 |
MCC: minimum cytotoxic concentration based on microscopic inspection of cell morphology.
EC50: 50% effective concentration, i.e. concentration producing 50% reduction in virus-induced cytopathicity, assessed by microscopy (mean values of two independent experiments).
When the SAR analysis of 5d is evaluated by comparison with the other compounds, it can be understood that the existence of the structure of the spirothiazolidinone moiety is significant in the activity against the mentioned virus. It can also be observed that the spirothiazolidinone structure of 5d is separated from 5b-c by the 4-hydroxyphenyl substituent at the 8-position, and thus it can be concluded that the corresponding substituent is significant in the activity of the compound.
Additionally, the 2-position of the spirothiazolidinone structure separates 5d from 6b-d, indicating that the nonsubstitution of position 2 is crucial for the antiviral activity against the Feline coronavirus.
The results also displayed that, the compounds bearing spirothiazolidinone structure did not show activity against DNA viruses, whereas compound 4c and 4d, which are carrying acyl-hydrazone moiety, showed activity against DNA viruses. The obtained result displayed the importance of the existence of the acyl-hydrazone residue for the activity against DNA viruses. Also, 4-phenylcyclohexyl and 4-(4-hydroxyphenyl)cyclohexyl derivatives of acyl-hydrazones have been found to further increase the activity against DNA viruses. Another hit compound, 6d, was somewhat active against Coxsackie B4 virus (EC50: 10 µg/mL and selectivity index ≥2 (Table 4 )).
Table 4.
Antiviral activity and cytotoxicity in VEROa cells.
| Compound | Cytotoxicity MCCb (µg/mL) |
Antiviral EC50c (µg/mL) | ||||
|---|---|---|---|---|---|---|
| Parainfluenza-3 virus | Reovirus-1 | Sindbis virus | Coxsackie B4 virus | Punto Toro virus | ||
| 4a | ≥100 | >100 | >100 | >100 | >100 | >100 |
| 4b | >100 | >100 | >100 | >100 | >100 | >100 |
| 4c | >100 | >100 | >100 | >100 | >100 | >100 |
| 4d | >100 | >100 | >100 | >100 | >100 | >100 |
| 5a | ≥20 | >100 | >100 | >100 | >100 | >100 |
| 5b | >100 | >100 | >100 | >100 | >100 | >100 |
| 5c | 20 | >100 | >100 | >100 | >100 | >100 |
| 5d | >100 | >100 | >100 | >100 | >100 | >100 |
| 6a | 100 | >100 | >100 | >100 | >100 | >100 |
| 6b | 100 | >100 | >100 | >100 | >100 | >100 |
| 6c | 20 | >100 | >100 | >100 | >100 | >100 |
| 6d | ≥20 | >100 | >100 | >100 | 10 | >100 |
| DS-5,000d | >100 | >100 | ≥100 | 60 | 84 | 60 |
| Ribavirin (µM) | >250 | 50 | >250 | >250 | >250 | 38 |
African Green monkey kidney VERO cells.
MCC: minimum cytotoxic concentration based on microscopic inspection of cell morphology.
EC50: 50% effective concentration, i.e. concentration producing 50% reduction in virus-induced cytopathicity, assessed by microscopy (mean values of two independent experiments).
DS-5,000: dextran sulphate MW 5,000; for this compound, concentrations are expressed in µg per mL.
3.2.2. Antimycobacterial activity
The antimycobacterial evaluation of compounds (4a-c, 5a-c) was performed. The results belonging in vitro primer analyses, that were performed against Mycobacterium tuberculosis H37Rv strain by using MABA in BACTEC 12B media, were given in Table 5 .
Table 5.
The antimycobacterial activity of the compounds.
| Compound | H37Rv Finding |
||
|---|---|---|---|
| Activity | lC50a (µg/mL) | MICb (µg/mL) | |
| 4a | Weak Active | >100 | 16,252 |
| 4b | Inactive | >100 | >100 |
| 4c | Weak Active | 49,242 | >100 |
| 5a | Active | 4,503 | 8,453 |
| 5b | Active | 1,446 | 1,566 |
| 5c | Active | 0,76 | 0,854 |
| Rifampin | Active | x | 0.125 |
IC50: The actual minimum inhibitory concentration required to inhibit the growth of 50% of H37Rv strain of M. Tuberculosis.
MIC: The actual minimum inhibitory concentration required to inhibit the growth of 90% of H37Rv strain of M. Tuberculosis.
According to the in vitro primer analyses, the compounds possessing any value where the MIC value was equal to less than 10 µg/mL were considered as active for antimicrobial activity and further analysis of the mentioned compounds were conducted. From this point forward compounds 4a-c were not detected as active and no further antimycobacterial activity was performed for them. In contrast to compounds 4a-c, compounds 5a-c were detected as active and further antimycobacterial activity of these compounds was conducted and the results were displayed in Table 6 .
Table 6.
The cytotoxicity of the compounds studied in VERO cells.
| Compound | lC50a (µg/mL) | MICb (µg/mL) | CC50c (µg/mL) | SId (CC50/MIC) |
|---|---|---|---|---|
| 5a | 4,503 | 8,453 | 24,074 | 2,8479 |
| 5b | 1,446 | 1,566 | 14,064 | 8,9808 |
| 5c | 0,76 | 0,854 | 11,935 | 13,975 |
| Rifampin | x | 0.125 | >100 | >800 |
IC50: The actual minimum inhibitory concentration required to inhibit the growth of 50% of H37Rv strain of M. Tuberculosis.
MIC: The actual minimum inhibitory concentration required to inhibit the growth of 90% of H37Rv strain of M. Tuberculosis.
CC50: 50% cytotoxic concentration against VERO cells in vitro
SI: Selectivity index, the ratio of CC50 to MIC
The obtained results displayed that, 4a-c, the acyl-hydrazone moiety containing imidazo[2,1-b]thiazole derivatives, showed no or weak antitubercular activity and the ring closure raised the antitubercular activity of the compounds as can be seen from the results related to the compounds 5a, 5b, and 5c.
The fact that the conversion of acyl-hydrazone derivatives to spirothiazolidinone derivatives by ring closure increased the activity, indicates the importance of the existence of spirothiazolidinone structure for antitubercular activity.
4. Conclusions
Antibiotic resistance is rising to high levels sharply all over the world and new kinds of resistance mechanisms are emerging worldwide. Infections such as pneumonia, tuberculosis, gonorrhea, blood poisoning are becoming harder to treat and current antibacterials are losing their effectivity day by day. Designing of new effectual compounds to deal with these resistant bacterias has become one of the most important issues today. Similarly, Mycobacterium tuberculosis remains a leading infectious cause of death worldwide today. Especially the evolution of multi-drug-resistant (MDR) strains of Mycobacterium tuberculosis, is the main reason for the increased incidence of tuberculosis, therefore, the development of new antimycobacterial agents has become an obligation. Another important is that needs new drug candidates are antiviral therapy and it is definitely necessary to design and develop new effective antiviral agents.
In the present study, in an attempt to find novel antiviral and antitubercular agents, 12 diverse derivatives of imidazo[2,1-b]thiazole were designed and synthesized. The compounds were screened for their antiviral and antitubercular activity. The antimycobacterial activities of the compounds were tested against the Mycobacterium tuberculosis H37Rv strain and in the test method, each value, in which the MIC value was equal to less than 10 µg/mL, was actively accepted for antimycobacterial activity. The MIC values of the compounds 5a, 5b, and 5c were determined as 8.453 µg/mL, 1.566 µg/mL and 0.854 µg/mL and the molecules were found as active. Also, compound 6d was found as effective for Coxsackie B4 virus. The antiviral activity and cytotoxicity of the compounds against Feline corona and Feline herpes viruses were investigated in CRFK cell cultures and in comparison with HHA, UDA and Ganciclovir references, compound 5d was found as highly effective. The findings revealed the promising antiviral and antitubercular activity of acyl-hydrazone, spirothiazolidinone and 2-methyl-substituted spirothiazolidinone derivatives of imidazo[2,1-b]thiazole and these derivatives could be an interesting starting point for further structural optimization to obtain new promising and more potent antiviral and antitubercular agents.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
We thank D. Joseph A. Maddry from the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF), National Institute of Allergy and Infectious Diseases Southern Research Institute, GWL Hansen’s Disease Center, Colorado State University, Birmingham, Alabama (USA) for the in vitro evaluation of antituberculosis activity.
This work was supported by Istanbul University Research Project (Project Numbers: T-8381, T-52074).
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discusses in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2019.103496.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Bertamino A., Musella S., Di Sarno V., Ostacolo C., Manfra M., Vanacore D., Stiuso P., Novellino E., Campiglia P., Gomez-Monterrey I.M. Eur. J. Med. Chem. 2015;102:106–114. doi: 10.1016/j.ejmech.2015.07.044. [DOI] [PubMed] [Google Scholar]
- 2.Parrino B., Carbone A., Ciancimino C., Spanò V., Montalbano A., Barraja P., Cirrincione G., Diana P., Sissi C., Palumbo M., Pinato O., Pennati M., Beretta G., Folini M., Matyus P., Balogh B., Zaffaroni N. Eur. J. Med. Chem. 2015;94:149–162. doi: 10.1016/j.ejmech.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Ulusoy-Güzeldemirci N., Küçükbasmacı Ö. Eur. J. Med. Chem. 2010;45:63–68. doi: 10.1016/j.ejmech.2009.09.024. [DOI] [PubMed] [Google Scholar]
- 4.Cascioferro S., Parrino B., Petri G.L., Cusimano M.G., Schillaci D., Di Sarno V., Musella S., Giovannetti E., Cirrincione G., Diana P. Eur. J. Med. Chem. 2019;167:200–210. doi: 10.1016/j.ejmech.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Andreani A., Granaiola M., Leoni A., Locatelli A., Morigi R., Rambaldi M. Eur. J. Med. Chem. 2001;36:743–746. doi: 10.1016/s0223-5234(01)01266-1. [DOI] [PubMed] [Google Scholar]
- 6.Çapan G., Ulusoy N., Ergenç N., Kiraz M. Monatsh. Chem. 1999;130:1399–1407. [Google Scholar]
- 7.Andreani A., Burnelli S., Granaiola M., Leoni A., Locatelli A., Morigi R., Rambaldi M., Varoli L., Calonghi N., Cappadone C., Farruggia G., Zini M., Stefanelli C., Masotti L., Radin N.S., Shoemaker R.H. J. Med. Chem. 2008;51:809–816. doi: 10.1021/jm701246g. [DOI] [PubMed] [Google Scholar]
- 8.Barradas J.S., Errea M.I., D’Accorso N.B., Sepúlveda C.S., Damonte E.B. Eur. J. Med. Chem. 2011;46:259–264. doi: 10.1016/j.ejmech.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Ulusoy-Güzeldemirci N., Pehlivan E., Naesens L. Marmara. Pharm. J. 2018;22(2):237–248. [Google Scholar]
- 10.Shetty N.S., Khazi I.A.M., Ahn C. Bull. Korean Chem. Soc. 2010;31(8):2337–2340. [Google Scholar]
- 11.Shahrasbi M., Movahed M.A., Dadras O.G., Daraei B., Zarghi A. Iran. J. Pharm. Sci. 2018;17(4):1288–1296. [PMC free article] [PubMed] [Google Scholar]
- 12.Bhongade B.A., Talath S., Gadad R.A., Gadad A.K. J. Saudi Chem. Soc. 2016;20:463–475. [Google Scholar]
- 13.Andreani A., Rambaldi M., Locatelli A., Cristoni A., Malandrino S., Pifferi G. Acta Pharm. Nord. 1992;4:93–96. [PubMed] [Google Scholar]
- 14.Andreani A., Rambaldi M., Leoni A., Locatelli A., Bossa R., Chiericozzi M., Galatulas I., Salvatore G. Eur. J. Med. Chem. 1996;31:383–387. doi: 10.1021/jm9509307. [DOI] [PubMed] [Google Scholar]
- 15.Andreani A., Rambaldi M., Bonazzi D. Arch. Pharm. (Weinheim) 1985;318:1003–1008. doi: 10.1002/ardp.19853181109. [DOI] [PubMed] [Google Scholar]
- 16.Andreani A., Rambaldi M., Mascellani G., Rugarli P. Eur. J. Med. Chem. 1987;22:19–22. [Google Scholar]
- 17.Andreani A., Rambaldi M., Leoni A., Locatelli A., Andreani F., Gehret J.C. Pharm. Acta Helv. 1996;71:247–252. [Google Scholar]
- 18.Andreani A., Rambaldi M., Carloni P., Greci L., Stipa P. J. Heterocycl. Chem. 1989;26(2):525–529. [Google Scholar]
- 19.Sun A., Chiang C.P., Chiou P.S., Wang J.T., Liu B.Y., Wu Y.C. J. Oral Pathol. Med. 1994;23:172–177. doi: 10.1111/j.1600-0714.1994.tb01108.x. [DOI] [PubMed] [Google Scholar]
- 20.P.A.J. Janssen, The Levamisole Story, in: Prog. Drug Res. Der Arzneimittelforschung/Progrés Des Rech. Pharm., Birkhäuser Basel, Basel, 1976, pp. 347–83. [DOI] [PubMed]
- 21.Popiolek L. Med. Chem. Res. 2017;26:287–301. doi: 10.1007/s00044-016-1756-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rollas S., Küçükgüzel Ş.G. Molecules. 2007;12:1910–1939. doi: 10.3390/12081910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vintonyak V.V., Warburg K., Kruse H., Grimme S., Hübel K., Rauh D., Waldmann H. Angew. Chem. Int. Ed. 2010;49(34):5902–5905. doi: 10.1002/anie.201002138. [DOI] [PubMed] [Google Scholar]
- 24.Şenkardeş S., Kaushik-Basu N., Durmaz İ., Manvar D., Basu A., Atalay R., Küçükgüzel Ş.G. Eur. J. Med. Chem. 2016;108:301–308. doi: 10.1016/j.ejmech.2015.10.041. [DOI] [PubMed] [Google Scholar]
- 25.Angelova V., Karabeliov V., Andreeva-Gateva P.A., Tchekalarova J. Drug Develop. Res. 2016;77(7):379–392. doi: 10.1002/ddr.21329. [DOI] [PubMed] [Google Scholar]
- 26.Mohareb R.M., El-Sharkawy K.A., Hussein M.M., El-Sehrawi H.M. J. Pharm. Sci. & Res. 2010;2(4):185–196. [Google Scholar]
- 27.Luo G., Colonno R., Krystal M. Virology. 1996;226:66–76. doi: 10.1006/viro.1996.0628. [DOI] [PubMed] [Google Scholar]
- 28.Cianci C., Yu K.L., Dischino D.D., Harte W., Deshpande M., Luo G., Colonno R.J., Meanwell N.A., Krystal M. J. Virol. 1999;73(3):1785–1794. doi: 10.1128/jvi.73.3.1785-1794.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robert J.F., Xicluna A., Panouse J.J. Eur. J. Med. Chem. 1975;10:59–64. [Google Scholar]
- 30.Harraga S., Nicod L., Drouhin J.P., Xicluna A., Panouse J.J., Seilles E., Robert J.F. Eur. J. Med. Chem. 1994;29:309–315. [Google Scholar]
- 31.Collings L.A., Franzblau S.G. Antimicrob. Agents Chemother. 1997;41(5):1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanderlinden E., Göktaş F., Cesur Z., Froeyen M., Reed M.L., Russell C.J., Cesur N., Naesens L. J. Virol. 2010;84(9):4277–4288. doi: 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akkurt M., Yalçın Ş.P., Ulusoy-Güzeldemirci N., Büyükgüngör O. Acta Crystallogr. E. 2008;64(5):810–811. doi: 10.1107/S1600536808009306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shamsuzzaman K.A.A.A., Baqi A., Ali M., Asif A., Mashrai H., Khanam A., Sherwani Z., Yaseen M. Owais. J. Mol. Struct. 2015;1085:104–114. [Google Scholar]
- 35.Ulusoy N., Forsch Arzneim. Drug Res. 2002;52(7):565–571. doi: 10.1055/s-0031-1299931. [DOI] [PubMed] [Google Scholar]
- 36.Kasimogullari B.O., Cesur Z. Molecules. 2004;9:894–910. doi: 10.3390/91000894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Güzel Ö., Terzioğlu N., Çapan G., Salman A. Arkivoc. 2006;12:98–110. [Google Scholar]
- 38.Güzel Ö., İlhan E., Salman A. Monatsh. Chem. 2006;137:795–801. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.