

Abstract
Purpose of Review
Fibrosis is a pathological feature of many human diseases that affect multiple organs. The development of anti-fibrotic therapies has been a difficult endeavor due to the complexity of signaling pathways associated with fibrogenic processes, complicating the identification and modulation of specific targets. Evidence suggests that ephrin ligands and Eph receptors are crucial signaling molecules that contribute to physiological wound repair and the development of tissue fibrosis. Here, we discuss recent advances in the understanding of ephrin and Eph signaling in tissue repair and fibrosis.
Recent Findings
Ephrin-B2 is implicated in fibrosis of multiple organs. Intercepting its signaling may help counteract fibrosis.
Summary
Ephrins and Eph receptors are candidate mediators of fibrosis. Ephrin-B2, in particular, promotes fibrogenic processes in multiple organs. Thus, therapeutic strategies targeting Ephrin-B2 signaling could yield new ways to treat organ fibrosis.
Keywords: Ephrins, Eph receptor, Ephrin-B2, Fibrosis
Introduction
An appropriate wound repair response is necessary to return injured tissues to homeostasis. However, maladaptive wound healing in response to chronic tissue injury can lead to fibrosis, a process characterized by excessive deposition of extracellular matrix (ECM) proteins including collagens. Fibrosis is a pathological feature of many human diseases and can occur in most organs including the lung, heart, brain, kidney, and skin. Fibrosis involves the impairment of multiple steps in the normal tissue repair response, including dysregulation of inflammation, angiogenesis, cell matrix deposition, cell migration, fibroblast activation, and persistence [1]. If left unchecked, the turnover of functional tissue towards an acellular fibrous connective tissue can result in organ failure [1]. For instance, lung fibrosis is a common and lethal complication of the autoimmune multi-organ fibrotic disorder systemic sclerosis (SSc) and is the hallmark feature of idiopathic pulmonary fibrosis (IPF) [1, 2]. In SSc, maladaptive tissue repair responses in the lung manifest as SSc-related interstitial lung disease (SSc-ILD) and pulmonary arterial hypertension (PAH). These pathologies contribute to a decline in lung function and increased mortality in patients with SSc [1, 3].
Fibrosis is ultimately a maladaptive response to organ damage; here, we briefly introduce the various stages of wound repair and discuss how dysregulation of these processes may contribute to organ fibrosis. Hemostasis, the vasoconstriction and clotting of damaged vessels, is initiated following an injury to reduce blood loss. In addition to regulating blood clot formation, thrombocytes release growth factors and cytokines that initiate tissue repair and recruit circulating white blood cells as part of the inflammatory response. During the proliferation phase, granulation tissue generation and epithelial cell proliferation aid in wound closure. Fibroblasts recruited to the injury niche transition into activated myofibroblasts, characterized by the expression of alpha smooth muscle actin (α-SMA), which confers a hypercontractile phenotype important for wound closure. Myofibroblasts are also responsible for ECM production at the site of injury. Meanwhile, angiogenesis is promoted to vascularize the newly formed tissue to provide nutrients and a route for cell infiltration [4, 5]. Wound maturation follows wound closure, wherein tissue remodeling is required to reorganize collagen structure, decrease inflammation, and promote the resolution of the wound healing response. Aberrant regulation at any of these stages can disrupt the normal wound repair program and may contribute to fibrosis [4, 6].
The intertwined nature of fibrotic signaling pathways has posed a real challenge in the identification of therapeutic targets to effectively attenuate fibrosis. Extensive research has shown that the cytokine transforming growth factor-beta (TGF-β) is a key mediator of fibrosis [7]. However, targeting TGF-β signaling in fibrosis has been complicated given the pleiotropic effects of TGF-β in multiple cell types [7]. Alternatively, targeting TGF-β-dependent pro-fibrotic effects in myofibroblasts is a safe strategy to mitigate organ fibrosis. Thus, targeting downstream mediators of TGF-β pro-fibrotic effects could selectively treat fibrosis without affecting homeostatic TGF-β functions. In this regard, recent studies have shown that the erythropoietin-producing human hepatocellular carcinoma (Eph) receptors and their Eph receptor-interacting (Ephrins) family of ligands may act downstream of TGF-β during tissue fibrogenesis. This review will discuss the contributions of ephrins and Eph receptors to various aspects of wound repair and fibrosis including inflammation, angiogenesis, fibroblast activation, and matrix deposition. In particular, Ephrin-B2 ligand will be highlighted as an important mediator of tissue fibrosis and a potential therapeutic target for anti-fibrotic therapy.
Ephrin/Eph Signaling
Ephrins are a diverse family of cell surface ligands classified by the manner in which they associate with the plasma membrane. Ephrin-A family members are attached by glycosylphosphatidylinositol anchors, whereas Ephrin-B ligands are single-pass transmembrane proteins (Fig. 1) [8]. Eph receptors, the largest subclass of receptor tyrosine kinases (RTK), are subdivided into EphA and EphB subclasses based on their ephrin ligand interactions [8]. Of note, ephrins and Ephs have the ability of modulate the activity of one another; however, interactions within the same subclasses are more common than between subclasses [9, 10].
Fig. 1.
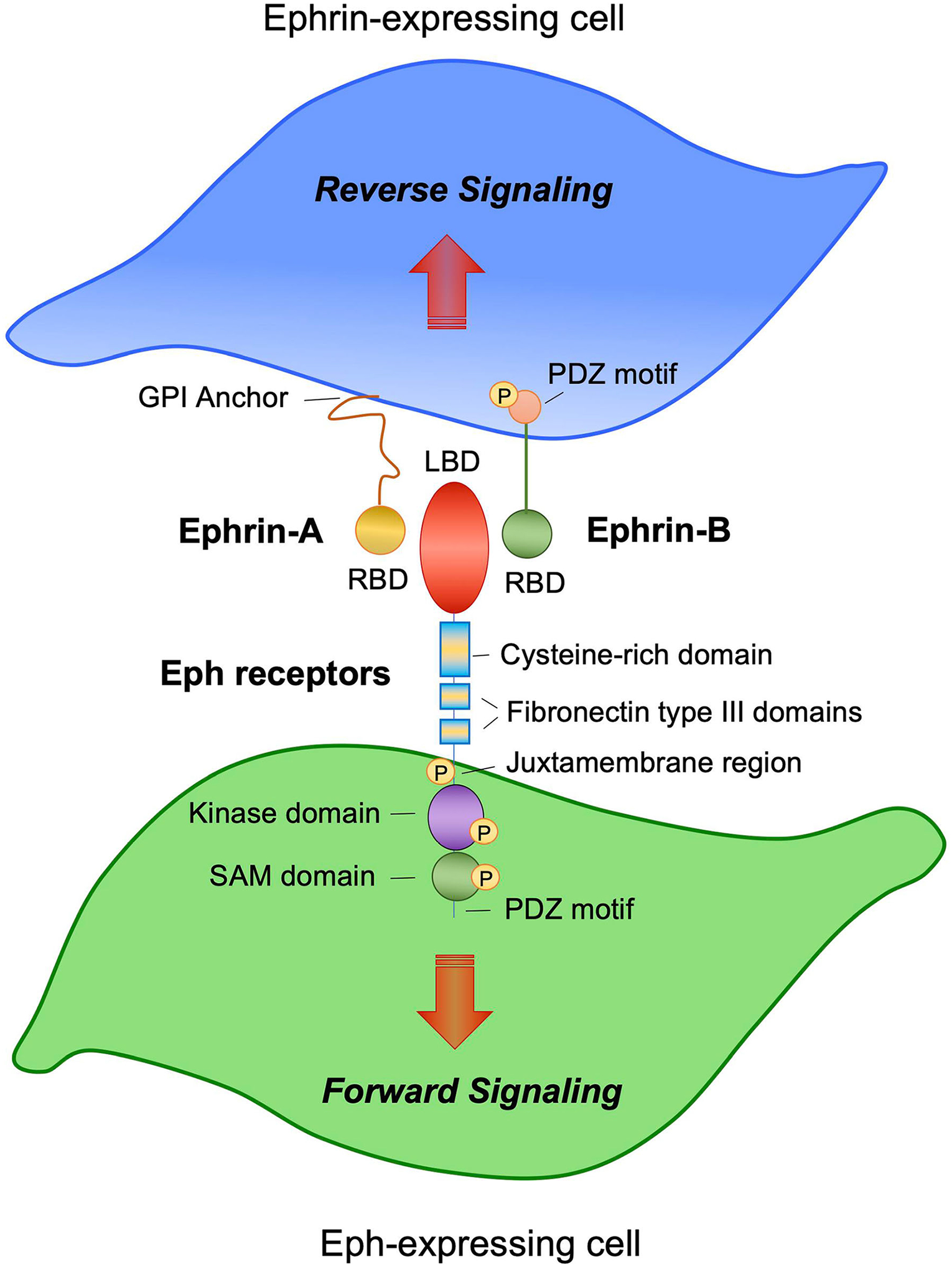
Ephrin/Eph receptor signaling. Schematic diagram showing an Ephrin-A- and Ephrin-B-expressing cell (top, blue) in contact with an Eph-expressing cell (bottom, green). Ephrin-A ligands have a short cytoplasmic tail and are glycosylphosphatidylinositol (GPI)-anchored to the cell membrane. Ephrin-B ligands have a long cytoplasmic region characterized by a PDZ-binding motif. Both Ephrin-A and Ephrin-B ligands express receptor-binding domains (RBD) at the N-terminal domain. EphA and -B receptors are characterized by an extracellular region that contains the ligand-binding domain (LBD) at the N-terminus. The LBD is connected to a cysteine-rich domain followed by two fibronectin type-III domains. The kinase domain is located intracellularly and is connected to the sterile-alpha motif (SAM) domain, followed by a PDZ-binding motif. Bidirectional signaling occurs between an ephrin-expressing cell and an Eph receptor-expressing cell. Forward signaling is initiated from ephrins and propagated into the Eph receptor-expressing cell. Reverse signaling is initiated by Eph receptors and is propagated into the ephrin-expressing cell
Ephrin-Eph interactions are unique due to the bidirectional signaling that follows, where ephrin binding initiates signaling cascades downstream of both the receptor (forward signaling) and ligand (reverse signaling) (Fig. 1) [8]. Ephrins and their receptors are capable of both trans (cell-cell interaction) and cis signaling (autocrine, within the same cell) [11]. As seen in classic RTK signaling, ephrin binding to Eph receptors mediates transautophosphorylation and downstream activation of multiple effectors that modulate cellular functions such as migration, repulsion, and morphogenesis [8].
Interestingly, Eph receptors are not limited to classic ligand-binding phosphorylation-dependent signaling. They also interact with other receptors at the plasma membrane to promote phosphorylation-independent signaling that results in distinct outcomes [11]. Both ephrins and Ephs have been shown to mediate signaling independently of one another [12], including by interacting with other receptors. For instance, Ephrin-B2 mediates endocytosis of members of the vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR) families [8, 13, 14], receptors involved in angiogenesis and wound repair (discussed below).
Ephrin/Eph receptor signaling was initially discovered to be essential for prenatal developmental processes including vasculogenesis, neurogenesis, and intestinal epithelial development [8]. Mouse knockouts of various individual Ephrin-B and EphB receptors are embryonically lethal, indicating their necessity for development and morphogenesis [15, 16]. However, inducible knockouts have been used to investigate the involvement of ephrins and Eph receptors in both postnatal tissue homeostasis and disease contexts, including their roles during wound repair and fibrosis [17].
Ephrin/Eph Signaling During Tissue Injury and Repair
Signaling by ephrin/Eph family members regulates cellular responses to tissue injury, helping to coordinate appropriate repair. Here, we describe the wound repair processes influenced by ephrin/Eph signaling in the context of various tissues and associated cells (summarized in Table 1).
Table 1.
Ephrins and Eph receptor signaling in wound healing
| Ephrin/Eph | Process | Organ | Cell type | Effect | Ref. |
|---|---|---|---|---|---|
| Ephrin-Al | Inflammation | Lung | Endothelial cells | Upregulates in response to inflammation | [18, 19] |
| Endothelial cells | Ephrin-Al treatment stimulates MCP-1 and CXCL1 secretion | [19] | |||
| Migration and adhesion | Comeal epithelium | Epithelial cells | Ephrin-Al mimetic decreases EphA2 expression and migration in scratch wound assay | [20] | |
| Angiogenesis | NA | Endothelial cells | Supports developmental vessel sprouting, formation, and survival | [21] | |
| Supports endothelial cell assembly and microvessel formation | [22, 23] | ||||
| Ephrin-A2 | Tissue remodeling | Skin | Epithelial cells | Efna2 knockout leads to increased collagen density within the dermis following surgical excision wounding and changes resulting scar morphology | [24] |
| Ephrin-Bl | Migration and adhesion | Skin | Keratinocytes | Upregulates following skin wounding along the leading edge of the wound | [25] |
| Increases in Ephiin-B 1 correlate with loosening of the adherens junctions and tight junctions between epithelial cells | [25] | ||||
| Increases in Ephrin-Bl lead to decreases in E-cadherin and claudin-1 | [25, 26] | ||||
| Double knockout with Ephrin-B2 leads to sustained decreases in adhesion proteins and loss of epithelial cell loosening following skin wounding | [25] | ||||
| Overexpression decreases adhesion and cell-cell contacts | [25] | ||||
| Intestine | Epithelial cells | ADAM 10 and Ephrin-Bl mediates E-cadherin cleavage | [26] | ||
| Ephrin-B2 | Migration and adhesion | Skin | Keratinocytes | Double knockout with Ephrin-B 1 leads to sustained decreases in adhesion proteins and loss of epithelial cell loosening following skin wounding | [25] |
| Overexpression decreases adhesion and cell-cell contacts | [25] | ||||
| Angiogenesis | Unspecified | Endothelial cells | Increases in response to hypoxia to drive angiogenesis | [27] | |
| Crucial for VEGF signaling and VEGFR2/3 intemalization/activation | [13, 28] | ||||
| EphA2 | Inflammation | Lung | Endothelial cells | Upregulates in response to inflammation | [18, 19] |
| EphA2 neutralizing antibody downregulates VCAM1 and E-selectin (monocyte recruitment) | [18] | ||||
| Endothelial cells | Epha2 knockouts do not upregulate MCP-1 and KC in response to inflammation (monocyte and neutrophil recruitment) | [19] | |||
| EphBl | Inflammation | Intestine | IEC-6 | EphBl antagonist increases MCP-1 and COX-2 | [29] |
| EphB4 | Angiogenesis | Unspecified | Endothelial cells | Interaction with Ephrin-B2 is crucial for angiogenesis | [28, 30, 31] |
ADAM10, a disintegrin and metalloproteinase domain-containing protein 10; COX2, cyclooxygenase 2; CXCL1, C-X-C motif chemokine ligand 1; Eph receptor, erythropoietin-producing human hepatocellular carcinoma receptors; IEC, intestinal epithelial cell; KC, keratinocyte chemoattractant; MCP-1, monocyte chemoattractant protein 1; VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor
Regulation of Vascular Permeability and Inflammation
Inflammation is a core component to the wound healing response and is necessary for wound resolution; however, chronic inflammation exacerbates disease pathology and contributes to the development and progression of fibrosis. In the wound healing program, inflammation is a tightly regulated process characterized by the accumulation of inflammatory cells and release of inflammatory mediators at the injury site. Previous studies show that EphA2 receptor signaling activated by Ephrin-A1 ligand regulates the secretion of inflammatory factors via the pulmonary endothelium in response to tissue injury [18, 19, 32–34]. Intratracheal instillation of either lipopolysaccharide (LPS) or bleomycin can induce lung injury and transient fibrosis in mice [35–37], albeit their inflammatory responses prior to the development of fibrosis are distinct. Lung injury induced by LPS or bleomycin rapidly elevates levels of both EphA2 receptor and Ephrin-A1 ligand in mouse lung tissue 24–48 h post-challenge [18, 19]. One probable source of Ephrin-A1 is the vascular endothelium, as levels of these ligands are increased in human aortic endothelial cells (HAECs), human coronary artery endothelial cell (HCAECs), and human umbilical vein endothelial cells (HUVECs) following pro-inflammatory stimulation with tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, or low-density lipoprotein (LDL) [34]. In the endothelium, Ephrin-A1/EphA2 signaling regulates the secretion of pro-inflammatory factors, such as monocyte chemoattractant protein 1 (MCP-1) and C-X-C motif chemokine ligand 1 (CXCL1) in vitro [19]. Accordingly, neutralizing EphA2 antibodies reduce neutrophil infiltration in a LPS-induced lung injury model [18]. EphA2 signaling also regulates inflammatory cell infiltration by modulating the expression of the endothelial adhesion proteins vascular cell adhesion molecule 1 (VCAM1) and E-selectin, both of which promote monocyte attachment and infiltration [34]. Together, Ephrin-A1/EphA2 signaling links endothelial barrier dysfunction to inflammation during lung injury and repair.
While the inflammatory effects of the Ephrin-A family of ligands have been well explored in vitro and in vivo, the role of Ephrin-B family ligands in regulating inflammation during tissue injury is poorly understood. Cultured intestinal epithelial cell-6 (IEC-6) cells express high levels of Ephrin-B1 and Ephrin-B2 ligands, and stimulation of reverse Ephrin-B signaling induces expression of pro-inflammatory effectors, including MCP-1 and cyclooxygenase-2 (COX-2), which regulate inflammation during intestinal repair [29]. Further studies investigating the role of Ephrin-B family ligands on inflammation during tissue repair are required.
Regulation of Tissue Re-epithelialization
During tissue repair, migration of epithelial cells from the wound edge over the denuded surface is required to close the wound, ultimately resulting in tissue re-epithelialization. Recent work demonstrates the important role for ephrin ligands and Eph receptors during this process. Mice subjected to skin punch biopsy have increased expression of Ephrin-B1 and multiple Eph receptors (Eph-B2, -B4, -A2, -A5) within the injured epidermis [25]. The exact role of the subclasses of ephrin ligands and Eph receptors in wound healing has yet to be deciphered, but evidence suggests that Ephrin-B1/EphB receptor signaling plays an important role in this cutaneous wound healing model. Ephrin-B1 transiently localizes to keratinocytes, a specialized skin epithelial cell, along the leading edge of the wound, whereas Ephrin-B2 expression remains diffuse throughout the epidermis before and after wounding [25]. In order to migrate, epithelial cells decrease expression of adhesion proteins enabling them to detach [25].
During tissue repair, increased Ephrin-B1 expression coincides with the loosening of epithelial cell tight junctions and adherens junctions, in addition to the downregulation of adhesion proteins E-cadherin and claudin-1 [25]. During intestinal repair, the cell surface protease a disintegrin and metalloproteinase 10 (ADAM10) mediates E-cadherin shedding to promote epithelial cell migration during intestinal repair [26]. In this mechanism, ADAM10 and EphB3 receptors interact within the intestinal epithelium, and activation of EphB3 by Ephrin-B1 ligands in trans increases ADAM10 activity to promote cleavage of E-Cadherin [26]. Accordingly, epithelial-specific double knockout of Efnb1 and Efnb2 in mice leads to sustained adhesion protein expression that interferes with the loosening of the epithelial cells, resulting in severe failure of re-epithelization during wound repair; however, total cellularity in wound tissue is not affected, suggesting that Ephrin-B1 and -B2 play a major role in cell migration but not proliferation [25].
Signaling of additional ephrin family members also positively regulates epithelial cell migration in vitro [20, 29, 38, 39]. For instance, treatment with soluble Ephrin-A1 mimetic leads to reduction in EphA2 receptor expression and epithelial cell migration in a scratch wound assay, indicating that an overabundance of Ephrin-A1 could interfere with wound closure [20]. Similarly, treating keratinocytes with recombinant Ephrin-A1 also decreases their migration in an EphA2-dependent manner [39]. However, cultured epithelial cells overexpressing Ephrin-B1 or Ephrin-B2 exhibit impaired E-cadherin and cell-cell contact, suggesting that expression of ephrin ligands and Eph receptors is fine-tuned to regulate wound healing [25]. While it is evident that epithelial expression of ephrin ligands and Eph receptors is crucial for epithelial function and may have a prominent role in wound healing, further research is required to clarify exact signaling pathways and functions of ephrins and Ephs in this regard.
Regulation of ECM Synthesis and Remodeling
The secretion of newly synthesized ECM proteins, such as proteoglycans and collagens, is an essential component of wound healing. ECM deposition by activated myofibroblasts in the wound bed provides a provisional matrix that not only provides structural integrity but also regulates multiple biological processes and cellular functions such as angiogenesis, inflammation, and re-epithelization [40]. During the remodeling phase of wound healing, myofibroblasts contract this provisional matrix to close the wound, leading to their apoptosis and subsequent termination of the tissue repair program [41]. Recent studies show that Eph/ephrin signaling plays a role in ECM deposition and remodeling during tissue repair. Of note, mice lacking Ephrin-A2 or Ephrin-A5 ligands show no wound healing phenotype and a normal rate of wound closure. While fibroblasts isolated from Efna2 and Efna5 knockout mice proliferate and migrate, an increased collagen density within the dermis in a surgical excision model of wound healing is observed, a phenotype absent in Efna2 and Efna5 double knockout mice [24]. Furthermore, both Efna2 knockout and Efna2/Efna5 double knockout mice exhibit changes in scar morphology following wounding [24]. Currently, there is scarce evidence implicating ephrin participation in the secretion of ECM molecules within the context of normal wound healing; thus, further research is needed to better understand how ephrin or Eph signaling mediates tissue remodeling processes under these circumstances.
Regulation of Angiogenesis
Angiogenesis is required for vascularization of granulation tissue to deliver nutrients and oxygen and permit inflammatory cell infiltration [42]. During this process, endothelial cells interact with cytokines and growth factors released at the injury site, and with the ECM to promote vessel growth [42]. Ephrin ligands and Eph receptor interactions, in particular Ephrin-A1/EphA2 and Ephrin-B2/EphB4, have prominent roles in angiogenesis [43]. Ephrin-A1/EphA2 signaling promotes VEGF-dependent angiogenesis by supporting vessel sprouting, vessel formation, and vessel survival [21]. Blockade of Ephrin-A1/EphA2 signaling with EphA2-Fc chimeric receptors abrogates corneal angiogenesis in vivo [21]. Evidence also suggests that Ephrin-A1 supports in vitro endothelial cell assembly and microvessel formation [22, 23]. However, the role of Ephrin-A1/EphA2 signaling in angiogenesis during tissue repair remains unknown.
On the contrary, Ephrin-B2/EphB4 signaling has been the dominant focus of studies investigating roles of ephrin/Eph signaling in angiogenesis. During injury, hypoxia drives angiogenesis through hypoxia-inducible factor (HIF)-1α activation and subsequent VEGF secretion [27]. In arteries, endothelial Ephrin-B2 expression is increased in response to hypoxia, driving angiogenesis and promoting arterial differentiation [27]. While VEGF is a master regulatory protein for angiogenesis, Ephrin-B2 is also crucial for both angiogenesis and VEGF signaling [13, 28, 30, 31]. Ephrin-B2 mediates the endocytosis and activation of VEGFR2 and VEGFR3, a significant event as VEGFR2 requires internalization to mediate endothelial wound healing [13, 28, 44, 45]. Thus, Ephrin-B2/EphB4 signaling is essential for angiogenesis.
The roles of ephrin and Eph receptors in the different stages of the normal wound healing response highlight the need for a better understanding of the contribution of ephrins and Eph receptors in pathological healing responses. Interest in dysregulated ephrin/Eph signaling is growing, particularly with respect to fibrosis, and various disease models highlight how ephrins contribute to pathology.
Ephrin/Eph Signaling in Tissue Fibrosis
Given the major role of ephrin/Eph receptor signaling during physiological tissue repair, it is not surprising that dysregulation in ephrin/Eph receptor signaling is implicated in the development of tissue fibrosis [46–49]. The role of ephrin/Eph signaling in tissue fibrogenesis has been studied in multiple mouse models using genetic and pharmacologic approaches, and targeting ephrin/Eph receptor signaling has recently emerged as a novel therapeutic strategy to combat tissue fibrosis. This section will highlight the role of ephrin/Eph receptor signaling in organ fibrosis and potential therapeutic strategies to mitigate fibrosis based on ephrin/Eph inhibition (Table 2).
Table 2.
Ephrins and Eph receptor signaling in fibrotic diseases
| Ephrin/Eph | Disease | Organ/tissue | Cell type | Effect | Ref. |
|---|---|---|---|---|---|
| Ephrin-A1 | Atherosclerosis | Blood vessels | Endothelial cells | Ephrin-Al expression increases in atherosclerotic ApoE mouse model and co-localizes with EphA2 surrounding atherosclerotic plaques | [50••] |
| EphA2 | Atherosclerosis | Blood vessels | Smooth muscle cells | Epha2 deletion exhibits smaller atherosclerotic plaques in ApoE mouse model at advanced stages of disease and also decreases in collagen and fibronectin in fibrotic regions | [34, 50••] |
| Cardiac fibrosis | Heart | Epha2 knockout reduces fibrosis nearly twofold in myocardial infarction hyperglycemic mouse model, but increases morbidity | [51] | ||
| EphA3 | Idiopathic pulmonary fibrosis | Lung | Epithelial cells | EphA3 localizes to cells within the fibrotic regions of IPF lung explants | [52] |
| EphA3-positive epithelial cell IV injection increases hydroxyproline content in lung tissue | [52] | ||||
| Ephrin-B2 | Cardiac fibrosis | Heart | Fibroblasts | Ephrin-B2 elevates in patients with heart failure compared with control patients | [53••] |
| In vivo and in vitro modulation of Ephrin-B2 indicates that it is important for myocardial myofibroblast activation and function | [53••] | ||||
| Idiopathic pulmonary fibrosis | Lung | Fibroblasts | Blockade of Ephrin-B2 forward signaling reduces collagen deposition in fibroblasts isolated from patients with IPF | [17] | |
| Soluble Ephiin-B2 is sufficient in facilitating pro-fibrotic effects in pulmonary fibroblasts | [17] | ||||
| Systemic sclerosis | Skin | Endothelial cells | Ephiin-B2 localizes to small vessels and vascular smooth muscle layers, with marked increase in endothelial cells of SSc patient skin | [49] | |
| Fibroblast-specific Ephrin-B2 knockout mice are protected from skin fibrosis in a mouse model of scleroderma | [17] | ||||
| Liver cirrhosis | Liver | Hepatic stellate cells | Stimulation of hepatic stellate cells with PDGF leads to upregulation of Ephrin-B2 and its EphB4 receptor, while the use of imatinib to inhibit PDGF receptors leads to decreased basal Ephrin-B2 expression | [46] | |
| Kidney fibrosis | Kidney | Endothelial cells | Inhibition of Ephrin-B2 PDZ domain-mediated reverse signaling increases kidney fibrosis in the unilateral ureteral obstruction model, which is indicated with increased myofibroblast activation and type I collagen deposition | [54] | |
| EphB2 | Liver fibrosis | Liver | Hepatic stellate cells | Malaria-induced and CCL4-induced liver fibrosis in ephB2 knockout mice have less collagen deposition in liver and less alpha-SMA expressing hepatic stellate cells | [55] |
| In the same models, Ephrin-B2 knockout reduces T GF-β and α-SMA expression in hepatic stellate cells and confers a protective phenotype against fibrosis | [55] | ||||
| EphB4 | Systemic sclerosis | Skin | Endothelial cells | EphB4 expression increases in the skin of SSc patients, localizing around small vessels and vascular smooth muscle layers | [49] |
ApoE, apolipoprotein E; α-SMA, alpha smooth muscle actin; Eph receptor, erythropoietin-producing human hepatocellular carcinoma receptors; PDGF, platelet-derived growth factor; SSc, systemic sclerosis; TGF-β, transforming growth factor-beta
Vascular fibrosis, characterized by reduced lumen diameter and arterial wall thickening, is prevalent in advanced stages of patients with atherosclerosis [34]. In this context, pathological EphA2 receptor signaling activated by Ephrin-A1 ligand in endothelial cells lining atherosclerotic plaques induces pro-inflammatory gene expression (VCAM-1, E-selectin) and stimulates monocyte adhesion [34]. In vivo, genetic deletion of EphA2 receptor in ApoE null mice fed a high fat diet have smaller atherosclerotic plaques and reduced plaque formation [50••]. In addition, the advanced plaques of these knockout mice have reduced total ECM, including collagen and fibronectin, indicating that genetic blockade of EphA2 receptor decreases matrix deposition and vascular fibrosis [50••]. However, targeting of EphA2 for anti-fibrotic therapy may require caution, as evidence suggests that Epha2 knockout mice are more prone to hyperglycemia-induced injury and show worse myocardial infarction and decreased survival [51]. Interestingly, while the myocardial infarct size of hyperglycemic Epha2 knockout mice is increased compared with wild-type mice, fibrosis within the mouse heart is reduced nearly two-fold. Although EphA2 has the potential to modulate fibrosis, consideration of patient comorbidities must be taken into consideration when developing therapies to inhibit EphA2. EphA2 receptor signaling has also been studied in lung injury. Epha2 knockout mice are protected from bleomycin-induced lung injury. Mechanistically, Ephrin-A1/EphA2 receptor signaling in the endothelium not only regulates vascular permeability but also the expression of pro-inflammatory cytokines such as CXCL1 and CCL2 in bleomycin-injured lungs [19].
In the lungs, epithelial cells surrounding fibroblastic foci of lung tissue explants from patients with IPF overexpress EphA3 receptor [52]. Further, infusion of EphA3-positive epithelial cells from IPF patients into immunodeficient mice is sufficient to drive a fibrotic response and increase the total hydroxyproline content of lung tissue, a surrogate marker of collagen deposition [52]. A clinical trial testing KB004, a neutralizing antibody of EphA3 receptor, for the treatment of glioblastoma shows therapeutic promise [56, 57]. In addition, administration of KB004 to a 78-year-old patient with relapsed acute myeloid leukemia (AML) gradually reduced reticulin and collagen deposition within the bone marrow [56]. Currently, an ongoing phase I clinical trial may shed further light into the safety and efficacy of anti-EphA3 therapy [58].
In the liver, fibrosis occurs after acute or chronic liver injury due to alcohol abuse, non-alcoholic steatohepatitis (NASH), or infection (hepatitis C virus or malaria). In mice, malaria infection increases EphB receptor mRNA and protein expression in hepatic stellate cells, Küpffer cells, inflammatory monocytes, and neutrophils [55]. Ephb2 knockout mice have decreased inflammation and collagen deposition within the liver of infected mice [55]. Mechanistically, EphB2 receptor signaling is required for activation of pro-fibrotic macrophages/Küpffer cells, which are the principal source of TGF-β and other profibrotic mediators [55]. This protective phenotype against fibrosis in Ephb2 knockout mice is replicated in the chemical-induced CCL4 model of liver fibrosis [55].
Among the ephrin/Eph family members, Ephrin-B2 ligand is a potent fibrogenic factor across multiple organs. Several recent studies implicate Ephrin-B2 in human fibrotic diseases such as scleroderma, IPF, and cardiac and liver fibrosis, suggesting a major role for Ephrin-B2 in tissue fibrogenesis, as discussed in detail below.
Ephrin-B2 as a Novel Target for Anti-Fibrotic Therapy
Ephrin-B2 in Cardiac Fibrosis
Increased expression of Ephrin-B2 is observed in the myocardium of patients with advanced heart failure, as well as in mouse models of myocardial infarction and cardiac hypertrophy induced by angiotensin II infusion, which is accompanied by myofibroblast activation and collagen fiber deposition [53••]. Of note, in vivo silencing of Ephrin-B2 in mice using a lentiviral vector ameliorates cardiac fibrosis and improves cardiac function [53••]. In vitro overexpression of Ephrin-B2 promotes cardiac fibroblast to myofibroblast activation, demonstrated by increased fibroblast proliferation, migration, and α-SMA expression, while Ephrin-B2 knockdown provides protection against hypoxia-mediated cardiac myofibroblast activation [53••]. Mechanistically, Ephrin-B2 signaling promotes activation of Stat3 and TGF-β/SMAD3 signaling pathways. Overall, Ephrin-B2 appears to have a prominent role in the development of cardiac fibrosis.
Ephrin-B2 in Skin Fibrosis
Both Ephrin-B2 ligand and EphB4 receptor are increased in the skin of patients with SSc [49]. Within the context of SSc skin, Ephrin-B2 largely localizes to small vessels and vascular smooth muscle layers, with a marked increase in endothelial cells; however, the exact roles of Ephrin-B2 and EphB4 in SSc progression remain unknown [49]. More recently, Ephrin-B2/EphB4 receptor signaling was shown to be required for the development of fibrosis in SSc by promoting myofibroblast activation and ECM deposition. In vivo, fibroblast-specific Efnb2 knockout mice are protected from skin fibrosis in a mouse model of SSc [17]. Together, Ephrin-B2 appears to drive fibrogenic responses during the development of skin fibrosis.
Ephrin-B2 in Lung Fibrosis
Ephrin-B2/EphB4 receptor signaling also contributes to fibrosis in lung disease. Blockade of Ephrin-B2 forward signaling reduces collagen deposition in fibroblasts isolated from patients with IPF. ADAM10 regulates the proteolytic shedding of the Ephrin-B2 ectodomain generating a soluble Ephrin-B2 (sEphrin-B2), which promotes myofibroblast activation and lung fibrosis via EphB4 receptor signaling. Mice genetically lacking fibroblast expression of Ephrin-B2 also exhibit significant protection from bleomycin-induced lung fibrosis. Interestingly, the small molecule ADAM10 inhibitor GX254023X also decreases the amount of sEphrin-B2 in mouse lung tissue and BAL and reduces bleomycin-induced lung fibrosis [17]. Increased sEphrin-B2 levels can also be detected in the BAL fluid and plasma of IPF patients compared healthy controls, suggesting ADAM10–sEphrin-B2 cleavage is likely upregulated in patients with IPF [17].
Ephrin-B2 in Kidney Fibrosis
While Ephrin-B2 ectodomain binding to EphB4 is required to activate “forward signaling”, Ephrin-B2 also signals through its intracellular domain via “reverse signaling”. In this regard, Ephrin-B2 reverse signaling has anti-fibrotic effects, and genetic inhibition of Ephrin-B2 reverse signaling results in augmented kidney fibrosis in the unilateral ureteral obstruction model [54]. The intracellular PDZ domain of Ephrin-B2 is activated in response to kidney injury and mice with dysfunctional PDZ-dependent Ephrin-B2 signaling (Ephrin-B2 ΔV mice) exhibit increased kidney fibrosis, demonstrated through increased myofibroblast proliferation and collagen deposition, while also impairing VEGFR2 internalization [54]. Thus, Ephrin-B2 forward and reverse signaling appear to have opposite effects during tissue fibrogenesis; however, the precise molecular mechanisms explaining this paradox remain poorly understood.
Ephrin-B2 in Liver Fibrosis
Hepatic stellate cells (HSCs) play major roles in the initiation and progression of liver fibrosis by secreting a myriad of fibrogenic molecules that drive ECM deposition and intrahepatic angiogenesis [59]. Thus, chronic activation of HSCs induces an aberrant architecture of the hepatic microvasculature in the cirrhotic liver. Ephrin-B2 signaling modulates HSC activation as well as their interaction with sinusoidal endothelial cells in vitro [46]. In this mechanism, PDGF signaling upregulates Ephrin-B2 expression in HSCs, which is required for HSC-induced capillary-tube formation. In vivo blockade of PDGF-Ephrin-B2 signaling with imatinib reduces pathological sinusoidal remodeling and portal hypertension in a BDL model of liver injury [46]. Further, Ephrin-B2 signaling has been shown to control angiogenesis by stimulation of VEGF production in HSCs [60]. Thus, Ephrin-B2 represents a novel target for the treatment of portal hypertension, a frequent and severe complication of liver cirrhosis.
Conclusions
Ephrin ligands and Eph receptors regulate fundamental biological processes involved in tissue fibrosis including cell migration, myofibroblast activation, angiogenesis, and tissue remodeling. A growing body of evidence has highlighted Ephrin-B2 as a key fibrogenic factor of the liver, skin, lungs, and heart in both diseased human tissues and in associated animal models. There is convincing evidence to suggest that Ephrin-B2 signaling plays a crucial role in promoting fibrogenic responses by inducing myofibroblast activation. Thus, interfering with the Ephrin-B2 signaling (including sEphrin-B2, Ephrin-B2 sheddase ADAM10, or its receptor EphB4) could represent a novel therapeutic strategy to mitigate organ fibrosis. Therapeutic strategies aimed at blocking Ephrin-B2 signaling have been developed for cancer treatment; however, their anti-fibrotic effects are yet to be explored.
Acknowledgments
BW is the recipient of the Training Graduate PhD Salary Award from The Arthritis Society (Canada). DL is supported in part by the NIH grant R01 HL147059-01, Start-up Package by Massachusetts General Hospital, Scleroderma Foundation New Investigator Grant, Scleroderma Research Foundation Investigator-Initiated Research Grant, American Thoracic Society Foundation/Pulmonary Fibrosis Foundation Research Grant, and Sponsored Research Grants from Boehringer Ingelheim, Unity Biotechnology, and Indalo Therapeutics. MK is supported in part by the Canada Research Chairs Program, Canadian Institute of Health Research, The Natural Sciences and Engineering Research Council of Canada (NSERC), Canadian Foundation for Innovation, The Krembil Foundation, Stem Cell Network, The Toronto General and Western Hospital Foundation, and The Arthritis Program, University Health Network.
Footnotes
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Papers of particular interest, published recently, have been highlighted as:
•• Of major importance
- 1.Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis—a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10:390–402. [DOI] [PubMed] [Google Scholar]
- 2.Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1:15002. [DOI] [PubMed] [Google Scholar]
- 3.Wells AU, Denton CP. Interstitial lung disease in connective tissue disease–mechanisms and management. Nat Rev Rheumatol. 2014;10(12):728–39. [DOI] [PubMed] [Google Scholar]
- 4.DiPietro LA. Angiogenesis and wound repair: when enough is enough. J Leukoc Biol. 2016;100(5):979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49(1):35–43. [DOI] [PubMed] [Google Scholar]
- 6.Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6(265):265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–38. [DOI] [PubMed] [Google Scholar]
- 8.Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5(9). 10.1101/cshperspect.a009159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai D, Huang Q, Nussinov R, Ma B. Promiscuous and specific recognition among ephrins and Eph receptors. Biochim Biophys Acta. 2014;1844(10):1729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Himanen JP, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, et al. Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci. 2004;7(5):501–9. [DOI] [PubMed] [Google Scholar]
- 11.Dravis C, Henkemeyer M. Ephrin-B reverse signaling controls septation events at the embryonic midline through separate tyrosine phosphorylation-independent signaling avenues. Dev Biol. 2011;355(1):138–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bochenek ML, Dickinson S, Astin JW, Adams RH, Nobes CD. Ephrin-B2 regulates endothelial cell morphology and motility independently of Eph-receptor binding. J Cell Sci. 2010;123(Pt 8): 1235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465(7297): 487–91. [DOI] [PubMed] [Google Scholar]
- 14.Nakayama A, Nakayama M, Turner CJ, Hoing S, Lepore JJ, Adams RH. Ephrin-B2 controls PDGFRbeta internalization and signaling. Genes Dev. 2013;27(23):2576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davy A, Aubin J, Soriano P. Ephrin-B1 forward and reverse signaling are required during mouse development. Genes Dev. 2004;18(5):572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen K, Bai H, Liu Y, Hoyle DL, Shen W-F, Wu L-Q, et al. EphB4 forward-signaling regulates cardiac progenitor development in mouse ES cells. J Cell Biochem. 2015;116(3):467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagares D, Ghassemi-Kakroodi P, Tremblay C, Santos A, Probst CK, Franklin A, et al. ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis. Nat Med. 2017;23(12):1405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong JY, Shin MH, Chung KS, Kim EY, Jung JY, Kang YA, et al. EphA2 receptor signaling mediates inflammatory responses in lipopolysaccharide-induced lung injury. Tuberc Respir Dis. 2015;78(3):218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carpenter TC, Schroeder W, Stenmark KR, Schmidt EP. Eph-A2 promotes permeability and inflammatory responses to bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2012;46(1):40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan N, Fatima A, Peng H, Bryar PJ, Lavker RM, Getsios S. EphA2/Ephrin-A1 signaling complexes restrict corneal epithelial cell migration. Invest Ophthalmol Vis Sci. 2012;53(2):936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, et al. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1(1):2–11. [PubMed] [Google Scholar]
- 22.Daniel TO, Stein E, Cerretti DP, St John PL, Robert B, Abrahamson DR. ELK and LERK-2 in developing kidney and microvascular endothelial assembly. Kidney Int Suppl. 1996;57:S73–81. [PubMed] [Google Scholar]
- 23.Dobrzanski P, Hunter K, Jones-Bolin S, Chang H, Robinson C, Pritchard S, et al. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res. 2004;64(3):910–9. [DOI] [PubMed] [Google Scholar]
- 24.Wijeratne D, Rodger J, Stevenson A, Wallace H, Prele CM, Wood FM, et al. Ephrin-A2 affects wound healing and scarring in a murine model of excisional injury. Burns. 2018. 10.1016/j.burns.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Nunan R, Campbell J, Mori R, Pitulescu ME, Jiang WG, Harding KG, et al. Ephrin-Bs drive junctional downregulation and actin stress fiber disassembly to enable wound re-epithelialization. Cell Rep. 2015;13(7):1380–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solanas G, Cortina C, Sevillano M, Batlle E. Cleavage of E-cadherin by ADAM10 mediates epithelial cell sorting downstream of EphB signalling. Nat Cell Biol. 2011;13:1100–7. [DOI] [PubMed] [Google Scholar]
- 27.Sohl M, Lanner F, Farnebo F. Sp1 mediate hypoxia induced ephrinB2 expression via a hypoxia-inducible factor independent mechanism. Biochem Biophys Res Commun. 2010;391(1):24–7. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465(7297):483–6. [DOI] [PubMed] [Google Scholar]
- 29.Hafner C, Meyer S, Hagen I, Becker B, Roesch A, Landthaler M, et al. Ephrin-B reverse signaling induces expression of wound healing associated genes in IEC-6 intestinal epithelial cells. World J Gastroenterol. 2005;11(29):4511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groppa E, Brkic S, Uccelli A, Wirth G, Korpisalo-Pirinen P, Filippova M, et al. EphrinB2/EphB4 signaling regulates non-sprouting angiogenesis by VEGF. EMBO Rep. 2018;19(5):e45054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erber R, Eichelsbacher U, Powajbo V, Korn T, Djonov V, Lin J, et al. EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J. 2006;25(3):628–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leem AY, Shin MH, Douglas IS, Song JH, Chung KS, Kim EY, et al. All-trans retinoic acid attenuates bleomycin-induced pulmonary fibrosis via downregulating EphA2-EphrinA1 signaling. Biochem Biophys Res Commun. 2017;491(3):721–6. [DOI] [PubMed] [Google Scholar]
- 33.Xiong Y, Li KX, Wei H, Jiao L, Yu SY, Zeng L. Eph/ephrin signalling serves a bidirectional role in lipopolysaccharide-induced intestinal injury. Mol Med Rep. 2018;18(2):2171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Funk SD, Yurdagul A Jr, Albert P, Traylor JG Jr, Jin L, Chen J, et al. EphA2 activation promotes the endothelial cell inflammatory response: a potential role in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(3):686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruscitti F, Ravanetti F, Essers J, Ridwan Y, Belenkov S, Vos W, et al. Longitudinal assessment of bleomycin-induced lung fibrosis by micro-CT correlates with histological evaluation in mice. Multidiscip Respir Med. 2017;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Limjunyawong N, Mitzner W, Horton MR. A mouse model of chronic idiopathic pulmonary fibrosis. Phys Rep. 2014;2(2): e00249–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Du S, Yang L, Chen Y, Huang W, Zhang R, et al. Rapid pulmonary fibrosis induced by acute lung injury via a lipopolysaccharide three-hit regimen. Innate Immun. 2009;15(3):143–54. [DOI] [PubMed] [Google Scholar]
- 38.Cho HJ, Hwang YS, Mood K, Ji YJ, Lim J, Morrison DK, et al. EphrinB1 interacts with CNK1 and promotes cell migration through c-Jun N-terminal kinase (JNK) activation. J Biol Chem. 2014;289(26):18556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ventrella R, Kaplan N, Hoover P, Perez White BE, Lavker RM, Getsios S. EphA2 transmembrane domain is uniquely required for keratinocyte migration by regulating Ephrin-A1 levels. J Investig Dermatol. 2018;138(10):2133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–21. [DOI] [PubMed] [Google Scholar]
- 41.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146(1):56–66. [PMC free article] [PubMed] [Google Scholar]
- 42.Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5(1):40–6. [DOI] [PubMed] [Google Scholar]
- 43.Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002;13(1):75–85. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HCA, et al. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15:249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santos SC, Miguel C, Domingues I, Calado A, Zhu Z, Wu Y, et al. VEGF and VEGFR-2 (KDR) internalization is required for endothelial recovery during wound healing. Exp Cell Res. 2007;313(8): 1561–74. [DOI] [PubMed] [Google Scholar]
- 46.Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135(2): 671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luzina IG, Atamas SP, Wise R, Wigley FM, Choi J, Xiao HQ, et al. Occurrence of an activated, profibrotic pattern of gene expression in lung CD8+ T cells from scleroderma patients. Arthritis Rheum. 2003;48(8):2262–74. [DOI] [PubMed] [Google Scholar]
- 48.Umeda N, Ozaki H, Hayashi H, Oshima K. Expression of ephrinB2 and its receptors on fibroproliferative membranes in ocular angiogenic diseases. Am J Ophthalmol. 2004;138(2):270–9. [DOI] [PubMed] [Google Scholar]
- 49.Avouac J, Clemessy M, Distler JH, Gasc JM, Ruiz B, Vacher-Lavenu MC, et al. Enhanced expression of ephrins and thrombospondins in the dermis of patients with early diffuse systemic sclerosis: potential contribution to perturbed angiogenesis and fibrosis. Rheumatology. 2011;50(8):1494–504. [DOI] [PubMed] [Google Scholar]
- 50.••.Finney AC, Funk SD, Green JM, Yurdagul A Jr, Rana MA, Pistorius R, et al. EphA2 expression regulates inflammation and fibroproliferative remodeling in atherosclerosis. Circulation. 2017;136(6):566–82 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides evidence for the role of EphA2 in atherosclerosis.
- 51.DuSablon A, Kent S, Coburn A, Virag J. EphA2-receptor deficiency exacerbates myocardial infarction and reduces survival in hyperglycemic mice. Cardiovasc Diabetol. 2014;13:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Habiel DM, Espindola MS, Jones IC, Coelho AL, Stripp B, Hogaboam CM. CCR10+ epithelial cells from idiopathic pulmonary fibrosis lungs drive remodeling. JCI Insight. 2018;3(16): e122211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.••.Su SA, Yang D, Wu Y, Xie Y, Zhu W, Cai Z, et al. EphrinB2 regulates cardiac fibrosis through modulating the interaction of Stat3 and TGF-beta/Smad3 signaling. Circ Res. 2017;121(6): 617–27 [DOI] [PubMed] [Google Scholar]; This study describes the role of EphrinB2-signaling in cardiac fibrosis.
- 54.Kida Y, Ieronimakis N, Schrimpf C, Reyes M, Duffield JS. EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol. 2013;24(4): 559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mimche PN, Brady LM, Bray CF, Lee CM, Thapa M, King TP, et al. The receptor tyrosine kinase EphB2 promotes hepatic fibrosis in mice. Hepatology. 2015;62(3):900–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swords RT, Greenberg PL, Wei AH, Durrant S, Advani AS, Hertzberg MS, et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: results from a phase 1 study. Leuk Res. 2016;50:123–31. [DOI] [PubMed] [Google Scholar]
- 57.Swords RT, Wei AH, Durrant S, Advani AS, Hertzberg MS, Lewis ID, et al. KB004, a novel non-fucosylated Humaneered® antibody, targeting EphA3, is active and well tolerated in a phase I/II study of advanced hematologic malignancies. Blood. 2014;124(21):3756. [Google Scholar]
- 58.A trial of KB004 in patients with glioblastoma. https://ClinicalTrials.gov/show/NCT03374943. Accessed Nov 2018.
- 59.Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131(11):1728–34. [DOI] [PubMed] [Google Scholar]
- 60.Das A, Shergill U, Thakur L, Sinha S, Urrutia R, Mukhopadhyay D, et al. Ephrin B2/EphB4 pathway in hepatic stellate cells stimulates Erk-dependent VEGF production and sinusoidal endothelial cell recruitment. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G908–15. [DOI] [PMC free article] [PubMed] [Google Scholar]