

Abstract
Persistent alterations of proopiomelanocortin (Pomc) and mu-opioid receptor (Oprm1) activity and stress responses after alcohol are critically involved in vulnerability to alcohol dependency. Gene transcriptional regulation altered by alcohol may play important roles. Mice with genome-wide deletion of neuronal Pomc enhancer1 (nPE1−/−), had hypothalamic-specific partial reductions of beta-endorphin and displayed lower alcohol consumption, compared to wildtype littermates (nPE1+/+). We used RNA-Seq to measure steady-state nuclear mRNA transcripts of opioid and stress genes in hypothalamus of nPE1+/+ and nPE1−/− mice after 1-day acute withdrawal from chronic excessive alcohol drinking or after water. nPE1−/− had lower basal Pomc and Pdyn (prodynorphin) levels compared to nPE1+/+, coupled with increased basal Oprm1 and Oprk1 (kappa-opioid receptor) levels, and low alcohol drinking increased Pomc and Pdyn to the basal levels of nPE1+/+ in the water group, without significant effects on Oprm1 and Oprk1. In nPE1+/+, excessive alcohol intake increased Pomc and Oprm1, with no effect on Pdyn or Oprk1. For stress genes, nPE1−/− had lowered basal Oxt (oxytocin) and Avp (arginine vasopressin) that were restored by low alcohol intake to basal levels of nPE1+/+. In nPE1+/+, excessive alcohol intake decreased Oxt and Avpi1 (AVP-induced protein1). Functionally examining the effect of pharmacological blockade of MOP-r, we found that naltrexone reduced excessive alcohol intake in nPE1+/+, but not nPE1−/−. Our results provide evidence relevant to the transcriptional profiling of the critical genes in mouse hypothalamus: enhanced opioid and reduced stress gene transcripts after acute withdrawal from excessive alcohol may contribute to altered reward and stress responses.
Keywords: RNA-Seq, alcohol, opioid, stress, nPE1 knockout, nuclear transcript
Introduction
The endogenous opioid systems are profoundly changed by alcohol. Specifically, for the proopiomelanocortin (POMC) in the hypothalamus, alcohol alters the Pomc gene expression levels after chronic alcohol consumption [1–3] or after prolonged withdrawal [4]. Since activation of mu-opioid receptor (MOP-r) by beta-endorphin (encoded by Pomc) is rewarding [5, 6] and modulates dopamine release [7], alcohol-induced beta-endorphin release [8–10] could play a role in the reinforcing effects of alcohol, its motivational behaviors and consumption. Indeed, numerous pharmacological studies provide consistent evidence that the MOP-r blockade with antagonist naltrexone or naloxone decreases alcohol reward, consumption, reinstatement of alcohol seeking and relapse-like drinking in rodents, as well as alcohol drinking, craving, and relapse episodes in human alcoholics, further indicating that the beta-endorphin/MOP-r is critically involved in the regulation of alcohol consumption [11]. Determining the specific role of hypothalamic POMC neurons in alcohol drinking behaviors, we recently used transgenic mice with hypothalamic-specific POMC deletion resulting in brain-specific beta-endorphin deficiency [12] and demonstrated that the hypothalamic-POMC deficient mice drink less alcohol [13]. Further, pharmacological blockade of MOP-r with naltrexone dose-dependently decreased intake in the wildtype mice but showed a blunted effect in the hypothalamic-POMC deficient mice [13]. Together, the above results suggest that beta-endorphin and MOP-r play a critical role in modulation of alcohol consumption, probably via a hypothalamic POMC neuron-mediated mechanism.
Results from both clinic and preclinical studies have demonstrated profound alterations of stress responsive systems after chronic alcohol abuse. Specifically, alcohol has direct or downstream effects on several hypothalamic stress-responsive systems, including arginine vasopressin (AVP) [11, 14, 15], oxytocin [16], corticotrophin releasing hormone (CRH) and their receptors [17], dynorphin and the kappa opioid receptors (KOP-r) [18, 19]. The studies in both humans and rodents also provide clear support for the importance of these stress responsive systems in the process of alcohol consumption with strong interactions with hypothalamic-pituitary-adrenal (HPA) hormones and their receptors (e.g., glucocorticoid receptors), especially after stress [20, 21]. Though acute exposure to alcohol profoundly activates the HPA axis, many alcoholics or rats develop HPA tolerance after chronic alcohol exposure [22, 23].
Based on the above background, we propose a hypothesis that gene transcriptional regulation in the hypothalamus at basal levels or altered by alcohol plays important roles in individual vulnerability to excessive alcohol drinking. RNA sequencing (RNA-Seq) provides relatively accurate, highly sensitive and reliable gene expression data, though this technology for quantification analysis is still in a developmental stage [24]. Therefore, high-throughput RNA-Seq of total RNAs was performed for comprehensive molecular profiling of the mouse hypothalamus to identify changes in genes expression after chronic excessive alcohol drinking. For this purpose, mice, subjected to 4-day drinking-in-the-dark (DID) paradigm followed by a 3-week chronic intermittent access (IA) drinking paradigm (two-bottle choice, 24-h access every other day), developed high alcohol consumption (15–25 g/kg/day) [13, 25]. As altered mRNA changes in the nuclear compartment are more direct and sensitive to the gene transcriptional activation than those in the cytoplasmic compartment [26–29], we analysed hypothalamic gene transcripts using nuclear RNAs, to determine transcriptional alterations after 1-day withdrawal from chronic IA drinking. Firstly, neuronal Pomc enhancer1 knockout (nPE1−/−) mice with hypothalamic-specific partial loss of Pomc transcriptional activity [12] was used to confirm that nPE1−/− mice, in comparison with nPE1+/+ mice, had lowered nuclear Pomc transcript levels in the hypothalamus. Then using the nPE1 knockout, we purposely mimic possible genetic variations in humans with less Pomc expression and/or function, and tested how the Pomc partial deficiency is involved in excessive alcohol drinking, the question that cannot be answered using the mice with complete POMC deletion [12, 13]. As naltrexone blocks MOP-r, we further examined whether naltrexone could reduce alcohol drinking in nPE1−/− mice as a genetic control for the effects of the MOP-r antagonist. As individual vulnerability to excessive alcohol drinking is a key feature of alcohol addiction, we tested whether the genetically determined vulnerability to excessive alcohol drinking between nPE1+/+ (high intake) and nPE1−/− (low intake) mice were associated with alterations of related transcriptome profile in the hypothalamus, specifically the opioid and stress genes. Finally, stress hormone corticosterone levels were determined to provide functional validation about the stress gene transcriptome profiling in the hypothalamus of both nPE1+/+ and nPE1−/− mice after acute alcohol withdrawal.
METHODS AND MATERIALS
ANIMALS
Male littermates with neuronal Pomc enhancer1 knockout (nPE1−/−) and wildtype (nPE1+/+) were screened by PCR analysis of genomic DNA extracted from mouse tail biopsies, as described previously [12]. The gene mutation was generated by homologous recombination in 129S6/SvEvTac Taffy ES cells to produce the chimeric founder mice, followed by ~16 generations of backcrossing onto the C57BL/6J strain. Specifically, in these transgenic mice, deletion of nPE1 in the context of intact nPE2 and Pomc pituitary enhancer regions and the proximal promoter reduces Pomc expression by 50–70% in the hypothalamic arcuate nucleus, without altering Pomc expression in pituitary cells. Hypothalamic content of beta-endorphin and melanocortin are reduced by approximately 70% in the mutant mice compared to wildtype controls [12].
Male nPE1−/− and nPE1+/+ littermates (9~10-week old) derived from heterozygous nPE1+/− parents were used for all the present experiments. All mice were given ad libitum access to food and water in a stress-minimized facility and housed in individual and ventilated cages fitted with steel lids and filter tops. Mice were placed on a 12-hour reverse light-dark cycle (lights off at 7:00 am). Animal care and experimental procedures were conducted according to the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources Commission on Life Sciences 1996). The experimental protocols were approved by the Institutional Animal Care and Use Committee of the Rockefeller University.
PROCEDURES
1. The drinking-in-the-dark (DID) procedure followed by chronic (3 weeks) intermittent access (IA) drinking procedure (Table S1).
In the DID model, after the beginning of the dark period, mice had access to alcohol drinking with limited time (4h/day) in their home cages, and with food available in a one-bottle paradigm with alcohol exposure every day for 4 days [30]. The basic paradigm with our modifications was as follows [13]: At the time when the mice started individual housing (1 week before the experiments), the water bottle was replaced with that with sipper tubes to acclimate the mice to the sipper tube. Beginning at 10:00 am (3 hours after lights off) the water bottle was replaced with an alcohol pipette that was fitted with a stainless-steel straight sipper tube (containing a ball bearing at the end to prevent alcohol leakage) and sealed with a rubber stopper. The alcohol tube was refilled with fresh alcohol solution, kept for 4 hours and then replaced with a water bottle. In all the experiments, 15% (v⁄v) alcohol solution was prepared by mixing alcohol with tap water to reach 15% alcohol concentration in tap water. Body weight was recorded every day, and alcohol intake value (i.e., g⁄kg) was recorded after 4 hours of alcohol access every day.
After the 4-day DID in the first week, the mice always had access to alcohol in the home cage for 3 weeks with food and water available in a two-bottle free choice paradigm, with alcohol drinking every other day. This IA model was like an earlier protocol [25], with some modifications [13]. The procedures were identical to the DID described above with the following exceptions: Beginning at 3 hours after lights off, both the 15% alcohol solution and water tubes were provided on home cages. The left ⁄right position of the tubes was randomly changed every other day to avoid the possible side preference. After 4, 8 and 24 hours of alcohol access, both alcohol and water intake values were recorded, and these data were used to calculate alcohol intake (i.e., g ⁄ kg) and relative preference for alcohol (i.e., alcohol intake ⁄ total fluid intake). Access to alcohol following the 3-week procedure led to high alcohol intake in the mice [13]. As with the above DID model, we purposely monitored alcohol drinking at the beginning of the dark period, the 4-hour time point. To evaluate alcohol drinking in the circadian active dark cycle, alcohol and water intake values were also recorded at 8- and 24-hour recording times.
In the experiment of transcriptome profiling and stress hormone (Table S1A), the water control groups for each genotype were run in parallel under identical procedures (e.g., two water bottles every other day during 3-week IA), but without alcohol available. As shown in Table S2, body weight data were recorded at several key time points for both genotypes.
2. RNA Extraction.
Mice in both alcohol and water groups were sacrificed 24 hours after the last IA session by decapitation with brief (10 s) CO2 exposure; the hypothalamus was dissected from the brain and frozen on dry ice immediately (Table S1A). The snap-frozen hypothalamus was fractionated into nuclear and cytoplasmic phases using the double-detergent lysis buffer and disposable tuberculin syringes with 22-gauge (0.40 mm id) needles as homogenizers [28]. Briefly, the hypothalamus was lysed by the addition of 0.5 ml/sample 0.3 M sucrose lysis buffer and layered over a 0.80-ml cushion of 0.4 M sucrose lysis buffer. Samples were centrifuged at 300 × g for 15 min at 4 °C. The supernatant (cytoplasmic fraction) was transferred to a fresh tube, treated with proteinase K (Boehringer Mannheim, Indianapolis, IN) for 1 h at 45 °C. The remaining 0.4-M sucrose cushion was removed, and the nuclear pellet was washed with 0.5 ml 0.4 M sucrose and centrifuged again. The supernatant was removed, and the nuclear pellet was treated with RNase-free DNase-1 (Worthington, Biochemicals, Freehold, NJ) for 5 min at 37 °C, followed by a 30 min proteinase K treatment at 45 °C. Finally, the nuclear RNA was added with Qiazol (Qiagen, Valencia, CA), and the total nuclear RNA was isolated using the miRNeasy kit (Qiagen), and the quality and quantity of nuclear RNA from each sample was determined using an Agilent 2100 Bioanalyzer. This method permits efficient lysis of the tissue as evidenced by absence of the cytoplasmic tRNA in the nuclear fraction, with minimal rupture of nuclei as indicated by the absence of DNA in the cytoplasmic fraction, even with very vigorous homogenization [31].
3. RNA-seq library preparation and sequencing.
RNA-seq library preparation and sequencing of samples isolated from mouse hypothalamus was performed by the Genomic Resource Center at the Rockefeller University. Hypothalamic RNA-seq libraries were prepared using Illumina’s TruSeq® Stranded Total RNA Library Prep Kit with Ribo-Zero following manufacturer protocol. Libraries were prepared with unique barcodes and pooled at equal molar ratios. Briefly, starting with 100 ng total nuclear RNA, the RNA was fragmented by incubating at 94°C for 8 minutes with divalent cations. The cleaved RNA fragments were copied into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA polymerase I and RNaseH. The double stranded cDNA fragments then had the addition of a single ‘A’ nucleotide to prevent self-ligation during the subsequent addition of the indexing adapters. PCR was then used to enrich only those DNA fragments that had adapter molecules on both ends to amplify the amount of DNA in the library. Libraries were validated using Agilent Tape Station High Sensitivity DNA kits and normalized. Libraries were multiplexed, 12 samples per lane and sequenced. Illumina NextSeq 500 sequencer using high output V2 reagents and NextSeq Control Software v1.4 to generate 75 bp paired end reads, following manufacture protocol.
4. RNA-seq data quality assessment and differential transcript analysis.
The fastq files were generated by configuring BclToFastq.pl from CASAVA v1.8.2 with the following parameters: --ignore-missing-stats, --ignore-missing-bcl, --ignore-missing-control, --positions-format, clocs, --fastq-cluster-count 350000000. They were then examined using FASTQC [32]. The reads were aligned to the mouse reference genome (version mm10) using STAR v2.3 [33] aligner with default parameters. The alignment results were then evaluated through qualimap v2.2 [https://academic.oup.com/bioinformatics/article/32/2/292/ 1744356] to ensure that all the samples had a consistent alignment rate, and no obvious 5’ or 3’ bias. Aligned reads were summarized through feature Counts [34] with the gene model from Ensembl (Mus_musculus.GRCm38.75.gtf) at gene level: specifically, the uniquely mapped reads (NH ‘tag’ in bam file) that overlapped with an exon (feature) by at least 1bp were counted and then the counts of all exons annotated to an Ensembl gene (meta features) were summed into a single number. Only protein coding genes were used for the downstream analysis of this study. Principal Component Analysis (PCA) was then applied to the normalized count of all the samples from the hypothalamus to detect potential outliers. As shown in Figure S1, two samples in groups A and B did not cluster with the rest of the samples under the same conditions. After careful review of the procedures (littermate, cohort, dissection, extraction, etc.), we noted that the individual variability was not attributable to any obvious technical issue, and could not exclude that this level of variation was not biological. Therefore, we analyzed all the samples so that we could present our data in an unbiased way. Different transcript Seq2 [35] was applied to the normalized counts to estimate the fold change between the samples from mice that had chronic alcohol drinking and those from water controls in each genotype, using negative binomial distribution.
The genes selected for analysis are opioid and stress genes (Table S3). The rationale for such selections came from our following experiments to provide pharmacological data using opioid receptor antagonist naltrexone (section 5) and neuroendocrine on stress hormone corticosterone levels (section 6).
5. Genotype effect on alcohol drinking with acute administration of naltrexone in male nPE1 mice.
The objective of this experiment was to determine whether a potential genotype difference after chronic excessive drinking with MOP-r blockade by naltrexone. For each dose of naltrexone (1 or 2 mg/kg), separate groups of male nPE1 mice were used. On the test day after the IA paradigm (Table S1B), alcohol was presented 10 min after a single injection of naltrexone or vehicle (saline), and then alcohol and water intake values were recorded.
6. Genotype effect on plasma corticosterone levels.
The objective of this experiment was to determine whether a potential genotype difference after chronic excessive drinking on stress hormone levels. At the time of decapitation (1 day after 3-week IA paradigm) (Table S1A), blood from each mouse was collected in EDTA-containing tubes, placed on ice, and spun in a centrifuge at 4 °C. Corticosterone levels were assayed using a rat corticosterone 125I kit from MP Biomedicals (Costa Mesa, CA). All values were determined in duplicate in a single assay.
7. Blood ethanol concentration (BEC).
In a separate set of experiment (Table S1C), mice were subjected to the 4-day DID paradigm followed by a 3-week IA drinking paradigm. In session 11, alcohol intake values were recorded after 4-hour alcohol drinking, and then plasma from each mouse was collected as described above. BEC levels were assayed with the EnzyChrom kit (BioAssay Systems). All values were determined in duplicate in a single assay.
8. Statistical analysis on data.
For behavioral data, group differences (alcohol intake or preference ratios were analyzed using two-way ANOVA for genotype (nPE1+/+ vs. nPE1−/−) and time (4, 8, 24 hour recording times, or 1, 4 days) or three-way ANOVA for genotype (nPE1+/+ vs. nPE1−/−), treatment (naltrexone vs. saline) and time (4, 8, 24 hour recording times) followed by Newman-Keuls post-hoc tests. For gene transcript or corticosterone data, group differences were analyzed using two-way ANOVA for genotype (nPE1+/+ vs. nPE1−/−) and treatment (alcohol vs. water) followed by Newman-Keuls post-hoc tests. To explore possible relationships between individual gene transcript level and vulnerability to alcohol drinking, the last 24-h alcohol intake and gene transcript were examined by linear regression. The accepted level of significance for all tests was p<0.05. All statistical analyses were performed using Statistica (version 5.5, StatSoft Inc, Tulsa, OK). Adjustment for multiple comparisons (false discovery rate, FDR) was performed for all the genes data.
RESULTS
1. Genetically determined differences between nPE1−/− and nPE1+/+ male mice in alcohol intake and preference during chronic alcohol drinking in DID and IA models.
On both day 1 and day 4 during the exposure to 4-day alcohol drinking in the DID model, nPE1−/− drank less alcohol than nPE1+/+ (Table 1A). Two-way ANOVA revealed significant effects of genotype [F(1,20)=48, p<0.00001] and time [F(1,20)=44, p<0.00005] on alcohol intake. Post hoc analysis showed that: (1) in nPE1+/+, there was significantly more alcohol intake on day 4 than that on day 1 [p<0.01]; (2) in nPE1−/−, there was significantly more alcohol intake on day 4 than that on day 1 [p<0.01]; and (3) nPE1−/− had significantly less alcohol intake than nPE1+/+ on both day 1 and day 4 [p<0.01 for both].
Table 1. Genotype differences on alcohol intake in 4-day drinking-in-the-dark (DID) model (A) and on alcohol intake and alcohol preference in chronic (3 weeks) intermittent access excessive drinking model (B) between male nPE1+/+ and nPE1−/− mice.
(A) in DID model, alcohol was presented 3 hours after the beginning of dark cycle, and alcohol intake was recorded after 4 hours of alcohol access for 4 days in the nPE1+/+ and nPE1−/− mice. Genotype difference: **p<0.01 vs. nPE1+/+ at the same day; Day difference: ++p<0.01 vs. the same genotype on day 1 by 2-way ANOVA with Newman-Keuls post-hoc tests (n=6 for each group); (B) in IA model, mice exposed to the 2-bottle “alcohol (15%) vs. water” choice regimen every other day for 3 weeks. Data are presented after 4, 8 and 24 hours of alcohol access in the session 1 and in the session 10 during 3 weeks of chronic IA excessive alcohol drinking. Genotype difference: *p<0.05 or **p<0.01 vs. nPE1+/+ at the same time point in the same session; Session difference: +p<0.05 or ++p<0.01 vs. the same genotype at the same time point in the session 1 by 3-way ANOVA with Newman-Keuls post-hoc tests (n=6 for each group). Data presented as mean ± SEM.
| Day 1 | Day 4 | |||
|---|---|---|---|---|
| Genotype | nPE1+/+ | nPE1−/− | nPE1+/+ | nPE1−/− |
| Alcohol intake | 4.1 ± 0.35 | 1.1 ± 0.38 ** | 5.8 ± 0.35 ++ | 3.9 ± 0.63 ** ++ |
| Session 1 | Session 10 | ||||
|---|---|---|---|---|---|
| nPE1+/+ | nPE1−/− | nPE1+/+ | nPE1−/− | ||
| Alcohol Intake g/kg | 4h | 3.9 ± 0.43 | 1.9 ± 0.28 | 5.6 ± 0.59 + | 2.7 ± 0.43 |
| 8h | 5.3 ± 0.51 | 2.3 ± 0.34 * | 9.7 ± 0.59 | 4.5 ± 0.74 ** | |
| 24h | 13 ± 2.3 | 4.9 ± 0.90 ** | 19 ± 2.0 ++ | 9.5 ± 1.2 ** ++ | |
| Alcohol preference ratio | 4h | 0.59 ± 0.10 | 0.38 ± 0.07 * | 0.86 ± 0.02 + | 0.63 ± 0.05 * ++ |
| 8h | 0.67 ± 0.05 | 0.34 ± 0.13 ** | 0.81 ± 0.04 | 0.60 ± 0.07 ++ | |
| 24h | 0.54 ±0.06 | 0.38 ± 0.06 ** | 0.73 ± 0.04 | 0.61 ± 0.06 + | |
The nPE1+/+ exposed to the IA model for 3 weeks showed alcohol intake averaging approximately 19 g/kg/day, with high preference ratio more than 0.8. In the nPE1−/−, however, alcohol intake increased over days, but did not reach high consumption (around 10 g/kg/day) with less preference ratio. As shown in Table 1B on alcohol intake, three-way ANOVA revealed significant effects of genotype [F(1,60)=79, p<0.000001], interaction between genotype × time [F(2,60)=13, p<0.0005]; and session [F(1,60)=37, p<0.000001]. Post hoc analysis showed that: (1) in session 1, nPE1−/− had significantly less alcohol intake at both 8 and 24 hours than nPE1+/+ [p<0.05 and p<0.01, respectively]; (2) in session 10, nPE1−/− had significantly less alcohol intake at 8 and 24 hours than nPE1+/+ [p<0.01 for both]; (3) at 4 and 24 hours, nPE1+/+ had significantly more alcohol intakes in session 10 than those in session 1 [p<0.05 and p<0.01, respectively]; and (4) at 24 hours, nPE1−/− had significantly more alcohol intake in session 10 than that in session 1 [p<0.01]. On alcohol preference, three-way ANOVA revealed significant effects of genotype [F(1,60)=45, p<0.00001], session [F(1,60)=85, p<0.000001] and interaction between genotype × session [F(2,60)=5.7, p<0.05]. Post hoc analysis showed that: (1) in session 1, nPE1−/− had significantly less preference at 4, 8 and 24 hours than nPE1+/+ [p<0.05, p<0.01 and p<0.01, respectively]; (2) in session 10, nPE1−/− had significantly less alcohol preference at 4 hours than nPE1+/+ [p<0.05]; (3) nPE1+/+ had significantly more preference in session 10 at 4 hours than that in session 1 [p<0.05]; and (4) nPE1−/− had significantly more preference in session 10 at 4, 8 and 24 hours than those in session 1 [p<0.01 for all]. As shown in Table 2, there was a significant effect of genotype on BEC levels [F(1,10)=8.5, p<0.01], which were associated with their differences in alcohol intake [F(1,10)=5.5, p<0.05] between the nPE1+/+ and nPE1−/− mice.
Table 2. Genotype difference in blood ethanol concentration (BEC) in male nPE1 mice.
After mice were subjected to the 4-day DID paradigm followed by a 3-week IA drinking paradigm, in session 11 after 4-hour alcohol drinking, blood from each mouse was collected for BEC levels. Genotype difference: *p<0.05 or **p<0.01 vs. nPE1+/+ mice by Student’s t-tests (n=6 for each group). Data presented as mean ± SEM.
| Genotype | nPE1+/+ (n=6) | nPE1−/− (n=6) |
|---|---|---|
| Alcohol intake, g/kg | 5.4 ± 0.49 | 2.9 ± 0.22* |
| BEC, mg/ml | 0.49 ± 0.03 | 0.24 ± 0.03** |
2. Genetically determined differences between nPE1−/− and nPE1+/+ male mice in opioid genes transcript levels after chronic alcohol drinking.
2.1. Pomc and Oprm1 [MOP-r gene].
For Pomc (Figure 1A), two-way ANOVA showed significant effects of alcohol [F(1,19)=18, p<0.001] and genotype [F(1,19)=4.8, p<0.05], without alcohol × genotype interaction. Newman-Keuls post-hoc tests just failed to show a significant difference between nPE1−/− and nPE1+/+ in water control groups (p=0.09). Increased Pomc levels were observed in nPE1+/+ after alcohol (post-hoc tests, alcohol vs. water in nPE1+/+, p<0.05) (FDR=0.12). Similarly, Pomc levels were significantly higher in nPE1−/− after alcohol than those after water (p<0.05) (FDR=0.08).
Figure 1.
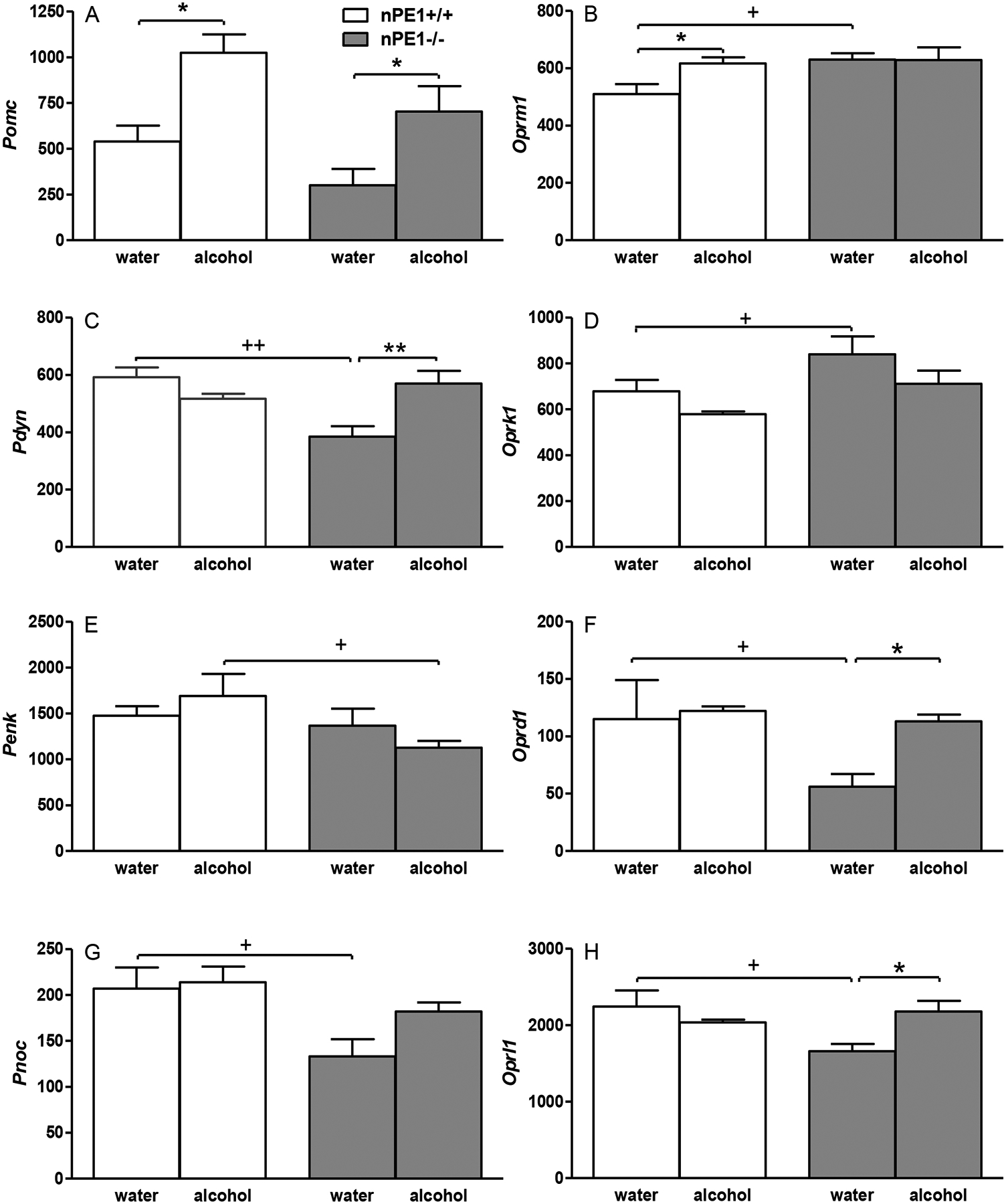
Genetically determined differences between nPE1+/+ and nPE1−/− male mice and effects of acute (1-day) withdrawal from chronic (4-week) excessive alcohol drinking on nuclear transcript levels (reads per kilobase per million per mapped reads) of opioid genes in the hypothalamus: (A) Pomc, (B) Oprm1, (C) Pdyn, (D) Oprk1, (E) Penk, (F) Oprd1, (G) Pnoc and (H) Oprl1. *p<0.05 or **p<0.01 vs. water control in the same genotype; +p<0.05 or ++p<0.01 vs. nPE1+/+ after the same treatment. n=5−6 for each group. Data presented as mean RPKM (reads per kilobase per million mapped reads) + SEM.
For Oprm1 (Figure 1B), two-way ANOVA showed a significant effect of genotype [F(1,19)=5.3, p<0.05] with no significant effect of alcohol [F(1,19)=3.5, p=0.08] or alcohol × genotype interaction. Although post-hoc tests failed to show a significant difference between nPE1−/− and nPE1+/+ in water groups (p=0.10), a planned comparison revealed that basal Oprm1 levels were significantly higher in nPE1−/− than nPE1+/+ (p<0.05) (FDR=0.08). Increased Oprm1 levels were observed in nPE1+/+ after alcohol (alcohol vs. water in nPE1+/+, p<0.05) (FDR=0.17).
2.2. Pdyn [prodynorphin gene] and Oprk1 [KOP-r gene].
For Pdyn (Figure 1C), two-way ANOVA showed significant effects of genotype [F(1,20)=5.3, p<0.05] and alcohol × genotype interaction [F(1,20)=14.9, p<0.001]. Post-hoc tests showed basal Pdyn levels were significantly lower in nPE1−/− than nPE1+/+ in water groups (p<0.01) (FDR=0.03). Increased Pdyn levels were observed in nPE1−/− mice after alcohol (alcohol vs. water in nPE1−/−, p<0.01) (FDR=0.05).
For Oprk1 (Figure 1D), two-way ANOVA showed significant effects of genotype [F(1,20)=7.3, p<0.05] with no significant effect of alcohol [F(1,20)=4.1, p=0.05]. Although post-hoc tests failed to show a significant difference between nPE1−/− and nPE1+/+ in water groups (p=0.10), a planned comparison revealed that basal Oprk1 level was significantly higher in nPE1−/− than that in nPE1+/+ (p<0.05) (FDR=0.08).
2.3. Penk [proenkephalin gene] and Oprd1 [DOP-r gene].
For Penk (Figure 1E), two-way ANOVA showed a significant effect of genotype [F(1,19)=5.5, p<0.05]. Compared with nPE1+/+, decreased Penk levels were observed in nPE1−/− mice after alcohol (alcohol nPE1−/− vs. alcohol nPE1+/+, p<0.05) (FDR=0.25).
For Oprd1 (Figure 1F), two-way ANOVA showed a significant effect of genotype [F(1,20)=4.8, p<0.05] with no significant effect of alcohol [F(1,20)=3.6, p=0.07]. Although post-hoc tests just failed to show a significant difference between nPE1−/− and nPE1+/+ in water groups (p=0.05), a planned comparison revealed that basal Oprd1 level was significantly lower in nPE1−/− than that in nPE1+/+ (p<0.05) (FDR=0.08). Increased Oprd1 levels were observed in nPE1−/− mice after alcohol (alcohol vs. water in nPE1−/−, p<0.05) (FDR=0.08).
2.4. Pnoc [pronociceptin gene] and Oprl1 [nociceptin receptor gene].
For Pnoc (Figure 1G), two-way ANOVA showed a significant effect of genotype [F(1,20)=9.9, p<0.01]. Post-hoc tests showed basal Pnoc level was significantly lower in nPE1−/− than that in nPE1+/+ in water groups (p<0.05) (FDR=0.08).
For Oprl1 (Figure 1H), two-way ANOVA showed a significant alcohol × genotype interaction [F(1,20)=7.7, p<0.05] with no significant effect of genotype [F(1,20)=2.9, p=0.09]. Post-hoc tests showed basal Oprl1 level was significantly lower in nPE1−/− than that in nPE1+/+ in water groups (p<0.05) (FDR=0.08). Increased Oprl1 levels were observed in nPE1−/− mice after alcohol (alcohol vs. water in nPE1−/−, p<0.05) (FDR=0.08).
3. Genetically determined differences between nPE1−/− and nPE1+/+ male mice in stress genes transcript levels after chronic alcohol drinking.
3.1. Oxt [oxytocin gene] and Oxtr [oxytocin receptor gene].
For Oxt (Figure 2A), two-way ANOVA showed significant effects of genotype [F(1,20)=4.8, p<0.05] and alcohol × genotype interaction [F(1,20)=9.8, p<0.01]. Post-hoc tests showed basal Oxt level was significantly lower in nPE1−/− than that in nPE1+/+ in water groups (p<0.01) (FDR=0.06). In nPE1+/+ mice, although post-hoc tests just failed to show a significant difference (p=0.08), a planned comparison revealed that Oxt levels after alcohol were significantly lower than the ones in water control (p<0.05) (FDR=0.18). In nPE1−/−, increased Oxt levels were observed after alcohol (alcohol vs. water in nPE1−/−, p<0.05) (FDR=0.10).
Figure 2.
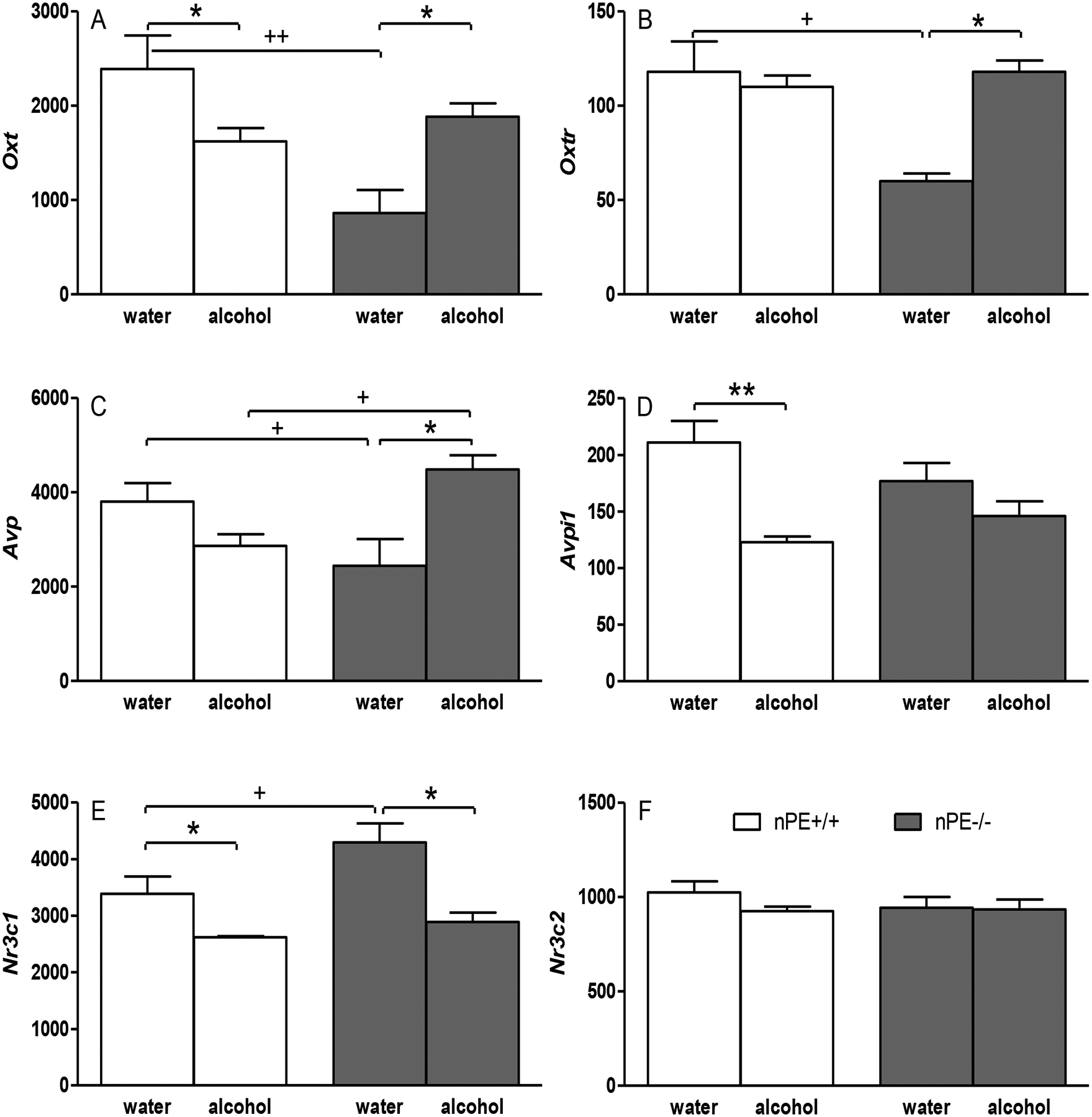
Genetically determined differences between nPE1+/+ and nPE1−/− male mice and effects of acute (1-day) withdrawal from chronic (4-week) excessive alcohol drinking on nuclear transcript levels of stress genes in the hypothalamus: (A) Oxt, (B) Oxtr, (C) Avp, (D) Avpi1, (E) Nr3c1 and (F) Nr3c2. *p<0.05 or **p<0.01 vs. water control in the same genotype; +p<0.05 or ++p<0.01 vs. nPE1+/+ after the same treatment. n=5−6 for each group. Data presented as mean RPKM (reads per kilobase per million mapped reads) + SEM.
For Oxtr (Figure 2B), two-way ANOVA showed a significant alcohol × genotype interaction [F(1,19)=5.3, p<0.05]. Post-hoc tests showed basal Oxtr level was significantly lower in nPE1−/− than that in nPE1+/+ in water groups (p<0.05) (FDR=0.09). In nPE1−/− mice, increased Oxtr levels were observed after alcohol (alcohol vs. water in nPE1−/−, p<0.05) (FDR=0.08).
3.2. Avp, Avpr1a [AVP type 1a receptor gene] and Avpi1 [AVP-induced protein 1 gene].
For Avp (Figure 2C), two-way ANOVA showed a significant alcohol × genotype interaction [F(1,20)=14, p<0.005]. Although post-hoc tests just failed to show a significant difference between nPE1−/− and nPE1+/+ in water groups (p=0.06), a planned comparison revealed that basal Avp level was significantly lower in nPE1−/− than that in nPE1+/+ (p<0.05) (FDR=0.08). In nPE1−/−, increased Avp levels were observed after alcohol (alcohol vs. water in nPE1−/−, p<0.01) (FDR=0.05). Compared with nPE1+/+ mice, increased Avp levels were observed in nPE1−/− mice after alcohol (alcohol nPE1−/− vs. alcohol nPE1+/+ mice, p<0.05) (FDR=0.25).
For Avpr1a (Table 3), two-way ANOVA showed a significant alcohol × genotype interaction [F(1,20)=4.7, p<0.05]. Although post-hoc tests failed to show a significant difference between alcohol and water in nPE1−/− (p=0.09), a planned comparison revealed that there was an increased Avpr1a levels after alcohol (p<0.05) (FDR=0.26).
Table 3.
Genetically determined differences between nPE1+/+ and nPE1−/− male mice and effects of acute (1-day) withdrawal from chronic (4-week) excessive alcohol drinking on nuclear transcript levels in the hypothalamus. Genotype difference: *p<0.05 vs. water control in the same genotype (n=6 for each group). Data presented as mean RPKM (reads per kilobase per million mapped reads) ± SEM.
| nPE1+/+ (n=6) | nPE1−/− (n=6) | |||
|---|---|---|---|---|
| Water | Alcohol | Water | Alcohol | |
| Avpr1a | 80 ± 10 | 74 ± 3 | 65 ± 5 | 85 ± 7 * |
| Crh | 70 ± 13 | 80 ± 8 | 56 ± 9 | 68 ± 9 |
| Crhr1 | 103 ± 24 | 87 ± 5 | 71 ± 5 | 88 ± 6 |
| Crhr2 | 61 ± 3 | 60 ± 4 | 48 ± 12 | 70 ± 7 |
| Crhbp | 178 ± 20 | 142 ± 6 | 154 ± 18 | 165 ± 17 |
| Fkbp5 | 325 ± 65 | 292 ± 14 | 231 ± 16 | 282 ± 17 |
For Avpi1 (Figure 2D), two-way ANOVA showed significant effects of alcohol [F(1,20)=17.5, p<0.0005] and alcohol × genotype interaction [F(1,20)=4.7, p<0.05]. In nPE1+/+, decreased Avpi1 levels were observed after alcohol (post-hoc tests, alcohol vs. water in nPE1+/+, p<0.005) (FDR=0.03).
3.3. Crh, Crhr1 [CRH type 1 receptor gene], Crhr2 [CRH type 2 receptor gene] and Crhbp [CRH binding protein gene].
For these genes, there was no significant effect of genotype, alcohol or their interaction (Table 3).
3.4. Nr3c1 [glucocorticoid receptor gene], Nr3c2 [mineralocorticoid receptor gene] and Fkbp5 [FK506 binding protein 5 gene].
For Nr3c1 (Figure 2E), two-way ANOVA showed a significant effect of alcohol [F(1,20)=14.8, p<0.001] with no significant effect of genotype [F(1,20)=3.8, p=0.06]. Post-hoc tests showed basal Nr3c1 level was significantly higher in nPE1−/− than that in nPE1+/+ in water groups (p<0.05) (FDR=0.09). In nPE1+/+, although post-hoc tests just failed to show a significant difference (p=0.08), a planned comparison revealed that Nr3c1 levels after alcohol were significantly lower than the ones in water control (p<0.05) (FDR=0.18). In nPE1−/− mice, decreased Nr3c1 levels were also observed after alcohol (alcohol vs. water in nPE1−/−, p<0.05) (FDR=0.05).
For Nr3c2 (Figure 2F) or Fkbp5 (Table 3), there was no significant effect of genotype, alcohol or their interaction.
4. Regression analysis.
Within the group of alcohol drinking mice, large individual differences in alcohol intake were observed. Then, linear regression between the last 24-hour alcohol intake and each nuclear mRNA transcript levels were further analyzed in both the nPE1+/+ and nPE1−/− mice, and correlation coefficient and p values were shown from Figure S2 to Figure S9.
5. Genetically determined differences between nPE1−/− and nPE1+/+ male mice in plasma corticosterone levels after chronic alcohol drinking.
Two-way ANOVA showed a significant effect of genotype [F(1,20)=38, p<0.0001] with no significant effect of alcohol [F(1,20)=3.9, p=0.06]. Post-hoc tests showed that basal plasma corticosterone level was significantly higher in nPE1−/− in water group (96±16 ng/ml, n=6) than that in nPE1+/+ in water groups (41±7 ng/ml, n=6) (p<0.005). Compared with the water control, nPE1+/+ had a decrease in plasma corticosterone levels after alcohol (14±2 ng/ml, n=6), but post-hoc tests just failed to show a significant difference (p=0.06). Compared with nPE1+/+ after alcohol, increased plasma corticosterone levels were observed in nPE1−/− after alcohol (84±10 ng/ml, n=6) (alcohol nPE1−/− vs. alcohol nPE1+/+, p<0.005).
6. Genetically determined differences between nPE1−/− and nPE1+/+ male mice in the effect of mu opioid receptor antagonist naltrexone on chronic alcohol drinking.
At 1 mg/kg naltrexone (Table 4A), there was no significant effect of this low-dose naltrexone on alcohol, water and alcohol preference ratio in either nPE1+/+ or nPE1−/− mice. Three-way ANOVA revealed significant effects of genotype [F(1,48)=48, p<0.0001] and interaction between genotype × time [F(2,48)=4.4, p<0.05] on alcohol intake. Post hoc analysis showed that nPE1−/− had significantly less alcohol intake at 8- and 24-hour time points than nPE1+/+ [p<0.05]. Also, three-way ANOVA revealed a significant effect of genotype [F(1,48)=34, p<0.001] only on alcohol preference.
Table 4. Genetically determined differences in the effect of mu opioid receptor antagonist naltrexone at 1 mg/kg (A) and 2 mg/kg (B) on alcohol drinking in nPE1 male mice.
The groups assigned as the vehicle- or naltrexone-treated mice in each genotypes had similar alcohol intake 24 hours before the test day (data not shown). On the test day, alcohol (15%) was presented 10 min after a single i.p. injection of naltrexone in saline or vehicle, and then alcohol and water intake values were recorded after 4, 8 and 24 hours of alcohol access. In these experiments, mice were assigned to one of four treatment groups: (1) nPE1+/+ with vehicle as control; (2) nPE1+/+ with naltrexone; (3) nPE1−/− with vehicle as control; and (4) nPE1−/− with naltrexone. Data are presented at all the 3 recording time points. Genotype difference: *p<0.05 vs. nPE1+/+ at the same time point after the same treatment; Naltrexone treatment difference: +p<0.05 vs. vehicle control in the same genotype at the same time point by 3-way ANOVA with Newman-Keuls post-hoc tests. Data presented as mean ± SEM.
| Genotype | nPE1+/+ (n = 5) | nPE1−/− (n = 5) | |||
|---|---|---|---|---|---|
| Treatment | vehicle | 1 mg/kg naltrexone | vehicle | 1 mg/kg naltrexone | |
| Alcohol intake g/kg | 4h | 5.4 ± 1.4 | 4.8 ± 1.5 | 2.6 ± 1.4 | 3.0 ± 0.41 |
| 8h | 9.8 ± 2.6 | 8.8 ± 2.2 | 4.2 ± 0.9 * | 3.5 ± 0.57 * | |
| 24h | 17.6 ± 2.4 | 18.1 ± 2.9 | 8.9 ± 2.2 * | 7.3 ± 2.1 * | |
| Alcohol preference | 4h | 0.80 ± 0.04 | 0.74 ± 0.11 | 0.59 ± 0.07 | 0.54 ± 0.15 |
| 8h | 0.71 ± 0.06 | 0.70 ± 0.05 | 0.57 ± 0.07 | 0.60 ± 0.11 | |
| 24h | 0.65 ± 0.05 | 0.62 ± 0.04 | 0.50 ± 0.03 | 0.55 ± 0.13 | |
| Genotype | nPE1+/+ (n = 5) | nPE1−/− (n = 5) | |||
| Treatment | vehicle | 2 mg/kg naltrexone | vehicle | 2 mg/kg naltrexone | |
| Alcohol intake g/kg | 4h | 6.1 ± 0.6 | 3.0 ± 0.5 + | 2.8 ± 1.2 | 2.4 ± 0.91 |
| 8h | 10.1 ± 2.1 | 6.8 ± 2.1 | 3.7 ± 1.0 * | 2.9 ± 0.60 * | |
| 24h | 18.0 ± 2.5 | 12.2 ± 1.3 + | 9.1 ± 1.2 * | 7.8 ± 1.8 * | |
| Alcohol preference | 4h | 0.82 ± 0.02 | 0.50 ± 0.06 | 0.61 ± 0.08 | 0.53 ± 0.10 |
| 8h | 0.70 ± 0.05 | 0.62 ± 0.04 | 0.56 ± 0.05 | 0.53 ± 0.15 | |
| 24h | 0.66 ± 0.05 | 0.64 ± 0.03 | 0.49 ±0.11 | 0.55 ± 0.12 | |
At 2 mg/kg, naltrexone reduced alcohol intake in nPE1+/+ only (Table 4B). Three-way ANOVA revealed significant effects of genotype [F(1,48)=43, p<0.0001], time [F(2,48)=42, p<0.0001], interaction between genotype × time [F(2,48)=5.8, p<0.01] and naltrexone at 2 mg/kg [F(1,48)=15, p<0.005] on alcohol intake. Post hoc analysis showed that (1) nPE1−/− had significantly less alcohol intake at 8- and 24-hour time points than nPE1+/+ [p<0.05]; and (2) naltrexone at 2 mg/kg significantly reduced alcohol intake at 4- and 24-hour time point in nPE1+/+ only [p<0.05]. Three-way ANOVA revealed significant effects of genotype [F(1,48)=25, p<0.01] and naltrexone at 2 mg/kg [F(1,48)=6.9, p<0.01] on alcohol preference.
DISCUSSION
Genetically determined differences in opioid genes transcript levels, alcohol consumption and the effect of naltrexone.
Using nuclear RNA-Seq with the Pomc enhancer1 deletion (nPE1−/−) mice with hypothalamic-specific partial loss of Pomc transcriptional activity, the present study confirmed the genotype difference: the nPE1−/− mice, in comparison with the nPE1+/+ mice, had lowered nuclear Pomc transcript levels in the hypothalamus (Figure 1A), as reported before at cytoplasmic Pomc mRNA levels [12]. Our preliminary data showed the Pomc mRNA levels in the nucleus accumbens were unaltered in the nPE1 knock male mice (Result S1). Of interest, the nPE1−/− mice displayed lower alcohol intake (~9 g/kg/day) and preference (~0.60 preference ratio), as compared with nPE1+/+ mice with higher levels of alcohol intake (~19 g/kg/day) and preference (~0.81 preference ratio) (Table 1 and Figure S2). Though chronic alcohol drinking for 4 weeks resulted in increases in the Pomc transcripts in both nPE1+/+ and nPE1−/− mice after acute 1-day withdrawal, Pomc in the nPE1−/− mice were only restored to basal levels of nPE1+/+ mice. Furthermore, chronic alcohol drinking also increased Oprm1 transcripts in nPE1+/+ mice, but not in nPE1−/− mice with already increased basal Opmr1 transcripts (Figure 1B). Activation of MOP-r by beta-endorphin produces rewarding effects [5, 6] and alcohol enhances beta-endorphin release and POMC biosynthesis in the hypothalamus [2, 3, 8], which play an important role in the reinforcing actions and motivational behaviors of alcohol drinking in rodents. Therefore, both alcohol-induced Pomc and Opmr1 transcripts for the ligand and receptor in nPE1+/+ mice may contribute to excessive alcohol drinking (Table 5A). In contrast, the lowered alcohol preference and intake in nPE1−/− mice may be attributed by the genetically-determined Pomc transcripts with lowered basal level and blunted response to alcohol.
Table 5.
Schematic diagram of genetically determined differences between nPE1+/+ and nPE1−/− male mice and effects of acute (1-day) withdrawal from chronic (4-week) excessive alcohol drinking on nuclear transcript levels of opioid genes (A) and stress genes (B) in the hypothalamus. “Increase”, “Decrease” or “No change [nc]” indicate the direction of changes from baseline in nPE1+/+ water group.
| Genotype | nPE1+/+ | nPE1−/− | ||
|---|---|---|---|---|
| Treatment | water | alcohol | water | alcohol |
| Pomc | baseline | increase | decrease | nc |
| Oprm1 | baseline | increase | increase | increase |
| Pdyn | baseline | nc | decrease | nc |
| Oprk1 | baseline | nc | increase | nc |
| Penk | baseline | nc | nc | nc |
| Oprd1 | baseline | nc | decrease | nc |
| Pnoc | baseline | nc | decrease | nc |
| Oprl1 | baseline | nc | decrease | nc |
| Genotype | nPE1+/+ | nPE1−/− | ||
| Treatment | water | alcohol | water | alcohol |
| Oxt | baseline | decrease | decrease | nc |
| Oxtr | baseline | nc | decrease | nc |
| Avp | baseline | nc | decrease | nc |
| Avpi1 | baseline | decrease | nc | nc |
| Avpr1a | baseline | nc | nc | nc |
| Nr3c1 | baseline | decrease | increase | nc |
| Nr3c2 | baseline | nc | nc | nc |
| Fkbp5 | baseline | nc | nc | nc |
| Crh | baseline | nc | nc | nc |
| Crhbp | baseline | nc | nc | nc |
| Crhr1 | baseline | nc | nc | nc |
| Crhr2 | baseline | nc | nc | nc |
We functionally examined the effect of pharmacological blockade of MOP-r and found that naltrexone dose-dependently reduced excessive alcohol intake and preference in nPE1+/+ mice, confirming that naltrexone reduced alcohol consumption in our mouse model (Table 4). Our data also suggest the possibility that chronic alcohol/acute withdrawal may cause beta-endorphin release [9, 10], which plays a functional role in enhancing nuclear Pomc transcript levels in nPE1+/+ mice (Figure 1A). Our result is consistent with previous studies showing that beta-endorphin, MOP-r and POMC neurons in the hypothalamus (the main brain region producing Pomc and beta-endorphin) contribute to alcohol consumption [13, 36–39]. Consistently, the pharmacological effect of naltrexone was blunted in nPE1−/− mice, though the same naltrexone dose significantly reduced alcohol drinking in nPE1+/+ mice (Table 4), further suggesting a lowered beta-endorphin tone resultant from decreased Pomc transcript levels in the nPE1−/− mice was involved in the lack of naltrexone response and low alcohol intake. In line with the result on drinking behavior, the observation of increased corticosterone levels in nPE1−/− mice indicates less beta-endorphin activity with neuronal POMC partial deficiency, as it is well known that the beta-endorphin/MOP-r plays an inhibitory role in HPA hormonal activity in both humans and rodents [11].
In contrast to the increases in both Pomc and Oprm1 transcripts (Figure 1A and 1B), neither Penk nor Oprd1 transcripts showed any changes after alcohol in nPE1+/+ mice (Figure 1E and 1F). Though enkephalins can also bind and activate MOP-r, the effect of naltrexone on alcohol drinking in nPE1+/+ mice with no change of Penk transcripts strongly indicates that Penk or enkephalins may not contribute much to the MOP-r mediated increase in alcohol drinking. Our results agree well with one early study demonstrating that alcohol consumption was not altered in Penk knockout mice [38]. The lack of significant effect by naltrexone in nPE1−/− mice was not due to its lowered basal alcohol intake or floor effect, as other compounds tested before (e.g., KOP-r or V1b antagonist) still significantly reduced alcohol intake in the nPE knockout mice [13]. Together, these findings suggest again that Pomc/beta-endorphin, but not Penk/enkephalins, acting on MOP-r, have a critical role in alcohol drinking.
Our study using nPE1 transgenic mice with region-specific POMC partial deficiency [12] further confirms that POMC neurons in the hypothalamus contribute to alcohol consumption. Earlier studies using beta-endorphin deficient mice showed inconsistent results by different groups [38, 40, 41]. The potential limitation of the global beta-endorphin knockout mouse model is that it did not allow for clarification of which specific regions of POMC cells (hypothalamus or other possible brain regions or pituitary) are involved in alcohol behaviors. As melanocortin (another neuropeptide derived from POMC) activates melanocortin 4 receptor (MC4R) and decreases alcohol and food consumption [3], the partial deficiency of Pomc/melanocortin in the nPE1 knockout mice may be also involved in their alcohol consumption and food intake, and further study is needed.
In rodents, KOP-r/dynorphin activation is associated with the negative reinforcement aspects of alcohol addictions, especially during acute withdrawal [11, 18, 42–44]. After chronic alcohol/acute withdrawal, there was a slight, but not significant, decrease in Pdyn and Oprk1 in nPE1+/+ (Figure 1C and 1D). In nPE1−/− mice, there were lower basal Pdyn transcript levels in the hypothalamus (Figure 1C), when compared with nPE1+/+ mice. Probably due to the dynorphin deficiency, the nPE1−/− mice showed a compensatory increase in basal Oprk1 transcripts (Figure 1D). In contrast to nPE1+/+, nPE1−/− after acute withdrawal from low alcohol intake restored the lowered Pdyn to basal levels of nPE1+/+ mice.
As orphanin FQ and its receptor in the hypothalamus have strong interactions with beta-endorphin/MOP-r and involved in alcohol related behaviors [45], we also examined their transcripts expression and found no change of either Pnoc or Oprl1 in nPE1+/+ mice after chronic alcohol/acute withdrawal (Figure 1G and 1H). Like Pdyn, however, acute withdrawal from low alcohol drinking in nPE1−/− mice revered the decreased Pnoc and Oprl1 to the basal levels of nPE1+/+ mice (Table 5A).
Genetically determined differences in stress genes transcript levels and corticosterone levels.
After acute withdrawal, oxytocin system was profoundly altered as Oxt transcript level was reduced in the hypothalamus of nPE1+/+ mice (Figure 2A) (Table 5B). Our finding is consistent with many studies demonstrating that there are alcohol-withdrawal related decreases in Oxt or Avp cytoplasmic mRNA and peptide levels in the hypothalamus (including the paraventricular nucleus) of mice, rats and humans [14, 46–49]. In several selectively bred alcohol drinking rat lines, there are lower basal levels of Avp mRNA in the hypothalamus of Indiana and Sardinian alcohol non-preferring rats, as compared with their alcohol preferring counterparts [48, 50]. Consistent to this notion, the present study showed a lower basal Avp transcript level in nPE1−/− mice, which could contribute to their lowered alcohol consumption and/or preference.
Both CRH/CRH1 receptor and AVP/V1b receptor systems are potent modulator of HPA axis and central stress responses. The present study found that the low Avp transcript levels in the hypothalamus were associated with low plasma corticosterone levels after acute withdrawal in nPE1+/+ mice. Different from Avp, there was no change of Crh, Crhr1, Crhr2 or Crhbp transcript levels in nPE1+/+ hypothalamus (Table 3), indicating that the AVP/V1b receptor system is specifically involved in the HPA modulation during acute alcohol withdrawal. Also, our results on plasma corticosterone levels after acute withdrawal confirm previous findings [51]. Our new observation of low Nr3c1 transcript levels in nPE1+/+ mice (Figure 2E) suggests a decreased glucocorticoid receptor expression and feedback activity mediated by the receptors. In contrast, nPE1−/− mice showed high basal levels of Nr3c1 and plasma corticosterone, indicating that upregulated HPA activity may lead to less alcohol intake in the nPE1−/− mice [11].
AVP-induced protein 1 was reported to function in MAP kinase activation, epithelial sodium channel down-regulation and cell cycling [52]. Here we unexpectedly observed a profound decrease of Avpi1 transcript levels in the nPE1+/+ hypothalamus after chronic alcohol drinking (Figure 2D), suggesting for the first time the potential interaction between the AVP-induced protein 1 and excessive alcohol drinking or withdrawal. Of interest, in a recent human genetic study, rs7913179 variant in the Avpi1 gene is found to associate with alcohol dependency in a genome-wide gene-by-alcohol dependence interaction analysis in European men [53]. Though the AVP/V1b receptor system plays important roles in alcohol drinking [11, 15], it seems unknown, however, whether and how the hypothalamic AVP-induced protein 1 is involved in alcohol drinking behaviors, and further study is needed.
Individual vulnerability to excessive alcohol drinking is a key feature of alcohol addiction. One focus of the present study was to explore whether individual differences in mouse vulnerability to excessive drinking may be related to genetically-determined individual differences in key neurochemical systems in the hypothalamus. For this purpose, we determined if the changes in individual opioid or stress gene expression were correlated with vulnerability to excessive alcohol intake between and within each genotype by regression analysis (Figure S2–Figure S9). There were positive correlations between Pomc, Penk, Oprd1, Nr3c1, Nr3c2 and Fkbp5 and a propensity for alcohol intake, and a negative correlation between the Avp and alcohol intake. Although caution should be used when interpreting the relationships between behavior and transcript levels, our study of nuclear transcriptome profiling in both the nPE1+/+ and nPE1−/− mice with relatively high and low alcohol drinking provide useful information about a potential role of individual variations in relation with alcohol intake.
Summary.
The comprehensive and accurate characterization of transcriptional activity represents an important step in the understanding of chronic alcohol exposure in which gene expression occurs in the hypothalamic neurons. Many studies, including ours, have found that chronic alcohol drinking resulted in persistent changes in cytoplasmic mRNA levels of opioid and stress genes in rodent hypothalamus [11,15,19]. In the present study, we hypothesized that alcohol-induced transcriptional regulation in the nucleus is the critical step for maintaining the persistent changes in cytoplasmic mRNA levels and then subsequent peptide activity that is critically involved in excessive alcohol drinking. Therefore, we investigated the effects of chronic alcohol and acute withdrawal on opioid and stress gene expressions at nuclear transcriptional activity in the hypothalamus. In this particular region, we observed a stimulatory effect on Pomc and Oprm1 opioid gene transcripts after acute withdrawal from chronic excessive drinking in the wildtype mice (Table 5A), with an inhibitory effect on stress gene transcripts (Oxt, Avpi1 and Nr3c1) (Table 5B), which may contribute to the persistent alterations of the rewarding effect and stress responses. Of interest, the opioid and stress gene transcription activities were differentially altered in the POMC-deficient mice with genetically-determined basal expression levels and low alcohol intake. The consequence of a new set point of the opioid and stress gene transcription activities in response to alcohol may play an important role in individual vulnerability to excessive alcohol drinking.
The altered transcriptional activity has been found to correlate with nuclear transcript levels both in the in vitro [26–28] and in vivo [27, 29] studies. Specifically, nuclear RNA quantity can reflect the levels at which transcriptional activity occurs in the cells for protein-coding genes induced by different stimuli [26–28]. Furthermore, several studies have demonstrated that altered cytoplasmic mRNA levels parallel the changes of nuclear transcript levels, as the accumulation of cytoplasmic mRNAs (e.g., Pomc) are mostly due to the increases in gene transcriptional activity in the nucleus [27, 28]. Therefore, nuclear RNA-Seq in the present study provides a representative description of nascent transcriptional activity of the opioid and stress genes in response to acute alcohol withdrawal. Therefore, nuclear RNA-Seq may recapitulate the transcript levels associated with activated neuronal transcriptome after acute withdrawal, enabling the identification of nascent and neuronal activity-associated mRNAs. Together, our results of the genetic, pharmacological and neuroendocrine analyses using nPE1 knockout mice, MOP-r antagonist naltrexone and stress hormone corticosterone together provide functional validation about the transcriptome profiling in the hypothalamus.
Supplementary Material
Acknowledgement:
NIH AA021970 (YZ), Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (MJK). No conflict of interest.
References
- [1].Angelogianni P & Gianoulakis C (1993) Chronic ethanol increases proopiomelanocortin gene expression in the rat hypothalamus. Neuroendocrinology 57, 106–114. [DOI] [PubMed] [Google Scholar]
- [2].Zhou Y, Colombo G, Niikura K, Carai MAM, Femenía T, García-Gutiérrez MS, Manzanares J, Ho A, Gessa GL & Kreek MJ (2013) Voluntary alcohol drinking enhances proopiomelanocortin (POMC) gene expression in nucleus accumbens shell and hypothalamus of Sardinian alcohol-preferring rats. Alcohol Clin Exp Res 37, E131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Olney JJ, Navarro M & Thiele TE (2014) Targeting central melanocortin receptors: a promising novel approach for treating alcohol abuse disorders. Front Neurosci 8, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rasmussen DD, Boldt BM, Wilkinson CW & Mitton DR (2002) Chronic daily ethanol and withdrawal: 3. Forebrain pro-opiomelanocortin gene expression and implications for dependence, relapse, and deprivation effect. Alcohol Clin Exp Res 26, 535–546. [PubMed] [Google Scholar]
- [5].Amalric M, Cline EJ, Martinez JL Jr, Bloom FE & Koob G,F (1987) Rewarding properties of beta-endorphin as measured by conditioned place preference. Psychopharmacology 91, 14–19. [DOI] [PubMed] [Google Scholar]
- [6].Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, Rakic P, Bechmann I, Cowley MA, Szigeti-Buck K, Dietrich MO, Gao XB, Diano S & Horvath TL (2015) Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 519, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Spanagel R, Heinz A, Bals-Kubik R & Shippenberg TS (1991) Beta-endorphin-induced locomotor stimulation and reinforcement are associated with an increase in dopamine release in the nucleus accumbens. Psychopharmacology 104, 51–56. [DOI] [PubMed] [Google Scholar]
- [8].Olive MF, Koenig HN, Nannini MA & Hodge CW (2001) Stimulation of endorphin neuro-transmission in the nucleus accumbens by ethanol, cocaine, and amphetamine. J Neurosci 21, RC184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marinelli PW, Quirion R & Gianoulakis C (2003) A microdialysis profile of beta-endorphin and catecholamines in the rat nucleus accumbens following alcohol administration. Psychopharmacology 169, 60–67. [DOI] [PubMed] [Google Scholar]
- [10].Roth-Deri I, Green-Sadan T & Yadid G (2008) Beta-endorphin and drug-induced reward and reinforcement. Prog Neurobiol 86, 1–21. [DOI] [PubMed] [Google Scholar]
- [11].Zhou Y & Kreek MJ (2018) Involvement of activated brain stress responsive systems in excessive and “relapse” alcohol drinking in rodent models: implications for therapeutics. J Pharmacol Exp Ther 366, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lam DD, de Souza FSJ, Nasif S, Yamashita M, López-Leal R, Otero-Corchon V, Meece K, Sampath H, Mercer AJ, Wardlaw SL, Rubinstein M & Low MJ (2015) Partially redundant enhancers cooperatively maintain Mammalian Pomc expression above a critical functional threshold. PLoS Genetics 11, e1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou Y, Rubinstein M, Low MJ & Kreek MJ (2017) Hypothalamic-specific proopiomelanocortin deficiency reduces alcohol drinking in male and female mice. Genes Brain Behav 16, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hoffman PL (1991) Chronic ethanol exposure uncouples vasopressin synthesis and secretion in rats. Neuropharmacology 30, 1245–1249. [DOI] [PubMed] [Google Scholar]
- [15].Harper KM, Knapp DJ, Criswell HE & Breese GR (2018) Vasopressin and alcohol: a multifaceted relationship. Psychopharmacology 235, 3363–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pedersen CA (2017) Oxytocin, Tolerance, and the Dark Side of Addiction. Int Rev Neurobiol 136, 239–274. [DOI] [PubMed] [Google Scholar]
- [17].Pomrenze MB, Fetterly TL, Winder DG & Messing RO (2017) The Corticotropin Releasing Factor Receptor 1 in Alcohol Use Disorder: Still a Valid Drug Target? Alcohol Clin Exp Res 41, 1986–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tunstall BJ, Carmack SA, Koob GF & Vendruscolo LF (2017) Dysregulation of Brain Stress Systems Mediates Compulsive Alcohol Drinking. Curr Opin Behav Sci 13, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Anderson RI & Becker HC (2017) Role of the Dynorphin/Kappa Opioid Receptor System in the Motivational Effects of Ethanol. Alcohol Clin Exp Res 41,1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Koob GF & Kreek MJ (2007) Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koob GF & Mason BJ (2016) Existing and Future Drugs for the Treatment of the Dark Side of Addiction. Annu Rev Pharmacol Toxicol 56, 299–322. [DOI] [PubMed] [Google Scholar]
- [22].Inder W, Joyce P, Ellis M, Evans M, Livesey J & Donald P (1995) Effects of alcoholism on the hypothalamic-pituitary-adrenal axis: interaction with endogenous opioid peptides. Clin Endocrinol 43, 283–290. [DOI] [PubMed] [Google Scholar]
- [23].Richardson HN, Lee SY, O’Dell LE, Koob GF & Rivier CL (2008) Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci 28, 1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Z, Gerstein M & Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hwa LS, Chu A, Levinson SA, Kayyali TM, DeBold JF & Miczek KA (2011) Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% alcohol. Alcohol Clin Exp Res 35, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mitchell JA, Clay I, Umlauf D, Chen CY, Moir CA, Eskiw CH, Schoenfelder S, Chakalova L, Nagano T & Fraser P (2012) Nuclear RNA sequencing of the mouse erythroid cell transcriptome. PLoS One 7, e49274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Grindberg RV, Yee-Greenbaum JL, McConnell MJ, Novotny M, O’Shaughnessy AL, Lambert GM, Araúzo-Bravo MJ, Lee J, Fishman M, Robbins GE, Lin X, Venepally P, Badger JH, Galbraith DW, Gage FH & Lasken RS (2013) RNA-sequencing from single nuclei. Proc Natl Acad Sci U S A 110, 19802–19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou Y & Lapingo C (2014) Modulation of pro-opiomelanocortin gene expression by ethanol in mouse anterior pituitary corticotrope tumor cell AtT20. Regul Pept 192–193, 6–14. [DOI] [PubMed] [Google Scholar]
- [29].Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, Parylak SL, Paquola ACM, Venepally P, Novotny M, O’Connor C, Fitzpatrick C, Erwin JA, Hsu JY, Husband D, McConnell MJ, Lasken R & Gage FH (2016) Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun 7, 11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rhodes JS, Best K, Belknap JK, Finn DA & Crabbe JC (2005) Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav 84, 53–63. [DOI] [PubMed] [Google Scholar]
- [31].Jakubowski M & Roberts JL (1992) Multiplex solution hybridization-ribonuclease protection assay for quantitation of different ribonucleic Acid transcripts from snap-frozen neuroendocrine tissues of individual animals. J Neuroendocrinol 4, 79–89. [DOI] [PubMed] [Google Scholar]
- [32].Andrews S (2010) FastQC: A quality control tool for high throughput sequence data. Available: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/
- [33].Dobin A, Davis C, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M & Gingeras T (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liao Y, Smyth GK & Shi W (2014) Feature Counts: an efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- [35].Love MI, Huber W & Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Roberts A, McDonald J, Heyser C, Kieffer B, Matthes H, Koob G & Gold L (2000) μ-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 293, 1002–1008. [PubMed] [Google Scholar]
- [37].Hall FS, Sora I & Uhl GR Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology 154, 43–49. [DOI] [PubMed] [Google Scholar]
- [38].Racz I, Schurmann B, Karpushova A, Reuter M, Cichon S, Montag C, Furst R, Schutz C, Franke PE, Strohmaier J, Wienker TF, Terenius L, Osby U, Gunnar A, Maier W, Bilkei-Gorzo A, Nothen M & Zimmer A (2008) The opioid peptides enkephalin and beta-endorphin in alcohol dependence. Biol Psychiatry 64, 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ben Hamida S, Boulos LJ, McNicholas M, Charbogne P & Kieffer BL (2019) Mu opioid receptors in GABAergic neurons of the forebrain promote alcohol reward and drinking. Addict Biol 24, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Grisel JE, Mogil JS, Grahame NJ, Rubinstein M, Belknap JK, Crabbe JC & Low MJ (1999) Ethanol oral self-administration is increased in mutant mice with decreased beta-endorphin expression. Brain Res 835, 62–67. [DOI] [PubMed] [Google Scholar]
- [41].Grahame NJ, Mosemiller AK, Low MJ & Froehlich JC (2000) Naltrexone and alcohol drinking in mice lacking beta-endorphin by site-directed mutagenesis. Pharmacol Biochem Behav 67, 759–766. [DOI] [PubMed] [Google Scholar]
- [42].D’Addario C, Caputi FF, Rimondini R, Gandolfi O, Del Borrello E, Candeletti S & Romualdi P (2013) Different alcohol exposures induce selective alterations on the expression of dynorphin and nociceptin systems related genes in rat brain. Addict Biol 13, 425–433. [DOI] [PubMed] [Google Scholar]
- [43].Kissler JL, Sirohi S, Reis DJ, Jansen HT, Quock RM, Smith DG & Walker BM (2014) The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biol Psychiatry 75, 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lalanne L, Ayranci G, Kieffer BL & Lutz PE (2014) The kappa opioid receptor: from addiction to depression, and back. Front Psychiatry 5, 170 eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Greenwald MK (2018) Anti-stress neuropharmacological mechanisms and targets for addiction treatment: A translational framework. Neurobiol Stress 9, 84–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Harding AJ, Halliday GM, Ng JL, Harper CG & Kril JJ (1996) Loss of vasopressin-immunoreactive neurons in alcoholics is dose-related and time-dependent. Neuroscience 72, 699–708. [DOI] [PubMed] [Google Scholar]
- [47].Silva SM, Madeira MD, Ruela C & Paula-Barbosa MM (2002) Prolonged alcohol intake leads to irreversible loss of vasopressin and oxytocin neurons in the paraventricular nucleus of the hypothalamus. Brain Res 925, 76–88. [DOI] [PubMed] [Google Scholar]
- [48].Zhou Y, Colombo G, Carai MAM, Ho A, Gessa GL & Kreek MJ (2011) Involvement of arginine vasopressin and V1b receptor in alcohol drinking in Sardinian alcohol-preferring rats. Alcohol Clin Exp Res 35, 1876–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hansson AC, Koopmann A, Uhrig S, Bühler S, Domi E, Kiessling E,, Ciccocioppo R, Froemke RC, Grinevich V, Kiefer F, Sommer WH, Vollstädt-Klein S & Spanagel R (2018) Oxytocin Reduces Alcohol Cue-Reactivity in Alcohol-Dependent Rats and Humans. Neuropsychopharmacology 43, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hwang BH, Froehlich JC, Hwang WS, Lumeng L & Li T-K (1998) More vasopressin mRNA in the paraventricular hypothalamic nucleus of alcohol-preferring rats and high alcohol-drinking rats selectively bred for high alcohol preference. Alcohol Clin Exp Res 22, 664–669. [DOI] [PubMed] [Google Scholar]
- [51].Zorrilla EP, Valdez GR & Weiss F (2001) Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology 158, 374–381. [DOI] [PubMed] [Google Scholar]
- [52].Nicod M, Michlig S, Flahaut M, Salinas M, Fowler JN, Horisberger JD, Rossier BC & Firsov D (2002) A novel vasopressin-induced transcript promotes MAP kinase activation and ENaC downregulation. EMBO J 21, 5109–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Polimanti R, Zhao H, Farrer LA, Kranzler HR & Gelernter J (2017) Ancestry-specific and sex-specific risk alleles identified in a genome-wide gene-by-alcohol dependence interaction study of risky sexual behaviors. Am J Med Genet B Neuropsychiatr Genet 174, 846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.