

Abstract
Remyelination is the phenomenon by which new myelin sheaths are generated around axons in the adult central nervous system (CNS). This follows the pathological loss of myelin in diseases like multiple sclerosis (MS). Remyelination can restore conduction properties to axons (thereby restoring neurological function) and is increasingly believed to exert a neuroprotective role on axons. Remyelination occurs in many MS lesions but becomes increasingly incomplete/inadequate and eventually fails in the majority of lesions and patients. Efforts to understand the causes for this failure of regeneration have fueled research into the biology of remyelination and the complex, interdependent cellular and molecular factors that regulate this process. Examination of the mechanisms of repair of experimental lesions has demonstrated that remyelination occurs in two major phases. The first consists of colonization of lesions by oligodendrocyte progenitor cells (OPCs), the second the differentiation of OPCs into myelinating oligodendrocytes that contact demyelinated axons to generate functional myelin sheaths. Several intracellular and extracellular molecules have been identified that mediate these two phases of repair. Theoretically, the repair of demyelinating lesions can be promoted by enhancing the intrinsic repair process (by providing one or more remyelination‐enhancing factors or via immunoglobulin therapy). Alternatively, endogenous repair can be bypassed by introducing myelinogenic cells into demyelinated areas; several cellular candidates have been identified that can mediate repair of experimental demyelinating lesions. Future challenges confronting therapeutic strategies to enhance remyelination will involve the translation of findings from basic science to clinical demyelinating disease.
I. What is Remyelination?
Remyelination is an example of spontaneous repair in the adult central nervous system (CNS), where new myelin sheaths are generated around demyelinated axons. This follows the pathological loss of myelin [as occurs in the demyelinating diseases, notably multiple sclerosis (MS)]. Remyelination provides an exception to a primary tenet of neuroscience, namely, that the adult CNS has a poor intrinsic capacity to repair following trauma, which is summarized in Cajal's famous postulate—“everything may die, nothing may regenerate” in the CNS (Cajal, 1928). The phenomenon of remyelination was documented in early morphological descriptions of brains from MS patients (Marburg, 1906). Plaques in freshly isolated CNS tissue are easily identifiable as distinct, discolored areas of myelin loss. In addition to such classical plaques, sharply delineated areas of reduced myelin density are observed, recognizable by evenly distributed, pale myelin staining that represent the Markschattenherde or shadow plaques (Schlesinger, 1909) (Fig. 1 ). While such observations were attributed initially to incomplete demyelination (due to ongoing evolution of lesions), there had been earlier suggestions that a process of repair could follow damage to myelin (Marburg, 1906). Despite this, the verification that spontaneous remyelination could follow myelin loss was not provided till 1961, when a pioneering ultrastructural study [using the method of cerebrospinal fluid (CSF) barbotage to induce demyelination] clearly demonstrated that demyelinated axons in the adult cat spinal cord can acquire new myelin sheaths (Bunge et al., 1961). This followed their earlier observations from light microscopic studies that myelin reappears at a time when animals show recovery from partial paralysis (Bunge 1958, Bunge 1957). The suggestion that the thin myelin lamellae observed in demyelinated lesions could represent areas of spontaneous remyelination came from an ultrastructural study of MS plaques (Perier and Gregoire, 1965). Furthermore, other studies proved that remyelination induced not only structural recovery but also allowed axons to regain conduction properties (Smith et al., 1979) and neurological function lost as a consequence of demyelination (Jeffery and Blakemore, 1997).
Fig. 1.
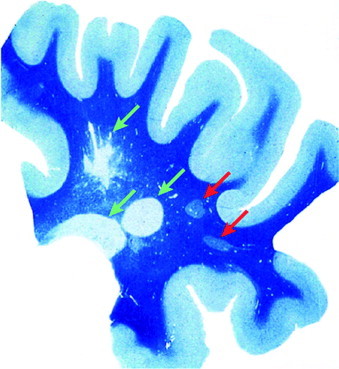
Section from the brain of an MS patient stained with Luxol fast blue to identify subcortical white matter (WM). Green arrows indicate three areas of myelin loss which represent foci of chronic demyelination. Red arrows indicate areas of pale myelin staining which represent the shadow plaques where remyelination has occurred. This illustrates the key point that spontaneous remyelination can occur efficiently in MS but that this process fails on many occasions, leaving axons chronically demyelinated. Reproduced with permission from Adams (1989).
Remyelination of large and medium diameter CNS axons is easily identifiable histologically, although unequivocal identification of remyelination of small diameter axons is generally more challenging or even impossible. Remyelinated axons display some distinguishing features, chief among which is a shortened length of the new internodes. Newly generated myelin sheaths are also thinner than normal; therefore, the relationship between myelin thickness and internodal length to axon diameter (that applies in normally myelinated axons) does not pertain in remyelinated axons, either in experimental lesions or MS shadow plaques. Despite this, remyelination allows axons to regain adequately secure conduction properties such that few conduction abnormalities can be recorded from remyelinated axons (this includes a restoration of latent periods of axons to normal) (Smith 1979, Smith 1981).
A. Remyelination in MS
Data on the extent and distribution of remyelination in MS are mainly obtained from postmortem tissue; an obvious problem with interpreting such data is that they represent a static picture of MS lesions and therefore provide few clues about the degree and pattern of repair of dynamic, evolving lesions that exist in the clinical disease. Clinically, magnetic resonance imaging (MRI) is generally considered to be the most sensitive method to detect alterations in the white matter (WM) of MS patients. While the identification of remyelination in such lesions has so far been largely unreliable, advances in multicontrast MRI will increasingly allow for the characterization and monitoring of remyelination in MS patients (Merkler et al., 2005).
The extent of remyelination observed between MS lesions is highly variable. This accompanies significant variations in plaque size with differences in patterns of demyelination and oligodendrocyte damage/loss. This may depend to some degree on the stage of progression of lesions but may also be related to variation in underlying pathogenic mechanisms (Lucchinetti et al., 2000). Generally, the degree of remyelination of MS lesions correlates directly with both numbers of oligodendrocytes (expressing myelin proteins or their mRNAs) and macrophages in lesions (Lucchinetti et al., 1999). Remyelination can occur rapidly and thin, short internodes can be detected in acute lesions within a few weeks of lesion genesis; remyelination can occur concomitant with tissue infiltration by macrophages (Ghatak 1989, Lassmann 1983, Prineas 1984, Prineas 1993) and occasionally myelin destruction (Lassmann 1997, Prineas 1993, Raine 1993). Conversely, some remyelinated lesions can undergo active demyelination (Prineas et al., 1993), resulting in new lesions overlapping remyelinated areas. Shadow plaques are more commonly observed in young patients with an active relapsing‐remitting course than in older patients with an inactive/chronic one (Prineas and Connell, 1979); chronic lesions from patients with long‐standing disease tend to show sparse remyelination, restricted to a thin rim‐bordering periplaque white matter (PPWM). Additionally, remyelination may be sparse in cases of primary progressive MS (Lassmann et al., 1997).
Remyelination of central axons can sometimes be accompanied by Schwann cell invasion of the CNS (Itoyama et al., 1983), which is more common near peripheral nerve root entry or exit zones in the spinal cord; this was found to be a prominent feature in some Japanese patients (Itoyama et al., 1985). It has conventionally been assumed that remyelinating Schwann cells originate from Schwann cells associated with spinal roots or nerves supplying the vasculature or meninges (Franklin and Blakemore, 1993). Schwann cell remyelination is generally prominent in areas of both astrocyte and oligodendrocyte loss, leading to the suggestion that death of astrocytes constituting the glia limitans allows Schwann cell entry into the CNS; reorganization of this structure would inhibit further influx. Challenging these views, a report suggests instead that remyelinating Schwann cells may be derived from CNS precursors; this awaits formal proof (Blakemore, 2005).
The extent to which remyelination is associated with differences in clinical outcome, disease severity, and recovery in MS is currently unclear. It is suggested that remyelination is unlikely to play a significant role in early remission (as conduction deficits persist during the early stages of remission), but may be important for later stages of functional recovery (Jones, 1993). Remyelination fails in most lesions in MS and while it is likely that numerous causal factors underlie progressive clinical deterioration in the disease, the inadequacy or failure of remyelination is likely to play a significant contributory role.
B. Why is Remyelination Important?
Permanent neurological dysfunction observed in MS has traditionally been attributed to demyelination. Recent clinical studies have questioned the validity of this assumption with the growing belief that permanent axon loss is the more likely pathological correlate of irreversible functional deficit (Fig. 2 ). Demyelinated axons may be vulnerable to damage (during the acute phase) and possibly damage plus atrophy (during the chronic phase) of MS (Ferguson 1997, Lovas 2000, Trapp 1998). While the causes for such axonal atrophy are unclear, demyelination may predispose axons to secondary injury by (1) multiple molecular cytotoxicity mechanisms, including ion channel dysfunction and mitochondrial damage, or (2) failure of local target‐derived neurotrophic support for myelinating oligodendrocytes. This implies that one of the principal functions of myelin is to protect axons from secondary insult (Rodriguez 2003, Waxman 2006). To date, there is no formal proof that remyelination does prevent axonal atrophy and loss. However, axon loss in experimental allergic encephalomyelitis (EAE) lesions is greater in demyelinated areas than remyelinated ones (Kornek et al., 2000). In addition, absence of proteolipid protein (PLP), a major myelin protein, results in progressive axonal degeneration in rodents and humans (Garbern 2002, Griffiths 1998, Edgar 2004). Mice lacking cyclic nucleotide phosphodiesterase (CNP), an enzyme expressed by oligodendrocytes, also show severe axonal degeneration with accumulation of amyloid precursor protein (APP). This is associated with a significant motor deficit that becomes manifest by about 4 months (Lappe‐Siefke et al., 2003). It is not clear how oligodendrocytes maintain axonal integrity. Oligodendroglia express NGF and BDNF, key neurotrophic factors that play a role in the maintenance and survival of neurons. OPCs and differentiated oligodendrocytes can also support neuronal survival through contact‐mediated/soluble mechanisms, providing potential mechanisms by which remyelination could prevent neuronal atrophy (Byravan 1994, Wilkins 2001).
Fig. 2.
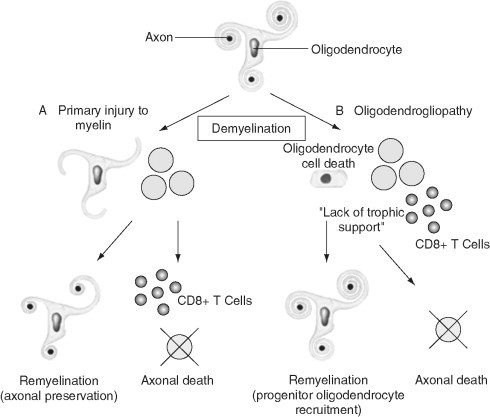
There are two options following demyelination in multiple sclerosis: either (A) remyelination can take place with axonal preservation or (B) axonal death may ensue, either due to a range of cytotoxicity mechanisms or due to failure of local neurotrophic support due to glial cell injury. Remyelination most likely occurs by the recruitment of OPCs into demyelinated areas that subsequently mature into myelinating cells. Modified with permission from Rodriguez (2003).
Secure electrical conduction in axons is made possible by a defined organization of cellular processes and domain‐specific ion channels at the nodes of Ranvier (reviewed by Sherman and Brophy, 2005). Remyelination supports the development and maturation of new nodes on central axons, which display an apparently normal ion channel distribution (Black 2006, Dugandzija‐Novakovic 1995, Sasaki 2006). These findings add support to the known role of remyelination in restoring conduction properties to axons. Strategies aimed at promoting CNS remyelination can therefore be predicted to both restore secure conduction (thereby promoting functional recovery) and protect axons in inflammatory lesions from secondary injury.
II. Experimental Remyelination
Several models of experimental demyelination are currently in use.
A. Autoimmune Models
Autoimmune models of demyelination are jointly referred to as EAE models. There are many variants but most models consist of immunizing genetically susceptible animals by direct injection of myelin antigens such as myelin basic protein (MBP), myelin‐associated glycoprotein (MAG), PLP, or myelin oligodendrocyte glycoprotein (MOG). Alternatively, T cells sensitized to myelin antigens can be transferred passively to animals (adoptive transfer). EAE lesions display many pathological features of MS lesions in terms of dissemination, demyelination, and infiltration by macrophages and T cells with significant similarities observed between the clinical course of EAE and MS (Kornek 2000, Storch 1998). The extent of axon loss in EAE lesions also correlates with chronic neurological deficit (Papadopoulos et al., 2006). Both acute and chronic forms of the disease can be induced in EAE models, as well as relapsing‐remitting and progressive forms (reviewed in Ercolini and Miller, 2006).
A targeted model of EAE has been described (Kerschensteiner et al., 2004), where subthreshold immunization of Lewis rats with myelin antigens is combined with injection of cytokines (like IFN‐γ and TNF‐α) at focal CNS sites. This creates single prototypic, inflammatory lesions at defined CNS foci. Such lesions share some pathological features of MS lesions and have the additional advantage of ease of lesion localization. Refined behavioral testing can also be carried out in such animals. In another model, antibody‐mediated demyelination can be produced by focally injecting anti‐galactocerebroside (GalC) antibodies plus complement into specific CNS sites (Keirstead et al., 1998).
B. Viral Demyelination
Some viruses cause CNS demyelination (reviewed in Ercolini and Miller, 2006); a widely studied model of demyelination is based on infection of mice with Theiler's murine encephalomyelitis virus (TMEV). When injected into the CNS of certain mouse strains, the virus induces demyelinating lesions associated with virus replication in oligodendrocytes. Low neurovirulent strains like DA and BeAn cause a biphasic disease in mice. There is some irrelevant matter which is not in, Disk file. The early phase consists of sublethal encephalitis while the later phase consists of viral infection of inflammatory cells, astrocytes and oligodendrocytes, multifocal demyelination, and axonal damage, depending on the mouse strain used (reviewed by Oleszak et al., 2004). Experimental demyelination at multiple foci can be induced by the A59 and JHM strains of mouse hepatitis virus (MHV), a coronavirus of mice, which is followed by extensive remyelination. The Semliki Forest virus (SMV) can also induce CNS demyelination in mice.
C. Toxin‐Mediated Demyelination
Many toxin‐mediated models of demyelination are used currently. One approach consists of systemically administering agents like cuprizone (a copper‐chelating agent) in the diets of laboratory mice. While this model does not have the disadvantages associated with direct toxin injection into the CNS (like CNS trauma, disruption of the blood–brain barrier, and an inflammatory response), the occurrence of demyelinating lesions may not be entirely reproducible with the doses used currently (Stidworthy et al., 2003). Other models consist of focal microinjections of toxins like ethidium bromide (EB) or lysolecithin at defined CNS foci (Fig. 3A –D). Injection of EB results in death of all nucleated cells at lesion sites, whereas lysolecithin‐induced lesions contain demyelinated axons present within an astrocytic environment. Toxin models have the advantage that highly reproducible demyelinating lesions can be created at defined CNS foci (Woodruff and Franklin, 1999).
Fig. 3.
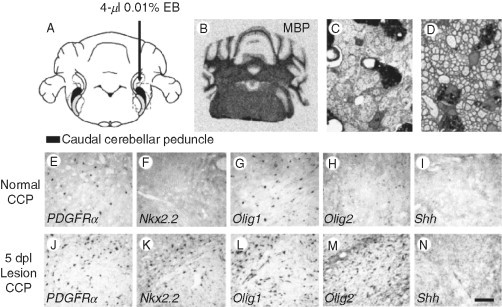
OPCs increase expression of Nkx2.2 mRNA and Olig2 mRNA in response to CNS demyelination. (A) Diagram to show focal demyelinating lesions in the caudal cerebellar peduncle (CCP), a large WM tract in adult rats, created by stereotaxic injection of ethidium bromide (EB). (B) This creates focally demyelinated areas, identifiable by absence of myelin basic protein (MBP) mRNA following in situ hybridization. (C) Toluidine blue‐stained resin section at 5 days postlesion showing demyelinated axons in transverse section and debris‐filled macrophages. (D) All demyelinated axons have been reinvested with thin myelin sheaths that are characteristic of CNS remyelination, at 4 weeks postlesion. (E–H) PDGFRα, Nkx2.2, Olig1, and Olig2 mRNA expression in normal WM detected by in situ hybridization. (J–M) The expression of the same mRNAs in areas of demyelination 5 days postlesion. Scale bar = 1.5 mm (A), 25 μm (C–D), and 100 μm (E–N). Modified with permission from Zhao et al. (2005).
D. Genetic Models of Demyelination
A number of myelin mutants exist where myelin is absent or appreciably reduced, with accompanying changes in myelin structure and oligodendrocyte number (Nave, 2001). Such mutants have proved invaluable for examining the biology of myelination and benefits of cell transplantation therapies. Myelin mutants can be divided into two categories: (1) the X‐linked mutants such as the myelin‐deficient (md) rat, shaking pup, and jimpy (jp) rat and (2) the autosomal recessive mutants such as the shiverer (shi) mouse, quaking (qk) mouse, and TAIEP rat. Of these, the X‐linked mutants form the biggest and most well‐characterized group.
Newer models to induce oligodendrocyte death include injection of viral proteins like the human endogenous retrovirus (HERV) glycoprotein–syncitin (such retroviral proteins are increasingly thought to play a role in the pathogenesis of MS). Alternatively, genetic strategies can be used to induce anatomical and lineage‐targeted cell depletion using Cre‐mediated gene recombination (Antony 2004, Brockschnieder 2004, Buch 2005, Christensen 2005). Each of these models has particular advantages and can mimic specific aspects of MS lesions, but no model is sufficient to mimic the range of pathological presentations of the disease. Immune models are generally considered to mimic the disease process and pathogenesis in MS better than the toxin models (although there is scarce evidence that the triggers for MS are similar to those inducing demyelination in these models). A drawback of immune‐mediated demyelination is the difficulty in separating cellular events associated with demyelination from repair mechanisms, as both can occur in parallel. In contrast, demyelination occurs as a single acute episode following toxin injection into the CNS, allowing demyelination to be temporally separated from regenerative events that occur in response to demyelination making toxin models particularly attractive for the study of remyelination biology. Oligodendrocyte apoptosis may be a major cause of pathology in some MS subgroups (Barnett and Prineas, 2004); toxin models causing oligodendrocyte death, and targeted oligodendrocyte depletion models do mimic this aspect of the disease.
A major factor to be considered when extrapolating experimental findings to the study of MS is that most models create small areas of demyelination which generally repair with high efficiency. This does not pertain in MS lesions which can be severalfold larger than experimental lesions; therefore, the temporal progression and dynamics of repair of experimental versus clinical lesions can be predicted to be significantly different, which may have implications for remyelination failure, as discussed later.
III. The Biology of Remyelination is Complex
A. Remyelination Occurs in Distinct Phases
Remyelination requires cell proliferation, which is highlighted by the finding that X‐irradiation of demyelinating lesions (a procedure that eliminates dividing cells, notably the oligodendrocyte progenitor cells or OPCs) is sufficient to abolish remyelination (Blakemore 1978, Chari 2006b). The origins of oligodendrocytes that undertake remyelination have been controversial in the past, with two cells proposed as putative sources (1) surviving oligodendrocytes in demyelinating lesions and (2) the OPCs. The OPCs are a group of ubiquitous, slowly dividing (potentially multipotent) neural progenitors that make up roughly 3–9% of the adult CNS cell population (reviewed in Polito and Reynolds, 2005). Most evidence suggests that mature oligodendrocytes are postmitotic and incapable of generating new oligodendrocytes (Crang 2004, Keirstead 1997). On the basis of a number of other studies, however, it is now widely accepted that new myelin sheaths are made by new oligodendrocytes derived from OPCs.
A scheme for remyelination has been proposed [based on observations on the repair of experimental (toxin‐induced) lesions], whereby remyelination occurs in two major phases. The first is a recruitment phase, where OPCs proliferate in order to populate demyelinated areas. OPCs subsequently differentiate into premyelinating oligodendrocytes which contact demyelinated axons and differentiate into mature, myelinating oligodendrocytes that form functional myelin sheaths—this constitutes the differentiation phase. The recruitment and differentiation phases are associated with distinct and timed patterns of expression of molecules, notable among which are a range of growth factors. While newly generated myelin (formed as a result of remyelination) is distinct from normal myelin (formed during development), experimental analyses show some striking similarities in both processes (reviewed in Franklin, 2002). Therefore, remyelination could be considered to be a recapitulation of the developmental myelination process (Franklin and Hinks, 1999), in keeping with the widely prevailing hypothesis that regenerative mechanisms recapitulate developmental events. Despite such demonstrable parallels, it should be kept in mind that developmental myelination occurs in a normal, ordered microenvironment. By contrast, remyelination occurs within a disrupted and pathological microenvironment dominated by macrophages and T cells that contains demyelinated axons within areas of astrocytosis, none of which pertain in development (Franklin, 2002).
For remyelination in be successful, it does appear critical for the molecular signaling environment in lesions to shift from one that initially promotes OPC recruitment/proliferation to one that favors differentiation (Franklin, 2002). Therefore, perturbations to the temporal progression of the individual stages of repair could significantly alter the outcome of repair processes, as discussed in Section IV.
B. Factors Regulating OPC Activation and Proliferation
OPCs are the major proliferative population of the adult CNS [accounting for 70% and 74% of bromodeoxyuridine (BrdU)‐incorporating cells in the spinal cord and cortex respectively]. Despite this, the OPCs are relatively quiescent cells that divide infrequently and have slow rates of division (Polito and Reynolds, 2005). In response to demyelination (and other CNS trauma), OPCs undergo a process of activation followed by rapid proliferation. OPC activation is associated with distinctive changes, including morphological alterations, increase in size, and upregulation of the cell surface chondroitin sulfate proteoglycan, NG2. They also demonstrate increased gene expression for transcription factors associated with developmental myelination. OPCs in WM normally express low levels of the bHLH transcription factor Olig2 and the homeodomain transcription factor Nkx2.2. However, striking increases in the expression of both Olig2 and Nkx2.2 (that is restricted to oligodendrocyte lineage cells) occur within acute demyelinating lesions (Fig. 3E–M) (Fancy et al., 2004). Coexpression of Olig2 and Nkx2.2 in OPCs appears necessary for these cells to differentiate into myelinating oligodendrocytes, so it has been hypothesized that expression of these two molecules constitutes a genetic switch that converts relatively quiescent OPCs into myelinating oligodendrocytes. Nkx2.2 levels show a rapid subsequent decrease during differentiation into oligodendrocytes. Therefore, the authors hypothesize that expression patterns of Nkx2.2 and Olig2 could enable a distinction to be made between OPCs actively participating in repair from OPCs that are quiescent. The authors further speculate that such a distinction could enable actively repairing lesions to be distinguished from the ones where remyelination failure has occurred.
Following activation, OPCs undergo rapid proliferation in order to populate demyelinated areas. Several factors have been identified, which alone or in combination can promote OPC proliferation in vivo and in vitro. These include platelet‐derived growth factor (PDGF), fibroblast growth factor 2 (FGF2), and glial growth factor 2 (GGF2). PDGF and FGF2 receptors are expressed on OPCs during their proliferative response to demyelination (Redwine and Armstrong, 1998). This accompanies increased expression of PDGF and FGF2 in areas of demyelination (Messersmith 2000, Woodruff 2004). PDGF‐A knockout mice show significantly reduced numbers of OPCs and oligodendrocytes accompanied by impaired genesis of myelin (Fruttiger et al., 1999). Conversely, PDGF‐A overexpression results in significant increases in OPC densities in demyelinating lesions (Woodruff et al., 2004). This study also examined if the increased availability of an OPC mitogen was associated with enhanced remyelination rates in old rats; no significant differences were observed between transgenic and control animals (despite increased OPC numbers in the former). This suggests that the OPC supply within lesions may not be rate limiting during remyelination. A recent study examining roles of individual signaling pathways has demonstrated distinctive roles in vivo for PDGF and FGF2 during remyelination. The authors suggest that PDGFRα signaling is likely to mainly regulate OPC proliferation (in response to demyelination), while FGF2 is a predominant inhibitor of OPC differentiation (Murtie et al., 2005). Systemically administered GGF2, a neuregulin expressed by neurons, has also been shown to promote OPC expansion, prevent OPC differentiation, and enhance remyelination of experimental EAE lesions (Marchionni et al., 1999). By contrast, direct administration of GGF into areas of toxin‐induced demyelination did not alter the extent of remyelination, suggesting that systemic GGF2 delivery may affect remyelination by altering the immune response rather than by direct effects on oligodendrocyte lineage cells (Penderis et al., 2003b).
C. Factors Regulating OPC Differentiation
Numerous extracellular and intracellular factors modulate the extent of OPC differentiation and myelin formation, but no coherent scheme has emerged so far on how the intricate process of progenitor differentiation into myelinating oligodendrocytes is regulated. Numerous transcription factors (and their regulatory molecules) mediate developmental myelination including Sox10, bHLH proteins, MyT1, Mash1, and Gtx among others; their roles in remyelination need to be elucidated (reviewed in Franklin, 2002). A key, nonredundant role for the transcription factor Olig1 has been reported in mediating remyelination (Arnett et al., 2004). Repair of toxin‐induced demyelinating lesions in both brain and spinal cord was significantly reduced in Olig1 knockout mice compared with controls. Recruitment of OPCs into lesions from knockout mice appeared normal, but these cells failed to differentiate into remyelinating oligodendrocytes. The subcellular localization of Olig1 could be a critical factor mediating differentiation; nuclear Olig1, expressed by OPCs at the margins of MS lesions, was suggested to be associated with cells actively engaged in repair.
As with OPC proliferation, the involvement of growth factors in regulating OPC differentiation has been extensively documented. Insulin‐like growth factor‐1(IGF‐1) is suggested to play a critical role in this process based on studies in transgenic mice overexpressing IGF‐1 where increases in myelin sheath thickness and numbers of myelinated axons (with increased numbers of oligodendrocytes) are observed; the expression of IGF in MS lesions is also consistent with the notion that this molecule plays a major role in the differentiation phase (Carson 1993, Gveric 1999, Luzi 2004, Ye 1995). Transforming growth factor β1 (TGFβ1) inhibits PDGF‐ and FGF2‐induced OPC proliferation and is suggested to be an important regulator of oligodendrocyte differentiation (McKinnon et al., 1993). The observed pattern and timing of expression of both IGF‐1 and TGFβ1 in demyelinating lesions supports the idea that these molecules regulate OPC differentiation during repair (Hinks 1999, Sim 2002). However, directly increasing levels of IGF‐1 mRNA using adenoviral‐mediated gene delivery was not sufficient to increase the extent of oligodendrocyte‐mediated remyelination of toxin‐induced lesions in old rats compared with young ones (O'Leary et al., 2002).
It is becoming increasingly clear that electrical activity in axons, axon‐derived signals, and neurotransmitter release all play important roles in the genesis, survival, and differentiation of oligodendrocytes and induction of developmental myelination (reviewed in Coman et al., 2005). Blocking or increasing the firing rates of neurons can respectively inhibit or enhance myelination (Demerens et al., 1996). Oligodendrocytes and OPCs express a range of ion channels and neurotransmitter receptors, the activation or inhibition of which can alter proliferation and differentiation of these cells and myelination (Gallo et al., 2001). OPCs are also thought to make close contacts with the nodes of Ranvier and synapses (Bergles 2000, Butt 1999) and are therefore well placed anatomically to respond to changes in axonal function in normal and pathological situations. These observations point to a clear link between electrical activity and myelination, but the potential effects of electrical activity and conduction block in demyelinated axons on the proliferation and differentiation of adult OPCs in surrounding tissue have yet to be established. Inhibitors of oligodendrocyte differentiation (like PSA‐NCAM, vitronectin, tenascins‐R, GGF2, and jagged) are all expressed by axons (reviewed in Coman et al., 2005). This lends support to the idea that axons could play key roles in the regulation of properties of oligodendrocyte lineage cells and myelination.
Several intracellular OPC differentiation factors have also been identified. The cyclin‐dependent kinase inhibitor p27Kip‐1 is found to accumulate in OPCs and on reaching a threshold appears to inhibit OPC proliferation and increased differentiation (Durand and Raff, 2000). p27Kip‐1 also appears to regulate both OPC responses to mitogens and the numbers of OPCs recruited to demyelinated areas—OPC numbers were significantly increased in p27Kip‐1 null mice compared with wild‐type controls (Crockett et al., 2005). Another recent study has demonstrated that a transmembrane CNS protein Lingo‐1 is expressed by oligodendrocytes and could play a critical role in their differentiation and myelination. The authors further suggest that this molecule could represent an important therapeutic target in demyelinating and dysmyelinating disorders (Mi et al., 2005).
D. Inflammation: A Double‐Edged Sword?
Inflammation undoubtedly contributes to the pathogenesis of demyelinating lesions in MS. However, there is considerable evidence to support the view that the inflammatory response provides an important stimulant for remyelination. Remyelination is frequently associated with a prominent inflammatory response in both experimental and MS lesions. Remyelination is also observed to occur most efficiently in early, active lesions where the inflammatory response is robust (Graca 1986, Ludwin 1980, Raine 1993). One study using Affymetrix microarrays to identify changes in gene expression during remyelination indicates that alterations in inflammatory genes represent the most prominent changes, emphasizing the role of the active inflammatory response (Arnett et al., 2003). Furthermore, several studies have demonstrated an impairment of remyelination in the absence of lymphocytes, MHC‐II, inflammatory cytokines, and macrophages (Arnett 2001, Arnett 2003, Bieber 2003, Kotter 2001, Kotter 2005, Mason 2001). This is supported by the findings that anti‐inflammatory drugs, such as minocycline and corticosteroids (such as dexamethasone, methylprednisolone, and prednisone), significantly delay remyelination of toxin‐induced demyelinating lesions (Chari 2006a, Li 2005, Triarhou 1986) (Fig. 4 ). Anti‐inflammatory agents appear to act mainly by inhibiting OPC differentiation, as colonization of lesions by OPCs was found to be essentially normal in corticosteroid (CS)‐treated animals. Such agents do not permanently impair the repair properties of myelinogenic cells as normal levels of remyelination can be found with increased survival times (following treatment withdrawal) (Chari et al., 2006a). While CS administration has numerous known benefits, these findings will have obvious implications for the use of anti‐inflammatory therapies in MS and raise the possibility that prolonged anti‐inflammatory treatment may predispose to enhanced axon loss in MS lesions (Chari et al., 2006a).
Fig. 4.
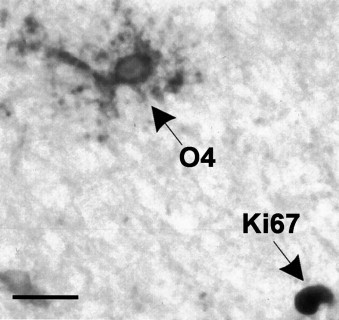
Chronic MS lesions contain O4‐positive OPCs that fail to bind the Ki67 antibody (a marker of cell proliferation), suggesting that these are a relatively quiescent population. Some lesions do contain some Ki67‐immunoreactive nuclei, such as the one indicated toward the lower right of the figure. Reproduced with permission from Wolswijk (1998).
How could inflammation influence remyelination? The exact mechanisms are unclear at present. Macrophages/microglia are suggested to produce molecules such as growth factors that mediate repair either directly or by activation of astrocytes to produce remyelination‐associated growth factors. The addition of peritoneal macrophages, in vitro, to fetal neuron glia cocultures increases the synthesis of myelin proteins like MBP, and increases myelin deposition (Loughlin 1997, Nathan 1987). Inflammation can also enhance the survival and migration of transplanted OPCs, in part, due to upregulation of multiple factors implicated in oligodendrocyte development (Setzu 2006, Tourbah 1997). A similar, remyelination‐promoting role has been suggested for T cells in demyelinating lesions (Bieber et al., 2003). Macrophages play a major role in myelin debris clearance with the suggestion that OPC maturation can be inhibited by contact with myelin (Miller, 1999). Indeed, introduction of purified myelin into demyelinating lesions in the brainstem causes a significant impairment of remyelination which is attributable to arrested OPC differentiation rather than impaired recruitment (Kotter et al., 2005). This impairment may be due to a physical restriction of contact between myelinating oligodendrocytes and demyelinated axons, or alternatively due to the presence of inhibitory molecules on myelin debris.
IV. What Causes Remyelination Failure in MS
Remyelination is regulated by complex mechanisms and the individual phases of repair are associated with distinct, timed patterns of expression of numerous molecules. Given the complexity of such regulatory processes and the prominent heterogeneity in pathogenesis and extent of cell survival in MS lesions, it is clear that the success of repair will be influenced by several interdependent variables. These include (1) OPC and oligodendrocyte availability in lesions, (2) astrocyte and macrophage presence in demyelinated areas, (3) the timed and coordinated expression of regulatory molecules, and (4) the ability of axons and OPCs/remyelinating oligodendrocytes to interact in a manner appropriate for successful remyelination to occur. These parameters are further influenced by age (Chari 2003a, Gilson 1993, Sim 2002), sex (Li et al., 2006), and possibly lesion size (Chari and Blakemore, 2002b). Therefore, a single causal factor in remyelination failure appears unlikely and the postulated causes for regenerative failure in MS remain largely speculative. Several issues must be considered.
First, given that OPCs are critical mediators of remyelination, the question of whether these cells are depleted in MS lesions is a crucial but unresolved one. Histopathological studies using markers like O4, NG2, and PDGFRα suggest that OPCs are present in chronic MS lesions in the brain and spinal cord in equal/higher numbers than periplaque WM (Chang 2000, Scolding 1998, Wolswijk 1998). Such OPCs were, however, suggested to be relatively quiescent based on the failure to label with a nuclear proliferation antigen (detected by the Ki‐67 antibody) (Fig. 5 ) and lack of oligodendrocytes in the center of MS lesions. It was suggested that these OPCs may be incapable of differentiation or alternatively that the lesion environment lacked factors to stimulate regenerative mechanisms (or contained factors that inhibited it) (Wolswijk, 1998). One study examining the specific possibility that remyelination fails due to local OPC destruction along with oligodendrocytes demonstrated that chronic lesions from spinal cords of MS patients contained significant numbers of O4 positive, GalC negative OPCs (Wolswijk, 2002). While interpreting such observations, it should be kept in mind that it is currently impossible to distinguish OPCs that survive in lesions from OPCs that colonize lesions from surrounding normal tissue. Therefore, an observed abundance of OPCs in lesions provides few clues as to their potential destruction during early stages of lesion development. This study further reported that with increasing age of subjects and duration of clinical symptoms (1) OPC densities declined significantly, (2) genesis of new oligodendrocytes appeared increasingly impaired, and (3) lesions did not appear to be significantly repopulated by OPCs (Wolswijk, 2002). Another study has reported that NG2 positive cells could not be detected in acute inflammatory lesions (although small numbers of lesions were examined in this study) (Chang et al., 2000). Support for the idea that OPCs could be killed in MS lesions comes from the demonstration that molecules like MOG and GalC (previously considered to be restricted to oligodendrocytes) are also present on OPCs (Li 2002, Shi 1998). Autoimmunity to MOG has been suggested to play an important role in the pathogenesis of MS lesions, so expression of such molecules on OPCs could, in theory, result in OPC destruction. The finding that patients with relapsing‐remitting MS synthesize antibodies to the OPC‐specific surface glycoprotein AN2 (which is absent in the CSF of patients with inactive disease) (Niehaus et al., 2000) lends support to this idea.
Fig. 5.
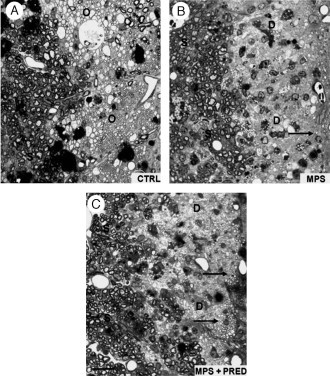
Corticosteroid (CS) treatment significantly reduces oligodendrocyte‐mediated remyelination of EB‐induced lesions in the spinal cord at 1 month. Treatment groups consisted of injection of high‐dose MPS with and without subsequent PRED taper, in order to mimic current CS treatment regimens used clinically to treat acute relapses in MS. (A) Extensive oligodendrocyte and Schwann cell‐mediated remyelination in saline‐injected control rats. (B, C) Reduced oligodendrocyte remyelination in CS‐treated rats with Schwann cell remyelination unaffected. Oligodendrocyte remyelination is indicated with arrows. MPS, methylprednisolone, PRED, prednisone. Modified with permission from Chari et al. (2006a).
If OPCs are significantly depleted along with oligodendrocytes in demyelinated areas, then the repopulation of lesions with repair‐mediating OPCs will clearly constitute the first step toward successful remyelination. An influential hypothesis regarding the failure of remyelination in MS has been the exhaustion of the OPC pool, which suggests that OPCs become depleted around lesions following remyelination. However, one study has demonstrated that focal areas of experimental OPC depletion can be fully repopulated by OPCs from surrounding normal tissue, a process that restores OPC densities to normal (Chari and Blakemore, 2002a). This is supported by the finding that repeated episodes of demyelination do not lead to depletion of the OPC pool or an impairment of remyelination (Penderis et al., 2003a). The repopulation of OPC‐depleted areas (by OPCs from adjoining, normal tissue) is a slow process [approximately 0.5 mm/week, a rate that declines further with age due to intrinsic changes in OPCs (Chari et al., 2003b)]. On the basis of such observations, it is clear that large areas of demyelination in MS could require several months to become completely repopulated by OPCs (assuming that tissue colonization rates for human OPCs are similar to that of rat OPCs). As acute inflammation appears to provide a major stimulus for OPC recruitment, it has been suggested that the abating of inflammation in demyelinated areas, with time, will reduce the drive for remyelination, thereby causing repair failure. In other words, remyelination failure may occur due to a “Temporal Mismatch” between the presence of a remyelination‐promoting environment and the arrival of OPCs in demyelinated areas (Chari 2002a, Chari 2002b). This would imply that a critical time window exists for the interaction of OPCs with demyelinated axons, outside of which the likelihood of successful remyelination will be significantly reduced. This hypothesis could provide an explanation for the finding that remyelination can be prominent at the margins of MS lesions but not the core (i.e., with increasing distance from lesion boundaries). It may also account for the finding that a relatively quiescent OPC population is present in chronic lesions, which does not appear to contribute to remyelination. Another study has suggested that the presence of dysfunctional OPCs may inhibit further colonization of tissue by OPCs from normal tissue (Chari et al., 2003b), which could potentially explain the failure of repopulation of lesions by OPCs from normal WM with disease progression in MS (Wolswijk, 2002).
In contrast, a study examining the distribution and morphologies of oligodendrocytes in chronic lesions found that most lesions contained oligodendrocytes that extended multiple processes and contacted axons, but failed to remyelinate them. Such axons were found to be dystrophic with multiple swellings. The authors concluded that remyelination failure could not be accounted for either by the absence of OPCs or their failure to generate oligodendrocytes, but instead suggested that demyelinated axons were themselves no longer receptive to remyelination (Chang et al., 2002). A recent review suggests that demyelination that induces major alterations in axonal and oligodendroglial signaling events could contribute to remyelination deficits. This may be linked to the reexpression of cell adhesion molecules like PSA‐NCAM on demyelinated axons in MS patients (which has an inhibitory effect on remyelination). The authors also speculate that modification of domain‐specific aggregations of nodal and perinodal proteins during demyelination (or disruption of axoglial interactions in these domains) could contribute to repair failure (Coman et al., 2005).
Reactive astrocytosis is usually evaluated by increased GFAP immunoreactivity and can be an early response to demyelination (Ayers et al., 2004). This accompanies inflammatory changes in MS lesions (Hemmer et al., 2002) which are characterized by a dense matrix of astrocytic processes termed a “glial scar.” Scarring astrocytes are widely believed to inhibit remyelination by ensheathing axons and restricting access of repair‐mediating cells to demyelinated axons (Groves 1993, Rosen 1989). Some studies do indicate that astrocytes may restrict migration of OPCs and inhibit remyelination (Blakemore 2002, Blakemore 2003). However, in the TAIEP mutant rat, the presence of chronically demyelinated axons and astrocytosis does not appear to represent a significant barrier to tissue colonization by OPCs (Foote and Blakemore, 2005). Other reports suggest that reactive astrocytes could be a major source of remyelination mediators in demyelinated areas (Franklin 1991, Frost 2003, Hinks 1999, Redwine 1998). Therefore, the exact roles played by reactive astrocytes during demyelination and repair are currently far from clear.
Notch‐jagged signaling pathways (consisting of Notch‐1 receptor and its ligand Jagged‐1) are powerful inhibitors of OPC differentiation in development. Lesions from brains of MS patients contain elevated levels of Notch‐1 and Jagged‐1 (John et al., 2002), suggesting the Notch pathway could potentially underlie the limited degree of remyelination observed in MS. More recently, it has been demonstrated that γ ‐secretase‐mediated inhibition of Notch signaling in oligodendrocytes in mice with EAE could significantly promote remyelination and reduce axonal damage in the CNS. This was accompanied by an enhancement of clinical recovery (Jurynczyk et al., 2005). Other molecules like the neuregulins (a family of ligands that exerts trophic effects on neurons and glia) appear necessary for normal oligodendrocyte development. Astrocyte‐derived neuregulin is dramatically reduced in both chronic and active lesions; this reduced expression is also suggested to contribute to the paucity of remyelination in MS lesions (Viehover et al., 2001). It has been demonstrated recently that the glycosaminoglycan hyaluronan accumulates in MS lesions and mice with induced EAE. Hyaluronan was found to inhibit remyelination of experimental demyelinating lesions; interestingly, OPCs were found to colonize such lesions but did not appear to mature into myelinating oligodendrocytes in areas of hyaluronan expression (Back et al., 2005). The identification of such molecules in demyelinated areas will aid in defining key therapeutic targets to enhance remyelination clinically.
Integrating some of the current hypotheses, the “Dysregulation Hypothesis” suggests that remyelination may fail because the complex signaling environment within lesions becomes inappropriately regulated or “dysregulated.” Therefore remyelination may fail because the precise coordination between the various cellular events necessary for successful remyelination is lost in demyelinated areas (Fig. 6 ) (Franklin, 2002). Any or all of the above factors can be predicted to contribute to such dysregulation.
Fig. 6.
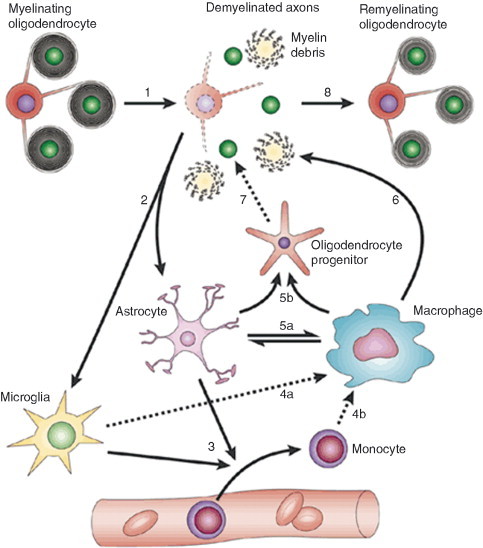
Diagram illustrating that remyelination involves a complex sequence of tightly coordinated events, the “dysregulation” of which will result in remyelination impairment. In response to a demyelinating insult, the myelinated axons undergo demyelination (1). This generates myelin debris. Demyelination causes activation of resident astrocytes and microglia (2). Activated astrocytes and microglia produce factors that act to recruit monocytes from the vasculature (3). Microglia (4a) and recruited monocytes (4b) differentiate into macrophages. Activated astrocytes and macrophages produce factors that can mutually activate each other (5a). As a result of such activation, both produce growth factors that act on OPCs to alter their proliferation and differentiation (5b). Macrophages remove myelin debris (6), a function that is beneficial for remyelination. Under the influence of factors produced by astrocytes and macrophages, recruited OPCs engage with demyelinated axons (7) and differentiate into remyelinating oligodendrocytes (8). Modified with permission from Franklin (2002).
V. MS Therapy: Strategies to Enhance Remyelination
With increasing evidence for a link between absence of myelin and the development of irreversible neurological deficits in MS, it is clear that achieving widespread remyelination of axons will constitute a major component of future therapies that aim to limit neurological disability and restore function in demyelinating disease (along with disease‐modifying and neuroprotective strategies). The strategies to enhance remyelination fall into two broad categories.
A. Strategies to Stimulate or Enhance Endogenous Remyelination
As remyelination in MS lesions appears to fail (or be incomplete), a potential strategy to repair MS lesions is to enhance the naturally occurring regenerative mechanisms. As detailed in previous sections, a number of molecules have been identified (including growth factors, transcription factors, and cell adhesion molecules), which represent potential therapeutic targets to enhance remyelination. Although this is attractive in principle, there are several challenges confronting such a strategy. (1) Given that the regulation of remyelination is a complex, multifactorial process, the identification of relevant molecules to enhance repair will be critical. This is complicated by the considerable redundancy in the regulatory mechanisms that mediate remyelination. (2) The extensive variability observed to occur in MS lesions raises important issues in relation to the nature, timing, and mode of delivery of therapeutic agents for individual patients. (3) Effective, targeted delivery of therapeutic agents to multifocal sites of demyelination may be essential in order to avoid the nonspecific side effects of systemic delivery—the use of new drug/biomolecule‐targeting strategies/technologies could prove very useful in this regard.
Antibody therapy: Interestingly, studies show that immunoglobulins (generally preparations of intact IgG molecules) can promote remyelination (Warrington et al., 2000 and reviewed in Trebst and Stangel, 2006). The mechanisms by which immunoglobulins enhance repair are not clear but the current postulated mechanisms can be divided into two categories—(1) Direct via an effect on OPCs (by modifying their proliferation, migration, and differentiation) and oligodendrocytes (by rescuing them from cell death) and (2) Indirect via an immunomodulatory role (by decreasing inflammatory damage, modifying cytokine release, and phagocytosis) in lesions.
B. Transplantation of Myelinogenic Cells
The alternative strategy to enhance remyelination is to introduce myelin‐forming cells into areas of demyelination. Several cell types (immature and adult) have been identified that achieve extensive remyelination (with restoration of saltatory conduction) in experimental demyelinating lesions and in myelin mutants. These include OPCs, Schwann cells, olfactory ensheathing cells (OECs) as well as various categories of stem cells [like neural stem cells (NSCs), embryonic stem cells (ESCs), and bone marrow‐derived stem cells (BMSCs)] and their derivatives—these have been the subject of a number of excellent reviews (Dubois‐Dalcq 2005, Duncan 2005, Ibrahim 2006, Keirstead 2005, Pluchino 2005, Pluchino 2005, Tepavcevic 2005) (Fig. 7 ). Numerous issues need to be considered when evaluating the clinical feasibility and benefits of cell transplantation‐based strategies in MS. These include (1) the possibility that transplanted cells may, in turn, become targets of the demyelinating pathology, (2) the ability of the transplanted population to proliferate and differentiate in sufficient numbers to mediate remyelination (myelination potential, in general, appears to be dependent on the degree of enrichment of immature cells in the cell preparation). ESCs are particularly attractive in this regard due to their capacity for unlimited expansion in vitro, broad differentiation potential (including differentiation into multipotent neural precursors and OPCs), and their amenability to genetic engineering (Keirstead 2005, Nistor 2005). Other studies have shown that supernumerary transplanted OPCs do not survive, proliferate, or migrate in the adult CNS (Franklin 1996, O'Leary 1997) and indicate that considerable depletion of the resident OPC population (>50%) is prerequisite to introducing significant numbers of OPCs into adult CNS tissue (Chari et al., 2006b). This may not represent a problem in lesions where OPC depletion occurs along with oligodendrocyte loss. However, as dysfunctional OPCs appear to inhibit tissue repopulation by new OPCs (Chari et al., 2003b), it remains to be established if transplant‐derived OPCs can, in fact, mediate repair of lesions that contain significant numbers of endogenous, dysfunctional OPCs. (3) Astrogliosis is a pathological hallmark of chronic MS lesions, so the ability of transplanted cells to survive within astrogliosed areas will be a key. OECs but not Schwann cells have been shown to survive in areas of astrocytosis (Barnett and Riddell, 2004), but it is not clear to what extent OECs can be expanded indefinitely in culture. OPCs, on the other hand, possess the ability to colonize areas of astrocytosis (with the caveat that prior depletion of the endogenous OPC population or the presence of an acute inflammatory environment is critical for transplant survival) (Foote and Blakemore, 2005). (4) As multifocal demyelination is another key feature of MS, the ability of transplanted cells to migrate within the CNS may be important unless it is feasible to introduce cells directly into lesions. Multipotent NSCs injected intravenously or intraventricularly can enter areas of demyelination, generate myelinating oligodendrocytes, and mediate functional recovery (possibly because these cells express a range of inflammatory molecules like adhesion molecules, cytokines, and chemokines). This “homing” behavior makes them attractive candidates to mediate targeted repair of multifocal lesions. In addition, such cells have been shown to exert significant anti‐inflammatory effects and reduce astrocytosis in demyelinated areas. Multipotent NSCs have been isolated from human subcortical WM; these cells can be maintained in vitro prior to transplantation and give rise to neurons and glia, when transplanted into the rodent CNS (Nunes et al., 2003). (5) Given that inflammation appears to be beneficial for remyelination and transplant survival, it will be important to establish the effects of common anti‐inflammatory and immunomodulatory treatments (used in MS treatment) on transplant survival and repair. (6) It is critical that the genetic stability and safety of the transplanted cells (including the elimination of pathogens and tumorigenic potential) be established prior to transplantation, (7) potential rejection of transplanted cells and the need for tissue‐type matching must be taken into account, and (8) a number of ongoing ethical and political issues relating to the derivation of cells for transplantation will have a serious impact on which therapies can be advanced to clinical trials. Additionally, there are many practical concerns when surgically introducing cells into neural tissue. These include (1) the consequences of disruption of the blood–brain barrier, (2) the possibility of inducing hemorrhage at the transplant site, (3) the potential expense associated with multiple surgical injections to introduce transplanted cells into multifocal lesions, (iv) the significant challenge associated with accurately targeting transplanted cells to inaccessible CNS lesions sites, and (5) the risk of inducing CSF occlusion or emboli when introducing cells intraventricularly or intravenously. It is clear, therefore, that the benefits of a range of therapeutic strategies can be demonstrated in experimental conditions. The translation of such approaches to the clinical setting will constitute a major objective for future studies.
Fig. 7.
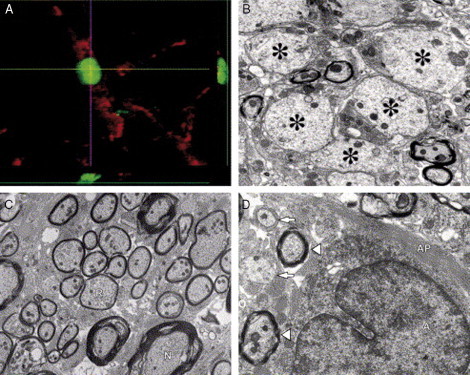
Transplantation of human ESC‐derived predifferentiated oligodendrocyte progenitors into a region of demyelination results in differentiation of ESCs into oligodendrocytes and remyelination. (A) Assessment of cell fate and differentiation following transplantation using double immunohistochemical staining for human nuclei (green) and the oligodendrocyte marker APC‐CC1 (red) confirms that transplanted cells survive and differentiate into mature oligodendrocytes. (B) Electron micrograph (EM) of the transplant environment 7 days postinjury. Asterisks indicate demyelinated axons in an astrogliosis‐free microenvironment. (C) EM of a rat spinal cord that was transplanted with human ESC‐derived oligodendrocyte progenitors 7 days postinjury. Oligodendrocyte‐remyelinated axons (R; with characteristically thin myelin sheaths) are evident among few normally myelinated axons (N). (D) EM of a spinal cord injury site 10 months postinjury, illustrating an astrocyte (A) with a large intermediate filament‐rich process (AP) extending to demyelinated axons (arrows) and myelinated axons (arrowheads), both surrounded by intermediate filament‐rich astrocytic processes. This illustrates the point that demyelinated axons in areas of chronic injury are ensheathed by astrocytic processes, which may prevent remyelination by endogenous or transplanted cells. Reproduced with permission from Keirstead (2005).
Acknowledgments
D.M.C. is supported by a Junior Research Fellowship from the Multiple Sclerosis Society of the United Kingdom and Northern Ireland. I am grateful to Professor Bill Blakemore and Professor Robin Franklin for their helpful comments on this chapter.
References
- Adams C.W.M. “A Colour Atlas of Multiple Sclerosis and Other Myelin Disorders.”. Wolfe Medical Publications; London: 1989. [Google Scholar]
- Antony J.M., van Marle G., Opii W., Butterfield D.A., Mallet F., Yong V.W., Wallace J.L., Deacon R.M., Warren K., Power C. Human endogenous retrovirus glycoprotein‐mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat. Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- Arnett H.A., Mason J., Marino M., Suzuki K., Matsushima G.K., Ting J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Arnett H.A., Wang Y., Matsushima G.K., Suzuki K., Ting J.P. Functional genomic analysis of remyelination reveals importance of inflammation in oligodendrocyte regeneration. J. Neurosci. 2003;23:9824–9832. doi: 10.1523/JNEUROSCI.23-30-09824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett H.A., Fancy S.P., Alberta J.A., Zhao C., Plant S.R., Kaing S., Raine C.S., Rowitch D.H., Franklin R.J., Stiles C.D. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;17:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Ayers M.M., Hazelwood L.J., Catmull D.V., Wang D., McKormack Q., Bernard C.C., Orian J.M. Early glial responses in murine models of multiple sclerosis. Neurochem. Int. 2004;45:409–419. doi: 10.1016/j.neuint.2003.08.018. [DOI] [PubMed] [Google Scholar]
- Back S.A., Tuohy T.M., Chen H., Wallingford N., Craig A., Struve J., Luo N.L., Banine F., Liu Y., Chang A., Trapp B.D., Bebo B.F. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Barnett M.H., Prineas J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Barnett S.C., Riddell J.S. Olfactory ensheathing cells (OECs) and the treatment of CNS injury: Advantages and possible caveats. J. Anat. 2004;204:57–67. doi: 10.1111/j.1469-7580.2004.00257.x. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles D.E., Bergles D.E., Roberts J.D., Somogyi P., Jahr C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- Bieber A.J., Kerr S., Rodriguez M. Efficient central nervous system remyelination requires T cells. Ann. Neurol. 2003;53:680–684. doi: 10.1002/ana.10578. [DOI] [PubMed] [Google Scholar]
- Black J.A., Waxman S.G., Smith K.J. Remyelination of dorsal column axons by endogenous Schwann cells restores the normal pattern of Nav1. 6 and Kv1. 2 at nodes of Ranvier. Brain. 2006;129:1319–1329. doi: 10.1093/brain/awl057. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F. The case for a central nervous system (CNS) origin for the Schwann cells that remyelinate CNS axons following concurrent loss of oligodendrocytes and astrocytes. Neuropathol. Appl. Neurobiol. 2005;31:1–10. doi: 10.1111/j.1365-2990.2005.00637.x. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F., Patterson R.C. Suppression of remyelination in the CNS by X‐irradiation. Acta Neuropathol. Berl. 1978;42:105–113. doi: 10.1007/BF00690975. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F., Chari D.M., Gilson J.M., Crang A.J. Modelling large areas of demyelination in the rat reveals the potential and possible limitations of transplanted glial cells for remyelination in the CNS. Glia. 2002;15:155–168. doi: 10.1002/glia.10067. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F., Gilson J.M., Crang A.J. The presence of astrocytes in areas of demyelination influences remyelination following transplantation of oligodendrocyte progenitors. Exp. Neurol. 2003;184:955–963. doi: 10.1016/S0014-4886(03)00347-9. [DOI] [PubMed] [Google Scholar]
- Brockschnieder D., Lappe‐Siefke C., Goebbels S., Boesl M.R., Nave K.A., Riethmacher D. Cell depletion due to diphtheria toxin fragment A after Cre‐mediated recombination. Mol. Cell. Biol. 2004;24:7636–7642. doi: 10.1128/MCB.24.17.7636-7642.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch T., Heppner F.L., Tertilt C., Heinen T.J., Kremer M., Wunderlich F.T., Jung S., Waisman A. A Cre‐inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- Bunge M.B., Bunge R.P., Ris H. Ultrastructural study of remyelination in an experimental lesion in adult cat spinal cord. J. Biophys. Biochem. Cytol. 1961;10:67–94. doi: 10.1083/jcb.10.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge R.P. Regenerative phenomena in the feline spinal cord in late stages of lesions inflicted by cerebrospinal fluid manipulation. Anat. Rec. 1958;130:279. [Google Scholar]
- Bunge R.P., Settlage P.H. Neurological lesions in cats following cerebrospinal fluid manipulation. J. Neuropathol. Exp. Neurol. 1957;16:471–491. doi: 10.1097/00005072-195710000-00003. [DOI] [PubMed] [Google Scholar]
- Butt A.M., Duncan A., Hornby M.F., Kirvell S.L., Hunter A., Levine J.M., Berry M. Cells expressing the NG2 antigen contact nodes of Ranvier in adult CNS white matter. Glia. 1999;26:84–91. [PubMed] [Google Scholar]
- Byravan S., Foster L.M., Phan T., Verity A.N., Campagnoni A.T. Murine oligodendroglial cells express nerve growth factor. Proc. Natl. Acad. Sci. USA. 1994;91:8812–8816. doi: 10.1073/pnas.91.19.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajal S.R. “Degeneration and Regeneration of the Central Nervous System,”. Humphrey Milford/Oxford University Press; London: 1928. pp. 734–760. [Google Scholar]
- Carson M.J., Behringer R.R., Brinster R.L., McMorris F.A. Insulin‐like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-o. [DOI] [PubMed] [Google Scholar]
- Chang A., Nishiyama A., Peterson J., Prineas J., Trapp B.D. NG2‐positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A., Tourtellotte W.W., Rudick R., Trapp B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Chari D.M., Blakemore W.F. Efficient recolonisation of progenitor‐depleted areas of the CNS by adult oligodendrocyte progenitor cells. Glia. 2002;37:307–313. [PubMed] [Google Scholar]
- Chari D.M., Blakemore W.F. New insights into remyelination failure in multiple sclerosis: Implications for glial cell transplantation. Mult. Scler. 2002;8:271–277. doi: 10.1191/1352458502ms842oa. Review. [DOI] [PubMed] [Google Scholar]
- Chari D.M., Crang A.J., Blakemore W.F. Decline in rate of colonization of oligodendrocyte progenitor cell (OPC)‐depleted tissue by adult OPCs with age. J. Neuropathol. Exp. Neurol. 2003;62:908–916. doi: 10.1093/jnen/62.9.908. [DOI] [PubMed] [Google Scholar]
- Chari D.M., Huang W.L., Blakemore W.F. Dysfunctional oligodendrocyte progenitor cell (OPC) populations may inhibit repopulation of OPC depleted tissue. J. Neurosci. Res. 2003;73:787–793. doi: 10.1002/jnr.10700. [DOI] [PubMed] [Google Scholar]
- Chari D.M., Zhao C., Kotter M.R., Blakemore W.F., Franklin R.J. Corticosteroids delay remyelination of experimental demyelination in the rodent central nervous system. J. Neurosci. Res. 2006;83:594–605. doi: 10.1002/jnr.20763. [DOI] [PubMed] [Google Scholar]
- Chari D.M., Gilson J.M., Franklin R.J., Blakemore W.F. Oligodendrocyte progenitor cell (OPC) transplantation is unlikely to offer a means of preventing X‐irradiation induced damage in the CNS. Exp. Neurol. 2006;198:145–153. doi: 10.1016/j.expneurol.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Christensen T. Association of human endogenous retroviruses with multiple sclerosis and possible interactions with herpes viruses. Rev. Med. Virol. 2005;15:179–211. doi: 10.1002/rmv.465. Review. [DOI] [PubMed] [Google Scholar]
- Coman I., Barbin G., Charles P., Zalc B., Lubetzki C. Axonal signals in central nervous system myelination, demyelination and remyelination. J. Neurol. Sci. 2005;233:67–71. doi: 10.1016/j.jns.2005.03.029. Review. [DOI] [PubMed] [Google Scholar]
- Crang A.J., Gilson J.M., Li W.W., Blakemore W.F. The remyelinating potential and in vitro differentiation of MOG‐expressing oligodendrocyte precursors isolated from the adult rat CNS. Eur. J. Neurosci. 2004;20:1445–1460. doi: 10.1111/j.1460-9568.2004.03606.x. [DOI] [PubMed] [Google Scholar]
- Crockett D.P., Burshteyn M., Garcia C., Muggironi M., Casaccia‐Bonnefil P. Number of oligodendrocyte progenitors recruited to the lesioned spinal cord is modulated by the levels of the cell cycle regulatory protein p27Kip‐1. Glia. 2005;49:301–308. doi: 10.1002/glia.20111. [DOI] [PubMed] [Google Scholar]
- Demerens C., Stankoff B., Logak M., Anglade P., Allinquant B., Couraud F., Zalc B., Lubetzki C. Induction of myelination in the central nervous system by electrical activity. Proc. Natl. Acad. Sci. USA. 1996;93:9887–9892. doi: 10.1073/pnas.93.18.9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois‐Dalcq M., Ffrench‐Constant C., Franklin R.J. Enhancing central nervous system remyelination in multiple sclerosis. Neuron. 2005;48:9–12. doi: 10.1016/j.neuron.2005.09.004. Review. [DOI] [PubMed] [Google Scholar]
- Dugandzija‐Novakovic S., Koszowski A.G., Levinson S.R., Shrager P. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J. Neurosci. 1995;15:492–503. doi: 10.1523/JNEUROSCI.15-01-00492.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan I.D. Remyelination and restoration of axonal function by glial cell transplantation. Ernst Schering Res. Found. Workshop. 2005;53:115–132. doi: 10.1007/3-540-27626-2_7. Review. [DOI] [PubMed] [Google Scholar]
- Durand B., Raff M. A cell‐intrinsic timer that operates during oligodendrocyte development. Bioessays. 2000;22:64–71. doi: 10.1002/(SICI)1521-1878(200001)22:1<64::AID-BIES11>3.0.CO;2-Q. Review. [DOI] [PubMed] [Google Scholar]
- Edgar J.M., McLaughlin M., Yool D., Zhang S.C., Fowler J.H., Montague P., Barrie J.A., McCulloch M.C., Duncan I.D., Garbern J., Nave K.A., Griffiths I.R. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercolini A.M., Miller S.D. Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J. Immunol. 2006;176:3293–3298. doi: 10.4049/jimmunol.176.6.3293. Review. [DOI] [PubMed] [Google Scholar]
- Fancy S.P., Zhao C., Franklin R.J. Increased expression of Nkx2. 2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004;27:247–254. doi: 10.1016/j.mcn.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Ferguson B., Matyszak M.K., Esiri M.M., Perry V.H. Axonal damage in multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Foote A.K., Blakemore W.F. Inflammation stimulates remyelination in areas of chronic demyelination. Brain. 2005;128:528–539. doi: 10.1093/brain/awh417. [DOI] [PubMed] [Google Scholar]
- Franklin R.J. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. Review. [DOI] [PubMed] [Google Scholar]
- Franklin R.J., Hinks G.L. Understanding CNS remyelination: Clues from developmental and regeneration biology. J. Neurosci. Res. 1999;58:207–213. Review. [PubMed] [Google Scholar]
- Franklin R.J., Bayley S.A., Blakemore W.F. Transplanted CG4 cells (an oligodendrocyte progenitor cell line) survive, migrate, and contribute to repair of areas of demyelination in X‐irradiated and damaged spinal cord but not in normal spinal cord. Exp. Neurol. 1996;137:263–276. doi: 10.1006/exnr.1996.0025. [DOI] [PubMed] [Google Scholar]
- Franklin R.J.M., Blakemore W.F. Requirements for Schwann cell migration within CNS environments: A viewpoint. Int. J. Dev. Neurosci. 1993;11:641–649. doi: 10.1016/0736-5748(93)90052-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin R.J.M., Crang A.J., Blakemore W.F. Transplanted type‐1 astrocytes facilitate repair of demyelinating lesions by host oligodendrocytes in adult rat spinal cord. J. Neurocytol. 1991;20:420–430. doi: 10.1007/BF01355538. [DOI] [PubMed] [Google Scholar]
- Frost E.E., Nielsen J.A., Le T.Q., Armstrong R.C. PDGF and FGF2 regulate oligodendrocyte progenitor responses to demyelination. J. Neurobiol. 2003;54:457–472. doi: 10.1002/neu.10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M., Karlsson L., Hall A.C., Abramsson A., Calver A.R., Bostrom H., Willetts K., Bertold C.H., Heath J.K., Betsholtz C., Richardson W.D. Defective oligodendrocyte development and severe hypomyelination in PDGF‐A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Gallo V., Ghiani C.A., Yuan X. The role of ion channels and neurotransmitter receptors in glial cell development. In: Jessen K.R., Richardson W.D., editors. “Glial Cell Development”. Oxford University Press; New York: 2001. pp. 109–130. [Google Scholar]
- Garbern J.Y., Yool D.A., Moore G.J., Wilds I.B., Faulk M.W., Klugmann M., Nave K.A., Sistermans E.A., van der Knaap M.S., Bird T.D., Shy M.E., Kamholz J.A. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length‐dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551–561. doi: 10.1093/brain/awf043. [DOI] [PubMed] [Google Scholar]
- Ghatak N.R., Leshner R.T., Price A.C., Felton W.L. Remyelination in the human central nervous system. J. Neuropathol. Exp. Neurol. 1989;48:507–518. doi: 10.1097/00005072-198909000-00002. [DOI] [PubMed] [Google Scholar]
- Gilson J., Blakemore W.F. Failure of remyelination in areas of demyelination produced in the spinal cord of old rats. Neuropathol. Appl. Neurobiol. 1993;19:173–181. doi: 10.1111/j.1365-2990.1993.tb00424.x. [DOI] [PubMed] [Google Scholar]
- Graca D.L., Blakemore W.F. Delayed remyelination in the rat spinal cord following ethidium bromide injection. Neuropathol. Appl. Neurobiol. 1986;12:593–605. doi: 10.1111/j.1365-2990.1986.tb00162.x. [DOI] [PubMed] [Google Scholar]
- Griffiths I.R., Klugmann M., Anderson T., Yool D., Thomson C., Schwab M.H., Schneider A., Zimmermann F., McCulloch M., Nadon N., Nave K.A. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Groves A.K., Barnett S.C., Franklin R.J., Crang A.J., Mayer M., Blakemore W.F., Noble M. Repair of demyelinated lesions by transplantation of purified O‐2A progenitor cells. Nature. 1993;362:453–455. doi: 10.1038/362453a0. [DOI] [PubMed] [Google Scholar]
- Gveric D., Cuzner M.L., Newcombe J. Insulin‐like growth factors and binding proteins in multiple sclerosis plaques. Neuropathol. Appl. Neurobiol. 1999;25:215–225. doi: 10.1046/j.1365-2990.1999.00187.x. [DOI] [PubMed] [Google Scholar]
- Hemmer B., Archelos J.J., Hartung H.P. New concepts in the immunopathogenesis of multiple sclerosis. Nat. Rev. Neurosci. 2002;3:291–301. doi: 10.1038/nrn784. Review. [DOI] [PubMed] [Google Scholar]
- Hinks G.L., Franklin R.J.M. Distinctive patterns of PDGF‐A, FGF‐2, IGF‐I, and TGF‐beta1 gene expression during remyelination of experimentally‐induced spinal cord demyelination. Mol. Cell. Neurosci. 1999;14:153–168. doi: 10.1006/mcne.1999.0771. [DOI] [PubMed] [Google Scholar]
- Ibrahim A., Li Y., Li D., Raisman G., El Masry W.S. Olfactory ensheathing cells: Ripples of an incoming tide? Lancet Neurol. 2006;5:453–457. doi: 10.1016/S1474-4422(06)70444-6. Review. [DOI] [PubMed] [Google Scholar]
- Itoyama Y., Webster H.D., Richardson E.P., Trapp B.D. Schwann cell remyelination of demyelinated axons in spinal cord multiple sclerosis lesions. Ann. Neurol. 1983;14:339–346. doi: 10.1002/ana.410140313. [DOI] [PubMed] [Google Scholar]
- Itoyama Y., Ohnishi A., Tateishi J., Kuroiwa Y., Webster H.D. Spinal cord multiple sclerosis lesions in Japanese patients: Schwann cell remyelination occurs in areas that lack glial fibrillary acidic protein (GFAP) Acta Neuropathol. (Berl.) 1985;65:217–223. doi: 10.1007/BF00687001. [DOI] [PubMed] [Google Scholar]
- Jeffery N.D., Blakemore W.F. Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain. 1997;120:27–37. doi: 10.1093/brain/120.1.27. [DOI] [PubMed] [Google Scholar]
- John G.R., Shankar S.L., Shafit‐Zagardo B., Massimi A., Lee S.C., Raine C.S., Brosnan C.F. Multiple sclerosis: Re‐expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- Jones S.J. Visual evoked potentials after optic neuritis. Effect of time interval, age and disease dissemination. J. Neurol. 1993;240:489–494. doi: 10.1007/BF00874118. [DOI] [PubMed] [Google Scholar]
- Jurynczyk M., Jurewicz A., Bielecki B., Raine C.S., Selmaj K. Inhibition of Notch signaling enhances tissue repair in an animal model of multiple sclerosis. J. Neuroimmunol. 2005;170:3–10. doi: 10.1016/j.jneuroim.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Keirstead H.S. Stem cells for the treatment of myelin loss. Trends Neurosci. 2005;28:677–683. doi: 10.1016/j.tins.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Keirstead H.S., Blakemore W.F. Identification of post‐mitotic oligodendrocytes incapable of remyelination within the demyelinated adult spinal cord. J. Neuropathol. Exp. Neurol. 1997;56:1191–1201. doi: 10.1097/00005072-199711000-00003. [DOI] [PubMed] [Google Scholar]
- Keirstead H.S., Levine J.M., Blakemore W.F. Response of the oligodendrocyte progenitor cell population (defined by NG2 labelling) to demyelination of the adult spinal cord. Glia. 1998;22:161–170. [PubMed] [Google Scholar]
- Kerschensteiner M., Stadelmann C., Buddeberg B.S., Merkler D., Bareyre F.M., Anthony D.C., Linington C., Bruck W., Schwab M.E. Targeting experimental autoimmune encephalomyelitis lesions to a predetermined axonal tract system allows for refined behavioral testing in an animal model of multiple sclerosis. Am. J. Pathol. 2004;164:1455–1469. doi: 10.1016/S0002-9440(10)63232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornek B., Storch M.K., Weissert R., Wallstroem E., Stefferl A., Olsson T., Linington C., Schmidbauer M., Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter M.R., Setzu A., Sim F.J., Van Rooijen N., Franklin R.J.M. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin‐induced demyelination. Glia. 2001;35:204–212. doi: 10.1002/glia.1085. [DOI] [PubMed] [Google Scholar]
- Kotter M.R., Zhao C., van Rooijen N., Franklin R.J. Macrophage‐depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005;18:166–175. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Lappe‐Siefke C., Goebbels S., Gravel M., Nicksch E., Lee J., Braun P.E., Griffiths I.R., Nave K.A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lassmann H. “Comparative Neuropathology of Chronic Experimental Allergic Encephalomyelitis and Multiple Sclerosis.”. Springer; Berlin: 1983. [PubMed] [Google Scholar]
- Lassmann H., Bruck W., Lucchinetti C., Rodriguez M. Remyelination in multiple sclerosis. Mult. Scler. 1997;3:133–136. doi: 10.1177/135245859700300213. Review. [DOI] [PubMed] [Google Scholar]
- Li G., Crang A.J., Rundle J.L., Blakemore W.F. Oligodendrocyte progenitor cells in the adult rat CNS express myelin oligodendrocyte glycoprotein (MOG) Brain pathol. 2002;12:463–471. doi: 10.1111/j.1750-3639.2002.tb00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.W., Penderis J., Zhao C., Schumacher M., Franklin R.J. Females remyelinate more efficiently than males following demyelination in the aged but not young adult CNS. Exp. Neurol. 2006;202:250–254. doi: 10.1016/j.expneurol.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Li W.W., Setzu A., Zhao C., Franklin R.J.M. Minocycline‐mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non‐immune model of demyelination. J. Neuroimmunol. 2005;158:58–66. doi: 10.1016/j.jneuroim.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Loughlin A.J., Copelman C.A., Hall A., Armer T., Young B.C., Landon D.N., Cuzner M.L. Myelination and remyelination of aggregate rat brain cell cultures enriched with macrophages. J. Neurosci. Res. 1997;47:384–392. [PubMed] [Google Scholar]
- Lovas G., Szilagyi N., Majtenyi K., Palkovits M., Komoly S. Axonal changes if chronically demyelinated cervical spinal cord plaques. Brain. 2000;123:308–317. doi: 10.1093/brain/123.2.308. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C., Bruck W., Parisi J., Scheithauer B., Rodriguez M., Lassmann H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain. 1999;122:2279–2295. doi: 10.1093/brain/122.12.2279. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C., Bruck W., Parisi J., Scheithauer B., Rodriguez M., Lassmann H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Ludwin S.K. Chronic demyelination inhibits remyelination in the central nervous system. Lab. Invest. 1980;43:382–387. [PubMed] [Google Scholar]
- Luzi P., Zaka M., Rao H.Z., Curtis M., Rafi M.A., Wenger D.A. Generation of transgenic mice expressing insulin‐like growth factor‐1 under the control of the myelin basic protein promoter: Increased myelination and potential for studies on the effects of increased IGF‐1 on experimentally and genetically induced demyelination. Neurochem. Res. 2004;29:881–889. doi: 10.1023/b:nere.0000021233.79076.72. [DOI] [PubMed] [Google Scholar]
- Marburg O. Die sogenannte ‘akute multiple Sklerose’ (Encephalomyelitis periaxialis scleroticans) J. Neurol. Psychiatry. 1906;27:213–312. [Google Scholar]
- Marchionni M.A., Cannella B., Hoban C., Gao Y.L., Garcia‐Arenas R., Lawson D., Happel E., Noel F., Tofilon P., Gwynne D., Raine C.S. Neuregulin in neuron/glial interactions in the central nervous system. GGF2 diminishes autoimmune demyelination, promotes oligodendrocyte progenitor expansion, and enhances remyelination. Adv. Exp. Med. Biol. 1999;468:283–295. [PubMed] [Google Scholar]
- Mason J.L., Suzuki K., Chaplin D.D., Matsushima G.K. Interleukin‐1beta promotes repair of the CNS. J. Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon R.D., Piras G., Ida J.A., Dubois‐Dalcq M. A role for TGF‐beta in oligodendrocyte differentiation. J. Cell Biol. 1993;121:1397–1407. doi: 10.1083/jcb.121.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkler D., Boretius S., Stadelmann C., Ernsting T., Michaelis T., Frahm J., Bruck W. Multicontrast MRI of remyelination in the central nervous system. NMR Biomed. 2005;18:395–403. doi: 10.1002/nbm.972. [DOI] [PubMed] [Google Scholar]
- Messersmith D.J., Murtie J.C., Le T.Q., Frost E.E., Armstrong R.C. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J. Neurosci. Res. 2000;62:241–256. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S., Miller R.H., Lee X., Scott M.L., Shulag‐Morskaya S., Shao Z., Chang J., Thill G., Levesque M., Zhang M., Hession C., Sah D. LINGO‐1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Miller R.H. Contact with central nervous system myelin inhibits oligodendrocyte progenitor maturation. Dev. Biol. 1999;216:359–368. doi: 10.1006/dbio.1999.9466. [DOI] [PubMed] [Google Scholar]
- Murtie J.C., Zhou Y.X., Le T.Q., Vana A.C., Armstrong R.C. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol. Dis. 2005;19:171–182. doi: 10.1016/j.nbd.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Nathan C.F. Secretory products of macrophages. J. Clin. Invest. 1987;79:319–326. doi: 10.1172/JCI112815. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave K.A. Myelin specific genes and their mutations in the mouse. In: Jessen K.R., Richardson W.D., editors. “Glial Cell Development”. Oxford University Press; Oxford, UK: 2001. pp. 177–208. [Google Scholar]
- Niehaus A., Shi J., Grzenkowski M., Diers‐Fenger M., Archelos J., Hartung H.P., Toyka K., Bruck W., Trotter J. Patients with active relapsing‐remitting multiple sclerosis synthesize antibodies recognizing oligodendrocyte progenitor cell surface protein: Implications for remyelination. Ann. Neurol. 2000;48:362–371. [PubMed] [Google Scholar]
- Nistor G.I., Totoiu M.O., Haque N., Carpenter M.K., Keirstead H.S. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia. 2005;49:385–396. doi: 10.1002/glia.20127. [DOI] [PubMed] [Google Scholar]
- Nunes M.C., Roy N.S., Keyoung H.M., Goodman R.R., McKhann G., Jiang L., Kang J., Nedergaard M., Goldman S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- O'Leary M.T., Blakemore W.F. Oligodendrocyte precursors survive poorly and do not migrate following transplantation into the normal adult central nervous system. J. Neurosci. Res. 1997;48:159–167. [PubMed] [Google Scholar]
- O'Leary M.T., Hinks G.L., Charlton H.M., Franklin R.J. Increasing local levels of IGF‐I mRNA expression using adenoviral vectors does not alter oligodendrocyte remyelination in the CNS of aged rats. Mol. Cell. Neurosci. 2002;19:32–42. doi: 10.1006/mcne.2001.1062. [DOI] [PubMed] [Google Scholar]
- Oleszak E.L., Chang J.R., Friedman H., Katset C.D., Platsoucas C.D. Theiler's virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004;17:174–207. doi: 10.1128/CMR.17.1.174-207.2004. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos D., Pham‐Dinh D., Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp. Neurol. 2006;197:373–385. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Penderis J., Shields S.A., Franklin R.J.M. Impaired remyelination and depletion of oligodendrocyte progenitors does not occur following repeated episodes of focal demyelination in the rat central nervous system. Brain. 2003;126:1382–1391. doi: 10.1093/brain/awg126. [DOI] [PubMed] [Google Scholar]
- Penderis J., Woodruff R.H., Lakatos A., Li W.W., Dunning M.D., Zhao C., Marchionni M., Franklin R.J. Increasing local levels of neuregulin (glial growth factor‐2) by direct infusion into areas of demyelination does not alter remyelination in the rat CNS. Eur. J. Neurosci. 2003;18:2253–2264. doi: 10.1046/j.1460-9568.2003.02969.x. [DOI] [PubMed] [Google Scholar]
- Perier O., Gregoire A. Electron microscopic features of multiple sclerosis lesions. Brain. 1965;88:937–952. doi: 10.1093/brain/88.5.937. [DOI] [PubMed] [Google Scholar]
- Pluchino S., Martino G. The therapeutic use of stem cells for myelin repair in autoimmune demyelinating disorders. J. Neurol. Sci. 2005;233:117–119. doi: 10.1016/j.jns.2005.03.026. Review. [DOI] [PubMed] [Google Scholar]
- Pluchino S., Zanotti L., Deleidi M., Martino G. Neural stem cells and their use as therapeutic tool in neurological disorders. Brain Res. Brain Res. Rev. 2005;48:211–219. doi: 10.1016/j.brainresrev.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Polito A., Reynolds R. NG2‐expressing cells as oligodendrocyte progenitors in the normal and demyelinated adult central nervous system. J. Anat. 2005;207:707–716. doi: 10.1111/j.1469-7580.2005.00454.x. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prineas J.W., Connell F. Remyelination in multiple sclerosis. Ann. Neurol. 1979;5:22–31. doi: 10.1002/ana.410050105. [DOI] [PubMed] [Google Scholar]
- Prineas J.W., Kwon E.E., Cho E.S., Sharer L.R. Continual breakdown and regeneration of myelin in progressive multiple sclerosis plaques. Ann. N. Y. Acad. Sci. 1984;436:11–32. doi: 10.1111/j.1749-6632.1984.tb14773.x. [DOI] [PubMed] [Google Scholar]
- Prineas J.W., Barnard R.O., Revesz T., Kwon E.E., Sharer L., Cho E.S. Multiple sclerosis. Pathology of recurrent lesions. Brain. 1993;116:681–693. doi: 10.1093/brain/116.3.681. [DOI] [PubMed] [Google Scholar]
- Raine C.S., Wu E. Multiple sclerosis: Remyelination in acute lesions. J. Neuropathol. Exp. Neurol. 1993;52:199–204. [PubMed] [Google Scholar]
- Redwine J.M., Armstrong R.C. In vivo proliferation of oligodendrocyte progenitors expressing PDGFalphaR during early remyelination. J. Neurobiol. 1998;37:413–428. doi: 10.1002/(sici)1097-4695(19981115)37:3<413::aid-neu7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Rodriguez M. A function of myelin is to protect axons from subsequent injury: Implications for deficits in multiple sclerosis. Brain. 2003;126:751–752. doi: 10.1093/brain/awg070. [DOI] [PubMed] [Google Scholar]
- Rosen C.L., Bunge R.P., Ard M.D., Wood P.M. Type 1 astrocytes inhibit myelination by adult rat oligodendrocytes in vitro. J. Neurosci. 1989;9:3371–3379. doi: 10.1523/JNEUROSCI.09-10-03371.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M., Black J.A., Lankford K.L., Tokuno H.A., Waxman S.G., Kocsis J.D. Molecular reconstruction of nodes of Ranvier after remyelination by transplanted olfactory ensheathing cells in the demyelinated spinal cord. J. Neurosci. 2006;26:1803–1812. doi: 10.1523/JNEUROSCI.3611-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger H. Zur Frage der akuten multiplen Sklerose und der encephalomyelitis disseminata im Kindesalter. Arb. Neurol. Inst. (Wien.) 1909;17:410–432. [Google Scholar]
- Scolding N., Franklin R., Stevens S., Heldin C.H., Compston A., Newcombe J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain. 1998;121:2221–2228. doi: 10.1093/brain/121.12.2221. [DOI] [PubMed] [Google Scholar]
- Setzu A., Lathia J.D., Zhao C., Wells K., Rao M.S., Ffrench‐Constant C., Franklin R.J.M. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia. 2006;54:297–303. doi: 10.1002/glia.20371. [DOI] [PubMed] [Google Scholar]
- Sherman D.L., Brophy P.J. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 2005;6:683–690. doi: 10.1038/nrn1743. [DOI] [PubMed] [Google Scholar]
- Shi J., Marinovich A., Barres B.A. Purification and characterization of adult oligodendrocyte precursor cells from the rat optic nerve. J. Neurosci. 1998;18:4627–4636. doi: 10.1523/JNEUROSCI.18-12-04627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim F.J., Zhao C., Penderis J., Franklin R.J.M. The age‐related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002;22:2451–2459. doi: 10.1523/JNEUROSCI.22-07-02451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.J., Blakemore W.F., McDonald W.I. Central remyelination restores secure conduction. Nature. 1979;280:395–396. doi: 10.1038/280395a0. [DOI] [PubMed] [Google Scholar]
- Smith K.J., Blakemore W.F., McDonald W.I. The restoration of conduction by central remyelination. Brain. 1981;104:383–404. doi: 10.1093/brain/104.2.383. [DOI] [PubMed] [Google Scholar]
- Stidworthy M.F., Genoud S., Suter U., Mantei N., Franklin R.J.M. Quantifying the early stages of remyelination following cuprizone‐induced demyelination. Brain Pathol. 2003;13:329–339. doi: 10.1111/j.1750-3639.2003.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch M.K., Stefferl A., Brehm U., Weissert R., Wallstrom E., Kerschensteiner M., Olsson T., Linington C., Lassmann H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998;8:681–694. doi: 10.1111/j.1750-3639.1998.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepavcevic V., Blakemore W.F. Glial grafting for demyelinating disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005;360:1775–1795. doi: 10.1098/rstb.2005.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourbah A., Linnington C., Bachelin C., Avellana‐Adalid V., Wekerle H., Baron‐Van Evercooren A. Inflammation promotes survival and migration of the CG4 oligodendrocyte progenitors transplanted in the spinal cord of both inflammatory and demyelinated EAE rats. J. Neurosci. Res. 1997;50:853–861. doi: 10.1002/(SICI)1097-4547(19971201)50:5<853::AID-JNR21>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Trapp B.D., Peterson J., Ransohoff R.M., Rudick R., Mork S., Bo L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Trebst C., Stangel M. Promotion of remyelination by immunoglobulins: Implications for the treatment of multiple sclerosis. Curr. Pharm. Des. 2006;12:241–249. doi: 10.2174/138161206775193118. [DOI] [PubMed] [Google Scholar]
- Triarhou L.C., Herndon R.M. The effect of dexamethasone on L‐alpha‐lysophosphatidyl choline (lysolecithin)‐induced demyelination of the rat spinal cord. Arch. Neurol. 1986;43:121–125. doi: 10.1001/archneur.1986.00520020015008. [DOI] [PubMed] [Google Scholar]
- Viehover A., Miller R.H., Park S.K., Fischbach G., Vartanian T. Neuregulin: An oligodendrocyte growth factor absent in active multiple sclerosis lesions. Dev. Neurosci. 2001;23:377–386. doi: 10.1159/000048721. [DOI] [PubMed] [Google Scholar]
- Warrington A.E., Asakura K., Bieber A.J., Ciric B., Van Keulen V., Kaveri S.V., Kyle R.A., Pease L.R., Rodriguez M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2000;97:6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman S.G. Ions, energy and axonal injury: Towards a molecular neurology of multiple sclerosis. Trends Mol. Med. 2006;12:192–195. doi: 10.1016/j.molmed.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Wilkins A., Chandran S., Compston A. A role for oligodendrocyte‐derived IGF‐1 in trophic support of cortical neurons. Glia. 2001;36:48–57. doi: 10.1002/glia.1094. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J. Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolswijk G. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain. 2002;125:338–349. doi: 10.1093/brain/awf031. [DOI] [PubMed] [Google Scholar]
- Woodruff R.H., Franklin R.J.M. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti‐galactocerebroside: A comparative study. Glia. 1999;25:216–228. doi: 10.1002/(sici)1098-1136(19990201)25:3<216::aid-glia2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Woodruff R.H., Fruttiger M., Richardson W.D., Franklin R.J.M. Platelet‐derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol. Cell. Neurosci. 2004;25:252–262. doi: 10.1016/j.mcn.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Ye P., Carson J., D'Ercole A.J. In vivo actions of insulin‐like growth factor‐I (IGF‐I) on brain myelination: Studies of IGF‐I and IGF binding protein‐1 (IGFBP‐1) transgenic mice. J. Neurosci. 1995;15:7344–7356. doi: 10.1523/JNEUROSCI.15-11-07344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Fancy S.P., Kotter M.R., Li W.W., Franklin R.J. Mechanisms of CNS remyelination—the key to therapeutic advances. J. Neurol. Sci. 2005;233:87–91. doi: 10.1016/j.jns.2005.03.008. [DOI] [PubMed] [Google Scholar]