

Abstract
Acute lung injury (ALI) is a leading cause of death in people infected with H5N1 avian influenza virus or the SARS-coronavirus. Imai et al. (2008) now report that ALI is triggered by the signaling of oxidized phospholipids through Toll-like receptor 4 (TLR4) and the adaptor protein TRIF. These findings provide insight into the molecular pathogenesis of ALI, a condition for which treatment options are currently very limited.
Main Text
Acute lung injury (ALI) affects more than 200,000 people in the US each year, with approximately 75,000 deaths, making it an important cause of morbidity, mortality, and health care expenditure (Rubenfeld et al., 2005). Bacterial and viral infections are important risk factors for ALI, but aspiration of gastric contents, major trauma, and repeated transfusions are additional risks. ALI is also a leading cause of death in people infected with H5N1 avian influenza virus or the coronavirus that causes SARS (severe acute respiratory syndrome). In this issue, Imai et al. (2008) report surprising insights from murine studies that provide a new perspective on the mechanisms contributing to ALI in humans.
The alveolar membrane of the lungs is the largest surface area of the body that is in continuous contact with the outside environment, and a complex set of defenses have evolved to protect it against inhaled particulates and microbes. The alveolar wall is a delicate structure, consisting of a thin alveolar epithelial layer, a basement membrane composed of collagens, glycoproteins, and glycosaminoglycans, and a thin endothelial cell layer. Surfactant phospholipids and associated proteins lining the alveolar surface are critical in reducing surface tension in alveolar fluid, so that alveoli do not collapse at low lung volumes. Cells called type II pneumocytes in the alveolar walls produce surfactant and actively transport sodium ions from the lumen to the interstitium, facilitating passive water movement from the alveoli to the interstitium and lymphatics in order to keep the airspaces dry. Acute damage to epithelial or endothelial cells in the alveolar membrane causes the clinical syndrome of ALI, in which the alveolar spaces fill with proteinaceous exudates, producing severe alterations in gas exchange, critical hypoxemia, and death in the absence of aggressive medical care. The hallmark findings of ALI include acute neutrophilic inflammation and an array of proinflammatory cytokines in the lungs, suggesting that activation of innate immunity is an initial event, whether or not overt infection is present. Activation of innate immune pathways combined with the physical stresses created by mechanical ventilation cause a synergistic increase in lung injury, but the mechanisms underlying ALI are not clear (Dos Santos and Slutsky, 2006).
In order to identify susceptibility factors for lung injury, Imai and colleagues screened several strains of mice using a simple model of lung injury, intratracheal instillation of 1.5 N hydrochloric acid (HCl), which approximates severe gastric acid aspiration. Surprisingly, mice with an inactivating mutation in Toll-like receptor 4 (TLR4) were protected from lung injury in this noninfectious model. TLR4 is the primary receptor for Gram-negative bacterial lipopolysaccharide (LPS) and also recognizes endogenous stimuli termed “alarmins” at sites of inflammation (Oppenheim et al., 2007). In macrophages, TLR4 signals via two different intracellular adaptor proteins, MyD88 and TRIF (TIR-domain-containing adaptor-inducing interferon-β), leading to two distinct intracellular signaling programs (Beutler, 2004). The MyD88 pathway causes rapid NF-κB activation and cytokine production. The TRIF pathway leads to the production of type I interferons via interferon regulatory factor 3 (IRF-3) and also causes delayed NF-κB activation via activation of TNF receptor-associated factor 6 (TRAF6) (Hoebe et al., 2003, Sato et al., 2003). Surprisingly, Imai and colleagues found that TRIF-deficient mice and mice lacking TRAF6 in myeloid cells were protected from HCl-induced injury, whereas MyD88 knockout mice were not, suggesting that the TRIF pathway, acting through TRAF6, is the major effector pathway in this noninfectious model. They also showed that TRIF-dependent lung injury is likely to be mediated by production of interleukin 6 (IL-6), as IL-6-deficient mice were also protected from injury.
The finding that the TLR4-TRIF pathway mediated injury in the absence of an infectious agent raised questions about the identity of the stimulus for TLR4, and the mechanism responsible for preferential activation of the TRIF pathway. The lung lavage fluid of HCl-treated mice contained oxidized phospholipids (OxPLs) detected by immunocytochemistry. An anti-OxPL antibody significantly reduced the proinflammatory activity of lung lavage fluid on lung macrophages in vitro. Intratracheal instillation of synthetically oxidized phospholipids caused lung inflammation in normal and surfactant-depleted mice, whereas mice lacking TLR4 were protected. The monoclonal antibody used to detect OxPL provided a clue to the specific OxPL responsible because it recognizes phospholipids containing oxidized phosphatidylcholine (e.g., 1-palmitoyl-2-arachidonoyl-phosphatidylcholine, OxPAPC). OxPAPC was shown to stimulate IL-6 production from lung macrophages via the TLR4-TRIF-TRAF6 pathway in vitro, independently of MyD88. These findings contrast sharply with signaling initiated by LPS, which occurs predominantly through TLR4-MyD88. In the complex inflammatory response initiated by HCl in the lungs, one might expect that TLR4 would be activated by several different endogenous stimuli; however, mice lacking TLR4, TRIF, or TRAF6 all resisted HCl- as well as OxPAPC-induced inflammation, supporting a role for OxPAPC as an important stimulus of TLR4 activation in this model.
Because patients infected with the influenza virus or SARS-coronavirus often develop severe lung injury, the authors looked for OxPL in the lungs of mice infected with an inactivated H5N1 avian influenza virus. As in the HCl injury model, immunohistochemical analysis identified OxPAPC in the lungs, but mice lacking TLR4 or TRIF had lung inflammation that was much less severe. Mice lacking the Ncf1 protein, which lack an active NADPH oxidase complex, were protected from viral lung inflammation and did not form OxPAPC in the airspaces, further supporting a key role for oxidation of phospholipids in the pathogenic pathway. High levels of OxPAPC were also detectable in the lungs of animals with experimental pulmonary infections due to Bacillus anthracis or Yersinia pestis, suggesting that OxPL-mediated lung injury is of more general significance. The relevance of this new mechanism for human lung injury was demonstrated by the observation that significant amounts of OxPAPC were present in lung tissue samples from two patients with lethal H5N1 avian influenza infection and nine patients with ALI following SARS-coronavirus infection.
These experimental results have surprising implications for understanding the pathogenesis of ALI. A central role of TLR4 in lung injury has been suspected because LPS is present in the lungs of many patients with ALI, whether or not overt bacterial infection is present (Martin et al., 1997). In addition, TLR4 is triggered by a number of different endogenous alarmins likely to be present in injured lungs (Oppenheim et al., 2007). Similarly, the formation of oxidized phospholipids in the lungs is not surprising given the intensely oxidative, neutrophil-rich environment in the lungs of patients with ALI (Sittipunt et al., 2001). However, the central role of TLR4 in acid-induced injury, the principal role of OxPL in triggering TLR4 signaling, the predominant role of the TRIF-TRAF6 signaling pathway ( Figure 1), and the general applicability of the findings to important respiratory viral infections are all unexpected. Paradoxically, the intravenous administration of OxPAPC protects mice from LPS-induced lung injury by protecting the endothelial barrier, suggesting that OxPAPC might have different effects in the alveolar and intravascular compartments (Nonas et al., 2006). These seemingly discrepant findings could be reconciled in part if systemic challenge with OxPAPC directly (via TLR4) or indirectly desensitizes the activation of circulating leukocytes.
Figure 1.
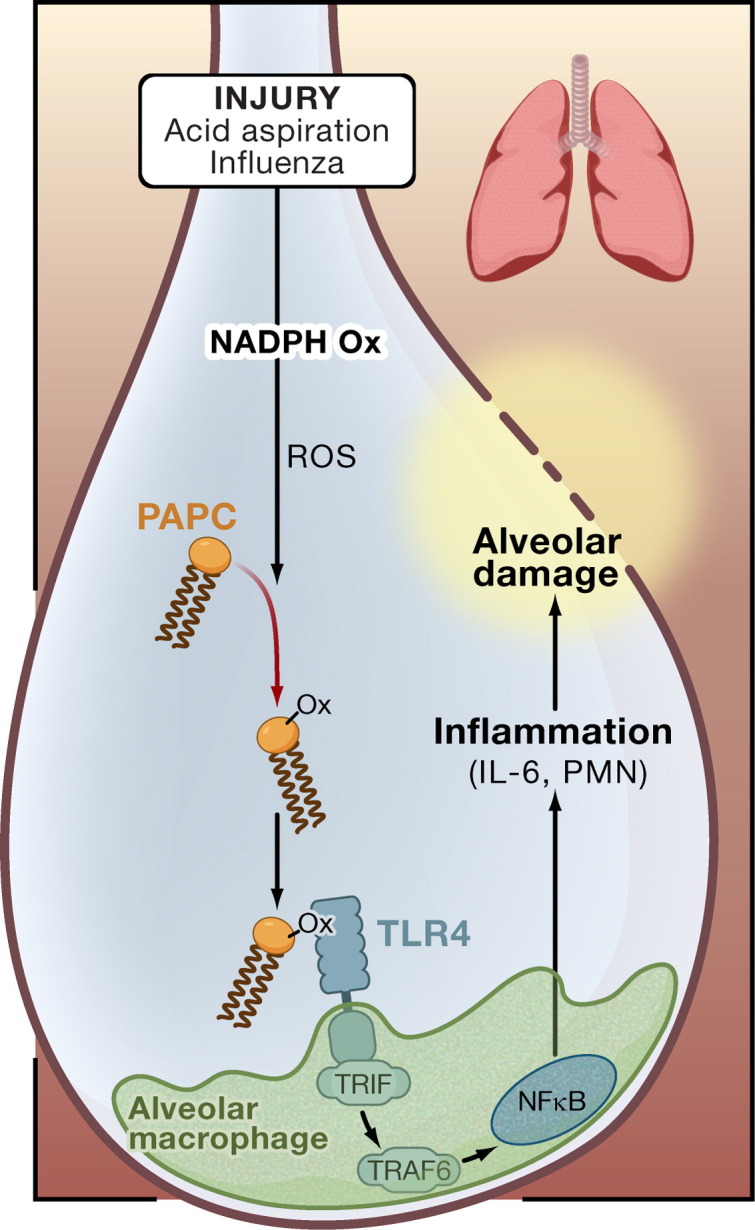
Oxidized Phospholipids and Acute Lung Injury
The work of Imai et al. (2008) provides evidence that acute lung injury involves oxidized phospholipids acting through Toll-like receptor 4 (TLR4). In this model, injury to the lungs through acid aspiration or viral infection leads to activation of NADPH oxidase (NADPH Ox) and production of reactive oxygen species (ROS), which oxidize 1-palmitoyl-2-arachidonoyl-phosphatidylcholine (PAPC, OxPAPC). OxPAPC activates TLR4 expressed by myeloid cells (an alveolar macrophage is shown), and the intracellular signal is transduced by the adaptor proteins TRIF and TRAF6, leading to interleukin 6 (IL-6) production, inflammation, and alveolar damage. PMN, polymorphonuclear leukocyte.
Given that the MyD88 pathway is critical to the host response to bacterial infections (Skerrett et al., 2007), the results of Imai and colleagues suggest that new strategies to modulate the TRIF-TRAF6 pathway, while leaving the MyD88 pathway largely intact, might be beneficial in some forms of ALI. Although the proximal event that creates the initial oxidative environment in the lungs remains unclear, neutrophil recruitment and activation are likely to be important because of the neutrophil's potent respiratory burst and because of the protection noted in Ncf1-deficient mice. Likewise, the key molecular “switch” that controls whether TRIF or MyD88 is activated by TLR4 remains a key unanswered question.
Almost 41 years after the clinical description of ALI, we have only one treatment that definitely improves survival, and this involves reducing the volume of air applied to the lungs during mechanical ventilation (Acute Respiratory Distress Syndrome Network, 2000). The work of Imai and colleagues points to potential molecular approaches that could further improve outcomes for this clinically important syndrome.
Acknowledgments
The authors are supported by the Medical Research Service of the Department of Veterans Affairs and by Grants HL081764, HL073996 (T.R.M.) and HL629063 (M.M.W.) from the NIH.
References
- Acute Respiratory Distress Syndrome Network N. Engl. J. Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- Beutler B. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Dos Santos C.C., Slutsky A.S. Annu. Rev. Physiol. 2006;68:585–618. doi: 10.1146/annurev.physiol.68.072304.113443. [DOI] [PubMed] [Google Scholar]
- Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S.O., Goode J., Lin P., Mann N., Mudd S., et al. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Imai Y., Slutsky A.S., Penninger J.M. Cell. 2008 this issue. [Google Scholar]
- Martin T.R., Rubenfeld G.D., Ruzinski J.T., Goodman R.B., Steinberg K.P., Leturcq D.J., Moriarty A.M., Raghu G., Baughman R.P., Hudson L.D. Am. J. Respir. Crit. Care Med. 1997;155:937–944. doi: 10.1164/ajrccm.155.3.9117029. [DOI] [PubMed] [Google Scholar]
- Nonas S., Miller I., Kawkitinarong K., Chatchavalvanich S., Gorshkova I., Bochkov V.N., Leitinger N., Natarajan V., Garcia J.G., Birukov K.G. Am. J. Respir. Crit. Care Med. 2006;173:1130–1138. doi: 10.1164/rccm.200511-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim J.J., Tewary P., de la Rosa G., Yang D. Adv. Exp. Med. Biol. 2007;601:185–194. doi: 10.1007/978-0-387-72005-0_19. [DOI] [PubMed] [Google Scholar]
- Rubenfeld G.D., Caldwell E., Peabody E., Weaver J., Martin D.P., Neff M., Stern E.J., Hudson L.D. N. Engl. J. Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- Sato S., Sugiyama M., Yamamoto M., Watanabe Y., Kawai T., Takeda K., Akira S. J. Immunol. 2003;171:4304–4310. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- Sittipunt C., Steinberg K.P., Ruzinski J.T., Myles C., Zhu S., Goodman R.B., Hudson L.D., Matalon S., Martin T.R. Am. J. Respir. Crit. Care Med. 2001;163:503–510. doi: 10.1164/ajrccm.163.2.2004187. [DOI] [PubMed] [Google Scholar]
- Skerrett S.J., Wilson C.B., Liggitt H.D., Hajjar A.M. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L312–L322. doi: 10.1152/ajplung.00250.2006. [DOI] [PubMed] [Google Scholar]