

Abstract
Background: Respiratory viral infection is known clinically to promote sensitization to antigen inhalation and the development of asthma. Objective: The purpose of this investigation was to determine whether influenza type A virus infection enhances inhalation sensitization and increases airway responsiveness in mice. Methods: Mice were infected by intranasal inoculation with influenza A viruses (strains: H1 N1 and H3 N2 ) or PBS. Animals were exposed to aerosols of ovalbumin on day 3. Two weeks after ovalbumin sensitization, mice were challenged with ovalbumin aerosols; 24 hours later, airway responsiveness (AR) to inhaled methacholine, levels of ovalbumin-specific IgE, and bronchoalveolar lavage fluid (BALF) were examined. Results: Neither influenza A virus (H1 N1 nor H3 N2 ) alone nor ovalbumin sensitization alone caused changes in AR or IgE. However, ovalbumin sensitization after inoculation with either influenza A virus increased AR and levels of ovalbumin-specific IgE. On BALF-cell analysis, ovalbumin sensitization after inoculation with influenza virus A increased the number of lymphocytes but not the number of eosinophils. No difference in AR or IgE levels was observed between the 2 strains of influenza A viruses. Immmunostaining of BALF cells showed an increase in T cells, especially CD8+ cells, with ovalbumin sensitization after inoculation with influenza virus A. Conclusion: Infection by influenza A virus enhances sensitization to inhaled antigens and airway responsiveness in mice by means of mechanisms including CD8+ cells and antigen-specific IgE. (J Allergy Clin Immunol 1998;102:732-40.)
Keywords: Virus infection, influenza A virus, IgE, airway hyperresponsiveness, CD8+cells, CD4+cells
Abbreviations: BAL: , Bronchoalveolar lavage; BALF: , Bronchoalveolar lavage fluid; FRC: , Functional residual capacity; H1 N1 : , Influenza virus A/PR/8/34; H3 N2 : , Influenza virus A/Guizhou-X; PD35 Mch: , Cumulative dose of inhaled methacholine at which airway resistance increases by 35%; PFU: , Plaque-forming unit; sRaw: , Specific airway resistance; sRawL : , sRaw of the lower airway
Clinically, asthma is exacerbated by respiratory viral infections. In older children and adults, rhinovirus, coronavirus, and influenza A virus infections cause wheezing.1, 2, 3 Airway hyperresponsiveness develops in subjects who have naturally acquired colds or who were infected with respiratory viruses transiently.4 Acute respiratory infections have also been implicated in the pathogenesis of asthma. It is known that children with respiratory virus infection later experience wheezing, airway hyperresponsiveness, and asthma.5, 6 From epidemiologic studies of respiratory infection7 and asthma and allergic rhinitis8 in the same area, Smith9 found that viral lower-respiratory infection in early life triggers the onset of asthma.
Although it is still unknown how viral infection induces asthma, respiratory viral infections may contribute to the development of IgE antibodies to environmental antigens. Influenza causes exfoliation of epithelial cells and increases permeability of the respiratory mucosal barrier to antigen penetration with the enhanced likelihood of IgE antibody sensitization. Respiratory virus infection has been proposed as a common trigger factor in the development of allergy in genetically susceptible children.10 Several studies of sensitization to allergens by inhalation alone are reported, and repeated exposure to allergen at a high concentration is needed.11 Sakamoto et al12 have shown that IgE antibody production in response to an inhaled antigen occurs in mice that have been infected with influenza A virus, but not in animals with no infection. Thus we hypothesized that a respiratory viral infection promotes sensitization to inhaled antigen and may cause asthma. To test this hypothesis, mice were infected with influenza type A virus; during the acute phase, animals were exposed to aerosols of ovalbumin. Two weeks after ovalbumin sensitization, mice were challenged with ovalbumin aerosols; 24 hours after challenge, airway responsiveness to methacholine, levels of ovalbumin-specific IgE, and bronchoalveolar lavage fluid were examined. We used 2 strains of influenza A viruses (A/PR/8/34 [H1 N1 ] and A/Guizhou-X [H3 N2 ]) to examine any difference in the effect of the strain of influenza A viruses on allergic sensitization.
METHODS
Animals
Six- to 11-week-old male BALB/c mice (Japan SLC, Shizuoka, Japan) were used. The animals were maintained on ovalbumin-free diets and kept under special pathogen-free conditions in a laminar flow container. Each group of animals was isolated. All experimental animals used in this study were maintained under a protocol approved by the Institutional Animal Care and Use Committee of the Yokohama City University School of Medicine.
Allergen
Ovalbumin (Sigma Chemical, St. Louis, Mo) was dissolved in PBS to a concentration of 0.1%. Ten milliliters of alum adjuvant (10 mg/mL) was added to 10 mL of 0.1% ovalbumin; the final concentration of ovalbumin was 0.05%. Aerosolized ovalbumin was used for sensitization and challenge.
Sensitization and challenge
Mice were sensitized by inhalation of aerosolized 0.05% ovalbumin or PBS with an ultrasonic nebulizer (NE-U1013; Omron, Tokyo, Japan). Aerosols of ovalbumin or PBS were exposed to animals in a plexiglass chamber (capacity, 23.5 L) for 30 minutes each day over a 5-day period. The inhalation challenge was an aerosol of 2% ovalbumin or PBS from the ultrasonic nebulizer for 30 minutes.
Viruses
Two mouse-adapted strains of influenza A, A/PR/8/34 (H1 N1 ) and A/Guizhou-X (H3 N2 ) (provided by Drs M. Tashiro and S. Tamura, Japan National Institute of Health, respectively), were used in the present study. The viruses were incubated in Madin-Darby canine kidney cell monolayers for 2 days, and then viruses were harvested. The virus titer was quantitated by plaque assay as described previously.13 The titer of the pool used in the present study was 1.5 × 106 plaque-forming units (PFU) in H1 N1 and 4.1 × 106 PFU in H3 N2 .
Animals were anesthetized with diethylene ether and inoculated intranasally with 50 L of the diluted virus solution. The virus stock solutions were diluted 10-fold from 10–1 to 10–6 with PBS.
Airway responsiveness
Nonspecific airway responsiveness was measured by inhalation of methacholine aerosols in unanesthetized, spontaneously breathing animals. Specific airway resistance (sRaw) was measured in a double plethysmograph apparatus consisting of a head chamber and chest chamber for small animals (Buxco, Troy, NY), according to the methods of Pennock et al.14 Briefly, pressure signals from the head and chest chambers represent nasal flow and thoracic flow, respectively. These 2 signals were displayed on an oscilloscope and used to calculate total respiratory duration (TT ) and the time lag of nasal flow (δt). sRaw was calculated according to the following formula:
sRaw = (1/ω) × PB × 1.36 × tanθ
where, ω is angular speed (= 2π × [1/TT ]), PB is atmospheric pressure in millimeters of mercury, and θ is a phase angle (= 2π × [δt/TT ]). The head and body chambers were warmed to 37°C before the measurement, and the air in the head chamber was saturated with water vapor by a pool of heated water at 37°C inside the box. The neck of the animal was placed through the bore of the rubber plate separating the 2 chambers and sealed tightly with clay to avoid air leak between the 2 chambers. The measurement of sRaw was performed only when the animals were quiet.
To examine whether sRaw by the double plethysmograph represents changes in airway resistance of the lower airway, we anesthetized animals with intraperitoneal injection of pentobarbital sodium (1 mg/animal) and inserted 7-mm long sterile plastic tube into the trachea. The tube was connected to the head chamber of the body plethysmograph, so that we were able to measure sRaw of the lower airway (sRawL ). Both sRaw and sRawL were measured after aerosol inhalation of either PBS or methacholine 12.5 mg/mL. We compared sRaw and sRawL in 5 nonsensitized and noninfected animals.
The concentration of methacholine was adjusted from 12.5 mg/mL to 49 μg/mL by serial 4-fold dilutions in PBS. Aerosols from a DeVilbiss 646 nebulizer (De Vilbiss, Somerset, Pa) were delivered to the animals by mask, which was put over the face; the animals were placed in the chest chamber, and the head chamber was disconnected. The animals were exposed to aerosols for 1 minute at each concentration; then the head chamber was connected, and sRaw was measured. Inhalation of methacholine was started from the concentration of 49 g/mL, and the concentration was increased until sRaw increases 35% or more of the baseline value. From the methacholine-sRaw curve, we calculated the cumulative dose (sum of all inhaled doses [PD35 Mch]) at which sRaw increases 35% from the baseline value after inhalation of PBS aerosols. One unit was defined as the dose inhaled at 1 mg/mL methacholine for 1 minute. When an animal’s sRaw did not increase more than 35% to a concentration of 12.5 mg/mL, all 5 doses from 0.049 to 12.5 units were summed and PD35 Mch was calculated to be 16.7 units.
Bronchoalveolar lavage (BAL)
Immediately after the assessment of airway responsiveness, the animals were anesthetized with pentobarbital sodium (40 mg/kg). PBS (0.6 mL) was instilled through the tracheal cannula and recovered by gentle manual aspiration. This lavage was repeated 4 times. The recovered fluid was immediately centrifuged (100g for 10 minutes at 4°C). The cell pellet was washed twice and resuspended in 1 mL of PBS. Cells were fixed to a slide by a cytocentrifuge at 100g (Cytobucket SC-2; Tomy Seiki, Tokyo, Japan) and stained with Diff-Quick (International Reagent, Kobe, Japan). Differential cell counts were performed by classification according to standard morphologic criteria of at least 200 cells.
Lymphocytes in BAL fluid (BALF) were analyzed for the total number of lymphocytes and for expression of lymphoid surface markers with anti-mouse monoclonal antibodies specific for CD4, CD8, and CD90 (PharMingen, San Diego, Calif), as previously described.15 Cells fixed to a slide were air dried, immersed in acetone solution cooled to –20°C for 10 minutes, and then stored at –80°C. The cells on the slide were incubated for 30 minutes in 0.3% H2 O2 in methanol and washed in distilled water and PBS for 5 minutes each. Incubation with primary antiserum diluted in PBS was done for 30 minutes. After being washed with PBS for 5 minutes, the cells on the slide were incubated with diluted biotinylated secondary antibody solution for 30 minutes. Then cells were treated with an avidin-peroxidase complex (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, Calif). Peroxidase activity was demonstrated by incubation in diaminobenzidine. We counted cells positive for CD90, CD4, and CD8.
IgE measurement
Before and at the end of the experiment, blood was drawn from the postorbital vein or the heart of each animal to measure ovalbumin-specific IgE, respectively. Ovalbumin-specific IgE antibody levels were measured by ELISA as follows16 : ELISA plates (96-well Immunolon II; Dynatech Laboratories, Alexandria, Va) were coated with 10 g/mL of ovalbumin. The plates were then blocked with PBS–1% BSA. Dilutions 1:10 of samples were incubated on these plates for 3 hours. Plates were washed and then overlaid with 1 g/mL of alkaline phosphatase-conjugated polyclonal anti-mouse IgE (PharMingen) for 3 hours and then washed. Plates were read in a microplate autoreader (Bio-Rad Laboratories, Hercules, Calif). The concentration of specific antibody bound to the plates was determined by comparison with a standard curve generated using high-titer antisera to ovalbumin.
Histology
The lungs were fixed by intratracheal instillation of buffered 20% formaldehyde solution. Lung sections were embedded in paraffin, sectioned, and stained with hematoxylin and eosin.
Protocol
Dose effects of viruses
To determine the dose of the 2 viruses (H1 N1 and H3 N2 ) required to cause moderate inflammation without apparent pneumonia, we inoculated intranasally with 50 L of the diluted virus solutions (from 10–1 to 10–6 ). Infection was verified by observing the pulmonary histology at 3, 7, or 14 days after inoculation. General locomotor activity, body weight, and survival rate were examined for 14 days. Five to 6 animals were used in each group.
Effect of influenza A viruses infection on sensitization of ovalbumin
To examine the effects of influenza A virus infection on inhalation sensitization by ovalbumin, we divided the animals into 6 groups (5 animals in each group): group 1, PBS inoculation, PBS sensitization, and PBS challenge; group 2, PBS inoculation, ovalbumin sensitization, and ovalbumin challenge; group 3, H1 N1 inoculation, PBS sensitization, and PBS challenge; group 4, H3 N2 inoculation, PBS sensitization, and PBS challenge; group 5, H1 N1 inoculation, ovalbumin sensitization, and ovalbumin challenge; and group 6, H3 N2 inoculation, ovalbumin sensitization, and ovalbumin challenge. From the dose effect study, sublethal dosages were determined: 1.5 × 102 PFU/mL and 4.1 × 103 PFU/mL for H1 N1 and H3 N2 , respectively. At first, we measured PD35 Mch and drew blood from the postorbital vein to determine IgE. On day 1, inoculation by virus or PBS was performed intranasally. On days 4 to 8, animals were sensitized with ovalbumin or PBS for 30 minutes daily. On day 22, the mice were challenged with aerosols of 2% ovalbumin or PBS for 5 minutes. Twenty-four hours after challenge, airway responsiveness, levels of ovalbumin-specific IgE, and BALF were analyzed, and the lung was removed and fixed.
Statistical analysis
All values are expressed as the mean ± SEM. Statistical comparison between groups was examined by 2-way analysis of variance with repeated measures (ANOVA), followed by a post-hoc comparison with the Newman-Keuls test. Two mean values were compared with the Wilcoxon matched pairs test. A P value less than .05 was considered significant.
RESULTS
Virus doses and acute phase responses
The body weight of animals inoculated with H1 N1 at dilutions of 10–1 to 10–3 decreased with time; however, with dilutions of 10–4 and 10–6 , weight increased (Fig 1).
Fig. 1.
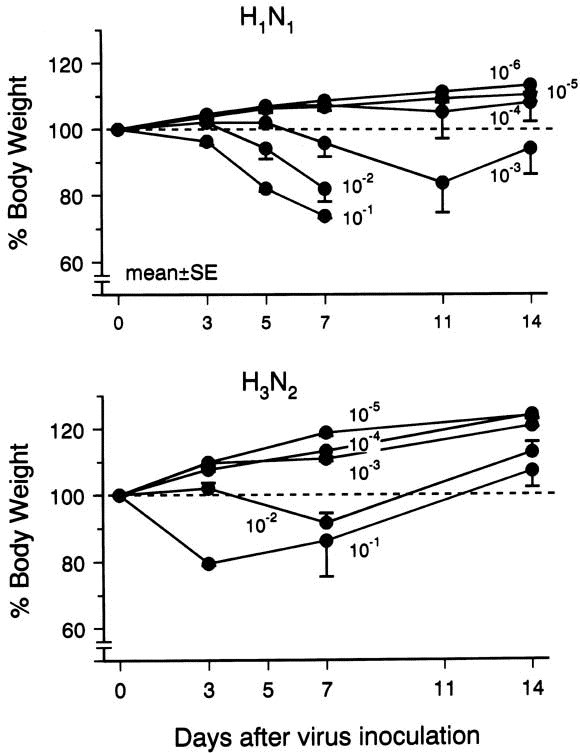
Body weight changes of mice are expressed as a percentage of the baseline value. Numbers represent dilution factors.
In the H3 N2 strain, body weight decreased with time at dilutions 10–1 and 10–2 ; but at 10–3 to 10-5 dilutions, the weight increased.
After being inoculated with H1 N1 virus at dilutions of 10–1 and 10–2 , all animals died from day 7 to day 14, and the histologic evidence showed apparent pneumonia. At a 10–3 dilution of H1 N1 virus, no animal died, and some animals had pneumonia. Animals inoculated with 10–4 dilution of H1 N1 virus showed mononuclear-cell infiltration in both the submucosal layer and perivascular area and loss of the epithelium of the airway on day 7 (Fig 2, B ); and little inflammation was observed on day 14. At dilutions of 10–5 and 10–6 , no apparent inflammation was observed. No animal died at any dilutions of H3 N2 virus. At dilutions of 10–1 and 10–2 of H3 N2 virus, mononuclear cell infiltration was significant, and some animals showed pneumonia; but at a dilution of 10–3 , loss of airway epithelial cells and mononuclear cell infiltration without pneumonia were observed (Fig 2, C ). Animals inoculated with a 10–4 dilution of H3 N2 virus showed little mononuclear cell infiltration.
Fig. 2.
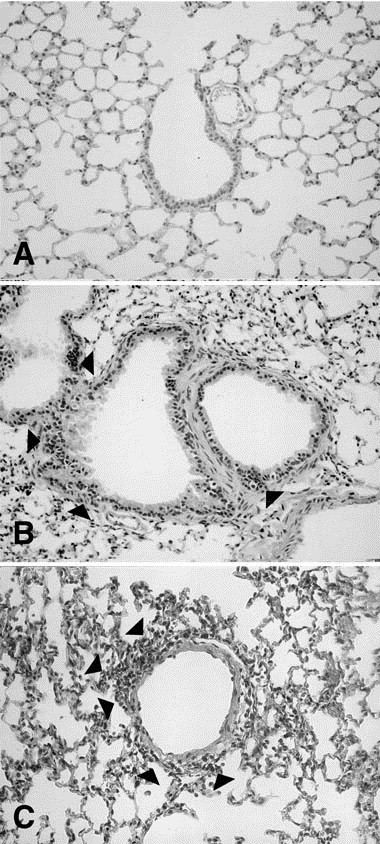
Microphotographs of the lung tissue. A, Inoculation with PBS (control animal). B, H1 N1 infection at a 10–4 dilution (1.5 × 102 PFU/mL) on day 7; mononuclear cell infiltration (arrowheads) in submucosal layer and loss of the epithelium of the airway were seen. C, H3 N2 infection at a 10–3 dilution (4.1 × 103 PFU/mL) on day 7; loss of epithelial cells of the airway and mononuclear cell infiltration (arrowheads) were diffusely observed. (Original magnification ×100.)
For the sensitization study, we chose the dilutions of H1 N1 and H3 N2 to be 10–4 and 10–3 (ie, 1.5 × 102 and 4.1 × 103 PFU/mL, respectively. No inflammatory change was observed in the control (Fig 2, A).
Airway resistance
A comparison of sRaw and sRawL for individual mice indicates a close correlation (r = 0.87, P < .01), although the absolute values of sRawL tended to be lower than those of sRaw at high level of airway resistance (Fig 3). This means that sRaw reflects approximately the response of the lower airway resistance.
Airway responsiveness
No difference in the baseline sRaw before inhalation of methacholine was observed either before inoculation or 24 hours after challenge among all groups. The threshold value of inhaled methacholine, PD35 Mch, measured 24 hours after challenge decreased in animals sensitized with ovalbumin after inoculation with either H1 N1 (group 5) or H3 N2 (group 6) compared with the respective baseline value (P < .01 for both; Fig 4).
Fig. 4.
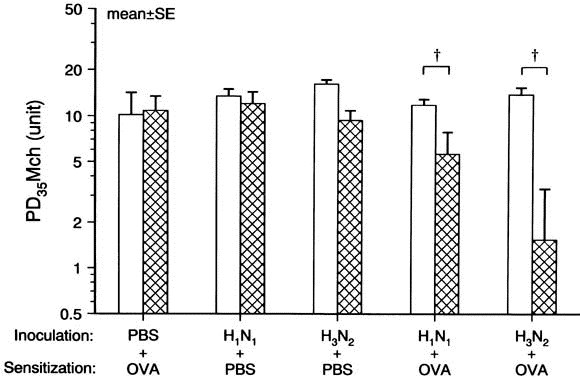
Airway responsiveness to inhaled methacholine (PD35Mch) . Influenza A virus inoculation with ovalbumin (OVA) sensitization increased PD35 Mch (P < .01 for H1 N1 and H3 N2 ); however, virus infection alone did not change it. Open and crosshatched columns represents the baseline and postchallenge values, respectively. P < .01 compared with the baseline value.
The decrease in PD35 Mch did not differ between H1 N1 and H3 N2 groups. However, neither ovalbumin sensitization alone (group 2) nor virus infection alone (groups 3 and 4) changed airway responsiveness.
IgE Levels
Levels of ovalbumin-specific IgE were increased 10-fold (from 9.5 ± 5.3 to 93 ± 43 arbitrary unit) on ovalbumin sensitization after inoculation with H1 N1 (group 5; P < .01), and 6-fold on ovalbumin sensitization after inoculation with H3 N2 compared with the baseline value (group 6; P < .01; Fig 5).
Fig. 5.
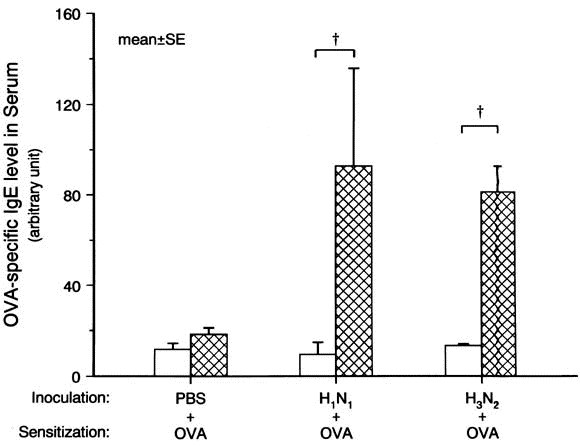
Changes in ovalbumin (OVA) -specific IgE level in serum. Influenza A–virus inoculation with ovalbumin sensitization increased ovalbumin-specific IgE level in serum (P < .01 for both H1 N1 and H3 N2 ). Open and crosshatched columns represent the baseline and postchallenge values, respectively. †P < .01 compared with the baseline value.
However, no changes in the level of ovalbumin-specific IgE occurred in animals inoculated with PBS and ovalbumin challenge (group 2).
Analysis of BALF
Total BALF cell number after ovalbumin sensitization and H3 N2 inoculation (group 6) was higher than in any other group; however, in group 5 there was no increase (Table I).
Table I.
Total cell count in bronchoalveolar lavage fluid (mean ± SEM)
| Group | Inoculation | Sensitization | Challenge | Total no. of cells (×104 )* |
|---|---|---|---|---|
| Group 1 | PBS | PBS | PBS | 5.7 ± 0.4 |
| Group 2 | PBS | OVA | OVA | 8.8 ± 1.0 |
| Group 3 | H1 N1 | PBS | PBS | 5.8 ± 1.0 |
| Group 4 | H3 N2 | PBS | PBS | 5.9 ± 0.8 |
| Group 5 | H1 N1 | OVA | OVA | 6.2 ± 0.9 |
| Group 6 | H3 N2 | OVA | OVA | 10.0 ± 0.9 ‡ |
| *Total cell number was significantly different among groups (P < .01 by ANOVA). †P < .05 compared with group 5. ‡P < .01 compared with group 1. | ||||
OVA , ovalbumin.
The percent alveolar macrophages decreased in a group of H3 N2 virus infection alone (P < .01) and in 2 groups of ovalbumin sensitization with virus infection (P < .01 for both groups; Fig 6).
Fig. 6.
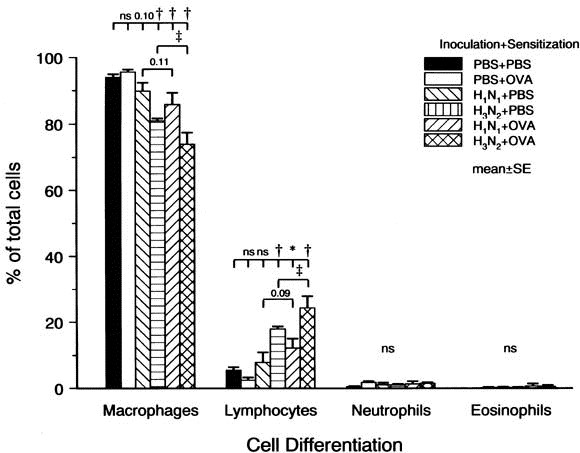
Cell differentiation in BALF. Sensitization after infection with influenza A viruses decreased alveolar macrophages and increased lymphocytes. Numbers represent P values. *P < .05. †P < .01 compared with the PBS groups. ‡P < .01 compared with H3 N2 alone.
Lymphocytes in BALF increased in a group of H3 N2 virus infection alone (P < .01) and 2 groups of ovalbumin sensitization with virus infection (groups 5 and 6) compared with a group of PBS inoculation with ovalbumin sensitization (P < .05 for group 5 and P < .01 for group 6) or a group of PBS inoculation with PBS sensitization (P < .01 for all). The increase in lymphocytes was greater in H3 N2 with ovalbumin sensitization (group 6) than in H3 N2 alone (group 4; P < .05). However, H1 N1 virus infection alone did not increase the percent lymphocytes. No change was observed in either percent neutrophils or percent eosinophils.
To study the T-cell population in BALF, BALF cells in H3 N2 -inoculated groups were immunostained for CD90, CD4, and CD8. Total T cells in H3 N2 inoculation alone (group 4) and in H3 N2 with ovalbumin sensitization (group 6) were 7.3% ± 2.2% and 7.9% ± 0.7%, respectively, and significantly higher than in the 2 PBS inoculation groups (groups 1 and 2; P < .01 for both; Fig 7).
Fig. 7.
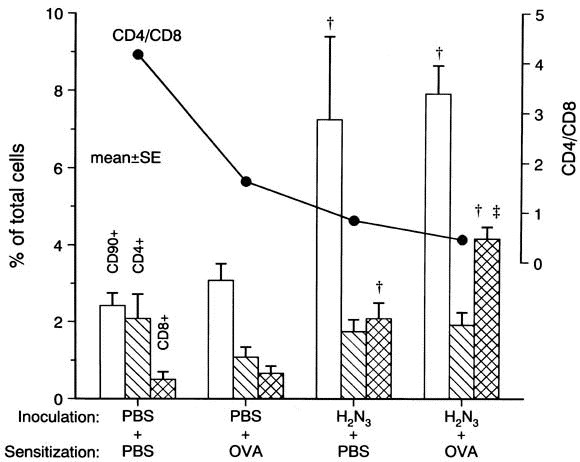
T cells in BALF. CD90+ cells and CD8+ cells were higher in H3 N2 inoculation groups than in PBS inoculation groups (P < .01 for both). CD8+ cells increased in virus inoculation with ovalbumin more than with PBS (P < .01). The ratio of CD4/CD8 decreased with inoculation by H3 N2 . Open , hatched , and crosshatched columns represent CD90+ , CD4+ , and CD8+ cells, respectively. †P < .01 compared with both PBS inoculation groups. ‡P < .01 compared with H3 N2 to PBS sensitization.
The number of CD4+ T cells did not change on virus infection and/or ovalbumin sensitization. However, virus infection with either PBS or ovalbumin sensitization increased CD8+ T cells in BALF (P < .01 for both), and the increase in ovalbumin sensitization with virus infection was significantly greater than that with PBS sensitization (P < .01). The CD4/CD8 ratio of BAL cells decreased from 4.17 to 0.84 in virus infection alone and to 0.46 in virus infection with ovalbumin sensitization.
Histologic examination
Histologic examination of the lungs was performed 24 hours after ovalbumin or PBS challenge (on day 23). In the animals inoculated with either H1 N1 or H3 N2 virus followed by PBS sensitization, no inflammatory cell infiltration was found (Fig 8, A ). However, in the virus-inoculated animals with ovalbumin sensitization (groups 5 and 6) mononuclear-cell infiltration was observed around bronchioli and blood vessels (Figs 8, B and C ). No difference in inflammation was observed between groups 5 and 6. Eosinophil infiltration was not detected in any group.
Fig. 8.
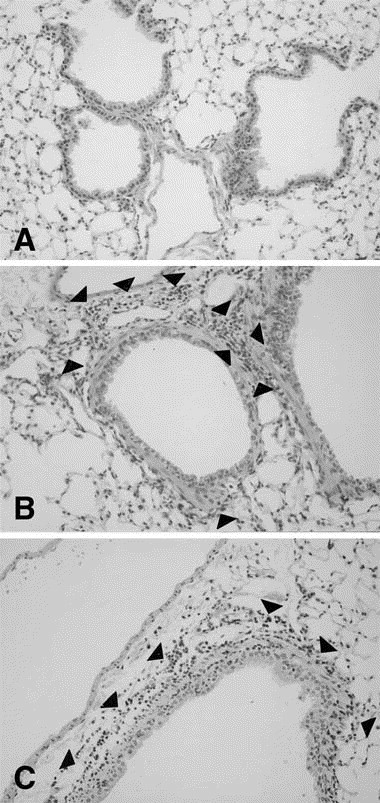
Microphotographs after ovalbumin (OVA) challenge. A, Inoculation with PBS and ovalbumin sensitization; no inflammatory cell was observed. B, Inoculation with H1 N1 and ovalbumin sensitization. C, Inoculation with H3 N2 and ovalbumin sensitization. Infiltration of mononuclear cells (arrowheads) were observed around bronchioli and blood vessels in both B and C. (Original magnification ×100.)
DISCUSSION
We have demonstrated that infection with influenza A viruses enhances inhalation sensitization to ovalbumin and airway responsiveness to methacholine in mice. No difference in airway responsiveness, ovalbumin-specific IgE, or cells in BALF was observed between the 2 strains of influenza A virus (H1 N1 and H3 N2 ). This is the first report that in mice, influenza A virus infection boosts sensitization by inhalation of a low-concentration antigen and increases airway responsiveness. Although in animal asthma models, subcutaneous or intraperitoneal injection is a common route of antigen immunization,17, 18 our inhalation sensitization technique may reflect more appropriately the actual route of sensitization in the development of human asthma. Our study suggests that viral infection is important for triggering sensitization to aeroallergen and the onset of asthma.
Influenza A virus infection
Influenza A viruses are the most important of the respiratory viruses with respect to morbidity and mortality rates in the world. We used 2 strains of influenza A virus, H1 N1 and H3 N2 , to examine differences in virus strain on the effects of allergen sensitization. In the present study, the highest dose of influenza viruses caused pneumonia and all animals died. With moderate doses of the viruses used for the allergen sensitization study, animals survived without pneumonia and showed infiltration of mononuclear cells into the airway wall and perivascular area 3 days after inoculation. Although in the present study, the virulence of viruses was stronger in strain H1 N1 than H3 N2 , the doses of the 2 strains used for sensitization caused comparable changes in histologic condition. These changes were similar to previous studies.19, 20 Sensitization by ovalbumin was performed during the acute phase of infection, 4 to 8 days after virus inoculation. Thus virus infection damages the airway epithelium so that changes in epithelial permeability may facilitate the access of allergens from the lumen to the airway wall.
Cells of BALF
Inflammation of the lung tissue in both influenza A viruses reached a maximum at 7 days and almost recovered at 14 days after virus inoculation. BAL was performed 22 days after virus inoculation, and the total cell number in groups with virus infection alone did not differ from that of groups inoculated with PBS. The increased cell number in the lung during the acute phase of influenza virus infection declined to normal values between 2 and 4 weeks after inoculation.21 The percent lymphocyte was increased in animals with virus infection. Circulating lymphocytes are reported to decrease during the acute phase of infection, and this decrease persists for 4 weeks after virus inoculation.22 Lymphocytes may accumulate in the lung during virus infection, in association with a decrease in the circulation. In an analysis of lymphocyte subsets, the T-cell population was found to increase with virus infection alone (group 4). In T-cell subsets in virus infection alone (group 4), CD8+ T cells increased but not CD4+ T cells, although no change in either CD4+ or CD8+ T cells was observed in the groups inoculated with PBS (groups 1 and 2). The CD4/CD8 ratio of BALF lymphocytes decreased in influenza virus infection, primarily because of an increase in CD8+ T cells. These changes in T-cell subsets were similar to previous studies of virus infection.21, 22, 23
During convalescence from respiratory syncytial virus infection, the circulating CD8+ T-cell number was lower in patients with wheezing than in those without wheezing; however, no difference in CD4+ T cells was seen.24 This suggests that CD8+ T cells may accumulate in the lung of patients who wheeze. Allergen sensitization has been considered to be controlled by CD4+ T cells. In animal models of inhalation sensitization with ovalbumin, T-cell subsets in the spleen are reported to change; CD8+ T cells increase and the CD4/CD8 ratio decreases after sensitization.25 Induction of a TH2 immune response to ovalbumin switches the virus-infected CD8+ T-cell response to IL-5 production.26 Thus in allergen sensitization after virus infection, CD8+ T cells increase in the lung. It is likely that allergen sensitization after virus infection is controlled directly by CD8+ T cells or by cytokines produced by these CD8+ T cells.
Eosinophils in groups of ovalbumin sensitization with virus infection were not increased in the lung tissue or BALF. In a BALB/c mouse model of sensitization by repeated exposure to ovalbumin, eosinophils were not observed in the airway, whereas the IgE level was increased.16 Sensitization by intraperitoneal injection alone in mice did not increase eosinophils in either lung tissue or BALF, and eosinophil peroxidase activity did not change after ovalbumin challenge, although increased airway responsiveness was observed.27 Thus, in terms of eosinophils, our results compare with other studies of mouse models of asthma.16, 27 In BALB/c mice, allergen inhalation seldom induces eosinophilia.
IgE Production
IgE production is reported to increase in mouse models of asthma.11, 16, 27 Our sensitization with 0.05% ovalbumin inhalation alone did not cause a significant production of IgE. Inhalation of 0.05% ovalbumin with influenza infection produced a high level of ovalbumin-specific IgE but no difference in IgE production between H1 N1 and H3 N2 strains. Sakamoto et al12 showed that 0.05% ovalbumin inhalation after influenza A virus infection generated ovalbumin-specific IgE measured by PCA. Therefore influenza A infection can amplify IgE production in exposure to aerosols of a low-concentration allergen, which alone cannot produce IgE. In a prospective study,10 allergic symptoms and immunologic evidence (such as IgE RAST for specific allergens) developed in children with 2 parents with allergies. Upper respiratory infection occurred in 10 of the 11 children 1 to 2 months before the onset of allergic sensitization. Why does virus infection enhance production of specific IgE by allergen inhalation? Respiratory syncytial virus–infected mice developed high ovalbumin-specific IgE in response to intranasal sensitization of ovalbumin; however, uninfected animals did not, suggesting that respiratory virus infection may change the immune response to antigens.28 In rats from which alveolar macrophages were eliminated, IgE production increased in response to inhaled allergen.29 Resident alveolar macrophages are reported to downregulate antigen that had cell functions of pulmonary dendritic cells.30 Our BALF analysis showed that infection with influenza A viruses increased lymphocytes and decreased the number of alveolar macrophages. It is likely that virus infection enhances antigen cell function, resulting in an increase in IgE production. Further, CD8+ T cells of influenza virus–infected mice can coexpress different combinations of TH1 and TH2 cytokines, IL-5, and IFN-γ.21 It is also reported that virus-specific CD8+ cells in the lung switch to IL-5 production.26 IL-5 upregulates IgE synthesis.31 Further, CD3– cells in the lung of influenza virus-infected mice express mRNA for IL-4, IL-6, and granulocyte-macrophage colony–stimulating factor.21 Thus influenza virus infection may cause a cell-population change in lung tissue, a functional change of T cells, and cytokine production that is not restricted to the T-cell population, resulting in an increase of IgE production.
Effects on airway responsiveness
In mice, measurements of airway function are now feasible, and we measured sRaw in unanesthetized animals because anesthesia affects the airway response.32 However, it has been questioned that in the airway resistance measurement that uses whole body plethysmography while the animals breathed through the nose, sRaw is dominated by the upper airway resistance.33 Recently, Hamelmann et al34 demonstrated that an index of airway resistance (Penh) by body plethysmography which includes both the upper and lower airway is a valid indicator of bronchoconstriction in inhalation challenge through the nose. We confirmed that sRaw reflects the change in the lower airway resistance (Fig 3). However, our absolute values of airway resistance calculated by assuming a functional residual capacity (FRC) of 1 mL are approximately 6 cm H2 O/mL/s in the baseline and higher than the previously reported values,34, 35 in which resistance was measured during mechanical ventilation after anesthesia was induced. Their methods of resistance measurement were different from ours. Their ventilation frequency and the magnitude of tidal ventilation were determined by each setting of a ventilator, and FRC was adjusted by controlling the end-expiratory pressure. Our mice breathed air spontaneously. Thus differences in method of resistance measurement, FRC, ventilation frequency, and tidal volume may affect the absolute values of resistance. Further studies may be needed for determination of precise values of airway resistance in mice.
Fig. 3.
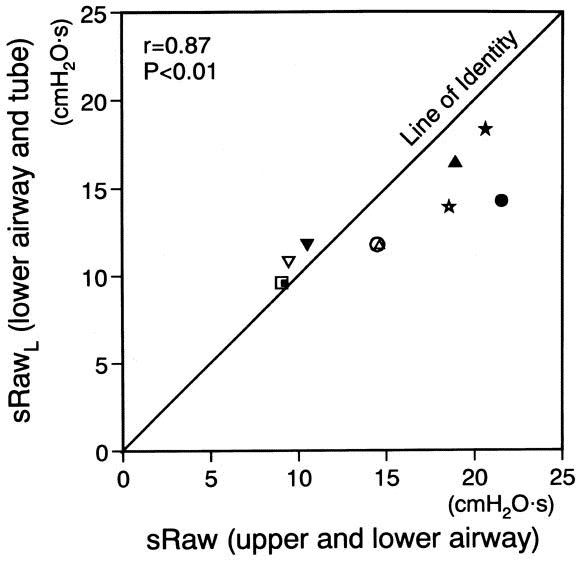
A comparison of total airway resistance (sRaw) and the lower airway resistance (sRawL) . sRaw and sRawL are closely correlated. Each symbol represents each individual (n = 5), and open and closed symbols express the values after inhalation of PBS and methacholine, respectively.
In BALB/c mice, airway responsiveness and IgE production increased when the animals were sensitized by inhalation of 1% ovalbumin alone.11, 16, 25 We used 0.05% ovalbumin for inhalation sensitization because we thought that the lower concentration of inhaled allergen may cause the weaker allergic responses. However, inhalation sensitization of 0.05% ovalbumin with PBS inoculation increased neither airway responsiveness nor IgE production. This lack of allergic response may be due to too low concentration of inhaled allergen.
In the ovalbumin sensitization group with influenza-virus infection, airway responsiveness was increased, although neither virus infection alone nor ovalbumin sensitization alone affected it. Airway responsiveness was measured 22 days after inoculation of viruses, when the BAL-cell number was not increased and inflammatory changes had recovered histologically. In our model, such airway inflammation may not cause an increase in airway responsiveness. Mechanically, airway diameter may relate to airway response. The baseline airway resistance did not differ among experimental groups. Therefore the increase in airway responsiveness may not be due to airway obstruction. Inflammation by eosinophils in the airway is an important cause of increased airway responsiveness.36 A relationship between eosinophil number in BALF and airway responsiveness is shown in mild asthma.37 In animals, an increase in airway responsiveness is reported to be associated with eosinophil infiltration of the lung tissue.38, 39 In our BALF, however, eosinophils were not found in ovalbumin sensitization with virus infection. In some asthma models of mice,16, 27 airway responsiveness was not associated with eosinophilia in lung tissue or BALF, as reflected in our results. This suggests that an increase in airway responsiveness may develop by means of mechanisms independent of eosinophil inflammation.27
When T cells of ovalbumin-sensitized animals were transferred to naive animals, an increase in airway responsiveness was induced in transferred animals.40 T cells, particularly CD8+ T cells, were increased in the group of ovalbumin sensitization with virus infection. Recently, it was reported that ovalbumin-sensitized animals depleted of CD8+ T cells did not develop airway hyperresponsiveness, although animals with ovalbumin sensitization alone had airway hyperresponsiveness.38 Transfer of CD8+ T cells restored the airway hyperresponsiveness. When virus-specific CD8+ cells are challenged by a different antigen, a TH2 -type immune response involving IL-5 production and eosinophilia occurs in the airway.26 As described earlier, virus-infected CD8+ T cells may enhance IgE production. Induction of allergen-specific IgE production is believed to be 1 of the elements contributing to increased airway responsiveness.41 In various animal models of asthma, airway responsiveness increases with the increase in IgE level.11, 16, 25, 27, 39 In this regard, our data compare with the previous studies. Therefore an increase in the IgE- and TH2 -type immune response of CD8+ T cells may contribute to the increase in airway responsiveness.
Footnotes
From the First Department of Internal Medicine, Yokohama City University School of Medicine.
Reprint requests: Shunsuke Suzuki, MD, First Department of Internal Medicine, Yokohama City University School of Medicine, 3-9 Fukuura, Kanazawa-ku, Yokohama 236, Japan.
0091-6749/98 $5.00 + 0 1/1/92476
References
- 1.Minor TE, Dick EC, Baker JW, Ouellette JJ, Cohen M, Reed CE. Rhinovirus and influenza type A infections as precipitants of asthma. Am Rev Respir Dis. 1976;113:149–153. doi: 10.1164/arrd.1976.113.2.149. [DOI] [PubMed] [Google Scholar]
- 2.Pattemore PK, Johnston SL, Bardin PG. Viruses as precipitants of asthma symptoms: I. Epidemiology. Clin Exp Allergy. 1992;22:325–336. doi: 10.1111/j.1365-2222.1992.tb03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemanske RF, Dick EC, Swenson CA, Vrtis RF, Busse WW. Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J Clin Invest. 1989;83:1–10. doi: 10.1172/JCI113843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zweiman B, Schoenwetter WF, Pappano JE, Jr, Tempest B, Hildreth EA. Patterns of allergic respiratory disease in children with a past history of bronchiolitis. J Allergy Clin Immunol. 1971;48:283–289. doi: 10.1016/0091-6749(71)90029-7. [DOI] [PubMed] [Google Scholar]
- 6.Gurwitz D, Mindorff C, Levison H. Increased incidence of bronchial reactivity in children with a history of bronchiolitis. J Pediatr. 1981;98:551–555. doi: 10.1016/s0022-3476(81)80758-5. [DOI] [PubMed] [Google Scholar]
- 7.Monto AS, Ullman BM. Acute respiratory illness in an American community. JAMA. 1974;227:164–169. [PubMed] [Google Scholar]
- 8.Broder I, Higgins MW, Mathews KP, Keller JB. Epidemiology of asthma and allergic rhinitis in a total community, Tecumseh, Michigan. J Allergy Clin Immunol. 1974;54:100–110. doi: 10.1016/0091-6749(74)90038-4. [DOI] [PubMed] [Google Scholar]
- 9.Smith JM. Epidemiology and natural history of asthma, allergic rhinitis, and atopic dermatitis (eczema) In: Middleton E Jr, Reed CE, Ellis EF, Adkinson NF Jr, Yunginger JW, editors. 3rd ed. CV Mosby; St. Louis: 1988. pp. 891–929. (Allergy: principles and practice.). [Google Scholar]
- 10.Frick OL, German DF, Mills J. Development of allergy in children: I. association with virus infections. J Allergy Clin Immunol. 1979;63:228–241. doi: 10.1016/0091-6749(79)90106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen GL, Renz H, Loader JE, Bradley KL, Gelfand EW. Airway response to electrical field stimulation in sensitized inbred mice: passive transfer to increased responsiveness with peribronchial lymph nodes. J Clin Invest. 1992;89:747–752. doi: 10.1172/JCI115651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakamoto M, Ida S, Takishima T. Effect of influenza virus infection on allergic sensitization to aerosolized ovalbumin in mice. J Immunol. 1984;132:2614–2617. [PubMed] [Google Scholar]
- 13.Lukacher AE, Braciale VL, Braciale TJ. In vivo effector function of influenza virus-specific cytotoxic T lymphocyte clones is highly specific. J Exp Med. 1984;160:814–826. doi: 10.1084/jem.160.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pennock BE, Cox CP, Rogers RM, Cain WA, Wells JH. A noninvasive technique for measurement of changes in specific airway resistance. J Appl Physiol. 1979;46:399–406. doi: 10.1152/jappl.1979.46.2.399. [DOI] [PubMed] [Google Scholar]
- 15.Mbow ML, Rutti B, Brossard M. Infiltration of CD4+ and CD8+ T cells, and expression of ICAM-1, Ia antigens, IL-1α and TNF-α in the skin lesion of BALB/c mice undergoing repeated infestations with nymphal Ixodes ricinus ticks. Immunology. 1994;82:596–602. [PMC free article] [PubMed] [Google Scholar]
- 16.Renz H, Smith HR, Henson JE, Ray BS, Irvin CG, Gelfand EW. Aerosolized antigen exposure without adjuvant causes increased IgE production and increased airway responsiveness in the mouse. J Allergy Clin Immunol. 1992;89:1127–1138. doi: 10.1016/0091-6749(92)90296-e. [DOI] [PubMed] [Google Scholar]
- 17.Holt PG, Vines J, Bilyk N. Effect of influenza virus infection on allergic sensitization to inhaled antigen in mice. Int Arch Allergy Appl Immunol. 1988;86:121–123. doi: 10.1159/000234617. [DOI] [PubMed] [Google Scholar]
- 18.Lebrec H, Sarlo K, Burleson GR. Effect of influenza virus infection on ovalbumin-specific IgE responses to inhaled antigen in the rat. J Toxicol Environ Health. 1996;49:619–630. doi: 10.1080/009841096160664. [DOI] [PubMed] [Google Scholar]
- 19.Azoulay-Dupuis E, Lambre CR, Soler P, Moreau J, Thibon M. Lung alterations in guinea-pig infected with influenza virus. J Comp Path. 1984;94:273–283. doi: 10.1016/0021-9975(84)90046-x. [DOI] [PubMed] [Google Scholar]
- 20.Folkerts G, Verheyen AKCP, Geuens GMA, Folkerts HF, Nijkamp FP. Virus-induced changes in airway responsiveness, morphology, and histamine levels in guinea pigs. Am Rev Respir Dis. 1993;147:1569–1577. doi: 10.1164/ajrccm/147.6_Pt_1.1569. [DOI] [PubMed] [Google Scholar]
- 21.Baumgarth N, Brown L, Jackson D, Kelso A. Novel features of the respiratory tract T-cell response to influenza virus infection: lung T cells increase expression of gamma interferon mRNA in vivo and maintain high levels of mRNA expression for interleukin-5 (IL-5) and IL-10. J Virol. 1994;68:7575–7581. doi: 10.1128/jvi.68.11.7575-7581.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dolin R, Richman DD, Murphy BR, Fauci AS. Cell-mediated immune responses in humans after induced infection with influenza A virus. J Infec Dis. 1977;135:714–719. doi: 10.1093/infdis/135.5.714. [DOI] [PubMed] [Google Scholar]
- 23.Baumgarth N, Kelso A. Functionally distinct T cells in three compartments of the respiratory tract after influenza virus infection. Eur J Immunol. 1996;226:2189–2197. doi: 10.1002/eji.1830260934. [DOI] [PubMed] [Google Scholar]
- 24.Welliver RC, Kaul TN, Sun M, Ogra PL. Defective regulation of immune responses in respiratory syncytial virus infection. J Immunol. 1984;133:1925–1930. [PubMed] [Google Scholar]
- 25.Renz H, Lack G, Saloga J, Schwinzer R, Bradley K, Loader J. Inhibition of IgE production and normalization of airways responsiveness by sensitized CD8 T cells in a mouse model of allergen-induced sensitization. J Immunol. 1994;152:351–360. [PubMed] [Google Scholar]
- 26.Coyle A, Erard F, Bertrand C, Walti S, Pircher H, Gros GL. Virus-specific CD8+ cells can switch to interleukin 5 production and induce airway eosinophilia. J Exp Med. 1995;181:1229–1233. doi: 10.1084/jem.181.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessel EM, Van Oosterhout AJM, Hofstra CL, De Bie JJ, Garssen J, Van Loveren H. Bronchoconstriction and airway hyperresponsiveness after ovalbumin inhalation in sensitized mice. Eur J Pharmacol. 1995;293:401–412. doi: 10.1016/0926-6917(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 28.Freihorst J, Piedra PA, Okamoto Y, Ogra PL. Effect of respiratory syncytial virus infection on the uptake of and immune response to other inhaled antigens. Proc Soc Exp Biol Med. 1988;188:191–197. doi: 10.3181/00379727-188-42727. [DOI] [PubMed] [Google Scholar]
- 29.Thepen T, McMenamin C, Girn B, Kraal G, Holt PG. Regulation of IgE production in pre-sensitized animals: in vivo elimination of alveolar macrophages preferentially increases IgE responses to inhaled antigen. Clin Exp Allergy. 1992;22:1107–1114. doi: 10.1111/j.1365-2222.1992.tb00137.x. [DOI] [PubMed] [Google Scholar]
- 30.Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, Kraal G. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med. 1993;177:397–407. doi: 10.1084/jem.177.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pene J. Regulatory role of cytokines and CD23 in the human IgE antibody synthesis. Int Arch Allergy Appl Immunol. 1989;90(suppl):32–40. doi: 10.1159/000235073. [DOI] [PubMed] [Google Scholar]
- 32.Holtzman MJ, Hahn HL, Sasaki K, Skoogh B-E, Graf PD, Fabbri LM. Selective effect of general anesthetics on reflex bronchoconstrictor responses in dogs. J Appl Physiol. 1982;53:126–133. doi: 10.1152/jappl.1982.53.1.126. [DOI] [PubMed] [Google Scholar]
- 33.Johns K, Sorkness R, Graziano F, Castleman W, Lemanske RF. Contribution of upper airways to antigen-induced late airway obstructive responses in guinea pigs. Am Rev Respir Dis. 1990;142:138–142. doi: 10.1164/ajrccm/142.1.138. [DOI] [PubMed] [Google Scholar]
- 34.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 35.Martin TR, Gerard NP, Galli SJ, Drazen JM. Pulmonary responses to bronchoconstrictor agonists in the mouse. J Appl Physiol. 1988;64:2318–2323. doi: 10.1152/jappl.1988.64.6.2318. [DOI] [PubMed] [Google Scholar]
- 36.Kay AB, Corrigan CJ. Eosinophils and neutrophils. Br Med Bull. 1992;48:51–64. doi: 10.1093/oxfordjournals.bmb.a072541. [DOI] [PubMed] [Google Scholar]
- 37.Taylor KJ, Luksza A. Peripheral blood eosinophil counts and bronchial responsiveness. Thorax. 1987;42:452–456. doi: 10.1136/thx.42.6.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamelmann E, Oshiba A, Paluh J, Bradley K, Loader J, Potter TA. Requirement for CD8+ T cells in the development of airway hyperresponsiveness in a murine model of airway sensitization. J Exp Med. 1996;186:1719–1729. doi: 10.1084/jem.183.4.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamelmann E, Oshiba A, Schwarze J, Bradley K, Loader J, Larsen GL. Allergen-specific IgE and IL-5 are essential for the development of airway hyperresponsiveness. Am J Respir Cell Mol Biol. 1997;16:674–682. doi: 10.1165/ajrcmb.16.6.9191469. [DOI] [PubMed] [Google Scholar]
- 40.Renz H, Bradley K, Saloga J, Loader J, Larsen GL, Gelfand EW. T cells expressing specific Vβ elements regulate immunoglobulin E production and airways responsiveness in vivo. J Exp Med. 1993;177:1175–1180. doi: 10.1084/jem.177.4.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broide DH, Gleich GJ, Cuomo AJ, Coburn DA, Federman EC, Schwartz LB. Evidence of ongoing mast cell and eosinophil degranulation in symptomatic asthma airway. J Allergy Clin Immunol. 1991;88:637–648. doi: 10.1016/0091-6749(91)90158-k. [DOI] [PubMed] [Google Scholar]