

Abstract
Virus replication can cause extensive rearrangement of host cell cytoskeletal and membrane compartments leading to the “cytopathic effect” that has been the hallmark of virus infection in tissue culture for many years. Recent studies are beginning to redefine these signs of viral infection in terms of specific effects of viruses on cellular processes. In this chapter, these concepts have been illustrated by describing the replication sites produced by many different viruses. In many cases, the cellular rearrangements caused during virus infection lead to the construction of sophisticated platforms in the cell that concentrate replicase proteins, virus genomes, and host proteins required for replication, and thereby increase the efficiency of replication. Interestingly, these same structures, called virus factories, virus inclusions, or virosomes, can recruit host components that are associated with cellular defences against infection and cell stress. It is possible that cellular defence pathways can be subverted by viruses to generate sites of replication. The recruitment of cellular membranes and cytoskeleton to generate virus replication sites can also benefit viruses in other ways. Disruption of cellular membranes can, for example, slow the transport of immunomodulatory proteins to the surface of infected cells and protect against innate and acquired immune responses, and rearrangements to cytoskeleton can facilitate virus release.
I. Introduction
Viruses are obligate intracellular parasites. Unlike their hosts, they cannot replicate by growth or division but use their genomes to redirect host cell processes to produce all the components needed to make new viruses. Virus replication and assembly are often confined within specific intracellular compartments called virus factories, viroplasm, or viral inclusions. These are thought to provide a physical platform to concentrate new genomes and proteins involved in replication and assembly, and this is likely to increase the efficiency of virus production. The formation of specialized sites of replication can involve extensive reorganization of cellular cytoskeleton and membrane compartments. This can lead to cell rounding and swelling and a “cytopathic effect” that has been documented for many years (Reissig 1956, Robbins 1950). Recent advances in microscopy, such as live cell imaging and tomography, combined with the power of reverse genetics, are now allowing the cytopathic effect to be redefined in terms of specific effects of viral proteins on specific cellular processes rather than an overwhelming assault on the cell in preparation for cell lysis.
There is considerable interest in understanding how virus infection leads to the large changes in cellular organization required to produce complex replication sites. In the simplest model, virus replication sites would form passively through self‐association of viral components and exclusion of host organelles. Viruses, however, require a considerable number of host proteins to facilitate replication, and there is increasing evidence that these are specifically transported to sites of replication. Host proteins may move to replication sites because they are actively recruited by binding to specific viral proteins. Alternatively, viruses may transport viral and host material to replication sites by subverting host defences against infection [reviewed by Kirkegaard 2004, Wileman 2006]. The large scale changes in cellular membrane and cytoskeletal organization, which occur during the formation of replication sites, can offer further benefit to viruses. Rearrangement of the cytoskeleton can, for example, facilitate virus release, and the block in the secretory pathway seen during infection with positive‐stranded RNA viruses can reduce release of inflammatory mediators and protect against innate and acquired immune responses. This is a broad subject of considerable interest to virologists and cell biologists, and we have benefited from excellent reviews that have been published (Mackenzie 2005, Novoa 2005). In writing this chapter, we have concentrated on describing sites of virus replication in the context of the cell in which its replication takes place. We have illustrated these concepts with reference to replication sites produced by many different viruses and, where possible, described how virus replication impacts on the functioning of the host cell.
II. Viroplasm, Virosomes, Factories, and Inclusions
Virus replication sites have been studied for many years and have evolved their own terminology. Early studies of poxvirus replication (Dales 1961, Morgan 1954) describe electron‐dense aggregates and amorphous material induced early during infection called viroplasm. Viroplasm has also been used to describe similar structures induced during infection with Poliovirus (Dales et al., 1965a). Viroplasm is often concentrated within perinuclear areas that exclude host organelles. Viroplasm is thought to indicate sites of virus replication, and concentrations of viroplasm have been called virosomes, or virus factories, to reflect an organelle involved in virus production. Virus infection also produces inclusion bodies. As a working definition, these can be considered to form later during infection. They can form virus factories once virus production has peaked, and/or at other sites in the cell they probably arise from an accumulation of viral proteins that do not become incorporated into viruses.
III. Membrane Rearrangements Occurring During the Replication of the Positive‐Stranded RNA Viruses
The positive‐stranded RNA viruses encode nonstructural proteins (NSP) that cause proliferation and modification of membranes of the host secretory pathway. The membranes are thought to provide a physical framework or “replication complex” that concentrates the cellular and viral components required for virus replication (Bienz 1987, Egger 2002, Froshauer 1988, Gazina 2002, Magliano 1998, Schlegel 1996, van der Meer 1998). Assembly of the replicase on membranes, rather than the cytosol, may also help viruses evade host defence pathways that monitor cells for double‐stranded RNA (dsRNA) intermediates indicative of virus replication. The replicase complexes of all the positive‐stranded RNA viruses contain an RNA‐dependent RNA polymerase (RdRp), a protein with NTPase and helicase activity, and in many cases a methyl transferase to cap viral RNA. These proteins are generated from the viral polyproteins by viral proteases, and are then targeted to membranes in ways that differ depending on virus family (Fig. 1 ).
Figure 1.
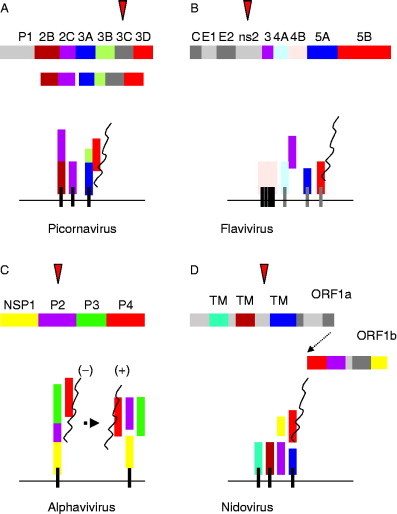
The replicase proteins of positive‐stranded RNA viruses are directed to membranes by NSP with membrane‐targeting information. (A) Picornavirus. The replication complex contains 3D, the RdRp (red), and 2C which has NTPase and helicase motifs (purple). The 3D polymerases do not have membrane‐targeting information but are synthesized as a 3ABCD precursor. 3ABCD is processed to 3AB by the 3C protease (red triangle) and a hydrophobic domain in 3A targets 3AB to the cytoplasmic face of ER membranes. 3AB binds directly to 3D and this targets the polymerase to the replication complex. The replication complex also requires 2BC and 2C proteins that are targeted to membranes via their own hydrophobic domains (black lines). (B) Flavivirues. The replication complex is encoded at the C‐terminus of a polyprotein that is processed by the NS2 protease (red triangle). NS5B is the RNA‐dependent polymerase (red), and NS3 acts as helicase (purple). NS4B is a polytopic membrane protein inserted into the ER cotranslationally. NS4A, 5A, and 5B have hydrophobic domains (gray lines) that allow posttranslational insertion into the cytoplasmic face of the ER membrane. NS3 is recruited into the complex by associating with NS4A. (C) Alphavirus. The NSP1234 polyprotein is processed by a protease activity in the C‐terminus of P2 (red triangle). The polyprotein is anchored to the cytoplasmic face of endosome and lysosome membranes by a hydrophobic region at the N‐terminus of P1. P1 also acts as the methyltransferase (yellow). P2 encodes the helicase (purple) and P4 is the RdRp (red). The P123 precursor associates with P4 and generates negative‐stranded RNA. Further processing produces a complex of separate P1, 2, 3, and 4 proteins that produce positive‐stranded RNA. (D) Nidoviruses. The Nidovirales order comprises the Arteriviridae, Coronaviridae, and Roniviridae families. The replicase gene is composed of two open reading frames termed ORF1a and ORF1b, both of which encode complex polyproteins. Arterivirus ORF1b encodes NSPs 9–12 including the RdRp (NSP9, red), helicase (NSP10, purple). The ORF1b reading frame lacks hydrophobic domains able to target the replicase to membranes. Proteins necessary for membrane targeting (brown and blue) are encoded by ORF1a (NSP2, 3, and 5). For the CoVs, for example, MHV and SARS‐CoV transmembrane domains are located in NSP3, 4, and 6, and helicase and polymerase proteins are NSP12 and 13, respectively. ORF1b also contains a methyltransferase (NSP16, yellow).
A. Regulation of membrane traffic in the early secretory pathway
Membrane rearrangements by the positive‐stranded RNA viruses arise from modifications of membrane compartments in the early secretory pathway. The secretory pathway is carefully regulated in cells, and subversion of this pathway by viruses involves interactions between viral proteins and the host proteins that control membrane traffic. For some viruses we are beginning to understand how this is achieved. It is therefore useful to review briefly what is known about the control of membrane traffic at the start of the secretory pathway. Membrane proteins and proteins secreted by cells are synthesized by ribosomes attached to the cytoplasmic face of the endoplasmic reticulum (ER). Proteins destined for transport to the Golgi apparatus, or the plasma membrane, are folded by chaperones and assembled in the lumen of the ER, and transport to the Golgi apparatus and beyond involves a series of transport vesicles. The formation of these vesicles is controlled by coat proteins that are recruited from the cytosol. They select cargos for transport into the secretory pathway and facilitate vesicle formation by inducing membrane curvature (Bonifacino and Glick, 2004).
Movement from the ER involves a coat made from COPII proteins that localize to specific domains of the ER called ER exit sites (ERES), or transitional ER. Vesicle budding from ERES requires the small GTPase, Sar1p. Binding of GTP to Sar1p translocates Sar1p from the cytosol onto ER membranes. Here, Sar1p‐GTP recruits cargo proteins into ERES and seeds polymerization of the COPII coat containing Sec13–Sec31p proteins and production of 60‐ to 80‐nm‐diameter vesicles. Movement of vesicles from the ER to the Golgi apparatus requires microtubules and the dynein/dynactin motor protein. The vesicles fuse with a series of membranes that lie between the ER and the Golgi apparatus called the ER‐Golgi intermediate compartment (ERGIC), or tubulovesicular structures, and specific fusion with ERGIC membranes is determined by a complex of proteins called transport protein particle 1 (TRAPP1). TRAPP1 proteins tether the vesicles on ERGIC and Golgi membranes, allowing interactions between vesicle and target SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) proteins to facilitate membrane fusion. The SNARE interactions are controlled by vesicle‐specific small GTPases called rab proteins (Fig. 2 ).
Figure 2.
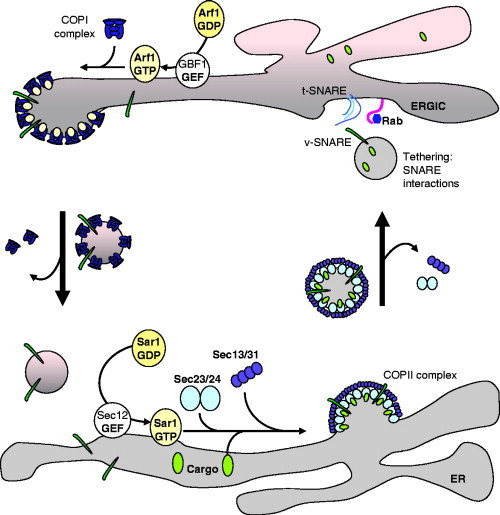
Protein trafficking in the early secretory pathway. 1. Anterograde transport from the ER to the ERGIC is mediated by COPII‐coated vesicles. Formation of COPII coats is regulated by the Sar1p‐GTPase. Binding of Sar1p to the ER requires binding of GTP and this is facilitated by the Sec12p‐GTP exchange protein. Sar1p‐GTP recruits the Sec23–Sec24p subcomplex (light blue) of the COPII coat and this recruits cargo proteins (light green) to ERES. The Sec23–Sec24p subcomplex then recruits the Sec13–Sec31p proteins (purple) that induce membrane curvature and formation of a vesicle. Hydrolysis of GTP on Sar1p by Sec23p results in coat disassembly. The vesicle docks with ERGIC membranes by binding tethering proteins and interactions between v‐SNAREs and t‐SNAREs results in vesicle fusion. 2. Retrograde transport from the ERGIC to the ER provides a pathway to retrieve proteins from the ERGIC and Golgi apparatus and is mediated by COPI‐coated vesicles. Formation of COPI coats is regulated by the Arf1‐GTPase. Binding of Arf1 to the ERGIC requires binding of GTP and this is facilitated by the GBF1 and BIG1/2 GTP exchange proteins. Arf1‐GTP recruits the COPI coat complex (dark blue), which induces membrane curvature and formation of a vesicle that returns to the ER.
Further sorting events in the ERGIC and early Golgi involve a second complex of coat proteins called COPI. The COPI complex contains seven proteins (α, β, β′, γ, δ, ɛ, and ζ COP proteins), which generate vesicles that take proteins from the ERGIC and Golgi apparatus back to the ER through a retrieval pathway (Fig. 2). The COPI proteins are recruited from the cytosol by the Arf1‐GTPase. Activation of Arf1 requires binding to GTP and is facilitated by GTP exchange protein, Arf‐GEF. Arf1‐GTP inititates coat assembly while hydrolysis of GTP by Arf1 leads to coat disassembly. This disassembly is stimulated by an Arf1‐GTP‐activating protein (Arf‐GAP) that promotes GTP hydrolysis by Arf1. A possible role for Arf1 in the generation of vesicles during picornavirus replication has been the focus of much work following the observation that Poliovirus replication is blocked by brefeldin‐A (BFA), a drug that inhibits the recruitment of Arf1 onto membranes (Maynell et al., 1992).
Membrane vesicles are also produced in cells in response to starvation. This pathway, known as autophagy, is used as a part of a quality control system that removes long‐lived proteins and damaged organelles from the cytoplasm and has been shown to provide a defence against intracellular pathogens (Deretic 2005, Kirkegaard 2004, Levine 2004, Shintani 2004). The origins of the membranes formed during autophagy are unclear but may be derived from the ER (Reggiori and Klionsky, 2005). Autophagy is suppressed by the target of rapamycin (TOR) kinase and is activated by conditions that lead to inactivation of TOR. This leads to the production of membrane crescents in the cytoplasm, called isolation membranes, which mature into double‐membraned vesicles of 500‐ to 1000‐nm diameter called autophagosomes. This maturation engulfs small quantities of cytoplasm, and any organelles or pathogens present at sites of autophagy become trapped within autophagosomes. The autophagosomes ultimately fuse with lysosomes resulting in degradation of their content. Autophagosomes are of interest because infection of cells with picornaviruses and coronaviruses (CoVs) can generate double‐membraned vesicles that may be related to autophagosomes.
In addition to supplying membrane and proteins to the secretory pathway, the ER acts as a major site of lipid synthesis. As a consequence, the ER contains a large quantity of membrane, and this is organized into a complex reticulum made from tubular and lamella structures (Borgese et al., 2006). The smooth ER increases in response to a buildup of ER membrane proteins and can be organized into lamellae or concentric whorls called organized smooth ER (OSER). Structures similar to OSER are also seen during virus replication.
B. Picornavirus replication induces numerous membrane vesicles
1. The picornavirus replicase
Picornaviruses are nonenveloped positive‐stranded RNA viruses. The genome encodes a large polyprotein that is processed to generate capsid proteins from the P1 region and nonstructural replicase proteins from the P2 and P3 regions. Picornavirus 3D contains the RdRp, while 2C has NTPase and helicase motifs. The 3D polymerase does not have membrane‐targeting information but is synthesized as a 3ABCD precursor. 3ABCD is processed to 3AB by the 3C protease, and a hydrophobic domain in 3A targets 3AB to the cytoplasmic face of the ER. 3D binds directly to 3AB, and this targets the polymerase to the replication complex. The 3D polymerase of Poliovirus is believed to self‐assemble into a large ordered array on membranes, which is critical for binding RNA and RNA elongation (Lyle et al., 2002). The replication complex also requires 2BC and 2C proteins that are targeted to membranes via their own hydrophobic domains (Fig. 1A).
2. Membrane rearrangements induced by picornaviruses provide sites for replication
The accumulation of large numbers of densely packed membrane vesicles in the cytoplasm is characteristic of a picornavirus infection (Bienz 1983, Bienz 1987, Cho 1994, Dales 1965a, Schlegel 1996, Stuart 1961, Suhy 2000). Studies have suggested that vesicles induced by Poliovirus are derived from the ER, either from COPII‐coated vesicles or from ER‐derived autophagic double‐membraned vacuoles (Bienz 1987, Jackson 2005, Rust 2001, Schlegel 1996, Suhy 2000). However, the detection of ER, Golgi, and lysosomal markers in membranes induced at later stages of infection by Poliovirus suggests that more than one organelle may contribute membranes to the replication complex (Schlegel et al., 1996). In interpreting these studies, it is important to consider if the vesicles observed are involved in replication, or if they represent a bystander response to virus infection. Evidence for a role of specific membranes in replication is provided by the presence of replicase proteins, or better still dsRNA or negative‐stranded intermediate viral RNA (Egger and Bienz, 2005). Examination of cells infected with Poliovirus for the first appearance of negative‐stranded RNA suggests that this initial stage of replication starts on the ER. This is consistent with high‐resolution immunofluorescence microscopy (Rust et al., 2001) showing the Poliovirus 2B protein associated with ERES containing the Sec13–Sec31p proteins of the COPII complex. These sites exclude resident ER proteins, suggesting colocalization of 2B with COPII‐coated transport vesicles. Replication complexes containing negative‐stranded RNA then move on microtubules to a perinuclear area to initiate synthesis of positive‐stranded RNA (Egger and Bienz, 2005).
3. Membrane rearrangements can be induced by expression of nonstructural proteins
Membrane rearrangements have been studied by expressing individual, or combinations, of picornavirus proteins in cells. Most of this work has involved studies of Poliovirus proteins, and membrane rearrangements are reported for the 2B, 2C, 2BC, 3A, and 3AB proteins. Poliovirus 2B causes fragmentation of the Golgi complex (Sandoval and Carrasco, 1997). The 2BC and 2C proteins lead to vesiculation and tubulation and sometimes myelin‐like swirls of ER‐derived membranes (Aldabe 1996, Cho 1994). Similar structures are induced by 2C and 2BC of hepatitis A virus (Teterina et al., 1997). Expression of the Poliovirus 3A protein causes swelling of ER cisternae (Doedens et al., 1997) and the disappearance of vesicles budding from the ER, while the 3AB protein also induces myelin‐like swirls of ER (Egger et al., 2000). The membrane rearrangements induced by expression of single proteins do not, however, mirror those observed in infected cells, and since myelin‐like modifications to the ER are also seen following overexpression of ER proteins [reviewed by Borgese et al. (2006)], their relevance to viral replication is unclear. Importantly for Poliovirus, it is a combination of 2BC and 3A protein expression that induces membrane structures morphologically similar to those seen in infected cells (Suhy et al., 2000).
4. Membrane rearrangements may vary between different picornavirus families
Gazina et al. (2002) have studied replication complexes formed by several different picornaviruses. Encephalomyocarditis virus (EMCV), parechovirus 1, and echovirus 11 induce clustered vesicles containing dsRNA in the perinuclear region of the cell. The precise nature of the vesicles varied with virus. Parechovirus 1 produced homogeneous vesicles of 70–100 nm, while membranes produced by EMCV and echovirus 11 were heterogeneous but more compact and associated with electron‐dense material. Differences for parechovirus 1 have also been reported by Krogerus et al. (2003) who suggest that replication may occur on membranes derived from the late Golgi rather than early ER and ERGIC compartments. All three viruses, however, cause loss of ribosomes from the ER and lack of visible Golgi apparatus. The COPI coat protein β‐COP was found to colocalize with echovirus 11 replication complexes, but not with replication complexes produced by EMCV, again suggesting that vesicles produced by different picornaviruses may differ. Infection with Foot‐and‐mouth disease virus (FMDV) also results in loss of ribosomes from the ER and an accumulation of heterogeneous vesicles to one side of the nucleus (Monaghan et al., 2004).
High‐pressure freezing can be used to increase the preservation of cellular ultrastructure during processing for electron microscopy. Such analysis of cells infected with Poliovirus shows that the vesicles have two membranes suggestive of autophagosomes (Jackson 2005, Suhy 2000). Double‐membraned structures containing electron‐dense material, and possibly viruses, were also revealed by the early work on Poliovirus (Dales et al., 1965a). High‐pressure freezing has been used to compare FMDV and Bovine enterovirus (BEV). BEV produced heterogeneous membrane clusters similar to the rosettes described for Poliovirus (Egger et al., 1996). Many of the vesicle membranes have high electron density suggestive of double membranes and lie adjacent to accumulations of virus‐like particles. Clusters of FMD viruses were also associated with vesicles and electron‐dense material, but there were fewer double‐membraned vesicles (Monaghan et al., 2004). Immunofluorescence analysis of Poliovirus vesicles shows colocalization of replicase protein 3A and autophagy marker LC3, suggesting assembly of the replicase on autophagosomes. Similar work suggesting the use of autophagosomes during replication of CoVs will be described below. For Poliovirus, expression of 3A and 2BC, which produces vesicles similar to those seen in infected cells (Suhy et al., 2000), can induce autophagy (Jackson et al., 2005), and inhibition of autophagy reduces yields of extracellular virus. The results suggest that the autophagy pathway may facilitate the release of Poliovirus from cells, and it will be interesting to see if this is true for other enteroviruses that are resistant to the low pH and proteases present in lysosomes and autophagosomes.
5. Vesicle coat proteins may play a role during picornavirus replication
Evidence that different members of the picornavirus family vary in the way that they interact with host membranes is provided by studies of virus sensitivity to BFA. BFA completely inhibits Poliovirus and echovirus 11 replication (Cuconati 1998, Gazina 2002, Irurzun 1992, Maynell 1992) and partially inhibits parechovirus 1 replication (Gazina et al., 2002) but not other picornaviruses such as EMCV (Gazina et al., 2002) or FMDV (Monaghan 2004, O'Donnell 2001). BFA prevents assembly of COPI coats and this has generated considerable interest in understanding how COPI and COPII coats contribute to formation of the replication complex, and how BFA inhibits picornavirus replication. In cells infected with the highly BFA‐sensitive virus echovirus 11, β‐COP was recruited into the replication complex; in contrast, the replication complex formed by the BFA‐resistant EMCV did not contain β‐COP. This correlation suggests that BFA‐sensitive viruses may require COPI coats for replication (Gazina 2002, Mackenzie 2005). Since COPII coats are resistant to BFA (Lippincott‐Schwartz 2000, Orci 1993, Ward 2001), it is suggested that COPII coats may provide the membranes for replication complexes formed by BFA‐insensitive viruses. The observation that Poliovirus replicase 2B protein is seen in ERES containing COPII proteins, but Poliovirus is sensitive to BFA, can be reconciled if this association of 2B with ERES is considered to be an early step in generation of membrane for the replication complex that precedes recruitment of COPI coat proteins. This is supported by work showing the movement of Poliovirus replication complexes containing negative‐stranded RNA from the ER to perinuclear sites (Egger and Bienz, 2005).
Direct evidence that COPI coat proteins are required for picornavirus replication comes from studies of Drosophila C virus (DCV). DCV is a positive‐stranded RNA dicistronic virus that is similar to Poliovirus and replicates in a cytoplasmic compartment containing virus‐induced membrane vesicles. A genome‐wide RNA silencing screen identified six (α, β, β′, γ, δ, and ζ) of the seven COPI coat proteins as essential for virus replication. Furthermore, the formation of virus‐induced vesicles required β‐COP, but not COPII protein, Sec23p. Notably, small interfering RNAs against α‐COP, but not Sec23p, also slowed Poliovirus replication (Cherry et al., 2006).
6. Arf proteins and Brefeldin‐A can modulate poliovirus and coxsackievirus replication
The formation of COPI‐coated vesicles is regulated by the Arf1‐GTPase. The observation that BFA inhibits the replication of enteroviruses such as Poliovirus, and also inhibits the function of the Arf1‐GTPase, provides a second link between virus replication and COPI coats. Arf proteins are regulated by Arf‐GEFs that facilitate binding of GTP by removing GDP, and by Arf‐GAPs that increase hydrolysis of GTP by Arfs. Arf1‐GEFs are inhibited by BFA, and BFA therefore reduces levels of Arf1‐GTP in cells. The GEFs affected by picornavirus infection are Golgi‐associated BFA‐resistant protein (GBF1) and BFA‐inhibited protein (BIG1/2). Work by Belov 2005, Belov 2007 indicates that infection of cells with Poliovirus increases intracellular Arf‐GTP levels fourfold, suggesting increased activity of Arf1‐GEFs or inhibition of Arf1‐GAP proteins. In the absence of virus, Arf1 is concentrated in the Golgi apparatus, but during infection with Poliovirus Arf1 staining fragments and colocalizes with replicase protein 2C. This suggests that infection leads to a redistribution of Arf proteins from the Golgi apparatus to the replication complex. The binding of Arf proteins to membranes is dynamic, with Arf‐GDP being released from membranes following hydrolysis of GTP. Cytosolic Arf1‐GDP would redistribute naturally to membranes enriched for the Arf1‐GEFs that facilitate loading of new GTP. Significantly, Poliovirus infection causes enrichment of GEFs in membranes containing replicase proteins, and this would provide a mechanism for increasing levels of Arf1‐GTP at sites of virus replication.
Translation of Poliovirus RNA on membranes in vitro provides an alternative means of studying the role of Arf proteins in virus replication. Replication is inhibited by BFA and peptides that function as competitive inhibitors of Arf (Cuconati et al., 1998), and for the most part, the assay mimics what is observed in infected cells. Translation in vitro leads to recruitment of Arf3 and Arf5 but not Arf6 (Belov et al., 2007) onto membranes. Suitable antibodies recognizing the ER‐associated Arf1 were not available for these experiments, so it is not known if Arf1 is also recruited to membranes during translation. Membrane recruitment of Arf proteins can be reconstituted by translation and expression of Poliovirus 3A or 3CD. Poliovirus proteins do not show intrinsic GEF activity, but 3A and 3CD will induce association of GBF1 and BIG1/2, respectively, with membranes in vitro. This raises the possibility that recruitment of 3A and 3CD to the replication complex during infection targets Arf‐GEF to virus‐induced membranes, which in turn increases local levels of Arf1‐GTP. This is thought to be necessary for replication because inhibition of Arf1‐GEF by BFA blocks replication, and replication can be rescued by overexpression of GBF1 (Belov et al., 2007). High levels of Arf1‐GTP would also increase recruitment of COPI proteins and be consistent with the work on DCV showing that COPI proteins are required for replication and vesicle production (Cherry et al., 2006). A Poliovirus 3A mutant with a serine insertion at position 16 is unable to cause translocation of Arf to membranes (Belov et al., 2005). Poliovirus carrying the 3A mutation does not, however, show defects in replication, suggesting that Arf1‐GEF recruitment to membranes by 3A is not essential for replication. It is possible that during infection the defect in 3A is compensated for by 3CD. Interestingly, a BFA‐insensitive Poliovirus with mutations in the 2C and 3A proteins (Crotty et al., 2004) induces vesicles and dispersal of the Golgi apparatus, which begs the questions, does this mutant use a different process for forming the replication complex, or do the mutations in 3A allow the proteins to compete with BFA for GBF1 recruitment?
The role of Arf proteins during coxsackievirus infection has also been studied. In common with Poliovirus, coxsackieviruses are enteroviruses and their replication is inhibited by BFA. Expression of coxsackievirus 3A causes loss of COPII coats from ERES, and an accumulation of 3A, COPII and a model secreted protein in both the ER, and tubular‐vesicular post‐ER structures containing ERGIC marker proteins. These effects closely resemble the effects of adding BFA to cells, suggesting coxsackievirus 3A may affect the function of Arf proteins. Coxsackievirus 3A affects the regulation of Arf proteins (Wessels et al., 2006b). Interestingly, the process differs to that described by Belov 2005, Belov 2007 for Poliovirus 3A translated in vitro. Expression of coxsackievirus 3A in cells caused loss of COPI and Arf1 from membranes, and there was redistribution of BIG1/2 and GBF1 from the Golgi apparatus into the cytoplasm. This suggests that coxsackievirus 3A reduces, rather than enhances, levels of Arf1‐GTP. Coxsackievirus 3A also caused redistribution of Arf1‐GAP to punctate structures suggestive of the ERGIC. A block in Arf1‐GEF activity, combined with recruitment of Arf1‐GAP, would reduce the levels of Arf‐GTP and inhibit membrane recruitment of COPI. Wessels et al. (2006a) examined the effects of the 3A proteins of other picornaviruses and found that only the 3A proteins of enteroviruses bound GEFs. Intriguingly, Wessels’ work contrasts with Belov in that they found the interaction of 3A with GEFs lead to a loss of Arf proteins from membranes. Why these differences are seen is, as yet, unknown but may be due to differences in cell type/methods used or differences in levels of 3A protein expression.
7. Picornavirus replication blocks protein secretion
Poliovirus and coxsackievirus slow protein movement through the secretory pathway (Doedens 1995, Wessels 2005). Expression of 2B, 2BC, and 3A individually were all able to slow secretion (Cornell 2006, Doedens 1995, Doedens 1997, van Kuppeveld 1997, Wessels 2005, Wessels 2006a), but for both viruses the 3A protein was found to have the greatest impact on ER‐to‐Golgi transport. Poliovirus infection, and the 3A protein expressed alone in cells, reduces surface expression of MHC class I, the TNF receptor, and secretion of β‐IFN, IL‐6, and IL‐8 (Choe 2005, Deitz 2000, Dodd 2001, Neznanov 2001), and this may offer an immune evasion strategy to the picornaviruses. This is consistent with the observation that the ability of the coxsackievirus 3A protein to slow secretion may be important for virulence (Wessels et al., 2006b) and has led to studies of the mechanism of action of 3A in blocking ER‐to‐Golgi transport.
Deletion analysis has identified residues in the unstructured N‐terminal region of Poliovirus and coxsackievirus 3A as important for the block in host protein secretion (Choe et al., 2005). An N‐terminal proline‐rich region (particularly Pro19) is important for coxsackievirus block in trafficking (Wessels et al., 2005). In Poliovirus, Lys9 appears important, and in the triple‐proline motif (positions 16–18), only the Pro18 is indispensable for inhibition of protein secretion (Choe et al., 2005). A serine insertion in 3A protein between Thr14 and Ser15, creating the 3A‐2 mutant virus (Berstein and Baltimore, 1988), was found to abolish the ER‐to‐Golgi inhibition of protein trafficking but has little effect on virus replication or membrane rearrangements (Dodd 2001, Doedens 1997). This important observation shows that the ability of 3A to inhibit protein secretion is separate from its role in membrane rearrangements and viral replication.
There is continuing interest in understanding how picornavirus proteins block secretion. Poliovirus 3A and 3CD, and coxsackievirus 3A, can interact with Arf‐GEF, but the downstream events are unclear. The recruitment of Arf‐GEF by Poliovirus 3A and 3CD would increase recruitment of Arf‐GTP to membranes of the replication complex. This would increase recruitment of COPI coat proteins into sites of virus replication and reduce the pool of COPI proteins available to the ERGIC and Golgi apparatus. Alternatively, inhibition of Arf‐GEF and recruitment of Arf‐GAP onto ERGIC membranes by enterovirus 3A would decrease membrane association of Arf‐GTP and again reduce recruitment of COPI onto ERGIC and Golgi membranes. Both mechanisms would reduce the formation of COPI vesicles, and as seen for BFA, block secretion. Poliovirus 3A also binds and inactivates L1S1, a component of the dynein–dynactin motor complex (Kondratova et al., 2005), which is required to move COPII‐derived vesicles from ERES to the ERGIC. As seen for expression of 3A, mutant L1S1 leads to disruption of the ER‐to‐Golgi traffic and reduction in plasma membrane receptors such as TNF receptor. It is possible that 3A may also slow ER‐to‐Golgi transport by binding L1S1.
a. Picornaviruses differ in the use of nonstructural proteins to block secretion
The ability of 3A to inhibit ER‐to‐Golgi trafficking has not been conserved in all picornaviruses (Choe 2005, Cornell 2006, Deitz 2000, Moffat 2005). For example FMDV infection leads to reduced surface expression of MHC class I (Sanz‐Parra et al., 1998), but the FMDV 3A protein does not inhibit ER‐to‐Golgi transport (Moffat et al., 2005). A lack of inhibition of secretion has also been reported for 3A proteins of human rhinovirus, hepatitis A, Theiler's virus, human enterovirus, and EMCV (Choe 2005, Wessels 2006a). The 3A protein of human rhinovirus is unable to bind GBF1, or inhibit COPI recruitment to membranes, and this may explain its inability to slow secretion. Importantly, studies on FMDV have shown that the 2BC protein, or a combination of the processed products, 2B and 2C, inhibits protein movement from the ER to the Golgi apparatus (Moffat 2005, Moffat 2007), and this may be similar for other picornaviruses with 3A proteins that do not block ER‐to‐Golgi transport.
A lack of effect of FMDV 3A on secretion does not result from an inability to bind membranes. FMDV 3A is recovered from postnuclear membrane fractions, and when expressed alone in cells it colocalizes with resident ER proteins. In common with 3A, picornavirus 2B, 2C, and 2BC proteins also contain membrane‐binding sequences. Sequence alignment of the 2B, 2C (2BC), and 3A proteins of different picornaviruses showed a high level of conservation between the 2C proteins, which contain an NTP‐binding site and predicted helicase motifs (Gorbalenya et al., 1990) but large variations in the sequences of the 2B and 3A proteins (Choe 2005, Moffat 2005), and these may explain their different abilities to block secretion. The FMDV 3A protein is, for example, much longer than 3A of enteroviruses, such as Poliovirus, and it does not contain the N‐terminal sequences thought important for Poliovirus 3A to block the secretory pathway.
The 2B protein of FMDV also locates to ER membranes but shows a more reticular pattern than the FMDV 3A protein (Moffat et al., 2005) and can be seen in punctate structures aligned along the ER suggestive of ERES (Fig. 3 ). This is similar to the 2B of Poliovirus that colocates with both Sec13p and Sec31p of the COPII coat. As expected, FMDV 2C is also membrane associated. When expressed in cells, 2C produces faint ER staining, but mainly locates to bright punctate structures in a perinuclear region close to β‐COP, reminiscent of Golgi staining. The β‐COP staining is, however, fragmented suggesting dispersal of the Golgi apparatus, and there is not complete colocalization since 2C structures negative for β‐COP protein can also be seen (Moffat et al., 2007). A similar location of FMDV NSP within the area of the cell occupied by the Golgi apparatus is seen in cells infected with FMDV, and again they do not colocalize with Golgi markers (Knox et al., 2005). The 2BC protein of FMDV is also recovered in postnuclear membrane fractions, but when expressed in cells, 2BC staining differs from that seen for the processed products, 2B and 2C (Fig. 3). FMDV 2BC locates to punctate cytoplasmic structures and larger structures surrounding the nucleus that contain ER markers suggesting swelling of the ER. 2BC shows partial overlap with luminal ER markers but, unlike Poliovirus 2BC, does not colocate with the COPII marker Sec13p. The ER markers also appeared punctate in cells expressing 2BC, suggesting disruption of the ER (Moffat et al., 2005). Interestingly, coexpression of 2B and 2C blocks secretion within post‐ER compartments, similar to those containing 2C. The site of block therefore seems to be determined by the subcellular location of 2C (Moffat et al., 2007) and is consistent with the observation that the block in the presence of 2B can be redirected to the ER, if 2C is tethered to the ER by an ER retention sequence.
Figure 3.
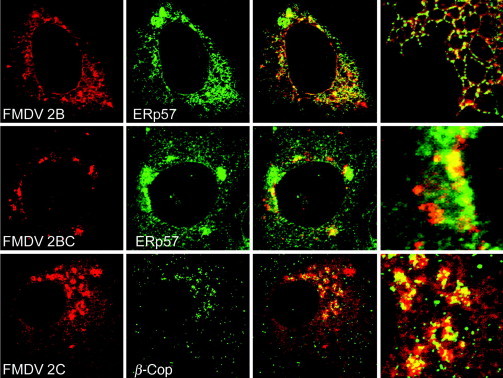
Subcellular location of Foot‐and‐mouth disease NSP encoded in the P2 region of the FMDV genome. Vero cells expressing FMDV 2B (top), 2BC (middle), or 2C (bottom) were fixed and permeabilized and processed for immunofluorescence. 2C and 2BC were located using antibodies specific for 2C (3F7) and 2B was located using an antibody raised against an epitope tag in 2B. Cells were counterstained using antibodies against ER luminal protein ERP57 (top and middle panels), or COPI protein β‐COP (bottom). Merged images are shown at higher magnification on the far left. See Moffat et al. (2005) for more details. Reprinted from Moffat et al. (2005) with permission from American Society for Microbiology.
C. Alphaviruses produce membrane invaginations and spherules
Sindbis virus (SbV) and Semliki Forest virus (SFV) are the best studied examples of alphavirus replication in mammalian cells [reviewed by Salonen et al. (2005)]. Early electron microscopy studies showed that vesicular structures called cytopathic vacuoles between 600‐ and 2000‐nm diameter, accumulated in infected cells. The vacuoles contained 50‐nm‐diameter vesicles called spherules, many of which were aligned along the inside face of the vacuole and attached by a neck to the limiting membrane. The neck was often seen connected to an electron‐dense matrix extending into the cytoplasm. The observation that the cytopathic vacuoles contained NSPs required for RNA replication, cofractionated with lysosomal enzymes, and could be labeled with endocytic markers (Froshauer et al., 1988), led to the conclusion that they are sites of viral replication derived from endosomes and lysosomes. In many cases, the vacuoles were also connected to the rough ER by filaments and granular material containing the RNA polymerase.
1. The alphavirus replicase is located Within invaginations in cellular membranes
Alphavirus NSPs are synthesized in the cytoplasm and bind to endosomes and lysosomes to generate a replication complex. The replicase proteins are synthesized as a polyprotein (P1234). The P4 domain is the RdRp while P2 has NTPase and helicase activities, and P1 is the methytransferase required to cap RNA (Fig. 1C). The P1234 polyprotein locates to endosome or lysosome membranes via an amphipathic peptide sequence in P1 (Salonen et al., 2003). At this stage the P4 polymerase is cleaved from the polyprotein and functions with the remaining P123 protein to generate negative‐stranded RNA. Interestingly, once the P123 is processed to individual NSPs, the polymerase preferentially produces positive‐stranded RNA. Expression of individual NSPs does not lead to the formation of a cytopathic vacuoles or spherules. Formation of spherules requires interactions between NSP P1, P3, and P4 and the P123 polyprotein precursor complex (Salonen et al., 2003).
Rubella virus is a member of the Togaviridae family within the Alphavirus genus. Cells infected with Rubella virus also contain vacuoles containing spherules and these colocalize with lysosomal markers, suggesting use of lysosomes for replication. A fibrous material connects the vacuoles to the ER (Lee 1994, Magliano 1998), again suggesting strong similarities with SFV and SbV. Members of the alphavirus superfamily share homologies between proteins required for RNA replication, and this extends to plant viruses. Alfalfa mosaic virus replicase proteins colocalize with the plant vacuole (van der Heijden et al., 2001), and Turnip yellow mosaic virus uses the chloroplast outer envelope as a site for replication. Replication of Tobacco mosaic virus, a tobamovirus, is dependent on Arabdopsis proteins TOM1 and TOM2A that are integral membrane proteins of the tonoplast (Hagiwara et al., 2003). The tonoplast is a membrane compartment within plants that surrounds the vacuole/lysosome, suggesting plant alphaviruses also use the endosome/lysosome system as a site of replication. Infection of plants with alphavirus‐like superfamily viruses can also induce the formation of spherules (Prod'homme et al., 2001). There is evidence that Tobacco mosaic virus also uses the ER as a site of replication because the replicase enzyme and viral RNA are located on the ER of infected cells, and infection causes major changes in ER morphology (Reichel and Beachy, 1998), including ER aggregation and formation of lamella structures.
Flock house virus replicates in spherules in the outer membrane of mitochondria. The RNA polymerase ( protein A) of Flock house virus is the only protein required for RNA replication and is targeted directly to the mitochondrial outer membrane by hydrophobic amino acids at the N‐terminus. This sequence contains a mitochondrial localization signal and transmembrane domain that leaves the bulk of the protein exposed to the cytoplasm (Miller and Ahlquist, 2002). Brome mosaic virus replicates in yeast and has been studied extensively. The 1a and 2a replicase proteins are produced from separate viral RNAs. The 1a protein contains a C‐terminal helicase domain and an N‐terminus required for RNA capping. 1a is targeted to the cytoplasmic face of ER membranes and recruits the 2a polymerase to the replication complex (Schwartz et al., 2002). Importantly, replication of Brome mosaic virus on the cytoplasmic face of the ER in yeast induces membrane invaginations of 50 nm that are very similar to the spherules produced in endosomes and lysosomes during alphavirus infection of mammalian cells.
2. Membrane invaginations and spherules induced by alphviruses share similarity with virus budding
It has been suggested that the active formation of spherules to separate viral RNA from host responses is analogous to the coordinated assembly of viral proteins, which leads to capsid assembly, genome packaging, and budding (Ahlquist 2006, Schwartz 2002). The Brome mosaic virus replication complex contains viral 1a and 2apol proteins within spherules. Expression of 1a alone produces a shell containing hundreds of copies of 1a on the inside of 50‐nm spherules. In a capsid assembly model (Schwartz et al., 2002), vesicles of uniform size would arise if the 1a protein first made a planar lattice with hexameric symmetry on membranes and achieved curvature by localized rearrangement of 1a into pentamers. Interestingly, the formation of spherules is dependent on the relative levels of 1a and 2apol. When levels of 2apol are high, the spherules are lost, and 1a and 2apol assemble into flat lamella structures associated with the ER (Schwartz et al., 2004). One explanation for a failure to achieve curvature is that high levels of 2apol may interfere with this hexamer to pentamer transition. This is supported by the observation that when domains that allow association of 1a and 2apol are deleted, the 2apol is unable to alter the structure of spherules formed by 1a. The correct ratio of 1a and 2apol is clearly important for replication complex assembly and may be maintained during infection through inhibition of translation initiation of the 2a RNA.
D. The Flaviviridae replicate in vesicular packets and membraneous webs
1. The Flavivirus Replicase
In the Flaviviridae family, which includes the Flavivirus, Pestivirus, and Hepacivirus genera, the RNA genome encodes a polyprotein precursor that is cleaved by viral proteases to produce structural proteins from the N‐terminal region. The replicase of the Flaviviridae is made from NSPs, NS5A, NS5B, NS4B, and NS3–4A, found at the C‐terminus. With the exception of the polytopic NS4B membrane protein, which is inserted cotranslationally into the ER, the membrane‐anchored components of the complex are inserted into the cytoplasmic face of the ER after translation (Fig. 1B). The NS5B is the RdRp, and a C‐terminal stretch of 21 hydrophobic amino acids directs NS5B to the cytoplasmic face of the ER (Dubuisson 2002, Moradpour 2004). The NS3 protein has NTPase/helicase activity. NS3 is not a membrane protein but is recruited to the complex through association with membrane‐anchored NS4A. NS5A is also membrane associated, and association is mediated via 31 amino acids at the N‐terminus that form an amphipathic α‐helix (Brass 2002, Elazar 2003).
2. Membranes used for flavivirus replication are provided by the trans‐Golgi network
Replication of flaviviruses (e.g., Dengue, West Nile, and Yellow Fever viruses) takes place in membrane invaginations. For historical reasons, these are called vesicular packets [reviewed in Mackenzie (2005)]. They are larger (80‐ to 100‐nm diameter) than the 50‐nm alphavirus spherules, and form from the limiting membrane of the trans‐Golgi network (TGN) (Uchil 2003, Westaway 1997b). Infection by Kunjin virus leads to unique membrane structures thought to be derived from both the early and late secretory pathways. These include convoluted membranes and paracrystalline arrays derived from the rough ER and ERGIC, and vesicle packets derived from the TGN (Mackenzie 1999, Ng 1987, Roosendaal 2006, Westaway 1997b). The detection of dsRNA and viral NSPs (NS1, NS2A, NS3, and NS4A) within the vesicle packets points strongly to this being the site of RNA replication (Mackenzie 1998, Westaway 1997b). The vesicle packets associate closely with the convoluted membranes and paracrystalline arrays, which are thought to be the sites of proteolytic processing of NS3 and NS2B (Westaway et al., 1997b). These modified membranes are linked with the ER, and ultrastructural studies have shown virions present in the ER, cytoplasmic vesicles, Golgi cisternae, and vacuoles. The results suggest that membranes containing the spherules responsible for replication may become associated with the ER to facilitate delivery of genomes to viruses, budding into early compartments of the secretory pathway (Mackenzie and Westaway, 2001).
3. Hepacivirus replication occurs in association with the ER
Hepatitis C virus (HCV) is closely related to the flaviviruses, and its importance as a human pathogen has generated great interest in its mechanism of replication. Until, recently infection models have not been available to study the replication complex of HCV, and the studies discussed here have focussed on the expression of the entire polyprotein from replicons (Egger 2002, Gosert 2003). However, the recent production of a HCV that replicates efficiently both in vivo and in cell culture (Lindenbach 2006, Wakita 2005, Zhong 2005) will expand the possibilities for studying and understanding the viral replication cycle. HCV replication is thought to occur on membranes derived from the ER as all studies of NSPs have found them localized to this organelle (Dubuisson 2002, Hugle 2001, Kim 1999, Wolk 2000). Studies have also identified a “membraneous web” of membrane vesicles of ∼85‐nm diameter associated with the ER and a population of irregular double‐membraned vesicles. The web resembled the “sponge‐like inclusions” seen in the liver of chimpanzees infected with HCV, suggesting it is physiologically relevant. Interestingly, the great majority of the NSP synthesized by full–length genomes or subgenomic replicons may not be involved in RNA replication (Quinkert et al., 2005). The bulk of the NSPs associated with membranes isolated from cells expressing replicons is sensitive to protease, while in vitro replicase activity is resistant to protease and nuclease activity (El‐Hage 2003, Quinkert 2005). The results suggest that replication of HCV takes place within membrane vesicles, rather than on the surface of the membraneous web. These vesicles may be associated with the membraneous web, but the similarity between HCV and the flaviviruses leaves open the possibility that the membrane invaginations responsible for replication may also form in the TGN but be closely associated with the ER.
4. Flavivirus nonstructural proteins can induce membrane rearrangements
Studies have investigated which viral proteins are responsible for membrane rearrangements seen in cells infected with flaviviruses. The NS4A of Kunjin virus induces the characteristic convoluted membranes and paracrystalline arrays seen in flavivirus infections. The NS4A‐B protein also causes membrane rearrangement, but the highly condensed structures seen in infected cells are not produced until the NS2B‐3 protease cleaves NS4A free from NS4B (Roosendaal et al., 2006). The NS4B then translocates to the nucleus (Westaway et al., 1997a). Interestingly, this contrasts with HCV where NS4B (and NS4A‐B) (Egger 2002, Konan 2003) rather than NS4A is able to induce the membranous structures.
5. Flaviviruses can modulate the secretory pathway
Flaviviruses have been found to upregulate cell surface expression of MHC class I and II in response to interferon (King 1988, Liu 1989, Lobigs 2004). This is not caused by effects of the NS4A or NS4B proteins on membrane traffic; instead flavivirus infection increases expression of the ER peptide transporter, TAP1. This increases the supply of peptides that are necessary for the folding and export of newly synthesized MHC proteins from the ER. Increased TAP expression is mediated by increased transcriptional activity of p53 and can be induced in liver HepG2 cells by expression of the HCV core/capsid protein alone (Herzer 2003, Momburg 2001).
While the capsid/core protein is able to increase cell surface expression of MHC class I through increase expression of TAP1, expression of the HCV polyprotein has been shown to slow the movement of proteins through the secretory pathway of host cells (Konan et al., 2003). The rate of delivery of MHC class I to the plasma membrane in cells infected with HCV was reduced three‐ to fivefold relative to cured control cells. Expression of the precursor NS4A‐B was found to reduce ER‐to‐Golgi traffic two‐ to threefold (Konan et al., 2003), while the other NS proteins of HCV including NS4A and NS4B, individually or combined, were unable to interfere with the trafficking pathway. NS4B alone induces a membraneous web in cells (Egger et al., 2002), and both NS4A‐B and NS4B induce, and locate to, clustered and aggregated membranes looking very similar to the membraneous web seen in cells expressing replicons. In addition to aggregated membranes, NS4A/B also induces, but does not colocalize with, swollen vesicular structures. Theses swollen vesicles have a similar morphology to the vesicles induced by the 3A protein of Poliovirus, which swells ER membranes and blocks secretion between the ER and the Golgi apparatus (Doedens et al., 1997). Konan et al. (2003) hypothesize that the NS4A/B could be functioning in a similar manner to Poliovirus 3A.
E. The Nidovirales replicate in association with double‐membraned vesicles
1. The Nidovirus replicase is generated from two polyproteins
The Nidovirales order comprises the Arteriviridae, Coronaviridae, and Roniviridae families. The replicase gene is composed of two open reading frames termed ORF1a and ORF1b. ORF1b is generated from a frameshift in 1a, and both reading frames encode complex polyproteins processed by viral proteases (Gorbalenya 2006, Ziebuhr 2006). The arterivirus ORF1b encodes NSPs 9–12, including the RdRp (NSP9) and helicase (NSP10). The ORF1b, however, lacks hydrophobic domains able to target the replicase to membranes. Interestingly, the hydrophobic domains necessary for membrane targeting are encoded by ORF1a in NSP2, 3, and 5, suggesting that ORF1a proteins produce a scaffold to locate the viral replication–transcription complex to membranes (Fig. 1D) (Pedersen 1999, van der Meer 1998). A similar strategy is used by CoV, for example mouse hepatitis virus (MHV) and severe acute respiratory syndrome‐CoV (SARS‐CoV) (Prentice 2004a, Prentice 2004b), where transmembrane domains are located in NSP3, 4, and 6, and helicase and polymerase proteins are NSP12 and 13, respectively, and NSP16 encodes the methytransferase. The Nidovirales have the largest coding capacity of the single‐stranded RNA viruses, and not all the 16 NSPs have been studied in detail. It is possible that other proteins encoded by ORFs1a and 1b, such as RNA processing enzymes, are incorporated into the replication complex.
2. Sites of arterivirus and CoV replication are separate from sites of envelopment and budding
Several studies have investigated the intracellular sites of replication of equine arterivirus (EAV), MHV, and SARS‐CoV. Such studies are difficult because during nidovirus infection, the processes of replication and envelopment occur on different membranes, and these may merge during encapsidation. Furthermore, late during infection cells infected with MHV can form syncitia. Newly synthesized MHV viral RNA has been found in perinuclear sites colocalized with the RdRp (Shi et al., 1999), and depending on whether human or murine cells were infected, these sites colocalized with Golgi or ER membranes, respectively. Similar studies in mouse L cells report that the polymerase and newly synthesized RNA locate to late endosomes and endocytic carrier vesicles (van der Meer et al., 1999). This discrepancy is in part reconciled by later work showing that the subcellular distribution of the replicase proteins can change during the course of infection, since replicase proteins move to sites of envelopment in the ERGIC (Bost et al., 2001). This is supported by the finding that individual replicase proteins distribute differently following cell membrane fractionation (Sims et al., 2000). Membrane fractionation has also been carried out by Gosert et al. (2002), who showed that several proteins encoded by ORF1a and b were associated with membranes, and when observed by immunogold electron microscopy, these were associated with rosettes of double‐membraned vesicles 200–350 nm in diameter. The role of these vesicles in viral RNA replication was confirmed by in situ hybridization of labeled riboprobes. Double‐membraned vesicles are also seen in cells infected with EAV (Pedersen et al., 1999). EAV replicase proteins accumulate in perinuclear regions containing ERGIC and ER markers and colocalize with newly synthesized viral RNA, again suggesting sites of genome replication. Notably, similar structures can be produced by expression of arterivirus ORF1a‐encoded proteins NSP2–7, which contain the membrane proteins thought to tether the replicase to membranes.
3. The double‐membraned vesicles induced by arteriviruses and CoVs may be related to autophagosomes
Double‐membraned vesicles are usually rare in cells but are induced during autophagy. A role for autophagy during MHV infection is suggested because autophagy is induced in cells infected with MHV. Furthermore, in cells lacking Atg5, a protein required for the formation of autophagosomes, there is a 99% reduction in virus yield and MHV fails to induce double‐membraned vesicles (Prentice et al., 2004a). Electron micrographs show that the double‐membraned vesicles induced by SARS‐CoV extend from the ER and can be labeled with antibodies specific for replicase proteins. This suggests that, in common with MHV, the vesicles are a site of replication (Snijder et al., 2006). Even though all SARS‐CoV replicase proteins tested colocalize to punctate structures that accumulate near the nucleus, there are conflicting reports about their relationship with autophagosomes. In monkey Vero cells, the replicase proteins colocalize with autophagosomes identified using antibodies against LC3 (Prentice et al., 2004a). However, when autophagosomes are identified by expression of GFP‐LC3, the replicase proteins do not colocalize with the GFP signal (Snijder et al., 2006). The vesicles induced by SARS‐CoV are smaller at 100‐ to 300‐nm diameter than autophagosomes (500–1000 nm) and are labeled with ER markers. This has lead Snijder and colleagues to suggest that they are virus‐induced extensions to the ER, rather than bona fide autophagosomes (Pedersen 1999, Snijder 2006). The precise origins of the membrane crescents that form at the start of autophagy are unclear, and a number of studies have suggested they may form from the ER. This makes it possible that the double‐membraned structures may be autophagosomes that have been modified by an accumulation of viral protein. Determining if autophagy is beneficial to SARS‐CoV replication will have to await studies in cells where key proteins in the autophagy pathway have been removed or suppressed by gene silencing.
IV. Virus Factories and Inclusion Bodies Generated by Large DNA Viruses
A. Cytoplasmic virus factories formed by large cytoplasmic DNA viruses
The asfiviruses, poxviruses, iridoviruses, and the phycodnaviruses are large DNA viruses encoding hundreds of proteins from genomes ranging between 150 and 350 kbp. A comparison of protein sequences encoded by these viruses has suggested that they should be grouped together in a family of viruses called the nucleocytoplasmic large DNA viruses (NCLDV) (Iyer et al., 2001). Sequence similarities are seen in the major capsid proteins, redox enzymes that maintain disulphide bonds in the cytosol, and proteins that regulate apoptosis; and the family has been extended to include the giant mimivirus isolated from the ameba Acanthamoeba polyphaga (La Scola et al., 2003). Even though these viruses infect a diverse range of hosts from different phyla, including vertebrates [poxviruses, African swine fever virus (ASFV)], arthropods (entomopox, ASFV, chloriridoviruses), amphibians and fish (Ranavirus, Megalocytivirus, and Lymphocystivirus genera of the Iridoviridae family), marine algae (phycodnaviruses), and protozoa (mimivirus), they all generate cytoplasmic factories as major sites of virus assembly and replication (illustrated in Fig. 4 ). The factories share many similarities with one another, again suggesting that this diverse group of viruses may be related and that the need to produce a virus factory in the cytoplasm was generated early in virus evolution.
Figure 4.
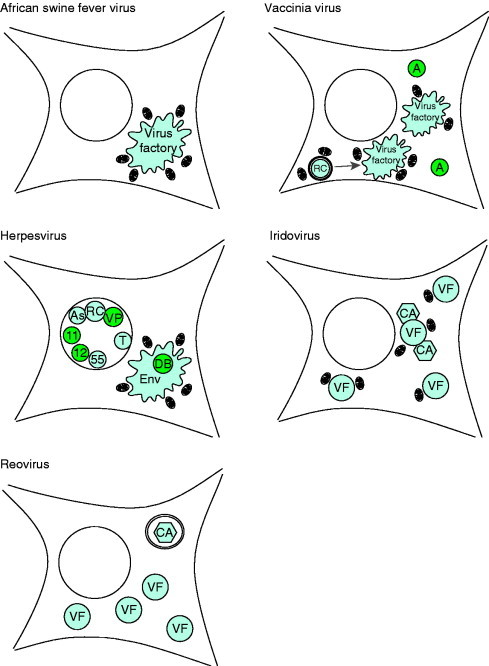
Schematics of inclusions induced during virus infection. ASFV induces single large perinuclear factories surrounded by mitochondria. Vaccinia virus induces multiple factories derived from membrane‐enclosed replication complexes (RC) both of which are associated with mitochondria. Certain poxviruses also induces electron‐dense A‐type inclusions (A). Human herpesvirus 1 induces capsid assembly sites, or assemblons (As), replication compartments (RC), inclusions of tegument proteins VP13/14 and VP22 (VP), and electron‐dense bodies of UL11 and UL12 gene products (11 and 12) in the nucleus. Human herpesvirus 2 also induces nuclear inclusions of UL55 gene product (55) and human herpesvirus 6 induces nuclear tegusomes (T). Herpesviruses induce cytoplasmic assembly sites where envelopment and some tegument are acquired (Env) in human herpesvirus 5, these sites include electron‐dense bodies (DB). Iridoviruses induce multiple cytoplasmic virus factories (VF) and crystalline arrays (CA), both of which associate with mitochondria. Reoviruses also induce multiple cytoplasmic virus factories (VF) and crystalline arrays (CA) that are enclosed within lysosomal membranes.
1. ASFV factories form next to the microtubule organizing center
ASFV is the sole member of the Asfivirus genus, family Asfarviridae but shares striking icosahedral similarity with the iridoviruses, phycodnaviruses, and mimivirus. ASFV is a large double‐stranded DNA (dsDNA) virus with a genome size ranging from 170 to 190 kbp. Gene expression is a regulated cascade and immediate early, early, early/late, intermediate, and true late gene types have been characterized to date. The virion has multiple concentric layers with an electron‐dense core at the center that contains the viral genome. A protein matrix surrounds the core, which in turn is enclosed by a lipid bilayer. Finally, the bilayer is surrounded by a protein capsid layer. ASFV can gain a third envelope when it buds from the plasma membrane at the tip of actin‐rich projections that resemble filopodia (Jouvenet et al., 2006). ASFV probably enters cells by receptor‐mediated endocytosis, but the steps following entry are poorly understood. It is possible that a viral core is delivered into the cytoplasm intact; alternatively, cores may dissociate in endosomes requiring some mechanism of genome delivery across the endosome membrane. Genome replication occurs both in the nucleus and cytoplasmic factories. Transfer to the nucleus may involve microtubule transport since late gene expression is inhibited by agents that depolymerize microtubules and the dominant‐negative dynein motor protein p50‐dynamitin (Alonso 2001, Heath 2001). ASFV does not produce nuclear inclusions analogous to those seen in herpesvirus and adenovirus infection, but there is evidence that small fragments of viral DNA are synthesized in the nucleus. The major site of ASFV DNA replication is, however, the virus factory (Rojo et al., 1999).
a. Cytoplasmic factories formed during ASFV infection are assembled at the microtubule organizing center
ASFV induces one principal factory in the cytoplasm during infection. Electron microscopy shows that the virus factory excludes obvious cellular organelles and contains mostly viral DNA, viral proteins, virus‐induced membranes, and partially and fully assembled virions (Table I ; Fig. 5A ; Brookes 1996, Moura Nunes 1975, Rouiller 1998). The mechanisms that target viral proteins, virus‐induced membranes, and viral DNA to the ASFV factories are poorly understood. Immunofluorescence staining for viral structural proteins generally reveals a strong signal at the factory and a weaker signal in the cytoplasm. The B602Lp protein (CAP80), which is a viral chaperone involved in folding and membrane recruitment of the major capsid protein, p73, is, for example, absent from the virus factories (Cobbold 2001, Epifano 2006). This suggests that p73 is synthesized and folded in the cytoplasm and then recruited to factories. Similarly, the viral dUTPase, which is necessary for efficient replication, is excluded from the viral factory (Oliveros et al., 1999). Since the bulk of viral DNA synthesis occurs in the factory (García‐Beato et al., 1992), it is not easy to explain how the viral dUTPase edits uracil from progeny viral genomes, without being present at the site of viral DNA synthesis and encapsidation. ASFV factories disperse when cells are incubated with drugs that depolymerize microtubules (Heath et al., 2001) suggesting their formation involves microtubule motors. This may involve dynein motor proteins since p50‐dynamitin, a dominant‐negative version of the dynein motor, prevents both late ASFV gene expression (Heath et al., 2001) and vimentin recruitment to factories (see below and Stefanovic et al., 2005). Yeast‐two‐hybrid screens and in vitro pull‐down experiments show that one ASFV structural protein, p54/j13Lp, interacts with dynein (Alonso et al., 2001). While direct binding of p54/j13Lp to the motor protein has not been observed in infected cells, it is possible that the protein is involved in transporting some viral proteins into factories. The protein locates to virus factories and deletion of the E183L gene encoding p54/j13Lp generates factories that lack viral membranes, the major capsid protein p73, and the polyprotein precursors (pp220, and pp62) of the viral matrix (Epifano 2006, Rodríguez 2004). P54/j13Lp is a membrane protein with the bulk of the protein, including the dynein‐binding motif, exposed to the cytosol. The p73 capsid protein and pp220 polyprotein associate with membranes before assembly into viruses (Cobbold 1998, Cobbold 1996, Heath 2003). If these membranes contain p54/j13Lp, it would provide a means of allowing recruitment to factories by retrograde transport along microtubules.
Table I.
Known contents of viral inclusions induced by different virus families. Each section includes a brief description of the viral inclusion and lists both viral and host-cell proteins confirmed to localize within, or associate with, the specified structure
| Asfarviridae, Asfivirus African swine fever virus | References |
| Cytoplasmic virus factory | |
| Appearance and contents of viral origina | |
| Viral membranes, assembling and complete particles, electron dense condensations, viral DNA, A224L IAP apoptosis inhibitor, A104R (5AR) DNA binding histone like, A137R p11.5, B119L Erv1p homologue, B438L p49, B646L p73 major capsid protein, CP2475L pp220 precursor to p150; p37; p34 and p14 CP530R pp62 precursor to p35 and p15, O61R p12 attachment, D117L (i1L) transmembrane, S273R (i6R) cysteine protease, H108R (j5R) membrane, E183L p54 ( j13L) dynein interacting, E199L ( j18L) membrane, E120R (k3R) p14.5 DNA binding necessary for viral exit from factory | Alcamí 1993, Alonso 2001, Andrés 1997, Andrés 2001, Borca 1996, Brookes 1998a, Brookes 1998b, Carrascosa 1986, Chacón 1995, Cobbold 1996, Galindo 2000, García‐Beato 1992, Heath 2001, Hingamp 1992, Jouvenet 2005, Jouvenet 2004, Martinez‐Pomares 1997, Moura Nunes 1975, Rodríguez 2006, Rouiller 1998, Sanz 1985, Simón‐Mateo 1997, Sun 1996, Vigário 1967 |
| Contents of cellular origin | |
| Ubiquitin, hsp70 chaperone, γ‐tubulin, Pericentrin, p21, mdm1 Surrounded by: ER membranes, vimentin, p230 Golgin, mitochondria, and tubulin. | Granja 2004, Heath 2001, Hingamp 1992, Jouvenet 2005, Netherton 2004, Netherton 2006, Rojo 1998, Rouiller 1998, Stefanovic 2005 |
| Poxviridae, Chordopoxvirinae, Orthopoxvirus Vaccinia virus | |
| Cytoplasmic A‐type inclusion | |
| Contentsb | |
| Electron dense, IMV, A26L (WR148 and WR149) myristylated | Patel et al., 1986 |
| Cytoplasmic B‐type inclusion, virosome, or virus factory | |
| Appearance and contents of viral originb | |
| Electron dense viroplasm, viral crescents, IV and IMV, viral DNA, A2.5L (WR121) redox, A3L (WR122) p4b core, A4L (WR123) p39 core, A9L (WR128) membrane, A10L (WR129) p4a core, A11R (WR130) phosphoprotein, A13L (WR132) membrane ERGIC, A14L (WR133) membrane ERGIC, A14.5L (WR134) membrane virulence, A15L (WR135) viroplasm/membrane association, A16L (WR136) cell‐fusion/entry, A17L (WR137) membrane assembly, A18R (WR138), A30L (WR153) viroplasm/membrane association, A35R (WR158) virulence, A40R (WR165) SUMO‐1 modified, A45R (WR171) virion superoxide dismutase homologue, B1R (WR183) protein kinase, D4R (WR109) uracil DNA glycolase, D8L (WR113) p32, D13L (WR118) p65 scaffold, E3L (WR059) dsRNA binding E5R (WR061) E8R (WR064) ER protein, surrounds virosome E10R (WR066), F10L (WR049) protein kinase viroplasm/membrane association, F17R (WR056) actin tail formation G7L (WR085) viroplasm/membrane association, H3L (WR101) p35 core membrane, H5R (WR103) transcription factor VLTF‐4, I3L (WR072) ssDNA binding, I4L (WR073) ribonucleotide reducatase large subunit, J1R (WR093) core viroplasm/membrane association, L1R (WR088) myristylated, L4R (WR091) p25K core; ssDNA/ssRNA binding, Ectromelia zinc finger binding protein (absent in Copenhagen, fragment in WR), Cowpox CP77 host range factor, WR011 E3‐ubiquitin ligase. | Almazán 2001, Beaud 1997, Betakova 2000, Chiu 2005, Cudmore 1996, da Fonseca 2000, Davis 1993, De Silva 2005, Domi 2000, Krijnse‐Locker 1996, Murcia‐Nicolas 1999, Nerenberg 2005, Ojeda 2006, Palacios 2005, Pedersen 2000, Reckmann 1997, Resch 2005, Risco 1999, Roper 2006, Salmons 1997, Senkevich 2002, Sodeik 1995, Szajner 2004a, Szajner 2004b, Szajner 2004c, Tolonen 2001, Vanslyke 1994, Welsch 2003, Wolffe 1995, Yeh 2000, Yuwen 1993 |
| Contents of cellular origin | |
| HMG20A viral genome binding protein, hSP90; transient association, Ubiquitin, ying‐yang 1 transcription factor, TBP transcription factor, SP1 transcription factor, RNA polymerase II, SUMO‐1, ERGIC‐53c Surrounded by: vimentin and mitochondria. | Broyles 1999, Dales 1961, Hsiao 2006, Hung 2002, Husain 2003, Nerenberg 2005, Oh 2005, Palacios 2005, Risco 2002, Wilton 1989 |
| Iridoviridae, Ranavirus | |
| Cytoplasmic virus factories | |
| Appearance and contentsf | |
| Electron lucent, virus, viral DNA, 108K early protein, 57K, 55K major capsid protein (ORF 90R in FV3), 38K, 17K, 16K Rana grylio virus dUTPase (ORF 63R in FV3). Surrounded by vimentin, rough ER, mitochondria and polysomes. | Chinchar 1984, Darlington 1966, Huang 2006, Murti 1983, Murti 1989, Zhao 2007 |
| Herpesviridae, alphaherpesvirinae, simplexvirus and varicellovirus | |
| Nuclear replication compartment | |
| Contents of viral origind | |
| UL3, UL4 virion, UL5 helicase‐primase, UL6 DNA cleavage/packaging, UL8 helicase‐primase, UL15 DNA packaging, UL17 tegument DNA packaging, UL18 DNA packaging, UL19 ICP5 major capsid protein, UL26.5 ICP35 DNA packaging, UL29 ICP8 single strand binding UL30 DNA polymerase, UL32 DNA packaging, UL33 DNA packaging, UL35 VP26 p12 capsid, UL42 65K DNA polymerase accessory, UL49 VP22 tegument, UL52 helicase‐primase UL54 ICP27 regulatory, α0 UL57 ICP0 transactivator, α4 ICP4 regulatory, α22 US1 ICP22 regulatory, US1.5 truncated, US1 regulatory. | Barnard 1997, de Bruyn Kops 1998, Everett 1994, Goodrich 1990, Jahedi 1999, Knipe 1987, Lamberti 1998, Leopardi 1997, Liptak 1996, Markovitz 2000, Olivo 1989, Randall 1986, Reynolds 2000, Taus 1998; Ward et al., 1996 |
| Contents of cellular origin | |
| RNA polymerase II, EAP ribosome component, proliferating cell antigen, retinoblastoma protein, p53, DNA ligase 1, DNA polymerase α, promyelocytic leukemia (PML), DNA‐PKcs, Ku86 nonhomologous end joining, Bloom syndrome gene product, breast cancer‐associated gene 1 protein, MSH2, Rad50, WRN RecQ helicase family member, BRG1 or BRM‐associated factor 155, brahma‐related gene‐1 protein, brahma protein, histone deacetylase 2, hSNF2H, mSin3a, TATA binding protein (TBP), TBP‐associated factors. | Leopardi 1997, Lukonis 1997, Quadt 2006, Taylor 2004, Wilcock 1991 |
| Nuclear sites of capsid assembly or assemblons | |
| Contents of viral origind | |
| UL7 (HHV‐2), UL14 (HHV‐2) tegument, UL16 capsid, UL19 ICP5 major capsid protein, UL26.5 ICP35 DNA packaging, UL27 DNA packaging, UL35 VP26 p12 capsid, UL38 VP19c capsid assembly, UL43.5, UL55. | de Bruyn Kops et al., 1998; Goshima 1998, Nalwanga 1996, Nozawa 2002, Wada 1999, Ward 1996a, Ward 1996b, Yamada 1998 |
| Contents of cellular origin | |
| Actin, myosin 5a actin motor | Feierbach et al., 2006 |
| Cytoplasmic assembly and envelopment site | |
| Contents of viral origind | |
| Membranes, vacuoles, capsids and enveloped virus UL19 (HHV‐2) VP5 major capsid protein UL27 (HHV‐2) gB VP7 UL36 (HHV‐2) ICP1–2, tegument UL46 (HHV‐2) tegument, UL48 (HHV‐2) tegument | Kato 2000, Murata 2000, Nozawa 2004, Watanabe 2000 |
| Contents of cellular origin | |
| Mitochondria, γ‐tubulin, hsp40 chaperone, hsp70 chaperone, GM130 Golgi marker | Murata 2000, Nozawa 2004 |
| Herpesviridae, Betaherpesvirinae, Cytomegalovirus Human herpesvirus 5 | |
| Cytoplasmic assembly sites | |
| Appearance and contentse | |
| Membranes, vacuoles, capsids, enveloped virus and dense bodies (see below) UL23 tegument, UL24 tegument, UL25, UL32 pp150, UL43 tegument, UL53, UL55 gB, UL73 gN, UL75 gH, UL80 p38, UL83 pp65–69 UL99 pp28, gp65. | Adair 2002, Battista 1999, Dal Monte 2002, Landini 1991, Pignatelli 2002, Sanchez 2000 |
| Cytoplasmic dense bodies | |
| Appearance and contentse | |
| Homogenous electron dense material, UL73 gN, UL83 p65–69. | Craighead 1972, Pignatelli 2002 |
| Herpesviridae, Betaherpesvirinae, Roseolovirus Human herpesvirus 6 | |
| Nuclear/cytoplasmic tegusome | |
| Appearance | |
| Enveloped nucleocapsids, virus with tegument in cytoplasmic invagination of nucleus | Roffman et al., 1990 |
| Adenoviridae, Mastadenovirus | |
| Nuclear small fibrillar masses, ssDNA accumulation sites or early replicative sites | |
| Appearance and contents | |
| Viral ssDNA replication, 72kDa ssDNA binding protein, viral RNA (early) | Puvion‐Dutilleul 1990, Puvion‐Dutilleul 1992 |
| Nuclear fibrillogranular matrix or peripheral replicative sites | |
| Appearance and contents | |
| Viral RNA (late), E1A oncogenic proteins, E4‐ORF3, 72kDa ssDNA‐binding protein, DNA polymerase, terminal protein, PML, splicesomes, sp100, hsp70, nuclear factor 1 | Bosher 1992, Carvalho 1995, Murti 1990, Puvion‐Dutilleul 1991, Puvion‐Dutilleul 1994 |
| Nuclear virus‐induced compact ring | |
| Appearance and contents | |
| Viral RNA (late), pIVa2 DNA packaging | Lutz et al., 1996 |
| Nuclear clear amorphous inclusion | |
| Appearance and contents | |
| pIX, PML, PKR, CK2α | Lutz 1996, Rosa‐Calatrava 2001, Rosa‐Calatrava 2003, Souquere‐Besse 2002 |
| Nuclear electron‐translucent area | |
| Appearance and contents | |
| Virus, protein crystals, pentons, hexons, fiber protein, pIX, L1 52 kDa, L1 55 kDa, PML, PKR, CK2β | Puvion‐Dutilleul 1995, Puvion‐Dutilleul 1999, Souquere‐Besse 2002 |
| Other pIVa2 positive nuclear structures induced during adenovirus 5 infection | |
| Name | |
| Nuclear irregular electron‐dense amorphous inclusion, Nuclear regular electron‐dense amorphous inclusion, nucleolus electron‐dense virus‐induced globules, nucleous irregular amorphous inclusion. | Lutz et al., 1996 |
| Reoviridae, Orthoreovirus | |
| Cytoplasmic virus factories | |
| Appearance and contents | |
| Filamentous or globular dependent on μ2, phase and electron dense, viral RNA, virus, σNS nonstructural, μ1 outer‐capsid, μ2 nonstructural, μNS nonstructural, λ1 core surface, λ2 core surface, λ3 RNA polymerase, σ2 core surface, σ3 structural, ubiquitin, microtubules, vimentin (association with). | Becker 2001, Becker 2003, Broering 2004, Cashdollar 1994, Dales 1965b, Miller 2004, Sharpe 1982, Silverstein 1970 |
| Reoviridae, rotavirus | |
| Cytoplasmic virus factories | |
| Appearance and contents | |
| Electron‐dense viroplasm, assembling and complete double‐shelled particles VP2, VP6, VP9, NSP2, NSP5, NSP6 | Altenburg 1980, González 2000, Petrie 1982, Petrie 1984, Silvestri 2004, Silvestri 2005 |
African swine fever virus gene nomenclature is based on that for the Badajoz 1971 vero adapted strain with that of the Malawi Lil 20/1 strain in parentheses.
Vaccinia virus gene nomenclature is based on that for the Copenhagen strain with that of the western reserve strain in parentheses.
One report places in ERGIC‐53 within the virosome (Risco et al., 2002), one report places it outside (Husain and Moss, 2003).
Open reading frames from human herpesvirus 1 (herpes simplex virus 1) unless specified otherwise.
Open reading frames from human herpesvirus 5 (human cytomegalovirus) unless specified.
Proteins specified by frog virus 3 unless indicated otherwise.
Figure 5.
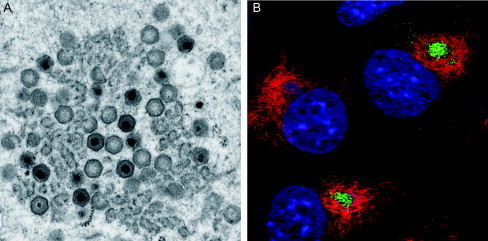
(A) Electron micrograph of an ASFV factory showing partially assembled, empty and fully mature capsids as well as electron‐dense viroplasm accumulating around viral membranes. Image courtesy of P. Hawes, J. Simpson, and P. Monaghan, Bioimaging Group, IAH‐Pirbright. (B) Confocal micrograph of ASFV‐infected cells immunolabeled with antimajor capsid protein (green) and vimentin (red) and stained with a DNA dye (blue). Note vimentin cages enclosing ASFV factories. Reprinted from Monaghan et al. (2003) with permission from Blackwell Publishing, Inc.
b. ASFV factories recruit intermediate filaments and resemble aggresomes
The formation and morphology of ASFV factories closely resemble the formation of aggresomes (Heath et al., 2001), a cellular response to accumulation of misfolded protein aggregates (Johnston et al., 1998). Aggresomes are microtubule‐dependent inclusions containing protein aggregates that form next to the microtubule‐organizing center (MTOC). Aggresomes recruit cellular components needed to deal with the problems associated with a buildup of aggregated misfolded protein. These include cellular chaperones and proteasomes to facilitate protein folding and/or degradation and mitochondria that may provide the ATP required for folding and proteolysis. The most striking structural changes seen during aggresome formation are the collapse of the intermediate filament protein, vimentin, into a cage surrounding the protein aggregates and the gross fragmentation of the Golgi apparatus. ASFV factory formation shows many similarities with this response to protein aggregation. Factory formation is preceded by clearance of cytoplasmic proteins from perinuclear areas around the MTOC. Vimentin then concentrates at the MTOC where it forms an aster aligned along microtubules (Stefanovic et al., 2005). Following the onset of virus DNA replication and synthesis of late structural proteins, the vimentin aster is rearranged into a cage around the factory (Fig. 5B; Heath 2001, Monaghan 2003, Stefanovic 2005). During this period, mitochondria and cellular chaperones are recruited to the factory (Heath 2001, Rojo 1998). Formation of vimentin cages in ASFV‐infected cells is linked to phosphorylation of vimentin at serine 82 by calcium calmodulin‐dependent protein kinase II (CamKinase II) (Stefanovic et al., 2005), and drugs that inhibit CamKinase II activity block late gene expression and vimentin rearrangement. As will be discussed for poxviruses and iridoviruses, the vimentin cage may form a physical scaffold within the factory, or act as a cage to prevent movement of viral components into the cytoplasm. Chaperones recruited to the factory may facilitate folding of viral structural proteins during assembly, as has been shown for other viruses. The proximity of mitochondria to viral factories may provide the ATP that is required for ASFV assembly (Cobbold et al., 2000) or be indicative of an antiviral response as mitochondria are effectors of apoptosis. Taken together these results suggest that a cellular response originally designed to deal with the buildup of protein aggregates in cells is used by ASFV to generate a site specialized for virus assembly. As will be described later, similarities between aggresomes and virus assembly sites are also seen for the iridoviruses and poxviruses.
Following the onset of ASFV DNA replication, the microtubule network becomes disorganized. Microtubules are partially excluded from virus factories and form bundles and concentric rings in the cytoplasm (Jouvenet and Wileman, 2005). ASFV infection leads to disassembly of γ‐tubulin and pericentrin from the centrosome, and the centrosome becomes less able to nucleate microtubules. At the same time microtubules are stabilized by acetylation (Jouvenet et al., 2004). Since pericentrin and γ‐tubulin play key roles in microtubule organization and nucleation at the MTOC, their loss from the centrosome, coupled with acetlylation of tubulin, may explain the rearrangement of microtubules induced by ASFV. The reasons for these profound effects on microtubules are not known but they may facilitate disruption of the virus factory allowing release of assembled viruses into the cytoplasm.
c. Membrane rearrangements caused by ASFV infection perturb the secretory pathway
Current models for ASFV envelopment in virus factories predict that viral membranes are obtained from the ER. The major structural proteins are recruited from the cytoplasm onto the cytoplasmic face of the ER, and after which protein–protein interactions between these, and possibly viral proteins targeted to the ER lumen, lead to constriction of ER cisternae and clearance of host proteins from the ER lumen prior to envelopment (Andrés 1998, Netherton 2004, Netherton 2006, Rouiller 1998). This is consistent with low levels of ER proteins observed at ASFV assembly sites by immunoelectronmicroscopy (Rouiller et al., 1998) and standard fluorescence microscopy where ER proteins appear to be actively excluded from areas of viral replication (Andrés 1998, Netherton 2004). In addition to effects on the ER, ASFV also affects the structure and function of later Golgi compartments of the secretory pathway (McCrossan 2001, Netherton 2006). Golgi structure is linked to microtubule organization and the changes seen during infection may in part be related to effects of ASFV infection on centrosome and microtubule function listed above. ASFV infection causes dispersal of ERGIC marker protein ERGIC‐53, the peripheral Golgi protein GM130, and late Golgi protein GalNac‐T2 transferase, suggesting disruption of ERGIC and Golgi membrane compartments. Most striking is the complete loss of the TGN. TGN loss is dependent on microtubules and involves dispersal of the TGN into separate vesicle populations containing either peripheral Golgi proteins or the integral membrane protein, TGN46. Not surprisingly, this dispersal slows the transport of proteins through the secretory pathway. ASFV slows the delivery of newly synthesized lysosomal enzymes to lysosomes (McCrossan et al., 2001), and in macrophages reduces transport of newly synthesized MHC class I to the plasma membrane (Netherton et al., 2006). Thus, in common with picornaviruses, disruption of the secretory pathway by ASFV has the potential to slow the transport of important immunomodulatory proteins to the surface of infected cells and may mask them from immune surveillance.
2. Poxviruses generate virus factories and inclusions
Poxviruses are large dsDNA viruses with genomes ranging from 130 to 375 kbp. Poxvirus gene expression follows the regulated cascade of other large dsDNA viruses with early, intermediate, and late transcripts described. Poxvirus progeny genomes are replicated exclusively in the cytoplasm in virus factories. The virus encodes all the enzymes necessary for transcription and replication of its genome. Genetic analysis has identified a minimum of five viral genes necessary for genome replication, these are A20R, B1R, D4R, D5R, and E9L encoding the DNA polymerase processivity factor, serine/threonine protein kinase, uracil DNA glycosylase, DNA‐independent nucleoside triphosphatase, and the DNA polymerase, respectively (De Silva 2005, Evans 1995, Millns 1994, Punjabi 2001, Rempel 1990, Sridhar 1983). Only the product of the D4R gene, encoding the viral DNA glycosylase, has been confirmed to localize to the site of genome synthesis (De Silva and Moss, 2005), and it would be interesting to discover the subcellular location of the other members of the minimum replicase. When viewed by electron microscopy, infectious virions have a striking brick‐shaped morphology, and different forms of virus are documented which vary in degree of complexity [for review, see Condit et al. (2006)]. The interior of all poxvirus particles contains the virus core which houses the viral genome. Cores are enveloped in virus factories to produce the intracellular mature virus (IMV), which is fully infectious. Additional envelope layers gained at the TGN give rise to intracellular enveloped viruses (IEV), which after budding through the plasma membrane form cell‐associated and extracellular enveloped viruses (CEV and EEV). Poxviruses induce two principal inclusions during infection, the A‐type inclusion that is nonreplicative and the B‐type inclusion where virus replication and assembly occur in the virus factory (Fig. 4; Kato et al., 1959).
a. Poxvirus A‐type inclusions contain the mature intracellular virus but not enveloped viruses
A‐type inclusions are cytoplasmic bodies of dense homogeneous matter that contain mature virus particles and are studded with polyribosomes (Fig. 6A ) (Ichihashi et al., 1971). A‐type inclusions are extremely rare in vaccinia, variola, and rabbit pox infections but are prominent in cowpox, ectromelia, fowlpox, and canarypox infections where they are also referred to as Downie, Marchal, Bollinger, and Burnet bodies, respectively (Kato et al., 1959). The major component of A‐type inclusions is the product of the A26L gene or its equivalents. In vaccinia, A26 is truncated and produces a protein of 92–94 kDa whereas the full‐length gene in cowpox encodes a protein of 160 kDa (Patel et al., 1986), both versions are myristylated (Martin et al., 1999). Immunfluorescence analysis of cells infected with Vaccinia virus with antibodies raised against A26 does reveal multiple A‐type inclusions in the cytoplasm, but they are much smaller than those seen in cells infected with cowpox, and do not contain virus particles (Patel et al., 1986). In cells infected with wild‐type cowpox, only IMV particles were observed within A‐type inclusions, but treatment with rifampicin, a drug that blocks poxvirus maturation at an early stage in morphogenesis, caused aberrant immature virus particles to integrate into the inclusions (Ichihashi et al., 1971). The factor necessary for occlusion of viral particles in A‐type inclusions has been identified as the 4c core protein (McKelvey 2002, Shida 1977, Ulaeto 1996). It has been hypothesized that 4c retains vaccinia virions within the cell as IMVs in A‐type inclusions preventing their transport to the TGN for envelopment and maturation to the IEV types of virion (McKelvey et al., 2002). A‐type inclusions are predicted to protect IMVs during transport between hosts akin to that of the polyhedra that occlude entomopox and baculoviruses (Rohrmann, 1986). Therefore, EEVs may be important for cell‐to‐cell spread, while IMVs (whether occluded or not) may be more important for host‐to‐host spread (McKelvey et al., 2002).
Figure 6.
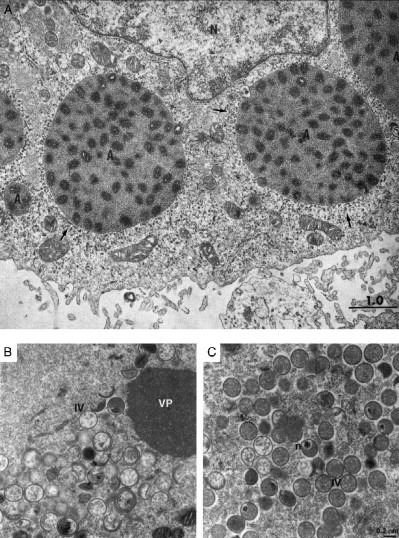
(A) Electron micrograph of A‐type inclusions from cowpox‐infected cells, showing intracellular mature virus in electron‐dense inclusions (A) surrounded by polyribosomes (arrows). Reprinted from Ichihashi et al. (1971) with permission from Elsevier. (B and C) Electron micrographs of factories of recombinant Vaccinia virus encoding the A15L gene under the control of the lac operon under nonpermissive (B) and permissive (C) conditions. Note empty immature virus particles (IV), viral crescents in an electron‐lucent environment, and a separate homogenous viroplasm (VP) in panel B and compare to wild‐type like conditions in panel C, which include immature virus with electron‐dense centers and particles containing nucleoids (n). Reprinted from Szajner et al. (2004a) with permission from Elsevier.
b. Poxvirus B‐type inclusions are factories and are the main sites of replication and assembly
B‐type inclusions originally called Guarnieri bodies (Guarnieri, 1893) are the primary replication centers of the poxviruses, now generally referred to as virosomes or virus factories (Fig. 6C). Electron microscopic analysis of B‐type inclusions revealed a granular matrix that was denser than the surrounding cellular material and in a defined area of the cytoplasm called viroplasm (Dales 1961, Higashi 1973). The factories also contain viral crescents consisting of membrane and viral proteins associated with viroplasm, spherical immature virus, and IMVs (Dales and Siminovitch, 1961). Factories are surrounded by mitochondria, increase in number and size during the replication cycle and can occupy the majority of the cytoplasm at late times of infection (Dales and Siminovitch, 1961).
The assembly and envelopment of Vaccinia virus within virus factories has been the subject of many studies and is discussed in papers and reviews (Griffiths 2001, Heuser 2005, Hollinshead 1999, Sodeik 2002). Here, we will review some of the early steps that lead up to the start of genome replication and factory production. These have also been described in a review (Schramm and Krijnse‐Locker, 2005). It is generally believed that infection results in the delivery of viral cores into the cytoplasm. Cores are seen associated with microtubules (Carter 2003, Mallardo 2001, Ploubidou 2000) and may use microtubules to reach perinuclear sites that will eventually house the virus factories. Viral cores can transcribe as many as 100 early mRNAs before the onset of DNA replication, and these early mRNAs appear in discrete foci that associate with microtubules, contain polyribosomes and other translational machinery. It is unlikely that foci involved in transcribing early RNAs mature into viral replication sites because they do not initiate DNA synthesis (Mallardo et al., 2002). It is likely that each infecting virus can induce its own replication center (Cairns, 1960), but it is not clear where in the cell the cores initiate DNA synthesis. It has been suggested that the onset of DNA synthesis may occur at peripheral sites and therefore precedes delivery to the perinuclear region of the cell. When cells are incubated with hydroxyurea to prevent the onset of viral DNA replication, it is possible to localize viral DNA released into the cytoplasm. Under these conditions, viral genomes are seen at several discrete sites that contain B1 protein kinase, E8 membrane protein, I3 ssDNA‐binding protein, and H5 late transcription factor (Domi 2000, Welsch 2003). After removal of hydroxyurea, these foci begin to make new viral DNA, showing that they are sites of DNA replication. Live cell imaging studies have shown that these initial sites of DNA replication form in the cell periphery and then move toward the nucleus where they coalesce into large structures (Schramm and Krijnse‐Locker, 2005).
Electron micrographs suggest that sites of DNA release from cores are intimately associated with ER membranes and become completely enclosed by them during the initial stages of DNA replication (Mallardo et al., 2002). This process is likely facilitated by the E8R gene product which is a membrane protein localized to the ER and early Golgi membranes, has DNA‐binding activity, and is able to capture viral genomes (Doglio 2002, Tolonen 2001). These ER‐enclosed genomes are short‐lived structures because they are not seen once viral crescents, IV and IMVs, appear in factories (Tolonen et al., 2001). The sites of DNA replication are also separate from the foci involved in transcribing early RNAs, and it is interesting to consider how the cores are separated from newly transcribed RNA. Viral cores and sites of RNA transcription both align on microtubules and partially colocalize with the L4 core DNA‐binding protein (Mallardo et al., 2001). The L4 protein is able to bind microtubules (Ploubidou et al., 2000) and may be involved in separating RNA from cores along microtubule tracks (Mallardo et al., 2001).
Inducible recombinants or temperature‐sensitive mutants grown under nonpermissive conditions can give further insight into the early stages of inclusion formation. Electron micrographic analysis of the factories formed under these conditions yield striking images of distinct inclusions of homogeneous electron‐dense viroplasm next to empty spherical immature virions (Fig. 6B and C) (Szajner 2001, Szajner 2003, Szajner 2004a). A seven‐protein complex comprising the gene products of the A15L, A30L, D2L, D3L, F10L G7L, and J1R open reading frames has been identified as being necessary for association of viral membranes with the viroplasm (Szajner et al., 2004a). Consistent with this role, all of these proteins are known to localize to the virus factory except D2 and D3 (Table I); however, these have been identified as core proteins (Dyster and Niles, 1991) so are likely to reside at viral assembly sites. Localization of D13L to the virus factory is sensitive to the antibiotic rifampicin (Miner and Hruby, 1989), and treatment with this drug induces irregular shaped viral membranes instead of the well‐defined hemispherical viral crescents seen in natural infection (Moss 1969, Pennington 1970). Therefore, it was suggested that D13L may act as a scaffold on which the viral membrane is shaped, allowing correct association with the viroplasm (Mohandas and Dales, 1995). Deep etch electron microscopy has confirmed this role for D13L, as it forms the honeycomb lattice identified as the outer coat of the viral membrane of immature virions (Heuser 2005, Szajner 2005). Interestingly, D13L shares a structural similarity with structural proteins from many other virus families, including those of the other large dsDNA viruses (Benson et al., 2004). It will be interesting to see if the structural similarities to D13L translate to functional similarities in the assembly strategies of other viruses.
c. Poxvirus infection recruits host proteins into factories and rearranges cellular organelles
Vaccinia virus recruits a number of cellular proteins to the viral factory. Ying‐Yang 1 (YY1), TBP, SP1 transcription factors, and RNA polymerase II are recruited from the nucleus to the factory (Broyles 1999, Oh 2005, Wilton 1989). YY1 is a nuclear transcription factor that can activate late viral promoters and although poxviruses encode most of the genes necessary for transcription, there is evidence that cellular factors may be required for intermediate and late gene expression (Lackner 2000, Rosales 1994, Wright 2001). The function of the other transcription factors in viral replication is unknown. They may be necessary for viral transcription like YY1, or perhaps they are sequestered into the factory to divert them from their normal roles in the nucleus, or their presence may represent an antiviral response by the cell. The presence of RNA polymerase II in the viral factory is a surprise because the virus encodes its own RNA polymerase activity which accounts for at least 9 ORFs and ∼7% of the genome capacity [Western Reserve (WR) strain]. Another cellular protein recruited from the nucleus to the cytoplasm is the HMG20A protein. This protein can bind the viral genome and has been implicated in host range restriction of Vaccinia virus in Chinese hamster ovary cells (Hsiao et al., 2006). During unproductive infection by Vaccinia virus, HMG20A is recruited from the nucleus to the factory where it binds viral DNA. If the cowpox host range gene CP77 is artificially introduced into Vaccinia virus then CP77 also enters the virus factory and binds to HMG20A; the cellular protein then dissociates from the viral genome and replication proceeds (Hsiao et al., 2006).
As seen for iridovirus and ASFV replication sites, vaccinia factories are surrounded by a vimentin cage (Risco 2002, Schepis 2006) and recruit molecular chaperones (Hung et al., 2002), suggesting similarity with aggresomes. Many proteins targeted to aggresomes are ubiqutinated, and most poxviruses encode a RING protein that is both a functional ubiquitin ligase and a virulence factor (Nerenberg et al., 2005). Exceptions to this are the two most common laboratory strains of vaccinia, Copenhagen and WR. The RING protein from the IHD‐W strain of vaccinia is capable of directing transfected tagged ubiquitin to WR virus factories (Nerenberg et al., 2005); however, it is unknown if native ubiquitin is localized to WR factories. The product of the A40R gene of vaccinia is tagged with the ubiquitin‐like protein SUMO‐1, and this modification is necessary for A40 targeting to viral factories, where it associates with ER membranes and may play a role in the formation of I3 sites (Palacios et al., 2005). It is not known if movement of SUMOlyated A40 and ubiquitinated protein is directed along microtubules in a manner analogous to HDAC6‐mediated targeting of misfolded proteins to aggresomes (Kawaguchi et al., 2003). As reported for ASFV (see above) and cells infected with herpes simplex virus (Avitabile et al., 1995), infection of cells with Vaccinia virus also leads to disruption of microtubule organization and centrosome function and dispersal of the Golgi apparatus (Ploubidou et al., 2000). Whether these are bystander effects of the production of virus factories close to the centrosome or induced deliberately to facilitate virus egress is not known. IMV exit from the factory and transport to envelopment sites at the TGN is nonetheless dependent on microtubules (Sanderson et al., 2000) and has been reported to be dependent on the A4L and A27L gene products (Sanderson 2000, Ward 2005). Following envelopment, the A35L and F12L gene products then regulate microtubule‐dependent movement of intracellular enveloped viruses from the TGN to the plasma membrane (Herrero‐Martínez 2005, Ward 2001).
3. The iridoviruses generate cytoplasmic factories and crystalline arrays
a. Iridoviruses
Iridoviruses are large dsDNA viruses with genomes ranging from 100 to 210 kbp in length encoding between 100 and 230 proteins (Williams et al., 2005). Much of the work on iridovirus replication has been carried out on the ranavirus frog virus 3 (FV3). FV3 genome synthesis occurs in the nucleus and cytoplasm. No nuclear inclusions have been reported during FV3 infection, and as such it is unclear how the nuclear replication stage is mediated. However, viral DNA is initially synthesized as units that are 1–2 genomes in length and then transported to the cytoplasm where multiple length concatemers are produced (Goorha, 1982).
b. Cytoplasmic factories formed during iridovirus infection resemble aggresomes
Infection induces two cytoplasmic inclusions. Viral factories form in the cytoplasm and become the major site of viral DNA replication. FV3 also induces large crystalline arrays of viral particles which give rise to the iridescent coloring of purified virus, and hosts, that are characteristic of iridovirus infections. Virus factories are electron lucent relative to the cytoplasm and contain viral membranes, partially assembled viruses, and are surrounded by rough ER membranes and polysomes. FV3 factories also resemble aggresomes since they recruit intermediate filaments (Fig. 7A ) and mitochondria, some of which show signs of damage (Darlington 1966, Granoff 1966, Huang 2006, Tripier 1977). Crystalline arrays of virus are associated with virus factories and can induce nuclear deformations that lead to kidney‐shaped nuclei similar to those seen in ASFV infection (Fig. 7B) and after aggresome formation (Darlington 1966, Heath 2001, Johnston 1998). As seen for ASFV and poxviruses, the intermediate filament vimentin plays an important role in replication (Murti and Goorha, 1983). Vimentin is phosphorylated during FV3 infection, prior to factory formation (Chen 1986, Willis 1979), and temperature‐sensitive mutants that are unable to phosphorylate vimentin do not form vimentin cages and are unable to proceed to late gene expression. Drug treatment with taxol or colchicine (Murti et al., 1988) showed that recruitment of vimentin to assembly sites requires dynamic, but not polymerizing microtubules, and microinjection of anti‐vimentin antibody prevented recruitment of vimentin to factories. This allowed intrusion of cell components into assembly sites and reduced virus growth by 70–80% (Murti et al., 1988). Vimentin may therefore provide a scaffold for iridovirus replication, maintaining a barrier between the cytoplasm and the contents of the virus factory. Consistent with this hypothesis is the observation that during infection, polyribosomes and most newly synthesized viral proteins associate with intermediate filaments (Murti and Goorha, 1989). FV3 factory formation may also be dependent on the early 108K protein, as it is recruited to factories in the absence of late protein synthesis (Chinchar et al., 1984).
Figure 7.
(A) Confocal micrograph of frog virus 3‐infected cell showing relationship between the major capsid protein (red), vimentin (green), and DNA (blue). Note multiple viral inclusions in the cytoplasm, each associated with an individual vimentin cage. Authors own. (B) Electron micrograph of a frog virus 3‐infected cell showing two crystalline arrays that appear to induce a kidney‐shaped nucleus (N). Reprinted from Darlington et al. (1966) with permission from Elsevier.
4. Phycodnavirus and mimivirus replicate in cytoplasmic factories
Phycodnaviruses and the recently described giant virus mimivirus (La Scola et al., 2003) induce replication complexes in the cytoplasm of infected ameba (Meints et al., 1986; Raoult et al., 2007). The factories of the phycodnavirus Paramecium bursaria Chlorella virus 1 (PBCV‐1) are electron translucent areas of the cytoplasm and contain viral membranes, electron‐dense viroplasm, and assembling viruses. Unlike many viral factories, a distinct order appears to be present in PBCV‐1 virosomes. The assembling viruses are arranged at the periphery of the virosome/factory, giving the appearance of a rosette (Meints et al., 1986). Phycodnavirus replication and factory formation are not affected by a wide range of pharmacological disruptors of the cytoskeleton, including microtubule depolymerization by nocodazole and taxol, and depolymerization of actin by cytochalasin D (Nietfeldt et al., 1992). In this way, they differ from factories formed by large DNA viruses such as ASFV, vaccinia, and FV3. The successful cultivation of algae in the laboratory has allowed studies of the intracellular sites of replication of large icosahedral MclaV‐1 and HincV‐1 viruses (Wolf 1998, Wolf 2000). These viruses produce a latent infection that becomes apparent once the algae produce reproductive organs that become host to millions of virus particles. Replication of these viruses begins in the nucleus, but the first evidence for virus assembly is provided by the appearance of electron‐dense bodies next to the nucleus at sites of breakdown in the nuclear envelope. Infection leads to stacking of ER cisternae that may provide membranes for virus envelopment. The dense bodies remain next to the nucleus in large inclusions, and take on the angular shape characteristic of capsid assembly seen for iridoviruses and ASFV. The nucleus eventually disintegrates, and the virus factory occupies most of the cytoplasm.
V. Herpesviruses Induce Nuclear Inclusions and Cytoplasmic Assembly Sites
A. Herpesviruses
Herpesviruses are large dsDNA viruses with genomes ranging in size from 120 to 250 kbp. Herpesvirus genes are expressed in a regulated cascade starting with the immediate early α genes, then early β genes, and finally two subsets of late γ genes, γ1 and γ2. Complete herpesvirus particles have four main layers, the core containing DNA, an icosahedral capsid, a poorly defined layer of protein called tegument, and finally the viral envelope containing several glycoproteins. Genome synthesis and packaging and capsid assembly occur in inclusions in the nucleus. Nucleocapsids then obtain tegument in either the nucleus or the cytoplasm, or both, and the viral envelope is acquired exclusively in the cytoplasm [see Mettenleiter 2002, Mettenleiter 2006 for more thorough analysis]. The transfer of virus from the nucleus to the cytoplasm and acquisition of tegument appears well defined for human herpesvirus 6 (HHV‐6) (Roffman et al., 1990) but is controversial for the alphaherpesviruses (Campadelli‐Fiume 2006, Mettenleiter 2006). The subcellular organization of herpesvirus replication complexes formed in the nucleus during the early stages of productive infection has been described in considerable detail. The inclusions function as sites of virus replication and contain the virally encoded proteins and host proteins needed for virus replication. Interestingly, nuclear inclusions formed during herpes virus replication also contain cellular proteins involved in the control of DNA damage and repair. These may be recruited into inclusions in response to virus genome replication, and whether they are beneficial or detrimental to virus replication is a subject of considerable interest [reviewed by Everett (2006)].
B. Herpesvirus replication generates inclusions in the nucleus
Herpesviruses enter the cell by fusing their envelopes with the plasma membrane, whereon the naked nucleocapsids migrate to nuclear pores, possibly along microtubules (Granzow 1997, Sodeik 1997) [reviewed by Smith and Enquist (2002)]. Nuclear inclusions housing herpesvirus DNA replication are globular and can occupy the majority of the nucleus (de Bruyn Kops 1988, Randall 1986, Taylor 2003). They are identified through the presence of the viral DNA‐binding protein encoded by the UL29 gene, which is also known as infected cell protein 8 (ICP8). A minimum set of seven genes, UL5, UL8, UL9, UL29, UL30, UL42, and UL52, has been identified as necessary for viral DNA replication (Challberg, 1991). A plasmid transfection system has shown in vitro these can form globular nuclear compartments that are sites of 5‐bromo‐2′‐deoxyuridine (BrdU) incorporation and visually are similar to those formed during infection (Lukonis 1997, Zhong 1997). Nuclear inclusions organizing viral DNA replication have been followed in real time by a recombinant virus expressing a GFP‐ICP8 fusion protein. Small inclusions merge with adjacent replication complexes and increase in size to form globular replication complexes, which eventually fill most of the nucleus (Randall 1986, Taylor 2003).
1. Nuclear inclusions associated with herpesvirus replication are linked to ND10/PML Bodies
Replication compartments are formed from a number of different discrete foci that are induced early in infection and whose interrelatedness is not fully understood. The initial stages of productive herpesvirus infection are, however, intimately linked with nuclear structures called ND10 bodies (illustrated in Fig. 8 ) [Ishov 1996, Maul 1996, review by Borden (2002)]. Live cell studies have shown that the immediate early regulatory protein ICP4, which binds viral DNA, forms discrete foci as early as 30‐min postinfection (Fig. 8A). These initially appear close to the nuclear envelope, possibly at sites where the genome first enters the nucleus following capsid disassembly at nuclear pores (Everett and Murray, 2005), and are then seen throughout the nucleus (Everett et al., 2004). ICP4 foci are seen juxtaposed to the ND10 marker promyelocytic leukemia protein (PML) some 60-min later. The early and late regulatory protein ICP27 is recruited to ICP4 foci 2‐h postinfection and facilitates efficient early gene expression (Everett et al., 2004). During the same period, the immediate early regulatory protein, ICP0, colocalizes with ND10 bodies, some of which are likely juxtaposed to ICP4 bodies (Everett et al., 2003). ICP0 mediates the ubiquitin and/or SUMO‐1‐targeted proteasomal degradation of ND10 components (Chelbi‐Alix 1999, Everett 2000, Everett 1994, Everett 2004). Finally, parental genomes localize to ICP4 foci (Everett and Murray, 2005), and the ICP4 foci enlarge into structures that resemble early ICP8 replication compartments (Everett 2005, Everett 2003). Formation of ICP8 replication compartments (Taylor and Knipe, 2004) is also known to involve redistribution of ND10 bodies (Burkham et al., 1998). The relationship between the early ICP4 structures associated with parental genome and the later ICP8 compartments associated with replication and production of progeny genome is not clear; however, ICP4 and ICP8 both localize to late replication compartments (Knipe et al., 1987). A description of the relative and temporal distribution of the two proteins at early times awaits live cell studies following both proteins simultaneously.
Figure 8.
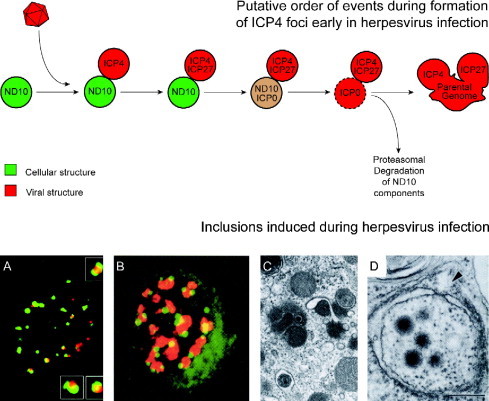
Schematic representing interaction of herpesvirus foci with ND10 bodies. (A) Cell expressing PML‐ECFP (green) and infected with human herpesvirus‐1‐encoding ICP4‐EYFP (red) 115‐min postinfection. Boxes show zoomed sections demonstrating juxtaposition of ND10 and ICP4 bodies early during virus infection. Reprinted from Everett et al. (2003) with permission from American Society for Microbiology. (B) Cell infected with human herpesvirus 2 showing assemblons immunolabeled with ICP35 (red) and UL55 inclusions (green). Note juxtaposition of the two compartments. Reprinted from Yamada et al. (1998) with permission from Society for General Microbiology. (C) Electron micrograph of human herpesvirus 5‐infected cell showing a section of a cytoplasmic assembly site. Note complete virus particle within a vacuole in bottom left‐hand corner, dense bodies in center of image, including one budding into a membrane. Reprinted from Craighead et al. (1972) with permission from American Society for Microbiology. (D) Electron micrograph of a tegusome within a nucleus of a human herpesvirus‐6‐infected cell, note apparent continuity between tegusome and cytoplasm (arrowed). Reprinted from Roffman et al. (1990) with permission from American Society for Microbiology.
2. Nuclear inclusions also form as sites of herpesvirus assembly: The assemblon
A second prominent nuclear inclusion induced by herpesvirus infection is the assemblon (Ward et al., 1996b). This is the site where capsid proteins accumulate and assemble into nucleocapsids (Fig. 8B). The assembly of herpesvirus nucleocapsids has been researched in great detail at the ultrastructural level facilitated by a cell‐free system for reconstituting the particles (Heymann 2003, Newcomb 1994, Newcomb 1996). The mature herpesvirus capsid is icosahedral with a T = 16 symmetry and is composed of 150 hexons and 11 pentons of the major capsid protein UL19. The place of the remaining penton is taken by a 12‐mer of the portal protein UL6, which by analogy with bacteriophage may be the site of genome entry. Nucleocapsids mature from fragile procapsids, through B capsids that lack DNA and contain the internal scaffold protein UL26.5, to C capsids that contain the viral genome.
The relationship between assemblons and sites of viral DNA replication has been a topic of some controversy as some reports show direct colocalization (Taus et al., 1998), whereas others have shown a proximity (Nalwanga 1996, Ward 1996b), similar to that seen between ND10 bodies and ICP4 foci during the initial stages of infection. Clearly, the DNA has to reach the capsid in order to complete assembly, and it is likely that the different results are indicative of the dynamic interactions between different herpesvirus nuclear inclusions. The DNA cleavage and packaging proteins encoded by the UL17 and UL32 genes are required for colocalization of viral DNA and capsids (Lamberti 1998, Taus 1998). Cells infected with a virus encoding a faulty UL32 gene exhibit nuclear localization of the capsid protein VP5 that is separate from replication sites (Lamberti and Weller, 1998). Similarly, in cells infected with mutants that lack functional UL17, the ICP8 protein fails to colocalize with ICP5 and ICP35 (Taus et al., 1998). Actin also plays an important role in the correct nuclear subcompartmentalization of viral proteins. Infection with HHV‐11 or suid herpesvirus‐12 causes actin filaments to assemble in the nucleus, prior to the accumulation of capsid proteins (Feierbach et al., 2006). Depolymerization of actin with latrunculin A inhibited correct nuclear compartmentalization of a representative capsid protein (VP26). VP26 colocalizes with the actin motor myosin Va (Feierbach et al., 2006), and capsid movement within the nucleus is inhibited by the myosin motor inhibitor 2,3‐butanedione monoxime (Forest et al., 2005). This suggests that the organization of nuclear inclusions involved in herpesvirus assembly is dependent on cellular actin filaments, and it will be interesting to see if the organization of inclusions housing viral DNA replication sites is similarly dependent.
Other inclusion bodies have been reported in the nucleus of cells infected with herpesvirus. The tegument proteins VP22 and VP13/14 localize to inclusion bodies that align closely but do not overlap ICP0/ND10/ICP8 pre‐replication complexes or assemblon inclusions (Hutchinson et al., 2002). UL55 also localizes to structures that overlap but are distinct from assemblons and DNA replication complexes (Fig. 8B) (Yamada et al., 1998). UL11 localizes to type IV and type V intranuclear dense bodies as well as virions and cytoplasmic ribbon structures (Baines et al., 1995). The alkaline DNase encoded by the UL12 gene localizes to discrete electron‐dense bodies within the nucleus that also contain B‐36 nucleolar protein (Lopez‐Iglesias 1988, Puvion‐Dutilleul 1986). It is unknown whether these different structures are related to each other, whether they are homogenous accumulations of the individual herpesvirus protein(s), or if they are simply dead‐end accumulations of protein.
3. Nuclear inclusions contain both viral and host proteins
A large number and variety of cellular proteins accumulate at nuclear sites of herpesvirus replication and assembly. A comprehensive proteomic analysis of ICP8 interacting proteins revealed more than 50 viral and cellular proteins that maybe recruited to DNA replication sites (Taylor and Knipe, 2004). A number of these interacting proteins were confirmed to localize to replication sites by microscopy experiments (Taylor and Knipe, 2004), and these as well as proteins identified in other studies (Leopardi 1997, Lukonis 1997, Quadt 2006, Wilcock 1991) reveal that at least 23 cellular proteins are known to localize to nuclear inclusions involved in DNA replication during herpesvirus infection (Table I). The functions of these proteins span the expected functions of nuclear genes, including DNA replication, transcription, chromatin remodeling, DNA repair, recombination, and nonhomologous end joining. Of particular importance is the recruitment of RNA polymerase II, which is required to transcribe the viral genome. RNA polymerase II is phosphorylated during viral infection by ICP22 and ICP27, and the latter modification is required for targeting to replication complexes (Dai‐Ju et al., 2006).
The role of all of these cellular genes in the viral replication cycle is poorly understood; however, cells deficient in WRN, a recQ helicase family member, produced reduced virus yields while cells lacking Ku86, part of a nonhomologous end‐joining protein complex, produced increased yields of virus (Taylor and Knipe, 2004). The implication therefore is that some cellular proteins may be actively recruited to replication complexes to aid viral replication, and some may be recruited by the cell as part of an antiviral response or sequestered by the virus in inclusions to subvert their antiviral nature. PML is induced by interferon, suggesting an antiviral role. Many of the genes shown to be required for recruitment of PML to viral pre‐replication sites are part of the minimal set of genes required to synthesize viral DNA. Recruitment of PML to viral replication sites is, for example, dependent on the viral DNA polymerase (UL30), the origin binding protein (UL9 gene) and the helicase–primase complex (UL5, UL8, and UL52) (Burkham et al., 2001). Recent evidence has suggested that this may be the reason why ICP0 causes dispersal of PML early in infection. PML knockdown by short interfering RNAs (siRNA) facilitates productive replication of ICP0 null mutants of herpesvirus (Everett 2004, Everett 2006); moreover, ICP0 null mutants are hypersensitive to interferon in a manner dependent on PML (Chee et al., 2003). This is of particular importance because ICP0 plays a role in determining whether herpesvirus induces a quiescent or a productive, lytic infection (Mossman and Smiley, 2002).
C. Cytoplasmic inclusions form during late stages of herpesvirus tegumentation: The cytoplasmic assembly compartment
The tegument layer of alphaherpesviruses is composed of at least 15 different proteins (Mettenleiter, 2002). US11, UL17, UL47, UL48, and UL49 are components of the tegument, and all are localized to the nucleus (if not exclusively) during the productive life cycle of the virus (Fuchs 2002, Hutchinson 2002, Kopp 2002, Roller 1992, Taus 1998). UL48 may play a role in egress from the nucleus, though this has not been unequivocally established (Mossman et al., 2000). Therefore, it is likely that some tegument proteins are acquired in or during viral egress from the nuclear inclusions. Recently, cytoplasmic aggresome‐like structures have been described in cells infected with HHV‐2.1 These contain the major capsid protein, tegument proteins, envelope glycoproteins, and markers for the Golgi complex (Nozawa et al., 2004). The latter finding is particularly interesting because herpesvirus envelopment involves membranes from the TGN (Mettenleiter 2006, Turcotte 2005). HHV‐53 is a betaherpesvirus and late during infection produces a juxtanuclear “assembly compartment” that again contains tegument proteins (pp150, pp28, and pp68), the major capsid protein, and viral envelope proteins (gB, gH, and gp65), suggesting a cytoplasmic site specialized for tegumentation and envelopment (Fig. 8C); (Adair 2002, Sanchez 2000). The precise role of the cytoplasmic assembly compartment is unclear. On the one hand, the concentration of glycoproteins and tegument proteins in one site may facilitate final stages of assembly prior to release from the cell. Interestingly, in common with aggresomes induced by ASFV and misfolded proteins, the cytoplasmic assembly compartment recruits chaperones and mitochondria and is dependent on microtubules and localizes to the microtubule organizing center.
At present, the assembly compartments are not considered to be bona fide aggresomes because they are not surrounded by a collapsed cage of intermediate filaments (Nozawa 2004, Sanchez 2000). It is nevertheless possible that these structures are related to aggresomes and are produced in response to a buildup of products resulting from nonproductive assembly pathways that occur late during infection. They may also contribute to the cytopathic effect seen in cells infected with HHV‐5. HHV‐5 infection results in cytomegaly characterized by increased cell size and intracellular water content. Cytomegaly and virus replication are both dependent on the presence of extracellular Na+, and infection results in sequestration of the plasma membrane Na‐K‐Cl‐cotransporter protein in large perinuclear structures that resemble the assembly compartment/viral aggresome (Maglova et al., 2004). Electron‐dense bodies can be seen by electron microscopy within the cytoplasmic assembly compartments induced during HHV‐5 infection (Craighead et al., 1972). Dense bodies are enveloped and obtain viral glycoproteins but do not contain DNA and are noninfectious. As can be seen in Fig. 8C, dense bodies bud into membranes and appear as oversized enveloped viral particles without a DNA containing core. Dense bodies exit the cell to become extracellular dense bodies (Craighead et al., 1972). Interestingly, HHV‐5 immediate early IE1 proteins also become associated with extracellular dense bodies despite no reported localization to their intracellular relations (Tsutsui and Yamazaki, 1991). Purified extracellular dense bodies are mostly composed of UL83 but have a full complement of viral glycoproteins (Irmiere and Gibson, 1983). The function of dense bodies remain unclear, and they may represent the end point of a nonproductive assembly pathway resulting from attempts to envelope capsids lacking genomes or may be used to deliver viral components to neighboring cells.
Interestingly, for human herpesvirus 6 (HHV‐6), the tegument layer appears to be acquired within a dedicated structure that has been dubbed the tegusome (Roffman et al., 1990). This work is based on electron microscopy of cells infected with HHV‐6 and shows tegusomes as intranuclear membrane compartments that abut the nuclear envelope (Fig. 8D). Tegusomes may be cytoplasmic invaginations of the nuclear envelope into the nucleus because they appear to contain ribosomes and are sometimes in continuity with the cytoplasm. Nucleocapsids appear to bud into the tegusome, capsids obtain a tegument layer, and then bud into cytoplasmic vacuoles where they acquire envelopes and exit the cell.
VI. Nuclear Inclusions Are Formed by Small DNA Viruses
A. Adenovirus
Adenovirus are medium‐sized, nonenveloped dsDNA viruses with genomes ranging from 26 to 45 kbp in length and virions of the order of 70–100 nm in diameter. Like other DNA viruses, they have an ordered cascade of transcripts, early, delayed early, and late types having been described. Adenovirus transcripts are spliced to generate multiple transcripts from a given transcriptional unit. Viral replication occurs in the nucleus, and adenovirus infection was utilized extensively as a model system for exploring different nuclear subcompartments. A productive infection of lytic adenovirus induces profound rearrangement of existing subcompartments and the induction of several new ones within the host nucleus. A study on the localization of the human adenovirus5 IVa2 protein described 10 distinct nuclear and nucleolar subcompartments induced or associated with virus replication (Lutz et al., 1996), and these are listed in Table I.
1. Structure and location of nuclear inclusions formed during adenovirus replication
Earlier studies carried out before markers for specific nuclear subcompartments were available have described the structures in terms of shape and location (see Table I). During the initial stage of infection, viral RNA (Puvion‐Dutilleul et al., 1992), single‐stranded DNA (ssDNA), and dsDNA (Puvion‐Dutilleul and Pichard, 1992) are all synthesized in small fibrillar regions termed early replication sites. By the intermediate stage of replication, the ssDNA is deposited in the center of these structures, while transcription and dsDNA synthesis occur on the outside and begin to form an inclusion. The inclusion has a characteristic doughnut shape, and has been called the fibrogranular network. At late stages of infection, dsDNA, viruses, and trace amounts of ssDNA appear in large viral inclusions (Besse 1994, Puvion‐Dutilleul 1992). Targeting of the initial replicon is dependent on a dCMP modification of the preterminal protein (pTP), which enables pTP to form a complex with the DNA polymerase and the genome (Temperley and Hay, 1992). PTP mediates targeting of the heterotrimeric complex to the nuclear matrix (Fredman and Engler, 1993), possibly through an interaction with CAD (carbamyl phosphate synthetase, aspartate transcarbamylase and dihydroorotase) (Angeletti and Engler, 1998). Transcription and splicing are mediated by host proteins and viral RNA, and non‐SNP RNA splicing factor, hnRNP C proteins, and RNA polymerase II all colocalize with viral RNA in nuclear inclusions. Splicing small nuclear ribonucleoproteins (snRNPs) colocalize with viral RNA but not replication foci (Pombo et al., 1994), and snRNPs then move to interchromatin granules late in infection, which is blocked by mutations in E4 (Bridge et al., 2003).
a. Rearrangement of host nuclear compartments during adenovirus replication
Like herpesvirus described above, adenovirus infection redistributes the components of ND10 bodies. Prior to infection, PML is associated with interchromatin granules but is redistributed to the fibrillogranular matrix within the nucleus along with SP100, another ND10 component (Carvalho et al., 1995). Later in infection, PML is redistributed once again from the fibrillogranular matrix to clear amorphous inclusions and protein crystals (Puvion‐Dutilleul et al., 1995). Another study reported that SP100 and NDP55, but not PML, were relocated from ND10 bodies to viral inclusions (Doucas et al., 1996). While this is confusing, it is clear that adenovirus employs multiple mechanisms to reorganize PML. The initial movement of PML, SP100, and NDP55 to the fibrillogranular matrix occurs prior to viral DNA synthesis and is dependent on the E4‐ORF3 11‐kDa protein (Carvalho 1995, Doucas 1996). It may also be mediated by E1A proteins that colocalize with PML (Carvalho et al., 1995). E1B‐55‐kDa protein also colocalizes with PML early on in infection, then associates with the periphery of replication centers; these interactions are mediated by the ORF6 protein of the E4 transcriptional unit (Lethbridge et al., 2003). Interestingly, E1B‐55K and E4‐ORF3 target the MRE11‐RAD50‐NBS1 (MRN) complex to aggresomes for degradation (Araujo 2005, Liu 2005). The MRN complex causes concatenation of viral DNA and inhibits packaging. Transport of MRN to the cytoplasm for degradation in aggresomes relieves this inhibition and facilitates production of infectious viruses. The later movement of PML from the fibrillogranular matrix to clear amorphous inclusions also appears important for replication. Movement is mediated by the IX gene product, and adenovirus encoding mutant IX do not create clear amorphous inclusions, have reduced growth, and are sensitive to PML overexpression (Rosa‐Calatrava et al., 2003). Interestingly, PKR is also redistributed to clear amorphous inclusions (Souquere‐Besse et al., 2002) during infection, as are pentons and hexons in the absence of the fiber gene; this suggests these structures may represent sites for sequestering excess viral proteins, and cellular proteins with potentially deleterious effects on the virus (Puvion‐Dutilleul et al., 1999).
B. Nuclear inclusions formed during polyomavirus and papillomavirus infection
Polyoma‐ and papillomaviruses are small double‐stranded tumorigenic DNA viruses with genomes of 5 and 8 kbp, respectively. Replication and assembly of these two viruses follow similar strategies, and both involve ND10 bodies. The VP1 capsid protein of human polyomavirus JC is targeted to ND10 domains by VP2, VP3, and agnoprotein where they are assembled into virions (Shishido‐Hara et al., 2004). A similar process occurs during papillomavirus infection where the minor capsid protein, L2, is responsible for targeting capsomeres of the major capsid protein, L1, to ND10 domains (Florin et al., 2002a). This process involves L2‐induced redistribution of ND10 bodies by targeting SP100 for proteasomal degradation. At this point the cellular Daxx protein is recruited (Florin et al., 2002b). Daax has multiple functions in the nucleus including transcriptional activation and modulating Fas‐mediated apoptosis [reviewed by Salomoni and Khelifi (2006)]. Its role in virus replication is at present unclear.
One characteristic of papillomavirus infections is the appearance of nuclear and cytoplasmic inclusions in cells contained within warts. The size and number of inclusions is dependent on the type of papillomavirus and the site of infection. Human papillomavirus 1 (HPV‐1), for example, induces many small inclusions while HPV‐4 induces one single inclusion that takes over most of the cytoplasm (Croissant et al., 1985). In vivo these structures label strongly with antiserum raised against E4 gene products which are the 17‐kDa E1∧E4 and 16‐kDa E4 proteins (Doorbar 1986, Rogel‐Gaillard 1993). Inclusions can be induced in certain cell types in vitro by expressing E4 gene products. HPV‐1 E4 staining reveals an initial association with the intermediate filament keratin and subsequent formation of inclusion bodies in the cytoplasm and nucleus (Roberts 2003, Rogel‐Gaillard 1993). The HPV‐1 cytoplasmic inclusions retain their association with keratin and appear to induce small cages surrounding E4 protein that are interconnected by keratin filaments (Roberts et al., 2003). The E4 gene gives rise to two proteins, the 17‐kDa E1∧E4 which can induce cytoplasmic and nuclear inclusions whereas the 16‐kDa E4 can induce inclusions solely in the cytoplasm (Rogel‐Gaillard et al., 1993). Interestingly, expression of E1∧E4 gene product from HPV‐16 induces the complete collapse of the keratin network, but not that of the microtubule or actin networks (Doorbar et al., 1991). It is unclear what the role of the inclusions is in viral replication or the pathology of infection. However, HPV‐1 E4 expression induces the redistribution of ND10 to the periphery of nuclear inclusions in cells in culture, and similar signals are seen in vivo (Roberts et al., 2003). The temporal and functional connection between E4 and L1 redistribution of PML is unknown.
VII. Virus Factories and Inclusions Formed by RNA Viruses
A. Reoviruses
Members of the Reoviridae family are dsRNA viruses with segmented genomes and include the clinically important rotavirus and orbiviruses that cause diseases in human and animals. Reoviruses are nonenveloped viruses with genome segments contained inside a virion ∼80 nm in diameter. The genome is encapsidated by two protein shells, an outer capsid and an inner core shell. The core contains the RdRp, capping enzymes, and the dsRNA genome segments [reviewed in Yue and Shatkin (1998) and Furuichi and Shatkin (2000)]. Viruses are taken up by receptor‐mediated endocytosis, the outer capsid is lost and the core is delivered into the cytoplasm. The core does not disassemble on entering cells and imports ribonucleoside triphosphates and S‐adenosyl‐l‐methionine from the cytosol to synthesize and then export viral mRNAs. In this way the core particle functions as a self‐contained transcriptional unit and as such represents the replication complex. Viral mRNA transcribed in the cytoplasm make viral proteins that eventually form large perinuclear inclusions, called virus factories that function as sites of further virus replication and assembly. The Reoviridae family contains 13 genera, and this chapter will concentrate on the two best characterized of these, the orthoreoviruses and rotaviruses.
1. Formation of factories during orthoreoviruses replication and assembly
a. The shape of orthoreovirus factories is determined by association with the cytoskeleton
Orthoreoviruses contain 10 genome segments which are classed by size and then numbered, that is L1 is large segment 1. Large segments encode λ genes, medium (M) segments encode μ genes, and small (S) segments encode σ genes. Virus replication occurs in the cytoplasm in virus factories, and the majority of the virus‐encoded proteins have been shown to localize completely or partially with factories (Table I). Early observations revealed that different strains of orthoreoviruses induced factories with different appearances; orthoreovirus type 1 Lang factories were filamentous while the factories of the Dearing isolate of orthoreovirus type 3 were globular (Fig. 9A and B ) (Parker et al., 2002). This difference maps precisely to a serine–proline switch at residue 208 of the μ2 core protein (Parker et al., 2002). Control of the localization of orthoreovirus factories reflects the degree of association μ2 has with the microtubule network. Filamentous virus μ2 stabilizes microtubules to a greater relative degree than globular virus μ2, and depolymerizing microtubules with nocodazole convert filamentous factories to globular ones.
Figure 9.
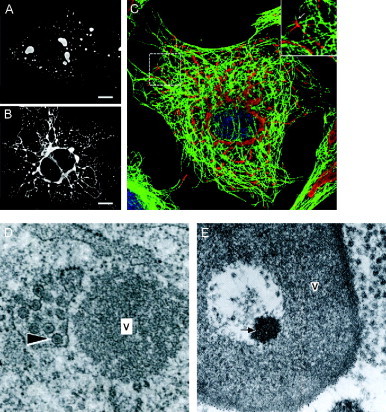
(A and B) Confocal images of orthoreovirus type 3 Dearing (A)‐ and type 1 Lang (B)‐infected cells labeled with showing difference between globular and filamentous types of viral factories. (C) Confocal image of an infected cell immunolabeled with (red) and α‐tubulin (green) showing relationship between filamentous factories and microtubules. Reprinted from Parker et al. (2002) with permission from American Society for Microbiology. (D) Electron micrograph showing rotavirus viroplasm (V) next to TLP within membranes derived from the ER (arrow). Reprinted from Petrie et al. (1984) with permission from Elsevier. (E) Doughnut‐shaped rotavirus factory labeled with anti‐NSP2 antibody showing electron‐lucent center with electron‐dense core (arrow) surrounded by viroplasm (V). Reprinted from Altenburg et al. (1980) with permission from Society for General Microbiology.
b. Virus nonstructural proteins determine orthoreovirus factory organization
Many of the events of orthoreovirus factory formation have been successfully reconstituted in vitro. A screen of orthoreovirus proteins revealed that μNS, σNS, and σ3 were the first viral proteins to localize with viral mRNA prior to the synthesis of progeny dsRNA (Antczak and Joklik, 1992). Subsequently, it was discovered that expression of the μNS protein of isolate Dearing in the absence of other viral proteins induced a phase‐dense structure that was indistinguishable in appearance from that observed during wild‐type infection (Broering et al., 2002). The shape of the artificial μNS inclusion could be altered from globular to filamentous by coexpressing a μ2 protein from a filamentous virus (Broering et al., 2002). Similar experiments showed that coexpression of λ1, λ2, and σ2 core surface proteins with μNS caused them to localize to the μNS inclusion (Broering et al., 2004). Furthermore, the shape of the μNS structure that the core proteins colocalized to could be altered to filamentous by coexpressing μ2 from a filamentous virus (Broering et al., 2004). μNS can also recruit σNS, but not σ3, to artificial inclusions (Becker et al., 2003), so other factors or conditions are necessary for complete assembly of an orthoreovirus factory. The precise domains involved in initiating factory formation are beginning to be elucidated. The minimal region of μNS necessary for inclusion like body formation in vitro is the region composed of 250 C‐terminal amino acids of the 721 residue proteins (Broering et al., 2005). Residues 1–11 of σNS are important for the interaction between σNS and RNA (Gillian and Nibert, 1998), and treatment with RNase dissociates a proportion of μNS from σNS in coimmunoprecipiation experiments (Miller et al., 2003). Interaction between μNS and μ2 is dependent on residues 1–40 or 41 of μNS (Broering et al., 2002) and residues 1–13 are necessary for interaction between μNS and σNS (Miller et al., 2003). It is likely that factory formation occurs through an interaction between μNS and a σNS‐RNA complex; this can then recruit μ2 that will determine the globular or filamentous localization of the factory and hence the localization of the other viral proteins.
Orthoreovirus factories are clearly intimately associated with the microtubule network (Fig. 9C) and have also been suggested to interact with intermediate filaments. Orthoreovirus type 3 infection induces a redistribution of vimentin and viral inclusions reported to contain filamentous structures (Sharpe et al., 1982). It will be interesting to see if the in vitro factories induced by μNS can also alter the distribution of the intermediate filament network. Orthoreovirus factories are also ubiquitinated, and interestingly the nature of the factory determined the degree of ubiquitination; globular factories are prone to contain more ubiquitinated protein than filamentous ones (Miller et al., 2004). Ubiquitination of orthoreovirus factories has been mapped to the μ2 protein but is independent of the filamentous/globular factory determinate of μ2, that is converting a filamentous factory to a globular factory does not lead to an increase in ubiquitinated μ2.
2. Formation of factories during rotavirus replication and assembly
a. Virus nonstructural proteins organize factory formation and virus assembly
Rotaviruses contain 11 genome segments of dsRNA and like the orthoreoviruses replicate in cytoplasmic factories. Rotavirus virions are composed of three protein layers. These are the core which contains the genome and polymerase, an inner capsid layer, and an outer capsid layer.The core and inner capsid layer comprise the double‐layered particle (DLP), while the addition of the third capsid layer forms the mature triple‐layered particle (TLP). The acquisition of the third capsid layer occurs after the virus buds into the ER, and in doing so obtains a transient envelope. Rotavirus factories are composed of electron‐dense viroplasm often in proximity to membranes derived from the ER (Fig. 9D) (Altenburg et al., 1980). Viroplasm contains high levels of NSP2 (Fig. 9E) and NSP5 which are thought to coordinate assembly of the factory and recruitment of structural proteins such as the inner core protein VP2 and viral polymerase VP1. The factory also contains double‐layered rotaviruses, whereas the ER membranes associated with the factory contain enveloped intermediates and TLP (arrowed in Fig. 9D). Virus factories grow in size and decrease in number during the course of infection as neighboring factories merge (Eichwald et al., 2004). Rotavirus factories appear to have an internal structure, as their centers occasionally appear more electron lucent than the periphery, giving a doughnut‐like appearance (Fig. 9E). Electron microscopy shows DLP at the periphery of the factory and this is (Altenburg et al., 1980) consistent with fluorescent microscopy showing that the nonstructural protein NSP2 localizes to the center of the virus factory, whereas NSP5 and inner capsid protein VP6 localize to the periphery (Eichwald 2004, González 2000). These different localizations could have functional relevance because VP6 binds the ER‐targeted NSP4 membrane protein and is implicated in the budding of DLPs into ER membranes associated with factories (Silvestri et al., 2005). Therefore, a localization to the exterior of the factory may represent an organized progression of virus maturation from the interior of the viroplasm to the exterior. However, things are probably not that straightforward because VP6 is also part of the viral RNA complex along with NSP2 (Aponte et al., 1996) which, as noted above, is localized to the center of the viroplasm.
Virus factory‐like structures can be introduced in vitro by coexpressing NSP2 and NSP5 (Fabbretti et al., 1999), and this is regulated by domains in the N‐ and C‐termini (Fabbretti et al., 1999) as well as the central portion of NSP5 (Eichwald et al., 2002). The process is also dependent on phosphorylation of NSP5, possibly by cellular casein kinase II (Eichwald et al., 2002). Structures similar to factories can also be induced by expressing the inner capsid protein VP6 in vitro (Nilsson et al., 1998). These structures look similar to factories in the electron microscope but lack electron‐lucent areas and DLPs. Interestingly, expression of VP6 of group A simian rotavirus SA‐11 induced globular structures, whereas expression of VP6 from group C porcine rotavirus Cowden/AmC‐1 induced filamentous structures (Nilsson et al., 1998) analogous to the difference between type 1 and type 3 orthoretroviruses. It is not clear if the difference in factory shape is solely determined by VP6 and if this involves differences in association of the factory components with microtubules.
b. Virus factories organize viral RNA replication and translation
The factory does provide the virus with a mechanism to organize viral RNAs. Positive‐stranded viral RNA is utilized as the template for synthesizing progeny dsRNA genomes and as mRNA for translating viral proteins. Interestingly, siRNA‐targeted degradation of NSP1 RNA blocks translation of the protein but does not block genome synthesis (Silvestri et al., 2004). Furthermore, RNA synthesis occurs in factories, but viral RNA transcribed in vitro and introduced to infected cells after infection does not localize to factories. The implication of these experiments are that the factory enables rotavirus to sort viral RNA into separate pools, one within the factory to act as a template for the RNA polymerase and genome replication, and the other outside the factory where it translated on ribosomes to make viral proteins. It likely that this organization allows the virus factory to protect dsRNA genomes from antiviral responses.
B. Inclusions formed during arenavirus infection
Arenaviruses are negative‐stranded RNA viruses that have two single‐stranded genome segments which are packaged into 60‐ to 200‐nm‐diameter enveloped virions. Lassa, Junín, and Manchupo viruses are responsible for emerging hemorraghic fevers in humans. Arenaviruses induce moderately electron‐dense inclusions in the cytoplasm that are composed of 20‐ to 25‐nm‐diameter granules identical to those seen within virus particles in the electron microscope (Murphy et al., 1970). The granules represent host ribosomes and between 2 and 10 are packaged into virions (Pedersen, 1979). The inclusions increase in size and density during infection until cytopathic effects are observed in cells (Buckley 1965, Buckley 1970) and stain positive for viral antigens (Young et al., 1987); however, it is unclear if they represent true virus factories. Arenavirus replication is believed to occur in the cytoplasm but also requires a nuclear step as limited growth is observed in enucleated cells (Banerjee et al., 1975). The viral Z protein may play a role in this as it is sufficient in vitro to shuttle PML from the nucleus to cytoplasmic inclusion bodies as occurs in vivo (Borden et al., 1998). N protein also localizes to discrete nuclear foci, as well as in the cytoplasm (Young et al., 1987), but the relationship to ND10 bodies and Z protein is unknown.
C. Inclusions formed during rabies virus infection
Rabies virus is a neurotropic lyssavirus of the rhabdovirus family. Rhabdovirus virions are bullet‐shaped 180 × 75 nm2 particles containing a single negative strand of RNA. Rabies induces two types of inclusion body in vitro, neither of which have been proven as replication sites. Negri bodies are induced by street rabies viruses in infected neurons of the brain (Negri, 1903) and are a good indicator for the presence of an infection site in tissue (Jackson et al., 2001). Different neuronal cell types appear to be more prone to Negri bodies (Jackson et al., 2001). Negri bodies contain innerbodies (Negri, 1909) and electron microscopic studies suggest the subcompartments may be cytoplasmic material engulfed by the coalescence of several smaller Negri bodies (Matsumoto, 1970). The role of Negri bodies in infection is poorly understood. Initial EM observations showed virions localized to some bodies in some cells (Matsumoto et al., 1974), and cytological staining show they contain genetic material, indicating they may be replication complexes. However, 3H‐thymidine or 3H‐uridine fail to label the structures, arguing against this conclusion (Matsumoto, 1970). Fixed (brain‐adapted laboratory strains) rabies can infect nonneuronal cell lines and in these cell types induce fuchsin‐stained cytoplasmic structures (FCPS) as well as Negri‐like bodies (Ni et al., 1996). FCPS increase in size during infection that correlates with cytopathic effects and are composed of rabies glycoprotein and matrix protein, whereas Negri bodies contain nucleocapsid (Ni et al., 1996).
VIII. Conclusions
This chapter has described the changes to cell architecture that are induced during virus replication. We have focused on viruses that induce new cellular structures, such as inclusion bodies, virus factories, or replication complexes, to concentrate virus and host factors necessary for replication and assembly. Much progress has been made in identifying which cellular components are used to generate these structures, and in some cases specific virus proteins have been identified that are able to induce them. Virus inclusions often result in rearrangement of cellular membrane compartments and/or cytoskeleton. The functions of these organelles are carefully regulated in cells, and it is a challenge for the future to determine how viruses disrupt them for use as sites of replication and assembly. Changes in cellular architecture may represent bystander responses to the stress associated with virus infection, and some viruses may replicate perfectly well without them. Alternatively, viruses may have evolved to target key stages in the regulatory pathways that control organelle structure and function to generate sites that are essential for replication and assembly. Given the coevolution of viruses with the cells that carry them, changes in cell structure induced during infection are likely to involve a combination of the two. It is also important to appreciate that many of the structures that have been studied to date have been generated by infecting tissue culture cells with attenuated viruses, often with disregard to the host range and tropism. It is possible that in the natural setting, changes in cell structure induced by viruses will be more subtle, particulary during persistent infections that occur without inflammation or cell lysis.
Footnotes
Human herpesvirus 1 is herpes simplex virus 1 and human herpesvirus 2 is herpes simplex virus 2.
Suid herpesvirus 1 is pseudorabiesvirus or Aujesky's disease virus.
Human herpesvirus 5 is human cytomegalovirus.
References
- Adair R., Douglas E.R., Maclean J.B., Graham S.Y., Aitken J.D., Jamieson F.E., Dargan D.J. The products of human cytomegalovirus genes UL23, UL24, UL43 and US22 are tegument components. J. Gen. Virol. 2002;83:1315–1324. doi: 10.1099/0022-1317-83-6-1315. [DOI] [PubMed] [Google Scholar]
- Ahlquist P. Parallels among positive‐strand RNA viruses, reverse‐transcribing viruses and double‐stranded RNA viruses. Nat. Rev. Microbiol. 2006;4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcamí A., Angulo A., Viñuela E. Mapping and sequence of the gene encoding the African swine fever virion protein of Mr 11500. J. Gen. Virol. 1993;74:2317–2324. doi: 10.1099/0022-1317-74-11-2317. [DOI] [PubMed] [Google Scholar]
- Aldabe R., Barco A., Carrasco L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 1996;271:23134–23137. doi: 10.1074/jbc.271.38.23134. [DOI] [PubMed] [Google Scholar]
- Almazán F., Tscharke D.C., Smith G.L. The vaccinia virus superoxide dismutase‐like protein (A45R) is a virion component that is nonessential for virus replication. J. Virol. 2001;75:7018–7029. doi: 10.1128/JVI.75.15.7018-7029.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso C., Miskin J., Hernáez B., Fernandez‐Zapatero P., Soto L., Cantó C., Rodríguez‐Crespo I., Dixon L., Escribano J.M. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light‐chain dynein. J. Virol. 2001;75:9819–9827. doi: 10.1128/JVI.75.20.9819-9827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenburg B.C., Graham D.Y., Estes M.K. Ultrastructural study of rotavirus replication in cultured cells. J. Gen. Virol. 1980;46:75–85. doi: 10.1099/0022-1317-46-1-75. [DOI] [PubMed] [Google Scholar]
- Andrés G., Simón‐Mateo C., Viñuela E. Assembly of African swine fever virus: Role of polyprotein pp220. J. Virol. 1997;71:2331–2341. doi: 10.1128/jvi.71.3.2331-2341.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés G., García‐Escudero R., Simón‐Mateo C., Viñuela E. African swine fever virus is enveloped by a two‐membraned collapsed cisterna derived from the endoplasmic reticulum. J. Virol. 1998;72:8988–9001. doi: 10.1128/jvi.72.11.8988-9001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés G., Alejo A., Simón‐Mateo C., Salas M.L. African swine fever virus protease, a new viral member of the SUMO‐1‐specific protease family. J. Biol. Chem. 2001;276:780–787. doi: 10.1074/jbc.M006844200. [DOI] [PubMed] [Google Scholar]
- Angeletti P.C., Engler J.A. Adenovirus preterminal protein binds to the CAD enzyme at active sites of viral DNA replication on the nuclear matrix. J. Virol. 1998;72:2896–2904. doi: 10.1128/jvi.72.4.2896-2904.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antczak J.B., Joklik W.K. Reovirus genome segment assortment into progeny genomes studied by the use of monoclonal antibodies directed against reovirus proteins. Virology. 1992;187:760–776. doi: 10.1016/0042-6822(92)90478-8. [DOI] [PubMed] [Google Scholar]
- Aponte C., Poncet D., Cohen J. Recovery and characterization of a replicase complex in rotavirus‐infected cells by using a monoclonal antibody against NSP2. J. Virol. 1996;70:985–991. doi: 10.1128/jvi.70.2.985-991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo F.D., Stracker T.H., Carson C.T., Lee D.V., Weitzman M.D. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 2005;79:11382–11391. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitabile E., Digaeta S., Torrisi M.R., Ward P.L., Roizman B., Campadellifiume G. Redistribution of microtubules and Golgi‐apparatus in herpes‐simplex virus‐infected cells and their role in viral exocytosis. J. Virol. 1995;69:7472–7482. doi: 10.1128/jvi.69.12.7472-7482.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines J.D., Jacob R.J., Simmerman L., Roizman B. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 1995;69:825–833. doi: 10.1128/jvi.69.2.825-833.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S.N., Buchmeier M., Rawls W.E. Requirement of cell nucleus for the replication of an arenavirus. Intervirology. 1975;6:190–196. doi: 10.1159/000149472. [DOI] [PubMed] [Google Scholar]
- Battista M.C., Bergamini G., Boccuni M.C., Campanini F., Ripalti A., Landini M.P. Expression and characterization of a novel structural protein of human cytomegalovirus, pUL25. J. Virol. 1999;73:3800–3809. doi: 10.1128/jvi.73.5.3800-3809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaud G., Beaud R. Preferential virosomal location of underphosphorylated H5R protein synthesized in vaccinia virus‐infected cells. J. Gen. Virol. 1997;78:3297–3302. doi: 10.1099/0022-1317-78-12-3297. [DOI] [PubMed] [Google Scholar]
- Becker M.M., Goral M.I., Hazelton P.R., Baer G.S., Rodgers S.E., Brown E.G., Coombs K.M., Dermody T.S. Reovirus σNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J. Virol. 2001;75:1459–1475. doi: 10.1128/JVI.75.3.1459-1475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker M.M., Peters T.R., Dermody T.S. Reovirus σNS and μNS proteins form cytoplasmic inclusion structures in the absence of viral infection. J. Virol. 2003;77:5948–5963. doi: 10.1128/JVI.77.10.5948-5963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Fogg M.H., Ehrenfeld E. Poliovirus proteins induce membrane association of GTPase ADP‐ribosylation factor. J. Virol. 2005;79:7207–7216. doi: 10.1128/JVI.79.11.7207-7216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Altan‐Bonnet N., Kovtunovych G., Jackson C.L., Lippincott‐Schwartz J., Ehrenfeld E. Hijacking components of the secretory pathway for replication of poliovirus RNA. J. Virol. 2006;81:558–567. doi: 10.1128/JVI.01820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson S.D., Bamford J.K.H., Bamford D.H., Burnett R.M. Does common architecture reveal a viral lineage spanning all three domains of life? Mol. Cell. 2004;16:673–685. doi: 10.1016/j.molcel.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Barnard E.C., Brown G., Stow N.D. Deletion mutants of the herpes simplex virus type 1 UL8 protein: Effect on DNA synthesis and ability to interact with and influence the intracellular localization of the UL5 and UL52 proteins. Virology. 1997;237:97–106. doi: 10.1006/viro.1997.8763. [DOI] [PubMed] [Google Scholar]
- Berstein H.D., Baltimore D. Poliovirus mutant that contains a cold‐sensitive defect in viral RNA synthesis. J. Virol. 1988;62:2922–2928. doi: 10.1128/jvi.62.8.2922-2928.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse S., Puvion‐Dutilleul F. High resolution localization of replicating viral genome in adenovirus‐infected HeLa cells. Eur. J. Cell. Biol. 1994;63:269–279. [PubMed] [Google Scholar]
- Betakova T., Wolffe E.J., Moss B. The vaccinia virus A14.5L gene encodes a hydrophobic 53‐amino‐acid virion membrane protein that enhances virulence in mice and is conserved among vertebrate poxviruses. J. Virol. 2000;74:4085–4092. doi: 10.1128/jvi.74.9.4085-4092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz K., Egger D., Rasser Y., Bossart W. Intracellular distribution of poliovirus proteins and the induction of virus‐specific cytoplasmic structures. Virology. 1983;131:39–48. doi: 10.1016/0042-6822(83)90531-7. [DOI] [PubMed] [Google Scholar]
- Bienz K., Egger D., Pasamontes L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus‐induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology. 1987;160:220–226. doi: 10.1016/0042-6822(87)90063-8. [DOI] [PubMed] [Google Scholar]
- Bonifacino J.S., Glick B.S. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Borca M.V., Irusta P.M., Kutish G.F., Carrillo C., Afonso C.L., Burrage T., Neilan J.G., Rock D.L. A structural DNA binding protein of African swine fever virus with similarity to bacterial histone‐like proteins. Arch. Virol. 1996;141:301–313. doi: 10.1007/BF01718401. [DOI] [PubMed] [Google Scholar]
- Borden K.L.B. Pondering the promyelocytic leukemia protein (PML) puzzle: Possible functions for PML nuclear bodies. Mol. Cell Biol. 2002;22:5259–5269. doi: 10.1128/MCB.22.15.5259-5269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden K.L.B., Campbell Dwyer E.J., Salvato M.S. An arenavirus RING (zinc‐binding) protein binds the oncoprotein promyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J. Virol. 1998;72:758–766. doi: 10.1128/jvi.72.1.758-766.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgese N., Francolini M., Snapp E. Endoplasmic reticulum architecture: Structures in flux. Curr. Opin. Cell Biol. 2006;18:358–364. doi: 10.1016/j.ceb.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosher J., Dawson A., Hay R.T. Nuclear factor I is specifically targeted to discrete subnuclear sites in adenovirus type 2‐infected cells. J. Virol. 1992;66:3140–3150. doi: 10.1128/jvi.66.5.3140-3150.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost A.G., Prentice E., Denison M.R. Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection. Virology. 2001;285:21–29. doi: 10.1006/viro.2001.0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass V., Bieck E., Montserret R., Wolk B., Hellings J.A., Blum H.E., Penin F., Moradpour D. An amino‐terminal amphipathic alpha‐helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2002;277:8130–8139. doi: 10.1074/jbc.M111289200. [DOI] [PubMed] [Google Scholar]
- Bridge E., Mattsson K., Aspegren A., Sengupta A. Adenovirus early region 4 promotes the localization of splicing factors and viral RNA in late‐phase interchromatin granule clusters. Virology. 2003;311:40–50. doi: 10.1016/s0042-6822(03)00189-2. [DOI] [PubMed] [Google Scholar]
- Broering T.J., Parker J.S., Joyce P.L., Kim J., Nibert M.L. Mammalian reovirus nonstructural protein μNS forms large inclusions and colocalizes with reovirus microtubule‐associated protein μ2 in transfected cells. J. Virol. 2002;76:8285–8297. doi: 10.1128/JVI.76.16.8285-8297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering T.J., Kim J., Miller C.L., Piggott C.D., Dinoso J.B., Nibert M.L., Parker J.S. Reovirus nonstructural protein μNS recruits viral core surface proteins and entering core particles to factory‐like inclusions. J. Virol. 2004;78:1882–1892. doi: 10.1128/JVI.78.4.1882-1892.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering T.J., Arnold M.M., Miller C.L., Hurt J.A., Joyce P.L., Nibert M.L. Carboxyl‐proximal regions of reovirus nonstructural protein μNS necessary and sufficient for forming factory‐like inclusions. J. Virol. 2005;79:6194–6206. doi: 10.1128/JVI.79.10.6194-6206.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes S.M., Dixon L.K., Parkhouse R.M.E. Assembly of African swine fever virus: Quantitative ultrastructural analysis in vitro and in vivo. Virology. 1996;224:84–92. doi: 10.1006/viro.1996.0509. [DOI] [PubMed] [Google Scholar]
- Brookes S.M., Hyatt A.D., Wise T., Parkhouse R.M.E. Intracellular virus DNA distribution and the acquisition of the nucleoprotein core during African swine fever virus particle assembly: Ultrastructural in situ hybridisation and DNase‐gold labelling. Virology. 1998;249:175–188. doi: 10.1006/viro.1998.9308. [DOI] [PubMed] [Google Scholar]
- Brookes S.M., Sun H., Dixon L.K., Parkhouse R.M.E. Characterization of African swine fever virion proteins j5R and j13L: Immuno‐localization in virus particles and assembly. J. Gen. Virol. 1998;79:1179–1188. doi: 10.1099/0022-1317-79-5-1179. [DOI] [PubMed] [Google Scholar]
- Broyles S.S., Liu X., Zhu M., Kremer M. Transcription factor YY1 is a vaccinia virus late promoter activator. J. Biol. Chem. 1999;274:35662–35667. doi: 10.1074/jbc.274.50.35662. [DOI] [PubMed] [Google Scholar]
- Buckley S.M. Junín and Tacaribe work in HeLa cells. Am. J. Trop. Med. Hyg. 1965;14:792–794. doi: 10.4269/ajtmh.1965.14.792. [DOI] [PubMed] [Google Scholar]
- Buckley S.M., Casals J. Lassa fever, a new virus disease of man from West Africa. 3. Isolation and characterization of the virus. Am. J. Trop. Med. Hyg. 1970;19:680–691. doi: 10.4269/ajtmh.1970.19.680. [DOI] [PubMed] [Google Scholar]
- Burkham J., Coen D.M., Weller S.K. ND10 protein PML is recruited to herpes simplex virus type 1 prereplicative sites and replication compartments in the presence of viral DNA polymerase. J. Virol. 1998;72:10100–10107. doi: 10.1128/jvi.72.12.10100-10107.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkham J., Coen D.M., Hwang C.B., Weller S.K. Interactions of herpes simplex virus type 1 with ND10 and recruitment of PML to replication compartments. J. Virol. 2001;75:2353–2367. doi: 10.1128/JVI.75.5.2353-2367.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. The initiation of vaccinia infection. Virology. 1960;11:603–623. doi: 10.1016/0042-6822(60)90103-3. [DOI] [PubMed] [Google Scholar]
- Campadelli‐Fiume G., Roizman B. The egress of herpesviruses from cells: The unanswered questions. J. Virol. 2006;80:6716–6717. doi: 10.1128/JVI.00386-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrascosa J.L., González P., Carrascosa A.L., Garciá‐Barenno B., Enjuanes L., Viñuela E. Localization of structural proteins in African swine fever virus particles by immunoelectron microscopy. J. Virol. 1986;58:377–384. doi: 10.1128/jvi.58.2.377-384.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter G.C., Rodger G., Murphy B.J., Law M., Krauss O., Hollinshead M., Smith G.L. Vaccinia virus cores are transported on microtubules. J. Gen. Virol. 2003;84:2443–2458. doi: 10.1099/vir.0.19271-0. [DOI] [PubMed] [Google Scholar]
- Carvalho T., Seeler J.S., Ohman K., Jordan P., Pettersson U., Akusjarvi G., Carmo‐Fonseca M., Dejean A. Targeting of adenovirus E1A and E4‐ORF3 proteins to nuclear matrix‐associated PML bodies. J. Cell Biol. 1995;131:45–56. doi: 10.1083/jcb.131.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashdollar L.W. Characterization and structural localization of the reovirus λ3 protein. Res. Virol. 1994;145:277–285. doi: 10.1016/s0923-2516(07)80032-x. [DOI] [PubMed] [Google Scholar]
- Chacón M.R., Almazán F., Nogal M.L., Viñuela E., Rodríguez J.F. The African swine fever virus IAP homolg is a late structural polypeptide. Virology. 1995;214:670–674. doi: 10.1006/viro.1995.0083. [DOI] [PubMed] [Google Scholar]
- Challberg M.D. Herpes simplex virus DNA replication. Semin. Virol. 1991;2:247. [Google Scholar]
- Chee A.V., Lopez P., Pandolfi P.P., Roizman B. Promyelocytic leukemia protein mediates interferon‐based anti‐herpes simplex virus 1 effects. J. Virol. 2003;77:7101–7105. doi: 10.1128/JVI.77.12.7101-7105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelbi‐Alix M.K., de Thé H. Herpes virus induced proteasome‐dependent degradation of the nuclear bodies‐associated PML and Sp100 proteins. Oncogene. 1999;18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- Chen M., Goorha R., Murti K.G. Interaction of frog virus 3 with the cytomatrix. IV. Phosphorylation of vimentin precedes the reorganization of intermediate filaments around the virus assembly sites. J. Gen. Virol. 1986;67:915–922. doi: 10.1099/0022-1317-67-5-915. [DOI] [PubMed] [Google Scholar]
- Cherry S., Kunte A., Wang H., Coyne C., Rawson R.B., Perrimon N. COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLoS Pathog. 2006;2:e102. doi: 10.1371/journal.ppat.0020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchar V.G., Goorha R., Granoff A. Early proteins are required for the formation of frog virus 3 assembly sites. Virology. 1984;135:148–156. doi: 10.1016/0042-6822(84)90125-9. [DOI] [PubMed] [Google Scholar]
- Chiu W.L., Szajner P., Moss B., Chang W. Effects of a temperature sensitivity mutation in the J1R protein component of a complex required for vaccinia virus assembly. J. Virol. 2005;79:8046–8056. doi: 10.1128/JVI.79.13.8046-8056.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M.W., Teterina N., Egger D., Bienz K., Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology. 1994;202:129–145. doi: 10.1006/viro.1994.1329. [DOI] [PubMed] [Google Scholar]
- Choe S.S., Dodd D.A., Kirkegaard K. Inhibition of cellular protein secretion by picornaviral 3A proteins. Virology. 2005;337:18–29. doi: 10.1016/j.virol.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Cobbold C., Wileman T. The major structural protein of African swine fever virus, p73, is packaged into large structures, indicative of viral capsid or matrix precursors, on the endoplasmic reticulum. J. Virol. 1998;72:5215–5223. doi: 10.1128/jvi.72.6.5215-5223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold C., Whittle J.T., Wileman T. Involvement of the endoplasmic reticulum in the assembly and envelopment of African swine fever virus. J. Virol. 1996;70:8382–8390. doi: 10.1128/jvi.70.12.8382-8390.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold C., Brookes S.M., Wileman T. Biochemical requirements of virus wrapping by the endoplasmic reticulum calcium store during envelopment of African swine fever virus. J. Virol. 2000;74:2151–2160. doi: 10.1128/jvi.74.5.2151-2160.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold C., Windsor M., Wileman T. A virally encoded chaperone specialized for folding of the major capsid protein of African swine fever virus. J. Virol. 2001;75:7221–7229. doi: 10.1128/JVI.75.16.7221-7229.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit R.C., Moussatche N., Traktman P., Karl Maramorosch A.J.S. Vol. 66. Academic Press; San Diego: 2006. In a nutshell: Structure and assembly of the vaccinia virion; pp. 31–124. (“Advances in Virus Research,”). [DOI] [PubMed] [Google Scholar]
- Cornell C.T., Kiosses W.B., Harkins S., Whitton J.L. Inhibition of protein trafficking by coxsackievirus b3: Multiple viral proteins target a single organelle. J. Virol. 2006;80:6637–6647. doi: 10.1128/JVI.02572-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craighead J.E., Kanich R.E., Almeida J.D. Nonviral microbodies with viral antigenicity produced in cytomegalovirus‐infected cells. J. Virol. 1972;10:766–775. doi: 10.1128/jvi.10.4.766-775.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croissant O., Breitburd F., Orth G. Specificity of cytopathic effect of cutaneous human papillomaviruses. Clin. Dermatol. 1985;3:43–55. doi: 10.1016/0738-081x(85)90048-3. [DOI] [PubMed] [Google Scholar]
- Crotty S., Saleh M.C., Gitlin L., Beske O., Andino R. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J. Virol. 2004;78:3378–3386. doi: 10.1128/JVI.78.7.3378-3386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuconati A., Molla A., Wimmer E. Brefeldin A inhibits cell‐free, de novo synthesis of poliovirus. J. Virol. 1998;72:6456–6464. doi: 10.1128/jvi.72.8.6456-6464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore S., Blasco R., Vincentelli R., Esteban M., Sodeik B., Griffiths G., Krijnse Locker J. A vaccinia virus core protein, p39, is membrane associated. J. Virol. 1996;70:6909–6921. doi: 10.1128/jvi.70.10.6909-6921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca F.G., Wolffe E.J., Weisberg A., Moss B. Characterization of the vaccinia virus H3L envelope protein: Topology and posttranslational membrane insertion via the C‐terminal hydrophobic tail. J. Virol. 2000;74:7508–7517. doi: 10.1128/jvi.74.16.7508-7517.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai‐Ju J.Q., Li L., Johnson L.A., Sandri‐Goldin R.M. ICP27 interacts with the C‐terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 2006;80:3567–3581. doi: 10.1128/JVI.80.7.3567-3581.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Monte P., Pignatelli S., Zini N., Maraldi N.M., Perret E., Prevost M.C., Landini M.P. Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. J. Gen. Virol. 2002;83:1005–1012. doi: 10.1099/0022-1317-83-5-1005. [DOI] [PubMed] [Google Scholar]
- Dales S., Siminovitch L. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S., Eggers H.J., Tamm I., Palade G.E. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- Dales S., Gomatos P.J., Hsu K.C. The uptake and development of reovirus in strain L cells followed with labeled viral ribonucleic acid and ferritin‐antibody conjugates. Virology. 1965;25:193–211. doi: 10.1016/0042-6822(65)90199-6. [DOI] [PubMed] [Google Scholar]
- Darlington R.W., Granoff A., Breeze D.C. Viruses and renal carcinoma of Rana pipiens: II. Ultrastructural studies and sequential development of virus isolated from normal and tumor tissue. Virology. 1966;29:149–156. doi: 10.1016/0042-6822(66)90204-2. [DOI] [PubMed] [Google Scholar]
- Davis R.E., Mathews C.K. Acidic C terminus of vaccinia virus DNA‐binding protein interacts with ribonucleotide reductase. Proc. Natl. Acad. Sci. USA. 1993;90:745–749. doi: 10.1073/pnas.90.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruyn Kops A., Knipe D.M. Formation of DNA replication structures in herpes virus‐infected cells requires a viral DNA binding protein. Cell. 1988;55:857–868. doi: 10.1016/0092-8674(88)90141-9. [DOI] [PubMed] [Google Scholar]
- de Bruyn Kops A., Uprichard S.L., Chen M., Knipe D.M. Comparison of the intranuclear distributions of herpes simplex virus proteins involved in various viral functions. Virology. 1998;252:162–178. doi: 10.1006/viro.1998.9450. [DOI] [PubMed] [Google Scholar]
- De Silva F.S., Moss B. Origin‐independent plasmid replication occurs in vaccinia virus cytoplasmic factories and requires all five known poxvirus replication factors. Virol J. 2005;2:23–34. doi: 10.1186/1743-422X-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitz S.B., Dodd D.A., Cooper S., Parham P., Kirkegaard K. MHC I‐dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA. 2000;97:13790–13795. doi: 10.1073/pnas.250483097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. Autophagy in innate and adaptive immunity. Trends Immunol. 2005;26:523–528. doi: 10.1016/j.it.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Dodd D.A., Giddings T.H., Jr., Kirkegaard K. Poliovirus 3A protein limits interleukin‐6 (IL‐6), IL‐8, and beta interferon secretion during viral infection. J. Virol. 2001;75:8158–8165. doi: 10.1128/JVI.75.17.8158-8165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens J.R., Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995;14:894–907. doi: 10.1002/j.1460-2075.1995.tb07071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens J.R., Giddings T.H., Jr., Kirkegaard K. Inhibition of endoplasmic reticulum‐to‐Golgi traffic by poliovirus protein 3A: Genetic and ultrastructural analysis. J. Virol. 1997;71:9054–9064. doi: 10.1128/jvi.71.12.9054-9064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doglio L., De Marco A., Schleich S., Roos N., Krijnse Locker J. The vaccinia virus E8R gene product: A viral membrane protein that is made early in infection and packaged into the virions’ core. J. Virol. 2002;76:9773–9786. doi: 10.1128/JVI.76.19.9773-9786.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domi A., Beaud G. The punctate sites of accumulation of vaccinia virus early proteins are precursors of sites of viral DNA synthesis. J. Gen. Virol. 2000;81:1231–1235. doi: 10.1099/0022-1317-81-5-1231. [DOI] [PubMed] [Google Scholar]
- Doorbar J., Campbell D., Grand R.J., Gallimore P.H. Identification of the human papilloma virus‐1a E4 gene products. EMBO J. 1986;5:355–362. doi: 10.1002/j.1460-2075.1986.tb04219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J., Ely S., Sterling J., McLean C., Crawford L. Specific interaction between HPV‐16 E1‐E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature. 1991;352:824–827. doi: 10.1038/352824a0. [DOI] [PubMed] [Google Scholar]
- Doucas V., Ishov A.M., Romo A., Juguilon H., Weitzman M.D., Evans R.M., Maul G.G. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 1996;10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- Dubuisson J., Penin F., Moradpour D. Interaction of hepatitis C virus proteins with host cell membranes and lipids. Trends Cell Biol. 2002;12:517–523. doi: 10.1016/s0962-8924(02)02383-8. [DOI] [PubMed] [Google Scholar]
- Dyster L.M., Niles E.G. Genetic and biochemical characterization of vaccinia virus genes D2L and D3R which encode virion structural proteins. Virology. 1991;182:455–467. doi: 10.1016/0042-6822(91)90586-z. [DOI] [PubMed] [Google Scholar]
- Egger D., Bienz K. Intracellular location and translocation of silent and active poliovirus replication complexes. J. Gen. Virol. 2005;86:707–718. doi: 10.1099/vir.0.80442-0. [DOI] [PubMed] [Google Scholar]
- Egger D., Pasamontes L., Bolten R., Boyko V., Bienz K. Reversible dissociation of the poliovirus replication complex: Functions and interactions of its components in viral RNA synthesis. J. Virol. 1996;70:8675–8683. doi: 10.1128/jvi.70.12.8675-8683.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger D., Teterina N., Ehrenfeld E., Bienz K. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 2000;74:6570–6580. doi: 10.1128/jvi.74.14.6570-6580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger D., Wolk B., Gosert R., Bianchi L., Blum H.E., Moradpour D., Bienz K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002;76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichwald C., Vascotto F., Fabbretti E., Burrone O.R. Rotavirus NSP5: Mapping phosphorylation sites and kinase activation and viroplasm localization domains. J. Virol. 2002;76:3461–3470. doi: 10.1128/JVI.76.7.3461-3470.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichwald C., Rodriguez J.F., Burrone O.R. Characterization of rotavirus NSP2/NSP5 interactions and the dynamics of viroplasm formation. J. Gen. Virol. 2004;85:625–634. doi: 10.1099/vir.0.19611-0. [DOI] [PubMed] [Google Scholar]
- El‐Hage N., Luo G. Replication of hepatitis C virus RNA occurs in a membrane‐bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 2003;84:2761–2769. doi: 10.1099/vir.0.19305-0. [DOI] [PubMed] [Google Scholar]
- Elazar M., Cheong K.H., Liu P., Greenberg H.B., Rice C.M., Glenn J.S. Amphipathic helix‐dependent localization of NS5A mediates hepatitis C virus RNA replication. J. Virol. 2003;77:6055–6061. doi: 10.1128/JVI.77.10.6055-6061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epifano C., Krijnse‐Locker J., Salas M.L., Rodríguez J.M., Salas J. The African swine fever virus non‐structural protein pB602L is required for the formation of the icosahedral capsid of the virus particle. J. Virol. 2006;80:12260–12270. doi: 10.1128/JVI.01323-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E., Klemperer N., Ghosh R., Traktman P. The vaccinia virus D5 protein, which is required for DNA replication, is a nucleic acid‐independent nucleoside triphosphatase. J. Virol. 1995;69:5353–5361. doi: 10.1128/jvi.69.9.5353-5361.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D. ICP0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 2000;74:9994–10005. doi: 10.1128/jvi.74.21.9994-10005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol. 2006;8:365–374. doi: 10.1111/j.1462-5822.2005.00677.x. [DOI] [PubMed] [Google Scholar]
- Everett R.D., Maul G.G. HSV‐1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994;13:5062–5069. doi: 10.1002/j.1460-2075.1994.tb06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D., Murray J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 2005;79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D., Sourvinos G., Orr A. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 2003;77:3680–3689. doi: 10.1128/JVI.77.6.3680-3689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D., Sourvinos G., Leiper C., Clements J.B., Orr A. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: Localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10‐like complexes. J. Virol. 2004;78:1903–1917. doi: 10.1128/JVI.78.4.1903-1917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D., Rechter S., Papior P., Tavalai N., Stamminger T., Orr A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006;80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbretti E., Afrikanova I., Vascotto F., Burrone O.R. Two non‐structural rotavirus proteins, NSP2 and NSP5, form viroplasm‐like structures in vivo. J. Gen. Virol. 1999;80:333–339. doi: 10.1099/0022-1317-80-2-333. [DOI] [PubMed] [Google Scholar]
- Feierbach B., Piccinotti S., Bisher M., Denk W., Enquist L.W. Alpha‐herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog. 2006;2:e85. doi: 10.1371/journal.ppat.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L., Sapp C., Streeck R.E., Sapp M. Assembly and translocation of papillomavirus capsid proteins. J. Virol. 2002;76:10009–10014. doi: 10.1128/JVI.76.19.10009-10014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin L., Schafer F., Sotlar K., Streeck R.E., Sapp M. Reorganization of nuclear domain 10 induced by papillomavirus capsid protein l2. Virology. 2002;295:97–107. doi: 10.1006/viro.2002.1360. [DOI] [PubMed] [Google Scholar]
- Forest T., Barnard S., Baines J.D. Active intranuclear movement of herpesvirus capsids. Nat. Cell. Biol. 2005;7:429–431. doi: 10.1038/ncb1243. [DOI] [PubMed] [Google Scholar]
- Fredman J.N., Engler J.A. Adenovirus precursor to terminal protein interacts with the nuclear matrix in vivo and in vitro. J. Virol. 1993;67:3384–3395. doi: 10.1128/jvi.67.6.3384-3395.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froshauer S., Kartenbeck J., Helenius A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell. Biol. 1988;107:2075–2086. doi: 10.1083/jcb.107.6.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W., Granzow H., Klupp B.G., Kopp M., Mettenleiter T.C. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 2002;76:6729–6742. doi: 10.1128/JVI.76.13.6729-6742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y., Shatkin A.J. Viral and cellular mRNA capping: Past and prospects. Adv. Virus Res. 2002;55:135–184. doi: 10.1016/S0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo I., Viñuela E., Carrascosa A.L. Characterization of the African swine fever virus protein p49: A new late structural polypeptide. J. Gen. Virol. 2000;81:59–65. doi: 10.1099/0022-1317-81-1-59. [DOI] [PubMed] [Google Scholar]
- García‐Beato R., Salas M.L., Viñuela E., Salas J. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology. 1992;188:637–649. doi: 10.1016/0042-6822(92)90518-t. [DOI] [PubMed] [Google Scholar]
- Gazina E.V., Mackenzie J.M., Gorrell R.J., Anderson D.A. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J. Virol. 2002;76:11113–11122. doi: 10.1128/JVI.76.21.11113-11122.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillian A.L., Nibert M.L. Amino terminus of reovirus nonstructural protein σNS is important for ssRNA binding and nucleoprotein complex formation. Virology. 1998;240:1–11. doi: 10.1006/viro.1997.8905. [DOI] [PubMed] [Google Scholar]
- González R.A., Espinosa R., Romero P., López S., Arias C.F. Relative localization of viroplasmic and endoplasmic reticulum‐resident rotavirus proteins in infected cells. Arch. Virol. 2000;145:1963–1973. doi: 10.1007/s007050070069. [DOI] [PubMed] [Google Scholar]
- Goodrich L.D., Schaffer P.A., Dorsky D.I., Crumpacker C.S., Parris D.S. Localization of the herpes simplex virus type 1 65‐kilodalton DNA‐binding protein and DNA polymerase in the presence and absence of viral DNA synthesis. J. Virol. 1990;64:5738–5749. doi: 10.1128/jvi.64.12.5738-5749.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goorha R. Frog virus 3 DNA replication occurs in two stages. J. Virol. 1982;43:519–528. doi: 10.1128/jvi.43.2.519-528.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Wolf Y.I. A new superfamily of putative NTP‐binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990;262:145–148. doi: 10.1016/0014-5793(90)80175-i. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006;117:17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double‐membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Egger D., Lohmann V., Bartenschlager R., Blum H.E., Bienz K., Moradpour D. Identification of the hepatitis C virus RNA replication complex in Huh‐7 cells harboring subgenomic replicons. J. Virol. 2003;77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima F., Daikoku T., Yamada H., Oshima S., Tsurumi T., Nishiyama Y. Subcellular localization of the US3 protein kinase of herpes simplex virus type 2. Arch. Virol. 1998;143:613–622. doi: 10.1007/s007050050317. [DOI] [PubMed] [Google Scholar]
- Granja A.G., Nogal M.L., Hurtado C., Salas J., Salas M.L., Carrascosa A.L., Revilla Y. Modulation of p53 cellular function and cell death by African swine fever virus. J. Virol. 2004;78:7165–7174. doi: 10.1128/JVI.78.13.7165-7174.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granoff A., Came P.E., Breeze D.C. Viruses and renal carcinoma of Rana pipiens. I. The isolation and properties of virus from normal and tumor tissue. Virology. 1966;29:133–148. doi: 10.1016/0042-6822(66)90203-0. [DOI] [PubMed] [Google Scholar]
- Granzow H., Weiland F., Jöns A., Klupp B.G., Karger A., Mettenleiter T.C. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: A reassessment. J. Virol. 1997;71:2072–2082. doi: 10.1128/jvi.71.3.2072-2082.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G., Roos N., Schleich S., Locker J.K. Structure and assembly of intracellular mature vaccinia virus: Thin‐section analyses. J. Virol. 2001;75:11056–11070. doi: 10.1128/JVI.75.22.11056-11070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri G. Recherches sur la pathologie et étiologie de l'infection vaccinique et varioleuse. Arch. Ital. de Biol. 1893;19:195. [Google Scholar]
- Hagiwara Y., Komoda K., Yamanaka T., Tamai A., Meshi T., Funada R., Tsuchiya T., Naito S., Ishikawa M. Subcellular localization of host and viral proteins associated with tobamovirus RNA replication. EMBO J. 2003;22:344–353. doi: 10.1093/emboj/cdg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath C.M., Windsor M., Wileman T. Aggresomes resemble sites specialized for virus assembly. J. Cell Biol. 2001;153:449–456. doi: 10.1083/jcb.153.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath C.M., Windsor M., Wileman T. Membrane association facilitates the correct processing of pp220 during production of the major matrix proteins of African swine fever virus. J. Virol. 2003;77:1682–1690. doi: 10.1128/JVI.77.3.1682-1690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero‐Martínez E., Roberts K.L., Hollinshead M., Smith G.L. Vaccinia virus intracellular enveloped virions move to the cell periphery on microtubules in the absence of the A36R protein. J. Gen. Virol. 2005;86:2961–2968. doi: 10.1099/vir.0.81260-0. [DOI] [PubMed] [Google Scholar]
- Herzer K., Falk C.S., Encke J., Eichhorst S.T., Ulsenheimer A., Seliger B., Krammer P.H. Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J. Virol. 2003;77:8299–8309. doi: 10.1128/JVI.77.15.8299-8309.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. Deep‐etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann J.B., Cheng N., Newcomb W.W., Trus B.L., Brown J.C., Steven A.C. Dynamics of herpes simplex virus capsid maturation visualized by time‐lapse cryo‐electron microscopy. Nat. Struct. Biol. 2003;10:334–341. doi: 10.1038/nsb922. [DOI] [PubMed] [Google Scholar]
- Higashi N. Electron microscopy of viruses in thin sections of cells grown in culture. Prog. Med. Virol. 1973;15:331–379. [PubMed] [Google Scholar]
- Hingamp P.M., Arnold J.E., Mayer R.J., Dixon L.K. A ubiquitin conjugating enzyme encoded by African swine fever virus. EMBO J. 1992;11:361–366. doi: 10.1002/j.1460-2075.1992.tb05058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead M., Vanderplasschen A., Smith G.L., Vaux D.J. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 1999;73:1503–1517. doi: 10.1128/jvi.73.2.1503-1517.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao J.C., Chao C.C., Young M.J., Chang Y.T., Cho E.C., Chang W. A poxvirus host range protein, CP77, binds to a cellular protein, HMG20A, and regulates its dissociation from the vaccinia virus genome in CHO‐K1 cells. J. Virol. 2006;80:7714–7728. doi: 10.1128/JVI.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.H., Huang Y.H., Yuan X.P., Zhang Q.Y. Electron microscopic examination of the viromatrix of Rana grylio virus in a fish cell line. J. Virol. Methods. 2006;133:117–123. doi: 10.1016/j.jviromet.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Hugle T., Fehrmann F., Bieck E., Kohara M., Krausslich H.G., Rice C.M., Blum H.E., Moradpour D. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology. 2001;284:70–81. doi: 10.1006/viro.2001.0873. [DOI] [PubMed] [Google Scholar]
- Hung J.J., Chung C.S., Chang W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J. Virol. 2002;76:1379–1390. doi: 10.1128/JVI.76.3.1379-1390.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain M., Moss B. Evidence against an essential role of COPII‐mediated cargo transport to the endoplasmic reticulum‐Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 2003;77:11754–11766. doi: 10.1128/JVI.77.21.11754-11766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson I., Whiteley A., Browne H., Elliott G. Sequential localization of two herpes simplex virus tegument proteins to punctate nuclear dots adjacent to ICP0 domains. J. Virol. 2002;76:10365–10373. doi: 10.1128/JVI.76.20.10365-10373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihashi Y., Matsumoto S., Dales S. Biogenesis of poxviruses: Role of A‐type inclusions and host cell membranes in virus dissemination. Virology. 1971;46:507–532. doi: 10.1016/0042-6822(71)90056-0. [DOI] [PubMed] [Google Scholar]
- Irmiere A., Gibson W. Isolation and characterization of a noninfectious virion‐like particle released from cells infected with human strains of cytomegalovirus. Virology. 1983;130:118–133. doi: 10.1016/0042-6822(83)90122-8. [DOI] [PubMed] [Google Scholar]
- Irurzun A., Perez L., Carrasco L. Involvement of membrane traffic in the replication of poliovirus genomes: Effects of brefeldin A. Virology. 1992;191:166–175. doi: 10.1016/0042-6822(92)90178-r. [DOI] [PubMed] [Google Scholar]
- Ishov A.M., Maul G.G. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 1996;134:815–826. doi: 10.1083/jcb.134.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L.M., Aravind L., Koonin E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001;75:11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A.C., Ye H., Ridaura‐Sanz C., Lopez‐Corella E. Quantitative study of the infection in brain neurons in human rabies. J. Med. Virol. 2001;65:614–618. [PubMed] [Google Scholar]
- Jackson W.T., Jr., Giddings T.H., Taylor M.P., Mulinyawe S., Rabinovitch M., Kopito R.R., Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahedi S., Markovitz N.S., Filatov F., Roizman B. Colocalization of the herpes simplex virus 1 UL4 protein with infected cell protein 22 in small, dense nuclear structures formed prior to onset of DNA synthesis. J. Virol. 1999;73:5132–5138. doi: 10.1128/jvi.73.6.5132-5136.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J.A., Ward C.L., Kopito R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N., Wileman T. African swine fever virus infection disrupts centrosome assembly and function. J. Gen. Virol. 2005;86:589–594. doi: 10.1099/vir.0.80623-0. [DOI] [PubMed] [Google Scholar]
- Jouvenet N., Monaghan P., Way M., Wileman T. Transport of African swine fever virus from assembly sites to the plasma membrane is dependent on microtubules and conventional kinesin. J. Virol. 2004;78:7990–8001. doi: 10.1128/JVI.78.15.7990-8001.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N., Windsor M., Rietdorf J., Hawes P., Monaghan P., Way M., Wileman T. African swine fever virus induces filopodia‐like projections at the plasma membrane. Cell. Microbiol. 2006;8:1803–1811. doi: 10.1111/j.1462-5822.2006.00750.x. [DOI] [PubMed] [Google Scholar]
- Kato S., Takahashi M., Kameyama S., Kamahora J. A study on the morphological and cyto‐immunological relationship between the inclusions of variola, cowpox, rabbitpox, vaccinia (variola origin) and vaccinia IHD and a consideration of the term “Guarnieri body.”. Biken's J. 1959;2:353. [Google Scholar]
- Kato K., Daikoku T., Goshima F., Kume H., Yamaki K., Nishiyama Y. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 2000;145:2149–2162. doi: 10.1007/s007050070045. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Kovacs J.J., McLaurin A., Vance J.M., Ito A., Yao T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kim J.E., Song W.K., Chung K.M., Back S.H., Jang S.K. Subcellular localization of hepatitis C viral proteins in mammalian cells. Arch. Virol. 1999;144:329–343. doi: 10.1007/s007050050507. [DOI] [PubMed] [Google Scholar]
- King N.J., Kesson A.M. Interferon‐independent increases in class I major histocompatibility complex antigen expression follow flavivirus infection. J. Gen. Virol. 1988;69:2535–2543. doi: 10.1099/0022-1317-69-10-2535. [DOI] [PubMed] [Google Scholar]
- Kirkegaard K., Taylor M.P., Jackson W.T. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004;2:301–314. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe D.M., Senechek D., Rice S.A., Smith J.L. Stages in the nuclear association of the herpes simplex virus transcriptional activator protein ICP4. J. Virol. 1987;61:276–284. doi: 10.1128/jvi.61.2.276-284.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C., Moffat K., Ali S., Ryan M., Wileman T. Foot‐and‐mouth disease virus replication sites form next to the nucleus and close to the Golgi apparatus, but exclude marker proteins associated with host membrane compartments. J. Gen. Virol. 2005;86:687–696. doi: 10.1099/vir.0.80208-0. [DOI] [PubMed] [Google Scholar]
- Konan K.V., Giddings T.H., Jr., Ikeda M., Li K., Lemon S.M., Kirkegaard K. Nonstructural protein precursor NS4A/B from hepatitis C virus alters function and ultrastructure of host secretory apparatus. J. Virol. 2003;77:7843–7855. doi: 10.1128/JVI.77.14.7843-7855.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratova A.A., Neznanov N., Kondratov R.V., Gudkov A.V. Poliovirus protein 3A binds and inactivates LIS1, causing block of membrane protein trafficking and deregulation of cell division. Cell Cycle. 2005;4:1403–1410. doi: 10.4161/cc.4.10.2041. [DOI] [PubMed] [Google Scholar]
- Kopp M., Klupp B.G., Granzow H., Fuchs W., Mettenleiter T.C. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: Role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 2002;76:8820–8833. doi: 10.1128/JVI.76.17.8820-8833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse‐Locker J., Schleich S., Rodriguez D., Goud B., Snijder E.J., Griffiths G. The role of a 21‐kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J. Biol. Chem. 1996;271:14950–14958. doi: 10.1074/jbc.271.25.14950. [DOI] [PubMed] [Google Scholar]
- Krogerus C., Egger D., Samuilova O., Hyypia T., Bienz K. Replication complex of human parechovirus 1. J. Virol. 2003;77:8512–8523. doi: 10.1128/JVI.77.15.8512-8523.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola B., Audic S., Robert C., Jungang L., de Lamballerie X., Drancourt M., Birtles R., Claverie J.‐M., Raoult D. A giant virus in amoebae. Science. 2003;299:2033. doi: 10.1126/science.1081867. [DOI] [PubMed] [Google Scholar]
- Lackner C.A., Condit R.C. Vaccinia virus gene A18R DNA helicase is a transcript release factor. J. Biol. Chem. 2000;275:1485–1494. doi: 10.1074/jbc.275.2.1485. [DOI] [PubMed] [Google Scholar]
- Lamberti C., Weller S.K. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 1998;72:2463–2473. doi: 10.1128/jvi.72.3.2463-2473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landini M.P., Severi B., Cenacchi G., Lazzarotto T., Lindenmeier W., Necker A. Human cytomegalovirus structural components: Intracellular and intraviral localization of p38. Virus Res. 1991;19:189–198. doi: 10.1016/0168-1702(91)90045-w. [DOI] [PubMed] [Google Scholar]
- Lee J.Y., Marshall J.A., Bowden D.S. Characterization of rubella virus replication complexes using antibodies to double‐stranded RNA. Virology. 1994;200:307–312. doi: 10.1006/viro.1994.1192. [DOI] [PubMed] [Google Scholar]
- Leopardi R., Ward P.L., Ogle W.O., Roizman B. Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the U(L)13 proteinkinase. J. Virol. 1997;71:1133–1139. doi: 10.1128/jvi.71.2.1133-1139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lethbridge K.J., Scott G.E., Leppard K.N. Nuclear matrix localization and SUMO‐1 modification of adenovirus type 5 E1b 55K protein are controlled by E4 Orf6 protein. J. Gen. Virol. 2003;84:259–268. doi: 10.1099/vir.0.18820-0. [DOI] [PubMed] [Google Scholar]
- Levine B., Klionsky D.J. Development by self‐digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Lindenbach B.D., Meuleman P., Ploss A., Vanwolleghem T., Syder A.J., McKeating J.A., Lanford R.E., Feinstone S.M., Major M.E., Leroux-Roels G., Rice C.M. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natal. Acad. Sci. USA. 2004;103:3805–3809. doi: 10.1073/pnas.0511218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott‐Schwartz J., Roberts T.H., Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol. 2000;16:557–589. doi: 10.1146/annurev.cellbio.16.1.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak L.M., Uprichard S.L., Knipe D.M. Functional order of assembly of herpes simplex virus DNA replication proteins into prereplicative site structures. J. Virol. 1996;70:1759–1767. doi: 10.1128/jvi.70.3.1759-1767.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., King N., Kesson A., Blanden R.V., Mullbacher A. Flavivirus infection up‐regulates the expression of class I and class II major histocompatibility antigens on and enhances T cell recognition of astrocytes in vitro. J. Neuroimmunol. 1989;21:157–168. doi: 10.1016/0165-5728(89)90171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Shevchenko A., Shevchenko A., Berk A.J. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 2005;79:14004–14016. doi: 10.1128/JVI.79.22.14004-14016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobigs M., Mullbacher A., Lee E. Evidence that a mechanism for efficient flavivirus budding upregulates MHC class I. Immunol. Cell Biol. 2004;82:184–188. doi: 10.1046/j.0818-9641.2004.01218.x. [DOI] [PubMed] [Google Scholar]
- Lopez‐Iglesias C., Puvion‐Dutilleul F., Cebrian J., Christensen M.E. Herpes simplex virus type 1‐induced modifications in the distribution of nucleolar B‐36 protein. Eur. J. Cell Biol. 1988;46:259–269. [PubMed] [Google Scholar]
- Lukonis C.J., Weller S.K. Formation of herpes simplex virus type 1 replication compartments by transfection: Requirements and localization to nuclear domain 10. J. Virol. 1997;71:2390–2399. doi: 10.1128/jvi.71.3.2390-2399.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukonis C.J., Burkham J., Weller S.K. Herpes simplex virus type 1 prereplicative sites are a heterogeneous population: Only a subset are likely to be precursors to replication compartments. J. Virol. 1997;71:4771–4781. doi: 10.1128/jvi.71.6.4771-4781.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz P., Puvion‐Dutilleul F., Lutz Y., Kedinger C. Nucleoplasmic and nucleolar distribution of the adenovirus IVa2 gene product. J. Virol. 1996;70:3449–3460. doi: 10.1128/jvi.70.6.3449-3460.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle J.M., Bullitt E., Bienz K., Kirkegaard K. Visualization and functional analysis of RNA‐dependent RNA polymerase lattices. Science. 2002;296:2218–2222. doi: 10.1126/science.1070585. [DOI] [PubMed] [Google Scholar]
- Mackenzie J. Wrapping things up about virus RNA replication. Traffic. 2005;6:967–977. doi: 10.1111/j.1600-0854.2005.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie J.M., Westaway E.G. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001;75:10787–10799. doi: 10.1128/JVI.75.22.10787-10799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie J.M., Khromykh A.A., Jones M.K., Westaway E.G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology. 1998;245:203–215. doi: 10.1006/viro.1998.9156. [DOI] [PubMed] [Google Scholar]
- Mackenzie J.M., Jones M.K., Westaway E.G. Markers for trans‐Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus‐infected cells. J. Virol. 1999;73:9555–9567. doi: 10.1128/jvi.73.11.9555-9567.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliano D., Marshall J.A., Bowden D.S., Vardaxis N., Meanger J., Lee J.Y. Rubella virus replication complexes are virus‐modified lysosomes. Virology. 1998;240:57–63. doi: 10.1006/viro.1997.8906. [DOI] [PubMed] [Google Scholar]
- Maglova L.M., Crowe W.E., Russell J.M. Perinuclear localization of Na‐K‐Cl‐cotransporter protein after human cytomegalovirus infection. Am. J. Physiol. Cell Physiol. 2004;286:C1324–C1334. doi: 10.1152/ajpcell.00404.2003. [DOI] [PubMed] [Google Scholar]
- Mallardo M., Schleich S., Krijnse Locker J. Microtubule‐dependent organization of vaccinia virus core‐derived early mRNAs into distinct cytoplasmic structures. Mol. Biol. Cell. 2001;12:3875–3891. doi: 10.1091/mbc.12.12.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallardo M., Leithe E., Schleich S., Roos N., Doglio L., Krijnse Locker J. Relationship between vaccinia virus intracellular cores, early mRNAs, and DNA replication sites. J. Virol. 2002;76:5167–5183. doi: 10.1128/JVI.76.10.5167-5183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovitz N.S., Roizman B. Small dense nuclear bodies are the site of localization of herpes simplex virus 1 U(L)3 and U(L)4 proteins and of ICP22 only when the latter protein is present. J. Virol. 2000;74:23–28. doi: 10.1128/jvi.74.1.523-528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K.H., Franke C.A., Hruby D.E. Novel acylation of poxvirus A‐type inclusion proteins. Virus Res. 1999;60:147–157. doi: 10.1016/s0168-1702(99)00013-1. [DOI] [PubMed] [Google Scholar]
- Martinez‐Pomares L., Simon‐Mateo C., Lopez‐Otin C., Viñuela E. Characterization of the African swine fever virus structural protein p14.5 a DNA binding protein. Virology. 1997;229:201–211. doi: 10.1006/viro.1996.8434. [DOI] [PubMed] [Google Scholar]
- Matsumoto S. Rabies virus. Adv. Virus Res. 1970;16:257–301. [PubMed] [Google Scholar]
- Matsumoto S., Schneider L.G., Kawai A., Yonezawa T. Further studies on the replication of rabies and rabies‐like viruses in organized cultures of mammalian neural tissues. J. Virol. 1974;14:981–996. doi: 10.1128/jvi.14.4.981-996.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul G.G., Ishov A.M., Everett R.D. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type‐1. Virology. 1996;217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- Maynell L.A., Kirkegaard K., Klymkowsky M.W. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 1992;66:1985–1994. doi: 10.1128/jvi.66.4.1985-1994.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan M., Windsor M., Ponnambalam S., Armstrong J., Wileman T. The trans Golgi network is lost from cells infected with African swine fever virus. J. Virol. 2001;75:11755–11765. doi: 10.1128/JVI.75.23.11755-11765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKelvey T.A., Andrews S.C., Miller S.E., Ray C.A., Pickup D.J. Identification of the orthopoxvirus p4c gene, which encodes a structural protein that directs intracellular mature virus particles into A‐type inclusions. J. Virol. 2002;76:11216–11225. doi: 10.1128/JVI.76.22.11216-11225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meints R.H., Lee K., Van Etten J.L. Assembly site of the virus PBCV‐1 in a Chlorella‐like green alga: Ultrastructural studies. Virology. 1986;154:240–245. doi: 10.1016/0042-6822(86)90448-4. [DOI] [PubMed] [Google Scholar]
- Mettenleiter T.C. Herpesvirus assembly and egress. J. Virol. 2002;76:1537–1547. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter T.C., Minson T. Egress of alphaherpes viruses. J. Virol. 2006;80:1610–1611. doi: 10.1128/JVI.80.3.1610-1612.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter T.C., Klupp B.G., Granzow H. Herpesvirus assembly: A tale of two membranes. Curr. Opin. Microbiol. 2006;9:423–429. doi: 10.1016/j.mib.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Miller D.J., Ahlquist P. Flock house virus RNA polymerase is a transmembrane protein with amino‐terminal sequences sufficient for mitochondrial localization and membrane insertion. J. Virol. 2002;76:9856–9867. doi: 10.1128/JVI.76.19.9856-9867.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.L., Broering T.J., Parker J.S., Arnold M.M., Nibert M.L. Reovirus σNS protein localizes to inclusions through an association requiring the μNS amino terminus. J. Virol. 2003;77:4566–4576. doi: 10.1128/JVI.77.8.4566-4576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.L., Parker J.S., Dinoso J.B., Piggott C.D., Perron M.J., Nibert M.L. Increased ubiquitination and other covariant phenotypes attributed to a strain‐ and temperature‐dependent defect of reovirus core protein μ2. J. Virol. 2004;78:10291–10302. doi: 10.1128/JVI.78.19.10291-10302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millns A.K., Carpenter M.S., DeLange A.M. The vaccinia virus‐encoded uracil DNA glycosylase has an essential role in viral DNA replication. Virology. 1994;198:504–513. doi: 10.1006/viro.1994.1061. [DOI] [PubMed] [Google Scholar]
- Miner J.N., Hruby D.E. Rifampicin prevents virosome localization of L65, an essential vaccinia virus polypeptide. Virology. 1989;170:227–237. doi: 10.1016/0042-6822(89)90370-x. [DOI] [PubMed] [Google Scholar]
- Moffat K., Howell G., Knox C., Belsham G.J., Monaghan P., Ryan M.D., Wileman T. Effects of foot‐and‐mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum‐to‐Golgi transport. J. Virol. 2005;79:4382–4395. doi: 10.1128/JVI.79.7.4382-4395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat K., Knox C., Howell G., Clark S.J., Yang Y.G., Belsham G.J., Ryan M., Wileman T. Inhibition of the secretory pathway by the Foot‐and‐Mouth disease virus 2BC protein is reproduced by co‐expression of 2B with 2C and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 2007;81:1129–1139. doi: 10.1128/JVI.00393-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas A.R., Dales S. Involvement of spicules in the formation of vaccinia virus envelopes elucidated by a conditional lethal mutant. Virology. 1995;214:494–502. doi: 10.1006/viro.1995.0060. [DOI] [PubMed] [Google Scholar]
- Momburg F., Mullbacher A., Lobigs M. Modulation of transporter associated with antigen processing (TAP)‐mediated peptide import into the endoplasmic reticulum by flavivirus infection. J. Virol. 2001;75:5663–5671. doi: 10.1128/JVI.75.12.5663-5671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan P., Cook H., Hawes P., Simpson J., Tomley F. High‐pressure freezing in the study of animal pathogens. J. Microsc. 2003;212:62–70. doi: 10.1046/j.1365-2818.2003.01245.x. [DOI] [PubMed] [Google Scholar]
- Monaghan P., Cook H., Jackson T., Ryan M., Wileman T. The ultrastructure of the developing replication site in foot‐and‐mouth disease virus‐infected BHK‐38 cells. J. Gen. Virol. 2004;85:933–946. doi: 10.1099/vir.0.19408-0. [DOI] [PubMed] [Google Scholar]
- Moradpour D., Brass V., Bieck E., Friebe P., Gosert R., Blum H.E., Bartenschlager R., Penin F., Lohmann V. Membrane association of the RNA‐dependent RNA polymerase is essential for hepatitis C virus RNA replication. J. Virol. 2004;78:13278–13284. doi: 10.1128/JVI.78.23.13278-13284.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan C., Ellison S.A., Rose H.M., Moore D.H. Structure and development of viruses observed in the electron microscope. II. Vaccinia and fowl pox viruses. J. Exp. Med. 1954;100:301. doi: 10.1084/jem.100.3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B., Rosenblum E.N., Katz E., Grimley P.M. Rifampicin: A specific inhibitor of vaccinia virus assembly. Nature. 1969;224:1280–1284. doi: 10.1038/2241280a0. [DOI] [PubMed] [Google Scholar]
- Mossman K.L., Smiley J.R. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon‐induced barriers to virus replication. J. Virol. 2002;76:1995–1998. doi: 10.1128/JVI.76.4.1995-1998.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman K.L., Sherburne R., Lavery C., Duncan J., Smiley J.R. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 2000;74:6287–6299. doi: 10.1128/jvi.74.14.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura Nunes J.F., Vigário J.D., Terrinha A.M. Ultrastructural study of African swine fever virus replication in cultures of swine bone marrow cells. Arch. Virol. 1975;49:59–66. doi: 10.1007/BF02175596. [DOI] [PubMed] [Google Scholar]
- Murata T., Goshima F., Daikoku T., Inagaki‐Ohara K., Takakuwa H., Kato K., Nishiyama Y. Mitochondrial distribution and function in herpes simplex virus‐infected cells. J. Gen. Virol. 2000;81:401–406. doi: 10.1099/0022-1317-81-2-401. [DOI] [PubMed] [Google Scholar]
- Murcia‐Nicolas A., Bolbach G., Blais J.C., Beaud G. Identification by mass spectroscopy of three major early proteins associated with virosomes in vaccinia virus‐infected cells. Virus Res. 1999;59:1–12. doi: 10.1016/s0168-1702(98)00114-2. [DOI] [PubMed] [Google Scholar]
- Murphy F.A., Webb P.A., Johnson K.M., Whitfield S.G., Chappell W.A. Arenoviruses in Vero cells: Ultrastructural studies. J. Virol. 1970;6:507–518. doi: 10.1128/jvi.6.4.507-518.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murti K., Goorha R. Interaction of frog virus‐3 with the cytoskeleton. I. Altered organization of microtubules, intermediate filaments, and microfilaments. J. Cell Biol. 1983;96:1248–1257. doi: 10.1083/jcb.96.5.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murti K., Goorha R. Synthesis of frog virus 3 proteins occurs on intermediate filament‐bound polyribosomes. Biol. Cell. 1989;65:205–214. [PubMed] [Google Scholar]
- Murti K.G., Goorha R., Klymkowsky M.W. A functional role for intermediate filaments in the formation of frog virus 3 assembly sites. Virology. 1988;162:264–269. doi: 10.1016/0042-6822(88)90420-5. [DOI] [PubMed] [Google Scholar]
- Murti K.G., Davis D.S., Kitchingman G.R. Localization of adenovirus‐encoded DNA replication proteins in the nucleus by immunogold electron microscopy. J. Gen. Virol. 1990;71:2847–2857. doi: 10.1099/0022-1317-71-12-2847. [DOI] [PubMed] [Google Scholar]
- Nalwanga D., Rempel S., Roizman B., Baines J.D. The UL 16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology. 1996;226:236–242. doi: 10.1006/viro.1996.0651. [DOI] [PubMed] [Google Scholar]
- Negri A. Beitrag zum Studium de Aetiologie der Tollwuth. Z. Hyg. Infektionskr. 1903;43:507–528. [Google Scholar]
- Negri A. Über die morphologie und den entwicklungszyklus des parasiten der tollwut. Z. Hyg. Infektionskr. 1909;63:421–443. [Google Scholar]
- Nerenberg B.T., Taylor J., Bartee E., Gouveia K., Barry M., Fruh K. The poxviral RING protein p28 is a ubiquitin ligase that targets ubiquitin to viral replication factories. J. Virol. 2005;79:597–601. doi: 10.1128/JVI.79.1.597-601.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton C., Rouiller I., Wileman T. The subcellular distribution of multigene family 110 proteins of African swine fever virus is determined by differences in C‐terminal KDEL endoplasmic reticulum retention motifs. J. Virol. 2004;78:3710–3721. doi: 10.1128/JVI.78.7.3710-3721.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton C.L., McCrossan M.C., Denyer M., Ponnambalam S., Armstrong J., Takamatsu H.H., Wileman T.E. African swine fever virus causes microtubule dependent dispersal of the trans‐Golgi network and slows delivery of membrane protein to the plasma membrane. J. Virol. 2006;80:11385–11392. doi: 10.1128/JVI.00439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Homa F.L., Thomsen D.R., Ye Z., Brown J.C. Cell‐free assembly of the herpes simplex virus capsid. J. Virol. 1994;68:6059–6063. doi: 10.1128/jvi.68.9.6059-6063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Homa F.L., Thomsen D.R., Booy F.P., Trus B.L., Steven A.C., Spencer J.V., Brown J.C. Assembly of the herpes simplex virus capsid: Characterization of intermediates observed during cell‐free capsid formation. J. Mol. Biol. 1996;263:432–446. doi: 10.1006/jmbi.1996.0587. [DOI] [PubMed] [Google Scholar]
- Neznanov N., Kondratova A., Chumakov K.M., Angres B., Zhumabayeva B., Agol V.I., Gudkov A.V. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)‐induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 2001;75:10409–10420. doi: 10.1128/JVI.75.21.10409-10420.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M.L. Ultrastructural studies of Kunjin virus‐infected Aedes albopictus cells. J. Gen. Virol. 1987;68:577–582. doi: 10.1099/0022-1317-68-2-577. [DOI] [PubMed] [Google Scholar]
- Ni Y., Iwatani Y., Morimoto K., Kawai A. Studies on unusual cytoplasmic structures which contain rabies virus envelope proteins. J. Gen. Virol. 1996;77:2137–2147. doi: 10.1099/0022-1317-77-9-2137. [DOI] [PubMed] [Google Scholar]
- Nietfeldt J.W., Lee K., Van Etten J.L. Chlorella virus PBCV‐1 replication is not affected by cytoskeletal disruptors. Intervirology. 1992;33:116–120. doi: 10.1159/000150240. [DOI] [PubMed] [Google Scholar]
- Nilsson M., von Bonsdorff C.H., Weclewicz K., Cohen J., Svensson L. Assembly of viroplasm and virus‐like particles of rotavirus by a Semliki Forest virus replicon. Virology. 1998;242:255–265. doi: 10.1006/viro.1997.8987. [DOI] [PubMed] [Google Scholar]
- Novoa R.R., Calderita G., Arranz R., Fontana J., Granzow H., Risco C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell. 2005;97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa N., Daikoku T., Yamauchi Y., Takakuwa H., Goshima F., Yoshikawa T., Nishiyama Y. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genus. 2002;24:257–266. doi: 10.1023/a:1015332716927. [DOI] [PubMed] [Google Scholar]
- Nozawa N., Yamauchi Y., Ohtsuka K., Kawaguchi Y., Nishiyama Y. Formation of aggresome‐like structures in herpes simplex virus type 2‐infected cells and a potential role in virus assembly. Exp. Cell Res. 2004;299:486–497. doi: 10.1016/j.yexcr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- O'Donnell V.K., Pacheco J.M., Henry T.M., Mason P.W. Subcellular distribution of the foot‐and‐mouth disease virus 3A protein in cells infected with viruses encoding wild‐type and bovine‐attenuated forms of 3A. Virology. 2001;287:151–162. doi: 10.1006/viro.2001.1035. [DOI] [PubMed] [Google Scholar]
- Oh J., Broyles S.S. Host cell nuclear proteins are recruited to cytoplasmic vaccinia virus replication complexes. J. Virol. 2005;79:12852–12860. doi: 10.1128/JVI.79.20.12852-12860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda S., Senkevich T.G., Moss B. Entry of vaccinia virus and cell‐cell fusion require a highly conserved cysteine‐rich membrane protein encoded by the A16L gene. J. Virol. 2006;80:51–61. doi: 10.1128/JVI.80.1.51-61.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros M., García‐Escudero R., Alejo A., Viñuela E., Salas M.L., Salas J. African Swine Fever Virus dUTPase is a highly specific enzyme required for efficient replication in Swine macrophages. J. Virol. 1999;73:8934–8943. doi: 10.1128/jvi.73.11.8934-8943.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivo P.D., Nelson N.J., Challberg M.D. Herpes simplex virus 1 gene products required for DNA replication: Identification and overexpression. J. Virol. 1989;63:196–204. doi: 10.1128/jvi.63.1.196-204.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L., Perrelet A., Ravazzola M., Wieland F.T., Schekman R., Rothman J.E. “BFA bodies”: A subcompartment of the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 1993;90:11089–11093. doi: 10.1073/pnas.90.23.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios S., Perez L.H., Welsch S., Schleich S., Chmielarska K., Melchior F., Locker J.K. Quantitative SUMO‐1 modification of a vaccinia virus protein is required for its specific localization and prevents its self‐association. Mol. Biol. Cell. 2005;16:2822–2835. doi: 10.1091/mbc.E04-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Broering T.J., Kim J., Higgins D.E., Nibert M.L. Reovirus core protein μ2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J. Virol. 2002;76:4483–4496. doi: 10.1128/JVI.76.9.4483-4496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D.D., Pickup D.J., Joklik W.K. Isolation of cowpox virus A‐type inclusions and characterization of their major protein component. Virology. 1986;149:174–189. doi: 10.1016/0042-6822(86)90119-4. [DOI] [PubMed] [Google Scholar]
- Pedersen I.R. Structural components and replication of arenaviruses. Adv. Virus. Res. 1979;24:277–330. doi: 10.1016/s0065-3527(08)60396-6. [DOI] [PubMed] [Google Scholar]
- Pedersen K.W., van der Meer Y., Roos N., Snijder E.J. Open reading frame 1a‐encoded subunits of the arterivirus replicase induce endoplasmic reticulum‐derived double‐membrane vesicles which carry the viral replication complex. J. Virol. 1999;73:2016–2026. doi: 10.1128/jvi.73.3.2016-2026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K., Snijder E.J., Schleich S., Roos N., Griffiths G., Locker J.K. Characterization of vaccinia virus intracellular cores: Implications for viral uncoating and core structure. J. Virol. 2000;74:3525–3536. doi: 10.1128/jvi.74.8.3525-3536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington T.H., Follett E.A., Szilagyi J.F. Events in vaccinia virus‐infected cells following the reversal of the antiviral action of rifampicin. J. Gen. Virol. 1970;9:225–237. doi: 10.1099/0022-1317-9-3-225. [DOI] [PubMed] [Google Scholar]
- Petrie B.L., Graham D.Y., Hanssen H., Estes M.K. Localization of rotavirus antigens in infected cells by ultrastructural immunocytochemistry. J. Gen. Virol. 1982;63:457–467. doi: 10.1099/0022-1317-63-2-457. [DOI] [PubMed] [Google Scholar]
- Petrie B.L., Greenberg H.B., Graham D.Y., Estes M.K. Ultrastructural localization of rotavirus antigens using colloidal gold. Virus Res. 1984;1:133–152. doi: 10.1016/0168-1702(84)90069-8. [DOI] [PubMed] [Google Scholar]
- Pignatelli S., Dal Monte P., Zini N., Valmori A., Maraldi N.M., Landini M.P. Immunoelectron microscopy analysis of HCMV gpUL73 (gN) localization. Arch. Virol. 2002;147:1247–1256. doi: 10.1007/s00705-002-0810-x. [DOI] [PubMed] [Google Scholar]
- Ploubidou A., Moreau V., Ashman K., Reckmann I., González C., Way M. Vaccinia virus infection disrupts microtubule organization and centrosome function. EMBO J. 2000;19:3932–3944. doi: 10.1093/emboj/19.15.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A., Ferreira J., Bridge E., Carmo‐Fonseca M. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J. 1994;13:5075–5085. doi: 10.1002/j.1460-2075.1994.tb06837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004;279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice E., McAuliffe J., Lu X., Subbarao K., Denison M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004;78:9977–9986. doi: 10.1128/JVI.78.18.9977-9986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prod'homme D., Le Panse S., Drugeon G., Jupin I. Detection and subcellular localization of the turnip yellow mosaic virus 66K replication protein in infected cells. Virology. 2001;281:88–101. doi: 10.1006/viro.2000.0769. [DOI] [PubMed] [Google Scholar]
- Punjabi A., Boyle K., DeMasi J., Grubisha O., Unger B., Khanna M., Traktman P. Clustered charge‐to‐alanine mutagenesis of the vaccinia virus A20 gene: Temperature‐sensitive mutants have a DNA‐minus phenotype and are defective in the production of processive DNA polymerase activity. J. Virol. 2001;75:12308–12318. doi: 10.1128/JVI.75.24.12308-12318.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F. Simultaneous detection of highly phosphorylated proteins and viral major DNA binding protein distribution in nuclei of adenovirus type 5‐infected HeLa cells. J. Histochem. Cytochem. 1991;39:669–680. doi: 10.1177/39.5.1707905. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Pichard E. Viral alkaline nuclease in intranuclear dense bodies induced by herpes simplex infection. Biol. Cell. 1986;58:15–22. doi: 10.1111/j.1768-322x.1986.tb00485.x. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Pichard E. Segregation of viral double‐stranded and single‐stranded DNA molecules in nuclei of adenovirus infected cells as revealed by electron microscope in situ hybridization. Biol. Cell. 1992;76:139–150. doi: 10.1016/0248-4900(92)90206-g. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Puvion E. Replicating single‐stranded adenovirus type 5 DNA molecules accumulate within well‐delimited intranuclear areas of lytically infected HeLa cells. Eur. J. Cell Biol. 1990;52:379–388. [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Roussev R., Puvion E. Distribution of viral RNA molecules during the adenovirus type 5 infectious cycle in HeLa cells. J. Struct. Biol. 1992;108:209–220. doi: 10.1016/1047-8477(92)90021-2. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Bachellerie J.P., Visa N., Puvion E. Rearrangements of intranuclear structures involved in RNA processing in response to adenovirus infection. J. Cell Sci. 1994;107:1457–1468. doi: 10.1242/jcs.107.6.1457. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Chelbi‐Alix M.K., Koken M., Quignon F., Puvion E., de Thé H. Adenovirus infection induces rearrangements in the intranuclear distribution of the nuclear body‐associated PML protein. Exp. Cell Res. 1995;218:9–16. doi: 10.1006/excr.1995.1125. [DOI] [PubMed] [Google Scholar]
- Puvion‐Dutilleul F., Legrand V., Mehtali M., Chelbi‐Alix M.K., de Thé H., Puvion E. Deletion of the fiber gene induces the storage of hexon and penton base proteins in PML/Sp100‐containing inclusions during adenovirus infection. Biol. Cell. 1999;91:617–628. [PubMed] [Google Scholar]
- Quadt I., Günther A.K., Voß D., Schelhaas M., Knebel‐Mörsdorf D. TATA‐binding protein and TBP‐associated factors during herpes simplex virus type 1 infection: Localization at viral DNA replication sites. Virus Res. 2006;115:207–213. doi: 10.1016/j.virusres.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Quinkert D., Bartenschlager R., Lohmann V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005;79:13594–13605. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall R.E., Dinwoodie N. Intranuclear localization of herpes simplex virus immediate‐early and delayed‐early proteins: Evidence that ICP 4 is associated with progeny virus DNA. J. Gen. Virol. 1986;67:2163–2177. doi: 10.1099/0022-1317-67-10-2163. [DOI] [PubMed] [Google Scholar]
- Reckmann I., Higley S., Way M. The vaccinia virus F17R protein interacts with actin. FEBS Lett. 1997;409:141–146. doi: 10.1016/s0014-5793(97)00450-x. [DOI] [PubMed] [Google Scholar]
- Reggiori F., Klionsky D.J. Autophagosomes: Biogenesis from scratch? Curr. Opin. Cell Biol. 2005;17:415–422. doi: 10.1016/j.ceb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Reichel C., Beachy R.N. Tobacco mosaic virus infection induces severe morphological changes of the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 1998;95:11169–11174. doi: 10.1073/pnas.95.19.11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissig M., Howes D.W., Melnick J.D. Sequence of morphological changes in epithelial cell cultures infected with poliovirus. J. Exp. Med. 1956;104:289–304. doi: 10.1084/jem.104.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel R.E., Anderson M.K., Evans E., Traktman P. Temperature‐sensitive vaccinia virus mutants identify a gene with an essential role in viral replication. J. Virol. 1990;64:574–583. doi: 10.1128/jvi.64.2.574-583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch W., Weisberg A.S., Moss B. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 2005;79:6598–6609. doi: 10.1128/JVI.79.11.6598-6609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A.E., Fan Y., Baines J.D. Characterization of the UL33 gene product of herpes simplex virus 1. Virology. 2000;266:310–318. doi: 10.1006/viro.1999.0090. [DOI] [PubMed] [Google Scholar]
- Risco C., Rodriguez J.R., Demkowicz W., Heljasvaara R., Carrascosa J.L., Esteban M., Rodriguez D. The vaccinia virus 39‐kDa protein forms a stable complex with the p4a/4a major core protein early in morphogenesis. Virology. 1999;265:375–386. doi: 10.1006/viro.1999.0046. [DOI] [PubMed] [Google Scholar]
- Risco C., Rodríguez J.R., López‐Iglesias C., Carrascosa J.L., Esteban M., Rodriguez D. Endoplasmic reticulum‐Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 2002;76:1839–1855. doi: 10.1128/JVI.76.4.1839-1855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins F.C., Enders J.F., Weller T.H. Cytopathogenic effect of poliomyelitis viruses ‘in vitro’ on human embryonic tissues. Proc. Soc. Exp. Biol. Med. 1950;75:370–374. doi: 10.3181/00379727-75-18202. [DOI] [PubMed] [Google Scholar]
- Roberts S., Hillman M.L., Knight G.L., Gallimore P.H. The ND10 component promyelocytic leukemia protein relocates to human papillomavirus type 1 E4 intranuclear inclusion bodies in cultured keratinocytes and in warts. J. Virol. 2003;77:673–684. doi: 10.1128/JVI.77.1.673-684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez J.M., García‐Escudero R., Salas M.L., Andrés G. African swine fever virus structural protein p54 is essential for the recruitment of envelope precursors to assembly sites. J. Virol. 2004;78:4299–4313. doi: 10.1128/JVI.78.8.4299-4313.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez I., Redrejo‐Rodríguez M., Rodríguez J.M., Alejo A., Salas J., Salas M.L. African swine fever virus pB119L protein is a flavin adenine dinucelotide‐linked sulfhydryl oxidase. J. Virol. 2006;80:3157–3166. doi: 10.1128/JVI.80.7.3157-3166.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffman E., Albert J.P., Goff J.P., Frenkel N. Putative site for the acquisition of human herpesvirus 6 virion tegument. J. Virol. 1990;64:6308–6313. doi: 10.1128/jvi.64.12.6308-6313.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogel‐Gaillard C., Pehau‐Arnaudet G., Breitburd F., Orth G. Cytopathic effect in human papillomavirus type 1‐induced inclusion warts: In vitro analysis of the contribution of two forms of the viral E4 protein. J. Investig. Dermatol. 1993;101:843–851. doi: 10.1111/1523-1747.ep12371705. [DOI] [PubMed] [Google Scholar]
- Rohrmann G.F. Polyhedrin structure. J. Gen. Virol. 1986;67:1499–1513. doi: 10.1099/0022-1317-67-8-1499. [DOI] [PubMed] [Google Scholar]
- Rojo G., Chamorro M., Salas M.L., Viñuela E., Cuezva J.M., Salas J. Migration of mitochondria to viral assembly sites in African swine fever virus‐infected cells. J. Virol. 1998;72:7583–7588. doi: 10.1128/jvi.72.9.7583-7588.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo G., García‐Beato R., Viñuela E., Salas M.L., Salas J. Replication of African swine fever virus DNA in infected cells. Virology. 1999;257:524–536. doi: 10.1006/viro.1999.9704. [DOI] [PubMed] [Google Scholar]
- Roller R.J., Roizman B. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 1992;66:3624–3632. doi: 10.1128/jvi.66.6.3624-3632.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosendaal J., Westaway E.G., Khromykh A., Mackenzie J.M. Regulated cleavages at the West Nile virus NS4A‐2K‐NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006;80:4623–4632. doi: 10.1128/JVI.80.9.4623-4632.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper R.L. Characterization of the vaccinia virus A35R protein and its role in virulence. J. Virol. 2006;80:306–313. doi: 10.1128/JVI.80.1.306-313.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa‐Calatrava M., Grave L., Puvion‐Dutilleul F., Chatton B., Kedinger C. Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J. Virol. 2001;75:7131–7141. doi: 10.1128/JVI.75.15.7131-7141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa‐Calatrava M., Puvion‐Dutilleul F., Lutz P., Dreyer D., de Thé H., Chatton B., Kedinger C. Adenovirus protein IX sequesters host‐cell promyelocytic leukaemia protein and contributes to efficient viral proliferation. EMBO Rep. 2003;4:969–975. doi: 10.1038/sj.embor.embor943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales R., Sutter G., Moss B. A cellular factor is required for transcription of vaccinia viral intermediate‐stage genes. Proc. Natl. Acad. Sci. USA. 1994;91:3794–3798. doi: 10.1073/pnas.91.9.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouiller I., Brookes S.M., Hyatt A.D., Windsor M., Wileman T. African swine fever virus is wrapped by the endoplasmic reticulum. J. Virol. 1998;72:2373–2387. doi: 10.1128/jvi.72.3.2373-2387.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust R.C., Landmann L., Gosert R., Tang B.L., Hong W., Hauri H.P., Egger D., Bienz K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001;75:9808–9818. doi: 10.1128/JVI.75.20.9808-9818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmons T., Kuhn A., Wylie F., Schleich S., Rodriguez J.R., Rodriguez D., Esteban M., Griffiths G., Locker J.K. Vaccinia virus membrane proteins p8 and p16 are cotranslationally inserted into the rough endoplasmic reticulum and retained in the intermediate compartment. J. Virol. 1997;71:7404–7420. doi: 10.1128/jvi.71.10.7404-7420.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomoni P., Khelifi A.F. Daxx: Death or survival protein? Trends Cell Biol. 2006;16:97–104. doi: 10.1016/j.tcb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Salonen A., Vasiljeva L., Merits A., Magden J., Jokitalo E., Kaariainen L. Properly folded nonstructural polyprotein directs the semliki forest virus replication complex to the endosomal compartment. J. Virol. 2003;77:1691–1702. doi: 10.1128/JVI.77.3.1691-1702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen A., Ahola T., Kaariainen L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005;285:139–173. doi: 10.1007/3-540-26764-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V., Greis K.D., Sztul E., Britt W.J. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: Characterization of a potential site of virus assembly. J. Virol. 2000;74:975–986. doi: 10.1128/jvi.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson C.M., Hollinshead M., Smith G.L. The vaccinia virus A27L protein is needed for the microtubule‐dependent transport of intracellular mature virus particles. J. Gen. Virol. 2000;81:47–58. doi: 10.1099/0022-1317-81-1-47. [DOI] [PubMed] [Google Scholar]
- Sandoval I.V., Carrasco L. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro‐090179. J. Virol. 1997;71:4679–4693. doi: 10.1128/jvi.71.6.4679-4693.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz‐Parra A., Sobrino F., Ley V. Infection with foot‐and‐mouth disease virus results in a rapid reduction of MHC class I surface expression. J. Gen. Virol. 1998;79:433–436. doi: 10.1099/0022-1317-79-3-433. [DOI] [PubMed] [Google Scholar]
- Sanz A., García‐Barreno B., Nogal M.L., Viñuela E., Enjuanes L. Monoclonal antibodies specific for African swine fever virus proteins. J. Virol. 1985;54:199–206. doi: 10.1128/jvi.54.1.199-206.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepis A., Schramm B., de Haan C.A.M., Krijnse‐Locker J. Vaccinia virus‐induced microtubule‐dependent cellular rearrangements. Traffic. 2006;7:308–323. doi: 10.1111/j.1600-0854.2005.00381.x. [DOI] [PubMed] [Google Scholar]
- Schlegel A., Giddings T.H., Jr., Ladinsky M.S., Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm B., Krijnse‐Locker J. Cytoplasmic organization of poxvirus DNA replication. Traffic. 2005;6:839–846. doi: 10.1111/j.1600-0854.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- Schwartz M., Chen J., Janda M., Sullivan M., den Boon J., Ahlquist P. A positive‐strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell. 2002;9:505–514. doi: 10.1016/s1097-2765(02)00474-4. [DOI] [PubMed] [Google Scholar]
- Schwartz M., Chen J., Lee W.M., Janda M., Ahlquist P. Alternate, virus‐induced membrane rearrangements support positive‐strand RNA virus genome replication. Proc. Natl. Acad. Sci. USA. 2004;101:11263–11268. doi: 10.1073/pnas.0404157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich T.G., White C.L., Weisberg A., Granek J.A., Wolffe E.J., Koonin E.V., Moss B. Expression of the vaccinia virus A2.5L redox protein is required for virion morphogenesis. Virology. 2002;300:296–303. doi: 10.1006/viro.2002.1608. [DOI] [PubMed] [Google Scholar]
- Sharpe A.H., Chen L.B., Fields B.N. The interaction of mammalian reoviruses with the cytoskeleton of monkey kidney CV‐1 cells. Virology. 1982;120:399–411. doi: 10.1016/0042-6822(82)90040-x. [DOI] [PubMed] [Google Scholar]
- Shi S.T., Schiller J.J., Kanjanahaluethai A., Baker S.C., Oh J.W., Lai M.M. Colocalization and membrane association of murine hepatitis virus gene 1 products and de novo‐synthesized viral RNA in infected cells. J. Virol. 1999;73:5957–5969. doi: 10.1128/jvi.73.7.5957-5969.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shida H., Tanabe K., Matsumoto S. Mechanism of virus occlusion into A‐type inclusion during poxvirus infection. Virology. 1977;76:217–233. doi: 10.1016/0042-6822(77)90298-7. [DOI] [PubMed] [Google Scholar]
- Shintani T., Klionsky D.J. Autophagy in health and disease: A double‐edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido‐Hara Y., Ichinose S., Higuchi K., Hara Y., Yasui K. Major and minor capsid proteins of human polyomavirus JC cooperatively accumulate to nuclear domain 10 for assembly into virions. J. Virol. 2004;78:9890–9903. doi: 10.1128/JVI.78.18.9890-9903.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein S.C., Schur P.H. Immunofluorescent localization of double‐stranded RNA in reovirus‐infected cells. Virology. 1970;41:564–566. doi: 10.1016/0042-6822(70)90178-9. [DOI] [PubMed] [Google Scholar]
- Silvestri L.S., Taraporewala Z.F., Patton J.T. Rotavirus replication: Plus‐sense templates for double‐stranded RNA synthesis are made in viroplasms. J. Virol. 2004;78:7763–7774. doi: 10.1128/JVI.78.14.7763-7774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri L.S., Tortorici M.A., Vasquez‐Del Carpio R., Patton J.T. Rotavirus glycoprotein NSP4 is a modulator of viral transcription in the infected cell. J. Virol. 2005;79:15165–15174. doi: 10.1128/JVI.79.24.15165-15174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón‐Mateo C., Andrés G., Almazán F., Viñuela E. Proteolytic processing in African swine fever virus: Evidence for a new structural polyprotein pp62. J. Virol. 1997;71:5799–5804. doi: 10.1128/jvi.71.8.5799-5804.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims A.C., Ostermann J., Denison M.R. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 2000;74:5647–5654. doi: 10.1128/jvi.74.12.5647-5654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.A., Enquist L.W. Break ins and break outs: Viral interactions with the cytoskeleton of mammalian cells. Annu. Rev. Cell Dev. Biol. 2002;18:135–161. doi: 10.1146/annurev.cellbio.18.012502.105920. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., van der Meer Y., Zevenhoven‐Dobbe J., Onderwater J.J., van der Meulen J., Koerten H.K., Mommaas A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006;80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik B., Krijnse‐Locker J. Assembly of vaccinia virus revisited: De novo membrane synthesis or acquisition from the host? Trends Microbiol. 2002;10:15–24. doi: 10.1016/s0966-842x(01)02256-9. [DOI] [PubMed] [Google Scholar]
- Sodeik B., Cudmore S., Ericsson M., Esteban M., Niles E.G., Griffiths G. Assembly of vaccinia virus: Incorporation of p14 and p32 into the membrane of the intracellular mature virus. J. Virol. 1995;69:3560–3574. doi: 10.1128/jvi.69.6.3560-3574.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik B., Ebersold M.W., Helenius A. Microtubule‐mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell. Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souquere‐Besse S., Pichard E., Filhol O., Legrand V., Rosa‐Calatrava M., Hovanessian A.G., Cochet C., Puvion‐Dutilleul F. Adenovirus infection targets the cellular protein kinase CK2 and RNA‐activated protein kinase (PKR) into viral inclusions of the cell nucleus. Microsc. Res. Tech. 2002;56:465–478. doi: 10.1002/jemt.10060. [DOI] [PubMed] [Google Scholar]
- Sridhar P., Condit R.C. Selection for temperature‐sensitive mutations in specific vaccinia virus genes: Isolation and characterization of a virus mutant which encodes a phosphonoacetic acid‐resistant, temperature‐sensitive DNA polymerase. Virology. 1983;128:444–457. doi: 10.1016/0042-6822(83)90269-6. [DOI] [PubMed] [Google Scholar]
- Stefanovic S., Windsor M., Nagata K.‐I., Inagaki M., Wileman T. Vimentin rearrangement during African swine fever virus infection involves retrograde transport along microtubules and phosphorylation of vimentin by calcium calmodulin kinase II. J. Virol. 2005;79:11766–11775. doi: 10.1128/JVI.79.18.11766-11775.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart J.D.C., Fogh J. Micromorphology of FL cells infected with polio and Coxsackie viruses. Virology. 1961;13:177–190. [Google Scholar]
- Suhy D.A., Giddings T.H., Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy‐like origin for virus‐induced vesicles. J. Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Jenson J., Dixon L.K., Parkhouse R.M.E. Characterization of the African swine fever virion protein j18L. J. Gen. Virol. 1996;77:941–946. doi: 10.1099/0022-1317-77-5-941. [DOI] [PubMed] [Google Scholar]
- Szajner P., Weisberg A.S., Wolffe E.J., Moss B. Vaccinia virus A30L protein is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 2001;75:5752–5761. doi: 10.1128/JVI.75.13.5752-5761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajner P., Jaffe H., Weisberg A.S., Moss B. Vaccinia virus G7L protein interacts with the A30L protein and is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 2003;77:3418–3429. doi: 10.1128/JVI.77.6.3418-3429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajner P., Jaffe H., Weisberg A.S., Moss B. A complex of seven vaccinia virus proteins conserved in all chordopoxviruses is required for the association of membranes and viroplasm to form immature virions. Virology. 2004;330:447–459. doi: 10.1016/j.virol.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Szajner P., Weisberg A.S., Moss B. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 2004;78:257–265. doi: 10.1128/JVI.78.1.257-265.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajner P., Weisberg A.S., Moss B. Physical and functional interactions between vaccinia virus F10 protein kinase and virion assembly proteins A30 and G7. J. Virol. 2004;78:266–274. doi: 10.1128/JVI.78.1.266-274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajner P., Weisberg A.S., Lebowitz J., Heuser J., Moss B. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 2005;170:971–981. doi: 10.1083/jcb.200504026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus N.S., Salmon B., Baines J.D. The herpes simplex virus 1 UL 17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology. 1998;252:115–125. doi: 10.1006/viro.1998.9439. [DOI] [PubMed] [Google Scholar]
- Taylor T.J., Knipe D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004;78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T.J., McNamee E.E., Day C., Knipe D.M. Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology. 2003;309:232–247. doi: 10.1016/s0042-6822(03)00107-7. [DOI] [PubMed] [Google Scholar]
- Temperley S.M., Hay R.T. Recognition of the adenovirus type 2 origin of DNA replication by the virally encoded DNA polymerase and preterminal proteins. EMBO J. 1992;11:761–768. doi: 10.1002/j.1460-2075.1992.tb05109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teterina N.L., Bienz K., Egger D., Gorbalenya A.E., Ehrenfeld E. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology. 1997;237:66–77. doi: 10.1006/viro.1997.8775. [DOI] [PubMed] [Google Scholar]
- Tolonen N., Doglio L., Schleich S., Krijnse Locker J. Vaccinia virus DNA replication occurs in endoplasmic reticulum‐enclosed cytoplasmic mini‐nuclei. Mol. Biol. Cell. 2001;12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripier F., Braunwald J., Markovic L., Kirn A. Frog virus 3 morphogenesis: Effect of temperature and metabolic inhibitors. J. Gen. Virol. 1977;37:39–52. doi: 10.1099/0022-1317-37-1-39. [DOI] [PubMed] [Google Scholar]
- Tsutsui Y., Yamazaki Y. Subcellular distribution of the major immediate early proteins of human cytomegalovirus changes during infection. J. Gen. Virol. 1991;72:1415–1419. doi: 10.1099/0022-1317-72-6-1415. [DOI] [PubMed] [Google Scholar]
- Turcotte S., Letellier J., Lippe R. Herpes simplex virus type 1 capsids transit by the trans‐Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 2005;79:8847–8860. doi: 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchil P.D., Satchidanandam V. Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double‐layered membrane compartments. J. Biol. Chem. 2003;278:24388–24398. doi: 10.1074/jbc.M301717200. [DOI] [PubMed] [Google Scholar]
- Ulaeto D., Grosenbach D., Hruby D.E. The vaccinia virus 4c and A‐type inclusion proteins are specific markers for the intracellular mature virus particle. J. Virol. 1996;70:3372–3377. doi: 10.1128/jvi.70.6.3372-3377.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden M.W., Carette J.E., Reinhoud P.J., Haegi A., Bol J.F. Alfalfa mosaic virus replicase proteins P1 and P2 interact and colocalize at the vacuolar membrane. J. Virol. 2001;75:1879–1887. doi: 10.1128/JVI.75.4.1879-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer Y., van Tol H., Locker J.K., Snijder E.J. ORF1a‐encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J. Virol. 1998;72:6689–6698. doi: 10.1128/jvi.72.8.6689-6698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer Y., Snijder E.J., Dobbe J.C., Schleich S., Denison M.R., Spaan W.J., Locker J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999;73:7641–7657. doi: 10.1128/jvi.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kuppeveld F.J., Melchers W.J., Kirkegaard K., Doedens J.R. Structure‐function analysis of Coxsackie B3 virus protein 2B. Virology. 1997;227:111–118. doi: 10.1006/viro.1996.8320. [DOI] [PubMed] [Google Scholar]
- Vanslyke J.K., Hruby D.E. Immunolocalization of vaccinia virus structural proteins during virion formation. Virology. 1994;198:624–635. doi: 10.1006/viro.1994.1074. [DOI] [PubMed] [Google Scholar]
- Vigário J.D., Relvas M.E., Ferraz F.P., Riberio J.M., Pereira C.G. Identification and localization of genetic material of African swine fever virus by autoradiography. Virology. 1967;33:173–175. doi: 10.1016/0042-6822(67)90108-0. [DOI] [PubMed] [Google Scholar]
- Wada K., Goshima F., Takakuwa H., Yamada H., Daikoku T., Nishiyama Y. Identification and characterization of the UL14 gene product of herpes simplex virus type 2. J. Gen. Virol. 1999;80:2423–2431. doi: 10.1099/0022-1317-80-9-2423. [DOI] [PubMed] [Google Scholar]
- Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H.G., Mizokami M., Bartenschlager R., Liang T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward B.M. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J. Virol. 2005;79:4755–4763. doi: 10.1128/JVI.79.8.4755-4763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward B.M., Moss B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 2001;75:11651–11663. doi: 10.1128/JVI.75.23.11651-11663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P.L., Barker D.E., Roizman B. A novel herpes simplex virus 1 gene, UL43.5, maps antisense to the UL43 gene and encodes a protein which colocalizes in nuclear structures with capsid proteins. J. Virol. 1996;70:2684–2690. doi: 10.1128/jvi.70.5.2684-2690.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P.L., Ogle W.O., Roizman B. Assemblons: Nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 1996;70:4623–4631. doi: 10.1128/jvi.70.7.4623-4631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward T.H., Polishchuk R.S., Caplan S., Hirschberg K., Lippincott‐Schwartz J. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 2001;155:557–570. doi: 10.1083/jcb.200107045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe D., Ushijima Y., Goshima F., Takakuwa H., Tomita Y., Nishiyama Y. Identification of nuclear export signal in UL37 protein of herpes simplex virus type 2. Biochem. Biophys. Res. Commun. 2000;276:1248–1254. doi: 10.1006/bbrc.2000.3600. [DOI] [PubMed] [Google Scholar]
- Welsch S., Doglio L., Schleich S., Krijnse Locker J. The vaccinia virus I3L gene product is localized to a complex endoplasmic reticulum‐associated structure that contains the viral parental DNA. J. Virol. 2003;77:6014–6028. doi: 10.1128/JVI.77.10.6014-6028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels E., Duijsings D., Notebaart R.A., Melchers W.J., van Kuppeveld F.J. A proline‐rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum‐to‐golgi transport. J. Virol. 2005;79:5163–5173. doi: 10.1128/JVI.79.8.5163-5173.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels E., Duijsings D., Lanke K.H., van Dooren S.H., Jackson C.L., Melchers W.J., van Kuppeveld F.J. Effects of picornavirus 3A proteins on protein transport and GBF1‐dependent COP‐I recruitment. J. Virol. 2006;80:11852–11860. doi: 10.1128/JVI.01225-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels E., Duijsings D., Niu T.K., Neumann S., Oorschot V.M., de Lange F., Lanke K.H., Klumperman J., Henke A., Jackson C.L., Melchers W.J., van Kuppeveld F.J. A viral protein that blocks Arf1‐mediated COP‐I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell. 2006;11:191–201. doi: 10.1016/j.devcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Westaway E.G., Khromykh A.A., Kenney M.T., Mackenzie J.M., Jones M.K. Proteins C and NS4B of the flavivirus Kunjin translocate independently into the nucleus. Virology. 1997;234:31–41. doi: 10.1006/viro.1997.8629. [DOI] [PubMed] [Google Scholar]
- Westaway E.G., Mackenzie J.M., Kenney M.T., Jones M.K., Khromykh A.A. Ultrastructure of Kunjin virus‐infected cells: Colocalization of NS1 and NS3 with double‐stranded RNA, and of NS2B with NS3, in virus‐induced membrane structures. J. Virol. 1997;71:6650–6661. doi: 10.1128/jvi.71.9.6650-6661.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock D., Lane D.P. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes‐infected cells. Nature. 1991;349:429–431. doi: 10.1038/349429a0. [DOI] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and autophagy generate sites of virus replication. Science. 2006;312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- Williams T., Barbosa‐Solomieu V., Chinchar V.G. A decade of advances in iridovirus research. Adv. Virus Res. 2005;65:173–248. doi: 10.1016/S0065-3527(05)65006-3. [DOI] [PubMed] [Google Scholar]
- Willis D.B., Goorha R., Granoff A. Macromolecular synthesis in cells infected by frog virus 3. XI. A ts mutant of frog virus 3 that is defective in late transcription. Virology. 1979;98:328–335. doi: 10.1016/0042-6822(79)90556-7. [DOI] [PubMed] [Google Scholar]
- Wilton S., Dales S. Relationship between RNA polymerase II and efficiency of vaccinia virus replication. J. Virol. 1989;63:1540–1548. doi: 10.1128/jvi.63.4.1540-1548.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S., Maier I., Katsaros C., Muller D.G. Virus assembly in Hincksia hincksiae (Ectocarpales, Phaeophyceae) an electron and fluorescence microscopic study. Protoplasma. 1998;203:153–167. [Google Scholar]
- Wolf S., Muller D.G., Maier I. Assembly of a large icosahedral DNA virus, MclaV‐1, in the marine alga Myriotrichia clavaeformis (Dictyosiphonales, Phaeophyceae) Eur. J. Phycol. 2000;35:163–171. [Google Scholar]
- Wolffe E.J., Vijaya S., Moss B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology. 1995;211:53–63. doi: 10.1006/viro.1995.1378. [DOI] [PubMed] [Google Scholar]
- Wolk B., Sansonno D., Krausslich H.G., Dammacco F., Rice C.M., Blum H.E., Moradpour D. Subcellular localization, stability, and trans‐cleavage competence of the hepatitis C virus NS3‐NS4A complex expressed in tetracycline‐regulated cell lines. J. Virol. 2000;74:2293–2304. doi: 10.1128/jvi.74.5.2293-2304.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C.F., Oswald B.W., Dellis S. Vaccinia virus late transcription is activated in vitro by cellular heterogeneous nuclear ribonucleoproteins. J. Biol. Chem. 2001;276:40680–40686. doi: 10.1074/jbc.M102399200. [DOI] [PubMed] [Google Scholar]
- Yamada H., Jiang Y.M., Oshima S., Daikoku T., Yamashita Y., Tsurumi T., Nishiyama Y. Characterization of the UL55 gene product of herpes simplex virus type 2. J. Gen. Virol. 1998;79:1989–1995. doi: 10.1099/0022-1317-79-8-1989. [DOI] [PubMed] [Google Scholar]
- Yeh W.W., Moss B., Wolffe E.J. The vaccinia virus A9L gene encodes a membrane protein required for an early step in virion morphogenesis. J. Virol. 2000;74:9701–9711. doi: 10.1128/jvi.74.20.9701-9711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P.R., Chanas A.C., Lee S.R., Gould E.A., Howard C.R. Localization of an arenavirus protein in the nuclei of infected cells. J. Gen. Virol. 1987;68:2465–2470. doi: 10.1099/0022-1317-68-9-2465. [DOI] [PubMed] [Google Scholar]
- Yue Z., Shatkin A.J. Enzymatic and control functions of reovirus structural proteins. Curr. Top. Microbiol. Immunol. 1998;233:31–56. doi: 10.1007/978-3-642-72092-5_2. [DOI] [PubMed] [Google Scholar]
- Yuwen H., Cox J.H., Yewdell J.W., Bennink J.R., Moss B. Nuclear localization of a double‐stranded RNA‐binding protein encoded by the vaccinia virus E3L gene. Virology. 1993;195:732–744. doi: 10.1006/viro.1993.1424. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Ke F., Gui J., Zhang Q. Characterization of an early gene encoding for dUTPase in Rana grylio virus. Virus Res. 2007;123(2):128–137. doi: 10.1016/j.virusres.2006.08.007. doi:10.1016/j.virusres. 2006.08.007. [DOI] [PubMed] [Google Scholar]
- Zhong L., Hayward G.S. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 1997;71:3146–3160. doi: 10.1128/jvi.71.4.3146-3160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J., Gastaminza P., Cheng G., Kapadia S., Kato T., Burton D.R., Wieland S.F., Uprichard S.L., Wakita T., Chisari F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA. 1997;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J. The coronavirus replicase: Insights into a sophisticated enzyme machinery. Adv. Exp. Med. Biol. 2006;581:3–11. doi: 10.1007/978-0-387-33012-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further Reading
- Raoult D., La Scola B., Birtles R. The discovery and characterization of mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 1986;45:95–102. doi: 10.1086/518608. [DOI] [PubMed] [Google Scholar]