

Abstract
T lymphocytes play a central role in the pathogenesis of multiple sclerosis (MS) (Zhang et al., 1992). Both CD4+ and CD8+ T cells have been demonstrated in MS lesions, with CD4+ T cells predominating in acute lesions and CD8+ T cells being observed more frequently in chronic lesions (Raine, 1994). Additionally, T cells are found in all four of the described histopathologic subtypes of MS (Lucchinetti et al., 2000). Activated myelin‐reactive CD4+ T cells are present in the blood and cerebrospinal fluid (CSF) of MS patients; in contrast, only nonactivated myelin‐reactive T cells are present in the blood of controls (Zhang et al., 1994). The success of several T‐cell‐targeted therapies in MS reinforces the importance of the role of the T cell in MS pathogenesis. Here, we outline basic concepts in CD4+ T‐cell immunology and summarize the current understanding of the role of CD4+ T cells in the pathogenesis of MS.
I. Overview of CD4+ T‐Cell Immunology
A. The T‐Cell Subset of Inflammatory Cells
T cells as well as B cells are critical components of the adaptive immune system. Both T cells and B cells are equipped with specific antigen recognition receptors, the former with the T‐cell receptor (TCR), while B cells secrete immunoglobulin (Ig) molecules. T cells originate and differentiate in the thymus. Every T cell that leaves the thymus is conferred with a unique specificity for recognizing antigens through its TCR. The TCR consists of two glycosylated polypeptide chains, the alpha (α) and beta (β) chains, which are linked by disulfide bonds. Each chain consists of variable (V), joining (J), and constant (C) regions closely resembling Ig chains. T cells that recognize self‐antigens with high affinity are either deleted or rendered tolerant within the thymus, through a process called central tolerance.
B. Subsets of T Cells
T cells may be divided into two groups on the basis of their expression of either the CD4+ or the CD8+ surface molecules. Functionally, CD4+ T cells are involved in delayed‐type hypersensitivity (DTH) responses and also provide help for B‐cell differentiation, and hence are termed T helper (Th) cells. In contrast, CD8+ T cells are involved in class I‐restricted lysis of antigen‐specific targets, and hence are termed cytotoxic T cells. The CD4 molecule binds to a nonpolymorphic site on the major histocompatibility complex (MHC) class II β chain that is expressed by antigen‐presenting cells (APCs). In contrast CD8 binds to the α‐3 domain of the MHC class I molecule expressed by most cell types. The MHC molecule serves to present antigen to the T cell via the TCR.
C. Activation of T Cells
Signaling through surface molecules by second messengers deliver signals for cell division to the nucleus. The CD3 molecule is part of the TCR complex, although the TCR interacts with the MHC–peptide complex on APCs, signals for the subsequent enactment of T‐cell activation and proliferation are delivered by the CD3 antigen. The cytoplasmic tail of the CD3 proteins contains one copy of a sequence motif important for signaling functions, called the immunoreceptor tyrosine‐based activation motif (ITAM). Phosphorylation of the ITAM initiates intracellular signaling events. The interaction of MHC–peptide complex with T cells, while necessary, is insufficient for T‐cell activation. Additional classes of molecules are involved in T‐cell antigen recognition, activation, intracellular signaling, adhesion, and trafficking of T cells to their target organs.
Two signals are required for T lymphocyte activation. According to this “two‐signal” model (Bretscher and Cohn, 1970), “signal 1” consists of the interaction of the TCR with antigen, presented by the MHC on the surface of APCs. “Signal 2” consists of the engagement of costimulatory receptors on the T cell by ligands present on the surface of APCs (Alegre 2001, Bretscher 1999). After contact with specific antigen–MHC complex and adequate costimulatory signals, T cells begin to proliferate, differentiate, and deliver a series of signals, enabling effector functions to other cells such as B cells and NK cells. T cells can thereby orchestrate the immune response.
Costimulatory molecules may deliver either a stimulatory (positive) or an inhibitory (negative) signal for T‐cell activation (Brunet et al., 1987). Examples of molecules delivering a positive costimulatory signal for T‐cell activation include the B7‐CD28 and CD40‐CD154 pathways. Examples of molecular pathways delivering a negative signal for T‐cell activation include B7‐CTLA4 and PD1‐PD ligand. The delicate balance between positive and negative regulatory signals can determine the outcome of a specific immune response. Importantly, in the absence of adequate costimulatory signals, T cells can die or become anergic in vitro, and thus, fail to initiate an effective immune response in vivo. Therefore, manipulation of costimulatory signals represents an important mechanism to inhibit immune‐activation.
D. Memory T Cells
On exposure to an antigen, antigen‐specific T cells proliferate and differentiate into effector T cells (Sprent and Surh, 2002). The vast majority of effector T cells undergo apoptosis as the immune response progresses, and the few lymphocytes that survive become long‐lived memory T cells (Dutton et al., 1998). Memory T cells are specific to the antigen encountered during the primary immune response and react rapidly and vigorously on reencounter with the same antigen. Functionally, in terms of activation requirements, memory T cells can be activated by lower concentrations of anti‐CD3 (Byrne et al., 1988), require less costimulation by anti‐CD28 (Kuiper et al., 1994), and readily secrete more effector cytokines (Bird 1998, Ehlers 1991, Lee 1990) than naive T‐cell counterparts, indicating a state of hyperresponsiveness.
E. Migration of T Cells
Molecules primarily involved in cell migration into tissues include chemokines, integrins, selectins, and matrix metalloproteinases (MMPs). Chemokines constitute a large family of chemoattractant peptides that regulate the vast spectrum of leukocyte migration events through interactions with chemokine receptors. The integrin family includes vascular cell adhesion molecule‐1 (VCAM‐1), intercellular adhesion molecule‐1 (ICAM‐1), and leukocyte function antigen 3 (LFA‐3), CD45, and CD2. The integrin family also mediates T‐cell adhesion, facilitates interaction with the APCs, as well mediates adhesion to nonhematopoietic cells such as endothelial cells and guides cell traffic. l‐Selectins facilitate the rolling of leucocytes along the surface of endothelial cells and function as a homing receptor to target peripheral lymphoid organs. The MMPs are a family of proteinases, secreted by inflammatory cells, which digest specific components of the extracellular matrix, thereby facilitating lymphocyte entry through basement membranes, including the blood–brain barrier (BBB).
F. T‐Cell Cytokine Production
The Th cells play a critical role in the orchestration of the immune response, in part, through the production of cytokines that provide secondary signals to other cells in the immune cascade. Two major types of Th cell responses have been described. Th1 cells produce IL‐2, TNF‐α, and interferon (IFN)‐γ, while Th2 cells produce IL‐4, IL‐5, IL‐10, and IL‐13. A Th3 cell that primarily secretes TGF‐β has been described in the context of oral tolerance to myelin antigens (Fukaura 1996, Hafler 1997) and in other immune‐mediated settings (Minguela et al., 1999). These subsets of T cells interregulate each other's development, with Th1 cytokines suppressing Th2 differentiation and vice versa (Fig. 1 ).
Fig. 1.
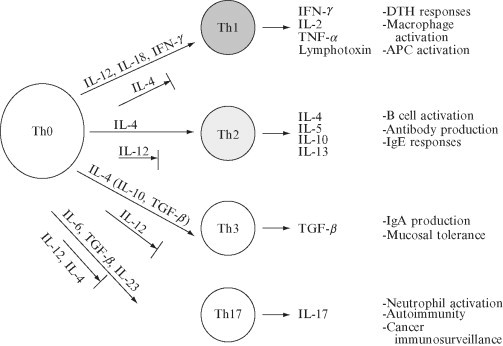
Factors that influence T helper cell differentiation.
A subset of T cells that predominantly produces IL‐17 has been described (Yao et al., 1995). These cells are believed to represent a distinct subset from IFN‐γ‐producing Th1 cells, evidenced by the dependence of ThIL‐17 cells on IL‐16 and TGF‐β for differentiation (Bettelli 2006, Mangan 2006, Veldhoen 2006) and IL‐23 for expansion (Aggarwal 2003, Langrish 2005), as opposed to Th1 cells which are dependent on IL‐12 and IL‐2, respectively, for differentiation and expansion. Both Th1 and Th2 cytokines have been shown to suppress the development of Th17 cells (Harrington 2005, Park 2005). An intriguing relationship between the generation of pathogenic Th17 and regulatory CD4+CD25+Foxp3+ cells has recently been demonstrated. TGF‐β is critical for the differentiation and generation of CD4+CD25+Foxp3+ regulatory T cells; however, if additionally exposed to IL‐6 during culture, these cells differentiate into Th17 cells (Bettelli et al., 2006).
Traditionally, Th cell subsets have been distinguished by their patterns of cytokine production, however identification of distinguishing surface molecule markers has been a major advance in the field. T‐cell‐, Ig‐, and mucin‐domain‐containing molecules (Tim) represent an important family of molecules which encode cell‐surface receptors involved in the regulation of Th1‐ and Th2‐cell‐mediated immunity. Tim‐3 is specifically expressed on Th1 cells and negatively regulates Th1 responses through interaction with the Tim‐3 ligand, galactin‐9, also expressed on CD4+ T cells (Monney 2002, Sabatos 2003, Zhu 2005). Tim‐2 is expressed on Th2 cells (Chakravarti et al., 2005) and appears to negatively regulate Th2 cell proliferation, although this has not been fully established. Tim‐1 is expressed on Th2 cells > Th1 cells and interacts with Tim‐4 on APCs to induce T‐cell proliferation (Meyers et al., 2005).
G. T‐Cell‐Signaling Pathways
Intracellular signaling mechanisms provide the link between the binding of the cytokine with its receptor and the effect of the cytokine on cellular function. The Janus kinase and signal transducer and activator of transcription (Jak/STAT) family of transducer/transcription‐activating factors plays a critical role in the signaling of many cytokine receptors. Cytokine binding to the specific receptor activates the Jak molecule associated with the receptor, causing phosphorylation of tyrosine residues, and binding of the Jak molecules to its receptor. This facilitates binding of STAT proteins to the phosphorylated receptor, which subsequently dissociates from the receptor, dimerizes, and activates transcription of genes containing specific cis‐regulatory STAT‐binding sequences. Different cytokine receptors are associated with different Jak/STAT proteins. The IL‐12 receptor is associated with Jak‐2 and STAT3 and STAT4 (Jacobson et al., 1995). The IL‐4 receptor is associated with Jak‐1–3 and STAT6 (Kaplan 1996, Takeda 1996) (Fig. 1). Mice deficient in STAT6 display a reduction in Th2 cytokine production, decreased IL‐4‐induced B‐cell proliferation and reduced IgE (Kaplan 1996, Takeda 1996). In contrast, STAT4 plays a pivotal role in Th1 immune responses. STAT4 is activated after IL‐12 interacts with the IL‐12 receptor, inducing transcription of IFN‐γ (Jacobson et al., 1995). Mice deficient in STAT4 lack IL‐12‐induced IFN‐γ production and Th1 differentiation (Kaplan 1996, Thierfelder 1996), and display a predominantly Th2 phenotype (Kaplan et al., 1996).
T‐bet is a Th1‐specific T box transcription factor that directly controls the expression of the hallmark Th1 cytokine, IFN‐γ, and IL‐12Rβ2 expression, thus facilitating Th1 cell differentiation (Szabo et al., 2000). The transcription factor c‐maf enhances IL‐4 production and represents an important step in the induction of Th2 cells (Ho et al., 1996). GATA‐3 was found to be an STAT6‐independent inducer of Th2 differentiation, and is located upstream of c‐maf, thus representing a master switch in Th2 development and commitment (Ouyang et al., 2000).
Th17 differentiation is independent of STAT4 and STAT6 signaling (Park et al., 2005); however, it has been demonstrated that STAT3 signaling is activated by both IL‐6 and IL‐23, and binds to IL‐17 gene promoters (Chen et al., 2006). SOCS‐3 is a major regulator of IL‐23‐mediated STAT3 phosphorylation and subsequent Th17 generation (Chen et al., 2006).
H. Regulatory T Cells
Several populations of regulatory or suppressor T cells have been described in humans. These include CD4+CD25+Foxp3 regulatory T cells (Baecher‐Allan 2001, Dieckmann 2001, Levings 2001, Stephens 2001, Yagi 2004), CD8+CD28− T cells (Koide and Engleman, 1990), IL‐10‐producing Th2 cells (Bacchetta et al., 1994), and TGF‐β‐producing Th3 cells (Kitani 2000, Roncarolo 2000). Regulatory T cells suppress T‐cell proliferation through a variety of mechanisms, including the production of immunosuppressive cytokines, or through T–T‐cell interactions. Several studies have demonstrated that these cells play an important role in the control of the immune response in multiple sclerosis (MS) and that the function of regulatory T cells may be enhanced by immunomodulatory therapies (Crucian 1995, Fukaura 1996, Hafler 1997, Karaszewski 1991, Viglietta 2004).
II. T‐Cell Immunologic Studies in MS
CD4+ T cells are believed to play a central role in the pathogenesis of MS. In this section, we summarize the prevailing theory of the pathogenesis of MS, and evidence for the role of CD4+ T cells from both human MS and animal models of disease.
A. Animal Models of MS
Much of the current understanding of the potential mechanistics and role of CD4+ T cells in MS comes from the animal models simulating features of MS. Experimental autoimmune encephalomyelitis (EAE) is an inflammatory central nervous system (CNS) demyelinating disease, and may be induced in several animal types via immunization with myelin proteins or peptides. Disease is primarily mediated by myelin‐reactive Th1 cells, which precipitate an inflammatory, demyelinating response within the CNS (Chitnis et al., 2001b). Transfer of myelin basic protein (MBP)‐specific T‐cell clones restricted to class II (Ia) antigens of the MHC into naive recipient animals causes a similar inflammatory demyelinating disease (Zamvil et al., 1985). EAE reproduces many of the clinical and immunologic aspects of MS, and has been widely used to study the mechanisms of CD4+ T‐cell priming and response to myelin components (Bettelli 1998, Chitnis 2001a) as well as to test potential therapies for MS (Aharoni 1999, Yednock 1992).
Theiler's murine encephalomyelitis virus‐induced demyelinating disease (TMEV‐IDD) model is a virally mediated model of CNS inflammatory demyelination, with some resemblance to MS and is induced by direct CNS infection of the neurotropic TMEV picornavirus, initially resulting in an immune‐mediated reaction primarily involving TMEV‐specific CD4 and CD8 T cells (Clatch 1986, Rodriguez 1996). However, during the chronic stages of disease, T‐cell reactivity to host myelin peptides has been observed, indicating epitope spreading has occurred, causing secondary T‐cell responses to myelin breakdown products, and resulting in a disseminated autoimmune response (Miller et al., 1997).
A summary of T‐cell immunology related to animal models of MS is beyond the scope of this chapter, and can be found elsewhere in this volume or in alternate sources (Chitnis 2003a, Chitnis 2003b); however, selected topics in MS that are illuminated by animal studies are discussed.
B. Molecular Mimicry and the Initiation of an Immune Response in MS
The prevailing theory of the etiology of MS is that of “molecular mimicry” whereby CD4+ T cells activated by a foreign antigen cross‐react with myelin antigens. Activated myelin‐reactive CD4+ T cells are present in the blood and cerebrospinal fluid (CSF) of MS patients; in contrast, only nonactivated myelin‐reactive T cells are present in the blood of controls (Zhang et al., 1994). Sequences in MBP has been shown to resemble several viral sequences, and in some cases, cross‐reactive T‐cell responses have been demonstrated. Although no pathogen has definitely been proven to be the cause of MS, it is conceivable that certain pathogens serve as molecular mimics to CNS components or play a role in the activation of myelin‐specific CD4+ T cells. Examples of cross‐reactive T cells with MBP antigens include human herpesvirus 6 (HHV‐6) (Tejada‐Simon et al., 2003), staphylococcal enterotoxin antigens (Zhang et al., 1995), coronavirus (Talbot et al., 1996), influenza virus hemagglutinin (Markovic‐Plese et al., 2005), and Epstein–Barr virus (EBV) (Lang et al., 2002). Proteolipid protein (PLP) shares common sequences with Haemophilus influenzae (Olson et al., 2001), while Semliki Forest virus (SFV) peptides mimic epitopes of myelin oligodendrocyte glycoprotein (MOG) (Mokhtarian et al., 1999).
These activated T cells are then thought to migrate to the CNS, where they undergo reactivation in response to nascent myelin antigens. The reactivation of T cells heralds an inflammatory response within the CNS, resulting in more tissue damage and release of secondary antigens. Subsequent T‐cell reactivity to secondary antigens is termed “epitope spreading.” Evidence of epitope spreading has been demonstrated in animal models of MS (McMahon 2005, Vanderlugt 1998), and may play an important role in the pathogenesis of the human disease.
Although this is the most widely accepted paradigm of MS pathogenesis, and is supported by evidence from studies discussed in this chapter, there still remain many unanswered questions, which include the identity of the initiating foreign cross‐reactive antigen(s), the identity of the initiating self‐antigen directed T‐cell response, and the nature of epitope spreading within the CNS. Moreover, the paucity of CD4+ T cells in certain pathological subtypes of MS questions whether alternate mechanisms may predominate in subsets of this heterogeneous disease.
C. CD4+ T Cells in the Peripheral Immune System of MS Patients
Because of the focus on myelin proteins and, in particular, MBP as a potential autoantigen in MS (Allegretta 1990, Chou 1992, Zhang 1994), considerable interest has developed in the role of T‐cell responses to MBP. Moreover, MS disease‐associated MHC class II allele, DRB1*1501 has been shown to be effective in presenting MBP peptide to T‐cell clones isolated from MS patients (Wucherpfennig 1995, Wucherpfennig 1997). Activated myelin‐reactive CD4+ T cells are present in the blood and CSF of MS patients; in contrast, only nonactivated myelin‐reactive T cells are present in the blood of controls (Zhang et al., 1994). Furthermore, MBP‐reactive T cells isolated from the CSF of MS patients display increased expression of the IL‐2 receptor (Zhang et al., 1994), consistent with a previously activated or memory phenotype. In MS patients, but not in healthy controls, these cells can be activated in the absence of CD28‐B7 costimulation, thus implying that they have been previously activated in vivo (Markovic‐Plese 2001, Scholz 1998). MBP‐reactive T cells from MS patients were found to be less responsive to CTLA‐4 blockade compared to those from healthy controls (Oliveira et al., 2003), signifying that in MS patients these T cells are not subject to the normal regulatory mechanisms. Although the contribution of MBP‐reactive T cells to the pathogenesis of MS is currently unknown, their differential phenotype and costimulatory requirements indicate a memory and potentially dysregulated cell population.
T‐cell reactivity to other myelin proteins and peptides in MS has been explored. Studies examining PLP T‐cell responses demonstrated T‐cell proliferation to certain epitopes (Pelfrey et al., 1994), with some differential reactivity when compared to controls (Markovic‐Plese 1995, Zhang 1994). T‐cell responses to recombinant MOG appeared to be similar in MS patients and healthy controls (Diaz‐Villoslada et al., 1999), although other studies demonstrated increased reactivity (Kerlero de Rosbo et al., 1993) or altered T‐cell properties (Van der Aa et al., 2003a) in MS patients. Other studies have examined T‐cell responses to myelin oligodendrocyte basic protein (MOBP) (Holz et al., 2000) or 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase (CNPase) (Muraro et al., 2002), with some reactivity demonstrated in T‐cell lines isolated from select MS patients. T‐cell reactivity to other CNS antigens has not been fully explored due to the technical difficulties with performing and interpreting the results of such assays. Many studies currently employ strategies to expand T cells using nonspecific methods or mixtures of myelin peptides.
Studies in patients with postinfectious encephalomyelitis or acute disseminated encephalomyelitis (ADEM) have consistently found robust T‐cell reactivity to myelin peptides in both the blood and CSF (Hafler 1987, Hemachudha 1988, Pohl‐Koppe 1998), and suggest an intriguing relationship in the pathophysiology of ADEM and MS.
D. TCR Repertoires in MS
Studies examining TCR repertoire in MS patients have demonstrated a bias for use of β chain variable region (Vβ) 5.2 and 5.3 (Kotzin 1991, Lozeron 1998, Oksenberg 1993), and this has led to the exploration of TCR Vβ 5.2/5.3‐targeted therapies. TCR vaccines employing TCR Vβ 5.2 peptides are thought to exert their effects by enhancing the function of regulatory T‐cell populations recognizing TCR determinants (Vandenbark 2005, Vandenbark 1996), and are currently undergoing pilot studies in MS. Phase II trials using antibodies specifically targeting the Vβ 5.2/5.3 sequence of the TCR have shown some success and no significant adverse effects (Killestein 2002, Olsson 2002). However, other groups (Gran 1998, Lozeron 1998, Musette 1996) have found predominant usage of other Vβ chains, indicative of potential limitations with TCR Vβ 5.2‐targeted therapies. Studies in twins demonstrated similar selection of TCR Vα chains in concordant twins in response to MBP, when compared to discordant twins or controls, suggesting a genetic basis for the evolution of self‐antigen T‐cell responses (Utz et al., 1993). MS patients treated with autologous hematopoietic stem cell transplantation demonstrated an increase in naive compared to memory CD4+ T cells, with increased TCR diversity indicative of broader clonal phenotypes 2 years following therapy (Muraro et al., 2005). A separate study ascertained similar findings and found that MBP‐reactive T cells demonstrated broader epitope recognition following reconstitution (Sun et al., 2004).
E. T Cells in MS Lesions
Pathologically, MS lesions are characterized by perivascular infiltrates of CD4+ and CD8+ T cells and macrophages (Prineas 1978, Traugott 1983b). CD4+ and CD8+ T cells and macrophages are also found toward the periphery of the lesion and in the normal appearing white matter (Traugott et al., 1983a). CD4+ T cells were shown to predominate in acute lesions, while CD8+ T cells were observed more frequently in chronic lesions (Raine, 1994). Similar densities of CD3+ T cells have been demonstrated in all four of the histopathologic subtypes of MS, although type III and IV lesions are additionally characterized by prominent oligodendrocyte degeneration (Lucchinetti et al., 1996). Attempts to isolate T‐cell clones from the brains of MS patients failed to show either MBP or PLP reactivity (Hafler et al., 1987). TCR analysis from MS lesions demonstrated a broad TCR Vα and Vβ repertoires in active lesions, while fewer TCR V genes were detected in chronic plaques and control samples (Wucherpfennig et al., 1992). Other studies demonstrated restricted TCR specificities, with rearranged Vβ 5.2 genes found in the brains of all patients who were HLA DRB1*1501, DQA1*0102, DQB1*0602, and DPB1*0401 positive, suggesting that MHC class II genotype may play a role in VDJ rearrangements in MS lesions (Oksenberg et al., 1993). T cells in parenchymal MS lesions lacked CCR7, indicating a differentiation of central‐memory T cells into effector memory cells presumably on restimulation by antigen within the CNS (Kivisakk et al., 2004).
In summary, multiple studies have demonstrated the presence of CD4+ T cells in MS lesions, arguing for a central role in MS pathogenesis. The lack of consensus regarding T‐cell specificities suggest a heterogeneity in T‐cell responses at the time of analysis, which may be a result of epitope spreading in chronic disease.
F. T‐Cell Activation and Costimulation
Costimulatory pathways deliver a positive or negative signal for T‐cell activation, and thus represent an important control step in the immune response. The CD28/CTLA‐4‐B7‐1/2 family of costimulatory molecules represents an important step in the activation of CD4+ T cells. Lesions in the CNS of patients with MS were found to be exclusively associated with the expression of B7‐1 in perivenular lymphocytes, while B7‐2 was expressed on macrophages in both MS and in other neurological diseases (Windhagen et al., 1995). Peripheral blood mononuclear cells (PBMCs), isolated from MS patients, showed increased expression of B7‐1 on both CD4+ and CD8+ cell patients with rapidly progressive disease, compared to those with stable disease, or normal controls (Mena and Rohowsky‐Kochan, 1999). In a separate study, B7‐1 expression localized to B cells was found to be increased during MS relapses, and treatment with IFN‐β 1b reduced the number of B7‐1‐expressing B cells but increased the number of B7‐2 monocytes (Genc et al., 1997). In total, these observations suggest that the B7‐CD28‐CTLA4 pathway is activated in MS, and that B7‐1, in particular, may play an important role in regulating disease activity.
Genetic polymorphisms of costimulatory molecules may contribute to disease susceptibility. Three CTLA‐4 gene polymorphisms were found in MS patients, but not in healthy controls (Ligers et al., 1999), however no association was found with disease course or severity (Masterman et al., 2002). In an Olmstead County study, two polymorphisms were associated with the presence of MS (Kantarci et al., 2003). The Canadian Collaborative Study found no association of CTLA‐4 polymorphisms with the disease course of MS (Dyment et al., 2002). The exon 1 A/G polymorphism was associated with the presence of oligoclonal bands in the CSF (Fukazawa et al., 1999). Thus, dysregulation of CTLA‐4 signaling may contribute to susceptibility to MS. A phase I safety study of CTLA4Ig (Repligen‐RG2077) as well as a multicenter study of CTLA4Ig (BMS‐188667) for MS are ongoing.
Interaction of CD40 on APCs with CD154 on T cells induces APC production of IL‐12, a major factor in Th1 cell differentiation (Kelsall et al., 1996). Expression of both CD40 and CD154 were increased in lesions from postmortem MS brains compared with controls, with CD40 found predominantly on macrophages and microglia, while CD154 colocalized with the CD4 T‐cell marker (Gerritse et al., 1996). Expression of CD154 was found to be higher in peripheral blood monocytes isolated from SPMS compared with RRMS or healthy controls (Filion 2003, Jensen 2001), and was reduced by IFN‐β treatment (Teleshova et al., 2000). PBMCs from SPMS patients produced more IL‐12 and IFN‐γ when restimulated in vitro, compared with healthy controls (Karni et al., 2002). In summary, these studies indicate that the CD40‐CD154 pathway is important for the regulation of Th1 cytokine production in MS. Clinical trials with an anti‐CD154 antibody (Biogen) in autoimmune disease such as ITP and lupus were terminated because of the occurrence of thromboembolic events. Another formulation of the antibody (IDEC pharmaceuticals) is under investigation. A phase I clinical trial in MS patients was recently performed with good safety data, and therapeutic effects are currently under investigation.
Programmed death‐1 (PD‐1) is expressed on T cells and is a negative regulator of T‐cell activation. PD‐1 polymorphism was shown to be a genetic modifier of the progression of MS, and this may relate to PD‐1‐mediated inhibition of T‐cell activation (Kroner et al., 2005).
The Tim family of molecules are important cell‐surface markers as well as costimulatory regulators of Th1/Th2 responses. In MS patients, CSF T‐cell clones demonstrated reduced levels of Tim‐3 and T‐bet and secreted higher amounts of IFN‐γ than did those from control subjects, indicating that Tim‐3 may represent an important regulator of Th1 responses in MS (Koguchi et al., 2006).
In summary, there is significant evidence that costimulatory molecules represent an important step in the control of T‐cell activation in MS and are viable therapeutic targets.
G. T‐Cell Cytokine Production in MS
In the context of MS, Th1 cytokines are thought to mediate disease, while Th2 cytokines are believed to play a protective role. However, as our understanding of the disease evolves, it is clear that this paradigm is not absolute. Moreover, evidence of a distinct lineage of Th17‐producing T cells has led to reevaluation of the role of Th1 cytokines in MS.
Th1 cytokines are predominantly found in the brains of MS patients, while a paucity of Th2 cytokines, in particular TGF‐β, and IL‐10 was observed (Cannella 1995, Hofman 1986, Woodroofe 1993). Studies using semiquantitative RT‐PCR and immunocytochemistry found increased expression of B7‐1 and IL12p40 in acute MS plaques, compared with samples isolated from inflammatory infarcts (Windhagen et al., 1995). IL‐6, IFN‐γ, and TNF‐α were expressed by cells located in the perivascular cuffs, suggesting that in acute MS lesions, inflammatory cells are the most important source of these cytokines (Woodroofe and Cuzner, 1993). Expression of TNF‐α was localized to macrophages, microglia, and astrocytes (Cannella 1995, Hofman 1989, Selmaj 1991) in chronic‐active lesions. A separate study found that IL‐2 was expressed predominantly in association with perivascular inflammatory cells examining acute MS lesions (Hofman et al., 1986). Contrary to the Th1/Th2 paradigm of MS, high levels of IL‐4 were expressed in both acute and chronic‐active MS lesions with no obvious correlation to the resolution of the lesion (Cannella and Raine, 1995b). Studies using RNA microarrays in MS brains at autopsy found increased transcripts of genes encoding for IL‐6, IL‐17, and IFN‐γ (Lock et al., 2002), indicating a potential role for both Th1 and Th17 cells in proinflammatory responses in MS.
Several studies have demonstrated enhanced production of the hallmark Th1 cytokine, IFN‐γ, from PBMCs restimulated ex vivo in MS patients compared to controls (Balashov 1997, Comabella 1998). Clinical attacks correlated with increased IFN‐γ production in vitro (Beck et al., 1988). In a similar study, IFN‐βwas found to blunt increased production of IFN‐γ during relapse (Becher et al., 1999). TCR‐mediated IFN‐γ and IL‐10 secretions are increased in relapsing‐remitting (RR) and secondary progressive (SP) patients, but not in primary progressive disease, suggesting a dysregulation of this signaling pathway in certain MS subtypes (Balashov et al., 2000). Interestingly, SPMS patients also exhibit seasonal variations of IFN‐γ production with increased expression in autumn and winter months compared with spring and summer months, which was not observed in normal controls (Balashov et al., 1998). A progressive course of MS was found to be significantly more frequent in carriers of the IFN‐γ receptor‐2 allele Arg64 (Schrijver et al., 2004). Administration of IFN‐γ to MS patients precipitated clinical attacks, confirming the role of IFN‐γ as a proinflammatory cytokine in MS (Panitch 1987a, Panitch 1987b).
Studies of the prototypic Th2 cytokine IL‐4 in MS are limited, IL‐4 was expressed in high levels in both acute‐ and chronic‐active MS lesions (Cannella and Raine, 1995a). High frequencies of T‐cell clones reactive to MBP‐ and PLP‐expressing IL‐4 were found in MS patients compared with untreated patients (Chou et al., 1992). Increased expression of IL‐4 secretion by CD3‐stimulated PBMCs was demonstrated in SPMS patients treated with cyclophosphamide/methylprednisolone compared with untreated patients (Smith et al., 1997). Thus, the role of IL‐4 in the pathogenesis of MS is unclear, however may be associated with responses to therapy.
Studies examining Th17 cell activity in MS found that dendritic cells from MS patients secrete elevated amounts of IL‐23 and express increased levels of IL‐23p19 mRNA, and are associated with increased T‐cell production of IL‐17 (Vaknin‐Dembinsky et al., 2006). A Japanese study examining cytokine expression found that CSF levels of IL‐17, IL‐8, and IL‐5 were significantly higher in opticospinal‐MS patients than in conventional RRMS patients, and may be associated with pronounced neutrophilic infiltrates typically found in the opticospinal disease variant (Ishizu et al., 2005).
H. Regulatory T Cells in MS
Alterations in the function of several populations of regulatory T cells have been demonstrated in MS. Induction of regulatory antigen‐specific Th3 cells through oral tolerance with myelin proteins has been described (Fukaura 1996, Hafler 1997). Deficiency in the CD8+CD28− subset of suppressor cells has been demonstrated in MS patients (Crucian et al., 1995). Moreover, increases in β‐adrenergic receptor density on CD8+CD28− cells were found in MS compared with controls (Karaszewski et al., 1991). Much attention has focused on the CD4+CD25+ subset of regulatory T cells. Induction of CD4+CD25+ T cells is controlled by the transcription regulator FOXP3 which also serves as a cell‐specific marker (Hori et al., 2003). Defects in the effector function of CD4+CD25+ regulatory T cells have been demonstrated in MS patients (Viglietta et al., 2004). Thus, defects in regulatory T‐cell populations may facilitate the development and/or progression of autoimmunity.
I. T‐Cell Migration in MS
T‐cell migration into the CNS in MS is believed to follow the sequence of capture, rolling, activation, adhesion strengthening, and finally transmigration through the BBB. The specifics of these events may vary depending on the region of the CNS, as well as the activation state of T cells and microvasculature.
Two processes appear to be important in T‐cell migration into the CNS: (1) migration of T cells from the blood into the CSF with interactions with dendritic elements on the luminal surface of the choroid plexus resulting in immunosurveillance and (2) T‐cell migration through inflamed endothelial BBB and interactions with perivascular APCs within the Virchow‐Robbins space (Engelhardt and Ransohoff, 2005). Evidence exists for both of these processes in MS, however the relative contribution of each is unclear, and may depend on the stage and subtype of disease.
Adhesion molecule ICAM‐1 is expressed on the inflamed BBB in MS, while LFA‐1 is expressed on infiltrating T cells, suggesting an important role for this pathway in T‐cell migration in MS (Bo et al., 1996). Inhibition of interactions between integrin molecule α4β1 present on the surface of T cells with VCAM‐1 present on endothelial cells of the BBB was shown to suppress the development of EAE (Yednock et al., 1992). This led to the development of an α4β1‐integrin antibody, natalizumab, which has been effective in reducing MS relapses (Miller et al., 2003). However, clinical trials with a natalizumab led to several cases of progressive multifocal leukoencephalopathy, resulting in the reevaluation of the role of such drugs in MS therapeutics (Kleinschmidt‐Demasters 2005, Langer‐Gould 2005, Van Assche 2005).
CXCR3 is postulated to be the major chemokine involved in the trafficking of T cells from the blood to the CSF for immunosurveillance in both normals and MS patients (Kivisakk et al., 2002). Studies of chemokine expression on peripheral blood T cells in MS patients found a positive correlation between CXCR3 expression on blood T cells (Eikelenboom et al., 2002) and CSF T cells (Sindern et al., 2002) on MRI measures of disease activity in MS patients.
T‐cell migration is an important step in the pathogenesis of MS, as confirmed by the success of α4β1‐integrin antibody therapy (natalizumab). However, effective blockade of CNS immunosurveillance has produced profound adverse effects, as evidenced by the development of PML in three treated patients. The risk of infection and emergence of tumors due to blockade of immunosurveillance must be balanced with therapeutic effect when utilizing such powerful therapeutics.
J. T‐Cell Interaction with Axons and Neurons
Much attention has focused on the presence of axonal damage in MS plaques as a substrate for chronic progressive disease (Trapp 1998, Trapp 1999). Although mediators of axonal damage include cytokines, complement, antibody, and nitric oxide, T cells may play a significant role both in neurodegeneration and in neuroprotection. The presence of CD8+ T cells, but not CD4+ T cells, in the MS lesion correlates with axonal damage (Bitsch et al., 2000). In the TMEV model of MS, demyelination but no axonal damage was found in the CNS of MHC class I‐deficient mice after infection with Theiler's virus (Rivera‐Quinones et al., 1998), suggesting that axonal damage is class I mediated. These mice also displayed a deficiency of CD8‐positive cells in their CNS lesions. Similarly, in vitro, T cell‐mediated neurotoxicity was dependent on IFN‐γ‐induced expression of MHC class I (Medana et al., 2001). Collectively, these studies suggest that class I expression in the CNS, particularly on neurons, may enhance a cytotoxic CD8+ T‐cell response.
In vitro coculture of human fetal neurons with OKT3‐activated CD4+ or CD8+ T cells has been found to produce apoptosis of neurons (Giuliani et al., 2003). This process required physical contact of the cells, as demonstrated by transwell experiments, and was not dependent on MHC I. Protection could be conferred by blocking CD40 on both T cells and neurons, and FasL on neurons. Attention has focused on the regulatory role that neurons may play on T cells. In a Lewis rat model of EAE, motoneurons were demonstrated to engulf T lymphocytes through a process consistent with emperipolesis (Smith et al., 2000). Neuronal production of TGF‐β has been shown to play a significant role in the induction of CD4+CD25+Foxp3+ regulatory T cells in a murine EAE model (Liu et al., 2006). These studies have important implications for understanding the T cell‐neuronal interactions in MS.
Although MBP‐reactive T cells have been implicated in the pathogenesis of MS (Zhang 1992, Zhang 1994), an intriguing study has shown that passive transfer of MBP‐reactive T cells resulted in protection of the retinal ganglion cell body after optic nerve crush injury in vivo (Moalem et al., 1999). Interestingly, transfer of T cells specific for other antigens was not protective. Furthermore, expression of mRNA for several nerve growth factors, including brain‐derived neurotrophic factor (BDNF), was upregulated after antigen‐activation of these T cells, while the injured nerve expressed mRNA for nerve growth factor receptors (Moalem et al., 2000). Two major types of Th cell responses have been described, based on cytokine secretion. Th1 cells produce the cytokines IFN‐γ and TNF‐α, while Th2 cells produce IL‐4, IL‐5, and IL‐10. EAE has been associated with a Th1 response, while Th2 cytokines are generally protective. In support of the above findings, a separate study demonstrated that the presence of myelin‐reactive Th2 cells conferred neuroprotection in organotypic hippocampal slice cultures (Wolf et al., 2002). These findings have several implications for MS. First, myelin‐reactive T cells may be neuroprotective under certain conditions including trauma, suggesting that the MS brain may be more susceptible to inflammation‐induced damage. Second, the local production of neurotrophic factors by inflammatory cells may be neuroprotective. However, in contrast to these findings, other studies have shown that MBP‐TCR transgenic mice sustained more CNS damage and inflammation following traumatic spinal cord injury, compared with wild‐type controls (Jones et al., 2002). T‐cell infiltrates were localized to areas of demyelination and axonal loss, suggesting that autoreactive T cells can trigger autoimmune demyelination in the setting of trauma under certain circumstances. Therefore, the therapeutic potential of neuroprotective T lymphocytes should be entertained only with caution.
Glatiramer acetate (GA) is an established treatment for RRMS. Its mechanism of action relies, in part, on the migration of Th2 cells into the CNS (Aharoni 2000, Aharoni 2002 ), where they presumably downregulate local inflammatory responses. Interestingly, these T cells have been shown to produce BDNF in the EAE model (Aharoni et al., 2003), and GA‐reactive T cells harvested from MS patients treated with GA produce BDNF on restimulation in vitro (Ziemssen et al., 2002). However, the role of BDNF in neuroprotection in MS or EAE has not been established. Thus, the role of this and other neurotrophic factors requires further exploration.
III. T‐Cell‐Targeted Therapies in MS
The success of several T cell‐targeted therapies in MS reinforces the importance of the role of the T cell in MS pathogenesis. Of the six approved therapies for MS, the effects of GA and natalizumab can be directly related to modulation of T‐cell function. The β‐interferons (IFN‐β1a and IFN‐β1b) modulate some T‐cell functions including T‐cell migration and Th1 cytokine production (Yong, 2002), while mitoxantrone is a cytotoxic agent that nonspecifically abrogates T‐ and B‐cell proliferation.
An altered peptide ligand (APL) may be defined as “any peptide that serves as a receptor ligand in which substitutions of a single or multiple amino acids lead to changes in the functional outcome of receptor signaling” (Bielekova and Martin, 2001). APLs have most commonly been used as TCR ligands to alter T‐cell responses to presumed immunogenic or target antigens presumably resulting in immune suppression or immune deviation as well as induction of a regulatory T‐cell population reactive to the APL itself, which then serves to downregulate the inflammatory disease process through bystander suppression.
Glatiramer acetate (GA; Copolymer‐1; Copaxone®) is an FDA approved therapy for the treatment of RRMS. GA is an APL that was originally developed to mimic MBP. It is composed of a random sequence of the amino acids glutamic acid, lysine, alanine, and tyrosine present in a specific molar ratio (0.14:0.34:0.43:0.09). Copaxone is administered by daily subcutaneous injection, and in a phase III clinical trial was found to reduce relapse frequency by 29%, as well as decrease the incidence of new gadolinium enhancing lesions on MRI (Ge 2000, Johnson 1995, Johnson 2001). Despite its crude resemblance to MBP, evidence from several studies showing GA stimulates several nonmyelin antigen T‐cell lines suggests that GA acts as a “universal” or degenerate T‐cell antigen (Duda et al., 2000). GA has been shown to inhibit responses to MBP‐specific T‐cell lines in vitro (Racke et al., 1992), and in vivo treatment with GA induces a hyporesponsiveness to this antigen (Schmied et al., 2003). Interestingly, GA‐reactive T‐cell lines isolated from both treated patients as well as untreated controls were found to cross‐react with a variety of peptides, suggesting degenerate antigenicity. In addition, Th2 cytokine deviation was noted in GA‐reactive T‐cell lines (Duda et al., 2000). In the EAE model, Th2‐producing GA‐reactive T cells were shown to accumulate in the CNS and attenuate disease (Aharoni et al., 2000). Thus, the principal mechanism of action of Copaxone may be the induction of Th2 responses, which exert bystander suppression of inflammation within the CNS.
APLs targeting MBP have been widely studied in MS because of the interest in this potential autoantigen. An APL to MBP87–99 peptide was shown to be effective in ameliorating disease in the EAE model (Karin et al., 1994). An initial phase I clinical trial tested four doses of an APL to MBP83–99 administered subcutaneously for 4 weeks demonstrated no safety concerns (Bielekova and Martin, 2001). Two phase II trials using MBP83–99 APLs were initiated: A small NIH‐based trial tested the highest dose of APL CGP77116 (50 mg) administered weekly for 9 months. Three of eight patients developed atypical MS exacerbations, characterized by a high gadolinium‐enhancing lesion load, tumefactive‐type lesion, or a flaccid paralysis with inflammatory involvement of the peripheral nervous system (Bielekova et al., 2000). 2/3 of exacerbations, correlated with enhanced reactivity to MBP (Bielekova et al., 2000). A second larger multicenter study testing three doses of APL NBI‐5788 (5, 20, or 50 mg) versus placebo in 144 total patients was terminated because of the occurrence of APL‐induced systemic hypersensitivity reactions in 9% of enrolled patients (Kappos et al., 2000). In both studies, enhanced ex vivo T‐cell responses to APL were observed following treatment. In patients who developed hypersensitivity reactions, enhanced Th2 responses to the APL could be demonstrated (Kappos et al., 2000). Further phase II studies investigating the effects of low‐dose APL (NBI‐5788) are currently undergoing safety evaluation.
T‐cell vaccination strategies attempt to eliminate pathogenic T cells through the enhancement of regulatory immune responses to autoreactive T cells. This approach requires the isolation of autoreactive T‐cell clones from the individual patient's blood or CSF, and subcutaneous reinjection in the form of an immunizing vaccine. Pilot trials of T‐cell vaccination with autologous MBP‐specific T cells from peripheral blood, in 28 RRMS and 26 SPMS patients, demonstrated a modest reduction in posttreatment relapse rate (Medaer 1995, Zhang 2002). In this study, the frequency of gadolinium‐enhancing lesions was largely unchanged posttreatment. A second pilot trial using autologous MBP and MOG‐reactive T‐cell vaccines in 20 RRMS nonresponders demonstrated a significant reduction in relapse rate (p = 0.026) as well as gadolinium enhancing and T2 lesion load (Achiron et al., 2004). In both studies, no serious adverse events were noted. In a small study utilizing myelin‐reactive CD4+ T cells derived from autologous CSF, no adverse effects were observed in any of the five treated patients (Van der Aa et al., 2003b). Further larger phase II trials using T‐cell vaccination are planned.
TCR vaccination strategies target TCR sequences believed to be critical in the immunopathogenesis of MS. In MS, Vβ 5.2/5.3+ has been identified as a dominant TCR variable region sequence involved in MBP T‐cell reactivity (Kotzin 1991, Lozeron 1998, Oksenberg 1993). TCR vaccines are thought to exert their effects by enhancing the function of regulatory T‐cell populations recognizing TCR determinants (Vandenbark 2005, Vandenbark 1996). TCR peptides derived from the Vβ 5.2 region of the TCR have been used as a vaccine in MS patients. In a double‐blind pilot study, 23 patients were treated with weekly to monthly injections of the peptide. All patients carried the HLADRB1*1501 allele. Enhanced T‐cell responses to the immunizing peptide correlated with clinical improvement (Bourdette 1994, Vandenbark 1996). T‐cell responses to MBP trended downward in responders. No major adverse events were observed in treated patients. ATM‐027 is an antibody specifically targeting the Vβ 5.2/5.3 sequence of TCR. Results from a multicenter phase II study in 47 MS patients treated with a run‐in regimen of ATM‐027 monthly for 6 months, showed no significant reduction in new gadolinium MRI lesions posttreatment despite a significant reduction in Vβ 5.2/5.3+ T cells (Killestein 2002, Olsson 2002). MS relapses occurred in three treated patients, however no other adverse events directly related to drug were observed. These negative results suggest that there is considerable variability in Vβ profiling in individual MS patients. An alternative explanation is that by the time the disease presents clinically, epitope spreading has occurred, negating the use of a single Vβ‐depleting agent.
CD4 is a cell‐surface marker of Th cells. In a randomized phase II double‐blind trial, an anti‐CD4 antibody (cM‐T412) was administered intravenously to 35 RRMS and SPMS patients (van Oosten et al., 1997). Administration of the antibody resulted in a rapid and sustained reduction in circulating CD4+ T cells. Infusion‐related side effects including nausea, fever, and tachycardia were limited to 24‐h postinfusion. After 9 months, treated patients demonstrated an approximately 40% reduction in relapse rate compared to placebo controls, however there was no significant change in the number of gadolinium‐enhancing lesions on MRI. Although anti‐CD4 therapy was effective in reducing relapse rate, lack of efficacy on primary MRI measures has led to questions regarding the effectiveness in MS.
The Jak/STAT family of transducer/transcription‐activating factors plays a critical role in the signaling of many cytokine receptors. 3‐Hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors such as atorvastatin has been shown to exert its protective effects in EAE through the induction of STAT6 phosphorylation and secretion of Th2 cytokines, with concomitant inhibition of STAT4 phosphorylation and secretion of Th1 cytokines (Youssef et al., 2002). A pilot clinical trial of simvastatin in MS showed positive results and a larger trial of Atorvastatin is under way.
Inhibition of molecular pathways involved in T‐cell migration has been effective in reducing MS relapses (Miller et al., 2003), however clinical trials with an α4‐integrin antibody led to several cases of progressive multifocal leukoencephalopathy, resulting in the reevaluation of the role of such drugs in MS therapeutics (Kleinschmidt‐Demasters 2005, Langer‐Gould 2005, Van Assche 2005).
IV. Conclusions
In conclusion, we have summarized the evidence for the central role of CD4+ T cells in the pathogenesis of MS, which represents the work of many teams of investigators over many years. Although much progress has been made in the field, which has led to important therapeutic advances, several questions remain unanswered, including the nature of the initiating T‐cell responses and the mechanisms of propagation of the disease within the CNS. The field of T‐cell biology remains an integral part of the MS question, and further advances will undoubtedly lead to improved treatment strategies for patients.
References
- Achiron A., Lavie G., Kishner I., Stern Y., Sarova‐Pinhas I., Ben‐Aharon T., Barak Y., Raz H., Lavie M., Barliya T., Faibel M., Cohen I.R., Mandel M. T cell vaccination in multiple sclerosis relapsing‐remitting nonresponders patients. Clin. Immunol. 2004;113:155–160. doi: 10.1016/j.clim.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Aggarwal S., Ghilardi N., Xie M.H., de Sauvage F.J., Gurney A.L. Interleukin‐23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin‐17. J. Biol. Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Aharoni R., Teitelbaum D., Arnon R., Sela M. Copolymer 1 acts against the immunodominant epitope 82–100 of myelin basic protein by T cell receptor antagonism in addition to major histocompatibility complex blocking. Proc. Natl. Acad. Sci. USA. 1999;96:634–639. doi: 10.1073/pnas.96.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R., Teitelbaum D., Leitner O., Meshorer A., Sela M., Arnon R. Specific Th2 cells accumulate in the central nervous system of mice protected against experimental autoimmune encephalomyelitis by copolymer 1. Proc. Natl. Acad. Sci. USA. 2000;97:11472–11477. doi: 10.1073/pnas.97.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R., Meshorer A., Sela M., Arnon R. Oral treatment of mice with copolymer 1 (glatiramer acetate) results in the accumulation of specific Th2 cells in the central nervous system. J. Neuroimmunol. 2002;126:58–68. doi: 10.1016/s0165-5728(02)00053-x. [DOI] [PubMed] [Google Scholar]
- Aharoni R., Kayhan B., Eilam R., Sela M., Arnon R. Glatiramer acetate‐specific T cells in the brain express T helper 2/3 cytokines and brain‐derived neurotrophic factor in situ. Proc. Natl. Acad. Sci. USA. 2003;100(24):14157–14162. doi: 10.1073/pnas.2336171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre M.L., Frauwirth K.A., Thompson C.B. T‐cell regulation by CD28 and CTLA‐4. Nat. Rev. Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- Allegretta M., Nicklas J.A., Sriram S., Albertini R.J. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science. 1990;247:718–721. doi: 10.1126/science.1689076. [DOI] [PubMed] [Google Scholar]
- Bacchetta R., Bigler M., Touraine J.L., Parkman R., Tovo P.A., Abrams J., de Waal Malefyt R., de Vries J.E., Roncarolo M.G. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J. Exp. Med. 1994;179:493–502. doi: 10.1084/jem.179.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baecher‐Allan C., Brown J.A., Freeman G.J., Hafler D.A. CD4+ CD25high regulatory cells in human peripheral blood. J. Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- Balashov K.E., Smith D.R., Khoury S.J., Hafler D.A., Weiner H.L. Increased interleukin 12 production in progressive multiple sclerosis: Induction by activated CD4+ T cells via CD40 ligand. Proc. Natl. Acad. Sci. USA. 1997;94:599–603. doi: 10.1073/pnas.94.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashov K.E., Olek M.J., Smith D.R., Khoury S.J., Weiner H.L. Seasonal variation of interferon‐gamma production in progressive multiple sclerosis. Ann. Neurol. 1998;44:824–828. doi: 10.1002/ana.410440519. [DOI] [PubMed] [Google Scholar]
- Balashov K.E., Comabella M., Ohashi T., Khoury S.J., Weiner H.L. Defective regulation of IFNγ and IL‐12 by endogenous IL‐10 in progressive MS. Neurology. 2000;55:192–198. doi: 10.1212/wnl.55.2.192. [DOI] [PubMed] [Google Scholar]
- Becher B., Giacomini P.S., Pelletier D., McCrea E., Prat A., Antel J.P. Interferon‐gamma secretion by peripheral blood T‐cell subsets in multiple sclerosis: Correlation with disease phase and interferon‐beta therapy. Ann. Neurol. 1999;45:247–250. [PubMed] [Google Scholar]
- Beck J., Rondot P., Catinot L., Falcoff E., Kirchner H., Wietzerbin J. Increased production of interferon gamma and tumor necrosis factor precedes clinical manifestation in multiple sclerosis: Do cytokines trigger off exacerbations? Acta Neurol. Scand. 1988;78:318–323. doi: 10.1111/j.1600-0404.1988.tb03663.x. [DOI] [PubMed] [Google Scholar]
- Bettelli E., Das M.P., Howard E.D., Weiner H.L., Sobel R.A., Kuchroo V.K. IL‐10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL‐10‐ and IL‐4‐deficient and transgenic mice. J. Immunol. 1998;161:3299–3306. [PubMed] [Google Scholar]
- Bettelli E., Carrier Y., Gao W., Korn T., Strom T.B., Oukka M., Weiner H.L., Kuchroo V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bielekova B., Martin R. Antigen‐specific immunomodulation via altered peptide ligands. J. Mol. Med. 2001;79:552–565. doi: 10.1007/s001090100259. [DOI] [PubMed] [Google Scholar]
- Bielekova B., Goodwin B., Richert N., Cortese I., Kondo T., Afshar G., Gran B., Eaton J., Antel J., Frank J.A., McFarland H.F., Martin R. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- Bird J.J., Brown D.R., Mullen A.C., Moskowitz N.H., Mahowald M.A., Sider J.R., Gajewski T.F., Wang C.R., Reiner S.L. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- Bitsch A., Schuchardt J., Bunkowski S., Kuhlmann T., Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Bo L., Peterson J.W., Mork S., Hoffman P.A., Gallatin W.M., Ransohoff R.M., Trapp B.D. Distribution of immunoglobulin superfamily members ICAM‐1, ‐2, ‐3, and the beta 2 integrin LFA‐1 in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 1996;55:1060–1072. [PubMed] [Google Scholar]
- Bourdette D.N., Whitham R.H., Chou Y.K., Morrison W.J., Atherton J., Kenny C., Liefeld D., Hashim G.A., Offner H., Vandenbark A.A. Immunity to TCR peptides in multiple sclerosis. I. Successful immunization of patients with synthetic Vβ 5.2 and Vβ 6.1 CDR2 peptides. J. Immunol. 1994;152:2510–2519. [PubMed] [Google Scholar]
- Bretscher P., Cohn M. A theory of self‐nonself discrimination. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- Bretscher P.A. A two‐step, two‐signal model for the primary activation of precursor helper T cells. Proc. Natl. Acad. Sci. USA. 1999;96:185–190. doi: 10.1073/pnas.96.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet J.F., Denizot F., Luciani M.F., Roux‐Dosseto M., Suzan M., Mattei M.G., Golstein P. A new member of the immunoglobulin superfamily—CTLA‐4. Nature. 1987;328:267–270. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- Byrne J.A., Butler J.L., Cooper M.D. Differential activation requirements for virgin and memory T cells. J. Immunol. 1988;141:3249–3257. [PubMed] [Google Scholar]
- Chakravarti S., Sabatos C.A., Xiao S., Illes Z., Cha E.K., Sobel R.A., Zheng X.X., Strom T.B., Kuchroo V.K. Tim‐2 regulates T helper type 2 responses and autoimmunity. J. Exp. Med. 2005;202:437–444. doi: 10.1084/jem.20050308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Laurence A., Kanno Y., Pacher‐Zavisin M., Zhu B.M., Tato C., Yoshimura A., Hennighausen L., O'Shea J.J. Selective regulatory function of Socs3 in the formation of IL‐17‐secreting T cells. Proc. Natl. Acad. Sci. USA. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis T., Khoury S.J. Cytokine shifts and tolerance in experimental autoimmune encephalomyelitis. Immunol. Res. 2003;28:223–239. doi: 10.1385/IR:28:3:223. [DOI] [PubMed] [Google Scholar]
- Chitnis T., Khoury S.J. Role of costimulatory pathways in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. J. Allergy Clin. Immunol. 2003;112:837–849. doi: 10.1016/j.jaci.2003.08.025. quiz 850. [DOI] [PubMed] [Google Scholar]
- Chitnis T., Najafian N., Abdallah K.A., Dong V., Yagita H., Sayegh M.H., Khoury S.J. CD28‐independent induction of experimental autoimmune encephalomyelitis. J. Clin. Invest. 2001;107:575–583. doi: 10.1172/JCI11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis T., Najafian N., Benou C., Salama A.D., Grusby M.J., Sayegh M.H., Khoury S.J. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J. Clin. Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y.K., Bourdette D.N., Offner H., Whitham R., Wang R.Y., Hashim G.A., Vandenbark A.A. Frequency of T cells specific for myelin basic protein and myelin proteolipid protein in blood and cerebrospinal fluid in multiple sclerosis. J. Neuroimmunol. 1992;38:105–113. doi: 10.1016/0165-5728(92)90095-3. [DOI] [PubMed] [Google Scholar]
- Clatch R.J., Lipton H.L., Miller S.D. Characterization of Theiler's murine encephalomyelitis virus (TMEV)‐specific delayed‐type hypersensitivity responses in TMEV‐induced demyelinating disease: Correlation with clinical signs. J. Immunol. 1986;136:920–927. [PubMed] [Google Scholar]
- Comabella M., Balashov K., Issazadeh S., Smith D., Weiner H.L., Khoury S.J. Elevated interleukin‐12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J. Clin. Invest. 1998;102:671–678. doi: 10.1172/JCI3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crucian B., Dunne P., Friedman H., Ragsdale R., Pross S., Widen R. Alterations in levels of CD28−/CD8+ suppressor cell precursor and CD45RO+/CD4+ memory T lymphocytes in the peripheral blood of multiple sclerosis patients. Clin. Diagn. Lab. Immunol. 1995;2:249–252. doi: 10.1128/cdli.2.2.249-252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Villoslada P., Shih A., Shao L., Genain C.P., Hauser S.L. Autoreactivity to myelin antigens: Myelin/oligodendrocyte glycoprotein is a prevalent autoantigen. J. Neuroimmunol. 1999;99:36–43. doi: 10.1016/s0165-5728(99)00099-5. [DOI] [PubMed] [Google Scholar]
- Dieckmann D., Plottner H., Berchtold S., Berger T., Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J. Exp. Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda P.W., Schmied M.C., Cook S.L., Krieger J.I., Hafler D.A. Glatiramer acetate (Copaxone) induces degenerate, Th2‐polarized immune responses in patients with multiple sclerosis. J. Clin. Invest. 2000;105:967–976. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton R.W., Bradley L.M., Swain S.L. T cell memory. Annu. Rev. Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- Dyment D.A., Steckley J.L., Willer C.J., Armstrong H., Sadovnick A.D., Risch N., Ebers G.C. No evidence to support CTLA‐4 as a susceptibility gene in MS families: The Canadian Collaborative Study. J. Neuroimmunol. 2002;123:193–198. doi: 10.1016/s0165-5728(01)00493-3. [DOI] [PubMed] [Google Scholar]
- Ehlers S., Smith K.A. Differentiation of T cell lymphokine gene expression: The in vitro acquisition of T cell memory. J. Exp. Med. 1991;173:25–36. doi: 10.1084/jem.173.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom M.J., Killestein J., Izeboud T., Kalkers N.F., van Lier R.A., Barkhof F., Uitdehaag B.M., Polman C.H. Chemokine receptor expression on T cells is related to new lesion development in multiple sclerosis. J. Neuroimmunol. 2002;133:225–232. doi: 10.1016/s0165-5728(02)00374-0. [DOI] [PubMed] [Google Scholar]
- Engelhardt B., Ransohoff R.M. The ins and outs of T‐lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Filion L.G., Matusevicius D., Graziani‐Bowering G.M., Kumar A., Freedman M.S. Monocyte‐derived IL12, CD86 (B7‐2) and CD40L expression in relapsing and progressive multiple sclerosis. Clin. Immunol. 2003;106:127–138. doi: 10.1016/s1521-6616(02)00028-1. [DOI] [PubMed] [Google Scholar]
- Fukaura H., Kent S.C., Pietrusewicz M.J., Khoury S.J., Weiner H.L., Hafler D.A. Induction of circulating myelin basic protein and proteolipid protein‐specific transforming growth factor‐beta1‐secreting Th3 T cells by oral administration of myelin in multiple sclerosis patients. J. Clin. Invest. 1996;98:70–77. doi: 10.1172/JCI118779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa T., Yanagawa T., Kikuchi S., Yabe I., Sasaki H., Hamada T., Miyasaka K., Gomi K., Tashiro K. CTLA‐4 gene polymorphism may modulate disease in Japanese multiple sclerosis patients. J. Neurol. Sci. 1999;171:49–55. doi: 10.1016/s0022-510x(99)00251-8. [DOI] [PubMed] [Google Scholar]
- Ge Y., Grossman R.I., Udupa J.K., Fulton J., Constantinescu C.S., Gonzales‐Scarano F., Babb J.S., Mannon L.J., Kolson D.L., Cohen J.A. Glatiramer acetate (Copaxone) treatment in relapsing‐remitting MS: Quantitative MR assessment. Neurology. 2000;54:813–817. doi: 10.1212/wnl.54.4.813. [DOI] [PubMed] [Google Scholar]
- Genc K., Dona D.L., Reder A.T. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta‐1b therapy. J. Clin. Invest. 1997;99:2664–2671. doi: 10.1172/JCI119455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerritse K., Laman J.D., Noelle R.J., Aruffo A., Ledbetter J.A., Boersma W.J., Claassen E. CD40‐CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani F., Goodyer C.G., Antel J.P., Yong V.W. Vulnerability of human neurons to T cell‐mediated cytotoxicity. J. Immunol. 2003;171:368–379. doi: 10.4049/jimmunol.171.1.368. [DOI] [PubMed] [Google Scholar]
- Gran B., Gestri D., Sottini A., Quiros Roldan E., Bettinardi A., Signorini S., Primi D., Ballerini C., Taiuti R., Amaducci L., Massacesi L. Detection of skewed T‐cell receptor V‐beta gene usage in the peripheral blood of patients with multiple sclerosis. J. Neuroimmunol. 1998;85:22–32. doi: 10.1016/s0165-5728(97)00250-6. [DOI] [PubMed] [Google Scholar]
- Hafler D.A., Benjamin D.S., Burks J., Weiner H.L. Myelin basic protein and proteolipid protein reactivity of brain‐ and cerebrospinal fluid‐derived T cell clones in multiple sclerosis and postinfectious encephalomyelitis. J. Immunol. 1987;139:68–72. [PubMed] [Google Scholar]
- Hafler D.A., Kent S.C., Pietrusewicz M.J., Khoury S.J., Weiner H.L., Fukaura H. Oral administration of myelin induces antigen‐specific TGF‐beta 1 secreting T cells in patients with multiple sclerosis. Ann. N. Y. Acad. Sci. 1997;835:120–231. doi: 10.1111/j.1749-6632.1997.tb48623.x. [DOI] [PubMed] [Google Scholar]
- Harrington L.E., Hatton R.D., Mangan P.R., Turner H., Murphy T.L., Murphy K.M., Weaver C.T. Interleukin 17‐producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Hemachudha T., Griffin D.E., Johnson R.T., Giffels J.J. Immunologic studies of patients with chronic encephalitis induced by post‐exposure Semple rabies vaccine. Neurology. 1988;38:42–44. doi: 10.1212/wnl.38.1.42. [DOI] [PubMed] [Google Scholar]
- Ho I.C., Hodge M.R., Rooney J.W., Glimcher L.H. The proto‐oncogene c‐maf is responsible for tissue‐specific expression of interleukin‐4. Cell. 1996;85:973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- Hofman F.M., von Hanwehr R.I., Dinarello C.A., Mizel S.B., Hinton D., Merrill J.E. Immunoregulatory molecules and IL 2 receptors identified in multiple sclerosis brain. J. Immunol. 1986;136:3239–3245. [PubMed] [Google Scholar]
- Hofman F.M., Hinton D.R., Johnson K., Merrill J.E. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med. 1989;170:607–612. doi: 10.1084/jem.170.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz A., Bielekova B., Martin R., Oldstone M.B. Myelin‐associated oligodendrocytic basic protein: Identification of an encephalitogenic epitope and association with multiple sclerosis. J. Immunol. 2000;164:1103–1109. doi: 10.4049/jimmunol.164.2.1103. [DOI] [PubMed] [Google Scholar]
- Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Ishizu T., Osoegawa M., Mei F.J., Kikuchi H., Tanaka M., Takakura Y., Minohara M., Murai H., Mihara F., Taniwaki T., Kira J. Intrathecal activation of the IL‐17/IL‐8 axis in opticospinal multiple sclerosis. Brain. 2005;128:988–1002. doi: 10.1093/brain/awh453. [DOI] [PubMed] [Google Scholar]
- Jacobson N.G., Szabo S.J., Weber‐Nordt R.M., Zhong Z., Schreiber R.D., Darnell J.E., Jr., Murphy K.M. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J. Exp. Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J., Krakauer M., Sellebjerg F. Increased T cell expression of CD154 (CD40‐ligand) in multiple sclerosis. Eur. J. Neurol. 2001;8:321–328. doi: 10.1046/j.1468-1331.2001.00232.x. [DOI] [PubMed] [Google Scholar]
- Johnson K.P., Brooks B.R., Cohen J.A., Ford C.C., Goldstein J., Lisak R.P., Myers L.W., Panitch H.S., Rose J.W., Schiffer R.B. Copolymer 1 reduces relapse rate and improves disability in relapsing‐remitting multiple sclerosis: Results of a phase III multicenter, double‐blind placebo‐controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- Johnson K.P., Brooks B.R., Cohen J.A., Ford C.C., Goldstein J., Lisak R.P., Myers L.W., Panitch H.S., Rose J.W., Schiffer R.B., Vollmer T., Weiner L.P. Copolymer 1 reduces relapse rate and improves disability in relapsing‐remitting multiple sclerosis: Results of a phase III multicenter, double‐blind, placebo‐controlled trial. 1995. Neurology. 2001;57:S16–S24. [PubMed] [Google Scholar]
- Jones T.B., Basso D.M., Sodhi A., Pan J.Z., Hart R.P., MacCallum R.C., Lee S., Whitacre C.C., Popovich P.G. Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: Implications for autoimmune vaccine therapy. J. Neurosci. 2002;22:2690–2700. doi: 10.1523/JNEUROSCI.22-07-02690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci O.H., Hebrink D.D., Achenbach S.J., Atkinson E.J., Waliszewska A., Buckle G., McMurray C.T., de Andrade M., Hafler D.A., Weinshenker B.G. CTLA4 is associated with susceptibility to multiple sclerosis. J. Neuroimmunol. 2003;134:133–141. doi: 10.1016/s0165-5728(02)00395-8. [DOI] [PubMed] [Google Scholar]
- Kaplan M.H., Sun Y.L., Hoey T., Grusby M.J. Impaired IL‐12 responses and enhanced development of Th2 cells in Stat4‐deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Kappos L., Comi G., Panitch H., Oger J., Antel J., Conlon P., Steinman L. Induction of a non‐encephalitogenic type 2 T helper‐cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo‐controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat. Med. 2000;6:1176–1182. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- Karaszewski J.W., Reder A.T., Anlar B., Kim W.C., Arnason B.G. Increased lymphocyte beta‐adrenergic receptor density in progressive multiple sclerosis is specific for the CD8+, CD28− suppressor cell. Ann. Neurol. 1991;30:42–47. doi: 10.1002/ana.410300109. [DOI] [PubMed] [Google Scholar]
- Karin N., Mitchell D.J., Brocke S., Ling N., Steinman L. Reversal of experimental autoimmune encephalomyelitis by a soluble peptide variant of a myelin basic protein epitope: T cell receptor antagonism and reduction of interferon gamma and tumor necrosis factor alpha production. J. Exp. Med. 1994;180:2227–2237. doi: 10.1084/jem.180.6.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni A., Koldzic D.N., Bharanidharan P., Khoury S.J., Weiner H.L. IL‐18 is linked to raised IFN‐gamma in multiple sclerosis and is induced by activated CD4+ T cells via CD40‐CD40 ligand interactions. J. Neuroimmunol. 2002;125:134–140. doi: 10.1016/s0165-5728(02)00018-8. [DOI] [PubMed] [Google Scholar]
- Kelsall B.L., Stuber E., Neurath M., Strober W. Interleukin‐12 production by dendritic cells. The role of CD40‐CD40L interactions in Th1 T‐cell responses. Ann. N. Y. Acad. Sci. 1996;795:116–126. doi: 10.1111/j.1749-6632.1996.tb52660.x. [DOI] [PubMed] [Google Scholar]
- Kerlero de Rosbo N., Milo R., Lees M.B., Burger D., Bernard C.C., Ben‐Nun A. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J. Clin. Invest. 1993;92:2602–2608. doi: 10.1172/JCI116875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killestein J., Olsson T., Wallstrom E., Svenningsson A., Khademi M., Blumhardt L.D., Fagius J., Hillert J., Landtblom A.M., Edenius C., Arfors L., Barkhof F. Antibody‐mediated suppression of Vβ5.2/5.3(+) T cells in multiple sclerosis: Results from an MRI‐monitored phase II clinical trial. Ann. Neurol. 2002;51:467–474. doi: 10.1002/ana.10146. [DOI] [PubMed] [Google Scholar]
- Kitani A., Chua K., Nakamura K., Strober W. Activated self‐MHC‐reactive T cells have the cytokine phenotype of Th3/T regulatory cell 1 T cells. J. Immunol. 2000;165:691–702. doi: 10.4049/jimmunol.165.2.691. [DOI] [PubMed] [Google Scholar]
- Kivisakk P., Trebst C., Liu Z., Tucky B.H., Sorensen T.L., Rudick R.A., Mack M., Ransohoff R.M. T‐cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: Implications for CNS trafficking. Clin. Exp. Immunol. 2002;129:510–518. doi: 10.1046/j.1365-2249.2002.01947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisakk P., Mahad D.J., Callahan M.K., Sikora K., Trebst C., Tucky B., Wujek J., Ravid R., Staugaitis S.M., Lassmann H., Ransohoff R.M. Expression of CCR7 in multiple sclerosis: Implications for CNS immunity. Ann. Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt‐Demasters B.K., Tyler K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta‐1a for multiple sclerosis. N. Engl. J. Med. 2005;353(4):369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- Koguchi K., Anderson D.E., Yang L., O'Connor K.C., Kuchroo V.K., Hafler D.A. Dysregulated T cell expression of TIM3 in multiple sclerosis. J. Exp. Med. 2006;203:1413–1418. doi: 10.1084/jem.20060210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide J., Engleman E.G. Differences in surface phenotype and mechanism of action between alloantigen‐specific CD8+ cytotoxic and suppressor T cell clones. J. Immunol. 1990;144:32–40. [PubMed] [Google Scholar]
- Kotzin B.L., Karuturi S., Chou Y.K., Lafferty J., Forrester J.M., Better M., Nedwin G.E., Offner H., Vandenbark A.A. Preferential T‐cell receptor beta‐chain variable gene use in myelin basic protein‐reactive T‐cell clones from patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA. 1991;88:9161–9165. doi: 10.1073/pnas.88.20.9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner A., Mehling M., Hemmer B., Rieckmann P., Toyka K.V., Maurer M., Wiendl H. A PD‐1 polymorphism is associated with disease progression in multiple sclerosis. Ann. Neurol. 2005;58:50–57. doi: 10.1002/ana.20514. [DOI] [PubMed] [Google Scholar]
- Kuiper H., Brouwer M., de Boer M., Parren P., van Lier R.A. Differences in responsiveness to CD3 stimulation between naive and memory CD4+ T cells cannot be overcome by CD28 costimulation. Eur. J. Immunol. 1994;24:1956–1960. doi: 10.1002/eji.1830240903. [DOI] [PubMed] [Google Scholar]
- Lang H.L., Jacobsen H., Ikemizu S., Andersson C., Harlos K., Madsen L., Hjorth P., Sondergaard L., Svejgaard A., Wucherpfennig K., Stuart D.I., Bell J.I. A functional and structural basis for TCR cross‐reactivity in multiple sclerosis. Nat. Immunol. 2002;3:940–943. doi: 10.1038/ni835. [DOI] [PubMed] [Google Scholar]
- Langer‐Gould A., Atlas S.W., Bollen A.W., Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005;353(4):375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- Langrish C.L., Chen Y., Blumenschein W.M., Mattson J., Basham B., Sedgwick J.D., McClanahan T., Kastelein R.A., Cua D.J. IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.T., Yin X.M., Vitetta E.S. Functional and ontogenetic analysis of murine CD45Rhi and CD45Rlo CD4+ T cells. J. Immunol. 1990;144:3288–3295. [PubMed] [Google Scholar]
- Levings M.K., Sangregorio R., Roncarolo M.G. Human CD25+ CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J. Exp. Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligers A., Xu C., Saarinen S., Hillert J., Olerup O. The CTLA‐4 gene is associated with multiple sclerosis. J. Neuroimmunol. 1999;97:182–190. doi: 10.1016/s0165-5728(99)00072-7. [DOI] [PubMed] [Google Scholar]
- Liu Y., Teige I., Birnir B., Issazadeh‐Navikas S. Neuron‐mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat. Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- Lock C., Hermans G., Pedotti R., Brendolan A., Schadt E., Garren H., Langer‐Gould A., Strober S., Cannella B., Allard J., Klonowski P., Austin A. Gene‐microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Lozeron P., Chabas D., Duprey B., Lyon‐Caen O., Liblau R. T cell receptor Vβ 5 and Vβ 17 clonal diversity in cerebrospinal fluid and peripheral blood lymphocytes of multiple sclerosis patients. Mult. Scler. 1998;4:154–161. doi: 10.1177/135245859800400313. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C.F., Bruck W., Rodriguez M., Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996;6:259–274. doi: 10.1111/j.1750-3639.1996.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan P.R., Harrington L.E., O'Quinn D.B., Helms W.S., Bullard D.C., Elson C.O., Hatton R.D., Wahl S.M., Schoeb T.R., Weaver C.T. Transforming growth factor‐beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Markovic‐Plese S., Fukaura H., Zhang J., al‐Sabbagh A., Southwood S., Sette A., Kuchroo V.K., Hafler D.A. T cell recognition of immunodominant and cryptic proteolipid protein epitopes in humans. J. Immunol. 1995;155:982–992. [PubMed] [Google Scholar]
- Markovic‐Plese S., Cortese I., Wandinger K.P., McFarland H.F., Martin R. CD4+ CD28‐costimulation‐independent T cells in multiple sclerosis. J. Clin. Invest. 2001;108:1185–1194. doi: 10.1172/JCI12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic‐Plese S., Hemmer B., Zhao Y., Simon R., Pinilla C., Martin R. High level of cross‐reactivity in influenza virus hemagglutinin‐specific CD4+ T‐cell response: Implications for the initiation of autoimmune response in multiple sclerosis. J. Neuroimmunol. 2005;169:31–38. doi: 10.1016/j.jneuroim.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Masterman T., Ligers A., Zhang Z., Hellgren D., Salter H., Anvret M., Hillert J. CTLA4 dimorphisms and the multiple sclerosis phenotype. J. Neuroimmunol. 2002;131:208–212. doi: 10.1016/s0165-5728(02)00274-6. [DOI] [PubMed] [Google Scholar]
- McMahon E.J., Bailey S.L., Castenada C.V., Waldner H., Miller S.D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- Medaer R., Stinissen P., Truyen L., Raus J., Zhang J. Depletion of myelin‐basic‐protein autoreactive T cells by T‐cell vaccination: Pilot trial in multiple sclerosis. Lancet. 1995;346:807–808. doi: 10.1016/s0140-6736(95)91622-9. [DOI] [PubMed] [Google Scholar]
- Medana I., Martinic M.A., Wekerle H., Neumann H. Transection of major histocompatibility complex class I‐induced neurites by cytotoxic T lymphocytes. Am. J. Pathol. 2001;159:809–815. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena E., Rohowsky‐Kochan C. Expression of costimulatory molecules on peripheral blood mononuclear cells in multiple sclerosis. Acta Neurol. Scand. 1999;100:92–96. doi: 10.1111/j.1600-0404.1999.tb01044.x. [DOI] [PubMed] [Google Scholar]
- Meyers J.H., Chakravarti S., Schlesinger D., Illes Z., Waldner H., Umetsu S.E., Kenny J., Zheng X.X., Umetsu D.T., DeKruyff R.H., Strom T.B., Kuchroo V.K. TIM‐4 is the ligand for TIM‐1, and the TIM‐1‐TIM‐4 interaction regulates T cell proliferation. Nat. Immunol. 2005;6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- Miller D.H., Khan O.A., Sheremata W.A., Blumhardt L.D., Rice G.P., Libonati M.A., Willmer‐Hulme A.J., Dalton C.M., Miszkiel K.A., O'Connor P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- Miller S.D., Vanderlugt C.L., Begolka W.S., Pao W., Yauch R.L., Neville K.L., Katz‐Levy Y., Carrizosa A., Kim B.S. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- Minguela A., Torio A., Marin L., Sanchez‐Bueno F., Garcia‐Alonso A.M., Ontanon J., Parrilla P., Alvarez‐Lopez M.R. Implication of Th1, Th2, and Th3 cytokines in liver graft acceptance. Transplant. Proc. 1999;31:519–520. doi: 10.1016/s0041-1345(98)02110-1. [DOI] [PubMed] [Google Scholar]
- Moalem G., Leibowitz‐Amit R., Yoles E., Mor F., Cohen I.R., Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat. Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- Moalem G., Gdalyahu A., Shani Y., Otten U., Lazarovici P., Cohen I.R., Schwartz M. Production of neurotrophins by activated T cells: Implications for neuroprotective autoimmunity. J. Autoimmun. 2000;15:331–345. doi: 10.1006/jaut.2000.0441. [DOI] [PubMed] [Google Scholar]
- Mokhtarian F., Zhang Z., Shi Y., Gonzales E., Sobel R.A. Molecular mimicry between a viral peptide and a myelin oligodendrocyte glycoprotein peptide induces autoimmune demyelinating disease in mice. J. Neuroimmunol. 1999;95:43–54. doi: 10.1016/s0165-5728(98)00254-9. [DOI] [PubMed] [Google Scholar]
- Monney L., Sabatos C.A., Gaglia J.L., Ryu A., Waldner H., Chernova T., Manning S., Greenfield E.A., Coyle A.J., Sobel R.A., Freeman G.J., Kuchroo V.K. Th1‐specific cell surface protein Tim‐3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- Muraro P.A., Kalbus M., Afshar G., McFarland H.F., Martin R. T cell response to 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase (CNPase) in multiple sclerosis patients. J. Neuroimmunol. 2002;130:233–242. doi: 10.1016/s0165-5728(02)00229-1. [DOI] [PubMed] [Google Scholar]
- Muraro P.A., Douek D.C., Packer A., Chung K., Guenaga F.J., Cassiani‐Ingoni R., Campbell C., Memon S., Nagle J.W., Hakim F.T., Gress R.E., McFarland H.F. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J. Exp. Med. 2005;201:805–816. doi: 10.1084/jem.20041679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musette P., Bequet D., Delarbre C., Gachelin G., Kourilsky P., Dormont D. Expansion of a recurrent Vβ 5.3+ T‐cell population in newly diagnosed and untreated HLA‐DR2 multiple sclerosis patients. Proc. Natl. Acad. Sci. USA. 1996;93:12461–12466. doi: 10.1073/pnas.93.22.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg J.R., Panzara M.A., Begovich A.B., Mitchell D., Erlich H.A., Murray R.S., Shimonkevitz R., Sherritt M., Rothbard J., Bernard C.C., Steinman L. Selection for T‐cell receptor V beta‐D beta‐J beta gene rearrangements with specificity for a myelin basic protein peptide in brain lesions of multiple sclerosis. Nature. 1993;362:68–70. doi: 10.1038/362068a0. [DOI] [PubMed] [Google Scholar]
- Oliveira E.M., Bar‐Or A., Waliszewska A.I., Cai G., Anderson D.E., Krieger J.I., Hafler D.A. CTLA‐4 dysregulation in the activation of myelin basic protein reactive T cells may distinguish patients with multiple sclerosis from healthy controls. J. Autoimmun. 2003;20:71–81. doi: 10.1016/s0896-8411(02)00106-3. [DOI] [PubMed] [Google Scholar]
- Olson J.K., Croxford J.L., Calenoff M.A., Dal Canto M.C., Miller S.D. A virus‐induced molecular mimicry model of multiple sclerosis. J. Clin. Invest. 2001;108:311–318. doi: 10.1172/JCI13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T., Edenius C., Ferm M., Samuelson P., Torrang A., Wallstrom E., Khademi M., Andersson M., Arfors L. Depletion of Vβ 5.2/5.3 T cells with a humanized antibody in patients with multiple sclerosis. Eur. J. Neurol. 2002;9:153–164. doi: 10.1046/j.1468-1331.2002.00370.x. [DOI] [PubMed] [Google Scholar]
- Ouyang W., Lohning M., Gao Z., Assenmacher M., Ranganath S., Radbruch A., Murphy K.M. Stat6‐independent GATA‐3 autoactivation directs IL‐4‐independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- Panitch H.S., Hirsch R.L., Haley A.S., Johnson K.P. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893–895. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- Panitch H.S., Hirsch R.L., Schindler J., Johnson K.P. Treatment of multiple sclerosis with gamma interferon: Exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- Park H., Li Z., Yang X.O., Chang S.H., Nurieva R., Wang Y.H., Wang Y., Hood L., Zhu Z., Tian Q., Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelfrey C.M., Trotter J.L., Tranquill L.R., McFarland H.F. Identification of a second T cell epitope of human proteolipid protein (residues 89–106) recognized by proliferative and cytolytic CD4+ T cells from multiple sclerosis patients. J. Neuroimmunol. 1994;53:153–161. doi: 10.1016/0165-5728(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Pohl‐Koppe A., Burchett S.K., Thiele E.A., Hafler D.A. Myelin basic protein reactive Th2 T cells are found in acute disseminated encephalomyelitis. J. Neuroimmunol. 1998;91:19–27. doi: 10.1016/s0165-5728(98)00125-8. [DOI] [PubMed] [Google Scholar]
- Prineas J.W., Wright R.G. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab. Invest. 1978;38:409–421. [PubMed] [Google Scholar]
- Racke M.K., Martin R., McFarland H., Fritz R.B. Copolymer‐1‐induced inhibition of antigen‐specific T cell activation: Interference with antigen presentation. J. Neuroimmunol. 1992;37:75–84. doi: 10.1016/0165-5728(92)90157-g. [DOI] [PubMed] [Google Scholar]
- Raine C.S. The Dale E. McFarlin Memorial Lecture: The immunology of the multiple sclerosis lesion. Ann. Neurol. 1994;36(Suppl.):S61–S72. doi: 10.1002/ana.410360716. [DOI] [PubMed] [Google Scholar]
- Rivera‐Quinones C., McGavern D., Schmelzer J.D., Hunter S.F., Low P.A., Rodriguez M. Absence of neurological deficits following extensive demyelination in a class I‐deficient murine model of multiple sclerosis. Nat. Med. 1998;4:187–193. doi: 10.1038/nm0298-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M., Pavelko K.D., Njenga M.K., Logan W.C., Wettstein P.J. The balance between persistent virus infection and immune cells determines demyelination. J. Immunol. 1996;157:5699–5709. [PubMed] [Google Scholar]
- Roncarolo M.G., Levings M.K. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr. Opin. Immunol. 2000;12:676–683. doi: 10.1016/s0952-7915(00)00162-x. [DOI] [PubMed] [Google Scholar]
- Sabatos C.A., Chakravarti S., Cha E., Schubart A., Sanchez‐Fueyo A., Zheng X.X., Coyle A.J., Strom T.B., Freeman G.J., Kuchroo V.K. Interaction of Tim‐3 and Tim‐3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003;4:1102–1110. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- Schmied M., Duda P.W., Krieger J.I., Trollmo C., Hafler D.A. In vitro evidence that subcutaneous administration of glatiramer acetate induces hyporesponsive T cells in patients with multiple sclerosis. Clin. Immunol. 2003;106:163–174. doi: 10.1016/s1521-6616(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Scholz C., Patton K.T., Anderson D.E., Freeman G.J., Hafler D.A. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J. Immunol. 1998;160:1532–1538. [PubMed] [Google Scholar]
- Schrijver H.M., Hooper‐Van Veen T., Van Belzen M.J., Crusius J.B., Pena A.S., Barkhof F., Polman C.H., Uitdehaag B.M. Polymorphisms in the genes encoding interferon‐gamma and interferon‐gamma receptors in multiple sclerosis. Eur. J. Immunogenet. 2004;31:133–140. doi: 10.1111/j.1365-2370.2004.00456.x. [DOI] [PubMed] [Google Scholar]
- Selmaj K., Raine C.S., Cannella B., Brosnan C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Invest. 1991;87:949–954. doi: 10.1172/JCI115102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindern E., Patzold T., Ossege L.M., Gisevius A., Malin J.P. Expression of chemokine receptor CXCR3 on cerebrospinal fluid T‐cells is related to active MRI lesion appearance in patients with relapsing‐remitting multiple sclerosis. J. Neuroimmunol. 2002;131:186–190. doi: 10.1016/s0165-5728(02)00263-1. [DOI] [PubMed] [Google Scholar]
- Smith D.R., Balashov K.E., Hafler D.A., Khoury S.J., Weiner H.L. Immune deviation following pulse cyclophosphamide/methylprednisolone treatment of multiple sclerosis: Increased interleukin‐4 production and associated eosinophilia. Ann. Neurol. 1997;42:313–318. doi: 10.1002/ana.410420307. [DOI] [PubMed] [Google Scholar]
- Smith T., Groom A., Zhu B., Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat. Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- Sprent J., Surh C.D. T cell memory. Annu. Rev. Immunol. 2002;20:551–579. doi: 10.1146/annurev.immunol.20.100101.151926. [DOI] [PubMed] [Google Scholar]
- Stephens L.A., Mottet C., Mason D., Powrie F. Human CD4+ CD25+ thymocytes and peripheral T cells have immune suppressive activity in vitro. Eur. J. Immunol. 2001;31:1247–1254. doi: 10.1002/1521-4141(200104)31:4<1247::aid-immu1247>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Sun W., Popat U., Hutton G., Zang Y.C., Krance R., Carrum G., Land G.A., Heslop H., Brenner M., Zhang J.Z. Characteristics of T‐cell receptor repertoire and myelin‐reactive T cells reconstituted from autologous haematopoietic stem‐cell grafts in multiple sclerosis. Brain. 2004;127:996–1008. doi: 10.1093/brain/awh117. [DOI] [PubMed] [Google Scholar]
- Szabo S.J., Kim S.T., Costa G.L., Zhang X., Fathman C.G., Glimcher L.H. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Takeda K., Kamanaka M., Tanaka T., Kishimoto T., Akira S. Impaired IL‐13‐mediated functions of macrophages in STAT6‐deficient mice. J. Immunol. 1996;157:3220–3222. [PubMed] [Google Scholar]
- Talbot P.J., Paquette J.S., Ciurli C., Antel J.P., Ouellet F. Myelin basic protein and human coronavirus 229E cross‐reactive T cells in multiple sclerosis. Ann. Neurol. 1996;39:233–240. doi: 10.1002/ana.410390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada‐Simon M.V., Zang Y.C., Hong J., Rivera V.M., Zhang J.Z. Cross‐reactivity with myelin basic protein and human herpesvirus‐6 in multiple sclerosis. Ann. Neurol. 2003;53:189–197. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- Teleshova N., Bao W., Kivisakk P., Ozenci V., Mustafa M., Link H. Elevated CD40 ligand expressing blood T‐cell levels in multiple sclerosis are reversed by interferon‐beta treatment. Scand. J. Immunol. 2000;51:312–320. doi: 10.1046/j.1365-3083.2000.00688.x. [DOI] [PubMed] [Google Scholar]
- Thierfelder W.E., van Deursen J.M., Yamamoto K., Tripp R.A., Sarawar S.R., Carson R.T., Sangster M.Y., Vignali D.A., Doherty P.C., Grosveld G.C., Ihle J.N. Requirement for Stat4 in interleukin‐12‐mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- Trapp B.D., Peterson J., Ransohoff R.M., Rudick R., Mork S., Bo L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Trapp B.D., Ransohoff R., Rudick R. Axonal pathology in multiple sclerosis: Relationship to neurologic disability. Curr. Opin. Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Traugott U., Reinherz E.L., Raine C.S. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia‐positive macrophages in lesions of different ages. J. Neuroimmunol. 1983;4:201–221. doi: 10.1016/0165-5728(83)90036-x. [DOI] [PubMed] [Google Scholar]
- Traugott U., Reinherz E.L., Raine C.S. Multiple sclerosis: Distribution of T cell subsets within active chronic lesions. Science. 1983;219:308–310. doi: 10.1126/science.6217550. [DOI] [PubMed] [Google Scholar]
- Utz U., Biddison W.E., McFarland H.F., McFarlin D.E., Flerlage M., Martin R. Skewed T‐cell receptor repertoire in genetically identical twins correlates with multiple sclerosis. Nature. 1993;364:243–247. doi: 10.1038/364243a0. [DOI] [PubMed] [Google Scholar]
- Vaknin‐Dembinsky A., Balashov K., Weiner H.L. IL‐23 is increased in dendritic cells in multiple sclerosis and down‐regulation of IL‐23 by antisense oligos increases dendritic cell IL‐10 production. J. Immunol. 2006;176:7768–7774. doi: 10.4049/jimmunol.176.12.7768. [DOI] [PubMed] [Google Scholar]
- van Oosten B.W., Lai M., Hodgkinson S., Barkhof F., Miller D.H., Moseley I.F., Thompson A.J., Rudge P., McDougall A., McLeod J.G., Ader H.J., Polman C.H. Treatment of multiple sclerosis with the monoclonal anti‐CD4 antibody cM‐T412: Results of a randomized, double‐blind, placebo‐controlled, MR‐monitored phase II trial. Neurology. 1997;49:351–357. doi: 10.1212/wnl.49.2.351. [DOI] [PubMed] [Google Scholar]
- Van Assche G., Van Ranst M., Sciot R., Dubois B., Vermeire S., Noman M., Verbeeck J., Geboes K., Robberecht W., Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- Van der Aa A., Hellings N., Bernard C.C., Raus J., Stinissen P. Functional properties of myelin oligodendrocyte glycoprotein‐reactive T cells in multiple sclerosis patients and controls. J. Neuroimmunol. 2003;137:164–176. doi: 10.1016/s0165-5728(03)00048-1. [DOI] [PubMed] [Google Scholar]
- Van der Aa A., Hellings N., Medaer R., Gelin G., Palmers Y., Raus J., Stinissen P. T cell vaccination in multiple sclerosis patients with autologous CSF‐derived activated T cells: Results from a pilot study. Clin. Exp. Immunol. 2003;131:155–168. doi: 10.1046/j.1365-2249.2003.02019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark A.A. TCR peptide vaccination in multiple sclerosis: Boosting a deficient natural regulatory network that may involve TCR‐specific CD4+ CD25+ Treg cells. Curr. Drug Targets Inflamm. Allergy. 2005;4:217–229. doi: 10.2174/1568010053586327. [DOI] [PubMed] [Google Scholar]
- Vandenbark A.A., Chou Y.K., Whitham R., Mass M., Buenafe A., Liefeld D., Kavanagh D., Cooper S., Hashim G.A., Offner H. Treatment of multiple sclerosis with T‐cell receptor peptides: Results of a double‐blind pilot trial. Nat. Med. 1996;2:1109–1115. doi: 10.1038/nm1096-1109. [DOI] [PubMed] [Google Scholar]
- Vanderlugt C.L., Begolka W.S., Neville K.L., Katz‐Levy Y., Howard L.M., Eagar T.N., Bluestone J.A., Miller S.D. The functional significance of epitope spreading and its regulation by co‐stimulatory molecules. Immunol. Rev. 1998;164:63–72. doi: 10.1111/j.1600-065x.1998.tb01208.x. [DOI] [PubMed] [Google Scholar]
- Veldhoen M., Hocking R.J., Atkins C.J., Locksley R.M., Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Viglietta V., Baecher‐Allan C., Weiner H.L., Hafler D.A. Loss of functional suppression by CD4+ CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhagen A., Newcombe J., Dangond F., Strand C., Woodroofe M.N., Cuzner M.L., Hafler D.A. Expression of costimulatory molecules B7–1 (CD80), B7–2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J. Exp. Med. 1995;182:1985–1996. doi: 10.1084/jem.182.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S.A., Fisher J., Bechmann I., Steiner B., Kwidzinski E., Nitsch R. Neuroprotection by T‐cells depends on their subtype and activation state. J. Neuroimmunol. 2002;133:72–80. doi: 10.1016/s0165-5728(02)00367-3. [DOI] [PubMed] [Google Scholar]
- Woodroofe M.N., Cuzner M.L. Cytokine mRNA expression in inflammatory multiple sclerosis lesions: Detection by non‐radioactive in situ hybridization. Cytokine. 1993;5:583–588. doi: 10.1016/s1043-4666(05)80008-0. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig K.W., Newcombe J., Li H., Keddy C., Cuzner M.L., Hafler D.A. T cell receptor V alpha‐V beta repertoire and cytokine gene expression in active multiple sclerosis lesions. J. Exp. Med. 1992;175:993–1002. doi: 10.1084/jem.175.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig K.W., Hafler D.A., Strominger J.L. Structure of human T‐cell receptors specific for an immunodominant myelin basic protein peptide: Positioning of T‐cell receptors on HLA‐DR2/peptide complexes. Proc. Natl. Acad. Sci. USA. 1995;92:8896–8900. doi: 10.1073/pnas.92.19.8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig K.W., Catz I., Hausmann S., Strominger J.L., Steinman L., Warren K.G. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLA‐DR2‐restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B‐cell and T‐cell epitopes. J. Clin. Invest. 1997;100:1114–1122. doi: 10.1172/JCI119622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H., Nomura T., Nakamura K., Yamazaki S., Kitawaki T., Hori S., Maeda M., Onodera M., Uchiyama T., Fujii S., Sakaguchi S. Crucial role of FOXP3 in the development and function of human CD25+ CD4+ regulatory T cells. Int. Immunol. 2004;16:1643–1656. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- Yao Z., Painter S.L., Fanslow W.C., Ulrich D., Macduff B.M., Spriggs M.K., Armitage R.J. Human IL‐17: A novel cytokine derived from T cells. J. Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- Yednock T.A., Cannon C., Fritz L.C., Sanchez‐Madrid F., Steinman L., Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- Yong V.W. Differential mechanisms of action of interferon‐beta and glatiramer aetate in MS. Neurology. 2002;59:802–808. doi: 10.1212/wnl.59.6.802. [DOI] [PubMed] [Google Scholar]
- Youssef S., Stuve O., Patarroyo J.C., Ruiz P.J., Radosevich J.L., Hur E.M., Bravo M., Mitchell D.J., Sobel R.A., Steinman L., Zamvil S.S. The HMG‐CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- Zamvil S., Nelson P., Trotter J., Mitchell D., Knobler R., Fritz R., Steinman L. T‐cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985;317:355–358. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- Zhang J., Weiner H.L., Hafler D.A. Autoreactive T cells in multiple sclerosis. Int. Rev. Immunol. 1992;9:183–201. doi: 10.3109/08830189209061790. [DOI] [PubMed] [Google Scholar]
- Zhang J., Markovic‐Plese S., Lacet B., Raus J., Weiner H.L., Hafler D.A. Increased frequency of interleukin 2‐responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J. Exp. Med. 1994;179:973–984. doi: 10.1084/jem.179.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Vandevyver C., Stinissen P., Mertens N., van den Berg‐Loonen E., Raus J. Activation and clonal expansion of human myelin basic protein‐reactive T cells by bacterial superantigens. J. Autoimmun. 1995;8:615–632. doi: 10.1016/0896-8411(95)90012-8. [DOI] [PubMed] [Google Scholar]
- Zhang J.Z., Rivera V.M., Tejada‐Simon M.V., Yang D., Hong J., Li S., Haykal H., Killian J., Zang Y.C. T cell vaccination in multiple sclerosis: Results of a preliminary study. J. Neurol. 2002;249:212–218. doi: 10.1007/pl00007867. [DOI] [PubMed] [Google Scholar]
- Zhu C., Anderson A.C., Schubart A., Xiong H., Imitola J., Khoury S.J., Zheng X.X., Strom T.B., Kuchroo V.K. The Tim‐3 ligand galectin‐9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- Ziemssen T., Kumpfel T., Klinkert W.E., Neuhaus O., Hohlfeld R. Glatiramer acetate‐specific T‐helper 1‐ and 2‐type cell lines produce BDNF: Implications for multiple sclerosis therapy. Brain‐derived neurotrophic factor. Brain. 2002;125:2381–2391. doi: 10.1093/brain/awf252. [DOI] [PubMed] [Google Scholar]
Further Reading
- Cannella B., Raine C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995;37:424–435. doi: 10.1002/ana.410370404. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C., Bruck W., Parisi J., Scheithauer B., Rodriguez M., Lassmann H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]