

Abstract
Over the last several years a wealth of transformative human–virus interaction discoveries have been produced using loss-of-function functional genomics. These insights have greatly expanded our understanding of how human pathogenic viruses exploit our cells to replicate. Two technologies have been at the forefront of this genetic revolution, RNA interference (RNAi) and random retroviral insertional mutagenesis using haploid cell lines (haploid cell screening), with the former technology largely predominating. Now the cutting edge gene editing of the CRISPR/Cas9 system has also been harnessed for large-scale functional genomics and is poised to possibly displace these earlier methods. Here we compare and contrast these three screening approaches for elucidating host–virus interactions, outline their key strengths and weaknesses including a comparison of an arrayed multiple orthologous RNAi reagent screen to a pooled CRISPR/Cas9 human rhinovirus 14–human cell interaction screen, and recount some notable insights made possible by each. We conclude with a brief perspective on what might lie ahead for the fast evolving field of human–virus functional genomics.
Keywords: Human–virus interactions, Genetic screening, siRNA, shRNA, RNA interference, CRISPR/Cas9, Haploid cells
1. Introduction
The burden imposed upon the health of the world's population by just three of the major pathogenic viruses is staggering, with nearly 300 million people chronically infected by either HIV-1 (36 million) or HBV (250 million), and another 5–6 million severe infections by influenza A virus (IAV) occurring transiently each year (Ortblad et al., 2013, Schweitzer et al., 2015) (http://www.who.int/immunization/topics/influenza/en/). Collectively these three viruses cause the deaths of over 2.5 million people annually. These infections arise because viruses must find and exploit the host's cellular resources and machinery to produce their progeny. Elucidating human pathogenic viral dependencies has been a longstanding pursuit of health science researchers whose goal is to use this knowledge to treat and cure infections. For decades, mammalian in vitro tissue culture systems have proved tremendously useful for studying host–virus interactions. Over this same period, loss-of-function genetic screening produced an impressive number of discoveries and illuminated gene and pathway function in multiple model systems. While loss-of-function genetic screening proved extremely valuable in model systems, such technologies did not exist for mammalian cells until the discovery and implementation of RNA interference (RNAi) (Fire et al., 1998). The initial technologic revolution of RNAi, and later the development of haploid cell screening, resulted in a wave of discoveries that shed new light on many vital human viral requirements (Brass et al., 2008, Hao et al., 2008, Krishnan et al., 2008, Randall et al., 2007, Sessions et al., 2009). The ascendance of CRISPR/Cas9 technologies, which can dramatically alter gene expression, has heralded a new era in mammalian in vitro genetic screening (Shalem, Sanjana, & Zhang, 2015). This review will discuss the available functional genomics strategies, highlight their strengths and weaknesses including a comparison of matched MORR RNAi and CRISRP/Cas9 screens, and provide some future perspectives on the use of mammalian in vitro genetics to elucidate human host–virus interactions.
2. Host–Virus Genetic Screens
The numbers of host–virus functional genomic screens using these technologies, particularly RNAi, have been increasing rapidly attesting to their innovative discovery power, generalizability and remarkable ease of use (Table 1 ). Drosophila cell in vitro RNAi screens were the first to detect novel host factor interactions for several human pathogens with the practical focus being on arboviruses, although an elegant approach using a recombinant virus also made it possible to screen for IAV dependency factors in this system (Arkov et al., 2008, Cherry et al., 2005, Hao et al., 2008). RNAi screens using human cells have now been done for the majority of major human pathogenic viruses (Table 1); these efforts have largely used arrayed siRNA libraries combined with high-throughput imaging or plate reader-based assays as readouts for viral replication. Collectively these works have identified multiple previously unappreciated dependencies for each virus, as well as host cell defense mechanisms. Recent publications covering viruses that have been functionally interrogated by multiple independent groups including HIV-1, IAV, and HCV have been discussed elsewhere in detail (Bushman et al., 2009, Hao et al., 2013, Stertz and Shaw, 2011, Zhu et al., 2014). In this work, we focus on the functional genomic screening technologies and provide a resource noting many of the published host–virus screens along with some of their key attributes.
Table 1.
Functional Genomic Screens for Elucidating Host–Viral Interactions
| Citation | Virus | Cell Line | Pooled/Arrayed | Library | Knockdown/Out Time | Challenge Time | Readout | Viral Dependency Factors | Viral Dependency Factor Selection Criteria | Viral Competitive or Restriction Factors | Viral Competitive or Restriction factors Selection Criteria | Main Candidates | Stage of Viral Lifecycle Impacted | Candidate Validation and Follow up Assays | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haploid cells | Carette et al. (2009) | Influenza virus (PR/8/34; H1N1) | Haploid human suspension cells KBM-7 |
Pooled | Haploid cell Insertional mutagenesis with lentiviral exon trap |
N/A | 2–3 weeks | Survival | Yes | Multiple independent integrations | No | N/A | CMAS; SLC35A2 | Entry | RT-PCR; immunofluorescence; complementation with cDNAs | |
| Carette et al. (2011) | rVSV-GP-Ebola virus | Haploid human adherent cells (HAP1) | Pooled | Haploid cell Insertional mutagenesis with lentiviral exon trap |
N/A | Unknown | Survival | Yes | Multiple independent integrations | No | N/A | NPC1, HOPS complex | Entry, viral fusion in lysosomal compartment | Complementation with cDNAs; test against related viruses; small-molecule U1866A and imipramine; immunofluorescence/electron microscopy viral entry assays; primary cell lines | ||
| Jae et al. (2013) | rVSV-GP-Lassa virus | HAP1 | Pooled | Haploid cell Insertional mutagenesis with lentiviral exon trap |
Gene-Trap | Unknown | Survival | Yes | Multiple independent integrations | No | N/A | TMEM5; B3GALNT2; B3GNT1; SLC35A1; SGK196 | Entry, presentation of laminin-binding carbohydrate | Null alleles TALENs; rescue cDNAs; analysis of know polymorphisms; flow cytometry; RT-PCR; clinical comparison | ||
| Kleinfelter et al. (2015) | rVSV-Andes virus-GP | HAP1 | Pooled | Haploid cell Insertional mutagenesis with lentiviral exon trap |
N/A | 8 days | Survival | Yes | Multiple independent integrations | No | N/A | S1P; S2P; SREBF2; SCAP; LSS; SQLE; ACAT2 | Entry | S1P CRISPR/Cas9 gene editing in U2OS; complementation with cDNA; small-molecule inhibitor | ||
| siRNA | ||||||||||||||||
| Haploid cell and siRNA | Petersen et al. (2014) | rVSV-Andes virus, either recombinant or pseudoparticles expressing Renilla luciferase | HAP1 | Pooled | Haploid cell Insertional mutagenesis with lentiviral exon trap |
N/A | 3 weeks | Survival | Yes | Multiple independent integrations | No | N/A | SCAP; S1P; S2P; SREBF2 | Entry | Functionally deficient cells S1P, S2P, or SCAP null CHO and SREBP2 KD HEK293T; TALEN-mediated gene disruption; small-molecule PF-429242 and mevastatin | |
| HEK29 | Arrayed | Ambion druggable genome library (9102 genes) (4 siRNAs/gene) (2 siRNAs/well) | 72 h | 24 h | Renilla luciferase expression | Yes | In both pools: Z score for infection <−1.5 (p < 0.009); viability <−2 | SREBF2 | Entry | 3 additional unique siRNAs screened with ANDV and VSV-G pseudoparticles; validated by 1 siRNA repeating finding two times. 105 candidate genes—33 validated—9 specific for ANDV | ||||||
| 210 dsRNAs; 112 genes reconfirmed | ||||||||||||||||
| Brass et al. (2008) | HIV-1-IIIB | TZM-bl | Arrayed | Dharmacon siARRAY siRNA library (21,121 siRNA pools) | 72 h | 48 h | % Infectivity (anti-HIV-1 p24) | Yes | Decreased Infectivity by ≥ 2 SDs; viability not decreased by > 2 SDs | No | N/A | RAB6A | Fusion | Subcellular localization; gene ontology (GO) biological processes analysis; Expression Genomic Institute of the Novartis Research Fund (GNF); individual shRNAs; individual siRNAs; infection with VSV-g; other cell lines Jurkat; qPCR | ||
| TNPO3 | Cytosolic post-RT–pre integration | |||||||||||||||
| MED28 | Transcription | |||||||||||||||
| Hao et al. (2008) | Influenza A virus Flu-VSV-G-GFP | DL1 | Arrayed | Ambion Drosophila RNAi library (13,071 genes) | 48 h | 24 h | Renilla luciferase activity | Yes | Inhibition > 2.4 SDs; Viability reduction Z score >-3 | Yes | Increase > 3 SDs; viability reduction Z score >−3 | COX6A1 | PB2/PB1-F2-mediated functions | RT-PCR; reagent redundancy; test human homologues, knockdown in HEK293 cells; individual siRNAs; small-molecule inhibitors; related viruses: WSN, H5N1 Influenza A/Indonesia/7/05, VSV, VACV | ||
| 176 candidate genes—110 confirmed | 123 candidate genes—11 genes confirmed | ATP6V0D1 | Fusion | |||||||||||||
| NXF1 | RNA export pathway | |||||||||||||||
| Krishnan et al. (2008) | West Nile virus WNV strain 2471 |
HeLa | Arrayed | Dharmacon siARRAY siRNA library (21,121 siRNA pools) | 72 h | 24 h | % Infectivity (viral E-proteins) | Yes | Infection reduction of > twofold | No | NA | CBLL1 | Entry | Individual siRNAs, small-molecule: MG132, cyclohexamide; colocalization; enrichment analysis using Panther; gene expression—microarray; protein interaction network | ||
| Dengue virus DENV New Guinea C strain |
30 h | 283 candidates | MCT4 | Replication phase | ||||||||||||
| Tai et al. (2009) | Hepatitis C virus Subgenomic genotype 1b replicon |
Huh7/Rep-Feo | Arrayed | Dharmacon siARRAY human genome siRNA library (21,094 genes) | 72 h | N/A | Viral replication (luciferase) | Yes | Replicon expression decreases by > 2 SDs | Yes | Increased replicon expression with threshold of q < 0.10 | PI1KA | Replication complex formation, generation of HCV nonstructural protein-associated membranes | Gene ontology; clustered; literature review; other cell line: OR6 replicon cell line, UHCVcon57.3; protein expression; Western blot; small-molecule Wortmannin, brefeldin A; reagent redundancy; shRNAs; localization studies; virus: HCV-JFH1 | ||
| 236 pools—186 replicated—96 confirmed | 13 pools | COPI-Coatomer | Early | |||||||||||||
| Hepcidin | Cellular translation | |||||||||||||||
| Li et al. (2009) | Hepatitis C virus JFH-1 | Huh 7.5.1 | Arrayed | Dharmacon siARRAY siRNA library; human genome (19,470 genes) | 72 h | 48 h | % Infectivity (HCV Core Antibody 6G7) | Yes | Infectivity < 50% plate mean; cell number > 50% of plate mean | Yes | Infectivity > 150% pf plate mean; cell number > 50% plate mean | RAB9p40 | Needed for both HCV and HIV | Individual siRNAs, enrichment analyses for molecular function and biological process according to Panther classification; network analyses interactome screens + HPRD; RT-PCR | ||
| 407 candidate pools | 114 candidate pools | |||||||||||||||
| Sessions et al. (2009) | Dengue virus DENV-S2 |
Dipteran cells | Arrayed | Genome-wide RNAi library DRSC 2.0 (22,632 dsRNAs) | 48 h | 72 h | Expression of envelope protein | Yes | Inhibited infection ≥ 1.5-fold with p < 0.05 | No | N/A | FLJ20254; TAZ; EXDL2; CNOT2 | RNA accumulation | Gene ontology; in vivo mosquito Ae. aegypti; validation of human homologue siRNAs in Huh-7 cells; other viruses: YFV 17D vaccine strain, Coxsackie B3 (strain 20; CB3); RT-qPCR | ||
| 218 candidate dsRNAs—rescreen 179 dsRNA—identified 118 dsRNA = 116 genes—111 novel | ||||||||||||||||
| Brass et al. (2009) | Influenza A virus A/Puerto Rico/8/34 | U2OS | Arrayed | Dharmacon siARRAY siRNA library; human genome (17,877 genes) | 72 h | 12 h | % Infectivity (anti-HA antibody) | Yes | < 55% infectivity; viability > 40% | Yes | > 200% infectivity; viability > 40% | IFITM3 | Early | Rescreened candidates; (GO) enrichment analysis; other cell lines primary lung fibroblasts, HeLa, A549, ChEFs, MDCKs; other viruses: HIV, PR8, H3N2 A/Udorn/72, A/Brisbane/59/07 H1N1, A/Uruguay/716/07 H3N2, A/Aichi/2/68 H3N2, MLV, VSV-G; pseudoparticles MLV with the following envelopes: H1, H3, H5, H7, MACH, MLVRescue construct; overexpression; Western blot; immunofluorescence | ||
| 312 pools | 22 pools | |||||||||||||||
| Shapira et al. (2009) | Influenza A virus IAV PR8 |
HBECs | Arrayed | Dharmacon SMARTpool | 72 h | 48 h | Viral particle production (reinfection); IFN production | Yes | Change > twofold less replication compared to median | Yes | Change > twofold more replication compared to median | WNT/p53 pathway | NS1 related | Pathway analysis; clustering of expression data; functional annotations; yeast 2 hybrid | ||
| Kolokoltsov, Saeed, Freiberg, Holbrook, and Davey (2009) | EBOV GP (Zaire)—pLENTI6-fluc | HEK293 | Arrayed | Kinase and phosphorylase subset of Ambion druggable genome (720 genes) | 48 h | 36 h | Luciferase expression | Yes | Decrease ≥ 3 × standard deviation | Yes | Increase ≥ 3 × standard deviation | PI3K | Membrane turnover | Verified in Vero cells; redundant siRNA activity analysis; Ingwnuity pathways knowledge base network analysis; small molecule: inhibitor drugs, KN-93, KN-92, LY294002 | ||
| CAMK2 | Transcription | |||||||||||||||
| Konig et al. (2010) | Influenza A virus Recombinant A/WSN/33 |
A549 | Arrayed | QIAGEN genome-wide (19,628 genes) | 48 h | 12, 24, 36 h | Luciferase activity | Yes | 2 siRNAs Luciferase reduction ≥ 35% | No | N/A | COPI coat complex | Entry | Reagent redundancy; viability; enrichment analysis; protein interactions; WT virus, clustering; pseudoparticles; GO analysis; STRING analysis; other virus IAV A/Hamburg/04/2009, A/Vietnam/1203/2004; lifecycle assays; localization assay | ||
| Karlas et al. (2010) | Influenza A virus IAV A/WSN/33 |
A549/293T | Arrayed | QIAGEN | 48 h | 24 h | Nuclear protein staining/luciferase | Yes | Robust Z score <−2 | No | N/A | CLK1 | Splicing viral mRNA | Reagent redundancy; viability assay; replication analysis; gene enrichment; network analysis; Western blot; lifecycle assay; RT-qPCR; small molecule: TG003; in vivo assay | ||
| Smith et al. (2010) | Human Papillomavirus Stable expressing HPV18LCR-Luc |
C33A/BE2/18LCR Clone 4 | Arrayed | Dharmacon human genome library (21,121 SMARTpools) | 72 h | N/A | Luciferase activity | No | N/A | Yes | Z score ≥ 2 | SMCX | E2-dependent transcriptional repression | Quantitative In-Cell Western; reagent redundancy; individual siRNAs; multiple different cell lines; protein interaction network; GO analysis; transient DNA transfections; immunoprecipitation; RT-qPCR | ||
| EP400 | ||||||||||||||||
| Brd4 | ||||||||||||||||
| Moser, Jones, Thompson, Coyne, and Cherry (2010) | Poxvirus | DL1 | Arrayed | Mini library Drosophila kinase and phosphate genes (440 genes) | 72 h | 48 h | % Infectivity (anti-B-gal antibody) | Yes | Robust Z score of <−2 | No | N/A | AMPK | Entry | Secondary dsRNAs; RT-PCR; mammalian cells—MEFs (null), U2OS; VSV control virus; Northern blot for virus; AMPK inhibitor Compound C; dextran uptake | ||
| 8 genes—7 validated | ||||||||||||||||
| Panda et al. (2011) | Vesicular Stomatitis virus VSV-eGFP |
HeLa | Arrayed | QIAGEN genome-wide siRNA library version 1 (22,909 genes) | 52 h | 18 h | Green fluorescence protein (GFP) intensity | Yes | > 5 SDs from mean | No | N/A | COPI; ARF1; GBF1 | Viral gene expression | RT-qPCR; cell viability; clustering/enrichment analysis; reagent redundancy; other viruses: HPIV3, LCMV; lifecycle assay | ||
| 233 genes | ||||||||||||||||
| Coyne et al. (2011) | Coxsackievirus B CVB |
HBMECs | Arrayed | Ambion druggable genome library (5492 genes) | 72 h | 14 h | % Infectivity (viral VP1 antigen) | Yes | Robust Z score < − 2; viability < 30% in cell number | Yes | Robust Z score > 2; viability < 30% in cell number | Akt1/Akt2 | Akt/MAPK signaling | 3 unique siRNAs; pathway enrichment; protein network analysis; microarray analysis; small-molecule Akt1/Akt2 inhibitor SH-6, TOR inhibitor rapamycin, ERK1/2 inhibitor FR180204; dominant negative mutant | ||
| CVB 144; PV 155; 38% confirmation; 46 validation overlap | CVB 31; PV 65; 38% confirmation; 17 validated overlap | MAP3K4; MAPK1 | ||||||||||||||
| Poliovirus PV | TLR8/IRK1 | Viral detection | ||||||||||||||
| ADCYs | cAMP mediated CREB-dependent transcription | |||||||||||||||
| Hussain, Leong, Ng, and Chu (2011) | HEV71 | RD cells | Arrayed | Dharmacon human genome siRNA endocytic and membrane trafficking genes subset library (119 genes) | 48 h | 12 h | Primary anti-HEV17 antibody | Yes | Viral antigen + cells < 50% of control | No | N/A | AP2A1; CLTC; CLTCL1 | Clathrin-mediated endocytosis | Dominant negative mutants; deconvolution of siRNAs; reagent redundancy; dosage-dependent KD; immunofluorescence entry assay; transmission electron microscopy entry assay; small molecule: Chlorpromazine, cytochalasin B, filipin, nystatin, methyl-B-cyclodextrin, EIPA | ||
| MAP4K2; PAK1; PIK3CG; PIK3C2G; ROCK1 | Signal transduction at viral entry | |||||||||||||||
| Liu et al. (2011) | HIV-189.6R | HeLa-CD4 | Arrayed | QIAGEN human whole genome siRNA Set V4.0 (19,121 genes) | 72 h | 48 h | % Infectivity (GFP expression) | No | N/A | Yes | GFP + Foci > 3 SDs from mean | PAF1 complex | Innate defense | Network pathway analysis (IPA); individual siRNAs; WT viral strains NL4-3, 89.6wt; mRNA levels; Western blot; cell lines MDMs, CD4 + T cells; qPCR | ||
| HIV-18.2N | 192 candidates—114 validated | SETDB1 | Preintegration | |||||||||||||
| Espeseth et al. (2011) | HXB2 HIV | HeLa P4/R5 | Arrayed | siRNA DNA repair factor library | 24 h | 48 h | β-galactoside activity | Yes | Inhibition > 40% | No | N/A | Base-excision repair pathway | Integration | cDNA rescue; lifecycle assays; qPCR; flow cytometry; GO annotation; cell line: murine embryonic fibroblasts (MEFs) | ||
| 41 siRNA pools | ||||||||||||||||
| Le Sommer, Barrows, Bradrick, Pearson, and Garcia-Blanco (2012) | Yellow Fever virus YF-17D |
Huh-7 | Arrayed | QIAGEN human genome library (22,909 genes) | 51 h | 42 h | % Infectivity (4G2 antibody) | Yes | Decrease % infection twofold | No | N/A | GRK2 | Entry | Individual siRNAs; comparison to WNV + DENV screens; Western blot; other cell lines: MEFs; other virus: DENV-NGC, HCV-JFH1; qRT-PCR; lifecycle assays | ||
| 395 hits—98 candidates | Genome amplification | |||||||||||||||
| Dziuba et al. (2012) | HIV-1 strain LAV | CD4 +/CCR5 +/CXCR4+TZM-bl | Arrayed | Dharmacon siRNA SMARTpool custom library of trapped genes | 48 h | 48 h | HIV-1 p24 capsid production | Yes | 50% inhibition | No | N/A | GTF2E1 | Tat-dependent gene transcription | Rescue experiment; infectivity of surviving clones; Western blot; individual siRNA; RT-PCR; ELISA; other viral strains: SF162, ADA, 89.6 HIV-1; pathway analysis | ||
| DHX8 | Release of spliced mRNA | |||||||||||||||
| UBA3 | Modification of HIV-1 proteins | |||||||||||||||
| KALRN; HAP1 | Protein trafficking | |||||||||||||||
| Arita, Wakita, and Shimizu (2012) | PV pseudovirus | HEK293 | Arrayed | Thermo Scientific human membrane trafficking gene library | 96 h | 7 hr | Luciferase activity | Yes | Strongest novel hit | No | N/A | VCP | Viral RNA replication | Rescue KD with mutant protein; immunofluorescence microscopy; immunoprecipitation; Western blot; two-hybrid assay; PLA; PV mutant resistant to KD | ||
| Mercer et al. (2012) | Vaccinia virus VACV-EGFP |
HeLa | Arrayed | QIAGEN druggable genome (7000 genes) | 72 h | 8 h | % Infectivity (GFP) | Yes | Median absolute deviation <−1.5 | No | N/A | Proteasome subunits | Late viral gene expression | Reagent redundancy; functional annotation clusters; protein interaction analysis; immunofluorescence; lifecycle assay; small molecules: MG132, UBEI-41, cytosine arabinoside; Western blot | ||
| Cullin 3 | vDNA replication | |||||||||||||||
| Ward et al. (2012) | Influenza A virus IAV A/WSN/33 |
HBEC30-KT | Arrayed | Dharmacon library (21,125 genes) | 48 h | 48 h | Luciferase assay | Yes | 3 SDs below mean | Yes | 3 SDs above mean | CDC2; CHEK1 | Viral production | Network analysis; comparison to other screens; literature review; plaque assay; small molecule: SB218078, 3-IPEHPC; Western blot; immunofluorescence; other cell line: A549 | ||
| 182 candidates | 53 candidates | |||||||||||||||
| Ooi, Stiles, Liu, Taylor, and Kielian (2013) | Sindbis virus SINV-Luc |
U2OS | Arrayed | Ambion Silencer human genome siRNA library V3 (21,687 genes) | 48 h | 24 h | Luciferase intensity | Yes | Robust Z score < − 3 | Yes | Robust Z score > 2 | FUZ | Viral uptake | Individual siRNAs; individual shRNAs; multicycle infectivity assay; other cell lines: HeLa, primary endothelial cells; other viruses: SFV, CHIKV, VSV, DENV; immunofluorescence lifecycle assays; fusion assay; endocytic pathway assay; quantigene analysis of mRNA | ||
| 400 genes | 59 genes | TSPAN9 | Viral fusion | |||||||||||||
| Sivan et al. (2013) | Vaccinia virus VACV IHD-J/GFP |
HeLa | Arrayed | Ambion Silencer Select human genome siRNA library (21,500 genes) | 48 h | 18 h | % Infectivity (GFP + cells) | Yes | <−1.5 median absolute deviation; < 50% reduction in cell number | Yes | <−1.5 median absolute deviation; < 50% reduction in cell number | NUP62 | Conversion of immature virion to mature virion | Gene network analysis (IPA); gene ontology (GO); common seed analysis; individual siRNAs; rescue experiment; Western blot; lifecycle evaluation; viral gene expression; TEM | ||
| Dharmacon siGENOME SMARTpool siRNA (18,120 genes) | 576 genes | 530 genes | ||||||||||||||
| Fusco et al. (2013) | Hepatitis C virus HCV-JFH1 |
Huh7.5.1 | Arrayed | Dharmacon siGENOME pooled siRNA library | 72 h | 48 h | % Infectivity (HCV anti-core antibody) | Yes | ≥ 3 × median absolute deviation | Yes | ≥ 3 × median absolute deviation | 12 interferon effector genes | Various | Western blot; qRT-PCR; shRNA KDs; overexpression; microarray analysis | ||
| Panda et al. (2013) | Sindbis virus SINV (HRsp) |
DL1 | Arrayed | Ambion Drosophila genome wide | 72 h | 36 h | % Infectivity (GFP) | Yes | Robust Z score < − 2; < 40% viability decrease | Yes | Robust Z score > 2; < 40% viability decrease | SEC61A | Entry/early stage | Gene ontology (GO) enrichment analysis; dsTE12H strain; independent dsRNAs; small-molecule Eeyarestatin 1, NH4Cl; Western blot analysis; in vivo assay; localization microscopy | ||
| 57 genes validated | 37 genes validated | VCP | ||||||||||||||
| Lavanya, Cuevas, Thomas, Cherry, and Ross (2013) | Junin virus GP pseudotyped Moloney Leukemia virus MLV-Lac-Z |
U2OS | Arrayed | Ambion druggable genome RNAi library | 72 h | 48 h | % Infectivity (anti-Lac-Z) | Yes | Robust Z score ≤ − 1.5; viability Z score decrease < 2 | Yes | Robust Z score ≥ 1.5; viability Z score decrease < 2 | CACNA2D2 | Entry | Independent siRNAs; luciferase assay; RT-qPCR; small molecules—U73122, U73343, BCECF-AM, BAPTAAM, gabapentin, nifedipine, verapamil, bafilomycin A; binding assay; in vivo assay C57BL/6 mice; molecular function (GO) analysis for enrichment; KD-related proteins | ||
| 89 genes | 13 genes | |||||||||||||||
| Hopkins et al. (2013) | Rift Vallety Fever virus RVFV (MP12) |
DL1 | Arrayed | Ambion genome-wide dsRNA library (13,073 genes) | 72 h | 30 h | % Infectivity (anti-RVFV N) | Yes | Robust Z score ≤ − 1.3; viability Z score > − 2 | Yes | Robust Z score ≥ 1.3; viability Z score > − 2 | Dcp2 | Decapping | Other RNA viruses DCV, SINV, LACV, VSV; colocalization; in vivo infectivity; Northern blot; RT-PCR; Aag-2 cells; Western blot | ||
| 7 validated genes | 124 validated genes | |||||||||||||||
| Zhu et al. (2014) | HIV-1-IIIB | P4-P5 MAGI cells | Arrayed | Ambion Silencer Select (21,584 siRNA pools) | 72 h | 48 h | % Infection (anti-p24 capsid antibody) | Yes | Infectivity ≤ 50%; viability ≥ 50% | Yes | Infectivity ≥ 200%; viability ≥ 50% | UMPS; ATIC; RRM | Pyrimidine and purine metabolism | MORR analysis; RIGER analysis; gene expression filtering; literature comparison; reagent redundancy; enrichment analysis ConsensusPathDB-human; microarray analysis; genome-wide enrichment of seed sequence matches (GESS); network analysis; lifecycle assays | ||
| Sigma esiRNA (15,300 siRNA pools) | THOC2 | Replication | ||||||||||||||
| COG complex | Glycosylation | |||||||||||||||
| Dharmacon SMARTpool RefSeq27, Revision Human 5 (4506 siRNA pools) | GOLGI49 | Entry | ||||||||||||||
| SEC13 | Nuclear | |||||||||||||||
| Yasunaga et al. (2014) | West Nile virus WNV |
DL1 | Arrayed | Ambion Drosophila library (13,071 genes) | 72 h | 48 h | % Infection (anti-WSN-NS1) | Yes | Robust Z score < − 2; Z score < − 2 | Yes | Robust Z score > 2; Z score < − 2 | dRUVBL1 | Antiviral | Repeat for validation with dsRNA against different region of gene; other viruses: WNV-KUN, DENV, SINV, VSV, RVFV MP12; functional annotation and clustering using DAVID bioinformatics resource; in vivo assay; Northern blot; RT-qPCR; small molecule: Leptomycin B, dichloroacetic acid, hexokinase II; other cell lines U2OS, Aag-2 | ||
| 376 genes | 161 genes | dXPO1 | Innate immune response | |||||||||||||
| Balistreri et al. (2014) | Semliki Forest virus SFV-ZsG |
HeLa | Arrayed | Dharmacon human ON-TARGET plus (4 pooled siRNAs/gene) | 72 h | 6 h | % Infection (Zoanthus species G, ZSG) viability (Hoechst) | No | N/A | Yes | Top hit | UPF1 | Early cytosolic | Specific validated shRNA; Western blot analysis; rescue with shRNA-resistant UPF1; immunofluorescence microscopy of viral components | ||
| Wen, Ding, Hunter, and Spearman (2014) | HIV-1 NL4-3-EGFP |
HeLa | Arrayed | Dharmacon-Thermo Fisher cellular membrane trafficking genes (140 genes) | 24 h | 48 h | Particle production in supernatants | Yes | Particle output < 50%; viability > 60% control | No | N/A | 24 genes overlap | Particle production | STRING—Search tool for retrieval of interacting genes; shRNA validation; Western blot analysis | ||
| Mason-Pfizer monkey virus pSARMX-EGFP + pTMO-Env | Cos-1 | 24 overlap hits; HIV-1 NL4-3 41 candidates (8 known); pSARMX 52 candidates |
||||||||||||||
| Kwon et al. (2014) | Dengue virus DENV2 (BR DEN2 01-01) |
Huh7 | Arrayed | Dharmacon siGENOME kinase library (G-003500-05) (779 genes) (4 siRNA/gene) (2 siRNAs/well) | 48 h | 48 h | % Infection (4G2 antibody) | Yes | − 2 standard deviations of mean | Yes | + 2 SDs from mean | SHPK | Macrophage polarization | 8 candidates—6 cherry picks; individual siRNAs; U937 DC-SIGN cell line; flow cytometry; gene expression analysis; qRT-PCR | ||
| ETNK2 | Entry/cellular trafficking | |||||||||||||||
| 22 candidates —6 cherry picks | 8 candidates —6 cherry picks | EIF2AK | Unfolded protein response | 22 candidates—16 cherry picks—6 validated; individual siRNAs; Western blot; flow cytometry; U937 DC-SIGN cell line; gene expression analysis; qRT-PCR | ||||||||||||
| SMAD7 | Prolong cell survival | |||||||||||||||
| Pohl, Edinger, and Stertz (2014) | Influenza A virus IAV VLP |
A549 | Arrayed | Custom library (169 siRNAs) | 48 h | 30 h | Renilla luciferase | Yes | 2 siRNA 50% reduction in infection, cell viability 70% | No | N/A | PEPD | Early endosomal block | Control VLPs (LASV and MLV); compare to previous screens; Western blotting; WT virus (A/WSN/33); strains: FPV/Dobson (H7N7), A/Hong Kong/68 (H3N2), A/Netherlands/602/2009 (H1N1), A/Panama/2007/99 (H3N2); WI38 primary cells; cell cycle assay; fusion assay; colocalization | ||
| 43 candidates—22 related to entry | ||||||||||||||||
| Beard et al. (2014) | Vaccinia virus VACV-A5eGFP |
HeLa | Arrayed | Dharmacon druggable genome siRNA SMARTpool library (6719 genes) (4 siRNAs/gene) | 48 h | 48 h | Infection (GFP fluorescence) | Yes | eGFP ≤ − 2 Z score; cell number > − 2 SDs from plate mean | Yes | eGFP ≥ 2 Z score; cell number > − 2 SDs from plate mean | AMPK | Regulation actin cytoskeleton | RT-PCR; individual siRNAs; comparison to known data; transcriptional profiling comparison; pathway analysis | ||
| 153 candidates—35 cherry picks—24 validated | 149 candidates—24 cherry picks—7 validated | Septins; MAZ; DNA replication/repair pathway | Unknown | |||||||||||||
| Lee, Burdeinick-Kerr, and Whelan (2014) | Vesicular Stomatitis virus rVSV-EGFP |
HeLa | Arrayed | Dharmacon SMARTpools (21,121 pools) | 48 h | 7 h | % Infectivity (EGFP +); EGFP intensity | Yes | > 3.0 SDs from mean for % infected or intensity; < 3.0 SDs alteration for viability | No | N/A | GPR149 | Entry | Individual siRNAs; Western blot; RNP cores | ||
| 405 candidates—305 confirmed—29 further evaluated | PSCA | Entry | ||||||||||||||
| Aydin et al. (2014) | Human Papillomavirus HPV16-GFP |
HeLa MZ | Arrayed | Qiagen druggable genome version 2 + siRNA#3 from Qiagen druggable genome version 3 (6979 genes) | 60 h | 36 h | % Infectivity (GFP) | Yes | Reduction in Z score > 3 | Yes | Increase in Z score > 3 | AURKB; ANAPC; INCENP | Mitosis regulators | Reagent redundancy; literature review; enrichment analysis; network analysis; lifecycle assay; other cell lines primary human keratinocytes; small molecules: aphidicolin, CPG74514A, NH4Cl; localization assays; immunofluorescence analysis | ||
| Schreiber et al. (2015) | Adeno-associated virus AAV9 CMV-Luc |
HeLa | Arrayed | SMARTpool siRNA library: Human siGENOME ubiquitin conjugation subsets #1 (89 genes), #2 (115 genes), and #3 (396 genes) | Unknown | 48 h | Luciferase expression | No | N/A | Yes | 10-fold increase | PHF5A; RAB40B; PRICKLE4 | Transduction efficiency | 12 candidate genes—3 confirmed hits: Verification with distinct siRNAs and lenti-shRNAs; rescue with PHF5A-HA-escape vector; small-molecule meayamycin B; immunoprecipitation | ||
| Sivan, Ormanoglu, Buehler, Martin, and Moss (2015) | Vaccinia virus VACV C7L−K1L −/+ GFP |
HeLa; BS-C-1 | Arrayed | Ambion Silencer Select genome siRNA library version 4 (~ 21,500 genes) (3 siRNA/gene) | Unknown | 18 h | % Infection (GFP) | No | N/A | Yes | 4 siRNAs > 3% GFP+ cells | SAMD9; WDR6; FTSJ1 | Unknown | Immunoprecipitation; CRISPR/Cas9; rescue of CRISPR; Western blotting | ||
| Arrayed | Dharmacon On-Target Plus SMARTpool siRNA (17,320 genes) (4 siRNAs pooled/gene) | |||||||||||||||
| de Wilde et al. (2015) | SARS-Coronavirus SARS-CoA-GFP | 293/ACE2 | Arrayed | Dharmacon ON-TARGET plus SMARTpool protein kinases siRNA library (779 genes) (4 siRNAs pooled/gene) | 48 h | 24 h | GFP expression | Yes | Proviral hits < 50% control; normalized viability > 0.85 | Yes | Antiviral hit > 150% control; normalized viability > 0.85 | PKR | Translation initiation | Individual siRNAs; Western blot; 90 candidates—mapped to cellular pathways | ||
| 90 candidates | 40 candidates | COPB2 | COPI-coatomer | Specific shRNAs; viral protein expression; KD of related/complex proteins; 40 candidates—mapped to cellular pathways | ||||||||||||
| PRKCι | Unknown | Small-molecule sodium aurothiomalate; 40 candidates—mapped to cellular pathways | ||||||||||||||
| Williams, Abbink, Jeang, and Lever (2015) | HIV-1 VSV-G pseudotyped |
HeLa | Arrayed | Library against 59 RNA helicases (3 siRNAs/gene) | Unknown | 96 h | Intracellular p24 capsid levels; infectious virion production; luciferase expression | Yes | Decrease all 3 parameters > 20% | No | N/A | DDX5; DDX10; DDX17; DDX28; DDX52 | Viral replication | Cherry picks screened with WT-HIV-1 (pLAI) virus; Western blot; cell viability | ||
| 48 candidates—42 repeat—8 cherry picks—5 confirm WT-HIV-1 | ||||||||||||||||
| Poenisch et al. (2015) | Hepatitis C virus JcR2a |
Huh7.5 Firefly luciferase | Arrayed | Ambion Silencer Select extended druggable genome library V3 (9102 genes) (3 siRNAs/gene) | 48 h | 72 h | Luciferase expression; production | Yes | <−2 Z score for 2/3 siRNAs | Yes | > 2 Z score for 2/3 siRNAs | HNRNPK | Entry/early replication | Meta-analysis with other studies; Dharmacon validation screen; pathway enrichment analysis; known to interact with virus core and related proteins; RT-qPCR; IF/subcellular localization | ||
| 78 candidates—40 validate | 29 candidates—16 validated | Production | ||||||||||||||
| 263 siRNA pools | 130 siRNA pools | |||||||||||||||
| Perreira et al., (2015) | Human Rhinovirus HRV14 |
HeLa-H1 | Arrayed | SMARTpool Dharmacon (21,121 pools, 3 oligos/pool) | 72 h | 14 h | % Infectivity (antibody to HRV14 V1 CA protein) | Yes | Infectivity < 50%; viability > 40% | Yes | Infectivity > 150%; viability > 40% | RNASEK | Entry | MORR analysis; RIGER analysis; gene expression filtering; pathway/complex enrichment analysis; other viral analysis IAV (X31H3N2) (WSN/33), DENV (2, 3, 4), YF17D, MLV-VSV, HIV-1-IIIB, MLV-CMV; lifecycle assay; mass spec; immunoprecipitation; acidification studies; immunofluorescence assay; cellular localization assay | ||
| Arrayed | Ambion Silencer Select (21,584 pools, 3 oligos/pool) | |||||||||||||||
| Arrayed | Sigma esiRNA (15,300 siRNA pools, complex pools) | |||||||||||||||
| Arrayed | Dharmacon RefSeq27 Revision Pools (4506 siRNA pools/4 oligos/pool) | |||||||||||||||
| shRNA | Yeung, Houzet, Yedavalli, and Jeang (2009) | HIV-1 NL4-3 | Jurkat | Pooled | SBI Feline immunodeficiency virus vector-based shRNA library (54,509 transcripts) | 1 week | 4 week | Survival | Yes | Survival | No | N/A | NRF1 | Entry—Affects co-receptor CXCR4 | Reagent redundancy; individual shRNAs; pathway analysis; qPCR; flow cytometry; lifecycle assay | |
| STXBP2 | Viral reverse transcription | |||||||||||||||
| PRDM2; NCOA2 | Transcription | |||||||||||||||
| EXOSC5 | Gag-trafficking | |||||||||||||||
| Su et al. (2013) | Influenza A virus IAV A/WSN/33 |
A549 | Pooled | TRC RNAi Consortium (81,925 shRNAs) (16,368 genes) | 5 days | 2 weeks | Survival | Yes | Survival with 2 unique shRNAs per gene | No | N/A | Itch | Exit endosomes | Western blot; immunofluorescence; RT-qPCR; cellular localization; ubiquitin assay; EST analysis; microarry analysis | ||
| 110 genes—38 selected | ||||||||||||||||
| Tran et al. (2013) | Influenza A virus IAV A/NY/55/2004 |
A549 | Pooled | 7 decode RNA GIPZ lentiviral positive screening library pools (Thermo) | 48 h | 72 h | Survival | Yes | Survival | No | N/A | TNFSF12-13; TNFSF13 | Late viral replication | Reagent redundancy; RT-qPCR; viability; lifecycle assay; immunofluorescence; flow cytometry; Western blot; other viruses: PR8 (H3N2), pandemic California (H1N1); GO analysis | ||
| 1256 candidates—127 selected | 20 confirmed | USP47 | Entry | |||||||||||||
| CRISPR/Cas9 | Ma et al. (2015) | West Nile virus WNV |
293FT | Pooled | Custom array library oligo pool—PCR amplified-cloned into plasmids—lentiviral vectors—transduced—transfected with Cas9 | Expansion time | 12 days | Survival | Yes | Multiple independent sgRNAs | No | No | EMC2 | WNV-induced death | sgRNA sequences amplified w/nested PCR + sequenced; Western blot; flow cytometry; other viruses WNV-NY99, SLEV | |
| 28,429 sgRNAs with reads more than 10 identified | EMC3 | |||||||||||||||
| SEL1L | ||||||||||||||||
We searched the literature for large-scale genetic screens using human viruses (or components of human viruses) and any of the three functional genomic screening strategies covered in this review. We then provided some of the major characteristics of each individual screen, including the virus, cell line, format, library, screen timelines, selection criteria, any main candidate focused upon, and the assays used for follow up and mechanistic validation if applicable. Not applicable (N/A).
3. RNAi Genetic Screening Technologies and Approaches
Nearing a decade ago the Nobel Prize winning discovery of RNAi in C. elegans and its mercurial extension into mammalian systems provided virologists and geneticists alike with a powerful new tool for detecting viral dependencies (Elbashir et al., 2001, Fire et al., 1998, Grishok and Mello, 2002). Academia and industry both quickly embraced RNAi and paired it with the contemporaneous completion of the genetic annotation of the entire human genome to create multiple large-scale libraries for functional genomic screening (Paddison et al., 2004, Root et al., 2006, Silva et al., 2005). Because the RNA-induced silencing complex (RISC) machinery's expression is ubiquitous, virtually all mammalian cell lines can carry out RNAi, permitting host–virus screens to be carried out with any tropic cell line and virus pairing (Elbashir et al., 2001). Two major types of RNAi libraries, pooled and arrayed, have been constructed and dictate the two methods of screening discussed below.
3.1. RNAi Pooled Screening
Retroviral expression of complex cDNA libraries in tissue culture cells predated the arrival of RNAi and was readily adapted to stably express short hairpin RNAs (shRNAs) that were subsequently processed into dsRNAs suitable for directing the destruction of target mRNAs by RISC. Three major pooled retroviral shRNA libraries were initially constructed, the Hannon–Elledge Open Biosystems shRNA library (Paddison et al., 2004, Silva et al., 2005), the RNAi Consortium (TRC) library (Root et al., 2006), both of which are lentiviral and have whole-genome coverage, and a smaller subgenomic gamma-retroviral library, the Bernards shRNA library (Berns et al., 2004), with additional libraries following (Boettcher & Hoheisel, 2010). While differing in their design (Hannon–Elledge-OB being comprised of microRNA-context shRNAs vs. TRC and Bernards being made up of simple shRNAs) these reagents all produce siRNAs resulting in alterations in target gene mRNA expression. Each gene is typically targeted by three or more distinct shRNAs resulting in library complexities of 100K + unique shRNAs. These pooled shRNA retroviral vectors are then packaged into complex populations of retroviruses (Fig. 1 ). A population of cells is transduced with the retroviral pools and then the cells are placed under selection to identify any modulations in viral replication conferred by the integrated provirus shRNA. For all pooled library screens, a key point is that each distinct shRNA vector should be over-represented by ≥ 1000-fold in the selected cell population to minimize bottle neck effects during the screening process; this tenet is also important for the pooled CRISRP/Cas9 screens to be discussed below.
Figure 1.
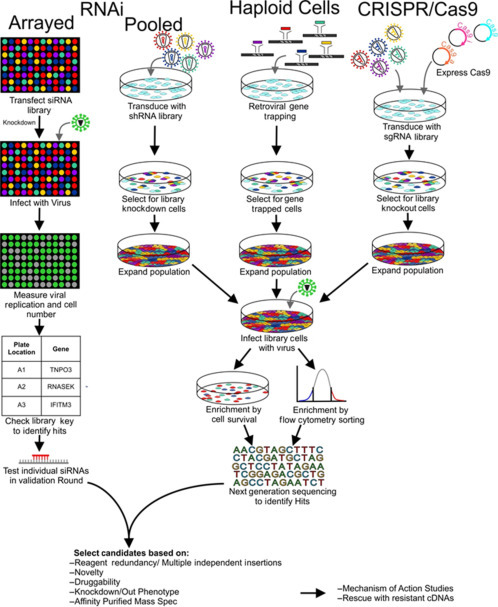
Functional genomic strategies for elucidating host–virus interactions. Schematic of the workflow for each of the three functional genomic screening strategies discussed in this review, RNAi (left) using either arrayed (siRNA) or pooled (shRNA) approaches, haploid cells with retroviral gene trapping (haploid cells, middle), and CRISPR/Cas9, using conventional catalytic (Cas9), CRISPR activators (CRISPRa, Cas9a), or CRISPR repressors (CRISPRi, Cas9i, right). Typical validation and mechanistic studies are outlined at bottom.
Pooled shRNA screens for host–virus interactions include an early effort to identify HIV-1 host factors required for replication in a T cell line, as well as two screens for IAV host factors (Su et al., 2013, Tran et al., 2013, Yeung et al., 2009). Advantages of pooled screening are its relative low cost and the higher knockdown efficiencies realized using retroviral transduction of cell types that are not readily transfected with siRNAs, e.g., primary cells or suspension cells. In addition longer term screening assays that may require weeks to run are best performed with stably expressed shRNA libraries since transient transfection of siRNAs in dividing cells peaks and falls quickly > 7 days posttransfection. The lack of published pooled shRNA screens for virus–host interactions is noticeable and likely stems from the limitations in readout when using a pooled strategy, as well as the issue of phenotypic penetration in the setting of partially decreased gene expression or hypomorphism. Two prevailing readouts have been used for pooled shRNA screening, flow cytometry-based sorting of cell populations, e.g., high and low expression of viral proteins or a fluorescent marker protein, as a surrogate for infection, as well as survival screens where a cytopathic virus destroys all of the cells that it can infect and spares any cells which are missing a critical host factor, with the survivors undergoing expansion and gene enrichment. The complete loss of gene expression (null phenotype) is unlikely to be achieved using RNAi, and in particular in a population of cells stably transduced with complex shRNA library. This stems from each cell in the screened population expressing only a single shRNA-expressing provirus. Even if a cell is transduced by more than one shRNA-expressing virus, it is highly improbable that both shRNAs will have the same target. It is difficult for a single proviral shRNA to have enough expression to efficiently deplete the mRNA for its intended target. Accordingly, a pooled shRNA screen using a cytopathic virus and cell survival as a means of gene enrichment might not find the host receptor for the virus because there will be some low level of receptor expression remaining (hypomorphism) that could render the cell susceptible to infection and death.
Detecting the shRNAs enriched for at the end of a pooled screen is done using next-gen sequencing technologies which specialize in short reads, combined with informatics programs such a bowtie to assign and quantitate the number of sequencing reads per shRNA in comparison to the starting population. Candidates are selected for follow up based on novelty and on the reagent redundancy principle which states that the likelihood of a gene being a true positive increases as the number of enriched orthologous shRNAs targeting that gene increases (Echeverri et al., 2006). For example, a gene targeted by three independent shRNAs that are enriched in the next-gen sequencing readout is more likely to be a true positive than a gene targeted by only one enriched shRNA. As we will see, the reagent redundancy principle is also important for selection of candidates using all of these functional genomic screening strategies, including the haploid cell screens (number of independent retroviral insertions) (Carette et al., 2009).
3.2. Arrayed RNAi Screening
The high-throughput transfection of arrayed cDNA libraries into mammalian cells for screening predates RNAi and this approach was readily emulated once large-scale arrayed RNAi reagents and appropriate transfection lipids were developed. Pioneering work defining human pathogen interactions was done first using insect cell lines and arrayed siRNA libraries targeting the Drosophila mRNA transcriptome (Cherry, 2011, Hao et al., 2008, Sessions et al., 2009). Advantages in using the Drosophila system are that the insect cells take up the siRNAs without the need for transfection reagents and that their simpler genetic repertoire may lack functional redundancies which could resist resolution in the more complex human system. Obvious shortcomings are that the findings in the fly cell screens require confirmation in human cells by targeting homologs and that there are human pathogenic viruses that cannot infect fly cells. Thus, a need arose for arrayed RNAi reagents for investigating human pathogenic cells using a human cell-based in vitro system. This need was addressed by four life sciences companies; Dharmacon, Ambion, Sigma, and Qiagen, which each introduced their own independently designed whole-genome siRNA libraries.
Methods for performing an arrayed siRNA library screen have been reviewed by us and others in detail elsewhere (Barrows et al., 2014, Chin and Brass, 2013, Panda and Cherry, 2015). Briefly, the project begins with optimizations of both siRNA transfection and infection conditions in the plate format chosen for the screen, with 384-well plates being strongly preferred due to lower amounts of siRNA library needed and the decreased costs and work load using this smaller scale. Once optimized the screen begins with the transfection of the arrayed library in either duplicate or triplicate (Fig. 1); this is usually done in a reverse transfection format with the siRNAs and lipid mixture added to the well first, followed by the cells added in suspension. Target mRNA depletion and decreased protein expression occurs over 1–4 days depending on assay conditions. The longer knockdown periods prior to viral challenge likely improve the observed phenotypes because of increased levels of target protein decay and the dilution effect of added cell divisions. The siRNA-transfected cells are then infected with virus for typically one or two viral lifecycles followed by an assessment of viral replication using either a microscope or plate reader. After the primary arrayed whole-genome screen, the individual siRNAs in the pools of select candidate genes are then rescreened individually in the validation round and the reagent redundancy principle used to select higher confidence genes for follow up.
Arrayed siRNA screening has several advantages over a pooled shRNA approach. For instance, employing an arrayed siRNA library permits shorter term transient transfection-based screens (Fig. 1; Table 2 ). Additionally the introduction of large effective concentrations of siRNAs into the cells using high efficiency lipid-mediated transfection improves target mRNA depletion producing enhanced phenotypic penetrance. Moreover, by depleting just one-gene-per-well an arrayed screen permits the selection of candidate genes based on more subtle gradations in phenotypes than when using pooled screening readouts. For instance using this format, readouts of viral protein expression, or the expression of a luciferase reporter gene, can be assessed with great sensitivity using high-throughput microscopes or plate readers. Having each gene targeted in its own designated well also creates a homogenously genetically altered population of cells that can be assessed using high content imaging, thus allowing cell biology phenotypes involved in host virus interactions (i.e., RNA virus replication complex morphology) to be screened for in great detail, something which is not possible using a pooled screening strategy. Last, using arrayed annotated libraries allows the immediately identification of which gene may underlie the observed phenotype. Disadvantages of using such an approach include the increased expense of having to purchase, array and maintain these large-scale resources, the analytical machinery needed to carry out and analyze the great number of plates produced by the screen, and the added costs for transfection and screening reagents. Finally, both the siRNA and shRNA screens have major limitations due to their high rates of false positives and false negatives; this last concern regarding the significant caveats of siRNA screening, as well as some corrective measures, are more fully discussed below.
Table 2.
Strengths and Weaknesses of Functional Genomic Screening Strategies for Human–Virus Interactions
| RNAi Arrayed (siRNA) | RNAi Pooled (shRNA) | Haploid Cells Pooled | CRISPR/Cas9 Pooled | |
|---|---|---|---|---|
| Strengths |
|
|
|
|
| Weaknesses |
|
|
|
|
The original Dharmacon arrayed human siRNA library, siGENOME, consists of pools of four 19-mer siRNAs (SMARTpools) designed against each of the 21,141 annotated human genes in RefSeq5–8, one gene per well. A later version, On-target-plus (OTP), was similarly constructed but with selective modification of some of the siRNA's base pairs with the intent of minimizing OTEs created by the first eight base pairs of the antisense, the seed sequence, or the sense-strand pairing with microRNA elements thereby unintentionally altering gene expression. Although useful, the antisense OTP reagents likely have a lower affinity for their intended targets which may explain their loss of efficacy compared to matched siGENOME reagents tested side-by-side for depletion of known positive controls (our unpublished data). An updated SMARTpool siGENOME library based on Refseq27 (Dharmacon 6–16) was constructed in a similar manner and has recently replaced the earlier library. An advantage of the SMARTpool library is that four siRNAs are available for validation round screening. A shortcoming is that the available siRNAs for reorder postscreening are continually changing over making it costly to order the exact siRNAs that scored in the original screen.
The Ambion Silencer Select library targets 21,584 genes using three siRNAs in an arrayed format, one siRNA per well with three total wells for each gene. The arrayed library can be readily converted to pools based on the way it is plated, with the same well on three matching plates (A, B, C) containing a different siRNA targeting the same gene. An advantage of individual siRNA arrayed screening is that candidate selection for follow up can be done immediately after the primary screen based on reagent redundancy, the disadvantage is that three times more reagents are needed to screen the individual siRNA arrayed Silencer Select library. Importantly, Silencer Select siRNAs mark a major advancement in siRNA design as they incorporate locked nucleic acids (LNAs) which increase antisense strand binding affinity to designed targets and inhibit sense-strand binding thereby decreasing OTEs (Puri et al., 2008). As with the SMARTpool library the three individual siRNAs available for the validation round are useful and Ambion maintains a consistent supply of the library oligos that can be reordered, with new potentially improved siRNAs being added without replacing the original library set.
Endonuclease processed siRNA (esiRNA) pools against most human genes are available individually as well as in genome-wide libraries from Sigma. esiRNA pools were originally developed by the Buckholz lab and consist of complex heterogeneous mixtures of overlapping siRNAs (18–25 base pairs in length) targeting the same mRNA sequence (Kittler et al., 2007). esiRNA pools are created using endoribonuclease to digestion of RNA transcribed in vitro from 200–400 base pair cDNA templates. Using this strategy concentration-dependent OTEs are anticipated to be less than using conventional siRNA pools or individual oligos. Since the pools cannot be deconvoluted into a few known components, validation is carried out using a distinct esiRNA pool against the same gene. While useful this approach is limited in terms of its level of reagent redundancy. Furthermore, although the relative concentrations of the individual esiRNA pools in the library are closely matched, the final sizes of the digested product vary leading to an induction of dsRNA-mediated antiviral response that precludes their use with some viruses which are vulnerable to such a defense, e.g., dengue virus.
3.3. RNAi Screening Problems and Some Solutions
RNAi screens are powerful and readily implemented discovery tools but suffer from shortcomings arising from their high levels of false negatives and false positives (OTEs) as can be seen when comparing the low concordance among the candidate genes detected in different screens using the same species of virus, e.g., HIV-1, HRV, or IAV (Booker et al., 2011, Bushman et al., 2009, Hao et al., 2013, Perreira et al., 2015, Zhu et al., 2014). To address these concerns, improvements in the design and synthesis of next-gen RNAi library reagents have been implemented including the elimination of siRNAs with seed sequences that are complementary to microRNA binding sites (Knott et al., 2014, Mohr and Perrimon, 2012, Petri and Meister, 2013). As noted, the seed sequences of the nontargeting siRNA sense strands have had their binding affinity decreased by selectively incorporating methylated or LNA nucleotides. Significant efforts have also been put into validating the siRNAs to find and remove ones that are ineffective and contribute to false negatives.
OTEs in particular must be rigorously controlled for by using reagent redundancy combined with complementation or rescue experiments and an assessment that target depletion and phenotype are proportional (Echeverri and Perrimon, 2006, Echeverri et al., 2006, Mohr and Perrimon, 2012). While a consistently low number of exact genes overlap across related siRNA screens, it is nonetheless clear that similar screens find bioinformatically related genes, e.g., genes that cluster in common pathways and complexes like the nuclear pore complex (NPC) with HIV-1 and the vacuolar ATPase (V-ATPase) for IAV or HRV (Bushman et al., 2009, Hao et al., 2013, Perreira et al., 2015, Stertz and Shaw, 2011, Zhu et al., 2014). With closer study it became readily apparent that this low level of saturation within the dataset of each primary screen was due to a high level of false negatives (Hao et al., 2013, Meier et al., 2014, Zhu et al., 2014). False negatives with RNAi may come about for several reasons including difficulty in targeting a protein (prolonged protein half-life or sufficient remaining catalytic activity), nonspecific toxicity of siRNAs, and plate edge effects. These interscreen comparisons also highlight the importance of a post hoc bioinformatic analysis across multiple related screens (meta-analysis) to provide a systems level understanding of viral dependencies. Additionally, candidate genes that score poorly in reagent redundancy validation assays, e.g., only confirming the phenotype with one of four possible siRNAs, are more likely to represent true positives if they physically or functionally interact with candidate genes that are members of enriched clusters. Consequently, bioinformatics can find useful associations that may save a potentially informative candidate gene from down selection.
RNAi screens have revealed the host cell requirements of many human viruses (Table 1), however, they are beset by false positives and false negatives. We reasoned that by using multiple orthologous RNAi reagents (MORR) in parallel we could take advantage of each large-scale reagent's best characteristics while minimizing their worst. With this in mind, we used MORR screens (Silencer Select, SMARTpool, and esiRNA libraries) to identify high-confidence HIV-1 dependency factors (HDFs) or HRV host factors (HRV-HFs) (Perreira et al., 2015, Zhu et al., 2014); these three libraries are > 90% orthologous based on a comparison of siRNA sequences. We then traditionally validated the candidates from each of the primary screens. In addition, we integrated the primary MORR datasets, and those of earlier studies in the case of HIV-1, by adapting an established analysis method, RNAi gene enrichment ranking (RIGER) (Luo et al., 2008). RIGER uses a weighted likelihood ratio to calculate a gene-specific enrichment score based on the rank distribution of each individual RNAi reagent across all of those screened. The RIGER enrichment score is expressed as a p value assigned to each gene which represents the likelihood that the gene plays a role in viral replication. By integrating the entire primary screen datasets RIGER also decreases false negatives created by the combination of hypomorphism and the use of absolute cutoffs for candidate selection. Both these projects represented two of the most comprehensive siRNA screening efforts to date and produced quantitatively integrated datasets for each virus which highly ranked both known viral dependency factors and previously unappreciated ones. To assess if MORR/RIGER improves the yield from the screen as compared to a more traditional screening approach, we assessed each respective dataset (RIGER (all screens integrated) and each of the individual MORR screens) for their enrichment of a set of annotated gene complexes or pathways. The annotated gene sets were selected because there was significant enrichment of their components across the individual screens (e.g., the NPC for HIV-1 or the 80S ribosome for HRV (Perreira et al., 2015). These comparative enrichment analyses quantitatively demonstrated that the MORR/RIGER approach produces a data set which is statistically better in its enrichment for expected host factors than any of the individual screens on their own. Since this approach is more sensitive and specific in finding known host factors, we conclude that it would also be the best method for detecting previously unappreciated host–virus interactions.
To further improve siRNA screening, we and others have decreased OTEs by using the method of gene expression filtering to remove candidates that are not found to be expressed in the cell line used for the screen based on either microarray assays or next-gen sequencing (Perreira et al., 2015, Zhu et al., 2014). OTEs in siRNA screens are also detected and removed using OTE identification programs, for instance, the genome-wide enrichment of seed sequence matches (GESS) method (Sigoillot et al., 2012). GESS is premised on the knowledge that OTEs are the result of siRNA seed sequences binding to mRNAs other than the intended target or by siRNAs inadvertently binding to microRNA sites. GESS detects prominent OTEs by searching for matches between the RefSeq mRNAs and the seed sequences of the siRNAs that confirm in the validation round. The negative control consists of a scrambled set of the validation round seed sequences. mRNAs that are more often complementary to the validation round siRNA seed sequences than the scrambled sequences are flagged as suspicious for being an OTE and removed from further evaluation. Collectively, MORR/RIGER screening combined with gene expression filtering, and OTE identification minimizes the caveats of RNAi screening thus improving its efficiency and yield.
4. Haploid Cell Genetic Screening Technology and Approach
The creation of haplo-insufficiencies using retroviral gene trapping has been and continues to be useful for mammalian genetic screening (Dziuba et al., 2012, Evans et al., 1997, Organ et al., 2004, von Melchner and Ruley, 1989); however, this approach is limited due to its inability to produce homozygous null mutations. This shortcoming was overcome through the introduction of a near-haploid cell line, KBM-7, for use in genetic screens where the remaining allele is inactivated using random retroviral insertion mutagenesis (Carette et al., 2009). KBM-7 cells originated from a 39-year-old gentleman with chronic myelogenous leukemia (CML) and were first reported by the McCredie lab (Andersson et al., 1987), with later isolation of a clonal population of near-haploid cells (2 copies of chromosome 8 and partial disomy of chromosome 15) by Kotecki, Reddy, and Cochran (1999). Haploid cell screens concerned with human–virus interactions have primarily been used in pooled screening approaches involving strong selective pressure by cytopathic viruses, either wild type or recombinant (Table 1). After transduction and selection for a retrovirally expressed selection marker, the cells are cultured to permit phenotypic penetrance via protein turnover and divisional dilution then infected with a cytopathic virus with the rolling infection leading to the destruction of any permissive cells (Fig. 1). The surviving cells are then expanded and the respective integration site of the proviruses are determined using PCR and next-gen sequencing. Genes which are found to have multiple independent insertions are selected as high-confidence candidates using a rationale similar to the reagent redundancy principle employed for selecting candidates in RNAi screens. While powerful, an acknowledged shortcoming of this approach is that it can only be done using a haploid cell line, which may not be readily infected by a human pathogen of interest, e.g., HBV. In an effort to overcome this limitation the KBM-7 cells were genetically reprogrammed, and while the result was not the desired induced pluripotent stem cell line, this work nevertheless gave rise to a more fibroblast like cell line, HAP1 (Carette et al., 2010), that demonstrates adherent growth as compared to the KBM-7 cells, which grow in suspension. The class of host factors predominantly found by the haploid cell screens to date is discussed below.
5. CRISPR/Cas9 Genetic Screening Technologies and Approaches
To defend themselves, bacteria and archaea employ an adaptive immune response using short guide RNAs (sgRNAs) to target and destroy the DNA of invading pathogens (Doudna & Charpentier, 2014). This protective response, known as the CRISPR/Cas9 system, has been adapted for genome editing and the regulation of gene expression in multiple model systems including genome-wide mammalian in vitro genetic screening (Cong et al., 2013, Doudna and Charpentier, 2014, Shalem et al., 2014, Wang et al., 2014). Because Cas9 acts on genomic DNA and not mRNA like RISC, this permits the generation of a permanent homozygous null phenotype. The CRISPR/Cas9 system works in all mammalian cells exogenously expressing Cas9, this combined with its gene targeting specificity make this approach more generalizable than haploid cell screens (Ran et al., 2013). Importantly, because Cas9 locates and binds to a determined DNA target via the complementary base pairing of a short guide RNA (sgRNA), a catalytically inactive Cas9 fused to an activation or repressor domain can bind a desired locus and modulate its gene expression, this capability is extremely powerful and has not been possible using RNAi or haploid cell-screening approaches (Gilbert et al., 2014, Qi et al., 2013) (Table 2). What's more, because a single integrated provirus expressing a sgRNA can, together with Cas9, permanently extinguish a gene's expression, it avoids the same mass action handicap that confronts a single shRNA-expressing provirus whose task is never completed as it must continually silence the products of ongoing transcription. It follows then that under pooled genetic screening conditions, where only one provirus is present per cell, CRISPR/Cas9 will produce greater phenotypic penetrance (Table 2). Several studies have found that while OTEs do occur using CRISPR/Cas9 they appear to be less prevalent than the levels of OTEs encountered with RNAi (Cho et al., 2014, Wang et al., 2015, Wu et al., 2014). Engineered Cas9 proteins with improved specificity also promise to make false positives even rarer (Slaymaker et al., 2016). In order to control for OTEs produced by inadvertent gene editing events the standard for validation of CRISPR/Cas9 results has become similar to RNAi's reagent redundancy principle with the results from two or more orthologous sgRNA against the same gene or two or more clones required. As with RNAi the most convincing confirmation is phenotypic restoration via the expression of a resistant cDNA.
CRISPR/Cas9 screens require the expression of Cas9 in the target cells (Fig. 1). Cas9 expression can be transient, inducible, or stable. If transient expression is chosen then the cells must already express the sgRNA library (Shalem et al., 2015, Wang et al., 2014). The exogenously expressed Cas9 can be either catalytically active and create null alleles, or a catalytically inactive protein fused to one of several transcription factor domains for activation or repression of the sgRNA-targeted locus (Gilbert et al., 2014, Qi et al., 2013). Pooled sgRNA retroviral vectors designed to target every human gene are then packaged into retroviruses and used to stably transduce the Cas9-expressing target cells at a high representation (goal of 1000-fold, Fig. 1). The transduced cells are placed under selection for two weeks to permit the phenotypic maturation. The gene-edited cells are then challenged with the virus of interest, with either cell survival or protein expression based selection or readout. The selected cells are expanded and the identities of enriched sgRNAs are obtained using next-gen sequencing of PCR products amplified from genomic DNA.
CRISPR/Cas9 promises to revolutionize genetic screening, however, due to its recent arrival published screens for host–virus interactions have been limited, but will likely expand greatly in short time. An early effort used CRISPR/Cas9 strategy to identify host factors that govern West Nile virus’ (WNV’s) cytopathic effect (Ma et al., 2015). An earlier WNV host factor arrayed siRNA screen had discovered a few hundred high-confidence candidates using viral protein expression (GFP transgene) as a readout (Krishnan et al., 2008). This much earlier siRNA screen was also stopped well before any cytopathic effect was appreciated. Not surprisingly the candidate gene overlap between the two efforts was small in part arising from the different endpoints, cell survival versus viral protein expression. Interestingly, the CRISPR/Cas9 screen found that the EMC complex, a conserved set of ER-associated proteins implicated in transmembrane protein expression and lipid trafficking was required for WNV's cytopathic effect but not its replication (Wideman, 2015).
6. Comparison of HRV-HF Screens: Arrayed MORR RNAi Versus Pooled CRISPR/Cas9
To date, RNAi screens have been the primary method used for human–virus loss-of-function genetic screens (Table 1). CRISPR/Cas9 is a newly arrived powerful functional genomic technology which can create homozygous null alleles for each human gene. We wished to compare these two approaches, arrayed MORR RNAi versus pooled CRISPR/Cas9, using the same screening platform involving a fully infectious cytopathic HRV strain, HRV14, and H1-HeLa cells that endogenously express the HRV host receptor, ICAM1. We first performed an image-based MORR/RIGER screen to find HRV14-HFs that modulate replication using viral V1 capsid (CA) expression as determined by an immunofluorescence readout (Fig. 2A). For the screens, we transfected a final concentration of each siRNA pool at 50 nM final concentration for 72 h then challenged the cells with HRV14 at an multiplicity of infection (moi) of 0.3 for 12 h at 33 °C. The replication cycle of HRV14 is approximately 8 h. To evaluate cell numbers the HeLa cell nuclear DNA was stained with Hoechst 33342. Magnified images of each well were captured in two wavelengths (FITC and DAPI) using a high-throughput microscope (ImageXpress Micro-XL, Molecular Devices) and the percent infected H1-HeLa cells calculated using image analysis software. These parallel efforts identified > 160 high-confidence candidates across the MORR screens using the Silencer Select, SMARTpool, and esiRNA libraries (Perreira et al., 2015). As seen with ours and others previous siRNA functional genomic screens, the number of exact genes identified across more than one primary screen dataset was low (Fig. 2B). Of interest is that in this instance the only factor that was different between the compared screens was the different siRNA libraries we used, demonstrating the marked influence of the targeting reagents in the observed lack of interscreen concordance. The primary screen candidates were traditionally validated using their respective deconvoluted individual siRNAs (Silencer Select pools with three siRNAs and SMARTpools with four siRNAs), or by retesting the esiRNA pools, in a manner identical to the primary screen (viral capsid expression). As is outlined above, we addressed the problems with siRNA screening by using these three libraries together with the RIGER analysis method to integrate all of the HRV-HF primary screen data sets; this permitted us to assign a numeric value for the likelihood that each gene was important for HRV replication (p value, Fig. 2C). KS, SBR, and WS represent three different RIGER methods; we found that the SBR and WS methods performed the best across multiple gene test sets (Fig. 2D). Our MORR screening approach was validated by the significant enrichment of multiple pathways and protein complexes in the respective screens (e.g., the 80S ribosome), as well as an improvement in these benchmarks when the datasets were integrated using RIGER (Fig. 2D) (Perreira et al., 2015). We also used gene expression filtering to remove candidates that were not expressed in the cells used for the screens, e.g., GRXCR1, whose net expression value is highlighted in red (Fig. 2C). The complete MORR/RIGER work flow extending from the primary screens through to top candidate evaluation is shown (Fig. 2G).
Figure 2.
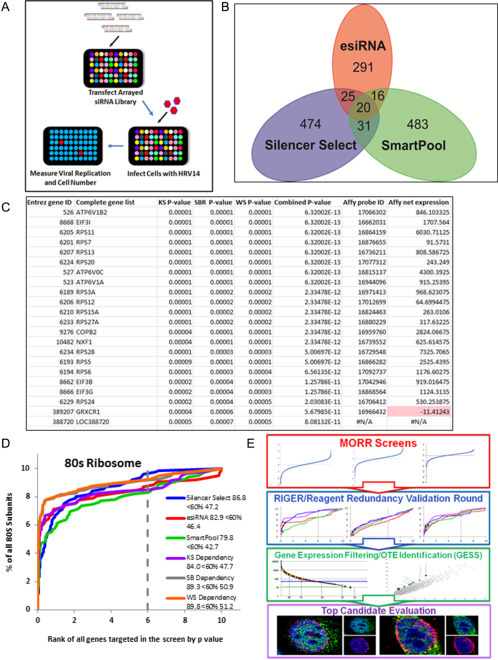
MORR/RIGER screen for HRV host factors. (A) The HRV-HF siRNA screen workflow showing the transfection of the arrayed MORR libraries, the challenge with HRV14 and the assessment of viral capsid expression and cell number using high-throughput imaging (Perreira et al., 2015). (B) The total number of primary screen candidates found in each of the MORR screens along with the number of exact genes that overlap across two or three of the screens is provided. (C) The ranked RIGER weighted sum (WS), second best (SB), and Kolmogorov–Smirnov (KS) analyses of the MORR HRV screen datasets with their respective individual and combined p values. The gene expression data (Affy net expression) is also given based on a microarray analysis of mRNA from the H1-HeLa cells used in the screen. The filled box indicates a gene, GRXCR1, whose expression was found to be below the lower cutoff for candidate selection and thus represents an OTE. (D) The RIGER analyses (WS, SB, and KS) and the individual MORR screen datasets were assessed by determining their respective levels of enrichment for an annotated list of 80S ribosome protein components. A numeric enrichment score was calculated by determining the area under the curve (AUC) produced by plotting the percent fraction of 80S component proteins (% of all 80S subunits) encountered moving from the lowest to highest p value on the ranked gene lists (rank of all genes targeted in the screen by p value). Numbers represent the percent enrichment of the total gene set at < 60% of the ranked gene list (Perreira et al., 2015). (E) A schematic of the workflow for the MORR/RIGER screening approach with the primary MORR screens, integrative RIGER analysis, and traditional reagent redundancy validation round shown. False positives are decreased using gene expression filtering and OTE identification using GESS (Sigoillot et al., 2012). This combined strategy minimizes both false positive and false negatives and is useful for identifying high-confidence HRV-HFs.
To compare screening strategies, as well as perform an orthologous investigation of HRV14’s human cell requirements, we next carried out a CRISPR/Cas9 screen using the exact same cell line and virus. We report this CRISPR/Cas9 HRV14 screen here for the first time. We stably expressed a human codon-optimized cDNA of S. pyogenes Cas9 in a population of HeLa-H1 cells (Fig. 3A) (Shalem et al., 2014). After selection with hygromycin, the cell population was tested for Cas9 expression by immunoblotting as well as the ability to satisfactorily extinguish the expression of the endogenous HRV14 receptor, ICAM1, and a provirus expressing green fluorescent protein (GFP) using a sgRNA against each respective target (Fig. 3B, data not shown). Next, we stably transduced the H1-HeLa-Cas9 cells at a moi of 0.2 with a complex lentiviral pool expressing the human GeCKO v.2 sgRNA library (Addgene #1000000049), which targets 19,052 genes in the human genome with six sgRNAs per gene across two half-libraries (library A and B) (Shalem et al., 2014). Libraries A and B each possess three unique sgRNA per gene and we used the two half-libraries to screen for HRV14-HFs independently. For each library, we plated 4 × 107 cells onto two 15-cm dishes to achieve a 600-fold representation of each sgRNA in the final cell population. We empirically determined this level of representation using a series of titration plates that were infected and processed side-by-side with the sgRNA library-expressing cells. We then selected the cells in puromycin for 11 days, a period of time which we had empirically determined to result in > 80% of cells losing expression of a sgRNA-targeted marker protein (GFP, Fig. 2B) The selected cells were then infected with HRV14 and cultured at 33 °C for ~ 7 days. To follow the progress of the infection, cytopathic effect (CPE) was monitored by eye using light microscopy. Control plates were run in parallel using the H1-HeLa-Cas9 cell parent population which does not contain the GeCKO library. About 7 days after infection the majority of cells, > 95%, had died. The remaining surviving cells were washed extensively and transferred to 37 °C with fresh medium.
Figure 3.
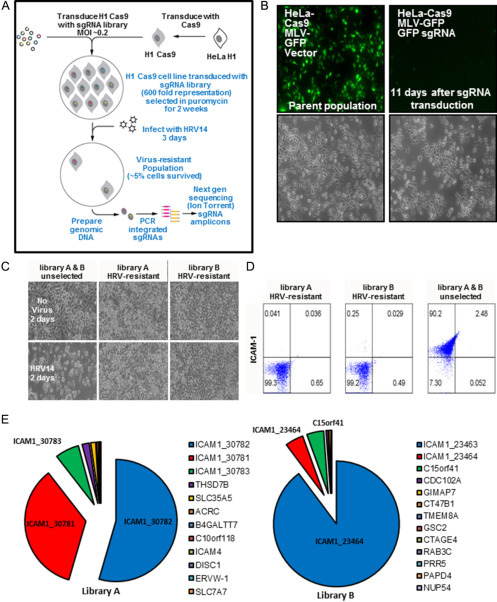
CRISPR/Cas9 screen for HRV host factors. (A) The HRV-HF CRISPR/Cas9 screen workflow showing the generation of the Cas9 expressing H1-HeLa cells containing the sgRNA libraries followed by their subsequent challenge with HRV14 and the assessment of the enriched sgRNAs using next-gen sequencing. (B) HeLa-H1-Cas9 cells were transduced with Moloney Leukemia virus (MLV)-GFP, then supra-transduced with either an empty vector control (parent population) or one expressing a sgRNA against GFP. The cells were selected for puromycin resistance and cultured for 11 days then fixed and imaged for GFP expression. Differential interference contrast (DIC) images are provided below. 4 × magnification. (C) DIC images of cells transduced with either library A or B that survived the HRV14 challenge were expanded and tested for their susceptibility to HRV14’s cytopathic effect over 2 days (bottom row) compared to the unselected parent cell population and the respective uninfected cell populations (top row). (D) Cells from (C) were fixed and immunostained for ICAM1 surface expression by flow cytometry. (E) A chart showing the relative proportion of total sequencing reads for the recovered sgRNAs from the HRV14 CRISPR/Cas9 pooled screen based upon the analysis of genomic DNA from the surviving cells from library A or B. Gene names are provided for each sgRNA with the associated numbers designating their unique identifying library number.
The surviving cells were expanded and genomic DNA prepared. No surviving cells were recovered from the control parental cell plates. Proviruses containing the sgRNA stably integrated into each of the surviving cells were amplified and identified from genomic DNA using PCR and next-gen sequencing using an Ion Torrent sequencer. Sequencing reads (reads) were trimmed at their sgRNA boundaries and mapped back to the complete sgRNA entries for both library A and B using Cutadapt, Bowtie2, and Samtools. This process allowed us to map and rank the frequency of 1153 unique reads from a total of 3,961,083 total reads. We also tested the expanded surviving cells for their susceptibility to HRV14 infection and found that the postscreen population of cells was highly resistant to viral CPE (Fig. 3C). Analysis of the resistant cell populations by flow cytometry showed the near complete absence of the HRV14 receptor, ICAM1, on the cell surface, which is in stark contrast to the pre-screen parent cell population (Fig. 3D). Similar to RNAi screens, we next used the reagent redundancy principle to select for candidate genes which had > 6 sequencing reads for two or more independent sgRNAs. Among the unique sgRNAs detected by next-gen sequencing only two genes presented with more than two independent sgRNAs, ICAM1 (five of six total sgRNAs recovered) and EXOC4 (two of six total sgRNAs, Fig. 3E). Of the 3.9 million total reads > 95% mapped to one of the five sgRNAs targeting ICAM1. Of these two candidates only ICAM1 overlapped with the MORR/RIGER screen HRV-HF candidate list (Fig. 4 ).
Figure 4.
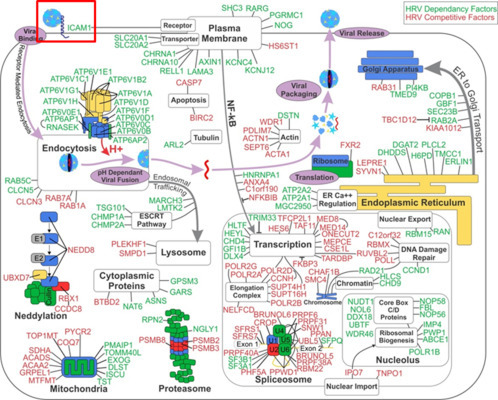
MORR and CRISPR/Cas9 HRV-HF screen candidate overlap. We used the RIGER analysis of the HRV-HF MORR screens to produce a speculative model cell showing the HRV lifecycle overlayed with where the top 164 high-confidence candidate HRV-HFs are likely to act based on available published data (Perreira et al., 2015). A single HRV-HF candidate, ICAM1, shared between the arrayed MORR/RIGER siRNA screen and the matched pooled CRISPR/Cas9 screen, is highlighted with a box.
The authors own all the figures included from published work (Perreira et al., 2015), under a creative commons license agreement with Cell Reports.
The comparison of these two screening approaches side-by-side, using the same cells and virus, raises an interesting point. The number of host factors found for HRV14 was far greater using the MORR/RIGER approach and is approaching a systems level understanding based on bioinformatic analyses and the near saturation of, or enrichment for, multiple complexes and pathways (Fig. 4) (Perreira et al., 2015). By comparison our matched pooled CRISPR/Cas9 screen for HRV-HFs yielded two high-confidence candidates based on reagent redundancy, ICAM1, the known receptor for HRV14, and EXOC4, a gene involved in exocyst targeting and vesicular transport (He & Guo, 2009). Given the known role of ICAM1 as the host receptor for most HRVs, these results point to entry as the major viral lifecycle stage interrogated by a pooled functional genomic screening approach using a population of randomly biallelic null cells infected by a cytopathic virus.
Our CRISPR/Cas9 screen results are not surprising given the predilection of earlier pooled haploid cell survival screens for finding viral entry-associated factors, including host receptors, genes required for receptor modification or endosomal trafficking (for example, the HOPS tethering complex, Table 1) (Carette et al., 2011). Therefore, while conventional catalytic CRISPR/Cas9 and haploid cell-screening technologies use different strategies for creating loss-of-function alleles, their shared method of screening complex pools of cells for survival likely leads to similar results. For an illustration, we note the IAV haploid cell screen and two additional haploid cell survival screens which identified the host receptors for Lassa virus and Ebola virus using similar pooled strategies to those being employed with CRISPR/Cas9 screens (Carette et al., 2009, Carette et al., 2011, Jae et al., 2013). Interestingly, the latter two haploid cell screens used identical recombinant vesicular stomatitis viruses (rVSVs) with the exception of their respective envelope proteins, Lassa virus or Ebola virus. Notably there was not a single candidate gene that was found in common between these two pooled screens, arguing that under such conditions only a total block to VSV entry can confer cell survival. A factor which may cause pooled screens to strongly enrich for entry-associated host factors is the intense selective pressure that the cells are subjected to as the levels of virus surge during the course of the screen. It is possible that even with the loss of a reasonably important postentry viral dependency factor that at such a high moi the overwhelming entry of so many viruses alone, even with some diminishment of their replication, would be sufficient to elicit apoptosis or exit from the cell cycle. This last notion is supported by two independently performed arrayed siRNA screens which respectively reported 301 and 72 high-confidence candidates necessary for VSV replication, many of which were involved in postentry phases of the viral lifecycle; none of these candidates were found in the rVSV-based haploid cell screens. Interestingly one of the screens found that coatomer (COP1) and the V-ATPase were required for VSV replication. COP1 and the V-ATPase are essential complexes which would be not be recovered in a haploid cell or CRISPR/Cas9 screen using cells with null phenotypes.
In the exemplary study by Petersen et al. for Arena virus (ANDV) host factors, the authors performed matching haploid cell and arrayed RNAi screens (Petersen et al., 2014). As with the Ebola and Lassa haploid cell screens above, the researchers engineered an rVSV which expressed the ANDV glycoprotein receptor (rVSV-ANDV) on its surface. One billion HAP1 cells were retrovirally mutagenized and screened for survival after infection with either rVSV-ANDV or a matched control virus, rVSV-G, which expressed the VSV-G receptor. After selection, the group expanded the surviving cells and used their pooled genomic DNA to identify 676 independent integrations sites. Of these sites, 37% occurred within four genes: regulatory element binding protein 2 (SREBF2), sterol regulatory element-binding protein cleavage-activating protein (SCAP), site 1 protease (S1P), and site 2 protease (S2P), all of which belong to the sterol regulatory element-binding protein pathway. A nearly identical haploid cell pooled screen was also completed by another group with similar results (Kleinfelter et al., 2015).
Petersen et al. also carried out a matched RNAi screen using an rVSV pseudoparticle (pp) which contains a luciferase transgene and expresses the ANDV glycoprotein on its surface. The VSV-ANDV pp was used to infect an arrayed panel of cells that had been previously transfected in a well by well manner with a first-generation subgenomic Ambion siRNA library targeting 9102 human genes. After VSV-ANDV pp challenge a plate reader was used to quantify pp replication based on relative light units (RLUs). Genes were selected as candidates if they met criteria for significantly decreasing RLUs as compared to the control with two or more unique siRNAs. Follow up involved an identical screen using additional orthologous siRNAs. Thirty three genes were ultimately selected as high-confidence candidates with only one, SREBF2, being shared in common with the companion haploid cell screen. Further mechanistic studies demonstrated that loss of the sterol regulatory element-binding protein pathway prevented ANDV glycoprotein-mediated entry. Given the greater number of high-confidence candidates found in the RNAi screen, it would be interesting to determine if they were also all acting at entry or were instead required for the early postentry replication and expression of the luciferase transgene within the rVSV genome. Therefore, as with the other haploid cell screen noted above, this approach excels at finding entry factors. In this instance the paired RNAi arm of the study showed itself to be more sensitive because it found more high-confidence host factors using viral replication (RLUs) and not survival as a readout.
While the haploid cell screens have been useful in defining host–virus interactions they predominantly select for host genes that play critical early roles in viral replication, e.g., the host receptor(s), proteins that modify receptors, or endosomal trafficking factors (Table 1, Table 2). Based on our experience using pooled CRISPR/Cas9 to screen for host factors required by cytopathic viruses (HRV and IAV, Fig. 3 and our unpublished data) it appears that this approach will produce similar results to those seen with the pooled haploid cell survival screens, with only very early factors associated with viral entry, or genes need for the expression or activity of such genes, being enriched for in the surviving cell populations. One approach for recovering a deeper set of viral host factors may lie in halting the cytopathic virus pooled screen at intermediate stages of CPE, however, in our experience screening with HRV using shifts to nonpermissive temperatures and incubation with neutralizing antibodies, the practical execution of this idea is difficult. An arrayed haploid cell or CRISPR/Cas9 approach would permit more subtle selection criteria to be used such as those employed with arrayed siRNA screens. With this in mind, recent efforts have resulted in 3396 clonal HAP1 cell populations being characterized and arrayed with each one lacking the expression of a single gene due to retroviral insertion (Petersen et al., 2014). Unfortunately, because retroviral insertion is a random process it is not possible to selectively inactivate one class of gene or pathway, making the assembly of specialty libraries a matter of hunt and peck. This expanding arrayed HAP1 null allele cell resource would allow detailed investigation of single clones or focused subsets of clones, although the long-term culturing of such large numbers of distinct cell lines simultaneously will present significant challenges. Similar concerns for whole-genome arrayed CRISPR/Cas9 cell lines or lentiviruses would also present similar hurdles. Price permitting, this limitation might be avoided using large-scale arrayed sgRNA oligos or gene blocks that can be introduced into cells in a one-gene-per-well manner via lipid-mediated transfection along with Cas9 mRNA; these are arrayed sgRNA libraries are presently on hand in smaller gene sets but will undoubtedly become available in druggable or whole-genome versions in the near future. Care will need to be taken to allow sufficient time to elapse posttransfection for the generation of biallelic null mutations and phenotypic maturation prior to screening.
How else might the sensitivity and yield of pooled screens using CRISPR/Cas9 or haploid cells be improved upon? One possibility is the use of less stringent selection criteria such as selecting cells from a pool based on their relative expression of a marker protein. An elegant example of such a strategy for gene enrichment using pooled screening was recently done using flow cytometry to sort cells based on their expression of tumor necrosis factor (Tnf), which is elaborated in primary dendritic cells (DCs) after exposure to the bacterial product, lipopolysaccharide (LPS) (Parnas et al., 2015). The DCs were transduced so as to express Cas9 together with a complex sgRNA library of 125,793 sgRNAs directed against 21,786 mouse genes (Sanjana, Shalem, & Zhang, 2014). The pooled screen was performed three times using > 60 million DCs stimulated with LPS. After LPS stimulation, the DCs were fixed, permeabilized, and immunostained for Tnf. Based on anti-Tnf antibody-associated immunofluorescence both high and low expressing Tnf populations were sorted using flow cytometry. The identities of the enriched sgRNAs were determined using PCR amplification of genomic DNA followed by next-gen sequencing. The authors arrived at > 100 high-confidence candidates, several of which were previously known to be involved in DC responses to LPS, thus validating their approach and demonstrating its sensitivity.
While most current CRISPR/Cas9 pooled screens lack sensitivity, they nonetheless appear to have fewer false positives than RNAi screens, lowering the work load and increasing the efficiency of validation (Table 2). In our HRV-HF CRISPR/Cas9 screen, we detected a number of single sgRNAs for multiple genes with the majority having < 6 reads. This may represent background PCR contamination or the facilitated carryover of phenotypically inconsequential sgRNAs by cells with intrinsic genetic resistance, e.g., cells that inherently lack ICAM1 expression. Therefore, all three genetic screening strategies benefit from the use of reagent redundancy, in the form of orthologous siRNAs and sgRNAs or multiple independent retroviral insertions, as a guiding principle for finding true positives.
To summarize, siRNA screens using arrayed one-gene-per-well format with moderate selection criteria, e.g., percent infected cells, permit the detection of a larger number of viral dependency factors, with the significant tradeoff being a greater number of false positives or OTEs. In contrast, pooled screens using cell survival as a readout as seen with the majority of haploid cell, and likely with additional CRISPR/Cas9 pooled screens to come, display limited sensitivity but excellent specificity in finding host genes that act very early in viral replication, for instance host factors needed for viral entry (ICAM1) (Table 1, Table 2). As can be seen in many of the arrayed siRNA screens, including our screens for HIV-1, HCV, and HRV14, host receptors and viral entry factors are also found with this approach, however, since these screens yield much greater lists of candidates, which include OTEs, any novel host receptors may not immediately jump to the fore. Therefore, given the currently available functional genomic strategies if the goal is to find viral entry factors (e.g., host receptors) with high specificity its best to use a pooled survival screen, but alternatively if the aim is to obtain with relative ease a more comprehensive set of host factors, albeit with more prevalent false positives, than an arrayed siRNA screen would be the preferred method.
7. Future Directions
While much has been learned about host–virus interactions there is still a great deal more to be achieved using functional genomic screens. Based on the greater adaptability of CRISPR/Cas9 for gene activation or inactivation/repression, all using a single sgRNA-expressing provirus, it seems likely that pooled shRNA screening will wane, given its comparatively poor phenotypic penetrance and greater burden of OTEs. Pooled haploid cell screens also appear vulnerable to displacement by CRISPR/Cas9 pooled approaches because of their dependence on only two transformed haploid cell lines, in conjunction with their more laborious identification of candidate genes. What's more, based on the established preference of retroviral insertion it is improbable that haploid cell screens will approach the saturation or representation produced with CRISPR/Cas9 methods.
The unique versatility of CRISPR/Cas9 technology to modulate gene expression using activation domain (CRISPRa) or repressor domain (CRISPRi) chimeras will assuredly give rise to many more notable discoveries. However, candidates found in screens using such synthetic transcription factors will need to be confirmed with rescue experiments given the questionable value of reagent redundancy approaches. This concern arises because of the potential for shared long distance OTEs being produced by orthologous sgRNAs designed against the same gene which will be binding relatively close to one another. Arrayed CRISRP/Cas9 screens using oligonucleotides (sgRNA and Cas9 mRNA) introduced into cells via lipid-mediated transfection may also rival or surpass established siRNA arrayed approaches, and while the current offerings of these reagents consist of smaller subgenomic gene sets it is anticipated that whole-genome versions will be commercially available shortly. That said, until the widespread implementation of arrayed CRISPR/Cas9 whole-genome screening, it seems likely that RNAi will continue to be the workhorse of functional genomic screening given its (i) first to market status, (ii) ease of use for arrayed screening, and (iii) high sensitivity and strong yields. However its prominent caveats increase the workload for validation substantially and may help to usher in an arrayed CRISPR/Cas9 screening era. We anticipate that approaches to minimize RNAi's problems, in combination with the expansion and adoption of CRISPR/Cas9 strategies, will continue to accelerate our understanding of human–virus interactions.
Acknowledgments
We thank S. Whelan, M.C. Smith, and J. Smith for helpful discussions, L. Brass and W. McDougall for their critical reading of the chapter, and University of Massachusetts Medical School colleagues (B. Hobbs, C. Barry, L. Benson, T. Brailey, and R. Fish). A.L.B. is grateful to Bill and Melinda Gates Foundation, the Burroughs Wellcome Fund, and the NIH (1R01AI091786) for their support.
References
- Andersson B.S., Beran M., Pathak S., Goodacre A., Barlogie B., McCredie K.B. Ph-positive chronic myeloid leukemia with near-haploid conversion in vivo and establishment of a continuously growing cell line with similar cytogenetic pattern. Cancer Genetics and Cytogenetics. 1987;24:335–343. doi: 10.1016/0165-4608(87)90116-6. [DOI] [PubMed] [Google Scholar]
- Arita M., Wakita T., Shimizu H. Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. Journal of Virology. 2012;86:5541–5553. doi: 10.1128/JVI.00114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkov K., Rosenbaum B., Christiansen L., Jonsson H., Munchow M. Treatment of suicidal patients: The Collaborative Assessment and Management of Suicidality. Ugeskrift for Laeger. 2008;170:149–153. [PubMed] [Google Scholar]
- Aydin I., Weber S., Snijder B., Samperio Ventayol P., Kuhbacher A., Becker M. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathogens. 2014;10:e1004162. doi: 10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balistreri G., Horvath P., Schweingruber C., Zund D., McInerney G., Merits A. The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host & Microbe. 2014;16:403–411. doi: 10.1016/j.chom.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Barrows N.J., Jamison S.F., Bradrick S.S., Le Sommer C., Kim S.Y., Pearson J. Functional genomics approach for the identification of human host factors supporting dengue viral propagation. Methods in Molecular Biology. 2014;1138:285–299. doi: 10.1007/978-1-4939-0348-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard P.M., Griffiths S.J., Gonzalez O., Haga I.R., Pechenick Jowers T., Reynolds D.K. A loss of function analysis of host factors influencing Vaccinia virus replication by RNA interference. PLoS One. 2014;9:e98431. doi: 10.1371/journal.pone.0098431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K., Hijmans E.M., Mullenders J., Brummelkamp T.R., Velds A., Heimerikx M. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- Boettcher M., Hoheisel J.D. Pooled RNAi screens—Technical and biological aspects. Current Genomics. 2010;11:162–167. doi: 10.2174/138920210791110988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker M., Samsonova A.A., Kwon Y., Flockhart I., Mohr S.E., Perrimon N. False negative rates in Drosophila cell-based RNAi screens: A case study. BMC Genomics. 2011;12:50. doi: 10.1186/1471-2164-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Brass A.L., Huang I.C., Benita Y., John S.P., Krishnan M.N., Feeley E.M. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman F.D., Malani N., Fernandes J., D'Orso I., Cagney G., Diamond T.L. Host cell factors in HIV replication: Meta-analysis of genome-wide studies. PLoS Pathogens. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J.E., Guimaraes C.P., Varadarajan M., Park A.S., Wuethrich I., Godarova A. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- Carette J.E., Pruszak J., Varadarajan M., Blomen V.A., Gokhale S., Camargo F.D. Generation of iPSCs from cultured human malignant cells. Blood. 2010;115:4039–4042. doi: 10.1182/blood-2009-07-231845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry S. RNAi screening for host factors involved in viral infection using Drosophila cells. Methods in Molecular Biology. 2011;721:375–382. doi: 10.1007/978-1-61779-037-9_23. [DOI] [PubMed] [Google Scholar]
- Cherry S., Doukas T., Armknecht S., Whelan S., Wang H., Sarnow P. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes & Development. 2005;19:445–452. doi: 10.1101/gad.1267905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C.R., Brass A.L. A genome wide RNA interference screening method to identify host factors that modulate influenza A virus replication. Methods. 2013;59:217–224. doi: 10.1016/j.ymeth.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Research. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne C.B., Bozym R., Morosky S.A., Hanna S.L., Mukherjee A., Tudor M. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host & Microbe. 2011;9:70–82. doi: 10.1016/j.chom.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., Wannee K.F., Scholte F.E., Goeman J.J., Ten Dijke P., Snijder E.J. A kinome-wide small interfering RNA screen identifies proviral and antiviral host factors in severe acute respiratory syndrome coronavirus replication, including double-stranded RNA-activated protein kinase and early secretory pathway proteins. Journal of Virology. 2015;89:8318–8333. doi: 10.1128/JVI.01029-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Dziuba N., Ferguson M.R., O'Brien W.A., Sanchez A., Prussia A.J., McDonald N.J. Identification of cellular proteins required for replication of human immunodeficiency virus type 1. AIDS Research and Human Retroviruses. 2012;28:1329–1339. doi: 10.1089/aid.2011.0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri C.J., Beachy P.A., Baum B., Boutros M., Buchholz F., Chanda S.K. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nature Methods. 2006;3:777–779. doi: 10.1038/nmeth1006-777. [DOI] [PubMed] [Google Scholar]
- Echeverri C.J., Perrimon N. High-throughput RNAi screening in cultured cells: A user's guide. Nature Reviews. Genetics. 2006;7:373–384. doi: 10.1038/nrg1836. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Espeseth A.S., Fishel R., Hazuda D., Huang Q., Xu M., Yoder K. siRNA screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS One. 2011;6:e17612. doi: 10.1371/journal.pone.0017612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M.J., Carlton M.B., Russ A.P. Gene trapping and functional genomics. Trends in Genetics. 1997;13:370–374. doi: 10.1016/s0168-9525(97)01240-7. [DOI] [PubMed] [Google Scholar]
- Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Fusco D.N., Brisac C., John S.P., Huang Y.W., Chin C.R., Xie T. A genetic screen identifies interferon-α effector genes required to suppress hepatitis C virus replication. Gastroenterology. 2013;144:1438–1449. doi: 10.1053/j.gastro.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A., Mello C.C. RNAi (Nematodes: Caenorhabditis elegans) Advances in Genetics. 2002;46:339–360. doi: 10.1016/s0065-2660(02)46012-9. [DOI] [PubMed] [Google Scholar]
- Hao L., He Q., Wang Z., Craven M., Newton M.A., Ahlquist P. Limited agreement of independent RNAi screens for virus-required host genes owes more to false-negative than false-positive factors. PLoS Computational Biology. 2013;9:e1003235. doi: 10.1371/journal.pcbi.1003235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L., Sakurai A., Watanabe T., Sorensen E., Nidom C.A., Newton M.A. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Guo W. The exocyst complex in polarized exocytosis. Current Opinion in Cell Biology. 2009;21:537–542. doi: 10.1016/j.ceb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins K.C., McLane L.M., Maqbool T., Panda D., Gordesky-Gold B., Cherry S. A genome-wide RNAi screen reveals that mRNA decapping restricts bunyaviral replication by limiting the pools of Dcp2-accessible targets for cap-snatching. Genes & Development. 2013;27:1511–1525. doi: 10.1101/gad.215384.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain K.M., Leong K.L., Ng M.M., Chu J.J. The essential role of clathrin-mediated endocytosis in the infectious entry of human enterovirus 71. The Journal of Biological Chemistry. 2011;286:309–321. doi: 10.1074/jbc.M110.168468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jae L.T., Raaben M., Riemersma M., van Beusekom E., Blomen V.A., Velds A. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science. 2013;340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlas A., Machuy N., Shin Y., Pleissner K.P., Artarini A., Heuer D. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- Kittler R., Surendranath V., Heninger A.K., Slabicki M., Theis M., Putz G. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nature Methods. 2007;4:337–344. doi: 10.1038/nmeth1025. [DOI] [PubMed] [Google Scholar]
- Kleinfelter L.M., Jangra R.K., Jae L.T., Herbert A.S., Mittler E., Stiles K.M. Haploid genetic screen reveals a profound and direct dependence on cholesterol for hantavirus membrane fusion. MBio. 2015;6:e00801. doi: 10.1128/mBio.00801-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott S.R., Maceli A.R., Erard N., Chang K., Marran K., Zhou X. A computational algorithm to predict shRNA potency. Molecular Cell. 2014;56:796–807. doi: 10.1016/j.molcel.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolokoltsov A.A., Saeed M.F., Freiberg A.N., Holbrook M.R., Davey R.A. Identification of novel cellular targets for therapeutic intervention against Ebola virus infection by siRNA screening. Drug Development Research. 2009;70:255–265. doi: 10.1002/ddr.20303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R., Stertz S., Zhou Y., Inoue A., Hoffmann H.H., Bhattacharyya S. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecki M., Reddy P.S., Cochran B.H. Isolation and characterization of a near-haploid human cell line. Experimental Cell Research. 1999;252:273–280. doi: 10.1006/excr.1999.4656. [DOI] [PubMed] [Google Scholar]
- Krishnan M.N., Ng A., Sukumaran B., Gilfoy F.D., Uchil P.D., Sultana H. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y.J., Heo J., Wong H.E., Cruz D.J., Velumani S., da Silva C.T. Kinome siRNA screen identifies novel cell-type specific dengue host target genes. Antiviral Research. 2014;110:20–30. doi: 10.1016/j.antiviral.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Lavanya M., Cuevas C.D., Thomas M., Cherry S., Ross S.R. siRNA screen for genes that affect Junin virus entry uncovers voltage-gated calcium channels as a therapeutic target. Science Translational Medicine. 2013;5:204ra131. doi: 10.1126/scitranslmed.3006827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Sommer C., Barrows N.J., Bradrick S.S., Pearson J.L., Garcia-Blanco M.A. G protein-coupled receptor kinase 2 promotes flaviviridae entry and replication. PLoS Neglected Tropical Diseases. 2012;6:e1820. doi: 10.1371/journal.pntd.0001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.S., Burdeinick-Kerr R., Whelan S.P. A genome-wide small interfering RNA screen identifies host factors required for vesicular stomatitis virus infection. Journal of Virology. 2014;88:8355–8360. doi: 10.1128/JVI.00642-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Brass A.L., Ng A., Hu Z., Xavier R.J., Liang T.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Oliveira N.M., Cheney K.M., Pade C., Dreja H., Bergin A.M. A whole genome screen for HIV restriction factors. Retrovirology. 2011;8:94. doi: 10.1186/1742-4690-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B., Cheung H.W., Subramanian A., Sharifnia T., Okamoto M., Yang X. Highly parallel identification of essential genes in cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Dang Y., Wu Y., Jia G., Anaya E., Zhang J. A CRISPR-based screen identifies genes essential for West-Nile-virus-induced cell death. Cell Reports. 2015;12:673–683. doi: 10.1016/j.celrep.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier R., Franceschini A., Horvath P., Tetard M., Mancini R., von Mering C. Genome-wide small interfering RNA screens reveal VAMP3 as a novel host factor required for Uukuniemi virus late penetration. Journal of Virology. 2014;88:8565–8578. doi: 10.1128/JVI.00388-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J., Snijder B., Sacher R., Burkard C., Bleck C.K., Stahlberg H. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Reports. 2012;2:1036–1047. doi: 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Mohr S.E., Perrimon N. RNAi screening: New approaches, understandings, and organisms. Wiley Interdisciplinary Reviews. RNA. 2012;3:145–158. doi: 10.1002/wrna.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T.S., Jones R.G., Thompson C.B., Coyne C.B., Cherry S. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathogens. 2010;6:e1000954. doi: 10.1371/journal.ppat.1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi Y.S., Stiles K.M., Liu C.Y., Taylor G.M., Kielian M. Genome-wide RNAi screen identifies novel host proteins required for alphavirus entry. PLoS Pathogens. 2013;9:e1003835. doi: 10.1371/journal.ppat.1003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organ E.L., Sheng J., Ruley H.E., Rubin D.H. Discovery of mammalian genes that participate in virus infection. BMC Cell Biology. 2004;5:41. doi: 10.1186/1471-2121-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortblad K.F., Lozano R., Murray C.J. The burden of HIV: Insights from the Global Burden of Disease Study 2010. AIDS. 2013;27:2003–2017. doi: 10.1097/QAD.0b013e328362ba67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison P.J., Silva J.M., Conklin D.S., Schlabach M., Li M., Aruleba S. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- Panda D., Cherry S. A genome-wide RNAi screening method to discover novel genes involved in virus infection. Methods. 2015;91:75–81. doi: 10.1016/j.ymeth.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D., Das A., Dinh P.X., Subramaniam S., Nayak D., Barrows N.J. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19036–19041. doi: 10.1073/pnas.1113643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D., Rose P.P., Hanna S.L., Gold B., Hopkins K.C., Lyde R.B. Genome-wide RNAi screen identifies SEC61A and VCP as conserved regulators of Sindbis virus entry. Cell Reports. 2013;5:1737–1748. doi: 10.1016/j.celrep.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O., Jovanovic M., Eisenhaure T.M., Herbst R.H., Dixit A., Ye C.J. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell. 2015;162:675–686. doi: 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreira J., Aker A., Savidis G., Chin C., McDougall W., Portmann J. RNASEK is a V-ATPase-associated factor required for endocytosis and the replication of rhinovirus, influenza A virus, and dengue virus. Cell Reports. 2015;12:850–863. doi: 10.1016/j.celrep.2015.06.076. [DOI] [PubMed] [Google Scholar]
- Petersen J., Drake M.J., Bruce E.A., Riblett A.M., Didigu C.A., Wilen C.B. The major cellular sterol regulatory pathway is required for Andes virus infection. PLoS Pathogens. 2014;10:e1003911. doi: 10.1371/journal.ppat.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri S., Meister G. siRNA design principles and off-target effects. Methods in Molecular Biology. 2013;986:59–71. doi: 10.1007/978-1-62703-311-4_4. [DOI] [PubMed] [Google Scholar]
- Poenisch M., Metz P., Blankenburg H., Ruggieri A., Lee J.Y., Rupp D. Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathogens. 2015;11:e1004573. doi: 10.1371/journal.ppat.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl M.O., Edinger T.O., Stertz S. Prolidase is required for early trafficking events during influenza A virus entry. Journal of Virology. 2014;88:11271–11283. doi: 10.1128/JVI.00800-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri N., Wang X., Varma R., Burnett C., Beauchamp L., Batten D.M. LNA incorporated siRNAs exhibit lower off-target effects compared to 2'-OMethoxy in cell phenotypic assays and microarray analysis. Nucleic Acids Symposium Series. 2008:25–26. doi: 10.1093/nass/nrn013. [DOI] [PubMed] [Google Scholar]
- Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall G., Panis M., Cooper J.D., Tellinghuisen T.L., Sukhodolets K.E., Pfeffer S. Cellular cofactors affecting hepatitis C virus infection and replication. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root D.E., Hacohen N., Hahn W.C., Lander E.S., Sabatini D.M. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nature Methods. 2006;3:715–719. doi: 10.1038/nmeth924. [DOI] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber C.A., Sakuma T., Izumiya Y., Holditch S.J., Hickey R.D., Bressin R.K. An siRNA screen identifies the U2 snRNP spliceosome as a host restriction factor for recombinant adeno-associated viruses. PLoS Pathogens. 2015;11:e1005082. doi: 10.1371/journal.ppat.1005082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer A., Horn J., Mikolajczyk R.T., Krause G., Ott J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet. 2015;386:1546–1555. doi: 10.1016/S0140-6736(15)61412-X. [DOI] [PubMed] [Google Scholar]
- Sessions O.M., Barrows N.J., Souza-Neto J.A., Robinson T.J., Hershey C.L., Rodgers M.A. Discovery of insect and human dengue virus host factors. Nature. 2009;458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nature Reviews. Genetics. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira S.D., Gat-Viks I., Shum B.O., Dricot A., de Grace M.M., Wu L. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigoillot F.D., Lyman S., Huckins J.F., Adamson B., Chung E., Quattrochi B. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nature Methods. 2012;9:363–366. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J.M., Li M.Z., Chang K., Ge W., Golding M.C., Rickles R.J. Second-generation shRNA libraries covering the mouse and human genomes. Nature Genetics. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- Sivan G., Martin S.E., Myers T.G., Buehler E., Szymczyk K.H., Ormanoglu P. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3519–3524. doi: 10.1073/pnas.1300708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan G., Ormanoglu P., Buehler E.C., Martin S.E., Moss B. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the Vaccinia virus K1L-C7L- mutant. MBio. 2015;6:e01122. doi: 10.1128/mBio.01122-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.A., White E.A., Sowa M.E., Powell M.L., Ottinger M., Harper J.W. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3752–3757. doi: 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz S., Shaw M.L. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes and Infection/Institut Pasteur. 2011;13:516–525. doi: 10.1016/j.micinf.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W.C., Chen Y.C., Tseng C.H., Hsu P.W., Tung K.F., Jeng K.S. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17516–17521. doi: 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai A.W., Benita Y., Peng L.F., Kim S.S., Sakamoto N., Xavier R.J. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host & Microbe. 2009;5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran A.T., Rahim M.N., Ranadheera C., Kroeker A., Cortens J.P., Opanubi K.J. Knockdown of specific host factors protects against influenza virus-induced cell death. Cell Death & Disease. 2013;4:e769. doi: 10.1038/cddis.2013.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Melchner H., Ruley H.E. Identification of cellular promoters by using a retrovirus promoter trap. Journal of Virology. 1989;63:3227–3233. doi: 10.1128/jvi.63.8.3227-3233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Wang Y., Wu X., Wang J., Wang Y., Qiu Z. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nature Biotechnology. 2015;33:175–178. doi: 10.1038/nbt.3127. [DOI] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward S.E., Kim H.S., Komurov K., Mendiratta S., Tsai P.L., Schmolke M. Host modulators of H1N1 cytopathogenicity. PLoS One. 2012;7:e39284. doi: 10.1371/journal.pone.0039284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X., Ding L., Hunter E., Spearman P. An siRNA screen of membrane trafficking genes highlights pathways common to HIV-1 and M-PMV virus assembly and release. PLoS One. 2014;9:e106151. doi: 10.1371/journal.pone.0106151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wideman J.G. The ubiquitous and ancient ER membrane protein complex (EMC): Tether or not? F1000Research. 2015;4:624. doi: 10.12688/f1000research.6944.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.A., Abbink T.E., Jeang K.T., Lever A.M. Identification of RNA helicases in human immunodeficiency virus 1 (HIV-1) replication—A targeted small interfering RNA library screen using pseudotyped and WT HIV-1. The Journal of General Virology. 2015;96:1484–1489. doi: 10.1099/vir.0.000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Scott D.A., Kriz A.J., Chiu A.C., Hsu P.D., Dadon D.B. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga A., Hanna S.L., Li J., Cho H., Rose P.P., Spiridigliozzi A. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathogens. 2014;10:e1003914. doi: 10.1371/journal.ppat.1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung M.L., Houzet L., Yedavalli V.S., Jeang K.T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. The Journal of Biological Chemistry. 2009;284:19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Davoli T., Perriera J.M., Chin C.R., Gaiha G.D., John S.P. Comprehensive identification of host modulators of HIV-1 replication using multiple orthologous RNAi reagents. Cell Reports. 2014;9:752–766. doi: 10.1016/j.celrep.2014.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]