

Abstract
Proteolytic enzymes constitute around 2% of the human genome and are involved in many stages of cell development from fertilization to death (apoptosis). The identification of many novel proteases from genome‐sequencing programs has suggested them as potential new therapeutic targets. In addition, several well‐characterized metallopeptidases were recently shown to possess new biological roles in neuroinflammation and neurodegeneration. As a result of these studies, metabolism of the neurotoxic and inflammatory amyloid peptide (Aβ) is considered as a physiologically relevant process with several metallopeptidases being suggested for the role of amyloid‐degrading enzymes. These include the neprilysin (NEP) family of metalloproteinases (including its homologue endothelin‐converting enzyme), insulin‐degrading enzyme, angiotensin‐converting enzyme, plasmin, and, possibly, some other enzymes. NEP also has a role in metabolism of sensory and inflammatory neuropeptides such as tachykinins and neurokinins. The existence of natural enzymatic mechanisms for removal of amyloid peptides has extended the therapeutic avenues in Alzheimer's disease (AD) and neurodegeneration. The proteolytic events underlying AD are highly compartmentalized in the cell and formation of amyloid peptide from its precursor molecule APP (amyloid precursor protein) takes place both within intracellular compartments and in the plasma membrane, especially in lipid raft domains. Degradation of amyloid peptide by metallopeptidases can also be both intra‐ and extracellular depending on the activity of membrane‐bound enzymes and their soluble partners. Soluble forms of proteases can be secreted or released from the cell surface through the activity of “sheddases”—another group of proteolytic enzymes involved in key cellular regulatory functions. The activity of proteases involved in amyloid metabolism depends on numerous factors (e.g., genetic, environmental, age), and some conditions (e.g., hypoxia and ischemia) shift the balance of amyloid metabolism toward accumulation of higher concentrations of Aβ. In this regard, regulation of the activity of amyloid‐degrading enzymes should be considered as a viable strategy in neuroprotection.
I. Introduction
Proteolysis represents one of the key processes underlying biological events from fertilization through development to death. In the human genome there are more than 500 proteases and homologues (almost 2% of the genome), many of whose physiological roles are yet to be identified and which may provide potential therapeutic targets in the treatment of human disease. At present there are 70 known human hereditary diseases caused by mutations in protease‐coding genes (http://www.uniovi.es/degradome/). Abnormal functioning of these genes are implicated in such pathologies as inflammatory diseases, cancer, cardiovascular diseases, and neurodegeneration. Understanding of the role of these enzymes and their evolutionary conservation is important for design of appropriate drugs and therapeutic strategies. Other eukaryotic species whose genomes have been deciphered to date also possess a very high number of protease and protease‐like genes which in the case of Drosophila melanogaster and mouse are even higher than in human (Rawlings et al., 2006). The number of species in which proteases have now been identified is approaching 3000 (http://merops.sanger.ac.uk/). Cysteine‐, serine‐, and metallopeptidases represent the major classes of peptidases; among these three classes, the metallopeptidase class is more consistently represented with the number of genes varying from 183 in Drosophila to 198 in mice (in human, 186). Two other classes have much higher diversity with the range in the number of genes for cysteine proteases from 76 in Drosophila to 153 in mice (Puente et al., 2003). For the serine peptidases this range is even more pronounced: from 100 in Caenorhabditis elegans to 309 in Drosophila. There are a smaller number of aspartic proteinases, although some are of key therapeutic importance. This more conservative representation of metallopeptidases in the genomes of various species may reflect roles of these enzymes in more generic cellular functions and their unique catalytic properties which required water and metal ions for activation of the proteolytic (hydrolytic) process, which were carefully preserved in the course of evolution.
The metallopeptidases can be divided into 12 clans according to the type and number of metal ions required for their activity (Rawlings and Barrett, 2004). This chapter will focus on two distinct zinc metallopeptidase families belonging to one of these clans, namely MA, whose representatives require zinc for their activity and contain one or two histidine residues in the zinc binding motif. The first of these metallopeptidase families is the M13 family represented by neprilysin [or neutral endopeptidase (NEP)] and the second is the M2 family represented by angiotensin‐converting enzyme [or peptidyl dipeptidase (ACE)]. Members of each of these families have served as important drug targets, particularly in cardiovascular disease and, more recently, have provided insight into mechanisms involved in neurodegeneration and neuroinflammation, especially from the point of view of processing of the amyloid precursor protein (APP) and its products in Alzheimer's disease (AD).
The discovery of ACE2 in our laboratory (Tipnis et al., 2000), as a result of genomics approaches to the identification of zinc metalloproteinases, together with its critical role in cardiac and lung development and function (Crackower 2002, Donoghue 2000) and as the cell surface receptor for the severe‐acute respiratory syndrome (SARS) Coronavirus (Li et al., 2003), emphasizes the validity of this strategy for identification of novel therapeutic targets. ACE2 was also suggested to play an important role in the brain renin–angiotensin system being widely expressed in various brain areas but restricted mostly to neuronal cells (Doobay et al., 2007).
II. The NEP Family
NEP, or neprilysin as it is now known, is a cell surface membrane‐bound glycoprotein and zinc peptidase also known as CD10 or common acute lymphoblastic leukemia antigen (CALLA). It was originally identified as a major antigen of renal membranes over 30 years ago and, at that time, was implicated in the metabolism of insulin. However, as found later, NEP degrades only the insulin B chain in vitro and not the intact insulin dimer, which suggests that NEP does not have any physiological role in degradation of insulin. Moreover, another zinc metallopeptidase, insulin‐degrading enzyme (IDE; insulysin), was subsequently discovered which appeared to fulfill the physiologically relevant function of insulin degradation. Whereas the highest concentrations of NEP are in the renal microvillar membrane, the first clues to its physiological roles came from studies on the metabolism of neuropeptides [especially the enkephalins and substance P (SP)] in the central nervous system where NEP is several orders of magnitude less abundant (Malfroy 1978, Matsas 1984, Relton 1983). It is now accepted that NEP functions to turn off neuropeptide signals at the synapse in an analogous fashion to the hydrolysis of acetylcholine by acetylcholinesterase at cholinergic synapses (see Turner and Tanzawa, 1997 for review). Evidence in vitro has clearly demonstrated that synaptic membranes efficiently degrade enkephalins and SP and that NEP was the primary enzyme responsible for these events (Matsas et al., 1983). Combined with the data obtained in vivo in rodents using potent and selective NEP inhibitors, such as phosphoramidon and thiorphan (Roques et al., 1980), key roles were established for NEP in the central nervous system. Subsequently, renal NEP was shown to be the principal enzyme inactivating the vasodilator, atrial natriuretic peptide (Kenny and Stephenson, 1988), which has led to much investment in the development of NEP inhibitors, either alone or in combination with ACE inhibitors (vasopeptidase inhibitors), as drugs in the treatment of hypertension, congestive heart failure, and renal disease (Bralet and Schwartz, 2001), although concerns over their safety have been raised (Quaschning, 2005).
NEP also plays important roles in other peripheral tissues such as in chemoreception and in potentiation of the response of the carotid body to hypoxia via degradation of its preferred substrate, SP (Kumar 1990, Kumar 2000). NEP was also detected in the liver, lungs, muscles, fat deposits, bones, the vertebrae, articular cartilages, and synoviae (Sales et al., 1991). In the bones, NEP plays a role in regulation of osteoblast and osteoclast metabolism mediated by both hormones and local bone peptide factors (Howell 1993, Ruchon 2000). In the skeletal muscles, NEP participates in regeneration of muscle fibers and there are data on its role associated with hereditary muscle disorders (Broccolini et al., 2006).
The cloning of NEP also revealed its identity with the CALLA or CD10 and has implicated NEP in cancer mechanisms (Letarte 1988, Tran‐Paterson 1989). For example, in human prostate cancer, NEP is dramatically downregulated (Papandreou et al., 1998) allowing mitogenic peptides such as bombesin and endothelin to drive androgen‐independent cell division in the prostate. The survey of the expression of NEP and the NEP homologue, endothelin‐converting enzyme 1 (ECE‐1), in a range of prostate cancers demonstrated that there is a certain balance between the levels of expression of NEP and ECE‐1 which determines the level of malignancy of the cells and that upregulation of ECE‐1 expression in metastatic cells may be indicative of its role in metastatic progression (Usmani et al., 2002). Later it was shown that NEP and ECE‐1 act as mediators of prostate cancer invasion via a stromal–epithelial interaction (Dawson et al., 2004). This and related observations have led to suggestions that the dysregulation of the balance of NEP and ECE can lead to the disease and selective reexpression of NEP in prostate cells may provide a novel approach to the treatment of prostate cancer. This is an excellent example where two homologous peptidases counterbalance each other's actions in physiology and pathology.
NEP cleaves a wide range of substrates including SP and other tachykinin peptides, in particular neurokinin B (Fig. 1 ), which makes it an important player in the arena of inflammation (Hooper and Turner, 1985). SP is released from sensory nerves inducing neurogenic inflammation and NEP, via SP degradation, limits its effects and those of other proinflammatory peptides. There are several studies reporting the role of NEP in peripheral inflammation, for example in the skin (Scholzen 2004, Scholzen 2001). Moreover, NEP but not ACE was shown to be the most important for CGRP degradation in human skin, and NEP inhibitors facilitated neurogenic inflammation in the skin (Kramer et al., 2005). NEP levels were also found to be significantly decreased in the plasma and monocytes of patients with juvenile idiopathic arthritis (Simonini et al., 2005). In the mouse model of intestinal inflammation caused by nematode infection NEP was demonstrated to downregulate the early onset of inflammation (Barbara et al., 2003). Inhibition of NEP was found to exacerbate both experimental pancreatitis and the associated lung injury (Day et al., 2005), and pretreatment with recombinant human NEP ameliorated this injury in NEP−/− transgenic mice (Lightner et al., 2002). In this connection, upregulation of NEP is considered as a potential therapeutic approach for pancreatitis‐associated lung injury, which can also be true in the case of other inflammatory diseases.
Fig. 1.

List of proinflammatory peptides cleaved by metalloproteinases.
Although the role of NEP in peripheral inflammation is well characterized, there are much less data on the role of NEP in inflammatory processes in the brain. However, the data on the localization and properties of NEP in the nervous system of insects provide evidence for an evolutionarily conserved role for NEP in the inactivation of tachykinin‐related peptides in the brain (Isaac 2003, Isaac 2002).
It was demonstrated that NEP might play an important role in the pathogenesis of AD due to its capacity to cleave the neurotoxic and inflammatory amyloid‐β (Aβ) peptide (Iwata et al., 2000) that is a primary trigger for the development of this disease (Hardy and Higgins, 1992). It is now well documented that Aβ is formed from a large precursor molecule called APP via two consecutive cleavages (Fig. 2 ). The first of these cleavages occurs in the extracellular or lumenal domain and is mediated by a membrane‐bound aspartic protease termed β‐secretase (BACE) (Vassar and Citron, 2000). It releases a large soluble fragment of sAPPβ and the residual membrane‐bound fragment. The latter is cleaved by a γ‐secretase protease complex, at residues 40–42 (termed γ‐site) or at residues 48–52 (termed ɛ‐site) within the transmembrane domain. The presenilin‐dependent γ‐secretase and γ‐site proteolytic activities are dependent on a multimeric complex of at least four different membrane proteins, including presenilin‐1 (PS1) or presenilin‐2 (PS2), nicastrin, Aph‐1, and Pen‐2 (Francis 2002, Yu 2000). In these complexes, the presenilins have been proposed as a novel type of transmembrane aspartic protease bearing the catalytic core of the γ‐secretase (Wolfe et al., 1999). Whereas the cleavage at the γ‐site generates Aβ, the subsequent cleavage at the ɛ‐site generates a cytosolic fragment referred to as ICD (Passer et al., 2000) or AICD (βAPP IntraCellular Domain). The exact role of AICD remains unclear but it has been suggested to act as a functional transcriptional regulator (Cao and Sudhof, 2001) in combination with the regulatory proteins Fe65 and the chromatin‐associated histone acetyltransferase, Tip60. There were reports that AICD and nicastrin regulate expression of NEP but this is controversial and still has to be proved experimentally in animal models (Hass 2005, Hebert 2006, Pardossi‐Piquard 2005, Pardossi‐Piquard 2006). An additional component of the γ‐secretase complex, termed TMP21, which is a member of the p24 cargo protein family, appears to differentially regulate γ‐secretase cleavage without affecting ɛ‐secretase activity (Chen et al., 2006). The proteolytic events involved in processing of APP are highly compartmentalized in the cell taking place both within intracellular compartments and in the plasma membrane. The rate limiting reaction of APP cleavage by β‐secretase (BACE) was shown to take place especially in lipid raft domains enriched with cholesterol and glycosphingolipids and targeting BACE to lipid rafts increased production of Aβ (Cordy et al., 2003). Depletion of cell cholesterol by lovastatin resulted in a decrease in both of sAPPβ and Aβ levels in the cell culture model. This explains the positive effect of statins on the development of AD pathology observed in patients with statin treatment (Sparks et al., 2006).
Fig. 2.
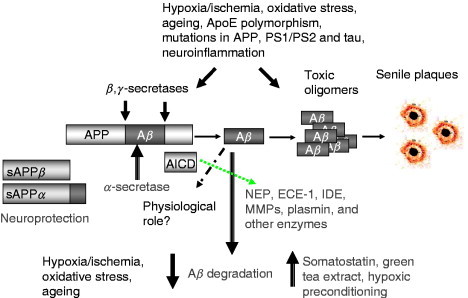
Amyloidogenic processing of APP and formation of amyloid (Aβ) peptide, fibrils, and plaques. Main avenues in neurodegeneration and neuroprotection. APP, amyloid precursor protein; sAPPα, soluble product of α‐secretase cleavage; sAPPβ, soluble product of β‐secretase cleavage; AICD, C‐terminal fragment of APP, product of γ‐secretase cleavage.
Under normal conditions, Aβ occurs as a soluble fragment, the concentration of which is normally tightly controlled below the threshold for its self‐aggregation into β sheet fibrils (Burdick et al., 1992). Until recently, production of Aβ in the brain and other tissues was thought to be an irreversible process, leading in the case of their disruption, to amyloidogenic diseases. However, in the last few years, neprilysin and several other proteases (IDE, ECE‐1 and ECE‐2, plasmin) have been found to be capable of degrading Aβin vitro and in vivo (for review see Carson 2002, Turner 2004). The sites of Aβ cleaved by known proteinases are shown in Fig. 3 . Pathological downregulation of these enzymes and, in particular of NEP, could predispose to accumulation of Aβ and the development of AD (Apelt 2003, Nalivaeva 2004). In particular, in mice deficient in NEP or ECE‐1, amyloid deposits are seen to deposit at significant levels in the brain (Eckman 2003, Hersh 2002). Furthermore, injection of amyloid‐β peptide into the brains of rodents significantly enhances the concentrations of NEP mRNA and protein, suggesting the operation of a regulatory feedback mechanism to protect neurons from toxic damage (Mohajeri et al., 2002). NEP levels appear to be reduced in high‐plaque‐bearing areas of human brain in AD and in cerebral amyloid angiopathy (Carpentier 2002, Yasojima 2001) but no association has been detected to date between polymorphisms in the NEP gene and AD (Lilius et al., 2003). Clinical data suggest that ischemia and stroke predispose to development of AD (Snowdon et al., 1997), and it was shown that hypoxia and ischemia lead to a decrease of NEP and ECE expression (Fisk 2006, Nalivaeva 2004). Thus, a possible therapeutic approach for treatment of AD might be a chronic upregulation of these proteinases (Fig. 4 ), either pharmacologically or through a gene therapy approach (Marr 2003, Saito 2005, Turner 2004).
Fig. 3.
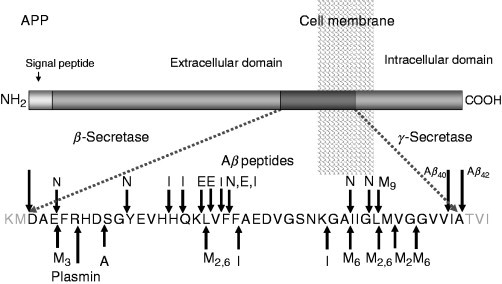
Cleavage sites of Aβ‐degrading enzymes. A, angiotensin‐converting enzyme; E, ECE‐1; I, IDE; N, NEP; M, matrix metalloproteinases (MMPs: M3, MMP‐3; M2 and M6, MMP‐2 and MMP‐6; M9, MMP‐9).
Fig. 4.
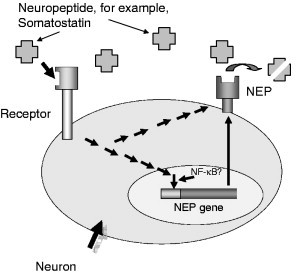
Scheme of upregulation of NEP gene and its possible involvement in neuroprotection [adapted from Saito et al. (2003)].
Maintenance of cellular concentrations of NEP, which is rather widely distributed in the body, is critical to peptide homeostasis and its up‐ or downregulation can lead to a range of pathological conditions including those of cardiovascular, neurodegenerative, and tumorigenic origins. NEP was, for quite some years, a lone mammalian zinc peptidase apparently mechanistically similar to the bacterial enzyme, thermolysin, both enzymes being potently inhibited by phosphoramidon. The structural solution of the catalytic domain of human NEP complexed with phosphoramidon (Oefner et al., 2000) reveals both similarities and differences with thermolysin, and the restricted access to the active site highlights why the enzyme acts exclusively as an oligopeptidase rather than a proteinase, unlike thermolysin.
A homologue of NEP, namely neprilysin 2 (NEP2), was discovered and in the brain shown to be restricted mainly to developing and differentiated fields of the CNS. Unlike NEP and ECE‐1, which are broadly expressed in the CNS and periphery, NEP2 was found to be almost exclusively expressed only in selected populations of neurons and in the spinal cord. The only peripheral areas where expression of NEP2 was detected were the pituitary and choroid plexuses. NEP2 was also found capable of degrading Aβ and its distinct localization from NEP suggests that, together with ECE‐1, it may be better poised to catabolize Aβ as it is more abundantly expressed in the areas relevant to AD pathology (Facchinetti 2003, Thomas 2005).
The human genome is now known to contain at least seven NEP‐related enzymes (summarized in Turner et al., 2001), of which the best characterized is ECE‐1, which catalyzes the final step in the biosynthesis of the potent vasoconstrictor peptide, endothelin‐1 (ET‐1; Matsumura 1990, Xu 1994). Several of the NEP‐like enzymes are, as yet, orphan peptidases with no recognized peptide substrates. Novel strategies are urgently needed to allow the identification of physiologically relevant substrates for such newly identified proteases.
III. The NEP Homologue ECE‐1
ECE‐1 was first purified from rat lung (Takahashi et al., 1993) but then was also found in a variety of tissues. It is most abundant in endothelial cells but is also expressed by exocrine cells, smooth muscle cells, neurons, and glia in the brain (Barnes 1999, Barnes 1997, Takahashi 1995). To date, four isoforms of human ECE‐1 differing only in a part of their N‐terminal cytoplasmic region but which cleave big ETs with similar efficiencies have been characterized: named ECE‐1a, ECE‐1b, ECE‐1c, and ECE‐1d (Schweizer 1997, Valdenaire 1999). Although the relative levels of the isoform mRNA species vary between human tissues, ECE‐1c mRNA is generally the predominant isoform message. There are distinct subcellular localizations for the four isoforms: whereas ECE‐1a, ECE‐1c, and ECE‐1d proteins are localized mainly at the cell surface, ECE‐1b was found to be intracellular and showed significant colocalization with a marker protein for the trans‐Golgi network (Schweizer et al., 1997). There are no significant differences in the catalytic properties between them, so it has been suggested that intracellular ECE‐1 localized in Golgi and vesicles might be involved in processing of big ET whereas cell surface ECE‐1 may metabolize other regulatory peptides (Turner et al., 1998).
ECE‐1 and its product ET‐1 have been shown to be involved in such inflammatory conditions as asthma (Zhang et al., 2004), chronic rhinitis with its expression in the nasal epithelium and mucosa being much higher in the case of rhinitis than in controls (Furukawa et al., 1996), and idiopathic pulmonary fibrosis (Saleh et al., 1997). In the latter case, an increased release of ET‐1 was accompanied by increased levels of ECE‐1 mRNA. Using normal bronchial epithelial cell culture, these authors have demonstrated that proinflammatory cytokines (IL‐1α, IL‐1β) induced a significant increase in ET‐1 release and mRNA expression, while TNF‐α stimulated expression of ECE‐1 mRNA. In the model of acute gastric infection caused by administration of Helicobacter pylori lipopolysaccharide, it was also shown that the levels of ET‐1 and expression of ECE‐1 were significantly increased in the gastric mucosa of infected cells on day 4 but then significantly reduced (down to 60%) by day 10 following reduced gastric inflammation (Slomiany et al., 2000). Developing this work further, the authors have discovered that ET‐1 has also an effect on leptin production in the gastric mucosa as a consequence of ET(A) receptor activation (Slomiany and Slomiany, 2005).
Together with ET‐1, ECE‐1 is abundantly present in human arteries and is involved in chronic inflammation in human atherosclerosis. Upregulation of the ECE‐1–ET‐1 system was shown to be closely linked to the presence of chronic inflammation at the very early stages of plaque evolution and, thus, has been suggested as a target in atherosclerosis therapy (Ihling 2001, Ihling 2004). Plasma levels of ECE‐1 were also shown to reflect the severity of ischemic complications after subarachnoid hemorrhage. The higher levels of plasma ECE‐1 resulted in reduced big ET‐1 and increased ET‐1/big ET‐1 ratio in patients who experienced symptomatic delayed cerebral ischemia, compared with other patients (Juvela, 2002). However, the levels of ECE‐1 expression in rat brain cortex hemispheres and hippocampus was found to be decreased after 15‐min global ischemia and restored to control levels after reperfusion (Nalivaeva et al., 2004), which might reflect an adaptive reaction of the brain to increased blood supply to the affected areas.
Although ECE‐1 has been regarded as a highly specific endopeptidase, it was demonstrated to be able to hydrolyze, apart from big ET‐1, a number of other biologically active peptides, such as bradykinin, SP, neurotensin, angiotensin I, and insulin B chain; it is not yet clear whether any of these, or other peptides, are physiological substrates of ECE‐1 (Hoang 1997, Johnson 1999). It was demonstrated that ECE‐1 can also degrade amyloid peptide Aβ, which made it an important player in the arena of AD (Eckman et al., 2001). Overexpression of ECE‐1 in Chinese hamster ovary cells, lacking endogenous ECE activity, was found to reduce extracellular Aβ concentration by up to 90% and this effect was completely abolished by treatment with a metalloproteinase inhibitor phosphoramidon. Recombinant soluble ECE‐1 was shown to hydrolyze synthetic Aβ40 and Aβ42 in vitro at multiple sites. Comparing ECE heterozygous and knockout mice, Eckman et al. (2003) showed that the concentration of Aβ peptides in the brain of these animals was elevated in comparison with control littermates in a gene‐dependent manner.
IV. The ACE Family
ACE was originally identified 50 years ago as a “hypertensin‐converting enzyme” (Skeggs et al., 1956) and its primary substrate was identified as angiotensin I, which it converts into the vasoconstrictor angiotensin II. In parallel, it inactivates the vasodilator and inflammatory peptide bradykinin. Hence, inhibition of ACE has a powerful effect in reduction of blood pressure and the enzyme has therefore been a major cardiovascular target for many years. The catalytic activity of ACE is primarily as a “peptidyl dipeptidase,” removing dipeptides from the C‐terminus of a susceptible peptide substrate. In the hydrolysis of some peptides (e.g., SP, luliberin), ACE can act as an endopeptidase, although with much lower catalytic efficiency (Turner and Hooper, 2002). Mammalian ACE exists as two distinct forms, arising from the use of alternative promoters. The simplest form is germinal or testicular ACE, which is essential for male fertility and which carries a single zinc‐binding and catalytic domain. Elsewhere in the body, the somatic form of ACE is duplicated and carries two active sites. The analysis of ACE X‐ray structures has revealed that ACE most closely resembles a neurotensin‐degrading zinc endopeptidase known as neurolysin rather than NEP, or carboxypeptidase A, on whose structure the design of ACE inhibitors was originally based (Hooper and Turner, 2003).
While seeking novel expressed sequence tags encoding zinc metallopeptidases, we identified and cloned the first human homologue of ACE (ACEH), which is now more commonly referred to as ACE2 (Tipnis et al., 2000). ACE2 is also a type I integral membrane peptidase showing 40% identity and 61% similarity with ACE and conserving the critical active site residues. It contains a single catalytic domain like testicular ACE, and it is most abundantly expressed in kidney, heart, and lung (Donoghue 2000, Tipnis 2000). Physiologically, the principal role of ACE2 is thought to be the conversion of angiotensin 2 to angiotensin (1–7), which opposes the actions of ACE2. Hence ACE and ACE2 act as counterbalances in metabolism in the renin–angiotensin system. Clues to the roles of ACE2 have come from the development of mice deficient in the ACE2 gene (Crackower et al., 2002). These mice have severe cardiac contractility defects, increased plasma angiotensin II levels, and an upregulation of cardiac hypoxia‐related genes, implicating ACE2 as an essential regulator of heart function. Intriguingly, a double knockout in mice of both the ACE and ACE2 genes is able to rescue the cardiac defect seen with the ACE2‐deficient mice (Crackower et al., 2002). ACE2 mRNA and protein levels are substantially reduced in the kidney in diabetic rats, suggesting that the enzyme may have a role in the development of diabetic complications (Tikellis et al., 2004). It has been shown that ACE2 is widely distributed throughout the brain but is mainly localized to the cytoplasm of neuronal cells in the brain and not present in glia. Moreover, ACE2 levels appear to be highly regulated by the renin–angiotensin system, suggesting its involvement in this system in the brain. ACE2 expression in the brain structures involved in the control of cardiovascular function suggests that it may have a role in the central regulation of blood pressure and hypertension (Doobay et al., 2007).
The most remarkable discovery in relation to ACE2 biology has been the demonstration that the enzyme functions as the receptor for the SARS virus (Li et al., 2003) and numerous subsequent studies have confirmed and extended this observation. Thus, the structure of the SARS Coronavirus spike receptor‐binding domain complexed with receptor has been determined (Li et al., 2005) and compounds blocking spike protein and ACE2 interaction discovered, for example emodin (Ho et al., 2007). ACE2 also appears to play a critical role in protection against acute lung injury from SARS infection, or other causes of acute respiratory distress syndrome (Imai et al., 2005).
One feature that both ACE (Hooper et al., 1987) and ACE2 (Lambert et al., 2005) share is the ability to be shed from the plasma membrane by cleavage within the juxtamembrane region releasing the bulk of the protein, including the intact catalytic domain, into the extracellular medium. This process is common to a growing number of membrane proteins of diverse characteristics (see Hooper et al., 1997 for review), an event which is generally receptor‐regulated and sensitive to inhibition by a group of hydroxamate metalloproteinase inhibitors such as batimastat. This has led to the identification of members of the ADAMs (a disintegrin and metalloproteinase) family of zinc proteinases, typified by tumor necrosis factor‐α converting enzyme (“TACE”; ADAM17) as candidate shedding enzymes (Allinson et al., 2004). Our data suggest that NEP might be also shed from the cell surface by a similar mechanism (Fisk, L., Nalivaeva, N. N., and Turner, A. J., unpublished data). However, the detailed molecular mechanism of this phenomenon has still to be elucidated.
ACE inhibitors have been shown to reduce development of diabetes, improve surrogate markers of inflammation, and reduce cardiovascular disease and renal disease (McFarlane et al., 2003). Increasing evidence indicates that systemic inflammation and neuroinflammation are central features in cerebrovascular disease and that hypertension, through the vasoactive peptides angiotensin and ET‐1, promotes and accelerates the atherosclerotic process via inflammatory mechanisms (Di Napoli and Papa, 2005). It was demonstrated that in humans the product of ACE activity angiotensin II induces IL‐6 production through a mineralocorticoid receptor‐dependent mechanism (Luther et al., 2007). This suggests that ACE inhibitors might also be considered as targets in neuroprotection.
An ACE polymorphism was demonstrated to be associated with AD in the Japanese population (Hu et al., 1999), and later it was found that ACE can indeed hydrolyze Aβin vitro and reduce accumulation of Aβ in cell cultures (Hu 2001, Oba 2005). However, Eckman et al. (2006) have demonstrated that in vivo ACE does not have a physiological role in clearing Aβ and it is cleaved in the brain mostly by NEP and ECE‐1 since ACE‐deficient mice did not demonstrate accumulation of Aβ while deficit of NEP or ECE‐1 activity resulted in additive increases in brain Aβ levels.
V. Ischemia/Hypoxia and Ageing as Factors Affecting Metalloproteinases
It is becoming more obvious that neurodegeneration and development of AD can be promoted by cardiovascular lesions, ischemia and stroke (Hofman 1997, Kalaria 2000). Indeed, the analysis of various autopsy series demonstrates that 60–90% of AD cases exhibit variable cerebrovascular pathology. Hypertension has also been suggested as a risk factor in development of AD (for review see Skoog and Gustafson, 2006). Taking into account that NEP and ECE‐1 are involved in vascular functions and can contribute to amyloid metabolism, we have analyzed levels of expression of ECE‐1 in the brain cortex of rats after 15‐min global ischemia and found a significant decrease of NEP and ECE‐1 protein levels in brain hemispheres and both hippocampi which returned to normal levels after 2‐h reperfusion (Nalivaeva et al., 2004). We have also demonstrated that NEP and ECE‐1 levels were lower in the cortex and striatum of rats subjected to prenatal hypoxia (7% O2, 3 h, 13th day of gestation) (Nalivaeva et al., 2003). Preconditioning to mild (15% O2) hypoxia before the episodes of acute hypoxia had a protective effect restoring the levels of NEP and ECE‐1 in rat brain structures analyzed during the first month after birth. Using human neuroblastoma NB7 cells, we have also demonstrated that hypoxia (1.0–2.5% O2) resulted in a decrease in expression of ECE‐1 (Fisk et al., 2006) and also in expression of NEP both at the protein and mRNA levels at and NEP activity (Fisk et al., 2007). Moreover, both hypoxia and ischemia resulted in an increased production of sAPPβ and reduced amount of sAPPα. These data allowed us to conclude that hypoxic and ischemic conditions in the brain might lead to a shift of amyloid metabolism toward formation of higher levels of Aβ due to an increased rate of β‐secretase reaction and reduced activity of such amyloid‐degrading enzymes as NEP and ECE‐1 (Fig. 4).
Analyzing the effect of chronic hypoxia (1%, 24 h) on expression of NEP estimated by the method of real‐time PCR in rat primary cortical neurons and astrocytes, we have found that NEP mRNA levels were downregulated (by 20%) under hypoxia in neurons and upregulated in astrocytes (Fig. 5 ). Previously, Apelt et al. (2003) reported NEP mRNA upregulation in amyloid plaque‐surrounding reactive astrocytes in transgenic Tg2576 mice that produce human amyloid‐β peptides from birth and develop amyloid‐β plaques which may suggest a role of plaque‐mediated astrogliosis in Aβ degradation. In our experiments, mRNA levels were also upregulated in hypoxic astrocytes, which might be an adaptive reaction of astrocytes to pathological conditions (Fisk et al., 2007).
Fig. 5.
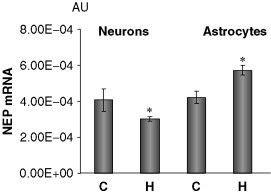
Effect of hypoxia on the level of NEP mRNA expression in primary cortical neurons and astrocytes. NEP mRNA detected by real‐time PCR (related to GAPDH mRNA). C, control; H, hypoxia (1% O2, 24 h, n = 12). *p < 0.05 compared to control.
It is important to note that expression of NEP with ageing was found to decrease in the cortex and hippocampus of rats, while its levels in the striatum (where amyloid deposits have not been reported in AD brains) were as high as at the end of the first month after birth (Nalivaeva et al., 2004). Decreased levels of another Aβ‐degrading enzyme, IDE, have also been reported in the brain of aged mice (Caccamo et al., 2005) and in rat brain structures (Nalivaeva‐Turner et al., 2006). Moreover, our experiments demonstrated that in the brain of rats with experimental type II diabetes IDE levels were lower than in control animals (Kochkina et al., 2006). These data suggest that age‐related deficit of amyloid‐degrading enzymes might be one of the factors leading to the development of the sporadic form of AD and upregulation of NEP and other Aβ‐degrading enzymes become one of the possible therapeutic targets in neurodegeneration and AD.
Several physiological and pharmaceutical ways of upregulation of the neprilysin gene have been suggested. One of the approaches, suggested by Saito et al. (2003), involves NEP substrates as molecules activating expression of the NEP gene via a positive feedback mechanism and as yet unknown signaling pathways (Fig. 4). Analyzing various NEP substrates in this experimental paradigm, these authors (Saito et al., 2005) were able to demonstrate that only somatostatin was capable to stimulate NEP activity in primary cortical neurons. Since somatostatin levels in the brain decrease with age these results indicate that age‐related downregulation of somatostatin expression could be one of the triggers for Aβ accumulation leading to late‐onset sporadic AD. In our experiments using human neuroblastoma cells expressing NEP and ECE‐1, we were not able to detect somatostatin‐dependent upregulation of NEP mRNA or activity while ECE‐1 was significantly upregulated by this peptide in a dose‐dependent manner (Fisk, L., Nalivaeva, N. N., and Turner, A. J., unpublished data).
Another physiological pathway of regulation of NEP gene has been suggested by Checler and colleagues who demonstrated that the C‐terminal product of APP cleavage by γ‐secretase (AICD) was able to transactivate the NEP gene promoter activating NEP expression and that presenilin molecules (PS1 and PS2) and the protein nicastrin in the γ‐secretase complex were essential for this effect (Pardossi‐Piquard 2005, Pardossi‐Piquard 2006).
Among chemical compounds that can upregulate NEP expression, green tea extract (EFLA85942) has been demonstrated as effective in human neuroblastoma SK‐N SH cells (Ayoub 2006, Melzig 2003). According to our observations, the active compound of the green tea extract, namely, polyphenol (–)‐epigallocatechin‐3‐gallate stimulates in a dose‐dependent manner both NEP expression at protein level and its activity in human neuroblastoma NB7 cells (Fisk, L., Nalivaeva, N. N., and Turner, A. J., unpublished data).
VI. Conclusions
In this chapter, several examples have demonstrated the role of metalloproteinases in neurodegeneration, neuroprotection, and neuroinflammation. This was mostly shown for NEP, ECE‐1, and ACE; however, there are other enzymes whose targeting might be beneficial for prevention of neurological disorders. Among them are the enzymes of amyloidogenic processing of APP by β‐secretase (and subsequently by γ‐secretase) and nonamyloidogenic APP processing by α‐secretase (for review see Cordy 2006, Neve 2003). Although both α‐ and β‐secretases produce neurotrophic fragments of APP (sAPPα and sAPPβ) and are believed to play a role in normal functioning of neuronal cells (Thornton 2006, Turner 2003), the α‐secretase pathway prevents production of Aβ and thus is regarded as neuroprotective. Downregulation of α‐secretase or upregulation of β‐secretase activity will lead to changes in the balance of production of Aβ toward its accumulation and there are emerging experimental data that this might be the case in ischemic or hypoxic brain (Nalivaeva 2004, Wen 2004). There are also some data that the same compounds can activate both α‐secretase and NEP activity, for example green tea extracts (Levites 2003, Melzig 2003), suggesting that neuroprotective therapies might target several points in pathogenic processes. However, there also might be situations when one compound upregulates one neuroprotective pathway and downregulates another. For example, the phorbol ester PMA was shown to activate the α‐secretase pathway (Zhu et al., 2001) but inhibit expression of ECE‐1 (Fisk et al., 2006). This observation implies the necessity of differential analysis of the effects of potentially effective neuroprotective drugs on various enzymes participating in amyloid metabolism.
Metalloproteinases represent important therapeutic targets not only in neurodegeneration but also in cardiovascular diseases and prostate cancer, and understanding their intricate interrelationship is still one of the most fascinating areas of modern molecular and cell biology.
References
- Allinson T., Parkin E., Turner A.J., Hooper N.M. The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. Eur. J. Biochem. 2004;271:2539–2547. doi: 10.1111/j.1432-1033.2004.04184.x. [DOI] [PubMed] [Google Scholar]
- Apelt J., Ach K., Schliebs R. Aging‐related down‐regulation of neprilysin, a putative β‐amyloid‐degrading enzyme, in transgenic Tg2576 Alzheimer‐like mouse brain is accompanied by an astroglial upregulation in the vicinity of β‐amyloid plaques. Neurosci. Lett. 2003;339:183–186. doi: 10.1016/s0304-3940(03)00030-2. [DOI] [PubMed] [Google Scholar]
- Ayoub S., Melzig M.F. Induction of neutral endopeptidase (NEP) activity of SK‐N‐SH cells by natural compounds from green tea. J. Pharm. Pharmacol. 2006;58:495–501. doi: 10.1211/jpp.58.4.0009. [DOI] [PubMed] [Google Scholar]
- Barbara G., De Giorgio R., Stanghellini V., Corinaldesi R., Cremon C., Gerard N., Gerard C., Grady E.F., Bunnett N.W., Blennerhassett P.A., Collins S.M. Neutral endopeptidase (EC 3. 4. 24. 11) downregulates the onset of intestinal inflammation in the nematode infected mouse. Gut. 2003;52:1457–1464. doi: 10.1136/gut.52.10.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes K., Turner A.J. Endothelin converting enzyme is located on β‐actin filaments in smooth muscle cells. Cardiovasc. Res. 1999;42:814–822. doi: 10.1016/s0008-6363(99)00009-7. [DOI] [PubMed] [Google Scholar]
- Barnes K., Walkden B.J., Wilkinson T.C., Turner A.J. Expression of endothelin‐converting enzyme in both neuroblastoma and glial cell lines and its localization in rat hippocampus. J. Neurochem. 1997;68:570–577. doi: 10.1046/j.1471-4159.1997.68020570.x. [DOI] [PubMed] [Google Scholar]
- Bralet J., Schwartz J.‐C. Vasopeptidase inhibitors: An emerging class of cardiovascular drugs. Trends Pharmacol. Sci. 2001;22:106–109. doi: 10.1016/s0165-6147(00)01644-8. [DOI] [PubMed] [Google Scholar]
- Broccolini A., Gidaro T., Morosetti R., Gliubizzi C., Servidei T., Pescatori M., Tonali P.A., Ricci E., Mirabella M. Neprilysin participates in skeletal muscle regeneration and is accumulated in abnormal muscle fibres of inclusion body myositis. J. Neurochem. 2006;96:777–789. doi: 10.1111/j.1471-4159.2005.03584.x. [DOI] [PubMed] [Google Scholar]
- Burdick D., Soreghan B., Kwon M., Kosmoski J., Knauer M., Henschen A., Yates J., Cotman C., Glabe C. Assembly and aggregation properties of synthetic Alzheimer's A4/amyloid peptide analogs. J. Biol. Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- Caccamo A., Oddo S., Sugarman M.C., Akbari Y., LaFerla F.M. Age‐ and region‐dependent alterations in Aβ‐degrading enzymes: Implications for Aβ‐induced disorders. Neurobiol. Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Cao X., Sudhof T.C. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Carpentier M., Robitaille Y., DesGroseillers L., Boileau G., Marcinkiewicz M. Declining expression of neprilysin in Alzheimer disease vasculature: Possible involvement in cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2002;61:849–856. doi: 10.1093/jnen/61.10.849. [DOI] [PubMed] [Google Scholar]
- Carson J.A., Turner A.J. β‐amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 2002;81:1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- Chen F., Hasegawa H., Schmitt‐Ulms G., Kawarai T., Bohm C., Katayama T., Gu Y., Sanjo N., Glista M., Rogaeva E., Wakutani Y., Pardossi‐Piquard R. TMP21 is a presenilin complex component that modulates γ‐secretase but not ɛ‐secretase activity. Nature. 2006;440:1208–1212. doi: 10.1038/nature04667. [DOI] [PubMed] [Google Scholar]
- Cordy J.M., Hussain I., Dingwall C., Hooper N.M., Turner A.J. Exclusively targeting β‐secretase to lipid rafts by GPI‐anchor addition up‐regulates beta‐site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordy J.M., Hooper N.M., Turner A.J. The involvement of lipid rafts in Alzheimer's disease. Mol. Membr. Biol. 2006;23:111–122. doi: 10.1080/09687860500496417. [DOI] [PubMed] [Google Scholar]
- Crackower M.A., Sarao R., Oudit G.Y., Yagil C., Kozieradzki I., Scanga S.E., Oliveira‐dos‐Santos A.J., da Costa J., Zhang L., Pei Y., Scholey J., Ferrario C.M. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:799–802. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- Dawson L.A., Maitland N.J., Turner A.J., Usmani B.A. Stromal‐epithelial interactions influence prostate cancer cell invasion by altering the balance of metallopeptidase expression. Br. J. Cancer. 2004;90:1577–1582. doi: 10.1038/sj.bjc.6601717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day A.L., Wick E., Jordan T.H., Jaffray C.E., Bunnett N.W., Grady E.F., Kirkwood K.S. Neutral endopeptidase determines the severity of pancreatitis‐associated lung injury. J. Surg. Res. 2005;128:21–27. doi: 10.1016/j.jss.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Di Napoli M., Papa F. Inflammation, blood pressure, and stroke: An opportunity to target primary prevention? Curr. Hypertens. Rep. 2005;7:44–51. doi: 10.1007/s11906-005-0054-8. [DOI] [PubMed] [Google Scholar]
- Doobay M.F., Talman L.S., Obr T.D., Tian X., Davisson R.L., Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin‐angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R373–R381. doi: 10.1152/ajpregu.00292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R.E., Acton S. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Eckman E.A., Reed D.K., Eckman C.B. Degradation of the Alzheimer's amyloid β peptide by endothelin‐converting enzyme. J. Biol. Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- Eckman E.A., Watson M., Marlow L., Sambamurti K., Eckman C.B. Alzheimer's disease β‐amyloid peptide is increased in mice deficient in endothelin‐converting enzyme. J. Biol. Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- Eckman E.A., Adams S.K., Troendle F.J., Stodola B.A., Kahn M.A., Fauq A.H., Xiao H.D., Bernstein K.E., Eckman C.B. Regulation of steady‐state β‐amyloid levels in the brain by neprilysin and endothelin‐converting enzyme but not angiotensin‐converting enzyme. J. Biol. Chem. 2006;281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- Facchinetti P., Rose C., Schwartz J.C., Ouimet T. Ontogeny, regional and cellular distribution of the novel metalloprotease neprilysin 2 in the rat: A comparison with neprilysin and endothelinconverting enzyme‐1. NeuroScience. 2003;118:627–639. doi: 10.1016/s0306-4522(02)01002-3. [DOI] [PubMed] [Google Scholar]
- Fisk L., Nalivaeva N.N., Turner A.J. Regulation of endothelin‐converting enzyme‐1 expression in human neuroblastoma cells. Exp. Biol. Med. 2006;231:1048–1053. [PubMed] [Google Scholar]
- Fisk L., Nalivaeva N.N., Boyle J.P., Peers C.S., Turner A.J. Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurons and astro cytes. Neurochem. Res. 2007 doi: 10.1007/s11064-007-9349-2. May 7, 2007 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Francis R., McGrath G., Zhang J., Ruddy D.A., Sym M., Apfeld J., Nicoll M., Maxwell M., Hai B., Ellis M.C., Parks A.L., Xu W. Aph‐1 and pen‐2 are required for Notch pathway signaling, γ‐secretase cleavage of APP, and presenilin protein accumulation. Dev. Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- Furukawa K., Saleh D., Bayan F., Emoto N., Kaw S., Yanagisawa M., Giaid A. Co‐expression of endothelin‐1 and endothelin‐converting enzyme‐1 in patients with chronic rhinitis. Am. J. Respir. Cell Mol. Biol. 1996;14:248–253. doi: 10.1165/ajrcmb.14.3.8845175. [DOI] [PubMed] [Google Scholar]
- Hardy J.A., Higgins G.A. Alzheimer's disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hass M.R., Yankner B.A. A γ‐secretase‐independent mechanism of signal transduction by the amyloid precursor protein. J. Biol. Chem. 2005;280:36895–36904. doi: 10.1074/jbc.M502861200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert S.S., Serneels L., Tolia A., Craessaerts K., Derks C., Filippov M.A., Muller U., De Strooper B. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 2006;7:739–745. doi: 10.1038/sj.embor.7400704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh L.B., Yu J., Zhu H., Gao J.T., Mucke L., Kindy M.S. Neprilysin regulation of amyloid deposition in Alzheimer's disease. J. Neurochem. 2002;81(Suppl. 1):67. [Google Scholar]
- Ho T.Y., Wu S.L., Chen J.C., Li C.C., Hsiang C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin‐converting enzyme 2 interaction. Antiviral Res. 2007;74:92–101. doi: 10.1016/j.antiviral.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang M.V., Turner A.J. Novel activity for endothelin‐converting enzyme: Hydrolysis of bradykinin. Biochem. J. 1997;327:23–26. doi: 10.1042/bj3270023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman A., Ott A., Breteler M.M., Bots M.L., Slooter A.J., Van Harskamp F., van Duijn C.N., Van Broeckhoven C., Grobbee D.E. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam Study. Lancet. 1997;349:151–154. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- Hooper N.M., Turner A.J. Neurokinin B is hydrolysed by synaptic membranes and by endopeptidase‐24.11 (enkephalinase) but not by angiotensin converting enzyme. FEBS Lett. 1985;190:133–136. doi: 10.1016/0014-5793(85)80443-9. [DOI] [PubMed] [Google Scholar]
- Hooper N.M., Turner A.J. An ACE structure. Nat. Struct. Biol. 2003;10:155–157. doi: 10.1038/nsb0303-155. [DOI] [PubMed] [Google Scholar]
- Hooper N.M., Keen J., Pappin D.J.C., Turner A.J. Pig kidney angiotensin converting enzyme. Purification and characterization of amphipathic and hydrophilic forms of the enzyme establishes C‐terminal anchorage to the plasma membrane. Biochem. J. 1987;247:85–93. doi: 10.1042/bj2470085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper N.M., Karran E.H., Turner A.J. Membrane protein secretases. Biochem. J. 1997;321:265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell S., Caswell A.M., Kenny A.J., Turner A.J. Membrane peptidases on human osteoblast‐like cells in culture: Hydrolysis of calcitonin and hormonal regulation of endopeptidase‐24.11. Biochem. J. 1993;290:159–164. doi: 10.1042/bj2900159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Miyatake F., Aizu Y., Nakagawa H., Nakamura S., Tamaoka A., Takahashi R., Urakami K., Shoji M. Angiotensin‐converting enzyme genotype is associated with Alzheimer disease in the Japanese population. Neurosci. Lett. 1999;277:65–67. doi: 10.1016/s0304-3940(99)00827-7. [DOI] [PubMed] [Google Scholar]
- Hu J., Igarashi A., Kamata M., Nakagawa H. Angiotensin‐converting enzyme degrades Alzheimer amyloid β‐peptide (Aβ); retards Aβ aggregation, deposition, fibril formation; and inhibits cytotoxicity. J. Biol. Chem. 2001;276:47863–47868. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- Ihling C., Szombathy T., Bohrmann B., Brockhaus M., Schaefer H.E., Loeffler B.M. Coexpression of endothelin‐converting enzyme‐1 and endothelin‐1 in different stages of human atherosclerosis. Circulation. 2001;104:864–869. doi: 10.1161/hc3301.094742. [DOI] [PubMed] [Google Scholar]
- Ihling C., Bohrmann B., Schaefer H.E., Technau‐Ihling K., Loeffler B.M. Endothelin‐1 and endothelin converting enzyme‐1 in human atherosclerosis–novel targets for pharmacotherapy in atherosclerosis. Curr. Vasc. Pharmacol. 2004;2:249–258. doi: 10.2174/1570161043385718. [DOI] [PubMed] [Google Scholar]
- Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong‐Poi H., Crackower M.A., Fukamizu A. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac R.E., Nassel D.R. Identification and localization of a neprilysin‐like activity that degrades tachykinin‐related peptides in the brain of the cockroach, Leucophaea maderae, and locust, Locusta migratoria. J. Comp. Neurol. 2003;457:57–66. doi: 10.1002/cne.10561. [DOI] [PubMed] [Google Scholar]
- Isaac R.E., Parkin E.T., Keen J.N., Nassel D.R., Siviter R.J., Shirras A.D. Inactivation of a tachykinin‐related peptide: Identification of four neuropeptide‐degrading enzymes in neuronal membranes of insects from four different orders. Peptides. 2002;23:725–733. doi: 10.1016/s0196-9781(01)00653-2. [DOI] [PubMed] [Google Scholar]
- Iwata N., Tsubuki S., Takaki Y., Watanabe K., Seikiguchi M., Hosoki E., Kawashima‐Morishima M., Lee H.‐J., Hama E., Sekine‐Aizawa Y., Saido T.C. Identification of the major Aβ1–42‐degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- Johnson G.D., Stevenson T., Ahn K. Hydrolysis of peptide hormones by endothelin‐converting enzyme‐1. A comparison with neprilysin. J. Biol. Chem. 1999;274:4053–4058. doi: 10.1074/jbc.274.7.4053. [DOI] [PubMed] [Google Scholar]
- Juvela S. Plasma endothelin and big endothelin concentrations and serum endothelin‐converting enzyme activity following aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2002;97:1287–1293. doi: 10.3171/jns.2002.97.6.1287. [DOI] [PubMed] [Google Scholar]
- Kalaria R.N. The role of cerebral ischemia in Alzheimer's disease. Neurobiol. Aging. 2000;21:321–330. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- Kenny A.J., Stephenson S.L. Role of endopeptidase‐24.11 in the inactivation of atrial natriuretic peptide. FEBS Lett. 1988;232:1–8. doi: 10.1016/0014-5793(88)80375-2. [DOI] [PubMed] [Google Scholar]
- Kochkina E.G., Plesneva S.A., Zhuravin I.A., Turner A.J., Nalivaeva N.N. Study of insulin‐degrading enzyme expression in the brain and peripheral organs of hypoxic and diabetic rats. 11th meeting of Czech and Slovak Neurochemical Society, Martin, Slovakia; 2006. p. 54. [Google Scholar]
- Kramer H.H., Schmidt K., Leis S., Schmelz M., Sommer C., Birklein F. Inhibition of neutral endopeptidase (NEP) facilitates neurogenic inflammation. Exp. Neurol. 2005;195:179–184. doi: 10.1016/j.expneurol.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Kumar G.K., Runold M., Ghai R.D., Cherniack N.S., Prabhakar N.R. Occurrence of neutral endopeptidase activity in the cat carotid body and its significance in chemoreception. Brain Res. 1990;517:341–343. doi: 10.1016/0006-8993(90)91047-k. [DOI] [PubMed] [Google Scholar]
- Kumar G.K., Yu R.K., Overholt J.L., Prabhakar N.R. Role of substance P in neutral endopeptidase modulation of hypoxic response of the carotid body. Adv. Exp. Med. Biol. 2000;475:705–713. [PubMed] [Google Scholar]
- Lambert D.W., Yarski M., Warner F.J., Thornhill P., Parkin E.T., Smith A.I., Hooper N.M., Turner A.J. Tumor necrosis factor‐alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe‐acute respiratory syndrome‐coronavirus (SARS‐CoV) receptor, angiotensin‐converting enzyme‐2 (ACE2) J. Biol. Chem. 2005;280:30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letarte M., Vera S., Tran R., Addis J.B., Onizuka R.J., Quackenbush E.J., Jongeneel C.V., McInnes R.R. Common acute lymphocytic leukemia antigen is identical to neutral endopeptidase. J. Exp. Med. 1988;168:1247–1253. doi: 10.1084/jem.168.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y., Amit T., Mandel S., Youdim M.B. Neuroprotection and neurorescue against Aβ toxicity and PKC‐dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (–)‐epigallocatechin‐3‐gallate. FASEB J. 2003;17:952–954. doi: 10.1096/fj.02-0881fje. [DOI] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightner A.M., Jordan T.H., Bunnett N.W., Grady E.F., Kirkwood K.S. Recombinant human neutral endopeptidase ameliorates pancreatic elastase‐induced lung injury. Surgery. 2002;132:193–199. doi: 10.1067/msy.2002.125309. [DOI] [PubMed] [Google Scholar]
- Lilius L., Forsell C., Axelman K., Winblad B., Graff C., Tjernberg L. No association between polymorphisms in the neprilysin promoter region and Swedish Alzheimer's disease patients. Neurosci. Lett. 2003;337:111–113. doi: 10.1016/s0304-3940(02)01300-9. [DOI] [PubMed] [Google Scholar]
- Luther J.M., Gainer J.V., Murphey L.J., Yu C., Vaughan D.E., Morrow J.D., Brown N.J. Angiotensin II induces interleukin‐6 in humans through a mineralocorticoid receptor‐dependent mechanism. Hypertension. 2007;48:1050–1057. doi: 10.1161/01.HYP.0000248135.97380.76. [DOI] [PubMed] [Google Scholar]
- Malfroy B., Swerts J.P., Guyon A., Roques B.P., Schwartz J.‐C. High‐affinity enkephalin‐degrading peptidase in brain is increased after morphine. Nature. 1978;276:523–526. doi: 10.1038/276523a0. [DOI] [PubMed] [Google Scholar]
- Marr R.A., Rockenstein E., Mukherjee A., Kindy M.S., Hersh L.B., Gage F.H., Verma I.M., Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsas R., Fulcher I.S., Kenny A.J., Turner A.J. Substance P and [Leu]enkephalin are hydrolyzed by an enzyme in pig caudate synaptic membranes that is identical with the endopeptidase of kidney microvilli. Proc. Natl. Acad. Sci. USA. 1983;80:3111–3115. doi: 10.1073/pnas.80.10.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsas R., Turner A.J., Kenny A.J. The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase‐24.11. Biochem. J. 1984;223:433–440. doi: 10.1042/bj2230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y., Ikegawa R., Takaoka M., Morimoto S. Conversion of porcine big endothelin to endothelin by an extract from the porcine aortic endothelial cells. Biochem. Biophys. Res. Commun. 1990;167:203–210. doi: 10.1016/0006-291x(90)91751-d. [DOI] [PubMed] [Google Scholar]
- McFarlane S.I., Kumar A., Sowers J.R. Mechanisms by which angiotensin‐converting enzyme inhibitors prevent diabetes and cardiovascular disease. Am. J. Cardiol. 2003;91:30H–37H. doi: 10.1016/s0002-9149(03)00432-6. [DOI] [PubMed] [Google Scholar]
- Melzig M.F., Janka M. Enhancement of neutral endopeptidase activity in SK‐N‐SH cells by green tea extract. Phytomedicine. 2003;10:494–498. doi: 10.1078/094471103322331449. [DOI] [PubMed] [Google Scholar]
- Mohajeri M.H., Wollmer M.A., Nitsch R.M. Aβ42‐induced increase in neprilysin is associated with prevention of amyloid plaque formation in vivo. J. Biol. Chem. 2002;277:35460–35465. doi: 10.1074/jbc.M202899200. [DOI] [PubMed] [Google Scholar]
- Nalivaeva N.N., Fisk L., Canet Aviles R.M., Plesneva S.A., Zhuravin I.A., Turner A.J. Effects of prenatal hypoxia on expression of amyloid precursor protein and metallopeptidases in the rat brain. Lett. Peptide Sci. 2003;10:455–462. [Google Scholar]
- Nalivaeva N.N., Fisk L., Kochkina E.G., Plesneva S.A., Zhuravin I.A., Babusikova E., Dobrota D., Turner A.J. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid‐degrading enzymes. Ann. NY Acad. Sci. 2004;1035:21–33. doi: 10.1196/annals.1332.002. [DOI] [PubMed] [Google Scholar]
- Nalivaeva‐Turner N.N., Fisk L., Kochkina E.G., Makova N., Plesneva S.A., Zhuravin I.A., Turner A.J. Developmental dynamics of amyloid‐degrading enzymes in the rat brain under normal and hypoxic conditions. Proceedings of the 5th FENS Forum, Vienna, Austria, July 8–12, 2006; 2006. A130.30. [Google Scholar]
- Neve R.L. A new wrestler in the battle between α‐ and β‐secretases for cleavage of APP. Trends Neurosci. 2003;26:461–463. doi: 10.1016/s0166-2236(03)00231-5. [DOI] [PubMed] [Google Scholar]
- Oba R., Igarashi A., Kamata M., Nagata K., Takano S., Nakagawa H. The N‐terminal active centre of human angiotensin‐converting enzyme degrades Alzheimer amyloid β‐peptide. Eur. J. Neurosci. 2005;21:733–740. doi: 10.1111/j.1460-9568.2005.03912.x. [DOI] [PubMed] [Google Scholar]
- Oefner C., D'Arcy A., Nennig M., Winkler F.K., Dale G.E. Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. J. Mol. Biol. 2000;296:341–349. doi: 10.1006/jmbi.1999.3492. [DOI] [PubMed] [Google Scholar]
- Papandreou C.N., Usmani B., Geng Y., Bogenrieder T., Freeman R., Wilk S., Finstad C.L., Reuter V.E., Powell C.T., Scheinberg D., Magill C., Scher H.I. Neutral endopeptidase 24.11 loss in metastatic human prostate cancer contributes to androgen‐independent progression. Nat. Med. 1998;4:50–57. doi: 10.1038/nm0198-050. [DOI] [PubMed] [Google Scholar]
- Pardossi‐Piquard R., Petit A., Kawarai T., Sunyach C., Alves da Costa C., Vincent B., Ring S., D'Adamio L., Shen J., Muller U., St. George‐Hyslop P., Checler F. Presenilin‐dependent transcriptional control of the Aβ‐degrading enzyme neprilysin by intracellular domains of APP and APLP. Neuron. 2005;46:541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Pardossi‐Piquard R., Dunys J., Yu G., St. George‐Hyslop P., Alves da Costa C., Checler F. Neprilysin activity and expression are controlled by nicastrin. J. Neurochem. 2006;97:1052–1056. doi: 10.1111/j.1471-4159.2006.03822.x. [DOI] [PubMed] [Google Scholar]
- Passer B., Pellegrini L., Russo C., Siegel R.M., Lenardo M.J., Schettini G., Bachmann M., Tabaton M., D'Adamio L. Generation of an apoptotic intracellular peptide by gamma‐secretase cleavage of Alzheimer's amyloid protein precursor. J. Alzheimers Dis. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- Puente X.S., Sanchez L.M., Overall C.M., Lopez‐Otin C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- Quaschning T. Vasopeptidase inhibition for blood pressure control: Emerging experience. Curr. Pharm. Des. 2005;11:3293–3299. doi: 10.2174/138161205774424708. [DOI] [PubMed] [Google Scholar]
- Rawlings N.D., Barrett A.J. Intruduction: Metallopeptidases and their clans. In: Barrett A.J., Rawlings N.D., Woessner J.F., editors. “Handbook of Proteolytic Enzymes”. Elsevier Academic Press; London: 2004. pp. 231–268. [Google Scholar]
- Rawlings N.D., Morton F.R., Barrett A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2006;34:D270–D272. doi: 10.1093/nar/gkj089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton J.M., Gee N.S., Matsas R., Turner A.J., Kenny A.J. Purification of endopeptidase‐24.11 (‘enkephalinase’) from pig brain by immunoadsorbent chromatography. Biochem. J. 1983;215:519–523. doi: 10.1042/bj2150519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roques B.P., Fournié‐Zaluski M.‐C., Soroca E., Lecomte J.‐M., Malfroy B., Llorens‐Cortes C., Schwartz J.‐C. The enkephalinase inhibitor thiorphan shows antinociceptive activity in mice. Nature. 1980;288:286–288. doi: 10.1038/288286a0. [DOI] [PubMed] [Google Scholar]
- Ruchon A.F., Marcinkiewicz M., Ellefsen K., Basak A., Aubin J., Crine P., Boileau G. Cellular localization of neprilysin in mouse bone tissue and putative role in hydrolysis of osteogenic peptides. J. Bone Miner. Res. 2000;15:1266–1274. doi: 10.1359/jbmr.2000.15.7.1266. [DOI] [PubMed] [Google Scholar]
- Saito T., Takaki Y., Iwata N., Trojanowski J., Saido T.C. Alzheimer's disease, neuropeptides, neuropeptidase, and amyloid‐β peptide metabolism. Sci. Aging Knowledge Environ. 2003;3:E1. doi: 10.1126/sageke.2003.3.pe1. [DOI] [PubMed] [Google Scholar]
- Saito T., Iwata N., Tsubuki S., Takaki Y., Takano J., Huang S.M., Suemoto T., Higuchi M., Saido T.C. Somatostatin regulates brain amyloid‐β peptide A42 through modulation of proteolytic degradation. Nat. Med. 2005;11:434–439. doi: 10.1038/nm1206. [DOI] [PubMed] [Google Scholar]
- Saleh D., Furukawa K., Tsao M.S., Maghazachi A., Corrin B., Yanagisawa M., Barnes P.J., Giaid A. Elevated expression of endothelin‐1 and endothelin‐converting enzyme‐1 in idiopathic pulmonary fibrosis: Possible involvement of proinflammatory cytokines. Am. J. Respir. Cell Mol. Biol. 1997;16:187–193. doi: 10.1165/ajrcmb.16.2.9032126. [DOI] [PubMed] [Google Scholar]
- Sales N., Dutriez I., Maziere B., Ottaviani M., Roques B.P. Neutral endopeptidase 24.11 in rat peripheral tissues: Comparative localization by ‘ex vivo’ and ‘in vitro’ autoradiography. Regul. Pept. 1991;33:209–222. doi: 10.1016/0167-0115(91)90215-3. [DOI] [PubMed] [Google Scholar]
- Scholzen T.E., Luger T.A. Neutral endopeptidase and angiotensin‐converting enzyme—key enzymes terminating the action of neuroendocrine mediators. Exp. Dermatol. 2004;13(Suppl. 4):22–26. doi: 10.1111/j.1600-0625.2004.00260.x. [DOI] [PubMed] [Google Scholar]
- Scholzen T.E., Steinhoff M., Bonaccorsi P., Klein R., Amadesi S., Geppetti P., Lu B., Gerard N.P., Olerud J.E., Luger T.A., Bunnett N.W., Grady E.F. Neutral endopeptidase terminates substance P‐induced inflammation in allergic contact dermatitis. J. Immunol. 2001;166:1285–1291. doi: 10.4049/jimmunol.166.2.1285. [DOI] [PubMed] [Google Scholar]
- Schweizer A., Valdenaire O., Nelböck P., Deuschle U., Dumas M., Edwards J.B., Stumpf J.G., Löffler B.M. Human endothelin converting enzyme (ECE‐1): Three isoforms with distinct subcellular localizations. Biochem. J. 1997;328:871–877. doi: 10.1042/bj3280871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonini G., Azzari C., Gelli A.M., Giani T., Calabri G.B., Leoncini G., Del Rosso A., Generini S., Cimaz R., Cerinic M.M., Falcini F. Neprilysin levels in plasma and synovial fluid of juvenile idiopathic arthritis patients. Rheumatol. Int. 2005;25:336–340. doi: 10.1007/s00296-004-0447-z. [DOI] [PubMed] [Google Scholar]
- Skeggs L.T., Kahn J.R., Shumway N.P.J. The preparation and function of the hypertensin‐converting enzyme. Exp. Med. 1956;103:295–299. doi: 10.1084/jem.103.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog I., Gustafson D. Update on hypertension and Alzheimer's disease. Neurol. Res. 2006;28:605–611. doi: 10.1179/016164106X130506. [DOI] [PubMed] [Google Scholar]
- Slomiany B.L., Slomiany A. Endothelin‐1‐dependent leptin induction in gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide. Biochem. Biophys. Res. Commun. 2005;336:1106–1111. doi: 10.1016/j.bbrc.2005.08.236. [DOI] [PubMed] [Google Scholar]
- Slomiany B.L., Piotrowski J., Slomiany A. Up‐regulation of endothelin‐converting enzyme‐1 in gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide. Biochem. Biophys. Res. Commun. 2000;267:801–805. doi: 10.1006/bbrc.1999.2037. [DOI] [PubMed] [Google Scholar]
- Snowdon D.A., Greiner L.H., Mortimer J.A., Riley K.P., Greiner P.A., Markesbery W.R. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- Sparks D.L., Sabbagh M., Connor D., Soares H., Lopez J., Stankovic G., Johnson‐Traver S., Ziolkowski C., Browne P. Statin therapy in Alzheimer's disease. Acta Neurol. Scand. Suppl. 2006;185:78–86. doi: 10.1111/j.1600-0404.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- Takahashi M., Matsushita Y., Iijima Y., Tanzawa K. Purification and characterization of endothelin converting enzyme from rat lung. J. Biol. Chem. 1993;268:21394–21398. [PubMed] [Google Scholar]
- Takahashi M., Fukuda K., Shimada K., Barnes K., Turner A.J., Ikeda M., Koike M., Yamamoto Y., Tanzawa K. Localization of rat endothelin converting enzyme to vascular endothelial cells and some secretory cells. Biochem. J. 1995;311:657–665. doi: 10.1042/bj3110657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J.E., Rylett C.M., Carhan A., Bland N.D., Bingham R.J., Shirras A.D., Turner A.J., Isaac R.E. Drosophila melanogaster NEP2 is a new soluble member of the neprilysin family of endopeptidases with implications for reproduction and renal function. Biochem. J. 2005;386:357–366. doi: 10.1042/BJ20041753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton E., Vink R., Blumbergs P.C., Van Den Heuvel C. Soluble amyloid precursor protein α reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006;1094:38–46. doi: 10.1016/j.brainres.2006.03.107. [DOI] [PubMed] [Google Scholar]
- Tikellis C., Johnston C.I., Forbes J.M., Burns W.C., Burrell L.M., Risvanis J., Cooper M.E. Characterization of renal angiotensin‐converting enzyme 2 in diabetic nephropathy. Hypertension. 2004;41:392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Tran‐Paterson R., Willard H.F., Letarte M. The common acute lymphoblastic leukemia antigen (neutral endopeptidase‐3.4.24.11) gene is located on human chromosome 3. Cancer Genet. Cytogenet. 1989;42:129–134. doi: 10.1016/0165-4608(89)90015-0. [DOI] [PubMed] [Google Scholar]
- Turner A.J., Hooper N.M. The angiotensin‐converting enzyme gene family: Genomics and pharmacology. Trends Pharmacol. Sci. 2002;23:177–183. doi: 10.1016/s0165-6147(00)01994-5. [DOI] [PubMed] [Google Scholar]
- Turner A.J., Tanzawa K. Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J. 1997;11:355–364. doi: 10.1096/fasebj.11.5.9141502. [DOI] [PubMed] [Google Scholar]
- Turner A.J., Barnes K., Schweizer A., Valdenaire O. Isoforms of endothelinconverting enzyme: Why and where? Trends Pharmacol. Sci. 1998;19:483–486. doi: 10.1016/s0165-6147(98)01251-6. [DOI] [PubMed] [Google Scholar]
- Turner A.J., Isaac R.E., Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: Genomics and function. Bioessays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Turner A.J., Fisk L., Nalivaeva N.N. Targeting amyloid‐degrading enzymes as therapeutic strategies in neurodegeneration. Ann. NY Acad. Sci. 2004;1035:1–20. doi: 10.1196/annals.1332.001. [DOI] [PubMed] [Google Scholar]
- Turner P.R., O'Connor K., Tate W.P., Abraham W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Usmani B.A., Harden B., Maitland N.J., Turner A.J. Differential expression of neutral endopeptidase‐24.11 (neprilysin) and endothelin‐converting enzyme in human prostate cancer cell lines. Clin. Sci. (Lond.) 2002;103(Suppl. 48):314S–317S. doi: 10.1042/CS103S314S. [DOI] [PubMed] [Google Scholar]
- Valdenaire O., Lepailleur‐Enouf D., Edidy G., Thouard A., Barrer A., Vranckx R., Tougard C., Michel J.B. A fourth isoform of endothelin‐converting enzyme (ECE‐1) is generated from an additional promoter. Eur. J. Biochem. 1999;264:341–349. doi: 10.1046/j.1432-1327.1999.00602.x. [DOI] [PubMed] [Google Scholar]
- Vassar R., Citron M. Aβ‐generating enzymes: Recent advances in β‐ and γ‐secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Wen Y., Onyewuchi O., Yang S., Liu R., Simpkins J.W. Increased β‐secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Wolfe M.S., Xia W., Ostaszewski B.L., Diehl T.S., Kimberly W.T., Selkoe D.J. Two transmembrane aspartates in presenilin‐1 required for presenilin endoproteolysis and γ‐secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Yasojima K., Akiyama H., McGeer E.G., McGeer P.L. Reduced neprilysin in high plaque areas of Alzheimer brain: A possible relationship to deficient degradation of β‐amyloid peptide. Neurosci. Lett. 2001;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- Xu D., Emoto N., Giaid A., Slaughter C., Kaw S., de Wit D., Yanagisawa M. ECE‐1: A membrane‐bound metalloprotease that catalyzes the proteolytic activation of big endothelin‐1. Cell. 1994;78:473–845. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- Yu G., Nishimura M., Arawaka S., Levitan D., Zhang L., Tandon A., Song Y.Q., Rogaeva E., Chen F., Kawarai T., Supala A., Levesque L. Nicastrin modulates presenilin‐mediated notch/glp‐1 signal transduction and APP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Adner M., Cardell L.O. Interleukin‐1β attenuates endothelin B receptor‐mediated airway contractions in a murine in vitro model of asthma: Roles of endothelin converting enzyme and mitogen‐activated protein kinase pathways. Clin. Exp. Allergy. 2004;34:1480–1487. doi: 10.1111/j.1365-2222.2004.02040.x. [DOI] [PubMed] [Google Scholar]
- Zhu G., Wang D., Lin Y.H., McMahon T., Koo E.H., Messing R.O. Protein kinase Cɛ suppresses Aβ production and promotes activation of α‐secretase. Biochem. Biophys. Res. Commun. 2001;285:997–1006. doi: 10.1006/bbrc.2001.5273. [DOI] [PubMed] [Google Scholar]