

Abstract
This chapter discusses the epidemiology of Parkinson's disease (PD). Classically, PD refers to progressive parkinsonism caused by loss of pigmented aminergic brainstem neurons without an identifiable cause, while parkinsonism refers simply to the syndrome of bradykinesia, resting tremor, rigidity and postural reflex impairment. Over nearly two centuries, Parkinson's clinical description has provided the framework for clinical investigations, including epidemiologic ones. Descriptions of PD were limited to selected clinical settings until the middle of the 20th century. Since then, epidemiologic approaches have been used not only to investigate the population distribution of PD, but also as a way to glean clues as to the cause of this “idiopathic” disorder. Because PD is relatively infrequent, a large base population must be surveyed to identify sufficient numbers of cases for a study. In some instances, PD cases can be identified through health service rosters within defined geographic areas or in enumerated populations. In others, cases of PD are sought independently of the health care system, such as through door-to-door surveys. While the latter approach is theoretically least likely to exclude cases, the time and cost involved are also greatest using this approach.
6.1. Introduction
6.1.1. Overview
Parkinson's disease (PD) was first described as ‘paralysis agitans’ by James Parkinson in 1817 (Parkinson, 1817). Classically, PD refers to progressive parkinsonism due to loss of pigmented aminergic brainstem neurons without an identifiable cause, while ‘parkinsonism’ refers simply to the syndrome of bradykinesia, resting tremor, rigidity and postural reflex impairment. Over nearly two centuries, Parkinson's clinical description has provided the framework for clinical investigations, including epidemiologic ones. More recently, investigations of a few families with genetic parkinsonism have revealed a wide range of clinical and pathological features in persons sharing a single disease‐causing mutation, calling into question the assumptions underlying the strict syndromic classification employed by most studies to date. Whether a similar heterogeneity applies to the common ‘idiopathic’ disorder is not known. Although our current clinicopathological definition of PD may merit revision, further work is needed to determine what an appropriate new definition should be, and the classical definitions will be used in this discussion.
Descriptions of PD were limited to selected clinical settings until the middle of the 20th century, when several population‐based epidemiologic studies were published (Kurland 1958, Gudmundsson 1967). Since then, epidemiologic approaches have been used not only to investigate the population distribution of PD, but also as a way to glean clues as to the cause of this ‘idiopathic’ disorder. Heredity, infection, toxicant exposure and multifactorial gene–environment interactions have been proposed. Although single genetic or environmental causes have been identified, these explain only a small proportion of all cases (Marras 2002, Korell 2005). This chapter will provide an overview of this work, beginning with descriptive studies, followed by studies investigating the determinants of PD. Because other chapters in this volume thoroughly address the genetics of PD, this topic will be considered briefly here.
6.1.2. Special considerations for epidemiologic studies
Epidemiologic investigations of PD must overcome practical challenges (Table 6.1 ). PD is relatively uncommon, and even studies of large populations will find relatively few cases. Therefore, the potential error in any single study may be significant. In analytic studies, this can be particularly problematic if the causes of disease differ across populations. Moreover, to the extent that the cause of PD is multifactorial, large populations will be necessary to test hypotheses involving multiple determinants.
Table 6.1.
Some challenges in the epidemiological study of Parkinson's disease
| No diagnostic test |
| • Examination by expert most reliable |
| • No criterion always predicts pathology |
| Long preclinical period |
| • Exposure may be years before signs |
| Late‐life disorder |
| • Risk factor identification retrospective |
| • Diagnostic accuracy in relatives poor |
| Relatively rare |
| • Large base population needed |
| No registries; not reported |
| • ‘True’ distribution of disease not certain |
Identification of the cases of PD within a community is typically not a trivial effort. Population‐based registries of PD are not common, and voluntary registries cannot be assumed to be representative. Because PD is relatively infrequent, a fairly large base population must be surveyed to identify sufficient numbers of cases for a study. In some instances, PD cases can be identified through health service rosters within defined geographic areas or in enumerated populations. In others, cases of PD are sought independently of the health care system, such as through door‐to‐door surveys. While the latter approach is theoretically least likely to exclude cases, the time and cost involved are also greatest using this approach.
Population surveys of PD are further complicated because there is no diagnostic test for PD. Clinical features remain the only way to diagnose PD during life. Clinical diagnostic accuracy can vary with the experience of the practitioner. Essential tremor, for example, may be confused with PD in up to 40% of diagnoses in some settings (Mutch et al., 1986). Conversely, actual cases of PD may be missed, particularly in older age groups, where slowness and tremor may be discounted as ‘normal’ or misdiagnosed as one of several other common disorders affecting this age group (for example, arthritis, stroke, dementia). A further difficulty is presented by persons with both parkinsonism and dementia, who may be classified as either primary disorder in different epidemiologic surveys. The common use of neuroleptics in institutionalized elderly, especially those with cognitive impairment, can further confound diagnosis in this age group.
Postmortem validation of clinical diagnosis, although ideal, is rarely available in a population‐based setting. First, because survival with PD typically involves many years or even decades, very long follow‐up is necessary. In addition, clinical diagnostic criteria do not perfectly predict ‘classic’ postmortem features of PD. Error rates of more than 20% were seen in one clinicopathological series by Hughes et al. (1993). The same authors later suggested that the use of standard clinical criteria (e.g. the UK PD brain bank criteria) improved accuracy of a clinical diagnosis in 100 patients (Hughes et al., 2001) in which 90 were shown to have idiopathic PD at postmortem and 10 had other parkinsonian syndromes. In a report published the following year (n = 143), Hughes et al. (2002) estimated that the positive predictive value of the clinical diagnosis for the whole group was 85.3%, with 122 cases correctly clinically diagnosed, 98.6% (72 out of 73) for idiopathic PD, and 71.4% (50 out of 70) for other parkinsonian syndromes. However, since autopsy is most likely performed when the clinical diagnosis is not certain, misclassification may be overestimated in published series (Maraganore et al., 1999). Nonetheless, in the few settings where postmortem validation of PD is possible, valuable insights can result (Ross et al., 2004).
The uncertainty of clinical diagnosis can be an important consideration in the design and critical analysis of epidemiologic studies of PD. Inclusion of those without disease and exclusion of those with disease can produce under‐ or overestimates of the distribution of PD. In either case‐control studies or family studies, including case subjects who do not actually have PD can obscure a causative association or even result in associations with factors determining a different disorder. In families, the mode of inheritance of a genetic defect can also be misinterpreted.
Positron emission tomography (PET) and single photon emission computed tomography (SPECT) are imaging techniques that detect and display the distribution of radiolabeled tracers within the body. Patients with PD show reduced tracer accumulation in the striatum contralateral to the affected limbs using markers of the presynaptic dopaminergic system in the very early stages of the disease (Marek 1996, Wenning 1998, Benamer 2003). Others have suggested that ultrasound may be a useful way of identifying nigral injury in PD (Berg et al., 2002). Although these approaches are promising, their abilities to distinguish normal from abnormal and to distinguish PD from other forms of parkinsonism have not yet been developed to the extent that any of these techniques can be used outside the research setting. In epidemiologic research, broad application of these techniques remains difficult, as they are not widely available. However, combining these tools with other, more easily characterized, potential biomarkers, such as olfactory testing, may be useful in prospective studies to identify those ‘at risk’ for developing parkinsonism, to identify those persons appropriate for interventions to protect against PD and to provide better methods for case definition in studies of genetic and environmental risk factors, where incorrect classification of cases and controls could significantly alter results (Siderowf 2005, Stiasny‐Kolster 2005).
Investigations of new cases of PD present additional uncertainties, because the definition of a new case is particularly problematic. Onset of the motor features of PD is insidious. It is commonly held that at least 50% of substantia nigra pars compacta cells are damaged before the symptoms of PD prompt medical attention (Bernheimer et al., 1973). It has long been observed in autopsy series that the pathologic changes of PD can be identified in the brains of persons who were not diagnosed during life; these ‘incidental Lewy body’ cases increase with increasing age of the population surveyed, and may represent clinically unrecognized PD (Gibb and Lees, 1991). More recently, improved neuropathological methods led to the proposal that neuropathologic injury in PD begins in lower brainstem and olfactory nuclei, and progresses through predictable stages over time, involving the substantia nigra and producing classical parkinsonism only relatively late, at stage 4 (Braak 2003a, Braak 2004). Lewy neurite pathology can also be seen in autonomic ganglia outside the central nervous system, leading to the further hypothesis that PD may begin outside the central nervous system, well before the classic signs of parkinsonism develop (Braak et al., 2003b). If this is correct, the true onset of the disease process may begin long before the neurological syndrome is diagnosed. This research hypothesis merits further investigation. To be effective, studies of risk or protective factors, as well as investigations of preventive therapies, may need to target time periods decades before the onset of disease.
6.2. Demographics
6.2.1. Incidence
Incidence, the number of new cases of a disorder diagnosed during a specific time interval within a defined population, provides the most complete description of the number of cases of disease, as this measurement is least affected by factors influencing survival. This is particularly important for a slowly progressive disorder such as PD. However, as discussed above, because the time of onset of PD is not easily determined, incidence can vary depending on the definition of disease, as well as by factors such as method of ascertainment and access to health care. Studies using more intensive ascertainment methods, such as in‐person screening, may find higher rates (de Lau et al., 2004). Crude estimates of incidence can also vary due to the age and gender distribution of the population studied, and crude rates must be compared with this in mind. For example, reported incidence of PD varies fourfold, from 4.5 to 19 per 100 000 population per year when the entire age spectrum of the population is considered (Rosati 1980, Harada 1983, Ashok 1986, Granieri 1991, Mayeux 1995, Sutcliffe 1995, Fall 1996, Kusumi 1996, Bower 1999, Kuopio 1999, MacDonald 2000, Twelves 2003, Van Den Eeden 2003). However, when studies using similar methods are compared, and rates are adjusted to a reference population, this range is markedly reduced (11.0–13.9/100 000 population per year; Van Den Eeden et al., 2003).
Age is a key determinant of PD incidence. In all populations studied, PD is very rare before age 50 (Kurland 1958, Brewis 1966, Rosati 1980, Ashok 1986, Granieri 1991, Wang 1991, Tanner 1992, Harada 1983, Mayeux 1995, Morens 1996a, Marras 2002, Van Den Eeden 2003, Korell 2005). PD incidence increases steadily in the sixth through the eighth decades in most populations, but a decrease in late life is seen in some studies. Whether this apparent decline in PD incidence is the result of methodologic challenges, such as the greater difficulty identifying and diagnosing PD in the very old (Bower et al., 2000), rather than an actual decline in disease frequency, is not certain. If the decline is real, a biological ‘window of vulnerability’ for PD may exist. Investigation of the determinants of this could provide important insights into the causes of PD.
PD incidence is also higher in men than in women in most populations studied, although gender‐specific differences show more variability worldwide than do differences associated with increasing age. In a large study in northern California, PD incidence in men was 91% higher than for women (19/100000 for men versus 9.9/100000 for women, age‐adjusted; Van Den Eeden et al., 2003).
Whether PD incidence varies by racial or ethnic group has been addressed in only a few studies. In a northern California population, estimated PD incidence, adjusted for age and gender to a comparison population, was highest in Hispanics (16.6/100 000), then non‐Hispanic whites (13.2/100 000), then Asians (11.3/100 000) and lowest in blacks (10.2/100 000; Van Den Eeden et al., 2003). When rates in other incidence studies were adjusted to the same comparison population, PD incidence in northern Manhattan was higher in blacks (18/100 000) than in whites (12.9/100 000) or ‘other’ (11.8/100 000), and PD incidence in men of Japanese and Okinawan descent in Honolulu was 13.1/100 000 (Mayeux 1995, Morens 1996a). Whether these variations within and across populations reflect real differences, or simply poor precision, resulting from small numbers of PD cases, will only be answered by additional race‐ and ethnicity‐specific studies.
Has PD incidence changed over time? Periodic fluctuation in incidence could result from any episodic exposure, such as an infectious process. A steady increase over the past several decades could implicate exposures increasingly present, such as those due to industrialization or lifestyle practices. Conversely, if PD incidence has remained stable over time, recent environmental factors are unlikely to be important causes of PD. Only two reports have addressed this possibility, with differing results. Reviews of the Mayo Clinic database from Olmsted County, Minnesota, found no change in age‐specific PD incidence between 1935 and 1990 (Rajput 1984, Rocca 2001). One limitation to this work is the small population size. Only 154 PD cases were incident in 15 years, resulting in poor precision of these estimates. In contrast, in southwestern Finland, based on a larger number of cases, estimated incidence of PD was increased in men, particularly those aged 60 and older, in 1992 as compared to 1971 (Kuopio et al., 1999). Although it is possible that these differences may reflect temporal changes in environmental exposures in Finland, but not in Minnesota, this cannot be determined from the published studies.
6.2.2. Prevalence
Because PD is relatively uncommon and associated with a long survival, estimates of prevalence are more easily obtained than estimates of incidence. PD prevalence is most commonly estimated through national or local reporting systems. In locations where health care is universally available, such methods provide a good estimate of correctly diagnosed cases, but exclude persons who have not sought medical care. Misclassification is dependent on the methods used to define cases, with those diagnosed by experts being likely the best estimates of PD in the population. Estimates of prevalence based on populations identified by other methods (such as participants in a hospital clinic) do not accurately reflect the general population of an area, since cultural, economic or other factors may influence case selection.
Ideally, both incidence and prevalence are determined by screening all members of entire populations defined by specific geographic or political boundaries. Door‐to‐door surveys of all households in an area, followed up by examination of individuals suspected of having parkinsonism based on the screen, is the generally the best measure of true prevalence. If cooperation is good, a door‐to‐door survey is the most likely means of identifying all cases of PD in a community. Typical community‐based and door‐to‐door prevalence studies utilize health professionals or, more often, trained survey assistants who screen the population using a set of questions designed to identify all individuals who may have the disease (Anca et al., 2002), even those who have not received medical attention (Tanner 1990, Mutch 1991, Duarte 1995, Giroud Benitez 2000). After the initial screening process, individuals suspected of having the disease are asked to have a careful evaluation performed by a neurologist and possibly ancillary diagnostic tests.
The estimated PD prevalence derived from medical care reporting systems in North America and Europe find rates between 100 and 200 cases/100 000 population, although rates in developing countries are reported to be as little as one‐tenth of these rates (Kurland 1958, Brewis 1966, Jenkins 1966, Marttila 1967, Kessler 1972a, Kessler 1972b, Rosati 1980, Nishitani 1981, Harada 1983, Sutcliffe 1985, Ashok 1986, Chalmanov 1986, Mutch 1986, Shi 1987, Okada 1990, Granieri 1991, Wang 1991, Caradoc‐Davies 1992, Mayeux 1992, Mayeux 1995, Tanner 1992, Morens 1996a, Hobson 2005). Table 6.2 shows examples of crude estimated prevalence from door‐to‐door studies (Li 1985, Schoenberg 1985, Schoenberg 1988, Bharucha 1988, Acosta 1989, Morgante 1992, Wang 1994). Recently, estimates as high as 780/100 000 have been reported in Sydney, Australia (Chan et al., 2005).
Table 6.2.
Parkinson's disease prevalence rates from selected door‐to‐door surveys
| Location of survey | Age groups screened | Crude prevalence/ 100 000 persons |
|---|---|---|
| Chinese cities (Li et al,. 1985) | > 50 years | 44.0 |
| Igbo‐ora, Nigeria (Schoenberg et al., 1988) | > 39 years | 58.6 |
| Kin‐Hu, Kinmen, Taiwan, ROC (Wang et al., 1994) | > 50 years | 170.0 |
| Sicily, Italy (Morgante et al., 1992) | > 12 years | 257.2 |
| Vejer de la Frontera, Cadiz, Spain (Acosta et al., 1989) | All ages | 270.0 |
| Copiah County, Mississippi, USA (Schoenberg et al., 1985) | > 39 years | 347.0 |
| Parsi community, Bombay, India (Bharucha et al., 1988) | All ages | 328.3 |
| Bankstown, Sydney, Australia (Chan et al., 2005) | ≥ 55 years | 780.0 |
Note: Studies listed should not be compared directly as the age and gender distributions of the underlying populations differ.
Comparison of prevalence studies worldwide suggests that PD may be more common in the developed world. Because there are many methodological differences among studies, as well as differences in culture and health care among countries, this observation must be viewed with caution. Importantly, crude prevalence rates cannot be compared directly, since different age groups were surveyed, and the age distribution in the underlying populations differ. Longevity increases the number of PD cases one can expect in a population. While age adjustment reduces differences across populations, the range of estimated PD prevalence remains broad. Comparisons of prevalence for PD from different populations can also reflect the relationship between prevalence and methodology used. For example, apparent age‐specific prevalence differences among studies may actually be quite similar when data are re‐evaluated according to the same diagnostic criteria (Anderson et al., 1998).
In one study, PD prevalence has been estimated at two time points. In southwestern Finland, PD prevalence appears to be increasing in men and in rural areas in 1992 as compared to 1971 (Kuopio et al., 1999). Because ascertainment methods were similar at both time points, this may reflect a true difference in prevalence in this region, possibly the result of changes in exposure to risk factors. Lending more credence to this is the observation that similar changes in incidence were observed, making it less likely that the prevalence changes are due to improved survival of persons with PD in 1992.
6.2.2.1. Age
Although PD is rare before age 40, after age 50 the prevalence rises almost exponentially (Kurland 1958, Brewis 1966, Jenkins 1966, Marttila 1967, Kessler 1972a, Kessler 1972b, Rosati 1980, Harada 1983, Li 1985, Schoenberg 1985, Schoenberg 1988, Sutcliffe 1985, Ashok 1986, Mutch 1986, Shi 1987, Acosta 1989, Mayeux 1992, Mayeux 1995, Morgante 1992, Tanner 1992, Wang 1994, Morens 1996a). By the eighth decade, estimated prevalence in European and North American populations is between 1000 and 3000 cases per 100 000 population. Although differences in the age distributions in these populations, diagnostic criteria, ascertainment methods, access to health care or disease survival rates may explain much of this variation, international variation in PD frequency is seen even after adjusting for many of these inconsistencies (Zhang and Roman, 1993). Risk factors, which may vary geographically, include both genetic differences in disease susceptibility and exposure to causative and protective environmental factors. Although PD is intimately related to aging, it has been well documented that its underlying process is distinct from natural aging (McGeer 1988, Fearnley 1991, Gibb 1991). An age‐determined process, such as an acquired defect in cellular metabolism, or a process requiring a long period of time to manifest – as might result from prolonged toxicant exposure or the cumulative effects of many individual injuries to nerve cells – might cause a similar pattern. It is also possible that both age‐related vulnerability and time‐dependent processes explain the late‐life preponderance of PD.
6.2.2.2. Gender
Men are diagnosed with PD about twice as often as women, irrespective of geographic location or race (Tanner 1996, Baldereschi 2000). This pattern is seen in both prevalence and incidence studies. In a meta‐analysis of seven incidence studies, men were found to have a 1.5‐times greater relative risk for PD than women (Wooten et al., 2004).
This increased risk in men may reflect biological differences between men and women, such as the effects of sex hormones or X‐chromosome‐linked susceptibility genes. Alternatively, culturally determined differences in male and female behavior, with associated differences in exposure to risk factors, could explain the pattern. The latter hypothesis is supported by a large Finnish study showing a dramatic increase in the male‐to‐female relative risk from 0.9 in 1971 to 1.9 in 1992 (Kuopio et al., 1999). Others have suggested that hormonal differences between men and women explain these differences, although the relationship does not appear to be a simple one (see also section 6.3.7). Further epidemiologic studies, along with experimental laboratory studies, will be necessary to determine whether men are at greater risk for PD.
6.2.2.3. Race
Although there are a surprising number of observations in the literature suggesting whites are at increased risk for PD, it has been thought that lower rates in non‐whites might be related to socioeconomic or cultural differences, leading to ascertainment bias (Kessler 1972a, Kessler 1972b, Tanner 1996). Nevertheless, two multiracial population‐based studies estimating the incidence of PD in upper West‐Side Manhattan (Mayeux et al., 1995) and in Northern California (Van Den Eeden et al., 2003) suggest racial differences in PD incidence. In the Manhattan study, African‐American women had lower rates, but African‐American men had higher rates than whites (Mayeux et al., 1995). The Northern California study, a much larger evaluation of PD incidence, showed a lower frequency of PD in both men and women of African or Asian descent than in non‐ Hispanic whites (Van Den Eeden et al., 2003). Results remain equivocal in both studies, however, as even in this large study the numbers of non‐whites were low and between‐group confidence intervals for race‐specific PD incidence overlapped.
If there are true differences in PD risk among groups defined by race or ethnicity, this may reflect differences in biologic susceptibility. For example, mutations in the LRRK2 gene account for about 2% of parkinsonism in northern European populations, but 15–20% in persons of Ashkenazi Jewish and North African origin (Lesage 2005, Ozelius 2006). Others have suggested that dermal melanin may protect against PD by trapping potential neurotoxins before they reach the brain (Mars and Larsson, 1999). Because dermal melanin is regularly sloughed with keratinized skin, persons with more dermal melanin may be protected from the passage of toxicant compounds into the central nervous system. Alternatively, differences in non‐genetic risk factors may explain differences among populations. For example, PD prevalence is high in the Inuit population of Greenland (Wermuth et al., 2004). This population is at risk for dietary and other exposures to persistent organic pollutants (Dewailly et al., 1999), agents suggested to be risk factors for PD.
6.2.3. Mortality
Studying mortality of PD based on information from death certificates is problematic because PD is a chronic disorder that is not the direct cause of death; thus, the frequency of the disease can be underestimated from such evaluations. Compared to persons of the same age and gender, mortality is increased approximately twofold among individuals with PD (Di Rocco 1996, Morens 1996a, Louis 1997, Morgante 2000). An observed north–south gradient of decreasing PD mortality (Lilienfeld et al., 1990), although possibly reflecting true differences in regional mortality, could also be an artifact of differential access to medical care or death certificate completion inconsistencies among physicians (Pressley et al., 2005).
Mortality in a clinical trial population may be affected by the influence of the health benefit obtained from participating in the study. After a 13‐year follow‐up, results from the DATATOP cohort study show that the mortality rate was similar to that of the general population and that PD did not affect survival differently across gender or age groups in a selected group of otherwise healthy clinical trial participants (Marras et al., 2005a). In other clinical trial populations, mortality has been higher than expected, however (Hely 1999, Lees 2001, Fall 2003). Among participants in the DATATOP study, severity, rate of worsening of parkinsonism and response to levodopa are related to survival (Marras et al., 2005b), suggesting that differences in these factors among studies may also account for the observed differences in PD‐related mortality among these studies.
6.3. Risk factors for Parkinson's disease
6.3.1. Introduction to epidemiologic clues
The demographic studies reviewed in the previous section may provide clues to the causes of PD (Table 6.3 ). Demographic differences in the frequency of PD, particularly differences in PD incidence, may be the result of ascertainment bias. Alternatively, differences in risk factors for PD among different demographic groups may explain these patterns. Some of these possibilities have been discussed above. Disease clusters, representing more than the expected number of new cases of PD at a certain time and/or in a certain place, are another type of pattern that may suggest a shared cause of disease and provide clues to the underlying etiology of all cases. An example is the cluster of parkinsonism in narcotics addicts caused by 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) exposure (Langston et al., 1983) (Fig. 6.1 ). MPTP induces parkinsonism that is similar to PD, with key symptoms that improve with levodopa treatment and a similar side‐effect profile. Differences include the more rapid onset of symptoms in MPTP‐induced parkinsonism than in PD, and possibly some differences in neuropathologic features as well. Although MPTP injection is clearly not a cause of most PD, investigation of this cluster provided an important animal model. Investigation of the mechanism of MPTP toxicity led to the hypothesis that toxicants may cause PD, and has focused interest on compounds with structural or functional similarities (Fig. 6.2 ). Other proposed clusters, such as the syndrome of motor neuron disease–parkinsonism–dementia in certain areas of the Western Pacific (Spencer, 1987) or several clusters in Canada (Kumar et al., 2004) have yet to reveal specific etiologic factors.
Table 6.3.
Factors associated with the risk of Parkinson's disease in one or more studies
| Factors directly associated |
| Increasing age |
| Male gender |
| White race |
| Drinking well water |
| Diet: animal fat, milk, iron |
| Obesity |
| Hysterectomy and/or supplemental estrogen |
| Midlife constipation |
| Rapid‐eye movement sleep disorder |
| Physical and emotional stress |
| Family history of Parkinson's disease |
| Rural residence |
| Pesticides |
| Farming |
| Teaching/health care work |
| Metals |
| Factors inversely associated |
| Smoking/tobacco |
| Caffeine intake |
| Non‐steroidal anti‐inflammatory drug use |
| Alcohol |
| Greater physical activity |
Fig.6.1.
1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridin (MPTP) and structurally related compounds. MPP, 1‐methyl‐4‐phenylpyridinium.
Fig. 6.2.
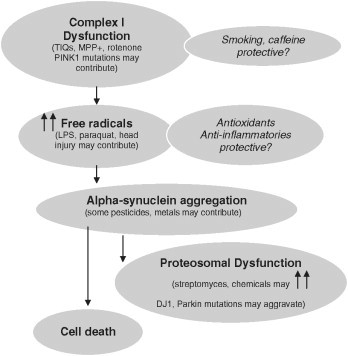
Epidemiologic clues and mechanisms of neuronal injury. TIQs, tetrahydroisoquinolines; MPP+, 1‐methyl‐4‐phenylpyridinium ion; LPS, lipopolysaccharide.
Familial clusters are generally interpreted to indicate a genetic cause for disease, but certain patterns within families, such as temporal clustering of disease, may be more suggestive of shared environmental risks. A number of case‐control studies have found increased PD risk if a first‐degree relative has PD (Semchuk 1993, Morano 1994, Payami 1994, Bonifati 1995, De Michele 1996, Marder 1996). Because persons with disease may be more aware of disease in relatives, these studies in part may reflect reporting bias. Elbaz et al. (2003a) showed evidence for family information bias whereby cases with PD are more likely to report a relative with PD than are control subjects, increasing the risk estimate by 133%. Studies in twins do not support a genetic cause for typical age at PD onset, although genetic factors appear to be increased in those with younger age at onset (Duvoisin 1981, Marsden 1987, Marttila 1988, Vieregge 1992, Tanner 1994, Wirdefeldt 2004).
Many proposed risks for PD will be reviewed here.
6.3.2. Single genes causing parkinsonism
Genetic defects responsible for parkinsonism have been identified in some families (Bonifati 1995, Polymeropoulos 1996, Polymeropoulos 1997, Hattori 1998, Kitada 1998, Paisan‐Ruiz 2004, Zimprich 2004). In many of these cases, the clinical features resemble typical PD. However, within affected families there are often clinical features that are unusual for PD. At present, mutations in at least five genes have been firmly associated with parkinsonism: (1) α‐synuclein (SNCA or PARK1; Polymeropoulos et al., 1997); (2) parkin (PRKN or PARK2; Kitada et al., 1998); (3) DJ‐1 (DJ1 or PARK7; Bonifati et al., 2003); (4) PTEN‐induced putative kinase I (PINK1 or PARK6; Polymeropoulos et al., 1997); and (5) leucine‐rich repeat kinase 2 or dardarin (LRRK2 or PARK8; Paisan‐Ruiz 2004, Zimprich 2004) (Table 6.4 ). PINK1 homozygous mutations have been reported to be an important cause of disease among Italian sporadic patients with early‐onset parkinsonism, whereas the role of single heterozygous mutations is less clear (Bonifati et al., 2005). The LRRK2 G2019S mutation is the most common pathogenic mutation linked to parkinsonism, accounting for 1–2% of cases, including cases of not only younger but also older age at disease onset (Kay et al., 2006). Other candidate PD loci have been proposed, including putative disease‐causing mutations in the ubiquitin carboxy‐terminal hydrolase L1 (UCHL1) (Leroy et al., 1998) and in a nuclear receptor of subfamily 4 (NR4A2 or NURRI) (Le et al., 2003). These candidates do not map to known PD linkage regions, but polymorphisms in both genes have been associated with PD in some case‐control studies (as reviewed by Bertram and Tanzi, 2005). The GSK3B polymorphism has been reported to alter transcription and splicing and interact with tau haplotypes to modify PD risk (Kwok et al., 2005).
Table 6.4.
Genes causing parkinsonism
| Locus/gene | Map position | Characteristics |
|---|---|---|
| Dominantly inherited | ||
| PARK1 (α‐synuclein) | 4q21–22 | Rare point mutations, duplication/triplication of normal gene |
| Atypical features, young onset | ||
| PARK8 (LRRK2/dardarin) | 12p11.2‐q13.1 | Sporadic and familial, heterogeneous signs and pathology |
| Old and young onset | ||
| Recessively inherited | ||
| PARK2 (parkin) | 6q25–27 | Many mutations, atypical, most onset < 30 years of age |
| PARK6 (PINK1) | 1p35–36 | Two mutations in three consanguineous families |
| PARK7 (DJ‐1) | 1p36 | Point mutation, deletion, few families, atypical |
| Uncertain inheritance | ||
| PARK5 (UCHL1) | 4p14 | |
Normal protein products of PARK1, 2, 5, 6 and 7 are all likely involved in protein degradation and/or cellular response to toxicant injury or oxidative stress.
From an epidemiologic perspective, the monogenic causes of PD appear to constitute a proportion of cases worldwide. However, investigation of the protein products of these genes can further our understanding of the process of nerve cell death in parkinsonism. Investigation of these forms has emphasized the role of key proteins (like α‐synuclein) and molecular pathways leading to neurodegeneration. Intriguingly, mitochondrial mechanisms, oxidative stress and protein clearance appear to be pathogenic in animal models derived both from toxicant and genetic forms of parkinsonism (Dawson 2003, DiMonte 2003).
6.3.3. Proposed environmental risk factors for Parkinson's disease
Risk factor investigation in PD is challenging, as the time of life most important to investigate is not known. It is likely that years, and possibly even decades, pass between the time of risk factor exposure and the clinical onset of parkinsonism. Case‐control studies are an efficient way to study proposed disease risk factors, particularly in relatively uncommon disorders, such as PD. Potential limitations to this design include biased recall, the lack of validation of exposure and, in prevalent studies, survivor bias. Prospective cohort studies, assessing risk factors in advance of disease, avoid many of the biases of case‐control studies, but risk factor investigation is limited to those selected for study and diagnostic accuracy may be less certain.
6.3.3.1. Rural living, farming, well water
Numerous studies worldwide have identified rural living, farming, gardening and drinking well water as risk factors for PD (Semchuk 1991, Butterfield 1993, Hubble 1993a, Morano 1994, Ferraz 1996, Gorell 1998, Marder 1998, Zorzon 2002, Korell 2005), but the results are somewhat inconsistent because of differences in the way the studies assessed the effects of rural living. Overall, risk of PD appears to be increased in rural dwellers – especially in the USA. Meta‐analysis results (Priyadarshi et al., 2000) also support that risk factors include farm living and use of well water and pesticides. Although the specific associations are varied, the consistency of the general finding is remarkable.
6.3.3.2. Pesticides
Pesticide exposure is associated with an increased risk of PD in many reports. A meta‐analysis of 19 published studies found a combined odds ratio (OR) of 1.94 (95% confidence interval (CI) 1.49–2.53) for pesticide exposure (Priyadarshi et al., 2000). However, the category of pesticides is very broad, and includes chemicals with many different mechanisms of action. Only a few studies have identified specific compounds or compound classes, including herbicides, insecticides, alkylated phosphates, organochlorines, wood preservatives, dieldrin and paraquat (Firestone 2005, Korell 2005).
Most of these studies have been limited by very broad measures of exposure. In many studies, the proportion of exposed persons was low, little was known about specific exposures and validation of exposure was not possible.
Gene–environment interaction may also be important, and those with impaired pesticide metabolism may be most vulnerable. A recent report (Elbaz et al., 2004) indicates an increased risk of PD with pesticide exposure in normal metabolizers, and about twofold increase in risk with pesticide exposure for CYP2D6 poor metabolizers, and no effect of the metabolizing status on risk for PD without pesticide exposure.
6.3.3.3. Metals
Iron has been shown to cause a higher susceptibility to oxidative stress in two ways. By depleting stores of glutathione, iron may have a role in the progression of parkinsonism associated with exposure to other chemicals that are metabolized to free radicals and/or contribute to the adverse effects of oxidative stress (Kaur et al., 2003). Also, since iron has a strong catalytic power to generate highly reactive hydroxyl radicals from iron (II) and hydrogen peroxide, increased levels of iron in the brain can increase oxidative stress (Fenton reaction). Excessive iron accumulation in the brain is also a potential risk for neuronal damage, which may be promoted by other triggering factors (Lan and Jiang, 1997). A combined high dietary intake of iron and manganese may increase the risk of developing PD (Powers et al., 2003). Dietary intake of manganese alone does not seem to have toxic effects, except among individuals with liver failure (Hauser et al., 1994). Although dietary intake is the main source of non‐occupational exposure to manganese, occupational exposure seems to be a more influential PD risk factor. Case‐control studies suggest that occupational exposure to metals (Gorell 2004, Racette 2005) may be at increased risk of PD, although cohort studies have not replicated this (Fryzek 2005, Fored 2006).
6.3.3.4. Polychlorinated biphenyls (PCBs)
PCBs are among the group of compounds classified as persistent environmental pollutants. In the USA, industrial use was common until 1977. Today, PCBs continue to cycle in the environment. Common sources of human exposure are fish and marine mammals, meat and dairy products. In laboratory studies, PCBs have been shown to reduce dopamine levels in the brain areas affected in PD (Seegal 1986, Chu 1996). The association between PCBs and PD has been studied in a blinded comparison of postmortem determinations of caudate PCB concentrations in PD patients and controls (Corrigan et al., 1998). PD brains had significantly higher levels of PCB congener 153, and several other congeners tended to be higher in PD caudate. Also increased in PD brains were the organochlorine pesticide dieldrin and the dichloro‐diphenyl‐trichloroethane (DDT) metabolite 1,1‐dichloro‐2,2‐bis(4‐chlorophenyl)ethene (DDE). In a previous investigation, frontal cortex of PD patients and controls did not show differences in PCB or organochlorine levels. This regional specificity lends indirect support to an association between PCBs and PD.
6.3.3.5. Occupation
Given the increasing evidence that environmental factors play a role in PD, there has been an increasing effort to identify occupational risk factors, but to date few have been identified. A higher frequency of PD has been reported among teachers and health care workers (Tsui et al., 1999). These findings were replicated in a case‐control study in twin pairs discordant for PD (Tanner et al., 2003), and in very large occupational mortality studies in the USA (Schulte et al., 1996) and the UK (Coggon et al., 1995). It has been suggested that an infectious etiology could explain the increased risk in these occupational groups. Alternatively, these associations could be related to some other unrecognized occupation‐associated risk factor, to premorbid personality characteristics predisposing to certain occupations (Menza, 2000) or to issues of study design, such as ascertainment bias or confounding by age or other factors. A higher frequency of PD has also been reported in carpenters and cleaners (Fall et al., 1999) and in workers chronically exposed to metals (Gorell et al., 2004). Welding has been proposed as a risk factor (Racette et al., 2005), but this finding is controversial (Fryzek et al., 2005), with recent results from a nationwide linkage study indicating no support for an association between welding and PD, or any other specific basal ganglia and movement disorders (Fored et al., 2006). Overall, results from the available studies are inconclusive, reported findings need confirmation and not all occupations have been evaluated. Differences within even one type of occupation make occupation groups heterogeneous and comparisons difficult.
Querying occupation in such a way that it triggers exposure‐specific questions as described by Stewart and Stewart (1994) may be more useful, but the potential misclassification of specific exposures will be appreciable and tend to bias effect measures toward the null – occupational exposures in community‐based studies are rare, which compounds the problem (Tielemans et al., 1999). Ideally, in addition to asking about occupation and exposure, there should be a quantitative measure in exposure‐response analyses – for most chronic diseases the exposure measure of choice is the level of exposure multiplied by the duration of exposure. The use of a single variable for primary lifetime occupation is problematic. Undoubtedly, most people work at a number of different jobs throughout their lives, and important associations may be missed or misclassified. A lifelong, job task‐specific occupational history has the potential to provide more complete information, and direct interviews may improve historical accuracy. Studies within specific at‐risk groups such as occupational cohorts can be important in clarifying whether there is a relationship between occupation and PD.
6.3.4. Diet, obesity, physical activity
6.3.4.1. Diet
Diet is a very difficult exposure to measure both because of its complexity and the fact that most individuals have qualitatively relatively similar diets (Willett, 1990). Despite these challenges, several dietary factors have been associated with PD.
Excess intake of dairy products has been associated with increased risk of PD in two large prospective cohorts (Chen 2002b, Park 2005). In a study of health professionals, whether the effect was due to calcium or milk could not be determined. Moreover, the risk was most marked in men, and not clearly observed in women. In the second study, PD incidence was more than twice as high in men drinking more than 16 ounces (approximately 450 grams) daily in midlife, compared to those who consumed no milk. This effect was independent of calcium. No women were included in this cohort. The reason for this association is unclear. One explanation is that milk may be a vehicle for potential neurotoxicants such as organochlorine pesticides or tetrahydroisoquinolines.
Other studies suggested different dietary risk factors for PD. PD risk was mildly raised in association with high dietary iron intake, but the risk markedly increased with high intake of both iron and manganese (Powers et al., 2003). Another study (Scheider et al., 1997) indicated increased risk with high vitamin C, carotenoids and sweet food, including fruit intake, but the number of cases studied was small (n = 57). Among those with PD, homocysteinemia has been indicated as a potentially reversible risk factor for depression or cognitive decline (O'Suilleabhain et al., 2004).
Studies of dietary antioxidant intake have been largely inconclusive. It is biologically plausible that dietary antioxidants may protect against nigral damage, analogous to their potential role in preventing heart disease and stroke (Rimm 1993, Kobbata 1991, Knekt 1996, Gale 1995). One prospective cohort study of 41 836 women indicated a significant protective effect seen for both vitamin C and manganese consumption; however, vitamin A intake was associated with an increased risk of PD (Cerhan et al., 1994). A sibpair study (Maher et al., 2002) reported a 3.2‐year older mean age at onset for affected siblings who reported taking multivitamins. Protective effects were proposed for B vitamins and folate, because of their shared pathways with homocysteine and ability to lessen oxidative stress (Duan et al., 2002). Comparison of two large prospective cohorts (Chen et al., 2004a) with 415 cases indicated PD risks did not differ in relation to dietary intakes of B vitamins and folate (relative risk 1.0 (95% CI 0.7–1.5) comparing the lowest to the highest intake quintile in men and 1.3 (95% CI 0.8–2.3) in women).
Dietary insufficiency has also been proposed as a risk factor for the development of PD, although evidence for this is indirect. In a 20–30‐year follow‐up of a cohort of ex‐Far‐East prisoners of war, who experienced severe dietary insufficiency between 1942 and 1945 (Gibberd and Simmonds, 1980), 24 PD cases were identified out of 4684 subjects, producing a crude prevalence rate of 512 per 100 000. This is particularly high considering the relatively young age of the cohort and the observation that 15 cases (63%) had disease onset under the age of 50 years. Emotional and physical stress has also been implicated in increased frequencies of PD in another study of prisoners of war (Page and Tanner, 2000), although a relationship to dietary insufficiency could not be determined.
Certain exotic dietary exposures have been proposed to cause atypical forms of parkinsonism, including ingestion of indigenous species from Guam (Spencer 1987, Murch 2004), or the British West Indies (Champy et al., 2005), although these reports are controversial.
6.3.4.2. Obesity
Conversely, oxidative stress may be increased by lipid consumption and higher caloric intake, and eating foods high in animal fat has been associated with increased risk of PD in several studies (Korell and Tanner, 2005). The link between measures of body composition and obesity and risk of PD is unclear. A large study in Japanese‐American men in Hawaii observed higher prevalence of PD with higher triceps skinfold thickness, subscapular skinfold thickness and body mass index (Abbott et al., 2002). A similar analysis in the Nurses' Health and the Health Professionals' study did not find an association between body mass index and risk of PD but, among never smokers, both waist circumference and waist–hip ratio showed significantly positive associations with PD risk as compared to smokers (Chen et al., 2004b).
6.3.4.3. Physical activity
Animal models have also been used to study the role of physical activity in PD. Results of studies of forced limb use in 6‐hydroxydopamine‐injected rats (Cohen et al., 2003) suggest that preinjury forced limb use can prevent the behavioral and neurochemical deficits. In treadmill tests of MPTP‐injected rats (Tillerson et al., 2003), exercise following the nigrostriatal damage ameliorated related motor symptoms and neurochemical deficits.
Physical activity in epidemiological studies includes cohort results by Chen et al. (2005a) that show either that higher levels of physical activity may lower the risk of PD in men, or that men predisposed to PD tend to avoid strenuous activity in their early adult years. A significantly lower level of physical activity was present before diagnosis (men, 12 years prior; women, 2–4 years prior), and there was a sustained decrease in physical activity after diagnosis. Case‐control studies, however, have shown inconsistent results. In one Chinese hospital‐based study investigating risk factors for classic‐onset versus young‐onset PD, the duration of exercise was substantially longer in the young‐onset group than in controls or in the classic‐onset PD group (Tsai et al., 2002). In another small study assessing the lifetime physical activity via sports/leisure activity and participation using visual analog scales, there was no difference in lifetime physical activity between cases and controls; however, there was a greater decline in activity after age 50 years in those with PD (Fertl et al., 1993). In a nested case‐control study of male Harvard students, moderate physical activity was associated with a lower risk of PD, but this association was not seen at higher levels of physical activity (Sasco et al., 1992).
6.3.5. Inflammation, infection, head trauma, non‐steroidal anti‐inflammatory drugs (NSAIDs)
6.3.5.1. Inflammation
Several lines of evidence support the idea that inflammation is involved in the pathogenesis of PD (Hirsch et al., 2003). Postmortem analyses showed gliosis and clustering of microglial cells around nerve cells in 3 subjects who had presented with MPTP‐induced parkinsonism 3–16 years earlier (Langston et al., 1999). In cell culture experiments injection of lipopolysaccharides (LPS), which activate glia, killed dopaminergic neurons in mixed neuron–glia but not in pure neuron cultures (Bronstein et al., 1995). In an animal model, a single intranigral injection of LPS damaged dopaminergic but not serotonergic or GABAergic neurons (Herrera 2000, Gao 2002). Application of dexamethasone before LPS injection prevented the loss of catecholaminergic content, tyrosine hydroxylase activity and immunostaining, and the microglia‐macrophage activation seen previously (Castano et al., 2002).
Human studies revealed elevated cytokine levels, which induce glia activation, in the brain and cerebrospinal fluid of PD patients compared to controls (Nagatsu et al., 2000). Additionally, increased expression of tumor necrosis factor‐α, interleukin‐β and interferon‐γ was observed in the substantia nigra of PD patients (Boka 1994, Hunot 1999). In recent epidemiologic studies, intake of NSAIDs was inversely associated with PD risk (Chen et al., 2003); more detailed analysis indicated that the association was significant for ibuprofen but not other NSAIDs (Chen et al., 2005b). Higher levels of uric acid, a potent antioxidant, during midlife were associated with a 40% reduced risk of PD in one prospective cohort (Davis et al., 1996). This observation was recently replicated in a nested case‐control study in the health professional prospective cohort study (Weisskopf et al., 2006, unpublished; see Ascherio et al., 2006 for abstract). However, uric acid levels can be increased by several agents inversely associated with PD, including alcohol, caffeine and aspirin, as well as by levodopa. Further studies are needed to determine whether this is a primary or secondary association.
6.3.5.2. Infections
The observation that encephalitis lethargica often resulted in parkinsonism during the influenza pandemic of the early 1900s suggested a possible infectious etiology for PD. Since that time, however, clinical and neuropathological criteria have clearly differentiated postencephalitic parkinsonism from typical idiopathic PD. Although subsequent studies have been unable to identify an infectious agent in PD (Marttila 1977, Wang 1993), a number of studies have continued to suggest that infection may play a role in idiopathic PD. As described previously, increased PD frequency in health care workers and teachers has been linked to infection (Schulte et al., 1996). One study observed elevated coronavirus antibody levels in the cerebrospinal fluid of PD patients, supporting a possible link of PD with coronavirus infections (Fazzini et al., 1992), a common cause of respiratory infections. Another study noticed reduced risk for PD associated with most viral childhood infections, especially measles (Sasco and Paffenbarger, 1985); both studies await replication. The soil pathogen Nocardia asteroides causes a levodopa‐responsive movement disorder and nigral degeneration in mice (Kobbata and Beaman, 1991), but a serologic case‐control study did not support its role in human PD (Hubble et al., 1995).
6.3.5.3. Head trauma
Previous head trauma has been associated with PD in numerous case‐control studies (Bharucha 1986, Tanner 1987, Stern 1991, Semchuk 1993, Van Den Eeden 2000, Bower 2003). Head injury can trigger an inflammatory cascade, or conceivably disrupt the blood–brain barrier, increasing risk of exposure to toxicants or infectious agents. In a sibpair study (Maher et al., 2002) and a study of twin pairs concordant for PD (Goldman et al., 2006), the sibling with younger‐onset PD was more likely to have sustained a head injury. In twins discordant for PD, a previous head injury with amnesia or loss of consciousness was associated with a nearly fourfold increased risk of PD. Significant head injury is rare, however, and there may be a latency up to 30 years between injury and PD diagnosis, minimizing the chance that disease‐related disability caused the injury (Factor 1991, Seidler 1996, Taylor 1999). Severity of head injury is likely to be important and there may be a dose effect; there is no association with PD and mild head injury without loss of consciousness. Nevertheless, medical record validation suggests that this is a real association, not explained by recall bias.
6.3.5.4. Non‐steroidal anti‐inflammatory drugs
Inflammatory mechanisms appear to contribute to neurodegeneration in PD, and animal studies suggest that NSAIDs have neuroprotective properties (McGeer and McGeer, 2004) by reducing general inflammation. Studies of Alzheimer's disease have shown that the regular use of NSAIDs may reduce the risk of Alzheimer's in humans (Breitner 2001, in t’ Veld 2001, McGeer 2004). The similarities in the pathogenetic background of PD and Alzheimer's disease and animal data suggesting that anti‐inflammatory drugs may protect against PD (Ferger et al., 1999) have encouraged investigation of the association between NSAID use and PD risk in humans. An inverse association of NSAID use with risk of PD has been observed in two prospective studies for non‐aspirin NSAIDs, as well as for aspirin (Abbott 2003, Chen 2003). Interestingly, in a cross‐sectional study of 1258 PD cases and 6638 controls from the General Practice Research Database, this inverse association was again observed for men, but not women, in whom non‐aspirin NSAID use was associated with a higher risk of PD (Hernan et al., 2006). Whether this reflects a characteristic of the study population or method, or a true gender difference in risk, will require studies in other populations.
6.3.6. Smoking, caffeine, alcohol
Although there are a number of health risks associated with smoking tobacco and drinking alcohol, cigarette, coffee and alcohol intakes are all inversely associated with risk for developing PD, suggesting they may be neuroprotective agents.
6.3.6.1. Smoking
Not smoking cigarettes is the most consistently observed risk factor for PD. An inverse association between cigarette smoking and PD has been observed in studies spanning more than 30 years, involving diverse populations and including several large prospective investigations (Doll 1994, Grandinetti 1994, Benedetti 2000, Willems‐Giesbergen 2000). A meta‐analysis (Hernan et al., 2002a) indicated a 40% reduced risk of PD in smokers. Three basic categories of smoking were evaluated: ever smoking, past smoking and current smoking behavior. Long duration (highest pack/year) correlated with dose, and smoking more than 5 years prior to PD onset was not protective; recent smoking appeared more protective. Other research suggests cigarette smoking, on average, appears to lower the risk of developing PD by about half (Sugita et al., 2001). This inverse association has been reported in nearly every population studied over more than 30 years (Quik, 2004), and a recent study in a population characterized by a high prevalence of occupational pesticide exposure confirms an inverse correlation between cigarette smoking and PD in this potentially ‘high‐risk’ group as well (Galanaud et al., 2005). One report suggests the inverse association of smoking and PD is only present in those with a specific monoamine oxidase‐B allele (Checkoway et al., 1998), although this was not replicated (Hernan et al., 2002b), and other single observations suggest other interactions of genes and smoking (Tan et al., 2002).
Non‐smoking behavior in people fated to develop PD may be the result of a lower reward of smoking due to low dopaminergic tone, a genetically conferred decreased propensity to smoke or a premorbid personality (Menza, 2000). Indeed, the personalities of those who have gone on to develop PD have been described as shy, cautious, inflexible, punctual and depressive (Hubble et al., 1993b); such persons may be less likely to smoke or drink. In contrast, indirect evidence against this theory derives from a study in twin pairs discordant for PD (Tanner et al., 2002). The twins without PD had smoked more than their brothers. Despite a high correlation for smoking in monozygotic twin pairs, this difference was more marked in the monozygotic pairs, known to be remarkably similar in personality. Similar results have been reported in other studies of twins (Bharucha et al., 1986) and siblings (Scott et al., 2005) discordant for PD.
If there is a biologic effect of smoking, whether this is due to nicotine or a combustion product is not known. Indirect evidence supporting a role for nicotine is provided by the observation that PD was less commonly reported among users of smokeless tobacco in a large prospective cohort (O'Reilly et al., 2005). Animal studies suggest that nicotine may protect against experimental parkinsonism (Janson 1993, Prasad 1994) (Table 6.5 ). Nicotine has been found to protect against transection‐induced and MPTP‐induced dopaminergic neuronal cell loss in rodent substantia nigra (Janson 1993, Prasad 1994). In addition, nicotine has antioxidant properties (Ferger et al., 1998), and increases striatal trophic factors (Maggio et al., 1997). Alternatively, smoking may afford indirect protection by inducing peripheral detoxifying enzymes, or by reducing bioactivation of protoxins. This latter hypothesis is supported by the observation that cigarette smoking reduces monoamine oxidase‐B activity in humans (Fowler et al., 1996).
Table 6.5.
Cigarette smoking and Parkinson's disease: some possible biologic mechanisms
| Nicotine |
| • Blocks nigral cell loss (hemitransection, MPTP) |
| • Increases growth factors |
| Cigarette smoke |
| • Reduces MAO‐B activity |
| • Gene–environment interaction of smoking and MAO‐B allele |
| • Complex mixture of combustion products – other actions? |
MPTP, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine; MAO‐B, monoamine oxidase B.
Assuming smoking is neuroprotective, one might expect it to delay the onset of PD and improve the course of the disease in people already affected. Neither hypothesis has yet been proven. Two studies compared clinical features and did not find differences between smokers and non‐smokers (Alves 2004, Papapetropoulos 2005). Although a study by Kuopio et al. (1999) reported the mean age at onset in ever‐smoking men was significantly higher than in never‐smoking men, results of four other studies assessing age at onset of PD in relation to smoking status (Haack 1981, Rajput 1987, Morens 1996b, Levy 2002, De Reuck 2005) revealed the same or a younger age of PD onset in smokers. Interestingly, however, in several prospective cohort studies, survival of those persons with PD who continue to smoke cigarettes appears to be similar to, or even somewhat better than, survival of non‐smokers with PD (Grandinetti 1994, Elbaz 2003b, Chen 2006), in contrast to the typically increased mortality observed in cigarette smokers. This tantalizing preliminary information suggests that some aspect of smoking may not only modify disease risk, but also improve survival once PD is manifest.
6.3.6.2. Coffee and caffeine
An inverse association of both coffee and caffeine consumption and PD has been reported in case‐control and cohort studies (Fall 1999, Benedetti 2000, Ross 2000, Ascherio 2001, Paganini‐Hill 2001). For example, a longitudinal study and two case‐control studies of incident PD cases provide provocative evidence that coffee drinking may be inversely associated with PD risk. A longitudinal study of Japanese‐American men indicated greater use of coffee was inversely associated with PD risk in a dose‐dependent fashion (Ross et al., 2000). A very provocative finding in the same cohort was that greater use of coffee was inversely associated with incidental Lewy bodies at postmortem (Ross et al., 1999). A similar dose‐dependent inverse association between coffee drinking and PD was observed in two prospective studies (Benedetti 2000, Willems‐Giesbergen 2000), and retrospectively an incident case‐control study in Northern California (Nelson et al., 1999). In each case, the inverse association between PD and coffee drinking continued to be observed in multivariate analyses adjusting for cigarette smoking, alcohol use and other potential confounders. Similar associations had previously been reported in a few case‐control studies of prevalent cases, but these results were inconsistent, and a dose–response gradient was not described (Tanner 1996, Checkoway 1998).
The effect of coffee appears to differ between men and women, with a direct dose–response association in men (higher consumption associated with lower risk) but a U‐shaped pattern in women, although fewer women have been studied. It has been suggested a potential interaction between hormone exposure, primarily estrogen, and caffeine consumption may mediate PD. In participants of the Cancer Prevention Study II, caffeine intake was associated with a significantly lower mortality of PD in men but not in women (Ascherio et al., 2004). In women, the association depended on estrogen use, with a relative risk for PD of 0.47 (95% CI 0.27–0.8) in caffeine consumers not using hormones and of 1.31 (95% CI 0.75–2.3) in hormone users. Caffeine may be neuroprotective through its antagonist action on the adenosine A2A‐receptor (Chen et al., 2002a), which in laboratory studies, modulates dopaminergic neurotransmission (Popoli 1991, Nehlig 1992) and protects against striatal dopamine loss caused by MPTP (Richardson 1997, Kanda 1998). A2A‐receptor antagonists are receiving increasing attention as potential treatments, in particular for on/off fluctuations and dyskinesia in combination with levodopa therapy (Xu et al., 2005), but also as a possible monotherapy in early‐stage PD because of positive results from animal studies and a small clinical trial (Hauser 2003, Jenner 2003).
6.3.6.3. Alcohol
Alcohol use has been found by some to be inversely associated with PD even after controlling for possible confounding by smoking (Hellenbrand 1996, Fall 1999, Paganini‐Hill 2001). A biologic explanation for this observation has not been articulated. One study found that fewer cases with PD had a diagnosis of alcoholism than controls (Benedetti et al., 2000). The variability across studies is great and, overall, the current evidence for an association between alcohol intake and risk of PD is weak. In the Nurses' Health and the Health Professionals' cohorts, no association between incidence of PD and overall alcohol consumption was observed (Hernan et al., 2003); however, an inverse association of beer (but not wine or liquor) consumption was seen. Comparison of alcoholics and non‐alcoholics in a large database found comparable PD incidence in both groups (Hernan et al., 2004). Interestingly, in a stratified analysis for men and women separately, male alcoholics had a significantly lower incidence of PD whereas female alcoholics had a twofold increased incidence. Low consumption of alcohol in PD has commonly been attributed to the reserved personality that has been observed prior to PD manifestation (Menza, 2000).
6.3.7. Gender
As noted above, men appear to be at greater risk of developing PD than are women. This could reflect an intrinsic difference in risk, such as might be due to an X‐chromosome‐linked genetic characteristic or a sex hormone‐related factor. Alternatively, gender‐determined differences in risk factor exposure may be the cause or a combination of biologic predisposition and differences in risk factors might explain this pattern.
Benedetti et al. (2001) used a population‐based case‐control method to determine whether reproductive factors may influence PD risk in women. Hysterectomy with or without an oophorectomy and early menopause were associated with increased risk of PD (OR = 3.36 and 2.18, respectively) and estrogen use after menopause was inversely associated with PD risk (OR = 0.47), although the latter two differences were not statistically significant. Several subsequent case‐control studies have similarly suggested that factors associated with estrogen deficiency such as hysterectomy and early menopause may increase PD risk (Currie 2004, Ragonese 2004). Recently, Popat et al. (2005) found that the association of postmenopausal hormone use with PD risk depended on the type of menopause. Among women with history of a hysterectomy with or without an oophorectomy, estrogen use alone was associated with a 2.6‐fold increased risk and the risk of PD increased with increasing duration of estrogen use. In contrast, among women with natural menopause, no increased risk of PD was observed with hormone use.
Gender may also determine the effects of risk or protective factors associated with PD. Women appear to have different risk profiles to at least some of the exposures linked to PD in men, as discussed in previous sections. Although the explanation for these differences is not known, investigation of the combined effects of risk factors may explain some of these differences. For example, in two prospective cohort studies, PD risk was influenced by the combined effects of caffeine consumption and supplemental estrogen use. Women using supplemental estrogens with low caffeine consumption were at a lower risk of PD, but this effect was attenuated or reversed in women who had a high caffeine consumption and were at higher risk of PD (Ascherio 2003, Ascherio 2004). Future studies including populations of women of sufficient size to allow the separate assessment of risk factors in women will be important to clarify the question of gender and PD risk.
6.4. Gene–environment interactions
Research in the area of gene–environment interactions is complicated in that multiple genes and various environmental factors may combine to determine the level of risk for PD in any one individual. As described previously, several environmental factors, including pesticide and chemical exposure, have been consistently shown to modify the risk for PD in epidemiologic studies. If PD results from a combination of genetic and environmental factors, then an interaction of genetic factors with certain exposures could result in a high level of disease risk. For example, increased risk from an environmental toxin could be influenced by the genetically determined level of activity of metabolizing enzymes. Few gene–environment interactions have been investigated. One case‐control study suggests that smoking history modifies the effect of family history on the risk for PD, such that the odds ratio is highest in those with a history of smoking and a family history of PD (OR 10.0; Elbaz et al., 2000). This is a surprising finding given the increasing body of evidence that smoking is negatively associated with the occurrence of PD.
Other interactions have also been reported, including possible interactions between monoamine oxidase‐B gene polymorphisms and smoking behavior. A reduced risk of PD with increasing number of pack‐years of smoking was found in the presence of the G allele, whereas PD risk decreased with increasing pack‐years smoked in the presence of the A allele (Checkoway et al., 1998). Interactions between xenobiotic metabolizing enzyme genotype and pesticide exposure in the risk of PD have also been studied (Menegon 1998, Taylor 2000). The association between pesticide exposure and PD may be modified by glutathione transferase P1 polymorphisms (Menegon et al., 1998). In a study of 96 patients and 95 controls, no overall difference in the distribution of glutathione S‐transferase (GST) P1 genotypes was found between cases and controls. In those with pesticide exposure, however, the GST P1 AA genotype was associated with the lowest risk for PD. Confirmation of each of these observations in additional populations may provide important clues to disease etiology.
Polymorphisms of many genes have been found to be associated with an increase or decrease in risk for PD in at least one or more studies. Unknown gene–gene or gene–environment interactions may produce misleading results if cases and controls are not appropriately matched, perhaps explaining some of the conflicting data seen in these studies. Whether the inconsistent results obtained to date are due to study design issues or to limited generalizability of the findings to different patient groups is not known.
6.5. The future of Parkinson's disease epidemiology
An emerging direction of epidemiologic research in PD also deserves mention. Recent work involves investigation of those ‘at risk’ for PD, before disease is manifest. A variety of disorders may precede formal diagnosis of PD, including olfactory dysfunction, rapid‐eye movement sleep behavior disorders, QT or rate‐corrected QT (QTc) interval prolongation on the electrocardiogram, adiposity and constipation. In vivo imaging of the dopamine transporter with (99mTc)TRODAT‐1 (TRODAT) and olfactory testing have both been proposed as potential biomarkers in PD, and impaired smell recognition correlated with lower TRODAT uptake (Siderowf et al., 2005). Rapid‐eye movement sleep behavior disorder is strongly predictive of PD, and RBD patients have been shown to have impaired olfactory function compared to controls (Stiasny‐Kolster et al., 2005). In addition to olfactory dysfunction and rapid‐eye movement sleep behavior disorders, a number of patients with PD and multiple system atrophy, have QT or QTc interval prolongation on the electrocardiogram. In one prospective cohort, these findings were highly predictive of PD incidence (LR White, personal communication). Although these QT or QTc interval abnormalities are likely related to autonomic dysfunction, the pathophysiology remains unknown (Deguchi et al., 2002). Other characteristics in midlife associated with increased PD risk include increased triceps skinfold thickness (Abbott et al., 2002) and constipation. Men with less than one bowel movement per day at midlife had a 4.1‐fold excess incidence of PD when compared with men with more frequent bowel movements (Abbott et al., 2001). Taken together, these observations suggest that PD may begin decades before nervous system symptoms are observed. PD may be first a disorder of the peripheral autonomic nervous system. If an environmental trigger is involved, the gastrointestinal tract or the olfactory epithelium may be portals of entry. This hypothesis is indirectly supported by neuropathologic findings, suggesting that nigral pathology is a relatively late event in the pathogenesis of PD (Braak et al., 2004). Further studies to identify those at risk will be essential in determining the causes of PD, and methods for its prevention.
In the next half‐century, the average age of individuals in both developed and developing countries is expected to show a progressive increase. In the USA alone, this phenomenon of population aging is predicted to result in a three‐ to fourfold increase in PD frequency, or several million persons with the disease. The impact of PD can also be expected to affect disease‐associated health expenditures, lost income and personal suffering. As described in this chapter, despite intensive research efforts during the past several decades, the cause (or causes) of typical PD remains unknown. Likely, PD will be understood to be multifactorial, and both genetic and environmental determinants will be important. For example, estimated lifetime penetrance in parkinsonism caused by LRRK2 in the Ashkenazi Jewish population is only about 30% (Ozelius et al., 2006). Both genetic and environmental factors may determine expression of this monogenic form of parkinsonism. Typical PD may similarly be due to many different combinations of genetic or environmental determinants. The investigation of possible gene–environment interaction in PD is just beginning. In the next decade, investigations involving careful characterization of genetic and environmental factors will be essential to defining the causes of PD.
Acknowledgments
Drs Chade and Kasten are Michael J. Fox Foundation Fellows at The Parkinson's Institute. Thank you to Jennifer Wright for editorial assistance.
References
- Abbott RD, Petrovitch H, White LR. Frequency of bowel movements and the future risk of Parkinson's disease. Neurology. 2001;57:456–462. doi: 10.1212/wnl.57.3.456. [DOI] [PubMed] [Google Scholar]
- Abbott RD, Ross GW, White LR. Midlife adiposity and the future risk of Parkinson's disease. Neurology. 2002;59:1051–1057. doi: 10.1212/wnl.59.7.1051. [DOI] [PubMed] [Google Scholar]
- Abbott RD, Ross GW, White LR. Environmental, life‐style, and physical precursors of clinical Parkinson's disease: recent findings from the Honolulu‐Asia Aging Study. J Neurol. 2003;250(Suppl 3):30–39. doi: 10.1007/s00415-003-1306-7. [DOI] [PubMed] [Google Scholar]
- Acosta J, Calderon E, Obeso JA. Prevalence of Parkinson's disease and essential tremor in a village in southern Spain. Neurology. 1989;39(Suppl 1):181. [Google Scholar]
- Alves G, Kurz M, Lie SA. Cigarette smoking in Parkinson's disease: influence on disease progression. Mov Disord. 2004;19:1087–1092. doi: 10.1002/mds.20117. [DOI] [PubMed] [Google Scholar]
- Anca M, Paleacu D, Shabtai H. Cross‐sectional study of the prevalence of Parkinson's Disease in the Kibbutz Movement in Israel. Neuroepidemiology. 2002;21:50–55. doi: 10.1159/000048614. [DOI] [PubMed] [Google Scholar]
- Anderson DW, Rocca WA, de Rijk MC. Case ascertainment uncertainties in prevalence surveys of Parkinson's Disease. Mov Disord. 1998;13:626–632. doi: 10.1002/mds.870130403. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Zhang SM, Hernan MA. Prospective study of caffeine consumption and risk of Parkinson's disease in men and women. Ann Neurol. 2001;50:56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Schwarzschild MA. Caffeine, postmenopausal estrogen, and risk of Parkinson's disease. Neurology. 2003;60(5):790–795. doi: 10.1212/01.wnl.0000046523.05125.87. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Weisskopf MG, O'Reilly EJ. Coffee consumption, gender, and Parkinson's disease mortality in the cancer prevention study II cohort: the modifying effects of estrogen. Am J Epidemiol. 2004;160(10):977–984. doi: 10.1093/aje/kwh312. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Weisskopf MG, Chen H. Uricemia and risk of Parkison's disease. Neurology. 2006;66(5 Suppl 4):A382. [Google Scholar]
- Ashok PP, Radhakrishnan K, Sridharan R. Parkinsonism in Benghazi, East Libya. Clin Neurol Neurosurg. 1986;88:109–113. doi: 10.1016/s0303-8467(86)80005-1. [DOI] [PubMed] [Google Scholar]
- Baldereschi M, Di Carlo A, Rocca WA. Parkinson's disease and parkinsonism in a longitudinal study: two‐fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology. 2000;55:1358–1363. doi: 10.1212/wnl.55.9.1358. [DOI] [PubMed] [Google Scholar]
- Benamer HT, Oertel WH, Patterson J. Prospective study of presynaptic dopaminergic imaging in patients with mild parkinsonism and tremor disorders: part 1. Baseline and 3‐month observations. Mov Disord. 2003;18:977–984. doi: 10.1002/mds.10482. [DOI] [PubMed] [Google Scholar]
- Benedetti MD, Bower JH, Maraganore DM. Smoking, alcohol, and coffee consumption preceding Parkinson's disease: a case‐control study. Neurology. 2000;55:1350–1358. doi: 10.1212/wnl.55.9.1350. [DOI] [PubMed] [Google Scholar]
- Benedetti MD, Maraganore DM, Bower JH. Hysterectomy, menopause, and estrogen use preceding Parkinson's disease: an exploratory case‐control study. Mov Disord. 2001;16(5):830–837. doi: 10.1002/mds.1170. [DOI] [PubMed] [Google Scholar]
- Berg D, Roggendorf W, Schroder U. Echogenicity of the substantia nigra: association with increased iron content and marker for susceptibility to nigrostriatal injury. Arch Neurol. 2002;59:999–1005. doi: 10.1001/archneur.59.6.999. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O. Brain dopamine and the syndromes of Parkinson and Huntington: clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–1457. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharucha NE, Stokes L, Schoenberg BS. A case‐control study of twin pairs discordant for Parkinson's disease: a search for environmental risk factors. Neurology. 1986;36:284–288. doi: 10.1212/wnl.36.2.284. [DOI] [PubMed] [Google Scholar]
- Bharucha NE, Bharucha EP, Bharucha AE. revalence of Parkinson's disease in the Parsi community of Bombay, India. Arch Neurol. 1988;45:1321–1323. doi: 10.1001/archneur.1988.00520360039008. [DOI] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett. 1994;172(1–2):151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Fabrizio E, Vanacore N. Familial Parkinson's disease: a clinical genetic analysis. Can J Neurol Sci. 1995;22(4):272–279. doi: 10.1017/s0317167100039469. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ. Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rohe CF, Breedveld GJ. Italian Parkinson Genetics Network. Early‐onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65(1):87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]
- Bower JH, Maraganore DM, McDonnell SK. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology. 1999;52:1214–1220. doi: 10.1212/wnl.52.6.1214. [DOI] [PubMed] [Google Scholar]
- Bower JH, Maraganore DM, McDonnell SK. Influence of strict, intermediate, and broad diagnostic criteria on the age and sex‐specific incidence of Parkinson's disease. Mov Disord. 2000;15:819–825. doi: 10.1002/1531-8257(200009)15:5<819::aid-mds1009>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Bower JH, Maraganore DM, Peterson BJ. Head trauma preceding PD: a case‐control study. Neurology. 2003;60:1610–1615. doi: 10.1212/01.wnl.0000068008.78394.2c. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Gai WP. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110(5):517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U. Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res. 2004;318(1):121–134. doi: 10.1007/s00441-004-0956-9. [Epub 2004 Aug 24] [DOI] [PubMed] [Google Scholar]
- Breitner JC, Zandi PP. Do nonsteroidal antiinflammatory drugs reduce the risk of Alzheimer's disease? N Engl J Med. 2001;345(21):1567–1568. doi: 10.1056/NEJM200111223452110. [DOI] [PubMed] [Google Scholar]
- Brewis M, Poskanzer DC, Rolland C. Neurological disease in an English city. Acta Neurol Scand. 1966;42(Suppl 24):9–89. [PubMed] [Google Scholar]
- Bronstein DM, Perez‐Otano I, Sun V. Glia‐dependent neurotoxicity and neuroprotection in mesencephalic cultures. Brain Res. 1995;704(1):112–116. doi: 10.1016/0006-8993(95)01189-7. [DOI] [PubMed] [Google Scholar]
- Butterfield PG, Valanis BG, Spencer PS. nvironmental antecedents of young‐onset Parkinson's disease. Neurology. 1993;43(6):1150–1158. doi: 10.1212/wnl.43.6.1150. [DOI] [PubMed] [Google Scholar]
- Caradoc‐Davies TH, Weatherall M, Dixon GS. s the prevalence of Parkinson's disease in New Zealand really changing? Acta Neurol Scand. 1992;86:40–44. doi: 10.1111/j.1600-0404.1992.tb08051.x. [DOI] [PubMed] [Google Scholar]
- Castano A, Herrera AJ, Cano J. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh‐TNF‐alpha, IL‐1beta and IFN‐gamma. J Neurochem. 2002;81(1):150–157. doi: 10.1046/j.1471-4159.2002.00799.x. [DOI] [PubMed] [Google Scholar]
- Cerhan JR, Wallace RB, Folsom AR. Antioxidant intake and risk of Parkinson's disease (PD) in older women. Am J Epidemiol. 1994;139:S65. [Google Scholar]
- Chalmanov VN. Epidemiological studies of parkinsonism in Sofia. Neuroepidemiology. 1986;5:171–177. doi: 10.1159/000110826. [DOI] [PubMed] [Google Scholar]
- Champy P, Melot A, Guerineau Eng V. Quantification of acetogenins in Annona muricata linked to atypical parkinsonism in Guadeloupe. Mov Disord. 2005;20(12):1629–1633. doi: 10.1002/mds.20632. [DOI] [PubMed] [Google Scholar]
- Chan DK, Cordato D, Karr M. Prevalence of Parkinson's disease in Sydney. Acta Neurol Scand. 2005;111:7–11. doi: 10.1111/j.1600-0404.2004.00348.x. [DOI] [PubMed] [Google Scholar]
- Checkoway H, Franklin GM, Costa‐Mallen P. A genetic polymorphism of MAO‐B modifies the association of cigarette smoking and Parkinson's disease. Neurology. 1998;50:1458–1461. doi: 10.1212/wnl.50.5.1458. [DOI] [PubMed] [Google Scholar]
- Checkoway H, Nelson L. Epidemiologic approaches to the study of Parkinson's Disease etiology. Epidemiology. 1999;10(3):327–336. [PubMed] [Google Scholar]
- Chen H, Steyn S, Staal R. 8‐(3‐Chlorostyryl)caffeine may attenuate MPTP neurotoxicity through dual actions of monoamine oxidase inhibition and A2A receptor antagonism. J Biol Chem. 2002;277(39):36040–36044. doi: 10.1074/jbc.M206830200. Epub 2002 Jul 18. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernan MA. Diet and Parkinson's disease: a potential role of dairy products in men. Ann Neurol. 2002;52:793–801. doi: 10.1002/ana.10381. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernan MA. Nonsteroidal anti‐inflammatory drugs and the risk of Parkinson disease. Arch Neurol. 2003;60:1059–1064. doi: 10.1001/archneur.60.8.1059. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Schwarzschild MA. Folate intake and risk of Parkinson's disease. Am J Epidemiol. 2004;160(4):368–375. doi: 10.1093/aje/kwh213. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Schwarzschild MA. Obesity and the risk of Parkinson's disease. Am J Epidemiol. 2004;159(6):547–555. doi: 10.1093/aje/kwh059. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Schwarzschild MA. Physical activity and the risk of Parkinson disease. Neurology. 2005;64(4):664–669. doi: 10.1212/01.WNL.0000151960.28687.93. [DOI] [PubMed] [Google Scholar]
- Chen H, Jacobs E, Schwarzschild MA. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol. 2005;58(6):963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Schwarzschild MA. Survival of Parkinson's disease patients in a large prospective cohort of male health professionals. Mov Disord. 2006;21(7):1002–1007. doi: 10.1002/mds.20881. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Chu I, Villeneuve DC, Yagminas A. Toxicity of 2,2′,4,4′,5,5′‐hexachlorobiphenyl in rats: effects following a 90‐day oral exposure. J Appl Toxicol. 1996;16(2):121–128. doi: 10.1002/(SICI)1099-1263(199603)16:2<121::AID-JAT320>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Coggon D, Inskip H, Winter P. Occupational mortality by cause of death. In: Drever F, editor. Occupational Health Decennial Supplement. Her Majesty's Stationery Office; London: 1995. pp. 62–76. [Google Scholar]
- Cohen AD, Tillerson JL, Smith AD. Neuroprotective effects of prior limb use in 6‐hydroxydopamine‐treated rats: possible role of GDNF. J Neurochem. 2003;85(2):299–305. doi: 10.1046/j.1471-4159.2003.01657.x. [DOI] [PubMed] [Google Scholar]
- Corrigan FM, Murray L, Wyatt CL. Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson's disease. Exp Neurol. 1998;150(2):339–342. doi: 10.1006/exnr.1998.6776. [DOI] [PubMed] [Google Scholar]
- Currie LJ, Harrison MB, Trugman JM. Postmenopausal estrogen use affects risk for Parkinson disease. Arch Neurol. 2004;61(6):886–888. doi: 10.1001/archneur.61.6.886. [DOI] [PubMed] [Google Scholar]
- Davis JW, Grandinetti A, Waslien CI. Observations on serum uric acid levels and the risk of idiopathic Parkinson's disease. Am J Epidemiol. 1996;144(5):480–484. doi: 10.1093/oxfordjournals.aje.a008954. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkisnon's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Deguchi K, Sasaki I, Tsukaguchi M. Abnormalities of rate‐corrected QT intervals in Parkinson's disease‐a comparison with multiple system atrophy and progressive supranuclear palsy. J Neurol Sci. 2002;199(1–2):31–37. doi: 10.1016/s0022-510x(02)00079-5. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Giesbergen PC, de Rijk MC. ncidence of parkinsonism and Parkinson disease in a general population. The Rotterdam Study. Neurology. 2004;63:1240–1244. doi: 10.1212/01.wnl.0000140706.52798.be. [DOI] [PubMed] [Google Scholar]
- De Michele G, Filla A, Volpe G. Environmental and genetic risk factors in Parkinson's disease: a case‐ control study in Southern Italy. Mov Disord. 1996;11(1):17–23. doi: 10.1002/mds.870110105. [DOI] [PubMed] [Google Scholar]
- De Reuck J, De Weweire M, Van Maele G. Comparison of age of onset and development of motor complications between smokers and non‐smokers in Parkinson's disease. J Neurol Sci. 2005;231(1–2):35–39. doi: 10.1016/j.jns.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Dewailly E, Mulvad G, Pedersen HS. Concentration of organochlorines in human brain, liver, and adipose tissue autopsy samples from Greenland. Environ Health Perspect. 1999;107(10):823–828. doi: 10.1289/ehp.99107823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMonte DA. The environment and parkinson's disease: is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol. 2003;2:531–538. doi: 10.1016/s1474-4422(03)00501-5. [DOI] [PubMed] [Google Scholar]
- Di Rocco A, Molinari SP, Kollmeier B. Parkinson's disease: progression and mortality in the L‐DOPA era. Adv Neurol. 1996;69:3–11. [PubMed] [Google Scholar]
- Doll R, Peto R, Wheatley K. Mortality in relation to smoking: 40 years' observations on male British doctors. BMJ. 1994;309:901–911. doi: 10.1136/bmj.309.6959.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Ladenheim B, Cutler RG. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson's disease. J Neurochem. 2002;80(1):101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- Duarte J, Claveria LE, de Pedro‐Cuesta J. creening Parkinson's disease: a validated questionnaire of high specificity and sensitivity. Mov Disord. 1995;10:643–649. doi: 10.1002/mds.870100518. [DOI] [PubMed] [Google Scholar]
- Duvoisin RC, Eldridge R, Williams A. Twin study of Parkinson disease. Neurology. 1981;31:77–80. doi: 10.1212/wnl.31.1.77. [DOI] [PubMed] [Google Scholar]
- Elbaz A, Manubens‐Bertran JM, Baldereschi M. Parkinson's disease, smoking, and family history. EUROPARKINSON Study Group. J Neurol. 2000;247(10):793–798. doi: 10.1007/s004150070095. [DOI] [PubMed] [Google Scholar]
- Elbaz A, McDonnell SK, Maraganore DM. Validity of family history data on PD: evidence for a family information bias. Neurology. 2003;61(1):11–17. doi: 10.1212/01.wnl.0000068007.58423.c2. [DOI] [PubMed] [Google Scholar]
- Elbaz A, Bower JH, Peterson BJ. Survival study of Parkinson disease in Olmsted County, Minnesota. Arch Neurol. 2003;60(1):91–96. doi: 10.1001/archneur.60.1.91. [DOI] [PubMed] [Google Scholar]
- Elbaz A, Levecque C, Clavel J. CYP2D6 polymorphism, pesticide exposure, and Parkinson's disease. Ann Neurol. 2004;55:430–434. doi: 10.1002/ana.20051. [DOI] [PubMed] [Google Scholar]
- Factor SA, Weiner WJ. Prior history of head trauma in Parkinson's disease. Mov Disord. 1991;6:225–229. doi: 10.1002/mds.870060306. [DOI] [PubMed] [Google Scholar]
- Fall PA, Axelson O, Fredriksson M. Age‐ standardized incidence and prevalence of Parkinson's disease in a Swedish community. J Clin Epidemiol. 1996;49(6):637–641. doi: 10.1016/0895-4356(96)00003-0. [DOI] [PubMed] [Google Scholar]
- Fall PA, Fredrikson M, Axelson O. Nutritional and occupational factors influencing the risk of Parkinson's disease: a case‐control study in southeastern Sweden. Mov Disord. 1999;14:28–37. doi: 10.1002/1531-8257(199901)14:1<28::aid-mds1007>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Fall PA, Saleh A, Fredrickson M. Survival time, mortality and cause of death in elderly patients with Parkinson's disease: a 9‐year follow‐up. Mov Disord. 2003;18:1312–1316. doi: 10.1002/mds.10537. [DOI] [PubMed] [Google Scholar]
- Fazzini E, Fleming J, Fahn S. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson's disease. Mov Disord. 1992;7(2):153–158. doi: 10.1002/mds.870070210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Ferger B, Spratt C, Earl CD. Effects of nicotine on hydroxyl free radical formation in vitro and on MPTP‐induced neurotoxicity in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:351–359. doi: 10.1007/pl00005264. [DOI] [PubMed] [Google Scholar]
- Ferger B, Teismann P, Earl CD. Salicylate protects against MPTP‐induced motor impairments in the dopaminergic neurotransmission at the striatal and nigral level in mice. Naunyn Schmiedebergs Arch Pharmacol. 1999;360(3):256–261. doi: 10.1007/s002109900079. [DOI] [PubMed] [Google Scholar]
- Ferraz HB, Andrade LA, Tumas V. Rural or urban living and Parkinson's disease. Arq Neuropsiquiatr. 1996;54(1):37–41. doi: 10.1590/s0004-282x1996000100006. [DOI] [PubMed] [Google Scholar]
- Fertl E, Doppelbauer A, Auff E. Physical activity and sports in patients suffering from Parkinson's disease in comparison with healthy seniors. J Neural Transm Park Dis Dement Sect. 1993;5(2):157–161. doi: 10.1007/BF02251206. [DOI] [PubMed] [Google Scholar]
- Firestone JA, Smith‐Weller T, Franklin G. Pesticides and risk of Parkinson disease: a population‐based case‐control study. Arch Neurol. 2005;62:91–95. doi: 10.1001/archneur.62.1.91. [DOI] [PubMed] [Google Scholar]
- Fored CM, Fryzek JP, Brandt L. Parkinson's disease and other basal ganglia or movement disorders in a large nationwide cohort of Swedish welders. Occup Environ Med. 2006;63(2):135–140. doi: 10.1136/oem.2005.022921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Wang GJ. Inhibition of monoamine oxidase B in the brains of smokers. Nature. 1996;379:733–736. doi: 10.1038/379733a0. [DOI] [PubMed] [Google Scholar]
- Fryzek JP, Hansen J, Cohen S. A cohort study of Parkinson's disease and other neurodegenerative disorders in Danish welders. J Occup Environ Med. 2005;47:466–472. doi: 10.1097/01.jom.0000161730.25913.bf. [DOI] [PubMed] [Google Scholar]
- Galanaud JP, Elbaz A, Clavel J. Cigarette smoking and Parkinson's disease: a case‐control study in a population characterized by a high prevalence of pesticide exposure. Mov Disord. 2005;20(2):181–189. doi: 10.1002/mds.20307. [DOI] [PubMed] [Google Scholar]
- Gale CR, Martyn CN, Winter PD. Vitamin C and risk of death from stroke and coronary heart disease in cohort of elderly people. BMJ. 1995;310:1563–1566. doi: 10.1136/bmj.310.6994.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B. Microglial activation‐mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. 2002;81(6):1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1991;54(5):388–396. doi: 10.1136/jnnp.54.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibberd FB, Simmonds JP. Neurological disease in ex‐Far‐East prisoners of war. Lancet. 1980;2(8186):135–137. doi: 10.1016/s0140-6736(80)90015-x. [DOI] [PubMed] [Google Scholar]
- Giroud Benitez JL, Collado‐Mesa F, Esteban EM. Prevalence of Parkinson disease in an urban area of the Ciudad de La Habana province, Cuba. Door‐to‐door population study. Neurologia. 2000;15(7):269–273. [PubMed] [Google Scholar]
- Granieri E, Carreras M, Casetta I. Parkinson's disease in Ferrara, Italy, 1967 through 1987. Arch Neurol. 1991;48:854–857. doi: 10.1001/archneur.1991.00530200096026. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Tanner CM, Oakes D. Head injury and Parkinson's disease risk in twins. Ann Neurol. 2006;60(1):65–72. doi: 10.1002/ana.20882. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA. The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50(5):1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Peterson EL, Rybicki BA. Multiple risk factors for Parkinson's disease. J Neurol Sci. 2004;217:169–174. doi: 10.1016/j.jns.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Grandinetti A, Morens DM, Reed D. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson's disease. Am J Epidemiol. 1994;139:1129–1138. doi: 10.1093/oxfordjournals.aje.a116960. [DOI] [PubMed] [Google Scholar]
- Gudmundsson KR. A clinical survey of parkinsonism in Iceland. Acta Neurol Scand. 1967;43(Suppl 33):1–61. [PubMed] [Google Scholar]
- Haack DG, Baumann RJ, McKean HE. Nicotine exposure and Parkinson disease. Am J Epidemiol. 1981;114(2):191–200. doi: 10.1093/oxfordjournals.aje.a113182. [DOI] [PubMed] [Google Scholar]
- Harada H, Nishikawa S, Takahashi K. Epidemiology of Parkinson's disease in a Japanese city. Arch Neurol. 1983;40:151–154. doi: 10.1001/archneur.1983.04050030045008. [DOI] [PubMed] [Google Scholar]
- Hattori N, Kitada T, Matsumine H. Molecular genetic analysis of a novel parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the parkin gene in affected individuals. Ann Neurol. 1998;44:935–941. doi: 10.1002/ana.410440612. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Zesiewicz TA, Rosemurgy AS. Manganese intoxication and chronic liver failure. Ann Neurol. 1994;36(6):871–875. doi: 10.1002/ana.410360611. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Hubble JP, Truong DD. Istradefylline US‐001 Study Group. Randomized trial of the adenosine A(2A) receptor antagonist istradefylline in advanced PD. Neurology. 2003;61(3):297–303. doi: 10.1212/01.wnl.0000081227.84197.0b. [DOI] [PubMed] [Google Scholar]
- Hellenbrand W, Boeing H, Robra BP. Diet and Parkinson's disease. II: a possible role for the past intake of specific nutrients. Results from a self‐administered food‐frequency questionnaire in a case‐control study. Neurology. 1996;47:644–650. doi: 10.1212/wnl.47.3.644. [DOI] [PubMed] [Google Scholar]
- Hely MA, Morris JG, Traficante R. The Sidney Multicentre Study of Parkinson's disease: progression and mortality at 10 years. J Neurol Neurosurg Psychiatry. 1999;67:300–307. doi: 10.1136/jnnp.67.3.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernan MA, Takkouche B, Caamano‐Isorna F. A meta‐analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Checkoway H, O'Brien R. MAOB intron 13 and COMT codon 158 polymorphisms, cigarette smoking, and the risk of PD. Neurology. 2002;58(9):1381–1387. doi: 10.1212/wnl.58.9.1381. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Chen H, Schwarzschild MA. Alcohol consumption and the incidence of Parkinson's disease. Ann Neurol. 2003;54(2):170–175. doi: 10.1002/ana.10611. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Logroscino G, Rodriguez LA. A prospective study of alcoholism and the risk of Parkinson's disease. J Neurol. 2004;251(Suppl 7):14–17. doi: 10.1007/s00415-004-1705-4. vII. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Logroscino G, Garcia Rodriguez LA. Nonsteroidal anti‐inflammatory drugs and the incidence of Parkinson disease. Neurology. 2006;66(7):1097–1099. doi: 10.1212/01.wnl.0000204446.82823.28. [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Castano A, Venero JL. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7(4):429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E. The role of glial reaction and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:214–228. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Hobson P, Gallacher J, Meara J. Cross‐sectional survey of Parkinson's disease and parkinsonism in a rural area of the United Kingdom. Mov Disord. 2005;20(8):995–998. doi: 10.1002/mds.20489. [DOI] [PubMed] [Google Scholar]
- Hubble JP, Cao T, Hassanein RE. Risk factors for Parkinson's disease. Neurology. 1993;43(9):1693–1697. doi: 10.1212/wnl.43.9.1693. [DOI] [PubMed] [Google Scholar]
- Hubble JP, Venkatesh R, Hassanein RE. Personality and depression in Parkinson's disease. J Nerv Ment Dis. 1993;181(11):657–662. doi: 10.1097/00005053-199311000-00001. [DOI] [PubMed] [Google Scholar]
- Hubble JP, Cao T, Kjelstrom JA. Nocardia species as an etiologic agent in Parkinson's disease: serological testing in a case‐control study. J Clin Microbiol. 1995;33(10):2768–2769. doi: 10.1128/jcm.33.10.2768-2769.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Blankson S. A clinicopathologic study of 100 cases of Parkinson's disease. Arch Neurol. 1993;50:140–148. doi: 10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology. 2001;57:1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Ben Shlomo Y. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Part 4):861–870. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- Hunot S, Dugas N, Faucheux B. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor‐alpha in glial cells. J Neurosci. 1999;19(9):3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in t’ Veld BA, Ruitenberg A, Hofman A. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345(21):1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Janson AM, Moller A. Chronic nicotine treatment counteracts nigral cell loss induced by a partial mesodiencephalic hemitransection: an analysis of the total number and mean volume of neurons and glia in substantia nigra of the male rat. Neuroscience. 1993;57:931–941. doi: 10.1016/0306-4522(93)90039-i. [DOI] [PubMed] [Google Scholar]
- Jenkins AC. Epidemiology of parkinsonism in Victoria. Med J Aust. 1966;2:497–502. doi: 10.5694/j.1326-5377.1966.tb97295.x. [DOI] [PubMed] [Google Scholar]
- Jenner P. A2A antagonists as novel non‐dopaminergic therapy for motor dysfunction in PD. Neurology. 2003;61(11 Suppl 6):S32–S38. doi: 10.1212/01.wnl.0000095209.59347.79. [DOI] [PubMed] [Google Scholar]
- Kanda T, Tashiro T, Kuwana Y. Adenosine A2A receptors modify motor function in MPTP‐treated common marmosets. Neuroreport. 1998;9(12):2857–2860. doi: 10.1097/00001756-199808240-00032. [DOI] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S. Genetic or pharmacological iron chelation prevents MPTP‐induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37(6):899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- Kay DM, Zabetian CP, Factor SA. Parkinson's disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov Disord. 2006;21(4):519–523. doi: 10.1002/mds.20751. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Kessler Epidemiologic studies of Parkinson's disease. II. A hospital‐based survey. Am J Epidemiol. 1972;95(4):308–318. doi: 10.1093/oxfordjournals.aje.a121399. [DOI] [PubMed] [Google Scholar]
- Kessler Epidemiologic studies of Parkinson's disease. III. A community‐based survey. Am J Epidemiol. 1972;96(4):242–254. doi: 10.1093/oxfordjournals.aje.a121455. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kobbata S, Beaman BL. L‐dopa‐responsive movement disorder caused by Nocardia asteroides localized in the brains of mice. Infect Immun. 1991;59(1):181–191. doi: 10.1128/iai.59.1.181-191.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korell M, Tanner CM. Epidemiology of Parkinson's Disease: an overview. In: Ebadi M, Pfeiffer RF, editors. Parkinson's Disease. CRC Press; New York: 2005. pp. 39–50. [Google Scholar]
- Knekt P, Reunanen A, Järvinen R. Antioxidant vitamin intake and coronary mortality in a longitudinal population study. Am J Epidemiol. 1994;139:1180–1189. doi: 10.1093/oxfordjournals.aje.a116964. [DOI] [PubMed] [Google Scholar]
- Knekt P, Järvinen R, Reunanen A. Flavonoid intake and coronary mortality in Finland: a cohort study. BMJ. 1996;312:478–481. doi: 10.1136/bmj.312.7029.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Calne SM, Schulzer M. Clustering of Parkinson disease: shared cause or coincidence? Arch Neurol. 2004;61(7):1057–1060. doi: 10.1001/archneur.61.7.1057. [DOI] [PubMed] [Google Scholar]
- Kuopio AM, Marttila RJ, Helenius H. Changing epidemiology of Parkinson's disease in southwestern Finland. Neurology. 1999;52:302–308. doi: 10.1212/wnl.52.2.302. [DOI] [PubMed] [Google Scholar]
- Kusumi M, Nakashima K, Harada H. Epidemiology of Parkinson's disease in Yonago City, Japan: comparison with a study carried out 12 years ago. Neuroepidemiology. 1996;15(4):201–207. doi: 10.1159/000109908. [DOI] [PubMed] [Google Scholar]
- Kurland LT. Epidemiology. Incidence, geographic distribution and genetic considerations. In: Field W, editor. Pathogenesis and Treatment of Parkinsonism. Charles C. Thomas; Springfield: 1958. pp. 5–43. [Google Scholar]
- Kwok JB, Hallupp M, Loy CT. GSK3B polymorphisms alter transcription and splicing in Parkinson's disease. Ann Neurol. 2005;58(6):829–839. doi: 10.1002/ana.20691. [DOI] [PubMed] [Google Scholar]
- Lan J, Jiang DH. Excessive iron accumulation in the brain: a possible potential risk of neurodegeneration in Parkinson's disease. J Neural Transm. 1997;104(6–7):649–660. doi: 10.1007/BF01291883. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW. Chronic parkinsonism in humans due to a product of meperidine‐analog synthesis. Science. 1983;219(4587):979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine exposure. Ann Neurol. 1999;46(4):598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Le WD, Xu P, Jankovic J. Mutations in NR4A2 associated with familial Parkinson's disease. Nat Genet. 2003;33:85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Katzenschlager R, Head J. Ten‐year follow‐up of three different initial treatments in de‐novo PD: a randomized trial. Neurology. 2001;57:1687–1694. doi: 10.1212/wnl.57.9.1687. [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G. The ubiquitin pathway in Parkinson's disease. Nature. 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Lesage S, Ibanez P, Lohmann E. French Parkinson's Disease Genetics Study Group G2019S LRRK2 mutation in French and North African families with Parkinson's disease. Ann Neurol. 2005;58(5):784–787. doi: 10.1002/ana.20636. [DOI] [PubMed] [Google Scholar]
- Levy G, Tang MX, Cote LJ. Do risk factors for Alzheimer's disease predict dementia in Parkinson's disease? An exploratory study. Mov Disord. 2002;17(2):250–257. doi: 10.1002/mds.10086. [DOI] [PubMed] [Google Scholar]
- Li SC, Schoenberg BS, Wang CC. A prevalence survey of Parkinson's disease and other movement disorders in the People's Republic of China. Arch Neurol. 1985;42:655–657. doi: 10.1001/archneur.1985.04060070045013. [DOI] [PubMed] [Google Scholar]
- Lilienfeld DE, Sekkor D, Simpson S. Parkinsonism death rates by race, sex and geography: a 1980s update. Neuroepidemiology. 1990;9(5):243–247. doi: 10.1159/000110780. [DOI] [PubMed] [Google Scholar]
- Louis ED, Marder K, Cote L. Mortality from Parkinson's disease. Arch Neurol. 1997;54(3):260–264. doi: 10.1001/archneur.1997.00550150024011. [DOI] [PubMed] [Google Scholar]
- MacDonald BK, Cockerell OC, Sander JW. The incidence and lifetime prevalence of neurological disorders in a prospective community‐based study in the UK. Brain. 2000;123(Pt 4):665–676. doi: 10.1093/brain/123.4.665. [DOI] [PubMed] [Google Scholar]
- Maggio R, Riva M, Vaglini F. Striatal increase of neurotrophic factors as a mechanism of nicotine protection in experimental parkinsonism. J Neural Transm. 1997;104:1113–1123. doi: 10.1007/BF01273324. [DOI] [PubMed] [Google Scholar]
- Maher NE, Golbe LI, Lazzarini AM. Epidemiologic study of 203 sibling pairs with Parkinson's disease: the GenePD study. Neurology. 2002;58(1):79–84. doi: 10.1212/wnl.58.1.79. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, Anderson DW, Bower JH. Autopsy patterns for Parkinson's disease and related disorders in Olmsted County, Minesota. Neurology. 1999;53(6):1342–1344. doi: 10.1212/wnl.53.6.1342. [DOI] [PubMed] [Google Scholar]
- Marder K, Tang MX, Mejia H. Risk of Parkinson's disease among first‐degree relatives: a community‐based study. Neurology. 1996;47:155–160. doi: 10.1212/wnl.47.1.155. [DOI] [PubMed] [Google Scholar]
- Marder K, Logroscino G, Alfaro B. Environmental risk factors for Parkinson's disease in an urban multiethnic community. Neurology. 1998;50(1):279–281. doi: 10.1212/wnl.50.1.279. [DOI] [PubMed] [Google Scholar]
- Marek KL, Seibyl JP, Zoghbi SS. [123I] beta‐CIT/SPECT imaging demonstrates bilateral loss of dopamine transporters in hemi‐Parkinson's disease. Neurology. 1996;46:231–237. doi: 10.1212/wnl.46.1.231. [DOI] [PubMed] [Google Scholar]
- Marras C, Tanner CM. Epidemiology of Parkinson's Disease. In: Watts RL, Koller WC, editors. Movement Disorders, Neurologic Principles and Practice. McGraw‐Hill; New York: 2002. pp. 117–195. [Google Scholar]
- Marras C, Goldman S, Smith A. Smell identification ability in twin pairs discordant for Parkinson's disease. Mov Disord. 2005;20(6):687–693. doi: 10.1002/mds.20389. [DOI] [PubMed] [Google Scholar]
- Marras C, McDermott MP, Rochon PA. Parkinson Study Group. Survival in Parkinson disease: thirteen‐year follow‐up of the DATATOP cohort. Neurology. 2005;64(1):87–93. doi: 10.1212/01.WNL.0000148603.44618.19. [DOI] [PubMed] [Google Scholar]
- Marsden CD. Twins and Parkinson's disease. J Neurol Neurosurg Psychiatry. 1987;50:105–106. doi: 10.1136/jnnp.50.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marttila RJ, Rinne UK. Epidemiology of Parkinson's disease in Finland. Acta Neurol Scand. 1967;43(Suppl 33):9–61. doi: 10.1111/j.1600-0404.1976.tb04328.x. [DOI] [PubMed] [Google Scholar]
- Marttila RJ, Arstila P, Nikoskelainen J. Viral antibodies in the sera from patients with Parkinson's disease. Eur Neurol. 1977;15:25–33. doi: 10.1159/000114785. [DOI] [PubMed] [Google Scholar]
- Marttila RJ, Kaprio J, Koshewvuo M. Parkinson's disease in a nationwide twin cohort. Neurology. 1988;38:1217–1219. doi: 10.1212/wnl.38.8.1217. [DOI] [PubMed] [Google Scholar]
- Mars U, Larsson BS. Pheomelanin as a binding site for drugs and chemicals. Pigment Cell Res. 1999;12(4):266–274. doi: 10.1111/j.1600-0749.1999.tb00760.x. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Denaro J, Hemenegildo N. A population‐based investigation of Parkinson's disease with and without dementia: relationships to age and gender. Arch Neurol. 1992;42:492–497. doi: 10.1001/archneur.1992.00530290076015. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Marder K, Cote LJ. The frequency of idiopathic Parkinson's disease by age, ethnic group, and sex in northern Manhattan, 1988–1993. Am J Epidemiol. 1995;142:820–827. doi: 10.1093/oxfordjournals.aje.a117721. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Akiyama H. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- Menegon A, Board PG, Blackburn AC. Parkinson's disease, pesticides, and glutathione transferase polymorphisms. Lancet. 1998;352(9137):1344–1346. doi: 10.1016/s0140-6736(98)03453-9. [DOI] [PubMed] [Google Scholar]
- Menza M. The personality associated with Parkinson's disease. Curr Psychiatry Rep. 2000;2:421–426. doi: 10.1007/s11920-000-0027-1. [DOI] [PubMed] [Google Scholar]
- Morano A, Jimenez‐Jimenez FJ, Molina JA. Risk‐factors for Parkinson's disease: case‐control study in the province of Caceres, Spain. Acta Neurol Scand. 1994;89(3):164–170. doi: 10.1111/j.1600-0404.1994.tb01655.x. [DOI] [PubMed] [Google Scholar]
- Morens DM, Davis JW, Grandinetti A. Epidemiologic observations on Parkinson's disease: incidence and mortality in a prospective study of middle‐aged men. Neurology. 1996;46:1044–1050. doi: 10.1212/wnl.46.4.1044. [DOI] [PubMed] [Google Scholar]
- Morens DM, Grandinetti A, Davis JW. Evidence against the operation of selective mortality in explaining the association between cigarette smoking and reduced occurrence of idiopathic Parkinson disease. Am J Epidemiol. 1996;144(4):400–404. doi: 10.1093/oxfordjournals.aje.a008941. [DOI] [PubMed] [Google Scholar]
- Morgante L, Rocca WA, Di Rosa AE. Prevalence of Parkinson's disease and other parkinsonisms: a door‐to‐door survey in three Sicilian municipalities. Neurology. 1992;42:1901–1907. doi: 10.1212/wnl.42.10.1901. [DOI] [PubMed] [Google Scholar]
- Morgante L, Salemi G, Meneghini F. Parkinson disease survival: a population‐based study. Arch Neurol. 2000;57(4):507–512. doi: 10.1001/archneur.57.4.507. [DOI] [PubMed] [Google Scholar]
- Murch SJ, Cox PA, Banack SA. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc Natl Acad Sci USA. 2004;101(33):12228–12231. doi: 10.1073/pnas.0404926101. [Epub 2004 Aug 4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch WJ, Dingwall‐Fordyce I, Downie AW. Parkinson's disease in a Scottish city. BMJ. 1986:534–536. doi: 10.1136/bmj.292.6519.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch WJ, Smith WC, Scott RF. A screening and alerting questionnaire for parkinsonism. Neuroepidemiology. 1991;10:150–156. doi: 10.1159/000110261. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H. Cytokines in Parkinson's disease. J Neural Transm. 2000;Suppl(58):143–151. [PubMed] [Google Scholar]
- Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical,metabolic and psychostimulant effects. Brain Res Brain Res Rev. 1992;17(2):139–170. doi: 10.1016/0165-0173(92)90012-b. [DOI] [PubMed] [Google Scholar]
- Nelson L, Van Den Eeden S, Tanner C. Vol. 52. 1999. Association of alcohol and tobacco consumption with Parkinson's disease: a population‐based study; pp. A538–A539. (Neurology). [Google Scholar]
- Nishitani H, Kuno A, Konishi T. Vol. 3. Government Press; Tokyo: 1981. Epidemiological study of Parkinson's disease: area survey and the follow‐up study; pp. 81–86. (Annual Report of the Research Committee of Degenerative Disorders. The Ministry of Health and Welfare of Japan). [Google Scholar]
- Okada K, Kobayashi S, Tsunematsu T. Prevalence of Parkinson's disease in Izumo City, Japan. Gerontology. 1990;36:340–344. doi: 10.1159/000213219. [DOI] [PubMed] [Google Scholar]
- O'Reilly EJ, McCullough ML, Chao A. Smokeless tobacco use and the risk of Parkinson's disease mortality. Mov Disord. 2005;20(10):1383–1384. doi: 10.1002/mds.20587. [DOI] [PubMed] [Google Scholar]
- O'Suilleabhain PE, Sung V, Hernandez C. Elevated plasma homocysteine level in patients with Parkinson disease: motor, affective, and cognitive associations. Arch Neurol. 2004;61(6):865–868. doi: 10.1001/archneur.61.6.865. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders‐Pullman R. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews [letter] N Engl J Med. 2006;354(4):424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Paganini‐Hill A. Risk factors for parkinson's disease: the leisure world cohort study. Neuroepidemiology. 2001;20:118–124. doi: 10.1159/000054770. [DOI] [PubMed] [Google Scholar]
- Page WF, Tanner CM. Parkinson's disease and motor‐neuron disease in former prisoners‐of‐war. Lancet. 2000;355(9206):843. doi: 10.1016/S0140-6736(05)72455-7. [DOI] [PubMed] [Google Scholar]
- Paisan‐Ruiz C, Jain S, Evans EW. Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos S, Singer C, Villar JM. Does cigarette smoking provide clinically significant neuroprotection among patients diagnosed with Parkinson's disease? Mov Disord. 2005;20(5):641–642. doi: 10.1002/mds.20433. [DOI] [PubMed] [Google Scholar]
- Park M, Ross GW, Petrovitch H. Consumption of milk and calcium in midlife and the future risk of Parkinson disease. Neurology. 2005;64(6):1047–1051. doi: 10.1212/01.WNL.0000154532.98495.BF. [DOI] [PubMed] [Google Scholar]
- Parkinson J. Sherwood. Neely and Jones; London: 1817. An Essay on the Shaking Palsy. [Google Scholar]
- Payami H, Larsen K, Bernard S. Increased risk of Parkinson's disease in parents and siblings of patients. Ann Neurol. 1994;36:659–661. doi: 10.1002/ana.410360417. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Higgins JJ, Golbe LI. Mapping of a gene for Parkinson's disease to chromosome 4q21‐q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E. Mutation in tha alpha‐synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Popat RA, Van Den Eeden SK, Tanner CM. Effect of reproductive factors and postmenopausal hormone use on the risk of Parkinson disease. Neurology. 2005;65(3):383–390. doi: 10.1212/01.wnl.0000171344.87802.94. [DOI] [PubMed] [Google Scholar]
- Popoli P, Caporali MG, Scotti de Carolis A. Akinesia due to catecholamine depletion in mice is prevented by caffeine. Further evidence for an involvement of adenosinergic system in the control of motility. J Pharm Pharmacol. 1991;43(4):280–281. doi: 10.1111/j.2042-7158.1991.tb06685.x. [DOI] [PubMed] [Google Scholar]
- Powers KM, Smith‐Weller T, Franklin GM. Parkinson's disease risks associated with dietary iron, manganese, and other nutrient intakes. Neurology. 2003;60(11):1761–1766. doi: 10.1212/01.wnl.0000068021.13945.7f. [DOI] [PubMed] [Google Scholar]
- Prasad C, Ikegami H, Shimizu I. Chronic nicotine intake decelerates aging of nigrostriatal dopaminergic neurons. Life Sci. 1994;54:1169–1184. doi: 10.1016/0024-3205(94)00839-6. [DOI] [PubMed] [Google Scholar]
- Pressley J, Tang M, Marder K. Disparities in the recording of Parkinson's disease on death certificates. Mov Disord. 2005;20(3):315–321. doi: 10.1002/mds.20339. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA. A meta‐analysis of Parkinson's disease and exposure to pesticides. Neurotoxicology. 2000;21:435–440. [PubMed] [Google Scholar]
- Quik M. Smoking, nicotine and Parkinson's disease. Trends Neurosci. 2004;27:561–568. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Racette BA, Tabbal SD, Jennings D. Prevalence of parkinsonism and relationship to exposure in a large sample of Alabama welders. Neurology. 2005;64:230–235. doi: 10.1212/01.WNL.0000149511.19487.44. [DOI] [PubMed] [Google Scholar]
- Ragonese P, D'Amelio M, Salemi G. Risk of Parkinson disease in women: effect of reproductive characteristics. Neurology. 2004;62(11):2010–2014. doi: 10.1212/wnl.62.11.2010. [DOI] [PubMed] [Google Scholar]
- Rajput AH, Offord KP, Beard CM. Epidemiology of parkinsonism: incidence, classification and mortality. Ann Neurol. 1984;16:278–282. doi: 10.1002/ana.410160303. [DOI] [PubMed] [Google Scholar]
- Rajput AH, Offord KP, Beard CM. A case‐control study of smoking habits, dementia, and other illnesses in idiopathic Parkinson's disease. Neurology. 1987;37(2):226–232. doi: 10.1212/wnl.37.2.226. [DOI] [PubMed] [Google Scholar]
- Richardson PJ, Kase H, Jenner PG. Adenosine A2A receptor antagonists as new agents for the treatment of Parkinson's disease. Trends Pharmacol Sci. 1997;18(9):338–344. doi: 10.1016/s0165-6147(97)01096-1. [DOI] [PubMed] [Google Scholar]
- Rimm EB, Stampfer MJ, Ascherio A. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328:1450–1456. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- Rocca WA, Bower JH, McDonnell SK. Time trends in the incidence of parkinsonisms in Olmsted County, Minnesota. Neurology. 2001;57:462–467. doi: 10.1212/wnl.57.3.462. [DOI] [PubMed] [Google Scholar]
- Rosati G, Granieri E, Pinna L. The risk of Parkinson's disease in Mediterranean people. Neurology. 1980;30:250–255. doi: 10.1212/wnl.30.3.250. [DOI] [PubMed] [Google Scholar]
- Ross G, White L, Petrovitch H. Lack of association to midlife smoking or coffee consumption with presence of lewy bodies in the locus ceruleus or substantia nigra at autopsy. Neurology. 1999;52(Suppl 2):A539. [Google Scholar]
- Ross GW, Abbott RD, Petrovitch H. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA. 2000;283:2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Ross GW, Petrovitch H, Abbott RD. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol. 2004;56(4):532–539. doi: 10.1002/ana.20226. [DOI] [PubMed] [Google Scholar]
- Sasco AJ, Paffenbarger RS., Jr Measles infection and Parkinson's disease. Am J Epidemiol. 1985;122(6):1017–1031. doi: 10.1093/oxfordjournals.aje.a114183. [DOI] [PubMed] [Google Scholar]
- Sasco AJ, Paffenbarger RS, Jr, Gendre I. The role of physical exercise in the occurrence of Parkinson's disease. Arch Neurol. 1992;49(4):360–365. doi: 10.1001/archneur.1992.00530280040020. [DOI] [PubMed] [Google Scholar]
- Scheider WL, Hershey LA, Vena JE. Dietary antioxidants and other dietary factors in the etiology of Parkinson's disease. Mov Disord. 1997;12(2):190–196. doi: 10.1002/mds.870120209. [DOI] [PubMed] [Google Scholar]
- Schoenberg BS, Anderson DW, Haerer AF. Prevalence of Parkinson's disease in the biracial population of Copiah County, Mississippi. Neurology. 1985;35:841–845. doi: 10.1212/wnl.35.6.841. [DOI] [PubMed] [Google Scholar]
- Schoenberg BS, Osuntokun BO, Adeuja AOG. Comparison of the prevalence of Parkinson's disease in black populations in the rural US and in rural Nigeria: door‐to‐door community studies. Neurology. 1988;38:645–646. doi: 10.1212/wnl.38.4.645. [DOI] [PubMed] [Google Scholar]
- Schulte PA, Burnett CA, Boeniger MF. Neurodegenerative diseases: occupational occurrence and potential risk factors, 1982 through 1991. Am J Public Health. 1996;86(9):1281–1288. doi: 10.2105/ajph.86.9.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WK, Zhang F, Stajich JM. Family‐based case‐control study of cigarette smoking and Parkinson disease. Neurology. 2005;64(3):442–447. doi: 10.1212/01.WNL.0000150905.93241.B2. [DOI] [PubMed] [Google Scholar]
- Seegal RF, Brosch KO, Bush B. Polychlorinated biphenyls produce regional alterations of dopamine metabolism in rat brain. Toxicol Lett. 1986;30:197–202. doi: 10.1016/0378-4274(86)90103-7. [DOI] [PubMed] [Google Scholar]
- Seidler A, Hellenbrand W, Robra BP. Possible environmental, occupational, and other etiologic factors for Parkinson's disease: a case‐control study in Germany. Neurology. 1996;46:1275–1284. doi: 10.1212/wnl.46.5.1275. [DOI] [PubMed] [Google Scholar]
- Semchuk KM, Love EJ, Lee RG. Parkinson's disease and exposure to rural environmental factors: a population based case‐control study. Can J Neurol Sci. 1991;18(3):279–286. doi: 10.1017/s0317167100031826. [DOI] [PubMed] [Google Scholar]
- Semchuk KM, Love E, Lee R. Parkinson's disease: a test of the multifactorial etiologic hypothesis. Neurology. 1993;43:1173–1180. doi: 10.1212/wnl.43.6.1173. [DOI] [PubMed] [Google Scholar]
- Shi Y. Study on the prevalence of Parkinson's disease in Hongkou District, Shanghai. Chinese J Epidemiol. 1987;4:205–209. [PubMed] [Google Scholar]
- Siderowf A, Newberg A, Chou KL. [99mTc]TRODAT‐1 SPECT imaging correlates with odor identification in early Parkinson disease. Neurology. 2005;64(10):1716–1720. doi: 10.1212/01.WNL.0000161874.52302.5D. [DOI] [PubMed] [Google Scholar]
- Spencer P. Guam ALS/Parkinsonism‐Dementia: a long‐latency neurotoxic disorder caused by “slow toxin(s)” in food? Can J Neurol Sci. 1987;14:347–357. doi: 10.1017/s0317167100037732. [DOI] [PubMed] [Google Scholar]
- Stern MB. Head trauma as a risk factor for Parkinson's disease. Mov Disord. 1991;6(2):95–97. doi: 10.1002/mds.870060202. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Stewart PA. Occupational case‐control studies: I. Collecting information on work histories and work‐related exposures. Am J Ind Med. 1994;26(3):297–312. doi: 10.1002/ajim.4700260304. [DOI] [PubMed] [Google Scholar]
- Stiasny‐Kolster K, Doerr Y, Moller JC. Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for alpha‐synucleinopathy demonstrated by dopamine transporter FP‐CIT‐SPECT. Brain. 2005;128(Pt 1):126–137. doi: 10.1093/brain/awh322. [Epub 2004 Nov 17] [DOI] [PubMed] [Google Scholar]
- Sugita M, Izuno T, Tatemichi M. Meta‐analysis for epidemiologic studies on the relationship between smoking and Parkinson's disease. J Epidemiol. 2001;11:87–94. doi: 10.2188/jea.11.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe RL, Meara JR. Parkinson's disease epidemiology in the Northampton District, England, 1992. Acta Neurol Scand. 1995;92(6):443–450. doi: 10.1111/j.1600-0404.1995.tb00478.x. [DOI] [PubMed] [Google Scholar]
- Sutcliffe RL, Prior R, Mawby B. Parkinson's disease in the district of the Northampton Health Authority, United Kingdom. Acta Neurol Scand. 1985;72:363–379. doi: 10.1111/j.1600-0404.1985.tb00886.x. [DOI] [PubMed] [Google Scholar]
- Tan EK, Chai A, Zhao Y. Mitochondrial complex I polymorphism and cigarette smoking in Parkinson's disease. Neurology. 2002;59(8):1288–1289. doi: 10.1212/01.wnl.0000031809.71668.a1. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Goldman SM. Epidemiology of movement disorders. Curr Opin Neurol. 1994;7:340–345. doi: 10.1097/00019052-199408000-00011. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Goldman SM. Epidemiology of Parkinson's disease. Neurol Clin. 1996;14:317–335. doi: 10.1016/S0733-8619(05)70259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Chen B, Wang WZ. Environmental factors in the etiology of Parkinson's disease. Can J Neurol Sci. 1987;14(3 Suppl):419–423. doi: 10.1017/s0317167100037835. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Gilley DW, Goetz CG. A brief screening questionnaire for parkinsonism. Ann Neurol. 1990;28:267–268. [Google Scholar]
- Tanner CM, Thelen JA, Offord KP. Parkinson's disease incidence in Olmsted County, MN: 1935 ‐ 1988. Neurology. 1992;42(Suppl 3):194. [Google Scholar]
- Tanner CM, Goldman SM, Aston DA. Smoking and Parkinson's disease in twins. Neurology. 2002;58(4):581–588. doi: 10.1212/wnl.58.4.581. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Goldman SM, Quinlan P. Occupation and Risk of Parkinson's Disease (PD): a preliminary investigation of standard occupational codes (SOC) in twins discordant for disease. Neurology. 2003;60(Suppl 1):A415. [Google Scholar]
- Taylor CA, Saint‐Hilaire MH, Cupples LA. Environmental, medical, and family history risk factors for Parkinson's disease: a New England‐based case control study. Am J Med Genet. 1999;88:742–749. [PubMed] [Google Scholar]
- Taylor MC, Le Couteur DG, Mellick GD. Paraoxonase polymorphisms, pesticide exposure and Parkinson's disease in a Caucasian population. J Neural Transm. 2000;107(8–9):979–983. doi: 10.1007/s007020070046. [DOI] [PubMed] [Google Scholar]
- Tielemans E, Heederik D, Burdorf A. Assessment of occupational exposures in a general population: comparison of different methods. Occup Environ Med. 1999;56(3):145–151. doi: 10.1136/oem.56.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reveron ME. Exercise induces behavioral recovery and attenuates neurochemical deficits in rodent models of Parkinson's disease. Neuroscience. 2003;119(3):899–911. doi: 10.1016/s0306-4522(03)00096-4. [DOI] [PubMed] [Google Scholar]
- Tsai CH, Lo SK, See LC. Environmental risk factors of young onset Parkinson's disease: a case‐control study. Clin Neurol Neurosurg. 2002;104(4):328–333. doi: 10.1016/s0303-8467(02)00027-6. [DOI] [PubMed] [Google Scholar]
- Tsui JK, Calne DB, Wang Y. Occupational risk factors in Parkinson's disease. Can J Public Health. 1999;90:334–337. doi: 10.1007/BF03404523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twelves D, Persins K, Counsell Systematic review of incidence studies of Parkinson's disease. Mov Disord. 2003;18(1):19–31. doi: 10.1002/mds.10305. [DOI] [PubMed] [Google Scholar]
- Van Den Eeden SK, Tanner C, Popat R. The risk of Parkinson's disease associated with head injury and depression: a population‐based case‐control study. Neurology. 2000;54(7, Suppl 3):A347. [Google Scholar]
- Van Den Eeden SK, Tanner CM, Bernstein AL. Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- Vieregge P, Schiffke KA, Friedrich HJ. Parkinson's disease in twins. Neurology. 1992;42:1453–1461. doi: 10.1212/wnl.42.8.1453. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Fuh JL, Liu CY. Parkinson's disease in Kin‐Hu, Kinmen: a community survey by neurologists. Neuroepidemiology. 1994;13:69–74. doi: 10.1159/000110361. [DOI] [PubMed] [Google Scholar]
- Wang WZ, Fang XH, Cheng XM. A case‐ control study on the environmental risk factors of Parkinson's disease in Tianjin, China. Neuroepidemiology. 1993;12:209–218. doi: 10.1159/000110319. [DOI] [PubMed] [Google Scholar]
- Wang Y, Collaborative Group of Neuroepidemiology of the PLA The incidence and prevalence of Parkinson's disease in the People's Republic of China. Chung‐Hua Liu Hsing Ping Hsueh Tsa Chih. Chinese Journal of Epidemiology. 1991;12:363–365. [PubMed] [Google Scholar]
- Wenning GK, Donnemiller E, Granata R. [123‐I]‐beta‐CIT and [123‐I]‐IBZM‐SPECT scanning in levodopanaive Parkinson's disease. Mov Disord. 1998;13:438–445. doi: 10.1002/mds.870130311. [DOI] [PubMed] [Google Scholar]
- Wermuth L, Bunger N, von Weitzel‐Mudersback P. Clinical characteristics of Parkinson's disease among Inuit in Greenland and inhabitants of the Faroe Islands and Als (Denmark) Mov Disord. 2004;19(7):821–824. doi: 10.1002/mds.20058. [DOI] [PubMed] [Google Scholar]
- Willems‐Giesbergen P, de Rijk M, van Swieten J. Smoking, alcohol, and coffee consumption and the risk of Parkinson's disease: results from the Rotterdam Study. Neurology. 2000;54:A347. [Google Scholar]
- Willett W. Overview of nutritional epidemiology. In: Willett W, editor. Nutritional Epidemiology. 1st edn. Oxford University Press; New York/Oxford: 1990. pp. 3–19. [Google Scholar]
- Wirdefeldt K, Gatz M, Schalling M. No evidence for heritability of Parkinson disease in Swedish twins. Neurology. 2004;63(2):305–311. doi: 10.1212/01.wnl.0000129841.30587.9d. [DOI] [PubMed] [Google Scholar]
- Wooten GF, Currie LJ, Bovbjerg VE. Are men at greater risk for Parkinson's disease than women? J Neurol Neurosurg Psychiatry. 2004;75(4):637–639. doi: 10.1136/jnnp.2003.020982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Bastia E, Schwarzschild M. Therapeutic potential of adenosine A(2A) receptor antagonists in Parkinson's disease. Pharmacol Ther. 2005;105(3):267–310. doi: 10.1016/j.pharmthera.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Roman G. Worldwide occurrence of Parkinson's disease: an updated review. Neuroepidemiology. 1993;12:195–208. doi: 10.1159/000110318. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P. Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zorzon M, Capus L, Pellegrino A. Familial and environmental risk factors in Parkinson's disease: a case‐control study in north‐east Italy. Acta Neurol Scand. 2002;105(2):77–82. doi: 10.1034/j.1600-0404.2002.1o040.x. [DOI] [PubMed] [Google Scholar]