

Abstract
Patients with resected stage II-III melanoma have approximately a 35% chance of death from their disease. A deeper understanding of the tumor immune microenvironment (TIME) is required to stratify patients and identify factors leading to therapy resistance. We previously identified that the melanoma immune profile (MIP), an interferon-based gene signature, and the ratio of CD8+ cytotoxic T lymphocytes (CTLs) to CD68+ macrophages both predict disease-specific survival (DSS). Here, we compared primary to metastatic tumors and found that the nuclei of tumor cells were significantly larger in metastases. The CTL/macrophage ratio was significantly different between primary tumors without distant metastatic recurrence (DMR) and metastases. Patients without DMR had higher degrees of clustering between tumor cells and CTLs, and between tumor cells and HLA-DR+ macrophages, but not HLA-DR- macrophages. The HLA-DR- subset co-expressed CD163+CSF1R+ at higher levels than CD68+HLA-DR+ macrophages, consistent with an M2 phenotype. Finally, combined transcriptomic and multiplex data revealed that densities of CD8 and M1 macrophages correlated with their respective cell phenotype signatures. Combination of the MIP signature with the CTL/macrophage ratio stratified patients into three risk groups that were predictive of DSS, highlighting the potential use of combination biomarkers for adjuvant therapy.
Introduction
Melanoma is the most aggressive and lethal form of skin cancer.(1) Even when completely surgically resected, tumors less than 5 millimeters in thickness are commonly lethal due to tumor seeding into the lymphatics and systemic circulation.(2,3) Once metastatic disease develops, survival rates range from 20% to 40% at 5 years with state-of-the-art immunotherapy.(4,5) While these rates are disappointing, they represent a vast improvement over survival rates at 5 years prior to immunotherapy which were under 5%.(6) Today, immunotherapy is given as the standard of care to virtually all patients diagnosed with metastatic melanoma.(7–9)
Early stage melanoma presents a more difficult clinical management challenge because the side effects of treatment are less justifiable in the absence of high risk of death. Under 5% of patients with stage I melanoma will die of disease, while 10-year survival rates range from 88% to 75% for stage II disease and from 88% to 24% for stage III disease.(2) Prediction of recurrence would thus greatly facilitate management of early stage disease.
Melanoma is an immunogenic tumor, and interactions between tumor cells and the tumor immune microenvironment (TIME) lead to tumor-driven modulation of immune response.(10) It has been hypothesized that the immune system is a key determinant of melanoma recurrence and metastasis. Tumor-infiltrating lymphocytes (TILs) have been studied extensively and shown to have positive prognostic value in melanoma.(11–13) However, TILs are not currently included in the American Joint Committee on Cancer (AJCC) staging system for melanoma, and their utility is limited by a lack of phenotypic, functional, and precise locational information.(10,14) Further, tumor-associated macrophages (TAMs) within the TIME have been shown to correlate with poor prognosis and survival in melanoma and many other cancers.(15,16) Thus, understanding the melanoma TIME is crucial for both the development of new therapeutic interventions, as well as for stratification of patients for adjuvant therapy.
In previous work, we defined and validated in two independent cohorts a 53-immune gene melanoma immune profile (MIP) predictive of non-progression using nanoString transcriptomic analysis.(17,18) MIP allows for stratification of patients with stage II-III melanoma into recurrence risk groups. Adding to this work, we utilized quantitative multiplex immunofluorescence (qmIF) to analyze the tumor microenvironment in patients with stage II-III melanoma and found that a high density of cytotoxic lymphocytes (CTLs) and a low density of macrophages in the stroma correlates with improved survival. Furthermore, we found that the distance between CTLs and macrophages is associated with poor survival,(19) highlighting the importance of both the composition and spatial localization of cells within the melanoma TIME.
We now expand our analysis of the TIME of primary stage II-III melanoma by combining nanoString and qmIF data. Using advanced statistical techniques, we analyze the clustering, cellular composition, and marker proteins of both primary and metastatic melanomas. In particular, to quantify the degree of spatial proximity among immune and tumor cells, we assess clustering using the pair correlation function (PCF). This technique was originally used in statistical physics to model particle interactions(20) and we recently adapted it to study differences in spatial structures in immunofluorescence imaging of glioblastomas.(21) The PCF computes the average number of pairs of two given cell types separated by a given distance (normalized by the number expected if all cells were randomly positioned), giving a robust, quantitative metric of spatial interactions. This more detailed characterization may serve to guide the development of targeted and immune therapies for melanoma.
Materials and Methods
Patients and samples
This study was approved by Columbia University Irving Medical Center’s (CUIMC) Institutional Review Board. A database of patients with stage II-III melanoma at CUIMC was retrospectively created and analyzed using qmIF on Mantra and RNA expression using nanoString for MIP (Table 1, Mantra: n=104 and MIP: n=78).(18,19) In order to correlate the results of qmIF with RNA expression, we analyzed the patients who overlap between the Mantra and the MIP cohorts (Table 1, Combination: n=53). Further, a sub-cohort of 13 patients from the Mantra cohort, named the Macrophage Survey sub-cohort (Supplemental Table ST1), was selected based on high density of CD68+HLA-DR- macrophages for additional phenotypic characterization. Finally, a database of patients with metastatic melanoma at CUIMC was created by screening the records of 86 patients with metastatic melanoma, of whom 57 were excluded due to lack of availability of solid tumor samples prior to immunotherapy treatment or insufficient clinical information. Tissue was requested for the 29 patients remaining, then slides were cut and reviewed with a dermatopathologist. A total of 9 patients were excluded at this stage due to lack of sufficient tumor or scattered tumor in the lymph node. The remaining 20 patients were stained for inForm analysis. 6 of these patients were excluded due to poor staining quality or tissue destruction, leaving a final total of 14 patients analyzable on inForm (Supplemental Table ST2).
Table 1. Patient characteristics of the MANTRA, MIP, and Combination cohorts.
The 104 patient samples in the Mantra cohort were analyzed using quantitative multiplex immunofluorescence (qmIF) on Mantra and the 81 patients samples in the Melanoma Immune Profile (MIP) cohort were analyzed using nanoString. The patients who overlap between the MANTRA and the MIP cohorts make up the Combination cohort (n=53).
| MANTRA | MIP | Combination | |
|---|---|---|---|
| Clinical characteristics | (n = 104) | (n = 81) | (n = 53) |
| Gender, n (%) | |||
| Male | 75 (72.1) | 62 (76.5) | 40 (75.5) |
| Female | 29 (27.9) | 19 (23.5) | 13 (24.5) |
| Age | |||
| Median, n (range) | 74.5 (22–96) | 67 (22–96) | 68 (22–96) |
| Location of tumor, n (%) | |||
| Trunk | 61 (58.7) | 48 (59.3) | 30 (56.6) |
| Extremity | 41 (39.4) | 32 (39.5) | 22 (41.5) |
| Unknown | 2 (1.9) | 1 (1.2) | 1 (1.9) |
| Stage, n (%) | |||
| II | 91 (87.5) | 63 (77.8) | 42 (72.9) |
| III | 13 (12.5) | 15 (18.5) | 11 (20.8) |
| Pathologic characteristics | |||
| Depth (mm) | |||
| Median, n (range) | 2.5 (0.6–26) | 2.7 (0.7–26) | 2.7 (0.7–11.4) |
| Ulceration, n (%) | |||
| Absent | 36 (34.6) | 32 (39.5) | 19 (35.8) |
| Present | 65 (62.5) | 45 (55.6) | 32 (60.4) |
| Unknown | 3 (2.9) | 4 (4.9) | 2 (3.8) |
| TILs, n (%) | |||
| Absent | 2 (1.9) | 3 (3.7) | 1 (1.9) |
| Non-brisk | 59 (56.8) | 50 (61.7) | 33 (62.3) |
| Brisk | 33 (31.7) | 22 (27.2) | 15 (28.3) |
| Unknown | 10 (9.6) | 6 (7.4) | 4 (7.5) |
| Outcome characteristics | |||
| Patient follow-up (months) | |||
| Median, n (range) | 45 (4–173) | 58 (5–187) | 62 (5–173) |
| Systemic recurrence, n (%) | |||
| Known recurrence | 16 (15.4) | 24 (29.6) | 16 (30.2) |
| No recurrence | 36 (34.6) | 50 (61.7) | 32 (60.4) |
| Unknown | 52 (50) | 7 (8.6) | 5 (9.4) |
| OS (months), n (%) | |||
| Alive (at least 2 years) | 31 (29.8) | 50 (61.7) | 28 (52.8) |
| Dead | 73 (70.2) | 31 (38.3) | 25 (47.2) |
| DSS (months), n (%) | |||
| Alive or NED at death | 42 (40.4) | 62 (76.5) | 39 (73.6) |
| Dead with melanoma | 22 (21.2) | 19 (23.5) | 14 (26.4) |
| Unknown | 40 (38.4) | 0 (0) | 0 (0) |
Abbreviations: DSS, disease-specific survival; NED, no evidence of disease; OS, overall survival; MIP, Melanoma Immune Profile
qmIF acquisition
QmIF imaging was obtained from 104 stage II-III primary melanomas from CUIMC in the Mantra cohort (Table 1).(19) 5-μm full section slides were stained using Opal qmIF for DAPI, CD3 (T lymphocytes; clone LN10; Leica; 1:200 dilution), CD8 (cytotoxic T lymphocytes, or CTLs; clone 4B11; Leica; ready to use: RTU), CD68 (macrophages; clone KP1; Biogenex; RTU), SOX10 (tumor; clone BC34; Biocare; 1:300), HLA-DR (antigen presentation and activation; clone LN-3; Abcam; 1:200 dilution), and Ki67 (proliferation; clone MIB1; Abcam; RTU). H&E slides were reviewed by a dermatopathologist to determine representative areas for multispectral image capture at 20x magnification using Mantra. Regions of interest (ROIs) were approximately 700×520 microns. Multispectral images were then analyzed using inForm software and inForm data from each patient was processed in separate software designed in RStudio (version 0.99.896; https://github.com/thmshrt/transform_essential). In this software, images were combined and analyzed to concatenate variables and determine density and distance of distinct phenotypes. Statistical analyses were then completed on GraphPad Prism, R Version 3.3.1, and Python 3.5. For visualization, intensity values of each of the 5 macrophage markers were first normalized to have a standard deviation of 1. These continuous values were processed with tSNE (perplexity = 30), a dimensionality reduction technique that places cells with similar marker profiles close to each other.(22) Binarized (+/−) expression (derived with thresholds using inForm) for each marker was then plotted onto the tSNE figures. Co-expression of markers with HLA-DR was assessed with the Chi squared test. In addition, the macrophage survey sub-cohort (n=13) was stained for DAPI, CD68, HLA-DR, CSF1R (inflammation; clone SP211; LifeSpan Biosciences; 1:750 dilution), CD163 (M2 macrophages; clone 10D6; BioCare Medical; 1:300 dilution), CD33 (myeloid-derived suppressor cells, or MDSCs; clone PWS44; Leica; RTU) and MHCI (antigen presentation; clone MEM-E/07; ThermoFisher; 1:400 dilution) and analyzed in the same way as above.
Spatial clustering analysis
To assess clustering in qmIF images, PCFs were computed using the spatstat R package.(23) This method compares the observed probability of two cell types being separated by a given distance to that expected by chance. A higher value for PCF indicates a greater degree of clustering at that radius, and the area under the curve (AUC) of the function can be used as an overall statistic of clustering. Inhomogeneous cross PCFs were calculated up to a radius of 50 microns, provided that there was a minimum of 10 immune cells in the image. One sample was excluded for lack of tumor cells. The isotropic edge correction and a normalization power of 2 were applied. The AUC for each PCF was computed as a summary statistic for quantifying the degree of clustering, and was compared across recurrence status with the Mann-Whitney U test. The PCFs are graphically displayed by the point-wise median across samples, with shaded 95% confidence intervals obtained via bootstrapping.
nanoString acquisition
For each of the 78 patients in the MIP cohort (Table 1, MIP), formalin-fixed paraffin-embedded (FFPE) blocks were cut in 10-μm sections to provide 250 mm2 of tissue. RNA extraction was performed and RNA was processed using a nanoString assay, which measures expression of 64 target genes (MIP (53 genes) plus 11 interferon-related and 17 housekeeping genes).(24) Following hybridization and purification of target-probe complexes, digital counts for each gene-specific target RNA were acquired on the nCounter detection analyzer and normalized in nSolver software (nanoString). MIP was computed for each patient in this cohort using the original signature’s gene coefficients, then was correlated with disease-specific survival (DSS).(17,18)
Transcriptomic Analysis
We assessed the similarity of each sample’s gene expression profile to the cellular subtype signatures from LM22, the reference standard for CIBERSORT.(25) A total of 20 genes overlapped between the nanoString panel and LM22, which was a not sufficient number of genes to deconvolute with the CIBERSORT algorithm.(25) Instead, we calculated the cosine similarity of the samples with each of the 22 cell types from LM22, thereby assessing the extent (from −1 to 1) that the matching genes from each sample resemble each cell type. For the creation of the heat map, raw gene expression values were log-transformed and scaled to have a mean of 0 and a standard deviation of 1. Hierarchical complete-linkage clustering was performed on both rows and columns with a cosine metric. We assessed enrichment of recurrence among this clustering by dividing the patients at the first branch point into the two largest clusters, followed by a Fisher Exact test. Differential expression was evaluated for all 63 genes with t-test with Benjamini-Hochberg correction, and visualized in a volcano plot. Top genes were noted if they had greater than +1.0 log2-fold change.
Prognostic prediction of CD8/CD68 alone and in combination with MIP
To assess the ability of the CD8/CD68 ratio to predict recurrence, we performed a Mann-Whitney U test and visualized the prediction using a receiver operating characteristic (ROC) curve. We tested the correlation of the top immune genes (>1 log2-fold change) with the CD8/CD68 ratio using Spearman correlation. We combined the CD8/CD68 ratio with MIP using the log-rank (Mantel-Cox) test to compare low- to medium-risk, low- to high-risk, and medium- to high-risk patients. Hazard Ratios (HR) and 95% Confidence Intervals (CI) calculated using the Mantel-Haenszel test.
Digital Spatial Profiling (DSP)
8 patients from the 104-patient Mantra cohort were selected based on CD68+HLA-DR- phenotype and CD8 expression (Supplemental Table ST3). Of this sub-cohort, 4 patients had a high density of stromal HLA-DR- macrophages and close proximity of HLA-DR- macrophages to CTLs, and 4 patients had a high CTL/macrophage ratio with low HLA-DR- macrophage density, as determined by previous qmIF. For DSP, FFPE slides were stained with antibodies conjugated to UV-photocleavable DNA barcodes and specific to 34 proteins. Twelve 600-micron ROIs with high macrophage density were selected for each patient. ROIs were selected in tumor/stroma areas containing less than 50% stroma. ROIs were analyzed using UV excitation, which releases DNA barcodes for downstream quantitation on the nanoString nCounter platform. Protein expression was compared between patients and statistical analyses were performed using a Mann-Whitney U test.
Patient populations
The Mantra cohort consists of 104 patients, 64 of whom had available disease-specific survival (DSS) data, defined as known cause and date of death and/or documented clinical follow-up.(19) The MIP cohort consisted of 81 patients, all of whom had available DSS.(18) The Combination cohort consisted of 53 patients, all of whom had available DSS. In the Mantra cohort, 15.4% of patients had known recurrence, 34.6% had no recurrence, and 50% had unknown recurrence status. In the MIP cohort, 29.6% of patients recurred, 61.7% did not recur, and 8.6% had unknown recurrence status. Finally, in the Combination cohort of 53 patients, 30.2% of patients recurred, 60.4% of patients didn’t recur, and 9.4% of patients had unknown recurrence status. The Combination cohort consisted of 75.5% males and 24.5% females, and the median patient age was 68. 56.6% of patients had a tumor on their trunk or head, and 72.9% of patients were stage II. Median tumor depth in this cohort was 2.7 mm, and ulceration was present in 60.4% of the cases. Median follow up was 62 months, and 25 of the patients died of any cause, with 14 having died from melanoma. The 13-patient Macrophage Survey sub-cohort stained for macrophage phenotypes consisted of 46.2% of patients with a high CD8/CD68 ratio, 38.5% of patients with a low HLA-DR- macrophage density, and 61.5% of patients with a large distance between CTLs and HLA-DR- macrophages (Supplemental Table ST1). The metastatic melanoma cohort consisted of 14 patients, of whom 1 had stage IIIC disease, 3 had stage IV M1a or M1b disease, 1 had stage IV M1c disease, 8 have stage IV M1d disease, and 1 had mucosal melanoma. The tissue origins of the biopsies of metastases used for staining were divided into 50% soft tissue or skin, 35.7% brain, 7.1% lung, and 7.1% lymph node (Supplemental Table ST2). Finally, the cohort used for DSP analysis consisted of 50% of patients with a high CD8/CD68 ratio, 50% of patients with a low HLA-DR- macrophage density, and 62.5% of patients with a large distance between CTLs and HLA-DR- macrophages (Supplemental Table ST3).
Results
QmIF analysis reveals distinct phenotypes of metastatic melanoma, primary melanoma tumors that did recur, and primary melanoma tumors that never recurred.
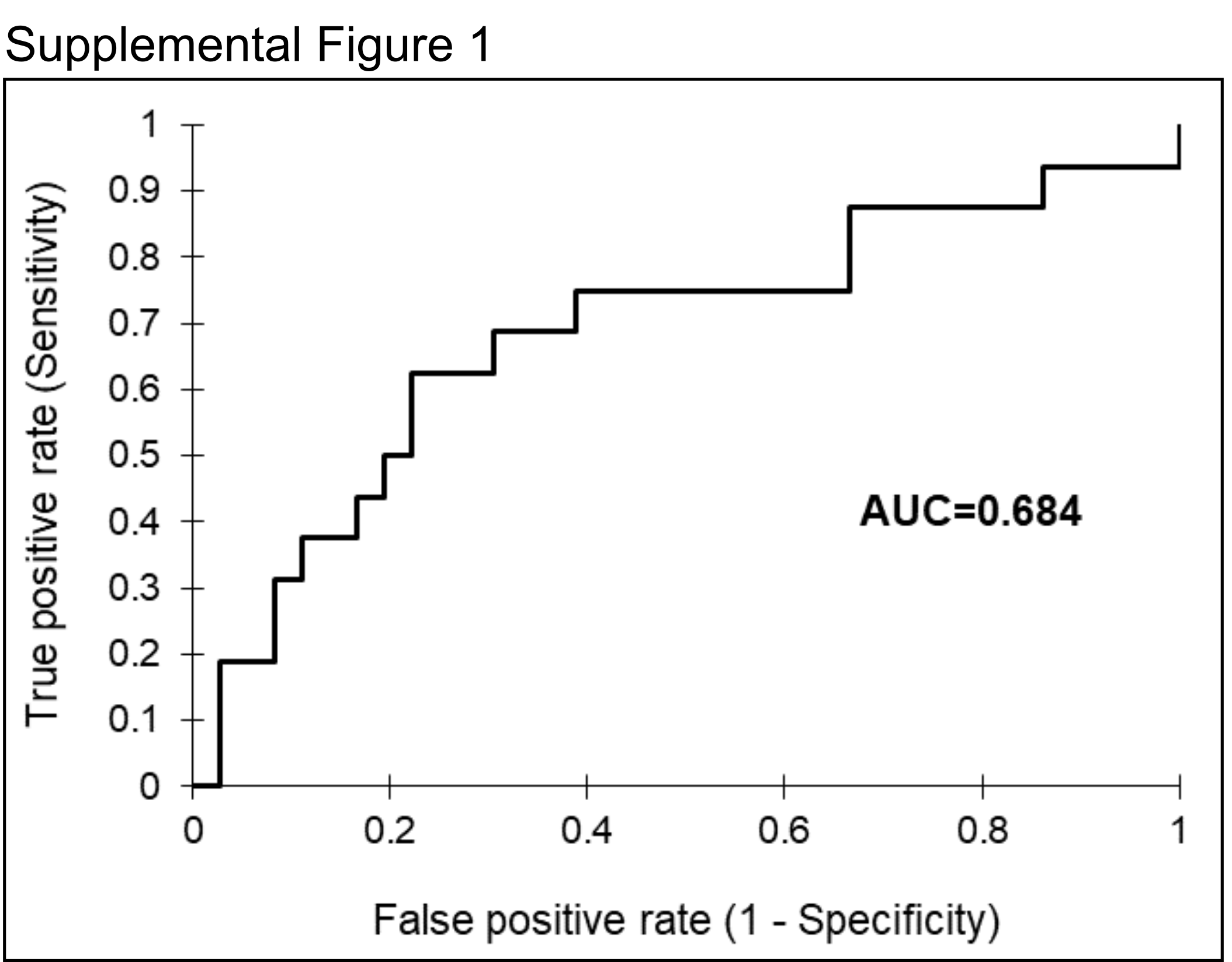
First, we evaluated the Mantra cohort for distant metastatic recurrence using the 52 patients with known recurrence status and found that, in addition to correlating with DSS and OS, as previously published, the CD8/CD68 ratio is also predictive of recurrence (n=52, AUC=0.684, p=0.0350 by Mann-Whitney U test, Supplemental Figure S1). Next, we applied qmIF to compare density of immune phenotypes in samples of primary (P) melanoma (stage II-III, Mantra cohort) to those with metastatic (M) lesions. We determined the proportions of tumor, macrophage, CD8+ T cells, and CD8- T cell as a fraction of the total number of cells as identified by DAPI in each sample, and concatenated these proportions with HLA-DR and Ki67 expression (Figure 1A). We further quantified and compared the densities of CD8+ T cells, macrophages, total immune cells (sum of CD3+ T cells plus CD68+ macrophages), and CD8/CD68 ratios in primary (P) and metastatic (M) patients (Figure 1B). As a whole, there was a greater proportion of immune cells in P samples compared to M samples (p = 0.0017, Mann-Whitney U test, Figure 1B), with specifically more CTLs in primary samples (p = 1.8e-6). Next, we separated the primary melanoma cohort into non-recurrent (NR, n=36) and recurrent (R, n=16) patients, finding that NR samples had a higher CD8/CD68 ratio relative to R patients (p = 0.024). Interestingly, there was no significant difference between the R and M samples (p = 0.52) (Figure 1B).
Figure 1. Comparison of cellular proportions and morphologies in primary and metastatic melanoma.
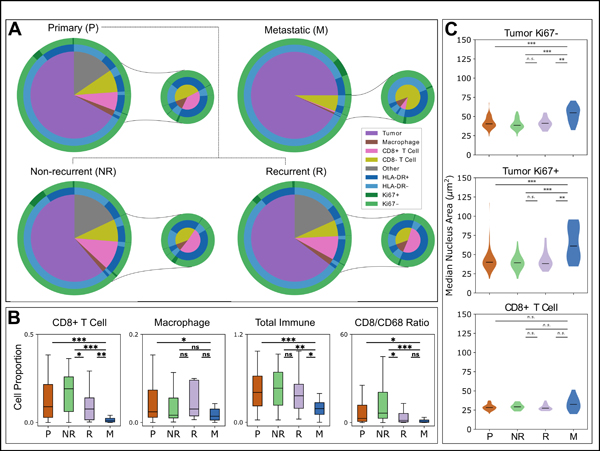
(A) All identified cells within the primary Mantra (n = 104) and metastatic (n = 14) cohorts, phenotyped via inForm. HLA-DR and Ki67 statuses are displayed, and proportions of phenotyped immune cells as a proportion of total cells as identified by DAPI are visualized in the smaller pies. The primary cohort is further distinguished into patients who were known to have recurrence (R, n = 16) or non-recurrence (NR, n = 36). (B) Comparison of cell proportions within the stromal compartments of tissue images, comparing all primary samples (P), known non-recurrent primaries (NR), recurrent primaries (R), and metastatic cases (M). “Total Immune” refers to the sum of all immune cells (CD3+ T cells and CD68+ macrophages), while CD8/CD68 ratio is the CD8+ T Cell (CTL) count divided by the CD68+ macrophage count. (C) Comparison of the sizes of nuclei within tumor Ki67-, tumor Ki67+, and CD8+ T cell populations between P, NR, R, and M patients. Statistical analyses performed using Mann-Whitney U test. ***, p<0.001; **, p<0.01; *, p<0.05; n.s., p≥0.05. Labels: all primary samples (P), known non-recurrent primaries (NR), recurrent primaries (R), and metastatic cases (M).
In further analysis we evaluated nuclear size by comparing nuclear areas and found that tumor cells in M samples had larger nuclear areas compared to their P counterparts (p < 0.001, Mann-Whitney U test, Figure 1C). This held true both for Ki67- and Ki67+ subpopulations. However, there were no significant differences in the sizes of the nuclei of CTLs between the cohorts (Figure 1C).
qmIF density analysis of immune cell phenotypes correlates with gene expression data
As a second element of our composite analysis of the TIME, we aimed to evaluate an interferon signature in our tumor samples from the 74 patients with known recurrence status in the MIP cohort while including 11 additional interferon-related genes for comparison. This was done for the purpose of correlating the gene expression data with qmIF data. We profiled RNA expression across 64 immune-related genes (53 genes from MIP and 11 additional interferon-related genes) and found that expression was associated with recurrence status using hierarchical clustering (p = 0.049, Fisher Exact test, Figure 2A). To identify the cellular source of this association, we assessed the degree of similarity between the transcriptomic signatures in our samples and the CIBERSORT cell reference signatures.(25) In evaluation of the signatures for macrophages of M1 and M2 polarity, we found that R patients had a decreased M1 signature (p = 0.042, Figure 2B top left) but an increased M2 signature (p = 0.048, Figure 2B, top right) compared to NR patients. We then compared the CIBERSORT signatures with findings from qmIF. We found that the CD8 T cell signature correlated with the proportion of CTLs in qmIF (p = 0.010 Figure 2B, bottom left) while the M1 signature correlated with density of HLA-DR+ macrophages (p = 0.022, bottom right).
Figure 2. Expression profiles of 64 immune-related genes.
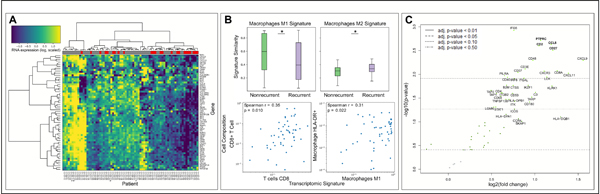
(A) Hierarchical clustering of (log-transformed) gene expression values across the 74 patients with known recurrence status from the MIP cohort, with relative levels of mRNA expression in each row and different patient samples in each column. Yellow indicates higher expression and blue indicates lower expression of each gene. Recurrence status of each patient is displayed in the top horizontal bar (recurrent patients are labeled in red; non-recurrent patients are labeled in grey). (B, top) Similarity of the expression profiles with CIBERSORT reference values was assessed to compare M1 (top left) and M2 (top right) macrophage signatures in non-recurrent and recurrent patients (p = 0.042 and 0.048, respectively). (B, bottom) Correlation plots for comparison of transcriptional similarity to CIBERSORT profiles (x-axis) with observed cellular compositions (y-axis). (C) Volcano plot of mRNA differential expression for all 64 genes. Genes to the right are higher in patients who did not die of melanoma. Statistical comparison in (A) performed using Mann-Whitney U test. Spearman correlation p-values in (B) evaluated using Fisher transformation. Differential expression in (C) evaluated using Benjamini-Hochberg (BH) correction.
Next, we evaluated differential expression of the 64 genes and found that nearly all genes were increased in patients who did not die of melanoma (Figure 2C). After filtering for genes that were both significant following Benjamini-Hochberg (BH) correction and also had greater than +1.0 log2-fold change in differential expression, we identified 9 genes, all of which are part of MIP (Table 2). We also found that the top 9 genes do not overlap with the 11 additional interferon-related genes and further that these 11 genes did not improve prediction accuracy. Lastly, we evaluated the correlation of these 9 genes with stromal density of CD3, CD3+CD8+, and CD68+, as well as the ratio of CD8/CD68 (Table 2). We found that CD3+ cells in the stroma correlated with CXCL9, CD8A, CXCR3, CXCL11, CCL5 and CD2 while CD3+CD8+ cells in the stroma correlated with the same genes minus CD2. Stromal CD68 cells and the CD8/CD68 ratio had no correlation with the top 9 genes from MIP.
Table 2. Top genes from MIP.
Top 9 immune genes that are significantly increased in patients with good DSS after Benjamini-Hochberg correction and correlation of these genes with qmIF data for stromal density of CD3, CD3+CD8+, and CD68+ as well as ratio of CD8/CD68. Values in bold are different from 0 with a significance level alpha=0.05 using Benjamini-Hochberg correction.
| Differential expression | Correlation with Density | |||||
|---|---|---|---|---|---|---|
| Gene | Fold Change | BH p value | CD8/CD68 Stroma | CD3+CD8+ Stroma | CD68+ Stroma | CD3+ Stroma |
| PTPRC | 1.07 | 0.0399 | 0.05 | 0.17 | 0.12 | 0.3 |
| CCL5 | 1.19 | 0.0399 | 0.21 | 0.36 | 0.03 | 0.41 |
| CD2 | 1.05 | 0.0399 | 0.07 | 0.24 | 0.14 | 0.36 |
| CD27 | 1.21 | 0.04 | 0.04 | 0.2 | 0.16 | 0.31 |
| CXCL9 | 1.56 | 0.0459 | 0.19 | 0.33 | 0.04 | 0.45 |
| CD8A | 1.26 | 0.0459 | 0.16 | 0.35 | 0.11 | 0.39 |
| CXCR3 | 1.11 | 0.0459 | 0.19 | 0.36 | 0.06 | 0.4 |
| CXCL11 | 1.39 | 0.0459 | 0.2 | 0.38 | 0.05 | 0.42 |
| LCK | 1.14 | 0.0459 | −0.03 | 0.12 | 0.21 | 0.24 |
Combining MIP and CD8/CD68 improves prediction of DSS
Given that genes from MIP did not directly correlate with the CD8/CD68 ratio, we chose to combine these two biomarkers to substratify the patients. We evaluated the ability of the combination of MIP and the CD8/CD68 ratio to predict DSS using the 53-patient Combination cohort (Table 1). We stratified patients into high, medium, and low-risk groups using their categorization as high or low risk in each of the data sets based on our previously published criteria.(18,19) High-risk patients had poor MIP and CD8/CD68 profiles; medium-risk patients had a poor MIP profile and a favorable CD8/CD68 profile; and low-risk patients had favorable MIP and CD8/CD68 profiles. We found that combining MIP and the CD8/CD68 ratio effectively distinguished low- and medium-risk patients from high-risk patients (p=0.003; HR 7.53 (95% CI: 1.98–28.74) and p=0.026; HR: 3.51 (95% CI: 1.16–10.61), respectively, Figure 3A and Supplemental Table ST4). Representative images of each of these risk groups are also shown in Figure 3B–D. Additionally, by creating a medium risk group we found that the combination biomarker improves on the previous biomarkers, MIP and CD8/CD68 ratio, individually (p=0.030, HR:3.84 (95% CI: 1.14–12.96) and p=0.044, HR: 3.04 (95% CI: 1.03–8.99), respectively) (Supplemental Table ST4). Finally, we found using univariable Cox analysis in medium- and high-risk patients (n=42) that the high-risk combination biomarker specifies poor prognosis (p=0.035, HR: 3.29 (95% CI: 1.09–9.96)), whereas standard predictors of prognosis were not found to be significant (Supplemental Table ST5).
Figure 3. Combination of MIP and CD8/CD68 ratio predicts DSS.
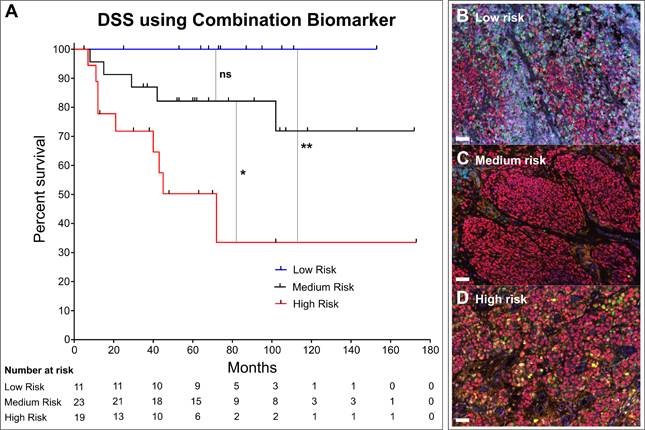
(A) KM curve for disease specific survival (DSS) after patients stratified into high-risk (low MIP + low CD8/CD68 ratio), medium-risk (low MIP + high CD8/CD68 ratio), and low-risk (high MIP + high CD8/CD68 ratio), groups. Log-rank (Mantel-Cox) test used to compare low- and medium-risk patients (p=0.1228), low- to high-risk (p=0.0031) and medium- to high-risk (p=0.0258). Multiplex images stained for DAPI (nuclei, blue), SOX10 (tumor, red), CD3 (T cells, cyan), CD8 (CTLs, magenta), CD68 (Macrophages, green), Ki67 (proliferation marker, yellow), HLA-DR (activation marker, orange) in (B) patients in low-risk combination group, (C) patients in medium-risk combination group, and (D) patients in high-risk combination group.
Clustering of tumor cells with CD8+T cells and HLA-DR+ macrophages is associated with non- recurrence
Next we extended the spatial analysis of qmIF by using PCF. We found that CD8+ T cells clustered more strongly with tumor cells in the non-recurrent samples compared to recurrent samples (p = 0.0086, Mann-Whitney U test, Figure 4A left). This effect was similar when comparing the clustering of tumor cells with HLA-DR+ macrophages (p = 0.016, Figure 4A right), but not with HLA-DR- macrophages (data not shown). We provide a representative depiction of the lowest and highest PCF values for each of the Tumor-CTL (Figure 4B left) and Tumor-Macrophage (Figure 4B right) interactions.
Figure 4. Pair correlation functions (PCF) reveal differences in clustering between tumor and immune cells.
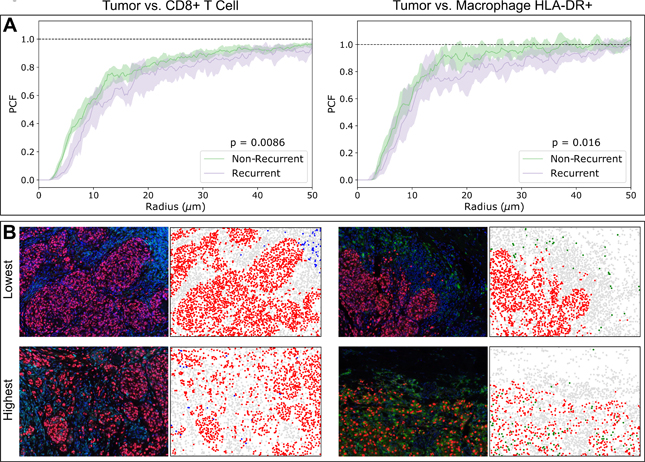
(A) Non-recurrent patients have higher degrees of clustering between tumor cells and CTLs as well as with HLA-DR+ macrophages. Shaded regions indicate 95% confidence intervals of the median PCF obtained via bootstrapping. (B) Representative false-color images with the 3 lowest and 3 highest PCF values for each of the Tumor-CTL and Tumor Macrophage interactions. To the right of each image is a rendition of its cell segmentation, depicting Tumor (Red), CTL (Blue), Macrophage (Green), and Other (Gray). Statistical analysis performed using Mann-Whitney U test on PCF areas under the curve (AUC).
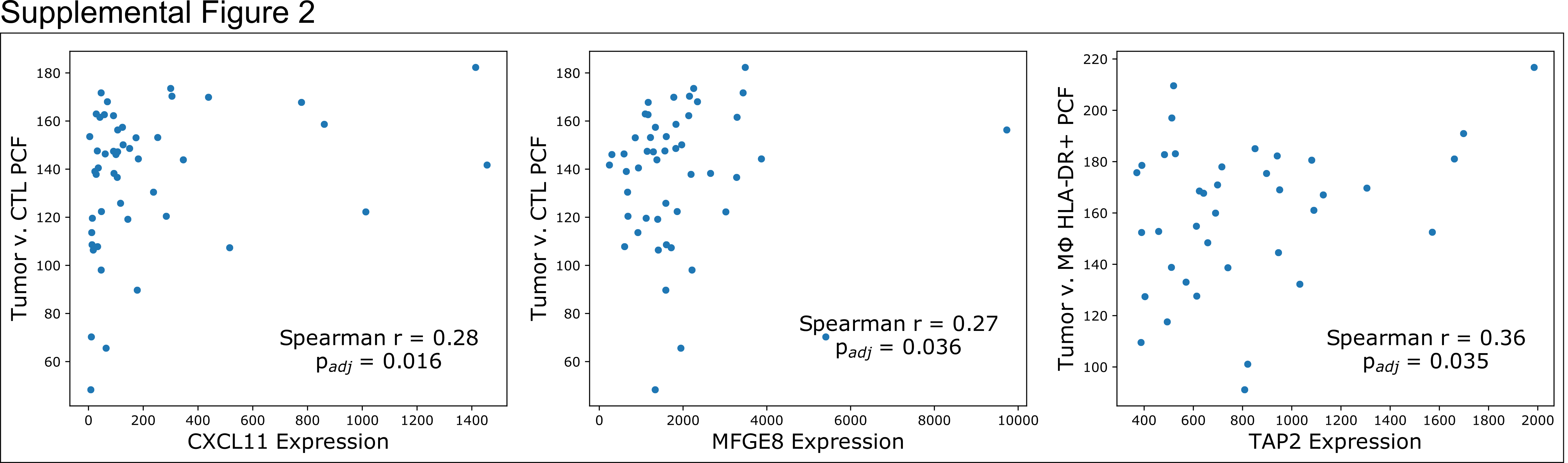
We next sought to compare the AUCs of these PCFs against gene expression, finding significant positive correlations between the tumor-CTL clustering with CXCL11 (p = 0.016, Spearman correlation, Bonferroni correction) and MFGE8 (p = 0.036), and between the tumor-macrophage clustering with TAP2 (p = 0.035) (Supplemental Figure S2).
Further phenotyping of HLA-DR- macrophages identifies this population as a predictor of an unfavorable TIME and immune suppression
Having previously found that high density of HLA-DR- macrophages predicts poor DSS,(19) we next investigated the properties of these macrophage subpopulations. We performed additional staining for CD68, HLA-DR, CD163, CSF1R, MHC1, and CD33 over a total of 5519 CD68+ macrophages identified across the 13 patients in the macrophage survey sub-cohort (Figure 5A). We visualized the concatenation of each of these markers using pie charts comparing HLA-DR- and HLA-DR+ macrophages (Figure 5B). We next visualized this data using T-distributed stochastic neighbor embedding (tSNE) to compare the expression of these markers on HLA-DR- and HLA-DR+ macrophages. HLA-DR expression was largely independent of CD163 (p=0.797, Figure 5C). However, HLA-DR expression was correlated with MHC1 expression (p = 1.4e-102, odds ratio 3.4, Chi-squared test), whereas lack of HLA-DR expression was correlated with CD33 expression (p = 1.7e-53, odds ratio 0.22, Figure 5C). To further study the cells expressing CSF1R and CD33, we evaluated the concatenation of CSF1R and CD33 with CD163, finding that HLA-DR- cells were more likely to also be CD163+CSF1R+ compared to HLA-DR+ cells (p<0.001, Figure 5C). Further, in comparison to HLA-DR+ cells, HLA-DR- cells were also more frequently CD163+CD33+ (p<0.001, Figure 5C).
Figure 5. Investigation of macrophage subpopulations.
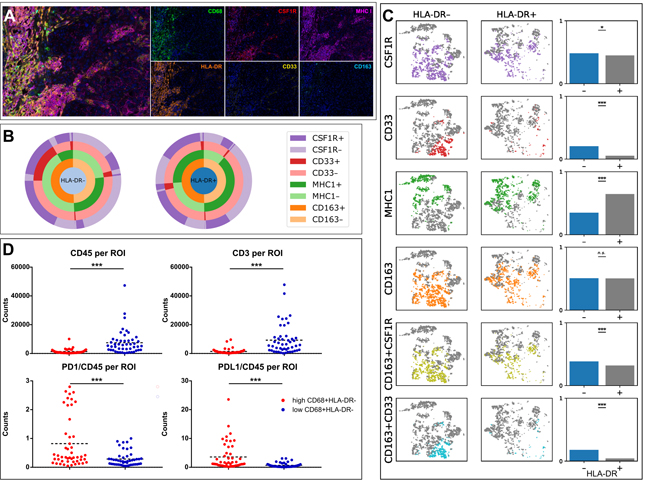
(A) Multiplex image of melanoma stained using qmIF for DAPI (blue), CD68 (green), HLA-DR (orange), CSF1R (red), CD163 (cyan), MHCI (magenta), and CD33 (yellow). (B) Macrophages were profiled for HLA-DR, CSF1R, CD33, MHC1, and CD163 expression in qmIF images obtained from 17 patients in the macrophage survey sub-cohort of primary melanomas. The pie charts assess the distribution of each marker within HLA-DR- and HLA-DR+ macrophage subpopulations. (C) T-distributed stochastic neighbor embedding (tSNE) was used to visually cluster macrophages based on the intensity values of all 5 profiled markers. Displayed are the subsets of this embedding that are HLA-DR- and HLA-DR+, and co-expression of each marker with HLA-DR is compared on the right. (D) Digital Spatial Profiling (DSP) analysis in 8 patients with primary melanoma from the DSP cohort. Displayed are plots showing CD45 and CD3 per region of interest (ROI) (top) and PD1 and PDL1 per CD45 comparing patients with high and low density of HLA-DR- macrophages. Statistical analysis performed using Chi squared test (C) or Mann-Whitney U test (D). ***, p<0.001; **, p< 0.01; *, p<0.05; n.s., p≥0.05.
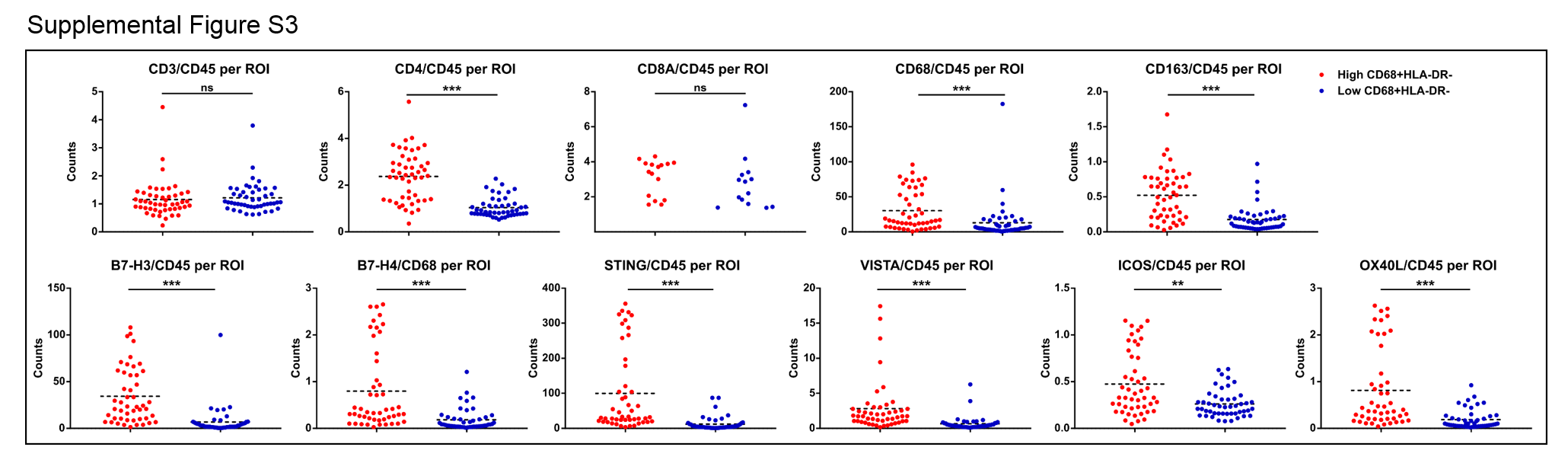
In addition to qmIF analysis, we further evaluated 8 primary melanoma cases using Digital Spatial Profiling (DSP). We found that the Regions of Interest (ROIs) from patients with higher stromal density of HLA-DR- macrophages had a lower immune infiltration overall as assessed by quantitation of CD45 per ROI (p<0.0001, Figure 5D). Additionally, CD3 per ROI was higher in patients with lower stromal density of HLA-DR- macrophages (p<0.0001, Figure 5D). Interestingly, patients with higher density of HLA-DR- macrophages also had a significantly higher ratio of CD4 to CD45 (p<0.0001), but a similar CD8A to CD45 ratio (Supplemental Figure S3). PDL1 and PD1 levels per CD45 were significantly higher in patients with higher HLA-DR- macrophage density (p<0.0001 and p=0.0002, respectively, Figure 5D).
Discussion
Innovations in digital pathology methods allow for quantitation of cell phenotypes and reproducible characterization of morphologic and genomic features, and provide new tools for clinical pathology.(18,19,26,27) These analyses enhance personalized medicine and allow for rapid development and implementation of new digital pathology-based biomarkers. Here we show that nuclear area and cellular clustering correlate with clinical features and propose a biomarker combining transcriptomic and qmIF data.
Current pathology methods such as TIL evaluation and tumor grading correlate with clinical outcome in melanoma, but are generally qualitative and therefore difficult to standardize.(2,13,14) In previous work, we quantitatively evaluated density and spatial relationships of immune infiltrates in primary melanoma specimens using qmIF, finding that the ratio of CD8 to CD68 can be used as a biomarker for DSS and OS in stage II-III primary melanoma.(19) In the current work we find that the CD8 to CD68 ratio also predicts recurrence in primary early stage melanoma. Further, we evaluate the same qmIF panel in metastatic cases and find that the CD8 to CD68 ratio in metastatic lesions (M) is similar to that in primary melanomas that distantly recur (R). Both M and R have a lower CD8 to CD68 ratio than non-recurrent primary melanomas (NR). This suggests that the TIME in primary melanomas with high metastatic potential is similar to that in metastatic lesions. These findings build on prior work showing that the ratio of CD8 to CD68 has potential as a favorable prognostic feature in primary melanoma tumors.(19) Phenotyping both lymphocytes and myeloid cells within the TIME can improve upon quantitation of lymphocyte populations alone.(28)
More precise phenotyping of immune cells and correlation of these findings with clinical features will likely lead to the development of more accurate biomarkers. Myeloid cells that are CD68+ are predominately macrophages and have been shown to be an unfavorable feature in several tumor types.(29) Macrophage populations, however, are notoriously diverse and can exert both pro-tumorigenic and pro-immune functions.(30,31) Previously, we distinguished HLA-DR- macrophages from HLA-DR+ macrophages as indicative of a poor prognosis.(19) HLA-DR expression indicates an activated state and is broadly associated with an M1, pro-immune, phenotype.(32,33) Here, we further characterize the HLA-DR- population as containing suppressive populations expressing CD163 and CSF1R. These findings suggest that CD68+HLA-DR- cells are suppressive, consistent with our hypothesis that these cells exert a pro-tumorigenic effect. As a future research direction, we hope to further explore the three-cell interaction between these macrophages, T cells, and tumor cells.
QmIF allows not only for calculation of the density of different immune cell phenotypes but also analysis of cell size and distances between cells.(34–36) First, we compare nuclear size in NR, R, and M lesions. Interestingly, we find that nuclei in metastatic lesions are larger than in primary lesions regardless of expression of the proliferation marker, Ki67. However, the size of the nuclei in primary lesions is similar regardless of recurrence status. From a clinical pathology perspective, findings suggest that in the absence of an epidermal component, increased nuclear size may favor a melanoma metastasis. However, larger studies are needed.
Distance between cells can be used to define spatial relationships of immune cells with tumor cells.(37) Previously, we found that simple nearest neighbor distance of CD8+ T cells to CD68+HLA-DR- macrophages predicted poor prognosis.(19) Here we further evaluate T cells and macrophages using clustering algorithms and find that even after accounting for variations in cell density there are patterns of positioning among immune and tumor cells that are related to prognosis. Specifically, clustering of tumor cells with CD8+ T cells correlates with non-recurrence. Additionally, although our prior study found that density of HLA-DR+ macrophages alone did not predict prognosis, we find that clustering of HLA-DR+ macrophages with tumor cells correlates with non-recurrence. The importance of spatial proximity of immune cells to each other and to tumor cells implies a mechanism by which their infiltration affects disease progression. These findings are consistent with the hypothesis that immune surveillance is operative in primary melanoma tumors and that proximity of tumor cells to immune cells of defined phenotypes impacts melanoma progression. Spatial location of cells may be impacted by local hypoxia or other features and future studies will be useful to further explore metabolic mechanisms underlying spatial distributions of immune cells within melanoma and other cancers.(38,39)
In addition to our qmIF analysis, we also previously defined and validated a transcriptomic 53 immune gene panel, MIP, predictive of recurrence in the same cohort of primary stage II-III melanoma patients.(17,18) Here, we evaluate transcriptomic profiling in addition to qmIF immune phenotyping and find consistency between the defined cell density and immune gene signatures. Interestingly, we find that density of CD8+ T cells and HLA-DR+ macrophages correlates with defined CIBERSORT signatures(25) for CD8 T cells and M1 macrophages, respectively.
Further, combining the CD8 to CD68 ratio with MIP allows for development of a composite biomarker that quantitatively assesses the entire specimen. This composite biomarker identifies three separate risk groups, most importantly a high risk population that may benefit from adjuvant studies of combination immunotherapies. Patient stratification is urgently needed in early stage melanoma because current protocols support treatment of all stage III patients despite the fact that 5 year survival is 83% in stage IIIB patients and 93% in stage IIIA patients. Meanwhile, survival rates in stage II populations are also generally favorable, ranging from 82% to 94%. Nonetheless melanoma, when it recurs is devastating. Therefore identifying high risk groups would facilitate application of combination immunotherapy in the adjuvant setting by allowing stratification for clinical trials. In addition, there is data to suggest that a favorable tumor immune microenvironment predisposes to response to PD1 blockade.(40) Therefore patients at intermediate risk might be more likely to respond to therapy both in the adjuvant setting and at recurrence while low risk patients could avoid immunotherapy entirely. Thus, our composite biomarker findings, while exploratory and based on small populations, suggests that further work to quantitatively characterize and correlate phenotypic and genomic features of the TIME using FFPE tissues is likely to improve personalized care of patients with early-stage melanoma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Statement of Significance.
Findings provide a deeper understanding of the tumor immune microenvironment; combining multiple modalities to stratify patients into risk groups, a critical step to improving the management of melanoma patients.
Acknowledgements
The authors of this publication were supported by the National Institutes of Health through Grant Numbers UH2CA21814901A1 (Y.M. Saenger) and KL2TR001874 (R.D. Gartrell-Corrado). A.X.C is funded by the Medical Scientist Training Program (T32GM007367). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Yvonne Saenger is also supported by funding from the Melanoma Research Alliance and by an Irving Assistant Professorship at Columbia University’s NIH/NCATS CTSA Program hub: UL1TR001873. Robyn Gartrell-Corrado is also supported by Swim Across America. R. Rabadán, A. Monod, and A. Chen have been supported by the NCI Center for Topology of Cancer Evolution and Heterogeneity (U54CA193313). A. Monod wishes to acknowledge the support of the New Frontiers in Research Fund--Fonds Nouvelles Frontières en Recherche (SSHRC-NFRF-FNFR, Government of Canada) NFRE-2018-00431. The funding sources had no role preparation of the manuscript or the decision to submit for publication.
R.D.G.-C. received grant support from nanoString for Digital Spatial Profiling and received honoraria and travel support from Northwest Biotherapeutics and PerkinElmer, respectively. E.M.R. received travel reimbursement from nanoString. Y.M.S. receives funding from Amgen and Regeneron. K.M.K. is currently employed at Genentech, Inc. R.R. is member of the SAB of AimedBio.
Footnotes
Conflicts of Interest
None of the above have impacted the contents of this manuscript.
References
- 1.Millet A, Martin AR, Ronco C, Rocchi S, Benhida R. Metastatic Melanoma: Insights Into the Evolution of the Treatments and Future Challenges. Medicinal research reviews 2017;37:98–148 [DOI] [PubMed] [Google Scholar]
- 2.Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA: a cancer journal for clinicians 2017;67:472–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Schuckmann LA, Hughes MCB, Ghiasvand R, Malt M, van der Pols JC, Beesley VL, et al. Risk of Melanoma Recurrence After Diagnosis of a High-Risk Primary Tumor. JAMA dermatology 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Annals of oncology : official journal of the European Society for Medical Oncology 2019;30:582–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogiers A, Boekhout A, Schwarze JK, Awada G, Blank CU, Neyns B. Long-Term Survival, Quality of Life, and Psychosocial Outcomes in Advanced Melanoma Patients Treated with Immune Checkpoint Inhibitors. Journal of oncology 2019;2019:5269062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman PB, Einhorn LH, Meyers ML, Saxman S, Destro AN, Panageas KS, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1999;17:2745–51 [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30 [DOI] [PubMed] [Google Scholar]
- 9.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. The New England journal of medicine 2015;372:2521–32 [DOI] [PubMed] [Google Scholar]
- 10.Camisaschi C, Vallacchi V, Castelli C, Rivoltini L, Rodolfo M. Immune cells in the melanoma microenvironment hold information for prediction of the risk of recurrence and response to treatment. Expert Rev Mol Diagn 2014;14:643–6 [DOI] [PubMed] [Google Scholar]
- 11.Lee N, Zakka LR, Mihm MC Jr., Schatton T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology 2016;48:177–87 [DOI] [PubMed] [Google Scholar]
- 12.Clemente CG, Mihm MC Jr., Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996;77:1303–10 [DOI] [PubMed] [Google Scholar]
- 13.Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:2678–83 [DOI] [PubMed] [Google Scholar]
- 14.Busam KJ, Antonescu CR, Marghoob AA, Nehal KS, Sachs DL, Shia J, et al. Histologic classification of tumor-infiltrating lymphocytes in primary cutaneous malignant melanoma. A study of interobserver agreement. American journal of clinical pathology 2001;115:856–60 [DOI] [PubMed] [Google Scholar]
- 15.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 2009;86:1065–73 [DOI] [PubMed] [Google Scholar]
- 16.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep 2017;19:203–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sivendran S, Chang R, Pham L, Phelps RG, Harcharik ST, Hall LD, et al. Dissection of immune gene networks in primary melanoma tumors critical for antitumor surveillance of patients with stage II-III resectable disease. J Invest Dermatol 2014;134:2202–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gartrell RD, Marks DK, Rizk EM, Bogardus M, Gerard CL, Barker LW, et al. Validation of Melanoma Immune Profile (MIP), a Prognostic Immune Gene Prediction Score for Stage II-III Melanoma. Clin Cancer Res 2019;25:2494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gartrell RD, Marks DK, Hart TD, Li G, Davari DR, Wu A, et al. Quantitative Analysis of Immune Infiltrates in Primary Melanoma. Cancer Immunol Res 2018;6:481–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson R A uniqueness theorem for fluid pair correlation functions. Physics Letters A 1974;49:197–8 [Google Scholar]
- 21.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nature medicine 2019;25:462–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 2011;12:2825–30 [Google Scholar]
- 23.Baddeley A, Turner R. spatstat: An R Package for Analyzing Spatial Point Patterns. 2005 2005;12:42 [Google Scholar]
- 24.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nature biotechnology 2008;26:317–25 [DOI] [PubMed] [Google Scholar]
- 25.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nature methods 2015;12:453–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerami P, Cook RW, Wilkinson J, Russell MC, Dhillon N, Amaria RN, et al. Development of a prognostic genetic signature to predict the metastatic risk associated with cutaneous melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21:175–83 [DOI] [PubMed] [Google Scholar]
- 27.Ferris LK, Gerami P, Skelsey MK, Peck G, Hren C, Gorman C, et al. Real-world performance and utility of a noninvasive gene expression assay to evaluate melanoma risk in pigmented lesions. Melanoma research 2018;28:478–82 [DOI] [PubMed] [Google Scholar]
- 28.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. The Journal of pathology 2014;232:199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rakaee M, Busund LR, Jamaly S, Paulsen EE, Richardsen E, Andersen S, et al. Prognostic Value of Macrophage Phenotypes in Resectable Non-Small Cell Lung Cancer Assessed by Multiplex Immunohistochemistry. Neoplasia 2019;21:282–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer science 2014;105:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends in immunology 2015;36:229–39 [DOI] [PubMed] [Google Scholar]
- 32.Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC cancer 2010;10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. British journal of cancer 2013;108:914–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014;70:46–58 [DOI] [PubMed] [Google Scholar]
- 35.Chang AY, Bhattacharya N, Mu J, Setiadi AF, Carcamo-Cavazos V, Lee GH, et al. Spatial organization of dendritic cells within tumor draining lymph nodes impacts clinical outcome in breast cancer patients. Journal of translational medicine 2013;11:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nature communications 2017;8:15095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nawaz S, Heindl A, Koelble K, Yuan Y. Beyond immune density: critical role of spatial heterogeneity in estrogen receptor-negative breast cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2015;28:766–77 [DOI] [PubMed] [Google Scholar]
- 38.Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015;7:277ra30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res 2014;2:598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maleki Vareki S High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 2018;6:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.