

Highlights
-
•
Fetal adverse events contribute to the onset of schizophrenia in adulthood.
-
•
Prenatal factors might enhance the risk of schizophrenia through inflammatory, oxidative and nitrosative stress pathways.
-
•
Fetal adversity can affect brain development, brain structure and functions in schizophrenia.
Keywords: Schizophrenia, Fetal programming, Prenatal, Perinatal, Stress, Infection, Diet, Inflammation, Oxidative stress, Neurodevelopment, Epigenetics
Abstract
Mounting evidence indicates that schizophrenia is associated with adverse intrauterine experiences. An adverse or suboptimal fetal environment can cause irreversible changes in brain that can subsequently exert long-lasting effects through resetting a diverse array of biological systems including endocrine, immune and nervous. It is evident from animal and imaging studies that subtle variations in the intrauterine environment can cause recognizable differences in brain structure and cognitive functions in the offspring. A wide variety of environmental factors may play a role in precipitating the emergent developmental dysregulation and the consequent evolution of psychiatric traits in early adulthood by inducing inflammatory, oxidative and nitrosative stress (IO&NS) pathways, mitochondrial dysfunction, apoptosis, and epigenetic dysregulation. However, the precise mechanisms behind such relationships and the specificity of the risk factors for schizophrenia remain exploratory. Considering the paucity of knowledge on fetal programming of schizophrenia, it is timely to consolidate the recent advances in the field and put forward an integrated overview of the mechanisms associated with fetal origin of schizophrenia.
1. Introduction
Epidemiological studies of birth and death records led to “Barker's hypothesis” almost 30 years ago, suggesting the influence of perturbed gestational milieu on the development of diseases later in life (Barker and Osmond, 1986). This generated a great deal of interest in the fetal origins of adult disorders. Although Barker's hypothesis was mainly based on the potential impact of gestational malnutrition to result in permanently altered organs function and structure, a growing body of evidence suggests that a range of maternal complications of pregnancy such as gestational diabetes mellitus, intrauterine growth restriction (IUGR), preeclampsia and maternal stress are associated with adult disorders (Barker and Clark, 1997, Cottrell and Seckl, 2009, Calkins and Devaskar, 2011).
Perturbation of developmental adaptive processes that are known to be involved in permanent changes in physiology, structure and metabolism or early life programming can adversely affect brain development, impacting both brain structure and function (Schlotz and Phillips, 2009). Findings from human and animal studies indicate that several environmental factors and physiological mechanisms involving endocrine as well as immune system during gestation play crucial role in early-life programming of later-life brain and behavior by long-term remodeling of the brain (Schlotz and Phillips, 2009, Bilbo and Schwarz, 2009, Auyeung et al., 2013). Evidence toward this notion was derived from neuropathological aberrations including enlarged cerebral ventricles, as well as changes in gray and white matter indicative of impaired neural development during gestation in serious mental illnesses such as schizophrenia (Harrison, 1999, Pantelis et al., 2005, De Peri et al., 2012). Importantly, structural brain changes during development of schizophrenia are seemingly determined by genetic components, altered expression of schizophrenia risk genes and epigenetic dysregulation (Lawrie et al., 2008, Miller et al., 2012, Stachowiak et al., 2013).
The developmental origin of schizophrenia might potentially be a result of prenatal exposure to a diversity of factors such as infection, stress, persistent organic pollutants, smoking and other substance use, maladaptive diet and developmental epigenetic changes (Brown et al., 2009, Kirkbride et al., 2012, Jacka et al., 2013). A two-hit model is one of the leading theories of schizophrenia pathogenesis. The two hit model predominantly implicating the neurodevelopmental theory is mainly based on the assumption that aberrant development during two critical time points (early brain development and adolescence) additively produces risk for schizophrenia-like symptoms. The “first hit” potentially occurs in utero. Although genetic components were primarily considered as the “first hit”, recent understanding suggests that along with genetic, environmental factors might also exert a similar function. Animal as well as human studies demonstrated that genetic susceptibility in combination with a developmental insults can prime an individual for a later event that ultimately increase risk for the onset of schizophrenia and this could possibly be mediated by known salient signaling pathways (Bayer et al., 1999, Maynard et al., 2001). The “first hit” may disrupt developing neuronal architecture, specific neural networks, establish an abnormal inflammatory response and account for premorbid signs and symptoms in individuals who later develop schizophrenia (Keshavan, 1999, Feigenson et al., 2014). Contextually, prenatal infection has become one of the best evidenced environmental risk factors of the “two-hit” hypothesis of schizophrenia as it increases the offspring's sensitivity to environmental challenges postnatally and renders them more vulnerable to the pathological effects of a second postnatal stimulus (Bayer et al., 1999, Maynard et al., 2001). This “second hit” due to stress, immune re-exposure or even peri-pubertal sex hormonal changes might potentially unmask and exacerbate the hitherto latent inflammatory processes with resultant onset of clinical symptoms (Meyer et al., 2011, Knickmeyer et al., 2010). Experimental support for the impact of a second hit is evident from animal studies, where prenatal immune activation elicited by gestational infection and subsequent exposure to stress, cannabinoid use, etc. during adolescence has been shown to unmask the latent neuropathological consequences of prenatal infection and lead to increased risk for the development of schizophrenia (Dalton et al., 2012, Giovanoli et al., 2013). During adolescence, events like excessive elimination of synapses and loss of plasticity might influence the development of the disorder (Fig. 1 ). Despite this understanding, precisely how the abnormal developmental trajectory of the brain is established during gestation and also how this is causally related to the manifestation of symptoms that appear early in adult life remains a challenging issue. Here we examine recent advances and discuss many emergent factors and mechanisms that might elucidate the mechanistic link between early life programming and adult manifestation of psychiatric conditions.
Fig. 1.
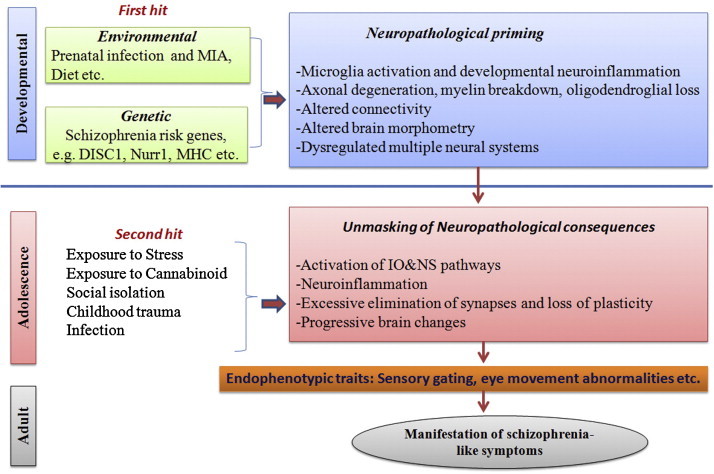
Depiction of ‘two-hit’ model of schizophrenia.
2. Neurodevelopmental origin of schizophrenia: a review of evidence
Disparate lines of evidence support the view that schizophrenia is a neurodevelopmental disorder. The primary reason is that the onset of schizophrenia has a cumulative age incidence distribution, or developmental function, that is nonlinear with a peak change in slope or acceleration that usually takes to occur during young adulthood. Given the plausibility of the existence of brain abnormalities in schizophrenia at the onset of the illness, it further seems reasonable to conceive schizophrenia as a neurodevelopmental disorder (Piper et al., 2012). Recent understanding suggests determining role of gene and environmental interactions in the neurodevelopmental trajectories of schizophrenia. The genetic and environmental factors cause structural and functional change not only during pre- and peri-natal periods but also in childhood and early adolescence (Ismail et al., 2000, Walker, 1994). Further support has been provided by epidemiological studies showing premorbid intellectual deficits dating back to early development, and neuropathological studies showing altered cerebral cytoarchitecture indicative of a developmental rather than acquired encephalopathy. The main neurodevelopmental hypotheses for schizophrenia set forth in the last 15 years are relatively restricted and share three assumptions (Woods, 1998). Firstly, the primary pathogenic defect is an early derangement of the orderly development of the central nervous system that occurs in the pre- or peri-natal period. Secondly, the period of active operation of the causative agent is of short duration, meaning that it is essentially static and lastly the behavioral consequences of this static process remain relatively latent until long after the primary pathogenic process has run its course.
In addition, it is now becoming evident that prenatal adversity induced neurodevelopmental disruption can have physical manifestations. Minor physical abnormalities, neurological soft signs and altered dermatoglyphics pattern represent important markers of disordered neurodevelopment in schizophrenia (Sivkov et al., 2009, Gabalda and Compton, 2010, Aksoy-Poyraz et al., 2011). Some of the minor physical abnormalities such as confluent eyebrows, hypertelorism, ear protrusion, low-set ears, palatal abnormalities, tongue furrows and so on represent a set of risk markers for schizophrenia (Xu et al., 2011). Neurological soft signs correlate with various neuro-cognitive and neuro-anatomical abnormalities in schizophrenia (Varambally et al., 2012). Neurological soft signs are understood as a manifestation of the “cerebello-thalamo-prefrontal” brain network model of schizophrenia (Zhao et al., 2014). Higher levels of neurological soft signs have been reported in healthy children who later develop schizophrenia and in first episode as well as antipsychotic-naïve schizophrenia patients (Leask et al., 2002, Dazzan and Murray, 2002, Varambally et al., 2006). Recent examinations of cortex morphology, a marker of brain development in first-episode schizophrenia patients with neurological soft signs revealed a lower global sulcal index (g-SI) in both hemispheres, a lower regional sulcal indexes (r-SI) in left dorsolateral prefrontal and right lateral occipital cortices (Gay et al., 2013). Taken together, these findings strongly suggest evidence of distinct neurodevelopmental pathways in schizophrenia patients with neurological soft signs. Although various findings including twin studies have highlighted the importance of genetic determination of neurological soft signs, minor physical abnormalities and dermatoglyphic patterns in schizophrenia (Niethammer et al., 2000, Fatjó-Vilas et al., 2008); the precise genetic determinant of these parameters are yet to be identified.
3. Different risk factors and mechanisms underlying developmental origin of schizophrenia
3.1. Exogenous risk factors
3.1.1. Prenatal teratogens
A range of teratogens and neurotoxic agents such as industrial chemicals (persistent organic pollutants and heavy metals), high levels of ionizing radiation, tobacco smoke, cocaine, alcohol and certain drugs affect early brain development (Grandjean and Landrigan, 2006). Multiple studies have demonstrated that chemical exposures during early development constitute a new potential class of risk factors for schizophrenia. Prenatal exposure to Pb2+ has been found to increase the risk of schizophrenia in the offspring (Opler et al., 2008). One plausible underlying neurobiological mechanism is the alteration of subunit composition of N-methyl-d-aspartate receptor (NMDAR) complexes with subsequent effects on calcium-sensitive signaling pathways involved in CREB phosphorylation (Toscano et al., 2002). Prenatal exposure to analgesics in the second trimester of pregnancy conferred a more than four-fold greater risk of schizophrenia (Sørensen et al., 2004). Aspirin taken during pregnancy interferes with the prostaglandin pathway, and exposure may influence the risk of schizophrenia in offspring (Gunawardana et al., 2011). Animal models of schizophrenia have demonstrated that rats exposed to cigarette smoke in the prenatal period exhibited a significant increase of the lipid peroxidation, protein oxidation and DNA damage in adult offspring (Fraga et al., 2011). These effects can lead to several neurochemical changes overlapping with the pathophysiology of schizophrenia. Maternal smoking through pregnancy increases the risk of later schizophrenia among offspring, conferring an increased severity of negative symptoms (Stathopoulou et al., 2013). Interestingly, some of the chemicals and drugs contribute to schizophrenia risk not only as a result of prenatal exposure but also an adolescent exposure. Cadmium and lead for example appear to be associated with both mood and psychotic disorders (Karim et al., 2006, Berk et al., 2014, Orisakwe, 2014). Pre-adolescence exposure to cannabis is shown to be associated with a twofold increase in the risk of schizophrenia (Van Os et al., 2002).
3.1.2. Prenatal nutrition
Compelling evidence suggests that prenatal malnutrition leads to long-term brain impairment and increased risk of schizophrenia (Butler et al., 1994, Brown and Susser, 2008, Victora et al., 2008). Studies conducted after in utero exposure to hunger/famine during Dutch Hunger Winter of 1944–1945 and 1959–1961 Chinese Famine have indicated a direct association between prenatal starvation and increased risk of schizophrenia (Hoek et al., 1998, St Clair et al., 2005). Prenatal exposure to famine affected brain morphology such as decreased intracranial volume, which is seen in schizophrenia (Hulshoff Pol et al., 2000). More recently, there is increasing evidence that poor quality diet rather than malnutrition per se is a risk factor. The western diet is calorie, fat and sugar rich and nutrient poor, and obesity is currently one of the major public health issues. There is now good prospective evidence in the depression literature that poor quality diet is a risk factor for depression (Quirk et al., 2013), and recent data that maternal prenatal diet impacts the risk of internalizing and externalizing diet in offspring (Jacka et al., 2013). The amount of information on the link between schizophrenia and diet is poorer, but such data is emerging (Dipasquale et al., 2013). There is emerging evidence of a link between gluten sensitivity and increased levels of antigliadin antibodies and schizophrenia (Jackson et al., 2014). What is known is that diet influences the pathways to neuroprogression in schizophrenia including inflammation and oxidative stress (Berk et al., 2013).
Maternal vitamin deficiency during early pregnancy has been associated with the risk of schizophrenia in the offspring. One of the widely studied vitamins in the context of neurodevelopmental disorders is vitamin D. Vitamin D is arguably the nutritionally related factor that is most deficient in western populations, and some data that this is particularly an issue in psychiatric cohort (Berk et al., 2008). There is now clear evidence that vitamin D is involved in brain development and being a potent pro-differentiation agent, vitamin D can influence brain functioning via many different pathways (Eyles et al., 2013). Transient prenatal vitamin D deficiency results in abnormal brain development, persistent changes in adult brain structure, neurotransmission, synaptic plasticity, neurochemistry, and behavior and several other biological pathways including oxidative phosphorylation, redox balance, cytoskeleton maintenance, calcium homeostasis, chaperoning and post-translational modifications (Eyles et al., 2007, Eyles et al., 2009, Eyles et al., 2013).
Several studies have demonstrated the immuno-modulatory properties of vitamin D and its implications in a number of autoimmune/inflammatory disorders (Fernandes de Abreu et al., 2009). Importantly, fetal vitamin D plays a crucial role in controlling placental inflammation (Liu et al., 2011a). Vitamin D influences Th1:Th2 balance by inhibiting Th1 pathway and promoting the Th2 pathway through down-regulation of pro-inflammatory cytokines and up-regulation of anti-inflammatory cytokines. Pro-inflammatory cytokine-induced alterations in cognition/behavior could therefore be linked to low levels of vitamin D. This is supported by recent findings that vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory cytokine production in rats (Briones and Darwish, 2012). In addition, developmental vitamin D3 deficiency induces persistent alterations in immune phenotype and function in adult offspring. Furthermore, when stimulated, lymphocytes from developmental vitamin D-deficient rats exhibited a pro-inflammatory phenotype (Harvey et al., 2010). Human population studies have also indicated a significant association between developmental vitamin D deficiency and risk of schizophrenia (McGrath et al., 2010).
3.1.3. Prenatal infection
Some of the strongest support toward the developmental etiology of schizophrenia has come from its association with prenatal infection (Boksa, 2010, Khandaker et al., 2013). Accumulating epidemiological data indicates that 38–46% of cases of schizophrenia may have an association with prenatal infection (Brown and Derkits, 2010). Maternal infection by Toxoplasma gondii, influenza, rubella, herpes simplex virus, and cytomegalovirus has consistently been associated with the risk of schizophrenia in adult offspring. Although some of these infectious agents, e.g. rubella, T. gondii, cytomegalovirus, etc. can cross the placenta and directly affect the developing fetus, the detrimental effects of infection is known to be elicited by activation of maternal immune system (Hsiao and Patterson, 2011). Converging evidence suggests that immuno-inflammatory responses together with downstream oxidative and nitrosative stress pathway are crucial in mediating prenatal infection induced neurodevelopmental abnormality (Anderson et al., 2013). The detrimental effects of redox dysregulation, excessive generation of reactive oxygen species (ROS)/reactive nitrogen species (RNS), etc. on developing brain after prenatal infection are quite well known (Do et al., 2009, Lante et al., 2007). Further, intrauterine infection/inflammation evokes the expression of inducible nitric oxide synthase (iNOS) and nitric oxide (NO) in the brain and leads to oligodendrocyte injury in the developing brain (Shen et al., 2007).
Prenatal immune activation leads to changes in the levels of multiple neurotransmitter systems (Winter et al., 2009). In this context, dopamine abnormalities are one of the most enduring etiological hypotheses of schizophrenia. Emerging findings from animal studies reveal that prenatal exposure to infection and/or inflammation has long-lasting effects on dopaminergic structures and functions (Aguilar-Valles et al., 2010, Eyles et al., 2012, Ozawa et al., 2006, Vuillermot et al., 2010, Zuckerman et al., 2003). In mice, a viral-like acute phase response during early/mid gestation causes a complex pattern of age-dependent structural abnormalities in the mesoaccumbal and nigrostriatal dopamine system (Vuillermot et al., 2010). Notably, cytokines are known to modulate dopaminergic neurotransmission and dopamine can also modulate immune response by influencing the cytokine network (Song et al., 1999, Ch Beck et al., 2004). Inflammation and oxidative/nitrosative stress strongly influence each other, and increased levels of serum oxidative stress markers and inflammatory cytokines have been reported in schizophrenia patients (Pedrini et al., 2012). Based on this understanding, it is likely that prenatal infection induced activation of IO&NS pathway will have an important downstream role in dopaminergic neurotoxicity in schizophrenia. A recent study has demonstrated increased protein oxidation in the dopamine-rich areas of the prefrontal cortex in schizophrenia patients, suggesting the importance of interactions between oxidative stress and dopamine in the pathophysiology of schizophrenia (Kim et al., 2014). Psychosis is hypothesized to be a hyperdopaminergic state, and models of increased dopamine transmission are linked to increases in ROS (Xie et al., 2014). Dopamine antagonists can block increased oxidative stress and apoptotic potential (Odaka et al., 2014). Additionally, dopamine metabolism itself, by generating its metabolites like dopamine-semiquinone and dopamine-quinone leads to the generation of ROS within CNS (Grima et al., 2003). Importantly, in neurodegenerative disorders quinone formation has been shown to be linked with inflammation and oxidative stress. Furthermore, oxidative stress is shown to inhibit the uptake of dopamine through post-translational modification of the dopamine transporter (Kim and Andreazza, 2012).
There is crosstalk between other transmitters germane to schizophrenia such as GABA and serotonin and inflammation (Wang et al., 2012). Prenatal immune activation in mouse with poly I:C is shown to cause maturation-dependent alterations in prefrontal GABAergic gene expression, implying long-term effect of prenatal immune insults on the GABAergic system (Richetto et al., 2014). Agents enhancing GABAergic transmission such as gabapentin down-regulate inflammatory and oxidative stress markers (Dias et al., 2014, Guseva et al., 2014, Zimmermann et al., 2014). Similarly, serotonergic agents cause down-regulation of inflammatory markers in some studies (Taraz et al., 2013, Leonard, 2014). Further relevant to schizophrenia pathogenesis, prenatal immune activation has shown to modulate hippocampal NMDAR function by interacting with stress and the stress hormone corticosterone at adolescence (Burt et al., 2013). Taken together, maternal infection induced IO&NS responses can adversely affect pre- and/or peri-natal outcomes and even lead to neurodegeneration or teratogenesis (Wells et al., 2009, Yuan et al., 2010), indicating that inflammation-induced effects may be associated with a direct damage by oxidative/nitrosative stress.
3.1.4. Prenatal stress
Prenatal stress-induced changes are found to confer high risk for diverse outcomes in offspring including schizophrenia (Malaspina et al., 2008). For example, gestational exposure to prenatal stress or stress hormones contributes to deficits in hippocampal structure and function, as well as the neurotransmitter and immune systems (Lemaire et al., 2000, Bellinger et al., 2008, Marques et al., 2013). The enhanced production of corticosterone or maternal exposure to exogenous glucocorticoids due to chronic exposures to stress has direct influence on fetal brain development and plasticity as well as programming of hypothalamic-pituitary-adrenocortical (HPA) axis (Koenig et al., 2005, Kapoor et al., 2006). Prenatal stress increases serotonin 2A and decreases mGlu2 expression in frontal cortex, suggesting schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors (Holloway et al., 2013). Prenatal stress also affects development of the hippocampal alpha 7 nicotinic acetyl choline receptor (nAChRs) in adult offspring (Schulz et al., 2013). It is interesting to note that repeated variable prenatal stress during critical period of fetal brain development reprograms the response of the HPA axis to acute stress and alters pre- and postsynaptic gene expression that might impact synaptic function in the offspring (Kinnunen et al., 2003). Studies have demonstrated that both down-regulation of glutamate decarboxylase 67 (GAD67) and maternal exposure to severe stress can increase the risk of schizophrenia in offspring. This is further supported by a study where heterozygous deletion in GAD67 enhances maternal and fetal stress vulnerability (Uchida et al., 2011).
3.1.5. Paternal age
Epidemiological studies have provided robust evidence of association between advanced paternal age and the enhanced risk of schizophrenia (McGrath et al., 2014, Brown et al., 2002, Miller et al., 2011). One of the contributing mechanisms that has consistently been implicated in paternal age associated risk of schizophrenia is de novo mutations, which are found to be more abundant in sperm of older men (Risch et al., 1987, Malaspina et al., 2002). In a comprehensive study on 78 families, an increase in two de novo mutations per year of advancing paternal age was observed, indicating a linear relationship between paternal age and the number of de novo mutations in offspring (Kong et al., 2012). Recently, “selfish spermatogonial selection” which means clonal expansion of germs cells carrying pathogenic mutations has been proposed to be a mechanism for association between advanced paternal age and the risk of schizophrenia (Goriely et al., 2013). The clonal expansion leads to relative enrichment of de novo mutations that essentially explain the effect of advanced paternal age on schizophrenia.
3.2. Endogenous risk factors
3.2.1. Obstetric events
Obstetric complications are well established predictors of risk of schizophrenia (Cannon et al., 2002a). Although the term “obstetric events” includes a wide range of complications, the most commonly found complications are fetal growth retardation, fetal hypoxia and prenatal complications.
Low birth weight and reduced length at birth are crude indicators of IUGR or abnormally slow fetal growth. Fetal growth restriction increases risk of later schizophrenia (Eide et al., 2013). Smaller head circumference and small gestational age have been associated with schizophrenia, indicating the implications of fetal growth restriction (McNeil et al., 2000). Although the underlying mechanism of growth restriction and risk of schizophrenia is poorly understood, accumulating evidence suggests that a combination of factors such as environment, placental, and genetic enable expression of a particular mental health outcome. Recent animal data suggests that IUGR leads to metabolic alterations in the fetal brain by influencing neuronal viability, inflammatory regulation, energy metabolism and oxidative stress pathways (van Vliet et al., 2013). Suggesting the importance of genetic effects on fetal growth restriction, it has been found that mothers with schizophrenia also have higher rates of low birth weight among offspring (Jablensky et al., 2005). However, in a study on same-sex twins discordant for schizophrenia, it was observed that within these twin pairs, low birth weight and smaller head circumference were significantly associated with later development of schizophrenia, indicating that the fetal growth restriction may be independent of familial factors (Nilsson et al., 2005).
Fetal hypoxia is an environmental risk factor of schizophrenia in the offspring. Hypoxia-associated obstetric complications and the increased risk of schizophrenia is a replicated finding (Zornberg et al., 2000, Dalman et al., 2001). It is evident that interactions between neuronal genes and molecular regulators of oxygen could lead to hypoxia induced neurodevelopmental abnormality and subsequent risk of neuropsychiatric disorders. Fetal hypoxia also appears to influence the severity of certain neuropathological attributes of schizophrenia, such as hippocampal and cortical gray matter reduction (Cannon et al., 2002b). Importantly, such changes in schizophrenia are proposed to be mediated by interactions between schizophrenia susceptibility genes and hypoxia related regulatory processes (Van Erp et al., 2002, Schmidt-Kastner et al., 2012). A large number of schizophrenia susceptibility genes viz. AKT1, BDNF, COMT, DNTBP1, NOTCH4, NRG1, PRODH, RELN, RGS4 are regulated by hypoxia (Schmidt-Kastner et al., 2006). Furthermore, genes regulated by hypoxia also interact with serious obstetric complications and influence schizophrenia risk (Nicodemus et al., 2008).
3.2.2. Gut microbiota and developmental immune modulation
Microbial colonization of gut is an evolutionary process, which influences both metabolic and immune functions, particularly during early neonatal life. However, recent findings indicate that microbial colonization or contact of the fetus with maternal gut microbiota may start in utero as bacteria from maternal gastrointestinal tract (GIT) have been detected in amniotic fluid, placental and fetal membranes (Hitti et al., 1997). Pregnancy is associated with a profound alteration of maternal gut microbiota, and the changes in gut microbiota composition have been associated with weight gain, altered biochemical and metabolic parameters during pregnancy (Santacruz et al., 2010, Koren et al., 2012). Obstetric complications, long known to increase risk for schizophrenia, may in theory be mediated through higher rates of cesarean section, which limits the infants accrual of a normal microbiota from the maternal genitourinary tract through birth (Boksa and El-Khodor, 2003). This has been hypothesized to also be a factor in autism (Curran et al., 2014). This implies the possible roles of the gut microbiota composition in mother's health during pregnancy as well as on maternal-fetal interactions, influencing the infant's health later in life.
The perinatal colonization of the gut by microbes contributes to developmental programming of gut homeostasis, angiogenesis and immune competence (Hooper and Gordon, 2001, Lundin et al., 2008). There are extensive interactions between the gut microbiota and the host immune system in the newborns and the development of the adaptive immune system is regulated by bacterial colonization of the gut. It is now evident that time variation in microbial colonization of the gut during early life shapes future immune system reactivity. For example, delayed colonization seems to exert permanent changes in the immune system (Hansen et al., 2012). A range of environmental factors including cesarean section, toxins, infectious agents, diet and stress could affect the gut microbiome during this critical developmental period. Disruption of the developing miocrobiome could lead to long-term changes in immune and psychological development. Furthermore, disturbances in the microbiota can also result in dysregulation of adaptive immune cells, intestinal inflammation and potentially enhance the host's susceptibility to immune-mediated diseases.
The gut microbiota communicates with the brain and affects normal brain development and subsequent adult behavior (Diaz et al., 2011). Gut–brain communication occurs through direct neuronal, immune-related signaling and hormonal pathways. Exposure to many of the afore-mentioned environmental factors during both the pre-and post-natal periods has been established as risk factors for disorders including schizophrenia and autism. One of the widely accepted underlying mechanisms that mediates the interaction between such environmental factors and host as well as gut–brain communication is through immunological signaling involving cytokines. Animal studies suggest that perturbation in the composition of gut microbiota influence the risk of depression and anxiety-like behaviors (Neufeld et al., 2011, Dinan and Cryan, 2013). In autistic children, there appears to be a distinct and less diverse gut microbe composition (Kang et al., 2013). A recent study highlighted the importance of gastrointestinal inflammation in schizophrenia pathology (Severance et al., 2012). Translocation of commensal microbiota such as Citrobacter koseri, Hafnia alvei, Pseudomonas aeruginosa, Pseudomonas putida, Klebsiella pneumonia and Morganella morganii across the gastrointestinal barrier can lead to an antibody response to bacterial translocation, and lysozyme production and consequently leads to persistent low-grade inflammation and increases the risk of depression (Maes et al., 2012, Maes et al., 2013). Bacterial translocation causes an imbalanced and activated innate immune state (Severance et al., 2013). Quality data linking these processes to schizophrenia is awaited.
3.2.3. Role of placenta in fetal brain development
There is a wide appreciation that perturbations in the maternal hormonal and nutrient environment have deleterious effects on the developing brain and subsequently influence susceptibility to a range of metabolic, neurodevelopmental and psychiatric disorders in adulthood (Fernandez-Twinn and Ozanne, 2010, Bale et al., 2010). Importantly, such effects can be transmitted to the fetus by changes in placental function (Jansson and Powell, 2007), thereby indicating a direct role of placenta in developmental programming of brain and behavior. Placental insufficiency causes intrauterine growth restriction (Barry et al., 2008). In mice, food deprivation for 24 h on days E12–E13 has been shown to affect placental gene expression, through which placenta provides protection to the fetal brain (Broad and Keverne, 2011). Interestingly, recent studies suggest that the placenta can convert maternal tryptophan into the neurotransmitter serotonin (5-hydroxytryptamine or 5-HT), which is essential for developing mouse forebrain at mid-gestation (Bonnin et al., 2011). This implies a significant role of tryptophan catabolites (TRYCATs) in the placenta in modulating fetal brain development and well as affecting long-term brain function (Bonnin and Levitt, 2012, Goeden et al., 2013). However, developmental disruption of 5-HT signaling in specific regions of fetal brain causes abnormal wiring of major axonal pathways, altered cell division and laminar organization in the neocortex (Vitalis and Parnavelas, 2003, Bonnin et al., 2007). Altered brain 5-HT signaling is consistently linked to several psychiatric disorders. Considering the significant role of 5-HT in essential neurodevelopmental processes, genetic or environmental perturbations directly affecting placental tryptophan metabolism can lead to neurodevelopmental abnormality, and therefore contribute to the developmental origin of diverse psychiatric disorders.
Prenatal exposure to maternal infection leads to increased expression of pro-inflammatory cytokines such as IL-6 and TNF-α in the placenta and also leads to fetal inflammation which in turn can cause organ damage and potentially downstream developmental deficiencies (Urakubo et al., 2001, Cardenas et al., 2010). Activation of the maternal immune system induces endocrine changes in the placenta, especially IL-6 dependent disruption of the growth hormone-insulin-like growth factor (GH-IGF) axis i.e. decreased levels of GH, IGFI and IGFBP3 (Hsiao and Patterson, 2011). Taken together, these observations suggest that the placenta might act as a central mediator of fetal programming of the TRYCAT pathway that may underlie developmental origin of adult psychiatric disorders including schizophrenia (Goeden et al., 2013).
3.2.4. Prenatal neuroendocrine pathway
Hormones are established environment-dependent coordinators of the developing “neuro-endocrine-immune network”. However, non-physiological concentrations of hormones during crucial phases of embryogenesis can act as ‘endogenous functional teratogens’. This is exemplified by fetal and neonatal hyper-insulinism in the offspring of diabetic mothers. Diabetes mellitus is the most common metabolic complication during gestation and is known to have neurodevelopmental sequelae. A number of studies have highlighted relationship between maternal diabetes and the risk of schizophrenia in the offspring (Van Lieshout and Voruganti, 2008). Importantly, the risk of schizophrenia in the offspring born to diabetic mothers could be induced by hyperglycemia and mediated by hypoxia, inflammation and oxidative stress.
Endocrine disruption as etiological component is theoretically implicated in schizophrenia. Estrogen is proposed to be a potential mediator of brain functions during development and adulthood. Estrogen protects brain cells against injury from oxidative stress, inflammation and apoptosis (Arevalo et al., 2010, Behl, 2002). Estrogen is relevant to schizophrenia due to its significant effects on synaptogenesis, neurogenesis, neuroendocrine and inflammatory processes. Recently, combinations of both estrogen and selective estrogen receptor modulators with antipsychotics have been shown to decrease positive and negative symptoms significantly in women with chronic schizophrenia (Kulkarni et al., 2010, Kulkarni et al., 2014, Ghafari et al., 2013). However, prenatal exposure to excess estrogen could increase the risk of schizophrenia in the offspring through certain mechanisms (Brown, 2011). For example, estrogen has been found to increase susceptibility to certain viral infections by altering innate and adaptive immune responses, such as by reducing CD4 T-cell responses and by inhibiting type 1 interferon production and dendritic cell maturation (Escribese et al., 2008, Pazos et al., 2012). Interestingly, many such viruses including borna disease virus, mumps, influenza, corona viruses, etc. are associated with increased risk of schizophrenia. A summary of prenatal endogenous and exogenous risk factors and/or mechanisms leading to psychopathology has been presented in Fig. 2 .
Fig. 2.
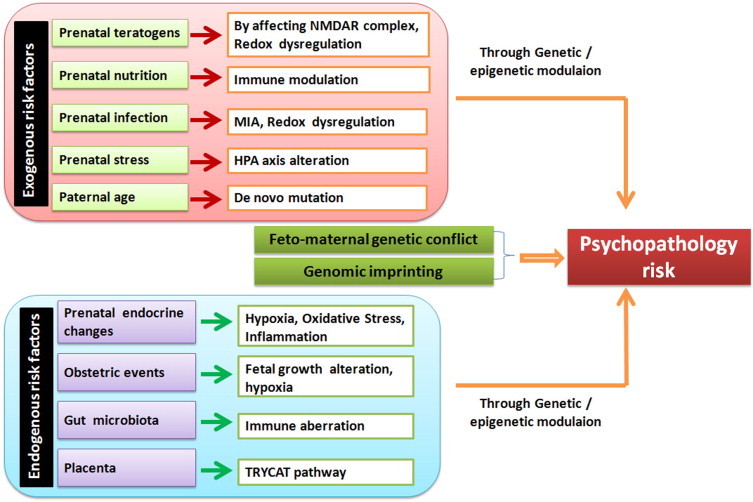
Summary of potential prenatal endogenous and exogenous risk factors and/or mechanisms leading to psychopathology.
3.3. Specificity of the risk factors of schizophrenia
Converging evidence from multiple animal studies indicate that prenatal infection can act as a “neurodevelopmental disease primer’, and this seems to be a shared environmental risk factor across a number of chronic mental illnesses (as reviewed in Meyer, 2014, Knuesel et al., 2014). It is now widely appreciated that activation of IO& NS pathway due to prenatal infection is one of the underlying mechanisms in the pathogenesis of various neuropsychiatric disorders like schizophrenia, autism, and bipolar disorder (Anderson et al., 2013; Anderson and Maes, 2014). Although the specificity of prenatal infection on subsequent disease or symptoms is yet to be precisely understood, it is likely to be influenced by the interactions between genetic and additional environmental factors. For example, major histocompatibility complex (MHC) molecules have been shown to play important roles during neurodevelopment (Chacon and Boulanger, 2013). Neurons exposed to MIA were found to have higher levels of MHC class I expression and lower synapse density (Elmer et al., 2013), suggesting that such effects of MIA on MHCI expression could affect activity-dependent plasticity and synaptic pruning during crucial phases of neurodevelopment. A plethora of genetic studies have consistently implicated MHC as a risk determinant of schizophrenia (Debnath et al., 2013); however, this has not been observed in bipolar disorder (Corvin and Morris, 2013). Furthermore, MHC is also found to be associated with various schizophrenia risk factors such as obstetric complications, season of birth, infection and so on (Debnath et al., 2013), thus implying that genetic components along with environmental factors might determine the specificity of disease pathogenesis.
4. Neurodevelopmental origin of schizophrenia: a unifying genetic–epigenetic–environmental pathway
4.1. Genomic imprinting
The expression of some genes in the human genome depends on whether they are located on the maternal or paternal chromosome. This is accomplished through a highly powerful epigenetic phenomenon called genomic imprinting, which is a significant form of gene regulation. Several of the imprinted genes that are expressed from only one of the parental chromosomes have novel roles in the normal placental development and fetal growth (Frost and Moore, 2010). Although most known imprinted genes are expressed during placentation, a considerable number are expressed primarily or entirely in the brain (Davies et al., 2008). Brain development is strongly influenced by the epigenetic regulation of imprinted genes, and there is increasing evidence that imprinted genes can influence neurogenetic and psychiatric manifestations by affecting neurodevelopmental processes (Isles and Wilkinson, 2000, Wilkinson et al., 2007). A recent family based trio study has suggested the occurrence of imprinting of schizophrenia candidate gene, GABRB2 that codes for GABA(A) receptor β(2) subunit (Pun et al., 2011). Evidence of imprinting in schizophrenia has also been observed in other chromosomal locus, such as DLK1-DIO3 region in chromosome 14q32 (Gardiner et al., 2012).
4.2. Feto-maternal genetic conflict
Fetal–maternal genetic conflict may potentially contribute to many disorders related to pregnancy including preeclampsia (Hiby et al., 2004). Interestingly, a combination of maternal/fetal genetic predisposition and environmental factors has been implicated as potential risk factors for preeclampsia (Roberts and Cooper, 2001). This may be based on MHC and Rh gene compatibility/incompatibility (Haig, 1997). With respect to HLA antigens, when there is a lack of maternal recognition i.e. if paternally derived HLAs do not differ from maternal HLAs, this may have an adverse effect on reproductive outcomes (Ober, 1998). HLA matching, in couples or between mother and fetus, increases prenatal or obstetric complications including preeclampsia, low birth weight, and can even lead to fetal loss (Ober et al., 1998).
The first indication of the importance of HLA matching in neurodevelopmental disorders came from a study showing that parents of children with autism are more likely to share at least one HLA-A, -B, or -C antigen in common compared to the parents of unaffected children (Stubbs et al., 1985). Recently, the significance of maternal–fetal genotype incompatibility at HLA loci in schizophrenia susceptibility has been described, in that maternal–fetal matching at the HLA-B locus increases fetal risk for developing schizophrenia (Palmer, 2010, Palmer et al., 2006). Maternal–fetal HLA-B matching may increase the risk of schizophrenia only in female offspring (Childs et al., 2011). Various roles of MHC molecules in neuronal development by in vitro and in vivo studies and risk of schizophrenia conferred by MHC variants have been well supported by genome wide association studies (Debnath et al., 2013, McAllister, 2013, Corvin and Morris, 2013). These observations led to the hypothesis that maternal–fetal genotype combinations at HLA loci, by modulating feto-maternal immune responses, can affect fetal neurodevelopment and subsequently enhance one's risk of developing schizophrenia.
4.3. Developmental epigenetic modifications
Environmental signals during prenatal life lead to adverse long-term effects. Although the mechanisms underlying such risk remain poorly understood, emerging studies both in animals and humans indicate that maternal exposure to infection, stress, drugs or toxins alter epigenetic programming in regulatory as well as growth-related genes (Gicquel et al., 2008). There is a growing recognition that prenatal epigenetic dysregulation due to such adverse in utero environments not only affect fetal brain development, but also predispose an individual to neurodevelopmental, behavioral and neurocognitive deficits later in life (Fagiolini et al., 2009, Petanjek and Kostović, 2012). A recent study has demonstrated that MIA causes epigenetic changes in adolescent mouse brain (Basil et al., 2014).
The impact of prenatal stress on the modification of epigenetic signatures during critical periods of fetal brain development has been elucidated by several recent studies. Exposure to gestational stress increases DNA methylation at the glucocorticoid receptor (GR) gene promoter and reduces methylation at the corticotrophin-releasing factor (CRF) gene promoter in hypothalamic tissue in adult male mice (Mueller and Bale, 2008). Prenatal stress down-regulates 11β-hydroxysteroid dehydrogenase type 2 (HSD11β2) that converts cortisone/corticosterone into inactive metabolites through DNA methylation in the placenta and fetal brain (Jensen et al., 2012). Further evidence suggests that the offspring of mice exposed to gestational stress have an altered transcriptomic brain profile of genes related to development, axonal guidance and neuropathology due to up-regulation and down-regulation of certain microRNA (miRNA) (Zucchi et al., 2013). In addition, prenatal stress through disruption of DNA methylation network affects GABAergic interneurons associated with schizophrenia-like phenotypes (Matrisciano et al., 2013).
Maternal cigarette smoking during pregnancy is a common hazard affecting key pathways crucial for proper fetal growth and development. Recent data suggests that maternal cigarette smoking during pregnancy can lead to alteration in DNA methylation and expression of microRNA (Knopik et al., 2012). Importantly, maternal smoking during pregnancy has been found to be associated with increased DNA methylation in a key gene involved in brain development, brain-derived neurotrophic factor (BDNF) in adolescent offspring (Toledo-Rodriguez et al., 2010).
Maternal diet during pregnancy can also affect brain development and function by modifying the fetal epigenome. Persistent epigenetic changes were observed in the offspring exposed to prenatal famine. Peri-conceptional exposure to famine during Dutch Hunger Winter was associated with reduced DNA methylation of Insulin-like growth factor 2 (IGF2), which is a key factor in human growth and development (Heijmans et al., 2008). Dietary intake of methyl-group (choline, methionine, and folate) containing nutrients during critical phases of prenatal development can alter the epigenomic profile of the developing offspring, thereby resulting in altered fetal and lifelong changes in gene expression. Unbalanced maternal diet during pregnancy alters DNA methylation in important genes controlling glucocorticoid function and fetal growth (Drake et al., 2012). Recently, an association between maternal prenatal nutrition and schizophrenia risk in the offspring via epigenetic effects has been highlighted (Kirkbride et al., 2012).
4.4. Gene–environment interactions
A large number of environmental factors have been proposed to cause neurodevelopmental abnormalities and confer enhanced risk of schizophrenia in offspring. It is now becoming apparent that genetic factors potentially modulate the adverse effects of environmental stressors on neurodevelopment during pre- and peri-natal periods as well as behavioral outcomes in the offspring (Cannon et al., 2003). Importantly, the impact of gene–environmental interactions on perinatal programming of schizophrenia is a critical issue in prenatal adversity induced changes in neurodevelopment. The genetic liability of perinatal environmental adversities has been exemplified by various studies. A recent study has shown how maternal cytomegalovirus infection influences the risk of schizophrenia in the offspring by interacting with the genotype of CTNNA3 gene of the progeny (Borglum et al., 2014). The additive or interactive effects of hypoxia with genetic factors in influencing liability to schizophrenia have been reported (Cannon et al., 2000). Animal studies have demonstrated that MIA during pregnancy with polyI:C of mice having mutation in DISC1, a consistently replicated risk gene of schizophrenia exacerbate schizophrenia-like phenotypes (Abazyan et al., 2010, Lipina et al., 2013). Another study has shown that prenatal immune activation in mice with mutations in Nurr1, a transcription factor crucial for dopaminergic development leads to neuropathological consequences, such as locomotor hyperactivity, deficits in sensorimotor gating, and attentional impairments (Vuillermot et al., 2012). Furthermore, recent animal studies demonstrated that combined effects of neonatal immune activation and mutant DISC1 on the risk of schizophrenia- like behavioral phenotypes in the offspring (Ibi et al., 2010). One of the important pathways that can be modulated by perinatal adverse conditions is HPA axis activity in offspring. Perinatal exposure to excess glucocorticoids can lead to a persistently altered HPA axis and subsequently enhance the risk of schizophrenia (Huang, 2011). Disturbed genome regulation due to adverse perinatal environmental conditions has been shown to contribute to schizophrenia risk and pathophysiology.
Recent observations indicate that prenatal infection increases the associated risk of schizophrenia if occurring in offspring with a family history of psychosis; this implies an interaction of genetic vulnerability with prenatal infection (Clarke et al., 2009). This could possibly be mediated by certain gene loci such as MHC, HLA-G, toll-like receptor (TLR) 3 and 4 that are known to have predominant expression during pregnancy (Debnath and Chaudhuri, 2006, Venkatasubramanian and Debnath, 2013). Taken together, such evidence suggests that fetal immune programming may have profound consequences on brain and behavior, and is associated with the adult presentation of schizophrenia.
5. Prenatal events and neuroprogression in schizophrenia
There is now strong evidence of pathological reorganization of the central nervous system along the course of severe mental disorder. Neuroimaging studies have demonstrated significant volume reduction of certain specific regions in the brain of schizophrenia patients, providing evidence of neuroprogressive processes including neurodegeneration and neuronal apoptosis (Ho et al., 2003). There is a wide appreciation that neuroprogression is partially mediated by IO&NS pathways, apoptosis and mitochondrial energy dysregulation (Berk et al., 2011, Anderson et al., 2013).
The generation of ROS and RNS has both physiologic and pathologic roles in the placenta, embryo and fetus; the developing brain is highly vulnerable to ROS and RNS. Amongst the several causative mechanisms of fetal origin of adult diseases, a significant role of oxidative and nitrosative stress (O&NS) in fetal programming is predominant (Thompson and Al-Hasan, 2012). It is now evident that a number of factors such as prenatal hypoxia, maternal under-and over-nutrition, excessive glucocorticoid exposure can induce O&NS process during pregnancy and subsequently lead to neuronal death/brain injury (Ikonomidou and Kaindl, 2011). In a recent study it was observed that chronic fetal hypoxia induced brain injury is associated with altered nitric oxide synthase activity (Dong et al., 2011).
It is now becoming evident that a range of prenatal adversities can lead to heightened maternal inflammation, which can contribute to placental hypoxia and O&NS process underlying altered fetal growth and development. For example, the effects of prenatal malnutrition have also been found to be mediated by pro-inflammatory factors (Shen et al., 2008). Fetal hypoxemia can cause fetal inflammatory response syndrome as well as fetal brain injury by up-regulating inflammatory cytokine cascade (Guo et al., 2010).
It is interesting to note that melatonin, an endogenously produced indoleamine from the pineal gland, acts as an antioxidant, free radical scavenger and anti-inflammatory molecule (Radogna et al., 2010). Importantly, melatonin has a role in redox modification in fetal programming and reverses oxidative stress during prenatal period. Further, melatonin influences epigenetic modifications associated with developmental programming (Korkmaz et al., 2012). Melatonin might play a pivotal role in epigenetic modifications induced by maternal stress, maternal under-nutrition or IUGR (Chen et al., 2013). As melatonin levels and melatonin circadian rhythm are significantly decreased in schizophrenia (Anderson and Maes, 2012), a role of melatonin in neuroprogressive pathways seems credible.
Molecular hydrogen is an odorless and tasteless gas, and this bioactive molecule has various biological attributes including anti-inflammatory, anti-apoptotic and anti-oxidative effects (Ohta, 2011). Animal studies have indicated that inhalation of hydrogen decreased acute lung inflammation and hydrogen enriched water decreased the production of ROS (Xie et al., 2012, Katakura et al., 2012). Importantly, a protective role of hydrogen on fetal brain injury during maternal hypoxia has also been recently highlighted (Liu et al., 2011b). Some intestinal bacteria, e.g. Escherichia coli can produce a remarkable amount of molecular hydrogen and under certain circumstances can suppress inflammation (Kajiya et al., 2009). Considering the above biological properties of molecular hydrogen and their relevance in neurodevelopment and function including neuroprogressive changes, implications of molecular hydrogen have been highlighted recently in bipolar disorder and schizophrenia (Ghanizadeh and Berk, 2013).
6. Implications: prediction and prevention
Understanding the pathophysiological basis of schizophrenia from the perspectives of MIA with fetal programming aberrations permits potential translational implications as well. Over the past few years multiple blood-based, imaging, neurophysiologic and neurocognitive biomarkers have been identified for schizophrenia (Chan et al., 2011a, Light et al., 2012, Zarogianni et al., 2013). However, there seems to be a considerable imprecision in the nosology of biomarkers that have been identified in schizophrenia. Recent conceptualization suggests that a classification of biomarkers based on six categories such as risk, diagnosis/trait, state or acuity, stage, treatment response and prognosis may be applicable to neuropsychiatric disorders (Davis et al., 2014). Several studies have consistently shown altered levels of markers of inflammation, oxidative stress, metabolism and hormonal status in the first onset schizophrenia patients and have suggested the potential implications of such risk biomarkers of schizophrenia (Guest et al., 2011, Perkins et al., 2014). Considering the ever growing support toward the neurodevelopmental origin of schizophrenia, identification of biomarkers bearing signatures of events from as early as embryonic development or birth would be of considerable interest in predicting risk as well as treatment responses. A recent methylome-wide association study examining blood based biomarkers in schizophrenia has shown changes in genes that reflect the effects of environmental insults related to hypoxia, infection etc. (Aberg et al., 2014), suggesting that pathogenic events might be preserved in the methylome. Such understanding essentially highlights the importance of environmental adversities on fetal epigenome, therefore, early epigenomic profiling might be one of the areas which could offer potential schizophrenia risk predicting biomarker. In addition, elevated levels of inflammatory marker such as maternal C-reactive protein, TNF-α and IL-8 have robustly been associated with increased risk of schizophrenia in adult offspring (Buka et al., 2001, Brown et al., 2004, Canetta et al., 2014). Such maternal inflammatory markers could also be considered as potential risk biomarker in predicting schizophrenia early in life. Maternal stress is an established risk factor of schizophrenia. Recently, O-GlcNAc transferase (OGT) has been demonstrated to be a placental biomarker of maternal stress (Howerton et al., 2013). This biomarker might also be useful in predicting gestational stress exposure and risk of schizophrenia in adult offspring.
Impaired sensory gating indicative of aberrant cerebral inhibition is a documented component of the pathogenesis of schizophrenia (Vlcek et al., 2014). Significantly, prenatal MIA can result in sensory gating aberrations (Romero et al., 2010). Hence, within the pathogenetic paradigm of aberrant fetal programming due to prenatal immune disturbances, certain biomarkers like P50, that are indicative of sensory gating deficits, especially in combination with other significant biomarkers like P85, P300, mismatch negativity and eye movement aberrations (smooth pursuit as well as antisaccades) (Bender et al., 2007), with subsequent analyses of markers for persistent immune activation (cytokines, lymphocyte functions (Drexhage et al., 2011) as well as microglia imaging using positron emission tomography (Doorduin et al., 2009) might facilitate reliable prediction of risk for schizophrenia.
Prevention of psychiatric disorders of neurodevelopmental origin such as schizophrenia is becoming a challenging issue (Jacka and Berk, 2014). Public health approaches including control of infectious diseases, improvements in obstetric and neonatal care and nutritional supplementation might be beneficial. For instance, choline, an essential nutrient, has been shown to improve immune parameters with associated adaptive modulation of cognitive functions (McCann et al., 2006, Lewis et al., 2014a, Lewis et al., 2014b). Choline plays a critical role in optimal neurodevelopment (Ueland, 2011). Interestingly, several maternal factors that elevate schizophrenia risk can lead to decreased availability of choline during fetal development (Zeisel, 2006). Recently, a randomized placebo-controlled clinical trial examined 100 healthy pregnant women with dietary phosphatidylcholine supplementation that was initiated in the second trimester until the time of delivery. After birth, infants were given 100 mg of phosphatidylcholine in an oral suspension once daily or placebo (Ross et al., 2013). In comparison with placebo-treated infants, choline-treated infants demonstrated better sensory gating as examined by P50 response (Ross et al., 2010). Choline supplementation was shown to enhance sensory gating measures in healthy adults as well (Knott et al., 2014). Availability of safe interventions like choline supplementation is worth exploring with large scale studies to ascertain potential preventive interventions.
7. Limitations of neurodevelopmental theory and future directions
The neurodevelopmental origin of schizophrenia although has become one of the most influential etiologic theories in recent times; however, there are certain limitations of this theory. Amongst the range of factors and mechanisms that are shown to alter the developmental trajectories, the association of certain risk factors/mechanisms remain weak and needs to be replicated. Various risk factors seem to mediate a large number of mechanisms that impair crucial phases of fetal development and result in a similar set of behavioral outcomes in the offspring. It is essential to classify the risk factors that confer predominant risk and delineate their mechanistic basis. The current understanding suggests that developmental modulation during embryogenesis by various factors and mechanisms could potentially predict behavioral outcomes in the offspring during adolescence or early adulthood and these precede the onset of illness. Early onset schizophrenia is a severe form of schizophrenia that occurs during childhood or adolescence and is often chronic and persistently debilitating. However, the frequency of early onset schizophrenia has been reported to be around 4% (Cannon et al., 1999). Although converging functional genomics suggests involvement of multiple genes in neurodevelopment, certain genes are found to confer susceptibility to various neurodevelopmental disorders. Furthermore, most of the risk genes have a variable pattern of expression and different effects at different developmental stages. Some of the genes exhibit preferential expression in the fetal brain and high gene expression occurring during fetal development can be reversed in early postnatal life (Colantuoni et al., 2011). In addition, progressive brain changes have also been reported in chronic adult patients with schizophrenia (Chan et al., 2011b). Therefore, it is important to study the effect of various risk factors including genes as well as neuroprogressive changes at different time points during the lifespan to gain more insights on the pathogenesis of schizophrenia rather than focussing emphatically on neurodevelopmental insults. The developmental neuroinflammation has also been contradicted by a recent study where neonates who later developed schizophrenia had unaltered levels of 17 inflammatory markers, thus refuting an association between neonatal inflammation and risk of schizophrenia (Nielsen et al., 2014).
Future research should pay more focus to establish ‘missing heritability’ aspects and disentangle the different effects of prenatal environmental and genetic exposures. The early-life epigenetic disruption also plays pivotal role in the neurodevelopmental origin of schizophrenia. This notion is experimentally supported by methylomic profiling of human brain tissue of schizophrenia patients, identifying disease-associated differential DNA methylation in the prefrontal cortex and discrete modules of co-methylated loci associated with the disorder that are significantly enriched for genes involved in neurodevelopmental processes (Pidsley et al., 2014). Improved understanding of the genetic as well as epigenetic contribution to environmental risk mediation will offer possible candidate for primary prevention and early intervention strategies. Combined studies of prenatal exposures and neurodevelopmental disorders with simultaneous evaluation of neural and immune systems might delineate the mechanisms enhancing individual vulnerability or resilience to neurodevelopmental disorders. The importance of early life programming as a target for prevention of mental disorders in the offspring has recently been highlighted by Lewis et al. (2014b) and several recommendations were made with respect to public health and clinical implications.
8. Conclusion
There is strong evidence that schizophrenia is a complex multi-factorial disorder and no one factor seems to be of solitary significance in the genesis of schizophrenia. Among the various etiological models tested so far, gene–environmental interactions appear to be most widely appreciated one in schizophrenia. The adverse effects of certain risk factors are found to be linked to genetic components, therefore, it is essential to identify specific genetic components that confer major risk to schizophrenia in combination with environmental stressors. The series of neurological insults rendered by an array of environmental factors during gestation reviewed above strongly level schizophrenia to a disorder of neurodevelopment and significantly highlight the implications of fetal programming of schizophrenia. Such compelling evidence calls upon researchers to translate these findings into interventions designed to prevent mental disorders.
Conflict of interest
The authors declare that they have no conflicts of interest to disclose germane to this paper.
Acknowledgement
MB is supported by a NHMRC Senior Principal Research Fellowship 1059660.
References
- Abazyan B., Nomura J., Kannan G., Ishizuka K., Tamashiro K.L., Nucifora F. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol. Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg K.A., McClay J.L., Nerella S., Clark S., Kumar G., Chen W. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014;71(March (3)):255–264. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Valles A., Flores C., Luheshi G.N. Prenatal inflammation induced hypoferremia alters dopamine function in the adult offspring in rat: relevance for schizophrenia. PLoS ONE. 2010;5:e10967. doi: 10.1371/journal.pone.0010967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy-Poyraz C., Poyraz B.Ç., Turan Ş., Arikan M.K. Minor physical anomalies and neurological soft signs in patients with schizophrenia and their siblings. Psychiatry Res. 2011;190:85–90. doi: 10.1016/j.psychres.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Anderson G., Berk M., Dodd S., Bechter K., Altamura A.C., Dell’osso B., Kanba S. Immuno-inflammatory, oxidative and nitrosative stress, and neuroprogressive pathways in the etiology, course and treatment of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;42:1–4. doi: 10.1016/j.pnpbp.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Anderson G., Maes M. Melatonin: an overlooked factor in schizophrenia and in the inhibition of anti-psychotic side effects. Metab. Brain Dis. 2012;27:113–119. doi: 10.1007/s11011-012-9307-9. [DOI] [PubMed] [Google Scholar]
- Anderson G., Maes M. Redox regulation and the autistic spectrum: role of tryptophan catabolites, immuno-inflammation, autoimmunity and the amygdala. Curr. Neuropharmacol. 2014;12(March (2)):148–167. doi: 10.2174/1570159X11666131120223757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo M.A., Santos-Galindo M., Bellini M.J., Azcoitia I., Garcia-Segura L.M. Actions of estrogens on glial cells: implications for neuroprotection. Biochim. Biophys. Acta. 2010;1800:1106–1112. doi: 10.1016/j.bbagen.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Auyeung B., Lombardo M.V., Baron-Cohen S. Prenatal and postnatal hormone effects on the human brain and cognition. Pflugers Arch. 2013;465:557–571. doi: 10.1007/s00424-013-1268-2. [DOI] [PubMed] [Google Scholar]
- Bale T.L., Baram T.Z., Brown A.S., Goldstein J.M., Insel T.R., McCarthy M.M., Nemeroff C.B., Reyes T.M., Simerly R.B., Susser E.S., Nestler E.J. Early life programming and neurodevelopmental disorders. Biol. Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D.J., Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barker D.J.P., Clark P.M. Fetal undernutrition and disease in later life. Rev. Reprod. 1997;2:105–112. doi: 10.1530/ror.0.0020105. [DOI] [PubMed] [Google Scholar]
- Barry J.S., Rozance P.J., Anthony R.V. An animal model of placental insufficiency-induced intrauterine growth restriction. Semin. Perinatol. 2008;32:225–230. doi: 10.1053/j.semperi.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Basil P., Li Q., Dempster E.L., Mill J., Sham P.C., Wong C.C., McAlonan G.M. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl. Psychiatry. 2014;4(September):e434. doi: 10.1038/tp.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer T.A., Falkai P., Maier W. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis”. J. Psychiatr. Res. 1999;33(November–December (6)):543–548. doi: 10.1016/s0022-3956(99)00039-4. [DOI] [PubMed] [Google Scholar]
- Behl C. Estrogen can protect neurons: modes of action. J. Steroid Biochem. Mol. Biol. 2002;83:1–5. doi: 10.1016/s0960-0760(02)00271-6. [DOI] [PubMed] [Google Scholar]
- Bellinger D.L., Lubahn C., Lorton D. Maternal and early life stress effects on immune function: relevance to immunotoxicology. J. Immunotoxicol. 2008;5:419–444. doi: 10.1080/15476910802483415. [DOI] [PubMed] [Google Scholar]
- Bender S., Weisbrod M., Resch F. Which perspectives can endophenotypes and biological markers offer in the early recognition of schizophrenia? J. Neural Transm. 2007;114(September (9)):1199–1215. doi: 10.1007/s00702-007-0742-4. [DOI] [PubMed] [Google Scholar]
- Berk M., Jacka F.N., Williams L.J., Ng F., Dodd S., Pasco J.A. Is this D vitamin to worry about? Vitamin D insufficiency in an inpatient sample. Aust. N. Z. J. Psychiatry. 2008;42:874–878. doi: 10.1080/00048670802345516. [DOI] [PubMed] [Google Scholar]
- Berk M., Kapczinski F., Andreazza A.C., Dean O.M., Giorlando F., Maes M. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 2011;35:804–817. doi: 10.1016/j.neubiorev.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Berk M., Williams L.J., Jacka F.N., O’Neil A., Pasco J.A., Moylan S. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013;11:200. doi: 10.1186/1741-7015-11-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk M., Williams L.J., Andreazza A.C., Pasco J.A., Dodd S., Jacka F.N., Moylan S., Reiner E.J., Magalhaes P.V. Pop, heavy metal and the blues: secondary analysis of persistent organic pollutants (POP), heavy metals and depressive symptoms in the NHANES National Epidemiological Survey. BMJ Open. 2014;4(July (7)):e005142. doi: 10.1136/bmjopen-2014-005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo S.D., Schwarz J.M. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front. Behav. Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boksa P., El-Khodor B.F. Birth insult interacts with stress at adulthood to alter dopaminergic function in animal models: possible implications for schizophrenia and other disorders. Neurosci. Biobehav. Rev. 2003;27(January–March (1–2)):91–101. doi: 10.1016/s0149-7634(03)00012-5. [DOI] [PubMed] [Google Scholar]
- Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav. Immun. 2010;24:881–897. doi: 10.1016/j.bbi.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Bonnin A., Torii M., Wang L., Rakic P., Levitt P. Serotonin modulates the response of embryonic thalamocortical axons to netrin-1. Nat. Neurosci. 2007;10:588–597. doi: 10.1038/nn1896. [DOI] [PubMed] [Google Scholar]
- Bonnin A., Goeden N., Chen K., Wilson M.L., King J., Shih J.C. A transient placental source of serotonin for the fetal forebrain. Nature. 2011;472:347–350. doi: 10.1038/nature09972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A., Levitt P. Placental source for 5-HT that tunes fetal brain development. Neuropsychopharmacology. 2012;37:299–300. doi: 10.1038/npp.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borglum A.D., Demontis D., Grove J., Pallesen J., Hollegaard M.V., Pedersen C.B. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol. Psychiatry. 2014;19(March (3)):325–333. doi: 10.1038/mp.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones T.L., Darwish H. Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J. Neuroinflamm. 2012;9:244. doi: 10.1186/1742-2094-9-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad K.D., Keverne E.B. Placental protection of the fetal brain during short-term food deprivation. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15237–15241. doi: 10.1073/pnas.1106022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.S., Jr. Association of increased prenatal estrogen with risk factors for schizophrenia. Schizophr. Bull. 2011;37:946–949. doi: 10.1093/schbul/sbp161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.S., Schaefer C.A., Wyatt R.J., Begg M.D., Goetz R., Bresnahan M.A., Harkavy-Friedman J., Gorman J.M., Malaspina D., Susser E.S. Paternal age and risk of schizophrenia in adult offspring. Am. J. Psychiatry. 2002;159(September (9)):1528–1533. doi: 10.1176/appi.ajp.159.9.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.S., Hooton J., Schaefer C.A., Zhang H., Petkova E., Babulas V., Perrin M., Gorman J.M., Susser E.S. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am. J. Psychiatry. 2004;161(May (5)):889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- Brown A.S., Susser E.S. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull. 2008;34:1054–1063. doi: 10.1093/schbul/sbn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.S., Vinogradov S., Kremen W.S., Poole J.H., Deicken R.F., Penner J.D. Prenatal exposure to maternal infection and executive dysfunction in adult schizophrenia. Am. J. Psychiatry. 2009;166:683–690. doi: 10.1176/appi.ajp.2008.08010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.S., Derkits E.J. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buka S.L., Tsuang M.T., Torrey E.F., Klebanoff M.A., Wagner R.L., Yolken R.H. Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav. Immun. 2001;15(December (4)):411–420. doi: 10.1006/brbi.2001.0644. [DOI] [PubMed] [Google Scholar]
- Burt M.A., Tse Y.C., Boksa P., Wong T.P. Prenatal immune activation interacts with stress and corticosterone exposure later in life to modulate N-methyl-d-aspartate receptor synaptic function and plasticity. Int. J. Neuropsychopharmacol. 2013;16:1835–1848. doi: 10.1017/S1461145713000229. [DOI] [PubMed] [Google Scholar]
- Butler P.D., Susser E.S., Brown A.S., Kaufmann C.A., Gorman J.M. Prenatal nutritional deprivation as a risk factor in schizophrenia: preclinical evidence. Neuropsychopharmacology. 1994;11:227–235. doi: 10.1038/sj.npp.1380109. [DOI] [PubMed] [Google Scholar]
- Calkins K., Devaskar S.U. Fetal origins of adult disease. Curr. Probl. Pediatr. Adolesc. Health Care. 2011;41:158–176. doi: 10.1016/j.cppeds.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canetta S., Sourander A., Surcel H.M., Hinkka-Yli-Salomäki S., Leiviskä J., Kellendonk C., McKeague I.W., Brown A.S. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am. J. Psychiatry. 2014;171(September (9)):960–968. doi: 10.1176/appi.ajp.2014.13121579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M., Jones P., Huttunen M.O., Tanskanen A., Huttunen T., Rabe-Hesketh S., Murray R.M. School performance in Finnish children and later development of schizophrenia. A population-based longitudinal study. Arch. Gen. Psychiatry. 1999;56:457–463. doi: 10.1001/archpsyc.56.5.457. [DOI] [PubMed] [Google Scholar]
- Cannon T.D., Rosso I.M., Hollister J.M., Bearden C.E., Sanchez L.E., Hadley T. A prospective cohort study of genetic and perinatal influences in the etiology of schizophrenia. Schizophr. Bull. 2000;26(2):351–366. doi: 10.1093/oxfordjournals.schbul.a033458. [DOI] [PubMed] [Google Scholar]
- Cannon M., Jones P.B., Murray R.M. Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry. 2002;159:1080–1092. doi: 10.1176/appi.ajp.159.7.1080. [DOI] [PubMed] [Google Scholar]
- Cannon T.D., van Erp T.G., Rosso I.M., Huttunen M., Lönnqvist J., Pirkola T., Salonen O., Valanne L., Poutanen V.P., Standertskjöld-Nordenstam C.G. Fetal hypoxia and structural brain abnormalities in schizophrenic patients, their siblings, and controls. Arch. Gen. Psychiatry. 2002;59:35–41. doi: 10.1001/archpsyc.59.1.35. [DOI] [PubMed] [Google Scholar]
- Cannon T.D., van Erp T.G., Bearden C.E., Loewy R. Early and late neurodevelopmental influences in the prodrome to schizophrenia: contributions of genes, environment, and their interactions. Schizophr. Bull. 2003;29:653–669. doi: 10.1093/oxfordjournals.schbul.a007037. [DOI] [PubMed] [Google Scholar]
- Cardenas I., Means R.E., Aldo P., Koga K., Lang S.M., Booth C.J. Viral infection of the placenta leads to fetal inflammation and sensitization to bacterial products predisposing to preterm labor. J. Immunol. 2010;185:1248–1257. doi: 10.4049/jimmunol.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon M.A., Boulanger L.M. MHC class I protein is expressed by neurons and neural progenitors in mid-gestation mouse brain. Mol. Cell. Neurosci. 2013;52(January):117–127. doi: 10.1016/j.mcn.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Ch Beck G., Brinkkoetter P., Hanusch C. Clinical review: immunomodulatory effects of dopamine in general inflammation. Crit. Care. 2004;8(6):485–491. doi: 10.1186/cc2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M.K., Guest P.C., Levin Y., Umrania Y., Schwarz E., Bahn S., Rahmoune H. Converging evidence of blood-based biomarkers for schizophrenia: an update. Int. Rev. Neurobiol. 2011;101:95–144. doi: 10.1016/B978-0-12-387718-5.00005-5. [DOI] [PubMed] [Google Scholar]
- Chan R.C., Di X., McAlonan G.M., Gong Q.Y. Brain anatomical abnormalities in high-risk individuals, first-episode, and chronic schizophrenia: an activation likelihood estimation meta-analysis of illness progression. Schizophr. Bull. 2011;37(January (1)):177–188. doi: 10.1093/schbul/sbp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.C., Sheen J.M., Tiao M.M., Tain Y.L., Huang L.T. Roles of melatonin in fetal programming in compromised pregnancies. Int. J. Mol. Sci. 2013;14:5380–5401. doi: 10.3390/ijms14035380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs E.J., Sobel E.M., Palmer C.G.S., Sinsheimer J.S. Detection of intergenerational genetic effects with application to HLA-B matching as a risk factor for schizophrenia. Hum. Hered. 2011;72:161–172. doi: 10.1159/000332051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M.C., Tanskanen A., Huttunen M., Whittaker J.C., Cannon M. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am. J. Psychiatry. 2009;166:1025–1030. doi: 10.1176/appi.ajp.2009.08010031. [DOI] [PubMed] [Google Scholar]
- Colantuoni C., Lipska B.K., Ye T., Hyde T.M., Tao R., Leek J.T. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvin A., Morris D.W. Genome-wide association studies: findings at the major histocompatibility complex locus in psychosis. Biol. Psychiatry. 2013;204(75):276–283. doi: 10.1016/j.biopsych.2013.09.018. [DOI] [PubMed] [Google Scholar]
- Cottrell E.C., Seckl J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009;3:19. doi: 10.3389/neuro.08.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran E.A., O’Neill S.M., Cryan J.F., Kenny L.C., Dinan T.G., Khashan A.S., Kearney P.M. Research review: birth by caesarean section and development of autism spectrum disorder and attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. J. Child Psychol. Psychiatry. 2014 doi: 10.1111/jcpp.12351. [DOI] [PubMed] [Google Scholar]
- Dalton V.S., Verdurand M., Walker A., Hodsgson D.M., Zavitsanou K. Synergistic effect between maternal infection and adolescent cannabinoid exposure on serotonin 5HT1A receptor binding in the hippocampus: testing the “two hit” hypothesis for the development of schizophrenia. ISRN Psychiatry. 2012;2012 doi: 10.5402/2012/451865. Article ID 451865, 9 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalman C., Thomas H.V., David A.S., Gentz J., Lewis G., Allebeck P. Signs of asphyxia at birth and risk of schizophrenia. Population-based case–control study. Br. J. Psychiatry. 2001;179:403–408. doi: 10.1192/bjp.179.5.403. [DOI] [PubMed] [Google Scholar]
- Davies W., Isles A.R., Humby T., Wilkinson L.S. What are imprinted genes doing in the brain? Epigenetics. 2008;2:201–206. doi: 10.4161/epi.2.4.5379. [DOI] [PubMed] [Google Scholar]
- Davis J., Maes M., Andreazza A., McGrath J.J., Tye S.J., Berk M. Towards a classification of biomarkers of neuropsychiatric disease: from encompass to compass. Mol. Psychiatry. 2014 doi: 10.1038/mp.2014.139. (in press) [DOI] [PubMed] [Google Scholar]
- Dazzan P., Murray R.M. Neurological soft signs in first-episode psychosis: a systematic review. Br. J. Psychiatry Suppl. 2002;43(September):s50–s57. doi: 10.1192/bjp.181.43.s50. [DOI] [PubMed] [Google Scholar]
- Debnath M., Chaudhuri T.K. The role of HLA-G in cytokine homeostasis during early pregnancy complicated with maternal infections: a novel etiopathological approach to the neurodevelopmental understanding of schizophrenia. Med. Hypotheses. 2006;66:286–293. doi: 10.1016/j.mehy.2005.06.033. [DOI] [PubMed] [Google Scholar]
- Debnath M., Cannon D.M., Venkatasubramanian G. Variation in the major histocompatibility complex [MHC] gene family in schizophrenia: associations and functional implications. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;42:49–62. doi: 10.1016/j.pnpbp.2012.07.009. [DOI] [PubMed] [Google Scholar]
- De Peri L., Crescini A., Deste G., Fusar-Poli P., Sacchetti E., Vita A. Brain structural abnormalities at the onset of schizophrenia and bipolar disorder: a meta-analysis of controlled magnetic resonance imaging studies. Curr. Pharm. Des. 2012;8:486–494. doi: 10.2174/138161212799316253. [DOI] [PubMed] [Google Scholar]
- Dias J.M., de Brito T.V., de Aguiar Magalhães D., da Silva Santos P.W., Batista J.A., do Nascimento Dias E.G., de Barros Fernandes H., Damasceno S.R., Silva R.O., Aragão K.S., Souza M.H., Medeiros J.V., Barbosa A.L. Gabapentin, a synthetic analogue of gamma aminobutyric acid, reverses systemic acute inflammation and oxidative stress in mice. Inflammation. 2014;37(October (5)):1826–1836. doi: 10.1007/s10753-014-9913-2. [DOI] [PubMed] [Google Scholar]
- Diaz H.R., Wang S., Anuar F., Qian Y., Björkholm B., Samuelsson A. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U. S. A. 2011;108:3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan T.G., Cryan J.F. Melancholic microbes: a link between gut microbiota and depression? Neurogastroenterol. Motil. 2013;25:713–719. doi: 10.1111/nmo.12198. [DOI] [PubMed] [Google Scholar]
- Dipasquale S., Pariante C.M., Dazzan P., Aguglia E., McGuire P., Mondelli V. The dietary pattern of patients with schizophrenia: a systematic review. J. Psychiatr. Res. 2013;47:197–207. doi: 10.1016/j.jpsychires.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Do K.Q., Cabungcal J.H., Frank A., Steullet P., Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr. Opin. Neurobiol. 2009;19:220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Doorduin J1, de Vries E.F., Willemsen A.T., de Groot J.C., Dierckx R.A., Klein H.C. Neuroinflammation in schizophrenia-related psychosis: a PET study. J. Nucl. Med. 2009;50(November (11)):1801–1807. doi: 10.2967/jnumed.109.066647. [DOI] [PubMed] [Google Scholar]
- Dong Y., Yu Z., Sun Y., Zhou H., Stites J., Newell K., Weiner C.P. Chronic fetal hypoxia produces selective brain injury associated with altered nitric oxide synthases. Am. J. Obstet. Gynecol. 2011;204(254):e16–e28. doi: 10.1016/j.ajog.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake A.J., McPherson R.C., Godfrey K.M., Cooper C., Lillycrop K.A., Hanson M.A. An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and foetal growth. Clin. Endocrinol. (Oxf.) 2012;77:808–815. doi: 10.1111/j.1365-2265.2012.04453.x. [DOI] [PubMed] [Google Scholar]
- Drexhage R.C., Hoogenboezem T.A., Cohen D., Versnel M.A., Nolen W.A., van Beveren N.J., Drexhage H.A. An activated set point of T-cell and monocyte inflammatory networks in recent-onset schizophrenia patients involves both pro- and anti-inflammatory forces. Int. J. Neuropsychopharmacol. 2011;14(July (6)):746–755. doi: 10.1017/S1461145710001653. [DOI] [PubMed] [Google Scholar]
- Eide M.G., Moster D., Irgens L.M., Reichborn-Kjennerud T., Stoltenberg C., Skjærven R. Degree of fetal growth restriction associated with schizophrenia risk in a national cohort. Psychol. Med. 2013;9:1–10. doi: 10.1017/S003329171200267X. [DOI] [PubMed] [Google Scholar]
- Elmer B.M., Estes M.L., Barrow S.L., McAllister A.K. MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J. Neurosci. 2013;33(August (34)):13791–13804. doi: 10.1523/JNEUROSCI.2366-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribese M.M., Kraus T., Rhee E., Fernandez-Sesma A., Lopez C.B., Moran T.M. Estrogen inhibits dendritic cell maturation to RNA viruses. Blood. 2008;112:4574–4584. doi: 10.1182/blood-2008-04-148692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles D., Almeras L., Benech P., Patatian A., Mackay-Sim A., McGrath J. Developmental vitamin D deficiency alters the expression of genes encoding mitochondrial, cytoskeletal and synaptic proteins in the adult rat brain. J. Steroid Biochem. Mol. Biol. 2007;103:538–545. doi: 10.1016/j.jsbmb.2006.12.096. [DOI] [PubMed] [Google Scholar]
- Eyles D.W., Feron F., Cui X., Kesby J.P., Harms L.H., Ko P., McGrath J.J., Burne T.H. Developmental vitamin D deficiency causes abnormal brain development. Psychoneuroendocrinology. 2009;34(December (Suppl. 1)):S247–S257. doi: 10.1016/j.psyneuen.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Eyles D., Feldon J., Meyer U. Schizophrenia: do all roads lead to dopamine or is this where they start? Evidence from tow epidemiologically informed developmental rodent models. Transl. Psychiatry. 2012;2:e81. doi: 10.1038/tp.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles D.W., Burne T.H., McGrath J.J. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front. Neuroendocrinol. 2013;34:47–64. doi: 10.1016/j.yfrne.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Fagiolini M., Jensen C.L., Champagne F.A. Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol. 2009;19:207–212. doi: 10.1016/j.conb.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatjó-Vilas M., Gourion D., Campanera S., Mouaffak F., Levy-Rueff M., Navarro M.E., Chayet M., Miret S., Krebs M.O., Fañanás L. New evidences of gene and environment interactions affecting prenatal neurodevelopment inschizophrenia-spectrum disorders: a family dermatoglyphic study. Schizophr. Res. 2008;103(August (1–3)):209–217. doi: 10.1016/j.schres.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Feigenson K.A., Kusnecov A.W., Silverstein S.M. Inflammation and the two-hit hypothesis of schizophrenia. Neurosci. Biobehav. Rev. 2014;38(January):72–93. doi: 10.1016/j.neubiorev.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Twinn D.S., Ozanne S.E. Early life nutrition and metabolic programming. Ann. N. Y. Acad. Sci. 2010;1212:78–96. doi: 10.1111/j.1749-6632.2010.05798.x. [DOI] [PubMed] [Google Scholar]
- Fernandes de Abreu D.A., Eyles D., Féron F. Vitamin D, a neuro-immunomofulator: implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology. 2009;34:S265–S277. doi: 10.1016/j.psyneuen.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Fraga D.B., Deroza P.F., Ghedim F.V., Steckert A.V., De Luca R.D., Silverio A. Prenatal exposure to cigarette smoke causes persistent changes in the oxidative balance and in DNA structural integrity in rats submitted to the animal model of schizophrenia. J. Psychiatr. Res. 2011;45:1497–1503. doi: 10.1016/j.jpsychires.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Frost J.M., Moore G.E. The importance of imprinting in the human placenta. PLoS Genet. 2010;6(7):e1001015. doi: 10.1371/journal.pgen.1001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabalda M.K., Compton M.T. Dermatoglyphic indices and minor physical anomalies in patients with schizophrenia and related disorders, their first-degree biological relatives, and non psychiatric controls. Psychiatry Res. 2010;178(July (2)):255–259. doi: 10.1016/j.psychres.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Gardiner E., Beveridge N.J., Wu J.Q., Carr V., Scott R.J., Tooney P.A., Cairns M.J. Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol. Psychiatry. 2012;17(July (8)):827–840. doi: 10.1038/mp.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay O., Plaze M., Oppenheim C., Mouchet-Mages S., Gaillard R., Olié J.P., Krebs M.O., Cachia A. Cortex morphology in first-episode psychosis patients with neurological soft signs. Schizophr. Bull. 2013;39(July (4)):820–829. doi: 10.1093/schbul/sbs083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafari E., Fararouie M., Shirazi H.G. Combination of estrogen and antipsychotics in the treatment of women with chronic schizophrenia: a double-blind, randomized, placebo-controlled clinical trial. Clin. Schizophr. Relat. Psychoses. 2013;6:172–176. [PubMed] [Google Scholar]
- Ghanizadeh A., Berk M. Molecular hydrogen: an overview of its neurobiological effects and therapeutic potential for bipolar disorder and schizophrenia. Med. Gas Res. 2013;3:11. doi: 10.1186/2045-9912-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gicquel C., El-Osta A., Le Bouc Y. Epigenetic regulation and fetal programming. Best Pract. Res. Clin. Endocrinol. Metab. 2008;22:1–16. doi: 10.1016/j.beem.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Giovanoli S., Engler H., Engler A., Richetto J., Voget M., Willi R. Stress in puberty unmasks latent neuropathological consequences of prenatal immuneactivation in mice. Science. 2013;339(Match (6123)):1095–1099. doi: 10.1126/science.1228261. [DOI] [PubMed] [Google Scholar]
- Goeden N., Velasquez J.C., Bonnin A. Placental tryptophan metabolism as a potential novel pathway for the developmental origins of mental diseases. Transl. Dev. Psychiatry. 2013;1:20593. [Google Scholar]
- Goriely A., McGrath J.J., Hultman C.M., Wilkie A.O., Malaspina D. “Selfish spermatogonial selection”: a novel mechanism for the association between advanced paternal age and neurodevelopmental disorders. Am. J. Psychiatry. 2013;170(June (6)):599–608. doi: 10.1176/appi.ajp.2013.12101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P., Landrigan P.J. Developmental neurotoxicity of industrial chemicals. Lancet. 2006;368:2167–2178. doi: 10.1016/S0140-6736(06)69665-7. [DOI] [PubMed] [Google Scholar]
- Grima G., Benz B., Parpura V. Dopamine-induced oxidative stress in neurons with glutathione deficit: implication for schizophrenia. Schizophr. Res. 2003;62:213–224. doi: 10.1016/s0920-9964(02)00405-x. [DOI] [PubMed] [Google Scholar]
- Guseva D., Holst K., Kaune B., Meier M., Keubler L., Glage S., Buettner M., Bleich A., Pabst O., Bachmann O., Ponimaskin E.G. Serotonin 5-HT7 receptor is critically involved in acute and chronic inflammation of the gastrointestinal tract. Inflamm. Bowel Dis. 2014;20(9):1516–1529. doi: 10.1097/MIB.0000000000000150. [DOI] [PubMed] [Google Scholar]
- Guest P.C., Schwarz E., Krishnamurthy D., Harris L.W., Leweke F.M. Altered levels of circulating insulin and other neuroendocrine hormones associated with the onset of schizophrenia. Psychoneuroendocrinology. 2011;36:1092–1096. doi: 10.1016/j.psyneuen.2010.12.018. [DOI] [PubMed] [Google Scholar]
- Gunawardana L., Zammit S., Lewis G., Gunnell D., Hollis C., Wolke D. Examining the association between maternal analgesic use during pregnancy and risk of psychotic symptoms during adolescence. Schizophr. Res. 2011;126:220–225. doi: 10.1016/j.schres.2010.10.021. [DOI] [PubMed] [Google Scholar]
- Guo R., Hou W., Dong Y., Yu Z., Stites J., Weiner C.P. Brain injury caused by chronic fetal hypoxemia is mediated by inflammatory cascade activation. Reprod. Sci. 2010;17:540–548. doi: 10.1177/1933719110364061. [DOI] [PubMed] [Google Scholar]
- Haig D. Maternal–fetal interactions and MHC polymorphism. J. Reprod. Immunol. 1997;35:101–109. doi: 10.1016/s0165-0378(97)00056-9. [DOI] [PubMed] [Google Scholar]
- Hansen C.H.F., Nielsen D.S., Kverka M., Zakostelska Z., Klimesova K. Patterns of early gut colonization shape future immune responses of the host. PLoS ONE. 2012;7(3):e34043. doi: 10.1371/journal.pone.0034043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P.J. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- Harvey L., Burne T.H., McGrath J.J., Eyles D.W. Developmental vitamin D3 deficiency induces alterations in immune organ morphology and function in adult offspring. J. Steroid Biochem. Mol. Biol. 2010;121:239–242. doi: 10.1016/j.jsbmb.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Heijmans B.T., Tobi E.W., Stein A.D., Putter H., Blauw G.J., Susser E.S. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiby S.E., Walker J.J., O’Shaughnessy K.M., Redman C.W.G., Carrington, Trowsdale J. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J. Exp. Med. 2004;200:957–965. doi: 10.1084/jem.20041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti J., Riley D.E., Krohn M.A., Hillier S.L., Agnew K.J., Krieger J.N. Broad-spectrum bacterial rDNA polymerase chain reaction assay for detecting amniotic fluid infection among women in premature labor. Clin. Infect. Dis. 1997;24:1228–1232. doi: 10.1086/513669. [DOI] [PubMed] [Google Scholar]
- Hoek H.W., Brown A.S., Susser E. The Dutch famine and schizophrenia spectrum disorders. Soc. Psychiatry Psychiatr. Epidemiol. 1998;33:373–379. doi: 10.1007/s001270050068. [DOI] [PubMed] [Google Scholar]
- Ho B.C., Andreasen N.C., Nopoulos P., Arndt S., Magnotta V., Flaum M. Progressive structural brain abnormalities and their relationship to clinical outcome: a longitudinal magnetic resonance imaging study early in schizophrenia. Arch. Gen. Psychiatry. 2003;60:585–594. doi: 10.1001/archpsyc.60.6.585. [DOI] [PubMed] [Google Scholar]
- Holloway T., Moreno J.L., Umali A., Rayannavar V., Hodes G.E., Russo S.J. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: role of maternal immune system. J. Neurosci. 2013;33:1088–1098. doi: 10.1523/JNEUROSCI.2331-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper L.V., Gordon J.I. Commensal host–bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- Howerton C.L., Morgan C.P., Fischer D.B., Bale T.L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. U. S. A. 2013;110(March (13)):5169–5174. doi: 10.1073/pnas.1300065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao E.Y., Patterson P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011;25:604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.T. The link between perinatal glucocorticoids exposure and psychiatric disorders. Pediatr. Res. 2011;69(May (5 Pt 2)):19R–25R. doi: 10.1203/PDR.0b013e318212c29b. [DOI] [PubMed] [Google Scholar]
- Hulshoff Pol H.E., Hoek H.W., Susser E., Brown A.S., Dingemans A., Schnack H.G. Prenatal exposure to famine and brain morphology in schizophrenia. Am. J. Psychiatry. 2000;157:1170–1172. doi: 10.1176/appi.ajp.157.7.1170. [DOI] [PubMed] [Google Scholar]
- Ibi D., Nagai T., Koike H., Kitahara Y., Mizoguchi H., Niwa M. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav. Brain Res. 2010;206(January (1)):32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C., Kaindl A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal. 2011;14:1535–1550. doi: 10.1089/ars.2010.3581. [DOI] [PubMed] [Google Scholar]
- Isles A.R., Wilkinson L.S. Imprinted genes, cognition and behaviour. Trends Cogn. Sci. 2000;4:309–318. doi: 10.1016/s1364-6613(00)01504-7. [DOI] [PubMed] [Google Scholar]
- Ismail B., Cantor-Graae E., McNeil T.F. Minor physical anomalies in schizophrenia: cognitive, neurological and other clinical correlates. J. Psychiatr. Res. 2000;34:45–56. doi: 10.1016/s0022-3956(99)00034-5. [DOI] [PubMed] [Google Scholar]
- Jablensky A.V., Morgan V., Zubrick S.R., Bower C., Yellachich L.A. Pregnancy, delivery, and neonatal complications in a population cohort of women with schizophrenia and major affective disorders. Am. J. Psychiatry. 2005;162:79–91. doi: 10.1176/appi.ajp.162.1.79. [DOI] [PubMed] [Google Scholar]
- Jacka F.N., Ystrom E., Brantsaeter A.L., Karevold E., Roth C., Haugen M. Maternal and early postnatal nutrition and mental health of offspring by age 5 years: a prospective cohort study. J. Am. Acad. Child Adolesc. Psychiatry. 2013;52:1038–1047. doi: 10.1016/j.jaac.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Jacka F.N., Berk M. Prevention of schizophrenia – will a broader prevention agenda support this aim? Schizophr. Bull. 2014;40(March (2)):237–239. doi: 10.1093/schbul/sbt202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson J., Eaton W., Cascella N., Fasano A., Santora D., Sullivan K. Gluten sensitivity and relationship to psychiatric symptoms in people with schizophrenia. Schizophr. Res. 2014;159:539–542. doi: 10.1016/j.schres.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson T., Powell T.L. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin. Sci. (Lond.) 2007;113:1–13. doi: 10.1042/CS20060339. [DOI] [PubMed] [Google Scholar]
- Jensen P.C., Monk C., Champagne F.A. Epigenetic effects of prenatal stress on 11b-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS ONE. 2012;7(6):e39791. doi: 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiya M., Sato K., Silva M.J., Ouhara K., Do P.M., Shanmugam K.T. Hydrogen from intestinal bacteria is protective for Concanavalin A-induced hepatitis. Biochem. Biophys. Res. Commun. 2009;386:316–321. doi: 10.1016/j.bbrc.2009.06.024. [DOI] [PubMed] [Google Scholar]
- Kang D.W., Park J.G., Ilhan Z.E., Wallstrom G., Labaer J., Adams J.B. Reduced incidence of prevotella and other fermenters in intestinal microflora of autistic children. PLOS ONE. 2013;8(7):e68322. doi: 10.1371/journal.pone.0068322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Dunn E., Kostaki A., Andrews M.H., Matthews S.G. Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J. Physiol. 2006;572:31–44. doi: 10.1113/jphysiol.2006.105254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim P., Hossain M.I., Nazmus Sadat A.F., Nahar Z., Md Khalid Hossain M.K., Hasnat A. Serum levels of cadmium, calcium, lead and iron in schizophrenic patients. Dhaka Univ. J. Pharm. Sci. 2006;5:9–13. [Google Scholar]
- Katakura M. Hydrogen-rich water inhibits glucose and alpha,beta-dicarbonyl compound-induced reactive oxygen species production in the SHR.Cg-Leprcp/NDmcr rat kidney. Med. Gas Res. 2012;2:18. doi: 10.1186/2045-9912-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavan M.S. Development, disease and degeneration in schizophrenia: a unitary pathophysiological model. J. Psychiatr. Res. 1999;33:513–521. doi: 10.1016/s0022-3956(99)00033-3. [DOI] [PubMed] [Google Scholar]
- Khandaker G.M., Zimbron J., Lewis G., Jones P.B. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychol. Med. 2013;43:239–257. doi: 10.1017/S0033291712000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.K., Andreazza A.C. The relationship between oxidative stress and post-translational modification of the dopamine transporter in bipolar disorder. Expert Rev. Neurother. 2012;12:849–859. doi: 10.1586/ern.12.64. [DOI] [PubMed] [Google Scholar]
- Kim H.K., Andreazza A.C., Yeung P.Y., Issacs-Trepanier C., Young L.T. Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia. J. Psychiatry Neurosci. 2014;39(July (4)):276–285. doi: 10.1503/jpn.130155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen A.K., Koenig J.I., Bilbe G. Repeated variable prenatal stress alters pre- and postsynaptic gene expression in the rat frontal pole. J. Neurochem. 2003;86:736–748. doi: 10.1046/j.1471-4159.2003.01873.x. [DOI] [PubMed] [Google Scholar]
- Kirkbride J.B., Susser E., Kundakovic M., Kresovich J.K., Smith G.D., Relton C.L. Prenatal nutrition, epigenetics and schizophrenia risk: can we test causal effects? Epigenomics. 2012;2:303–315. doi: 10.2217/epi.12.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig J.I., Elmer G.I., Shepard P.D., Lee P.R., Mayo C., Joy B. Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav. Brain Res. 2005;156:251–261. doi: 10.1016/j.bbr.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Kong A., Frigge M.L., Masson G., Besenbacher S., Sulem P., Magnusson G., Gudjonsson S.A., Sigurdsson A., Jonasdottir A., Jonasdottir A., Wong W.S., Sigurdsson G., Walters G.B., Steinberg S., Helgason H., Thorleifsson G., Gudbjartsson D.F., Helgason A., Magnusson O.T., Thorsteinsdottir U., Stefansson K. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488(August (7412)):471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O., Goodrich J.K., Cullender T.C., Spor A., Laitinen K., Bäckhed H.K. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz A., Rosales-Corral S., Reiter R.J. Gene regulation by melatonin linked to epigenetic phenomena. Gene. 2012;503:1–11. doi: 10.1016/j.gene.2012.04.040. [DOI] [PubMed] [Google Scholar]
- Knickmeyer R.C., Styner M., Short S.J., Lubach G.R., Kang C., Hamer R. Maturational trajectories of cortical brain development through the pubertal transition: unique species and sex differences in the monkey revealed through structural magnetic resonance imaging. Cereb. Cortex. 2010;20:1053–1063. doi: 10.1093/cercor/bhp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopik V.S., Maccani M.A., Francazio S., McGeary J.E. The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev. Psychopathol. 2012;24(4):1377–1390. doi: 10.1017/S0954579412000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott V., Smith D., de la Salle S., Impey D., Choueiry J., Beaudry E., Smith M., Saghir S., Ilivitsky V., Labelle A. CDP-choline: effects of the procholine supplement on sensory gating and executive function in healthy volunteers stratified for low, medium and high P50 suppression. J. Psychopharmacol. 2014;28(December (12)):1095–1108. doi: 10.1177/0269881114553254. [DOI] [PubMed] [Google Scholar]
- Knuesel I., Chicha L., Britschgi M., Schobel S.A., Bodmer M., Hellings J.A., Toovey S., Prinssen E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014;10(November (11)):643–660. doi: 10.1038/nrneurol.2014.187. [DOI] [PubMed] [Google Scholar]
- Kulkarni J., Gurvich C., Lee S.J., Gilbert H., Gavrilidis E., de Castella A., Berk M. Piloting the effective therapeutic dose of adjunctive selective estrogen receptor modulator treatment in postmenopausal women with schizophrenia. Psychoneuroendocrinology. 2010;35:1142–1147. doi: 10.1016/j.psyneuen.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Kulkarni J., Gavrilidis E., Wang W., Worsley R., Fitzgerald P.B., Gurvich C., Van Rheenen T., Berk M., Burger H. Estradiol for treatment-resistant schizophrenia: a large-scale randomized-controlled trial in women of child-bearing age. Mol. Psychiatry. 2014 doi: 10.1038/mp.2014.33. (in press) [DOI] [PubMed] [Google Scholar]
- Lante F., Meunier J., Guiramand J., Maurice T., Cavalier M. Neurodevelopmental damage after prenatal infection: role of oxidative stress in the fetal brain. Free Radic. Biol. Med. 2007;42:1231–1245. doi: 10.1016/j.freeradbiomed.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Lawrie S.M., McIntosh A.M., Hall J., Owens D.G., Johnstone E.C. Brain structure and function changes during the development of schizophrenia: the evidence from studies of subjects at increased genetic risk. Schizophr. Bull. 2008;34:330–340. doi: 10.1093/schbul/sbm158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask S.J., Done D.J., Crow T.J. Adult psychosis, common childhood infections and neurological soft signs in a national birth cohort. Br. J. Psychiatry. 2002;181:837–892. doi: 10.1192/bjp.181.5.387. [DOI] [PubMed] [Google Scholar]
- Lemaire V., Koehl M., Le Moal M., Abrous D.N. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11032–11037. doi: 10.1073/pnas.97.20.11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard B.E. Impact of inflammation on neurotransmitter changes in major depression: an insight into the action of antidepressants. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2014;48(January):261–267. doi: 10.1016/j.pnpbp.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Lewis J.E., Melillo A.B., Tiozzo E., Chen L., Leonard S., Howell M., Diaz J., Gonzalez K., Woolger J.M., Konefal J., Paterson E., Barnes D. A double-blind, randomized clinical trial of dietary supplementation on cognitive and immune functioning in healthy older adults. BMC Complement Altern. Med. 2014;14(February):43. doi: 10.1186/1472-6882-14-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A.J., Galbally M., Gannon T., Symeonides C. Early life programming as a target for prevention of child and adolescent mental disorders. BMC Med. 2014;12(February (1)):33. doi: 10.1186/1741-7015-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light G.A., Swerdlow N.R., Rissling A.J., Radant A., Sugar C.A. Characterization of neurophysiologic and neurocognitive biomarkers for use in genomic and clinical outcome studies of schizophrenia. PLoS ONE. 2012;7(7):e39434. doi: 10.1371/journal.pone.0039434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N.Q., Kaplan A.T., Lagishetty V., Ouyang Y.B., Ouyang Y., Simmons C.F. Vitamin D and the regulation of placental inflammation. J. Immunol. 2011;186:5968–5974. doi: 10.4049/jimmunol.1003332. [DOI] [PubMed] [Google Scholar]
- Liu W., Chen O., Chen C., Wu B., Tang J., Zhang J.H. Protective effects of hydrogen on fetal brain injury during maternal hypoxia. Acta Neurochir. 2011;111:307–311. doi: 10.1007/978-3-7091-0693-8_51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipina T.V., Zai C., Hlousek D., Roder J.C., Wong A.H. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J. Neurosci. 2013;33(May (18)):7654–7666. doi: 10.1523/JNEUROSCI.0091-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin A., Bok C.M., Aronsson L., Björkholm B., Gustafsson J.A., Pott S. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell. Microbiol. 2008;10:1093–1103. doi: 10.1111/j.1462-5822.2007.01108.x. [DOI] [PubMed] [Google Scholar]
- Maes M., Kubera M., Leunis J.C., Berk M. Increased IgA and IgM responses against gut commensals in chronic depression: further evidence for increased bacterial translocation or leaky gut. J. Affect. Disord. 2012;141:55–62. doi: 10.1016/j.jad.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Maes M., Kubera M., Leunis J.C., Berk M., Geffard M., Bosmans E. In depression, bacterial translocation may drive inflammatory responses, oxidative and nitrosative stress (O&NS), and autoimmune responses directed against O&NS-damaged neoepitopes. Acta Psychiatr. Scand. 2013;127:344–354. doi: 10.1111/j.1600-0447.2012.01908.x. [DOI] [PubMed] [Google Scholar]
- Malaspina D., Corcoran C., Fahim C., Berman A., Harkavy-Friedman J., Yale S. Paternal age and sporadic schizophrenia: evidence for de novo mutations. Am. J. Med. Genet. 2002;114(April (3)):299–303. doi: 10.1002/ajmg.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina D., Corcoran C., Kleinhaus K.R., Perrin M.C., Fennig S. Acute maternal stress in pregnancy and schizophrenia in offspring: a cohort prospective study. BMC Psychiatry. 2008;8:71. doi: 10.1186/1471-244X-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques A.H., O’Connor T.G., Roth C., Susser E., Bjørke-Monsen A.L. The influence of maternal prenatal and early childhood nutrition and maternal prenatal stress on offspring immune system development and neurodevelopmental disorders. Front. Neurosci. 2013;7:120. doi: 10.3389/fnins.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisciano F., Tueting P., Dalal I., Kadriu B., Grayson D.R., Davis J.M., Nicoletti F., Guidotti A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68(May):184–194. doi: 10.1016/j.neuropharm.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard T.M., Sikich L., Lieberman J.A., LaMantia A.S. Neural development, cell–cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr. Bull. 2001;27(3):457–476. doi: 10.1093/oxfordjournals.schbul.a006887. [DOI] [PubMed] [Google Scholar]
- McAllister A.K. Major histocompatibility complex I in brain development and schizophrenia. Biol. Psychiatry. 2014;75(4):262–268. doi: 10.1016/j.biopsych.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann J.C., Hudes M., Ames B.N. An overview of evidence for a causal relationship between dietary availability of choline during development andcognitive function in offspring. Neurosci. Biobehav. Rev. 2006;30(5):696–712. doi: 10.1016/j.neubiorev.2005.12.003. [DOI] [PubMed] [Google Scholar]
- McGrath J.J., Gurne T.H., Feron F., Mackay-Sim A., Eyles D.W. Developmental vitamin D deficiency and risk of schizophrenia: a 10-year update. Schizophr. Bull. 2010;36:1073–1078. doi: 10.1093/schbul/sbq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J.J., Petersen L., Agerbo E., Mors O., Mortensen P.B., Pedersen C.B. A comprehensive assessment of parental age and psychiatric disorders. JAMA Psychiatry. 2014;71(3):301–309. doi: 10.1001/jamapsychiatry.2013.4081. [DOI] [PubMed] [Google Scholar]
- McNeil T.F., Cantor-Graae E., Ismail B. Obstetric complications and congenital malformation in schizophrenia. Brain Res. Brain Res. Rev. 2000;31:166–178. doi: 10.1016/s0165-0173(99)00034-x. [DOI] [PubMed] [Google Scholar]
- Meyer U., Feldon J., Dammann O. Schizophrenia and autism: both shared and disorder specific pathogenesis via perinatal inflammation? Pediatr. Res. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry. 2014;75(February (4)):307–315. doi: 10.1016/j.biopsych.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Miller B., Messias E., Miettunen J. Meta-analysis of paternal age and schizophrenia risk in male versus female offspring. Schizophr. Bull. 2011;37:1039–1047. doi: 10.1093/schbul/sbq011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.H., Zeier Z., Xi L., Lanz T.A., Deng S., Strathmann J. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc. Natl. Acad. Sci. U. S. A. 2012;109:3125–3130. doi: 10.1073/pnas.1113793109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B.R., Bale T.L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld K.A., Kang N., Bienenstock J., Foster J.A. Effects of intestinal microbiota on anxiety-like behavior. Commun. Integr. Biol. 2011;4:492–494. doi: 10.4161/cib.4.4.15702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodemus K.K., Marenco S., Batten A.J., Vakkalanka R., Egan M.F., Straub R.E. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol. Psychiatry. 2008;13:873–877. doi: 10.1038/sj.mp.4002153. [DOI] [PubMed] [Google Scholar]
- Nielsen P.R., Agerbo E., Skogstrand K., Hougaard D.M., Meyer U., Mortensen P.B. Neonatal levels of inflammatory markers and later risk of schizophrenia. Biol. Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.07.013. (in press) [DOI] [PubMed] [Google Scholar]
- Niethammer R., Weisbrod M., Schiesser S., Grothe J., Maier S., Peter U. Genetic influence on laterality in schizophrenia? A twin study of neurological soft signs. Am. J. Psychiatry. 2000;157(February (2)):272–274. doi: 10.1176/appi.ajp.157.2.272. [DOI] [PubMed] [Google Scholar]
- Nilsson E., Stålberg G., Lichtenstein P., Cnattingius S., Olausson P.O., Hultman C.M. Fetal growth restriction and schizophrenia: a Swedish twin study. Twin Res. Hum. Genet. 2005;8:402–408. doi: 10.1375/1832427054936727. [DOI] [PubMed] [Google Scholar]
- Ohta S. Recent progress toward hydrogen medicine: potential of molecular hydrogen for preventive and therapeutic applications. Curr. Pharm. Des. 2011;17:2241–2252. doi: 10.2174/138161211797052664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C. HLA and pregnancy: the paradox of the fetal allograft. Am. J. Hum. Genet. 1998;62:1–5. doi: 10.1086/301692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C., Hyslop T., Elias S., Weitkamp L.R., Hauck W.W. Human leukocyte antigen matching and fetal loss: results of a 10 year prospective study. Hum. Reprod. 1998;13:33–38. doi: 10.1093/humrep/13.1.33. [DOI] [PubMed] [Google Scholar]
- Odaka H., Numakawa T., Adachi N., Ooshima Y., Nakajima S., Katanuma Y. Cabergoline, dopamine D2 receptor agonist, prevents neuronal cell death under oxidative stress via reducing excitotoxicity. PLOS ONE. 2014;9(June (6)):e99271. doi: 10.1371/journal.pone.0099271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opler M.G., Buka S.L., Groeger J., McKeague I., Wei C., Factor-Litvak P. Prenatal exposure to lead, delta-aminolevulinic acid, and schizophrenia: further evidence. Environ. Health Perspect. 2008;116:1586–1590. doi: 10.1289/ehp.10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orisakwe O.E. The role of lead and cadmium in psychiatry. N. Am. J. Med. Sci. 2014;6(August (8)):370–376. doi: 10.4103/1947-2714.139283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K., Hashimoto K., Kishimoto T., Shimizu E., Ishikura H., Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol. Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Palmer C.G.S., Hsieh H.J., Reed E.F. HLA-B maternal–fetal genotype matching increases risk of schizophrenia. Am. J. Hum. Genet. 2006;79:710–715. doi: 10.1086/507829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C.G. Evidence for maternal–fetal genotype incompatibility as a risk factor for schizophrenia. J. Biomed. Biotechnol. 2010;2010:576318. doi: 10.1155/2010/576318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelis C., Yücel M., Wood S.J., Velakoulis D., Sun D., Berger G. Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophr. Bull. 2005;31:672–696. doi: 10.1093/schbul/sbi034. [DOI] [PubMed] [Google Scholar]
- Pazos M.A., Kraus T.A., Muñoz-Fontela C., Moran T.M. Estrogen mediates innate and adaptive immune alterations to influenza infection in pregnant mice. PLoS ONE. 2012;7(7):e40502. doi: 10.1371/journal.pone.0040502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrini M., Massuda R., Fries G.R., de Bittencourt Pasquali M.A., Schnorr C.E. Similarities in serum oxidative stress markers and inflammatory cytokines in patients with overt schizophrenia at early and late stages of chronicity. J. Psychiatr. Res. 2012;46(June (6)):819–824. doi: 10.1016/j.jpsychires.2012.03.019. [DOI] [PubMed] [Google Scholar]
- Perkins D.O., Jeffries C.D., Addington J., Bearden C.E., Cadenhead K.S., Cannon T.D. Towards a psychosis risk blood diagnostic for persons experiencing high-risk symptoms: preliminary results from the NAPLS project. Schizophr. Bull. 2014 doi: 10.1093/schbul/sbu099. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petanjek Z., Kostović I. Epigenetic regulation of fetal brain development and neurocognitive outcome. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11062–11063. doi: 10.1073/pnas.1208085109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R., Viana J., Hannon E., Spiers H.H., Troakes C., Al-Saraj S. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 2014;15(October (10)):483. doi: 10.1186/s13059-014-0483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M., Beneyto M., Burne T.H., Eyles D.W., Lewis D.A., McGrath J.J. The neurodevelopmental hypothesis of schizophrenia: convergent clues from epidemiology and neuropathology. Psychiatr. Clin. North Am. 2012;35:571–584. doi: 10.1016/j.psc.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Pun F.W., Zhao C., Lo W.S., Ng S.K., Tsang S.Y., Nimgaonkar V., Chung W.S., Ungvari G.S., Xue H. Imprinting in the schizophrenia candidate gene GABRB2 encoding GABA(A) receptor β(2) subunit. Mol. Psychiatry. 2011;16(May (5)):557–568. doi: 10.1038/mp.2010.47. [DOI] [PubMed] [Google Scholar]
- Quirk S.E., Williams L.J., O’Neil A., Pasco J.A., Jacka F.N., Housden S. The association between diet quality, dietary patterns and depression in adults: a systematic review. BMC Psychiatry. 2013;13:175. doi: 10.1186/1471-244X-13-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radogna F., Diederich M., Ghibelli L. Melatonin: a pleiotropic molecule regulating inflammation. Biochem. Pharmacol. 2010;80:1844–1852. doi: 10.1016/j.bcp.2010.07.041. [DOI] [PubMed] [Google Scholar]
- Richetto J., Calabrese F., Riva M.A., Meyer U. Prenatal immune activation induces maturation-dependent alterations in the prefrontal GABAergic transcriptome. Schizophr. Bull. 2014;40(March (2)):351–361. doi: 10.1093/schbul/sbs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N., Reich E.W., Wishnick M.M., McCarthy J.G. Spontaneous mutation and parental age in humans. Am. J. Hum. Genet. 1987;41(August (2)):218–248. [PMC free article] [PubMed] [Google Scholar]
- Roberts J.M., Cooper D.W. Pathogenesis and genetics of pre-eclampsia. Lancet. 2001;357:53–56. doi: 10.1016/s0140-6736(00)03577-7. [DOI] [PubMed] [Google Scholar]
- Romero E1, Guaza C., Castellano B., Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol. Psychiatry. 2010;15(April (4)):372–383. doi: 10.1038/mp.2008.44. [DOI] [PubMed] [Google Scholar]
- Ross R.G., Stevens K.E., Proctor W.R., Leonard S., Kisley M.A., Hunter S.K. Research review: cholinergic mechanisms, early brain development, and risk for schizophrenia. J. Child. Psychol. Psychiatry. 2010;51(5):535–549. doi: 10.1111/j.1469-7610.2009.02187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R.G., Hunter S.K., McCarthy L., Beuler J., Hutchison A.K., Wagner B.D. Perinatal choline effects on neonatal pathophysiology related to later schizophrenia risk. Am. J. Psychiatry. 2013;170(March (3)):290–298. doi: 10.1176/appi.ajp.2012.12070940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz A., Collado M.C., García-Valdés L., Segura M.T., Martín-Lagos J.A., Anjos T. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br. J. Nutr. 2010;104:83–92. doi: 10.1017/S0007114510000176. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R., van Os J., W M Steinbusch H., Schmitz C. Gene regulation by hypoxia and the neurodevelopmental origin of schizophrenia. Schizophr. Res. 2006;84:253–271. doi: 10.1016/j.schres.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R., van Os J., Esquivel G., Steinbusch H.W., Rutten B.P. An environmental analysis of genes associated with schizophrenia: hypoxia and vascular factors as interacting elements in the neurodevelopmental model. Mol. Psychiatry. 2012;17:1194–1205. doi: 10.1038/mp.2011.183. [DOI] [PubMed] [Google Scholar]
- Schlotz W., Phillips D.I. Fetal origins of mental health: evidence and mechanisms. Brain Behav. Immun. 2009;23:905–916. doi: 10.1016/j.bbi.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Schulz K.M., Andrud K.M., Burke M.B., Pearson J.N., Kreisler A.D., Stevens K.E. The effects of prenatal stress on Alpha4 Beta2 and Alpha7 hippocampal nicotinic acetylcholine receptor levels in adult offspring. Dev. Neurobiol. 2013;73:806–814. doi: 10.1002/dneu.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance E.G., Alaedini A., Yang S., Halling M., Gressitt K.L., Stallings C.R. Gastrointestinal inflammation and associated immune activation in schizophrenia. Schizophr. Res. 2012;138:48–53. doi: 10.1016/j.schres.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance E.G., Gressitt K.L., Stallings C.R., Origoni A.E., Khushalani S., Leweke F.M. Discordant patterns of bacterial translocation markers and implications for innate immune imbalances inschizophrenia. Schizophr. Res. 2013;148:130–137. doi: 10.1016/j.schres.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Yu H.M., Yuan T.M., Gu W.Z., Wu Y.D. Intrauterine infection induced oligodendrocyte injury and inducible nitric oxide synthase expression in the developing rat brain. J. Perinat. Med. 2007;35(3):203–209. doi: 10.1515/JPM.2007.058. [DOI] [PubMed] [Google Scholar]
- Shen Q., Li Z.Q., Sun Y., Wang T., Wan C.L., Li X.W. The role of pro-inflammatory factors in mediating the effects on the fetus of prenatal undernutrition: implications for schizophrenia. Schizophr. Res. 2008;99:48–55. doi: 10.1016/j.schres.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Sivkov S.T., Akabaliev V.H., Kaleva N.N. Comparative dermatoglyphic study of schizophrenic patients: evidence of the neurodevelopmental model of schizophrenia. Folia Med. (Plovdiv) 2009;51:25–30. [PubMed] [Google Scholar]
- Song C., Merali Z., Anisman H. Variations of nucleus accumbens dopamine and serotonin following systemic interleukin-1, interleukin-2 or interleukin-6 treatment. Neuroscience. 1999;88:823–836. doi: 10.1016/s0306-4522(98)00271-1. [DOI] [PubMed] [Google Scholar]
- Sørensen H.J., Mortensen E.L., Reinisch J.M., Mednick S.A. Association between prenatal exposure to analgesics and risk of schizophrenia. Br. J. Psychiatry. 2004;185:366–371. doi: 10.1192/bjp.185.5.366. [DOI] [PubMed] [Google Scholar]
- Stachowiak M.K., Kucinski A., Curl R., Syposs C., Yang Y., Narla S. Schizophrenia: a neurodevelopmental disorder – integrative genomic hypothesis and therapeutic implications from a transgenic mouse model. Schizophr. Res. 2013;143:367–376. doi: 10.1016/j.schres.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Stathopoulou A., Beratis I.N., Beratis S. Prenatal tobacco smoke exposure, risk of schizophrenia, and severity of positive/negative symptoms. Schizophr. Res. 2013;148(August (1–3)):105–110. doi: 10.1016/j.schres.2013.04.031. [DOI] [PubMed] [Google Scholar]
- St Clair D., Xu M., Wang P., Yu Y., Fang Y., Zhang F. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA. 2005;294:557–562. doi: 10.1001/jama.294.5.557. [DOI] [PubMed] [Google Scholar]
- Stubbs E.G., Ritvo E.R., Mason-Brothers A. Autism and shared parental HLA antigens. J. Am. Acad. Child Psychiatry. 1985;24:182–185. doi: 10.1016/s0002-7138(09)60445-3. [DOI] [PubMed] [Google Scholar]
- Taraz M., Khatami M.R., Dashti-Khavidaki S., Akhonzadeh S., Noorbala A.A., Ghaeli P., Taraz S. Sertraline decreases serum level of interleukin-6 (IL-6) in hemodialysis patients with depression: results of a randomized double-blind, placebo-controlled clinical trial. Int. Immunopharmacol. 2013;17(November (3)):917–923. doi: 10.1016/j.intimp.2013.09.020. [DOI] [PubMed] [Google Scholar]
- Thompson L.P., Al-Hasan Y. Impact of oxidative stress in fetal programming. J. Pregnancy. 2012;2012 doi: 10.1155/2012/582748. Article ID 582748, 8 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Rodriguez M., Lotfipour S., Leonard G., Perron M., Richer L., Veillette S. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am. J. Med. Genet. B: Neuropsychiatr. Genet. 2010;153B(October (7)):1350–1354. doi: 10.1002/ajmg.b.31109. [DOI] [PubMed] [Google Scholar]
- Toscano C.D., Hashemzadeh-Gargari H., McGlothan J.L., Guilarte T.R. Developmental Pb2+ exposure alters NMDAR subtypes and reduces CREB phosphorylation in the rat brain. Brain Res. Dev. Brain Res. 2002;139:217–226. doi: 10.1016/s0165-3806(02)00569-2. [DOI] [PubMed] [Google Scholar]
- Uchida T., Oki Y., Yanagawa Y., Fukuda A. A heterozygous deletion in the glutamate decarboxylase 67 gene enhances maternal and fetal stress vulnerability. Neurosci. Res. 2011;69:276–282. doi: 10.1016/j.neures.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Urakubo A., Jarskog L.F., Lieberman J.A., Gilmore J.H. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr. Res. 2001;47:27–36. doi: 10.1016/s0920-9964(00)00032-3. [DOI] [PubMed] [Google Scholar]
- Ueland P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2011;34(1):3–15. doi: 10.1007/s10545-010-9088-4. [DOI] [PubMed] [Google Scholar]
- Van Erp T.G., Saleh P.A., Rosso I.M. Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorder, their unaffected siblings, and healthy unrelated volunteers. Am. J. Psychiatry. 2002;159:1514–1520. doi: 10.1176/appi.ajp.159.9.1514. [DOI] [PubMed] [Google Scholar]
- Van Os J., Bak M., Hanssen M., Bijl R.V., de Graaf R., Verdoux H. Cannabis use and psychosis: a longitudinal population-based study. Am. J. Epidemiol. 2002;156(4):19–27. doi: 10.1093/aje/kwf043. [DOI] [PubMed] [Google Scholar]
- Van Lieshout R.J., Voruganti L.P. Diabetes mellitus during pregnancy and increased risk of schizophrenia in offspring: a review of the evidence and putative mechanisms. J. Psychiatry Neurosci. 2008;33:395–404. [PMC free article] [PubMed] [Google Scholar]
- van Vliet E., Eixarch E., Illa M., Arbat-Plana A., González-Tendero A. Metabolomics reveals metabolic alterations by intrauterine growth restriction in the fetal rabbit brain. PLOS ONE. 2013;8(5):e64545. doi: 10.1371/journal.pone.0064545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S., Venkatasubramanian G., Thirthalli J., Janakiramaiah N., Gangadhar B.N. Cerebellar and other neurological soft signs in antipsychotic-naïve schizophrenia. Acta Psychiatr. Scand. 2006;114(November (5)):352–356. doi: 10.1111/j.1600-0447.2006.00837.x. [DOI] [PubMed] [Google Scholar]
- Varambally S., Venkatasubramanian G., Gangadhar B.N. Neurological soft signs in schizophrenia – the past, the present and the future. Indian J. Psychiatry. 2012;54:73–80. doi: 10.4103/0019-5545.94653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubramanian G., Debnath M. The TRIPS (Toll-like receptors in immuno-inflammatory pathogenesis) hypothesis: a novel postulate to understand schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;44:301–311. doi: 10.1016/j.pnpbp.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Victora C.G., Adair L., Fall C., Hallal P.C., Martorell R., Richter L. Maternal and child undernutrition: consequences for adult health and human capital. Lancet. 2008;371:340–357. doi: 10.1016/S0140-6736(07)61692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis T., Parnavelas J.G. The role of serotonin in early cortical development. Dev. Neurosci. 2003;25:245–256. doi: 10.1159/000072272. [DOI] [PubMed] [Google Scholar]
- Vlcek P., Bob P., Raboch J. Sensory disturbances, inhibitory deficits, and the P50 wave in schizophrenia. Neuropsychiatr. Dis. Treat. 2014;10(July):1309–1315. doi: 10.2147/NDT.S64219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S., Weber L., Feldon J., Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J. Neurosci. 2010;30:1270–1287. doi: 10.1523/JNEUROSCI.5408-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S., Joodmardi E., Perlmann T., Ögren S.O., Feldon J., Meyer U. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J. Neurosci. 2012;32:436–451. doi: 10.1523/JNEUROSCI.4831-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker E. Developmentally moderated expressions of the neuropathology underlying schizophrenia. Schizophr. Bull. 1994;20:453–480. doi: 10.1093/schbul/20.3.453. [DOI] [PubMed] [Google Scholar]
- Wang D.S., Zurek A.A., Lecker I., Yu J., Abramian A.M., Avramescu S. Memory deficits induced by inflammation are regulated by α5-subunit-containing GABAA receptors. Cell Rep. 2012;2(September (3)):488–496. doi: 10.1016/j.celrep.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells P.G., McCallum G.P., Chen C.S., Henderson J.T., Lee C.J., Perstin J. Oxidative stress in developmental origins of disease: teratogenesis, neurodevelopmental deficits, and cancer. Toxicol. Sci. 2009;108:4–18. doi: 10.1093/toxsci/kfn263. [DOI] [PubMed] [Google Scholar]
- Wilkinson L.S., Davies W., Isles A.R. Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci. 2007;8:832–843. doi: 10.1038/nrn2235. [DOI] [PubMed] [Google Scholar]
- Winter C., Djodari-Irani A., Sohr R., Morgenstern R., Feldon J., Juckel G. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int. J. Neuropsychopharmacol. 2009;12:513–524. doi: 10.1017/S1461145708009206. [DOI] [PubMed] [Google Scholar]
- Woods B.T. Is schizophrenia a progressive neurodevelopmental disorder? Toward a unitary pathogenetic mechanism. Am. J. Psychiatry. 1998;155(December (12)):1661–1670. doi: 10.1176/ajp.155.12.1661. [DOI] [PubMed] [Google Scholar]
- Xie K., Yu Y., Huang Y., Zheng L., Li J., Chen H. Molecular hydrogen ameliorates lipopolysaccharide-induced acute lung injury in mice through reducing inflammation and apoptosis. Shock. 2012;37:548–555. doi: 10.1097/SHK.0b013e31824ddc81. [DOI] [PubMed] [Google Scholar]
- Xie B., Lin F., Peng L., Ullah K., Wu H., Qing H., Deng Y. Methylglyoxal increases dopamine level and leads to oxidative stress in SH-SY5Y cells. Acta Biochim. Biophys. Sin. (Shanghai) 2014;46(November (11)):950–956. doi: 10.1093/abbs/gmu094. [DOI] [PubMed] [Google Scholar]
- Xu T., Chan R.C.K., Compton M.T. Minor physical anomalies in patients with schizophrenia, unaffected first-degree relatives, and healthy controls: a meta-analysis. PLoS ONE. 2011;6(9):e24129. doi: 10.1371/journal.pone.0024129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T.M., Sun Y., Zhan C.Y., Yu H.M. Intrauterine infection/inflammation and perinatal brain damage: role of glial cells and Toll-like receptor signaling. J. Neuroimmunol. 2010;29:16–25. doi: 10.1016/j.jneuroim.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Zarogianni E1, Moorhead T.W., Lawrie S.M. Towards the identification of imaging biomarkers in schizophrenia, using multivariate pattern classification at a single-subject level. Neuroimage Clin. 2013;3(September):279–289. doi: 10.1016/j.nicl.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel S.H. Choline: critical role during fetal development and dietary requirements in adults. Annu. Rev. Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Li Z., Huang J., Yan C., Dazzan P., Pantelis C. Neurological soft signs are not “soft” in brain structure and functional networks: evidence from ALE meta-analysis. Schizophr. Bull. 2014;40(3):626–641. doi: 10.1093/schbul/sbt063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann K., van Phi V.D., Brase A., Phi-van L. Inhibition of serotonin transporter expression by C/EBPβ in LPS-activated macrophage cells (HD11) Innate Immun. 2014 doi: 10.1177/1753425914547434. pii:1753425914547434. [DOI] [PubMed] [Google Scholar]
- Zornberg G.L., Buka S.L., Tsuang M.T. Hypoxic-ischemia-related fetal/neonatal complications and risk of schizophrenia and other nonaffective psychoses: a 19-year longitudinal study. Am. J. Psychiatry. 2000;157:196–202. doi: 10.1176/appi.ajp.157.2.196. [DOI] [PubMed] [Google Scholar]
- Zucchi F.C.R., Yao Y., Ward I.D., Ilnytskyy Y., Olson D.M. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PLOS ONE. 2013;8(2):e56967. doi: 10.1371/journal.pone.0056967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman L., Rehavi M., Nachman R., Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]