

Abstract
The molecular analysis of disease pathogenesis in cattle has been limited by the lack of availability of tools to analyze both host and pathogen responses. These limitations are disappearing with the advent of methodologies such as microarrays that facilitate rapid characterization of global gene expression at the level of individual cells and tissues. The present review focuses on the use of microarray technologies to investigate the functional pathogenomics of infectious disease in cattle. We discuss a number of unique issues that must be addressed when designing both in vitro and in vivo model systems to analyze host responses to a specific pathogen. Furthermore, comparative functional genomic strategies are discussed that can be used to address questions regarding host responses that are either common to a variety of pathogens or unique to individual pathogens. These strategies can also be applied to investigations of cell signaling pathways and the analyses of innate immune responses. Microarray analyses of both host and pathogen responses hold substantial promise for the generation of databases that can be used in the future to address a wide variety of questions. A critical component limiting these comparative analyses will be the quality of the databases and the complete functional annotation of the bovine genome. These limitations are discussed with an indication of future developments that will accelerate the validation of data generated when completing a molecular characterization of disease pathogenesis in cattle.
Keywords: Bovine coronavirus, Bovine herpesvirus-1, Bovine rotavirus, CpG ODN, Functional genomics, Microarray, Microflora, Toll-like receptors
1. Introduction
Cattle are an economically important species with a wide variety of acute and chronic diseases that significantly impact on animal production and welfare. Models have been developed for many bovine infectious diseases and these models have been used to investigate host–pathogen interactions and immune responses in cattle. Many of these disease models also have substantial relevance to human infections but a paucity of research tools have limited the molecular characterization of disease pathogenesis and host responses to pathogens (Hein and Griebel, 2003). A variety of molecular and biological techniques now hold substantial promise for enhancing the scope of infectious disease research in cattle and providing the basis for effective interspecies comparison of disease pathogenesis. The present review focuses on the application of microarray technology to the investigation of disease pathogenesis in cattle. Future investigations with microbial microarrays will also be critical to understanding the evolutionary balance between host and pathogen.
A variety of tools have been developed over the last 10 years to facilitate large-scale analyses of gene expression at the level of individual cells, tissues, or whole organisms. The most commonly used tools at the present time include oligonucleotide microarrays (Fodor et al., 1993, Southern and Maskos, 1994), cDNA microarrays (Schena et al., 1995), and serial analysis of gene expression (Velculescu et al., 1995). Microarray technology has developed rapidly and numerous textbooks and reviews have been published addressing the critical issues of microarray experimental design (Yang et al., 2002), data analyses (Butte, 2002), and the application of microarray technology to investigate normal physiology (Barlow and Lockhart, 2002) and disease pathogenesis (Zamvil and Steinman, 2002). Microarrays have been developed for a wide variety of microbial pathogens, but the application of this technology to functional genomic studies in mammals has been limited primarily to mice and humans. A variety of commercial microarrays are now available for these two species and microarrays representing specific cell signaling pathways or biological functions are being used as routine tools to address hypotheses in basic research and clinical trials. However, there has been a substantial delay in the application of microarray technologies to the investigation of biological questions in species of veterinary importance.
A relatively small number of bovine microarray studies have been published during the last three years. These investigations used a variety of approaches to analyze gene expression in a wide variety of tissues, including PBMC (Yao et al., 2001, Burton et al., 2001, Hernandez et al., 2003, Coussens et al., 2002, Coussens et al., 2003, Coussens et al., 2004, Tao et al., 2004), PMN (Madsen et al., 2004), macrophages (Weiss et al., 2004), spleen (Band et al., 2002), brain (Band et al., 2002), placenta (Band et al., 2002), reproductive tissues (Robert et al., 2002; Dalbies-Tran and Mermillod, 2003, Bauersachs et al., 2003, Bauersachs et al., 2004, Ishiwata et al., 2003, Sirard et al., 2003, Evans et al., 2004), mammary gland (Suchyta et al., 2003a, Suchyta et al., 2003b), liver (Herath et al., 2004), and muscle (Reverter et al., 2003, Sudre et al., 2003). Microarray analyses of gene expression were used primarily to address a variety of questions relating to normal physiological processes, such as cell differentiation, pregnancy, lactation, and parturition. The majority of these studies were conducted using custom designed bovine cDNA microarrays but a number investigations explored the practicality of cross-species hybridization with human micro- or macroarrays (Robert et al., 2002; Dalbies-Tran and Mermillod, 2003, Sudre et al., 2003; and Hernandez et al., 2003). Cross-species hybridization was explored as a potential approach to circumvent a number of the problems associated with custom designed bovine cDNA microarrays, which included either relatively small numbers of well-annotated ESTs (Tao et al., 2004, Coussens et al., 2002) or much larger EST numbers with limited annotation (Ishiwata et al., 2003, Reverter et al., 2003, Bauersachs et al., 2004, Weiss et al., 2004). Thus, there is a substantial need to develop high-density bovine cDNA or oligomicroarrays for functional genomic studies in cattle.
The present review will focus on issues arising from the use of microarrays to characterize host–pathogen interactions. Presently, few microarray studies have been published which specifically focus on bovine responses to infectious agents (Coussens et al., 2002, Coussens et al., 2003, Coussens et al., 2004, Weiss et al., 2004). Band et al. (2002) developed a 3800 gene bovine microarray which was subsequently expanded to include a total of 7884 ESTs and is commercially available (http://www.pyxisgenomics.com). The annotation available for this microarray has been substantially improved using ProbeLynx (Roche et al., 2004) but the utility of this microarray is still limited by the presence of unannotated ESTs and while EST selection represented abroad range of cell function there was not a biased selection for genes associated with immune functions.
2. Experimental design for microarray investigations
Disease pathogenesis reflects a complex interaction between host, pathogen, and environmental factors that influence both host and pathogen responses. The potential for environmental factors to significantly alter host responses has always been a major concern for scientific investigators and a variety of experimental procedures have been used to control this variable. When using very sensitive methodology, such as microarray analyses, it is even more critical to control for the effects of experimental manipulation. This can apply to both isolated cells and the whole animal.
2.1. Cell isolation
Purified cell populations provide a unique opportunity to directly correlate changes in gene expression to specific cell populations, but when isolating or purifying cell populations steps should be taken to minimize the impact on gene expression. For example, culturing cells for 24–36 h prior to conducting an experiment will equilibrate gene expression (Lafleur et al., 2001). To understand the effects of cell isolation, bovine primary monocytes were purified by using anti-CD14 or anti-CD172a monoclonal antibodies (mAb) combined with magnetic activated cell sorting (MACS). This isolation methodology was selected since it provides rapid isolation of highly purified monocytes (>99.5%) and cell function and viability are reportedly preserved (Miltenyi Biotech, Instructions for Use: MiniMACS and MidiMACS). CD14 was selected as a target molecule because it lacks a transmembrane domain and mAb binding should not initiate cell signaling. In contrast, the CD172a molecule is an integral membrane protein, but it has been used extensively as a target for bovine monocyte isolation.
RNA was extracted from MACS purified monocytes immediately following isolation (Time 0) and after culture in medium (Aim-V + 2% FBS) for 4, 12, and 36 h. Microarray analysis (three biological and two technical replicates) was performed for each culture interval relative to Time 0. Bovine monocytes isolated with anti-CD172a or anti-CD14 showed extensive gene activation after 4 h in culture, but gene expression decreased sharply over time (Table 1 ). Monocytes isolated with anti-CD14 had 30% less gene activation at 4 h, but the number of differentially expressed genes did not decline to the same level as observed with CD172a selected monocytes cultured for 24 and 36 h. Thus, MACS isolation had a significant impact on monocyte gene expression and this effect was not eliminated by changing the surface molecule used for cell selection. Furthermore, a comparison of differentially expressed genes (Table 1) revealed no over-lap in the gene expression patterns for CD14+ and CD172A+ cells. This observation indicates that time-matched and cell specific controls may be advisable within each experiment.
Table 1.
Cell isolation influences gene expression changes
RNA collected from monocytes following culture for 0, 4, 12, and 36 h.
Differentially expressed genes (p < 0.05 and 2.0-fold change) were identified with a bovine microarray (7884 ESTs) relative to time 0.
MACS purified monocytes (>99.5% CD14+ or CD172a+) were isolated from the blood of three 6–8-month-old castrated male calves.
2.2. Animal models and commensal microflora
The surgical preparation of multiple, sterile intestinal “loops” has been used as a model for the analysis of mucosal immune responses (Gerdts et al., 2001) and to analyze differential gene expression induced by bovine rotavirus infection (Tatlow et al., 2000). Inflammation associated with the surgical creation of intestinal “loops” is expected to have a significant impact on gene expression. Furthermore, removal of microflora during “loop” preparation may also have an impact on host gene transcription. There is increasing evidence that both the mucosal immune system and mucosal epithelial cells have evolved in response to the presence of commensal microflora (Cummings et al., 2003; de Vrese and Schrezenmeir, 2002, Macpherson and Uhr, 2004) and these interactions may influence inflammation and disease in the gastrointestinal tract (Macpherson and Uhr, 2004, Miniello et al., 2003).
Before using the intestinal “loop” model for functional genomic analyses of host responses to enteric pathogens, we investigated the effects of surgical manipulation and the elimination of commensal microflora. RNA was isolated from intestinal “loop” tissue (Sample) and adjacent, intact intestine (Control) at 48 h (n = 3) and 12 days (n = 3) post-surgery. These within animal comparisons revealed that 2.3% (180/7884) of ESTs were significantly (p < 0.05 and 1.5-fold change) altered in expression levels at 48 h post-surgery and approximately 80% (143/180) of these genes were up-regulated (Aich et al., 2005). Differential gene expression revealed that a variety of cell processes, including apoptosis, cell cycle, and cell differentiation, had been significantly changed. The absence of genes involved in inflammation and cell recruitment may reflect the low abundance of relevant immune genes on the Pyxis microarray and, consequently, the effects of surgery on host gene expression may be under-estimated for the small intestine. Therefore, a similar experiment was repeated in young calves (n = 3) but this time the intestinal “loops” were either flushed with saline containing antibiotics (−microflora) or left intact (+microflora) and tissues were collected 24 h later. Microarray analyses of these paired tissue samples identified 5.1% (406/7884) genes with altered expression (p < 0.05 and 1.5-fold change) following antibiotic treatment to eliminate microflora. Thus, each experimental manipulation significantly altered host gene expression, confirming the importance of within animal controls.
The importance of commensal microflora for normal development and function of the mucosal epithelium and immune system was further investigated. To address this question, host responses to an enteric viral infection was analyzed in the presence or absence of commensal microflora. Again, paired intestinal “loops” (−microflora and +microflora) were prepared in young calves (n = 3) and both “loops” were injected with bovine coronavirus. Microarray analyses revealed that removal of ingesta and commensal microflora had a marked impact on host responses at 24 h post-infection (Fig. 1 A). Only 196 of the differentially expressed genes (p < 0.2) were common to both experimental systems and a more stringent analysis (p < 0.05) of the data reduced this number to 63. Furthermore, over 4-fold more genes were differentially expressed in the small intestine when coronavirus infection occurred in the presence of ingesta and commensal microflora. These observations provide substantial evidence for a significant interaction between intestinal microflora and the mucosal epithelium and immune system. Thus, surgical manipulations and microflora must be carefully considered when designing and performing functional genomic analyses of host–pathogen interactions at mucosal surfaces.
Fig. 1.
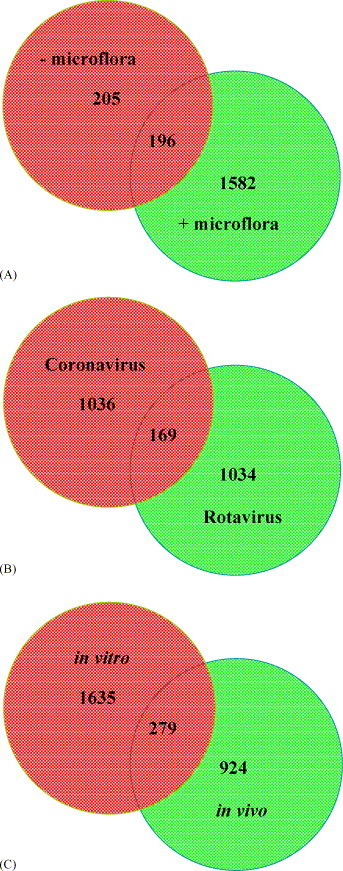
A Venn diagram showing the number of differentially expressed genes for studies on (A) effect of commensal microflora on host responses to enteric viral infection observed 24 h after corona infection in antibiotic flushed (−microflora) or in “loops” containing ingesta (+microflora), (B) comparative gene expression analyses between in vivo bovine rotavirus and coronavirus infections in young calves, (C) comparative gene expression analyses between in vivo and in vitro bovine rotavirus infections. The numbers in circles indicate the altered genes (p < 0.2 and 1.2-fold change) for each condition and those at the intersection indicate the genes common between conditions.
3. Host responses to pathogens
3.1. Enteric infections
Over 90% of all bovine pathogens invade through mucosal surfaces and the immune responses that contribute to either pathology or protection are unknown for most diseases. Tools, such as microarrays, have the potential to provide significant insight into the molecules that regulate the induction and differentiation of mucosal immune responses. Microarrays should facilitate the identification of host responses that are conserved among pathogens and help identify those responses unique to each pathogen.
This strategy was used to identify host immune responses to bovine rotaviruses and coronaviruses using the intestinal “loop” model. This model system can minimize effects due to genetic variability within an outbred population by comparing uninfected and infected intestinal tissues collected from the same animal. Data presented in Fig. 1B indicates that both RNA viruses induce differential expression of approximately 13% of the genes on the array, but even with lenient stringency (p < 0.2 and 1.2-fold change) over 90% of the differentially expressed genes were unique to each pathogen. Preliminary validation studies of these genes using quantitative real-time PCR (Table 2 ) indicate that approximately 75% of the genes tested by PCR concur with the microarray results and this validation efficiency remains relatively constant despite reducing the stringency of the significance cutoffs used for microarray analysis. This suggests that using a reduced stringency for initial microarray analysis may be an advantageous approach to maximize the identification of potential gene targets for further biological investigation.
Table 2.
Validation of microarray data with quantitative real time-PCR (qRT-PCR)
| Experimentsa | Differentially expressed genes |
|
|---|---|---|
| Microarrayb | Validated/selectedc (qRT-PCR) | |
| Effect of surgery | 180 (p < 0.05) | 17/25 |
| Bovine rotavirus | 1203 (p < 0.2) | 42/48 |
| Bovine coronavirus | 1205 (p < 0.2) | 18/26 |
Intestinal “loop” experiments with gene expression compared within the same animal.
The number of differentially expressed genes identified following microarray analyses (1.5-fold change) is indicated with the significance level in brackets.
The numerator represents the number of genes validated by qRT-PCR following selection of gene targets by microarray analysis (denominator).
The relation of microarray data to biology is complicated when using RNA isolated from tissues. For example, both rotavirus and coronavirus infection result in substantial destruction of mucosal epithelial cells. In addition, the immune cell populations in the intestine can change rapidly in response to infection. Thus, apparent changes in gene expression may simply reflect the altered abundance of mucosal epithelial cells or changes in the relative cellular composition of the mucosal immune system. One approach to tackling the limitations of studying gene expression in complex tissues is to study in parallel differential gene expression in a specific cell population in an in vitro model. Using this combined approach, a primary bovine intestinal epithelial cell line was developed to assess bovine rotavirus infection. Primary jejunal epithelial cell cultures were infected with bovine rotavirus and RNA was isolated at 16 h post-infection. Microarray analyses revealed that a total of 1914 genes were differentially expressed (p < 0.2 and 1.2-fold change) in the epithelial cells following viral infection (Fig. 1C; in vitro). More importantly, 279 of the differentially expressed genes were common to the set of genes identified in bovine rotavirus infected intestinal “loops” (Fig. 1C; in vivo). This is important since the in vitro model not only provides independent confirmation of the in vivo observations, but also provides evidence to link expression of specific genes to mucosal epithelial cells. Thus, a combination of in vitro and in vivo models can provide powerful tools to enhance functional genomic analyses and associate gene expression with specific cell types. Furthermore, the comparative analysis of gene expression induced by two or more enteric pathogens provides a strategy for the identification of conserved mucosal responses that may be of broad relevance to disease protection. These analyses should also reveal innate immune responses that play a pivotal role in determining the outcome of host–pathogen interactions.
3.2. Respiratory infections
A diverse number of viral and bacterial pathogens have been associated with bovine respiratory disease. Furthermore, the epidemiology of bovine respiratory disease in North America has been linked to environmental and nutritional changes, as well as husbandry procedures, with an increased incidence of respiratory disease at the time of weaning. These observations have implicated stress as a significant factor that can compromise host defenses and increase disease susceptibility. To date, it has been difficult to quantify stress responses and determine what adaptation period is required to eliminate the effects of stress when studying host responses to infectious disease. Microarray analyses, however, may be able to provide a more global view of the physiological responses influenced by stress.
The interplay between stress and disease resistance was investigated by using microarray analyses to profile gene expression in peripheral blood mononuclear cells (PBMC). Two groups of calves were housed in a single pen with one group of suckling calves (n = 8) removed from their dams 24 h prior to bovine herpesvirus-1 (BHV-1) infection (abrupt weaning) and a second group removed from their dams and fed hay and grain for 2 weeks prior to viral challenge (pre-conditioned). RNA was extracted from PBMC collected prior to infection and 4 days following BHV-1 respiratory infection and microarray analysis performed relative to a reference RNA (5 μg RNA aliquots pooled from all PBMC samples). Microarray analyses revealed that both groups of calves shared common transcriptional responses (22 ESTs; p < 0.05 and 1.5-fold change) on day 4 post-infection but. it was also apparent that abrupt weaning and pre-conditioning had a marked effect on host responses to a respiratory infection. There was almost 3-fold more changes in gene expression (39 ESTs) (p < 0.05 and 1.5-fold change) in calves that experienced the stress of abrupt weaning and transport immediately prior to infection. Thus, to minimize variability in gene expression it is critical that changes in management or diet be carefully controlled when performing functional genomic experiments. It is also apparent that microarray analyses may provide a powerful tool to determine how stress can compromise innate immunity and disease resistance.
4. Host responses to pathogen-associated molecules
Innate immunity plays a critical role in host responses to infectious disease and there is increasing evidence that innate immunity also influences adaptive immune responses. Furthermore, it has become apparent that the activation of many innate immune responses is regulated by pattern recognition receptors (PRR) that recognize specific structural motifs of pathogen-associated molecules. An important family of PRR is the toll-like receptors (TLR) which recognize a wide variety of viral, bacterial and fungal molecules. Understanding the role of TLR in the regulation of innate immunity is critical for a full understanding of host responses to infectious disease. Microarray analysis of differential gene expression following exposure of dendritic cells to specific pathogens and pathogen-associated molecules has been an effective approach to understanding the induction of both innate and adaptive immune responses (Huang et al., 2001).
Lipopolysaccharide (LPS) and non-methylated CpG motifs in bacterial DNA are pathogen-associated molecules which activate innate immune responses through interactions with TLR4 and TLR9, respectively. LPS triggers monocyte activation by binding to LPS-binding protein (LBP) and this complex associates with CD14. The LPS–CD14 complex then binds to TLR4 which initiates the intracellular signaling that leads to an inflammatory response (Hemmi et al., 2000). Synthetic CpG oligodeoxynucleotides (CpG ODN) can be used as TLR9 ligands to activate immune cells, including NK cells, monocytes, macrophages, dendritic cells and B cells (Krieg et al., 1995, Takeshita et al., 2001). To determine how these cells contribute to the induction of innate immunity it is important to understand the consequences of TLR signaling for each leukocyte population.
To identify common and unique TLR signaling pathways that may result in the activation of unique cellular functions we used bovine microarrays to profile gene expression in bovine monocytes following LPS (TLR4) and class B CpG ODN (TLR9) stimulation. Microarray analyses identified a large number of transcriptionally activated genes following LPS or CpG ODN stimulation. Approximately 30 common genes were up-regulated in the presence of LPS or CpG ODN but the majority of the differentially expressed genes were unique to each of the stimuli. Thus, microarrays provide an effective tool to analyze cell signaling in response to specific ligands and to determine how families of related molecules can induce distinct cellular responses.
5. Future analyses of disease pathogenesis
The advent of the microarray, with its high throughput analyses has revolutionized the analysis of whole cell gene expression and disease studies (Ball et al., 2002, DeRisi et al., 1996, DeRisi et al., 2000, McKendry et al., 2002, Schena, 1996, Schena et al., 1996, Schena et al., 1998, Stears et al., 2003). Traditional microarray hybridization techniques are based on fluorescence, but this technique is limited by low sensitivity, high background signal, quenching, and photobleaching (Ball et al., 2002, Pan et al., 2002, Wang et al., 2002). Emerging techniques such as Resonance Light Scattering (RLS) and Near Infrared Imaging are gaining popularity in both research and clinical application of microarray detection due to increased sensitivity and robustness of data (Bao et al., 2002, Waddell et al., 2000). The RLS technique has several advantages over fluorescence based techniques for microarray studies, including increased sensitivity, better signal to noise ratio, no photobleaching (so arrays can be repeatedly scanned), and no need for dye swapping. RLS was used throughout our microarray analyses and resulted in a substantially increased detection of differentially expressed genes when compared to fluorescence (Fig. 2 ). Thus, there is substantial capacity to improve functional genomic analyses at the level of microarray detection of differentially expressed genes, especially when RNA is of limited quantity.
Fig. 2.
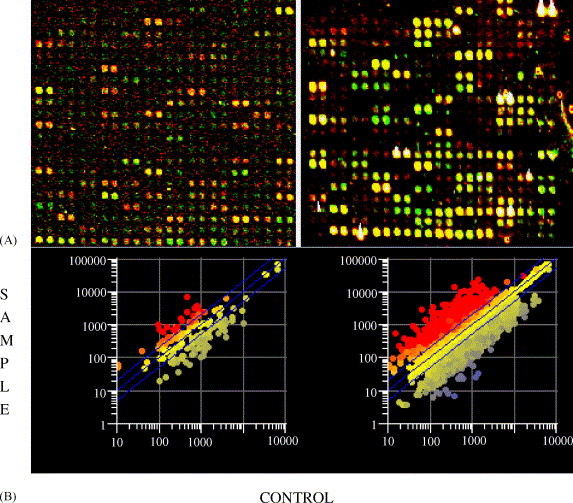
Comparison of fluorescence and RLS microarray technology. (A) Partial Tiff images of the same microarray grids after scanning hybridized bovine microarrays are shown in pseudo colors. Fluorescence scanned arrays were hybridized with Cy3/Cy5 dye labeled ‘control’ and ‘sample’ cDNA, respectively (left panel) and RLS scanned arrays were hybridized with silver/gold labeled ‘control’ and ‘sample’ cDNA, respectively (right panel). The amount of total RNA used for fluorescence and RLS techniques was 5 and 1 μg, respectively. (B) Scatter plot of processed and normalized intensities for gene expression as observed in fluorescence (left panel) and RLS analysis (right panel). Lines above and below the center line denote the limits for 2-fold up or down regulation of gene expression and points between these lines are considered unchanged between ‘Control’ and ‘Sample’.
Molecular diagnostic tests typically analyze key protein, DNA, or RNA markers to identify specific diseases. Immunohistochemistry, Western blotting, DNA sequencing, Southern and Northern blotting, and (quantitative) polymerase chain reaction are molecular techniques that have been effectively used in clinical diagnostic laboratories. However, molecular tests that involve the simultaneous analysis of tens to hundreds of markers within a clinical sample will soon be required to allow maximum translation of post-genomic research to patient care. This is particularly important today for the rapid identification of biomarkers and potential therapeutic targets for emerging diseases such as SARS and West Nile. However, because of the inherent variability in microarray technologies a global standard may be needed to facilitate the comparison of results among laboratories. This will also be critical if we are to realize the huge biological potential of performing interspecies comparative functional genomics.
While microarrays represent a powerful technology to interrogate bovine gene expression in response to disease pathogenesis, the quality of the microarray analysis depends heavily upon accurate, high quality annotation of the array probes. Annotation should include both assessment of the specificity of a given probe sequence for a given transcript (to detect cases of cross-hybridization), as well as high quality information about the transcripts themselves. This annotation is critical in order to extract meaningful biological data from microarray signal intensity measurements. However, in the absence of a complete bovine genome sequence, the availability of accurate gene prediction and annotation data is limited.
ProbeLynx (Roche et al., 2004) is one bioinformatics tool that has been developed to tackle the need for improved annotation. ProbeLynx is a microarray annotation update tool which allows researchers to continuously assess the specificity of their probes and ensure up-to-date gene annotations by using the latest sequence and annotation data available for a particular species. By exploiting the TIGR Gene Index and Eukaryotic Gene Orthologs (EGO) databases (Quackenbush et al., 2001), ProbeLynx allows users to map their probe sequences to predicted bovine transcript sequences and use interspecies information to link putative orthologs to uncharacterized transcript sequences for inference of gene function. This integration of resources makes ProbeLynx a powerful tool for bovine microarray researchers, as it ensures up-to-date, accurate links between probe sequences and the latest, enhanced gene annotation information for biological interpretation. In the future, additional integration of meta-data in probe annotations including protein subcellular localization predictions or pathway information (for both signaling and metabolic networks), will enable improved higher order bioinformatics analyses. Furthermore, a complete bovine genome sequence will be critical to future bioinformatic and laboratory studies in the identification of gene regulatory sequences. The integrated use of high-throughput analyses of protein and metabolite profiles may also provide an effective approach to accelerate validation of gene prediction and gene expression as well as to predict the dynamics of systems in a more definitive way.
The continuous development of bioinformatics approaches for improved array annotation combined with new data analysis tools that enable cross-species comparisons (Hokamp et al., 2004) will greatly enhance the extraction of biological information from bovine microarrays and advance our understanding of disease pathogenesis in cattle.
Acknowledgements
This manuscript is published with permission of the Director of VIDO as journal series #376 and the work was supported by a grant from Genome Canada with funding provided by Pyxis Genomics (USA) and Inimex Pharmaceuticals Inc. (Canada) through Genome Prairie (Canada). HLW is supported by a post-doctoral Fellowship (Saskatchewan Health Research Foundation, Canada). FSLB is a Michael Smith Foundation for Health Research Scholar, and LAB is a holder of the Canada Research Chair in Vaccinology.
References
- Aich, P., Wilson, H.L., Rawlyk, N., Jalal, S., Kaushik, R.S., Begg, A.A., Potter, A., Babiuk, L.A., Abrahamsen, M.S., Griebel, P., 2005. Microarray analysis of gene expression following preparation of sterile intestinal “loops” in calves. Can. J. Anim. Sci., in press.
- Ball C.A., Sherlock G., Parkinson H., Rocca-Sera P., Brooksbank C., Causton H.C., Cavalieri D., Gaasterland T., Hingamp P., Holstege F., Ringwald M., Spellman P., Stoeckert C.J., Jr., Stewart J.E., Taylor R., Brazma A., Quackenbush J. Standards for microarray data. Science. 2002;298(5593):539. doi: 10.1126/science.298.5593.539b. [DOI] [PubMed] [Google Scholar]
- Band M.R., Olmstead C., Everts R.E., Liu Z.L., Lewin H.A. A 3800 gene microarray for cattle functional genomics: comparison of gene expression in spleen, placenta, and brain. Anim. Biotechnol. 2002;13(1):163–172. doi: 10.1081/ABIO-120005779. [DOI] [PubMed] [Google Scholar]
- Bao P., Frutos A.G., Greef C., Lahiri J., Muller U., Peterson T.C., Warden L., Xie X. High-sensitivity detection of DNA hybridization on microarrays using resonance light scattering. Anal. Chem. 2002;74(8):1792–1797. doi: 10.1021/ac0111964. [DOI] [PubMed] [Google Scholar]
- Barlow C., Lockhart D.J. DNA arrays and neurobiology—what's new and what's next? Curr. Opin. Neurobiol. 2002;12(5):554–561. doi: 10.1016/s0959-4388(02)00353-7. [DOI] [PubMed] [Google Scholar]
- Bauersachs S., Blum H., Mallok S., Wenigerkind H., Rief S., Prelle K., Wolf E. Regulation of ipsilateral and contralateral bovine oviduct epithelial cell function in the postovulation period: a transcriptomics approach. Biol. Reprod. 2003;68:1170–1177. doi: 10.1095/biolreprod.102.010660. [DOI] [PubMed] [Google Scholar]
- Bauersachs S., Rehfeld S., Ulbrich S.E., Mallok S., Prelle K., Wenigerkind H., Einspanier R., Blum H., Wolf E. Monitoring gene expression changes in bovine oviduct epithelial cells during the oestrus cycle. J. Mol. Endocrinol. 2004;32(2):449–466. doi: 10.1677/jme.0.0320449. [DOI] [PubMed] [Google Scholar]
- Burton J.L., Madsen S.A., Yao J., Sipkovsky S.S., Coussens P.M. An immunogenomics approach to understanding periparturient immunosuppression and mastitis susceptibility in dairy cows. Acta Vet. Scand. 2001;42(3):407–424. [PubMed] [Google Scholar]
- Butte A. The use and analysis of microarray data. Nat. Rev. Drug. Discov. 2002;1(12):951–960. doi: 10.1038/nrd961. [DOI] [PubMed] [Google Scholar]
- Coussens P.M., Colvin C.J., Wiersma K., Abouzied A., Sipkovsky S. Gene expression profiling of peripheral blood mononuclear cells from cattle infected with Mycobacterium paratuberculosis. Infect. Immun. 2002;70(10):5494–5502. doi: 10.1128/IAI.70.10.5494-5502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens P.M., Colvin C.J., Rosa G.J.M., Laspiur J.P., Elftman M.D. Evidence for a novel gene expression program in peripheral blood mononuclear cells from Mycobacterium avium subsp. paratuberculosis-infected cattle. Infect. Immun. 2003;71(11):6487–6498. doi: 10.1128/IAI.71.11.6487-6498.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens P.M., Jeffers A., Colvin C. Rapid and transient activation of gene expression in peripheral blood mononuclear cells from Johne's disease positive cows exposed to Mycobacterium paratuberculosis in vitro. Microb. Pathog. 2004;36:93–108. doi: 10.1016/j.micpath.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Cummings J.H., Macfarlane G.T., Macfarlane S. Intestinal bacteria and ulcerative colitis. Curr. Issues Intest. Microbiol. 2003;4(1):9–20. [PubMed] [Google Scholar]
- Dalbies-Tran R., Mermillod P. Use of heterologous complementary DNA array screening to analyze bovine oocyte transcriptome and its evolution during in vitro maturation. Biol. Reprod. 2003;68:252–261. doi: 10.1095/biolreprod.102.007872. [DOI] [PubMed] [Google Scholar]
- DeRisi J., Penland L., Brown P.O., Bittner M.L., Meltzer P.S., Ray M., Chen Y., Su Y.A., Trent J.M. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 1996;14(4):457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- DeRisi J., van den Hazel B., Marc P., Balzi E., Brown P., Jacq C., Goffeau A. Genome microarray analysis of transcriptional activation in multidrug resistance yeast mutants. FEBS Lett. 2000;470(2):156–160. doi: 10.1016/s0014-5793(00)01294-1. [DOI] [PubMed] [Google Scholar]
- de Vrese M., Schrezenmeir J. Probiotics and non-intestinal infectious conditions. Br. J. Nutr. 2002;88(Suppl. 1):S59–S66. doi: 10.1079/BJN2002630. [DOI] [PubMed] [Google Scholar]
- Evans A.C., Ireland J.L., Winn M.E., Lonergan P., Smith G.W., Coussens P.M., Ireland J.J. Identification of genes involved in apoptosis and dominant follicle development during follicular waves in cattle. Biol. Reprod. 2004;70(5):1475–1484. doi: 10.1095/biolreprod.103.025114. [DOI] [PubMed] [Google Scholar]
- Fodor S.P., Rava R.P., Huang X.C., Pease A.C., Holmes C.P., Adams C.L. Multiplexed biochemical assays with biological chips. Nature. 1993;364(6437):555–556. doi: 10.1038/364555a0. [DOI] [PubMed] [Google Scholar]
- Gerdts V., Uwiera R.R., Mutwiri G.K., Wilson D.J., Bowersock T., Kidane A., Babiuk L.A., Griebel P.J. Multiple intestinal ‘loops’ provide an in vivo model to analyse multiple mucosal immune responses. J. Immunol. Methods. 2001;256(1–2):19–33. doi: 10.1016/s0022-1759(01)00429-x. [DOI] [PubMed] [Google Scholar]
- Hein W.R., Griebel P.J. A road less travelled: large animal models in immunological research. Nat. Rev. Immunol. 2003;3(1):79–84. doi: 10.1038/nri977. [DOI] [PubMed] [Google Scholar]
- Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Herath C.B., Shiojima S., Ishiwata H., Katsuma S., Kadowaki T., Ushizawa K., Imai K., Takahashi T., Hirasawa A., Tsujimoto G., Hashizume K. Pregnancy-associated changes in genome-wide gene expression profiles in the liver of cow throughout pregnancy. Biochem. Biophys. Res. Commun. 2004;313(3):666–680. doi: 10.1016/j.bbrc.2003.11.151. [DOI] [PubMed] [Google Scholar]
- Hernandez A., Karrow N., Mallard B.A. Evaluation of immune responses of cattle as a means to identify high and low responders and use of a human microarray to differentiate gene expression. Gene Sel. Evol. 2003;35(Suppl. 1):S67–S81. doi: 10.1186/1297-9686-35-S1-S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokamp K., Roche F.M., Acab M., Rousseau M.E., Kuo B., Goode D., Aeschliman D., Bryan J., Babiuk L.A., Hancock R.E., Brinkman F.S. ArrayPipe: a flexible processing pipeline for microarray data. Nucleic Acids Res. 2004;32(Web Server issue):W457–W459. doi: 10.1093/nar/gkh446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Liu D., Majewski P., Schulte L.C., Korn J.M., Young R.A., Lander E.S., Hacohen N. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294(5543):870–875. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- Ishiwata H., Katsuma S., Kizakai K., Patel O.V., Nakano H., Takahashi T., Imai K., Hirasawa A., Shojima A., Ikawa H., Suzuki Y., Tsujimoto G., Izaike Y., Todoroki J., Hashizume K. Characterization of gene expression profiles in early bovine pregnancy using a custom cDNA microarray. Mol. Reprod. Dev. 2003;65(1):9–18. doi: 10.1002/mrd.10292. [DOI] [PubMed] [Google Scholar]
- Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., Koretzky G.A., Klinman D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374(6522):546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- Lafleur R.L., Malazdrewich C., Jeyaseelan S., Bleifield E., Abrahamsen M.S., Maheswaran S.K. Lipopolysaccharide enhances cytolysis and inflammatory cytokine induction in bovine alveolar macrophages exposed to Pasteurella (Mannheimia) haemolytica leukotoxin. Microb. Pathog. 2001;30(6):347–357. doi: 10.1006/mpat.2000.0438. [DOI] [PubMed] [Google Scholar]
- Macpherson A.J., Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303(5664):1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- Madsen S.A., Chang L.C., Hickey M.C., Rosa G.J., Coussens P.M., Burton J.L. Microarray analysis of gene expression in blood neutrophils of parturient cows. Physiol. Genomics. 2004;16(2):212–221. doi: 10.1152/physiolgenomics.00121.2003. [DOI] [PubMed] [Google Scholar]
- McKendry R., Zhang J., Arntz Y., Strunz T., Hegner M., Lang H.P., Baller M.K., Certa U., Meyer E., Guntherodt H.J., Gerber C. Multiple label-free biodetection and quantitative DNA-binding assays on a nanomechanical cantilever array. Proc. Natl. Acad. Sci. USA. 2002;99(15):9783–9788. doi: 10.1073/pnas.152330199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniello V.L., Moro G.E., Armenio L. Prebiotics in infant milk formulas: new perspectives. Acta Paediatr. Suppl. 2003;91(441):68–76. doi: 10.1111/j.1651-2227.2003.tb00649.x. [DOI] [PubMed] [Google Scholar]
- Pan K.H., Lih C.J., Cohen S.N. Analysis of DNA microarrays using algorithms that employ rule-based expert knowledge. Proc. Natl. Acad. Sci. USA. 2002;99(4):2118–2123. doi: 10.1073/pnas.251687398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J., Cho J., Lee D., Liang F., Holt I., Karamycheva S., Parvizi B., Pertea G., Sultana R., White J. The TIGR Gene Indices: analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 2001;29:159–164. doi: 10.1093/nar/29.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverter A., Byrne K.A., Brucet H.L., Wang Y.H., Dairymple B.P., Lehnert S.A. A mixture model-based cluster analysis of DNA microarray gene expression data on Brahman and Brahman composite steers fed high-, medium-, and low-quality diets. J. Anim. Sci. 2003;81(8):1900–1901. doi: 10.2527/2003.8181900x. [DOI] [PubMed] [Google Scholar]
- Robert C., Hue I., McGraw S., Gagne D., Sirard M.-A. Quantification of Cyclin B1 and p34cdc2 in bovine cumulus–oocyte complexes and expression mapping of genes involved in the cell cycle by complementary DNA macroarrays. Biol. Reprod. 2002;67:1456–1464. doi: 10.1095/biolreprod.102.002147. [DOI] [PubMed] [Google Scholar]
- Roche F.M., Hokamp K., Acab M., Babiuk L.A., Hancock R.E., Brinkman F.S. ProbeLynx: a tool for updating the association of microarray probes to genes. Nucleic Acids Res. 2004;32(Web Server issue):W471–W474. doi: 10.1093/nar/gkh452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M. Genome analysis with gene expression microarrays. Bioessays. 1996;18(5):427–431. doi: 10.1002/bies.950180513. [DOI] [PubMed] [Google Scholar]
- Schena M., Heller R.A., Theriault T.P., Konrad K., Lachenmeier E., Davis R.W. Microarrays: biotechnology's discovery platform for functional genomics. Trends Biotechnol. 1998;16(7):301–306. doi: 10.1016/s0167-7799(98)01219-0. [DOI] [PubMed] [Google Scholar]
- Schena M., Shalon D., Davis R.W., Brown P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schena M., Shalon D., Heller R., Chai A., Brown P.O., Davis R.W. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc. Natl. Acad. Sci. USA. 1996;93(20):10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirard M.A., Dufort I., Coenen K., Tremblay K., Massicotte L., Robert C. The use of genomics and proteomics to understand oocyte and early embryo functions in farm animals. Reprod. Suppl. 2003;61:117–129. [PubMed] [Google Scholar]
- Southern E.M., Maskos U. Parallel synthesis and analysis of large numbers of related chemical compounds: applications to oligonucleotides. J. Biotechnol. 1994;35(2–3):217–227. doi: 10.1016/0168-1656(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Stears R.L., Martinsky T., Schena M. Trends in microarray analysis. Nat. Med. 2003;9(1):140–145. doi: 10.1038/nm0103-140. [DOI] [PubMed] [Google Scholar]
- Suchyta S.P., Sipkovsky S., Halgren R.G., Kruska R., Elftman M., Weber-Nielsen M., Vandehaar M.J., Xiao L., Tempelman R.J., Coussens P.M. Bovine mammary gene expression profiling using cDNA microarray enhanced mammary-specific transcripts. Physiol. Genomics. 2003;161(1):8–18. doi: 10.1152/physiolgenomics.00028.2003. [DOI] [PubMed] [Google Scholar]
- Suchyta S.P., Sipkovsky S., Kruska R., Jeffers A., McNulty A., Coussens M.J., Tempelman R.J., Halgren R.G., Saama P.M., Bauman D.E., Boisclair Y.R., Burton J.L., Collier R.J., DePeters E.J., Ferris T.A., Lucy M.C., McGuire M.A., Medrano J.F., Overton T.R., Smith T.P., Smith G.W., Sonstegard T.S., Spain J.N., Spiers D.E., Yao J., Coussens P.M. Development and testing of a high-density cDNA microarray resource for cattle. Physiol. Genomics. 2003;15(2):158–164. doi: 10.1152/physiolgenomics.00094.2003. [DOI] [PubMed] [Google Scholar]
- Sudre K., Leroux C., Pietu G., Cassar-Malek I., Petit E., Listrat A., Auffray C., Picard B., Martin P., Hocquette J.-F. Transciptome analysis of two bovine muscles during ontogensis. J. Biochem. 2003;133:745–756. doi: 10.1093/jb/mvg096. [DOI] [PubMed] [Google Scholar]
- Takeshita F., Leifer C.A., Gursel I., Ishii K.J., Takeshita S., Gursel M., Klinman D.M. Cutting edge: role of toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 2001;167(7):3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- Tao W., Mallard B., Karrow N., Bridle B. Construction and application of a bovine immune-endocrine cDNA microarray. Vet. Immunol. Immunopathol. 2004;101:1–17. doi: 10.1016/j.vetimm.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Tatlow D., Brownlie R., Babiuk L.A., Griebel P. Differential display analysis of gene expression during the induction of mucosal immunity. Immunogenetics. 2000;52(1–2):73–80. doi: 10.1007/s002510000253. [DOI] [PubMed] [Google Scholar]
- Velculescu V.E., Zhang L., Vogelstein B., Kinzler K.W. Serial analysis of gene expression. Science. 1995;270(5235):484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Waddell E., Wang Y., Stryjewski W., McWhorter S., Henry A.C., Evans D., McCarley R.L., Soper S.A. High-resolution near-infrared imaging of DNA microarrays with time-resolved acquisition of fluorescence lifetimes. Anal. Chem. 2000;72(24):5907–5917. doi: 10.1021/ac0009705. [DOI] [PubMed] [Google Scholar]
- Wang D., Coscoy L., Zylberberg M., Avila P.C., Boushey H.A., Ganem D., DeRisi J.L. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. USA. 2002;99(24):15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss D.J., Evanson O.A., Deng M., Abrahamsen M.S. Gene expression and antimicrobial activity of bovine macrophages in response to Mycobacterium avium subsp. paratuberculosis. Vet. Pathol. 2004;41(4):326–337. doi: 10.1354/vp.41-4-326. [DOI] [PubMed] [Google Scholar]
- Yang, I.V., Chen, E., Hasseman, J.P., Liang, W., Frank, B.C., Wang, S., Sharov, V., Saeed, A.I., White, J., Li, J., Lee, N.H., Yeatman, T.J., Quackenbush, J., 2002. Within the fold: assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 3 (11), research0062. [DOI] [PMC free article] [PubMed]
- Yao J., Burton J.L., Saama P., Sipkovsky S., Coussens P.M. Generation of EST and cDNA microarray resources for the study of bovine immunobiology. Acta Vet. Scand. 2001;42(3):391–405. [PubMed] [Google Scholar]
- Zamvil S.S., Steinman L. Cholesterol-lowering statins possess anti-inflammatory activity that might be useful for treatment of MS. Neurology. 2002;59(7):970–971. doi: 10.1212/wnl.59.7.970. [DOI] [PubMed] [Google Scholar]