

Highlights
-
•
MASP-1 and MASP-2 are central players of the lectin pathway of complement.
-
•
MASP1 and MASP2 gene polymorphisms regulate protein serum levels and activity.
-
•
MASP deficiencies are associated with increased infection susceptibility.
-
•
MASP polymorphisms and serum levels are associated with disease progression.
Keywords: MASP-1, MASP-2, MASP-3, MAp44, MAp19, Complement system
Abstract
The lectin pathway of the complement system has a pivotal role in the defense against infectious organisms. After binding of mannan-binding lectin (MBL), ficolins or collectin 11 to carbohydrates or acetylated residues on pathogen surfaces, dimers of MBL-associated serine proteases 1 and 2 (MASP-1 and MASP-2) activate a proteolytic cascade, which culminates in the formation of the membrane attack complex and pathogen lysis. Alternative splicing of the pre-mRNA encoding MASP-1 results in two other products, MASP-3 and MAp44, which regulate activation of the cascade. A similar mechanism allows the gene encoding MASP-2 to produce the truncated MAp19 protein. Polymorphisms in MASP1 and MASP2 genes are associated with protein serum levels and functional activity. Since the first report of a MASP deficiency in 2003, deficiencies in lectin pathway proteins have been associated with recurrent infections and several polymorphisms were associated with the susceptibility or protection to infectious diseases. In this review, we summarize the findings on the role of MASP polymorphisms and serum levels in bacterial, viral and protozoan infectious diseases.
1. Introduction
The complement system is one of the major effectors of the innate immune system and an important bridge between innate and adaptative immunity (Ricklin et al., 2010, Walport, 2001). Complement clears immune complexes and cell debris and elicits immediate, highly efficient and tightly regulated inflammatory and cytolytic immune responses to infectious organisms including bacteria, viruses and protozoan parasites (Dunkelberger and Song, 2010). The complement system comprises more than 50 plasma and membrane-associated proteins (Kjaer et al., 2013). Complement activation leads to a proteolytic cascade that culminates with the recruitment of inflammatory cells, phagocytosis and cell lysis (Kjaer et al., 2013). It produces anaphylatoxins (C3a, C4a, C5a), which are potent proinflammatory mediators, and opsonins (C3b and C4b), which cover the pathogen's surface, mediating phagocytosis. The cascade culminates with assembly of the membrane attack complex (MAC) on the cell membrane, forming pores that lead to cell lysis (Dunkelberger and Song, 2010).
Complement can be activated through the classical, alternative and lectin pathways (Fig. 1 ). Independently of the initiation pathway, proteolytic cascades converge towards activation of the major component C3, with subsequent assembly of MAC (Chen et al., 2010). Activation of the classical pathway is initiated on immune complexes by the binding of the C1 complex mainly to IgM or IgG, but also to apoptotic cells, pentraxins and pathogens. On the other hand, the activation of the alternative pathway occurs by spontaneous hydrolysis of C3 in plasma. The lectin pathway is initiated by the binding of pattern recognition molecules (PRMs) to carbohydrates or acetylated residues present on the surface of microorganisms (known as PAMPs or pathogen-associated molecular patterns) or on aberrant glycocalyx patterns of apoptotic, necrotic or malignant cells (known as DAMPs or damage-associated molecular patterns) (Ricklin et al., 2010).
Fig. 1.
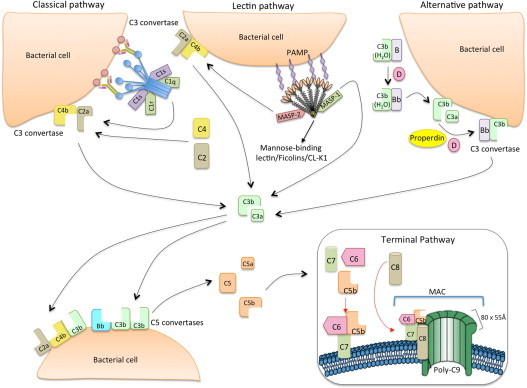
The classical, lectin and alternative pathways of complement activation. The classical pathway is initiated via binding of C1 complex (which consists of C1q, C1r and C1s molecules) through its recognition molecule C1q to target molecules on the surface of pathogens. Subsequently, C1s cleaves C4, which binds covalently to the pathogen surface, and then cleaves C2, leading to the formation of C4b2a complex, the C3 convertase of the classical pathway. Activation of the lectin pathway occurs through binding of mannose-binding lectin (MBL) oligomers, ficolin oligomers or collectin heteromers (CL-K1 + CL-L1), complexed with MBL-associated serine proteases 1 and 2 homodimers (MASP-1 and MASP-2, respectively), to various carbohydrates or acetylated groups on the surface of pathogens (PAMPs: pathogen associated molecular patterns). Like C1s, MASP-2 leads to the formation of the C3 convertase, C4b2a, but its activation is dependent on MASP-1. MASP-1 also cleaves C2. Activation of the alternative pathway depends on spontaneous low-grade hydrolysis of C3 in plasma leading to the formation of C3b. This C3b binds Factor B (homologous to C2) to form a C3bB complex. The cleavage of Factor B by Factor D form the alternative pathway C3 convertase, C3bBb. Properdin stabilizes this complex. The C3 convertases cleave C3 to C3b, which bind covalently next to the site of complement activation (opsonization). This amplifies the cascade and mediates phagocytosis, as well as adaptive immune responses. The addition of additional C3b molecules to the C3 convertase forms C5 convertases (C3bBbC3b for the alternative pathway or C4bC2aC3b for both classical and lectin pathways). This C3b acts as a binding site for C5 and initiate the assembly of the membrane-attack complex (MAC) by cleavage of C5–C5a and C5b. Whereas C5a acts as a potent anaphylatoxin, C5b forms a complex with C6 and C7, which is inserted in the cell membrane. Thereafter, C8 and 10 – 18 C9 molecules (80 × 55 Å each) bind to this complex, resulting in a fully functional MAC (C5b-9). The three pathways converge to this common terminal pathway, culminating with cell lysis and death (Abbas et al., 2012).
2. Lectin pathway
The lectin pathway was discovered by Matsushita and Fujita in 1992, a landmark report on the mechanism of the lectin pathway activation by MBL and MASPs (Matsushita and Fujita, 1992). In fact, different PRMs activate the lectin pathway: oligomers of mannose-binding lectin (MBL), heteromers of collectins 10 and 11 (COLEC10, alias collectin liver 1 or CL-L1 and COLEC11, alias collectin kidney 1 or CL-K1) and oligomers of either Ficolin-1, 2 or 3 (also called M-, L- and H-ficolin, respectively) (Henriksen et al., 2013, Holmskov et al., 2003, Ma et al., 2013). They recognize PAMPs, highly conserved structures present in several microorganisms, such as lipoteichoic acid of Gram-positive bacteria, endotoxin or lipopolysaccharide of Gram-negative bacteria, β-glucan of fungi (Zhu et al., 2005), glycoproteins of viruses (Tarr et al., 2012), protozoa and multicellular parasites (reviewed by (Messias-Reason and Boldt, 2008). They also bind DAMPs, altered carbohydrate patterns on the surface of apoptotic, necrotic and malignant cells (Ren et al., 2014). The dimers, trimers and/or higher oligomers of trimeric structural subunits of these PRMs form complexes with homodimers of serine proteases (known as “MBL-associated” serine proteases or MASPs), which are initially in the form of proenzymes (zymogens) (Kjaer et al., 2013). After binding of these PRMs to their targets, MASPs are cleaved and activated (Dahl et al., 2001, Degn et al., 2009, Héja et al., 2012b). Although MASP-2 autoactivates, being long thought to be the central activator of the lectin pathway (Ambrus et al., 2003, Gál et al., 2005, Matsushita et al., 2000), different authors provided recent evidence that MASP-1, which also autoactivates, has a significant role in activating MASP-2 (Degn et al., 2012, Héja et al., 2012b, Kjaer et al., 2013).
Activation was initially supposed to occur through conformational changes similar to those undergone by the C1 complex of the classical pathway (Gingras et al., 2011). More recently, the proteolytic cascade was proposed to initiate through MASP-1–MASP-2 cross-activation. This occurs with comparable efficiency within co-complexes comprised of a MASP-1 homodimer, a MASP-2 homodimer and a pentamer or higher-order oligomer of PRMs, or by clustering and juxtaposition of PRM/MASP-1 and PRM/MASP-2 complexes on ligand surfaces, where the PRMs may not be identical (MBL and FCNs, for example) (Degn et al., 2014). Although MASP heterodimerization (e.g. MASP-1 + MASP-2, MASP-1 + MASP-3) is possible in vitro, its physiological importance is uncertain (Degn et al., 2013, Paréj et al., 2014). The observation that around 70% of MBL circulate in blood as trimers of trimeric subunits (MBL3 × 3) associated with MASP-1 or MAp19, or as tetramers of trimeric subunits (MBL4 × 3) associated with MASP-2 or MASP-3 (Dahl et al., 2001) implies a major role for intercomplex cross-activation in the initiation of the complement cascade. This is also supported by images of 450 kDa MBL/MASP-1 complexes obtained with small-angle X-ray scattering and electron microscopy (Kjaer et al., 2015). Non-active complexes may be formed with the truncated proteins MAp44 (also called MAP-1) (Degn et al., 2009, Skjoedt et al., 2010a) and MAp19 (also known as sMAP) (Stover et al., 1999a, Stover et al., 1999b, Takahashi et al., 1999), which lack the serine protease domain and thus functional activity. Once associated with MBL, MASP-1 and 2 seem not to exchange, nor are they competitively replaced by MASP-3 and MAp19 (Laursen et al., 2012). Nevertheless, they can be displaced by MAp44 (Degn et al., 2013).
Proteolytic activity is acquired by cleavage of the MASP Arg Ile bond of the activation peptide, also called “linker peptide”. This cleavage generates two chains covalently linked by a disulfide bond: a heavy chain (alias “A chain”) and a light chain (alias “B chain”) (Sato et al., 1994). The heavy chain comprises the linker and the CUB1-EGF-CUB2-CCP1-CCP2 regulatory domains, i.e. the N-terminal CUB (C1r/C1s, Uegf and bone morphogenetic protein-1) domain of about 110 amino acid residues (CUB1), followed by an epidermal growth factor (EGF)-like domain of the Ca2+-binding type, of approximately 50 amino acid residues, a second CUB domain (CUB2) and two contiguous complement control protein modules (CCP1 and CCP2). The light chain constitutes the chymotrypsin-like serine protease (SP) domain (reviewed by (Kjaer et al., 2013)). The CUB1-EGF domains are responsible for Ca2+-dependent dimerization of MASP polypeptides, yet CUB1-EGF-CUB2 are involved in the Ca2+-dependent association of MASP dimers with PRMs (Degn et al., 2009, Thielens et al., 2001, Wallis and Dodd, 2000).
3. MASPs
3.1. MASP1 gene
MASP-1 was identified in the early 90s as a bactericidal factor with structural similarities to C1s, reactive to Salmonella typhimurium (Takada et al., 1993). The MASP1 gene is located on chromosome 3q27-8 (Takada et al., 1995) and contains 18 exons, which encode three proteins by pre-mRNA alternative splicing: MASP-1, MASP-3 and MAp44 (Fig. 2 ). Exon 1 is not translated and exons 2–11 encode the CUB1-EGF-CUB2-CCP1-CCP2 regulatory domains. The first eight exons are common to all three proteins, meaning that they share CUB1-EGF-CUB2-CCP1. Exon 9 is unique to MAp44. Thus, MAp44 lacks the second CCP domain and the entire SP domain (Degn et al., 2009, Skjoedt et al., 2010a). The SP domains of MASP-3 and MASP-1 are encoded by exon 12 and exons 13–18, respectively. This explains why both proteins have different SP domains and also differ in the preceding 15 amino acid residues. Interestingly, the sequence encoding the upstream MASP-3 SP domain has no introns and its active serine residue is encoded by an AGY codon, as in MASP-2, C1r and C1s. This reflects a common origin for the unique SP exon, presumably by retrotransposition of a sequence encoding a MASP-3-like factor B encoding protease in ascidians, after the divergence of urochordates, and subsequent gene duplication events. Instead, the active serine of MASP-1 is encoded by a TCN codon, as typical for chymotrypsin genes, turning the MASP-1 encoding sequence representative of the most ancestral serine protease gene of the lectin pathway, appearing as early as in cnidaria (Kimura et al., 2009, Nonaka and Miyazawa, 2001).
Fig. 2.
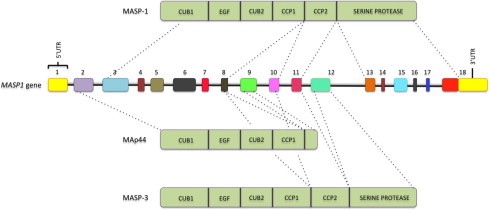
MASP1 gene and MASP-1, MAp44, and MASP-3 proteins. Exons are numbered and not depicted to scale. MASP: mannose-binding lectin associated serine protease. MAp44: mannose-binding lectin associated protein of 44 kDa. CUB: C1r/C1s, Uegf, and bone morphogenetic protein; EGF: epidermal growth factor; CCP: complement control protein.
At least 12 MASP1 gene single nucleotide polymorphism (SNP), some of them strongly linked, are associated with protein serum levels (Table 1 ). Interestingly, contrasting values of MASP-1, MASP-3 and MAp44 serum levels were found associated with SNPs located near the alternative exons 9 (exclusive to MAp44) and 12 (exclusive to MASP-3), probably due to mutually alternative splicing mechanisms. For example, homozygosity for the rs3774275 SNP in intron 8 (G/G) was associated with increased MASP-1 and MAp44 but decreased MASP-3 concentrations. Five haplotypes comprised by the rs35089177, rs7625133 and rs72549254 SNPs in the upstream regulatory region seem also to play a role. The most common haplotype (TAG) occurs with 54% frequency in the Danish population, being associated with the highest levels of MASP-1 and MAp44 and lowest levels of MASP-3 (Ammitzbøll et al., 2013).
Table 1.
MASP1 single nucleotide polymorphisms associated with protein levels.
| dbSNP | Gene region | HGVS name | Reference: NP_001870 | Protein domain | Protein levelsa, b | MAF (%) #/1000 genomes project (all populations) |
|---|---|---|---|---|---|---|
| rs190590338 | Promoter | 3:g.187294096G>A | n.a. | – | G/A: 35% higher MASP-1 | 1.64/<1 |
| rs75284004 | Promoter | 3:g.187293111A>G | n.a. | – | A/G: 16% lower MASP-3 | 3.21/2.0 |
| rs35089177 | Promoter | 3:g.187293050T>A | n.a. | – |
T/A: 10% lower MASP-1, A/A: 15% lower MASP-1, A/A: 14% lower MAp44 |
31.2/31.0 |
| rs7625133 | Promoter | 3:g.187292593A>C | n.a. | – |
A/C: 10% lower MAp44, C/C: 34% lower MAp44 |
7.6/3.0 |
| rs72549254 | Intron 01 | 3:g.187291624G>A | n.a. | 5′ UTR | A/A: 29% lower MAp44 | 15.0/16.0 |
| rs710469 | Intron 02 | 3:g.1872667308G>A | n.a. | CUB1 | A/A: on-admission high MASP-3 levels in critically ill children | 46.5/47.0 |
| rs3774275 | Intron 08 | 3:g.187247480A>G | n.a. | CCP1 |
A/G: 10% higher MASP-1, G/G: 26% lower MASP-3, A/G: 16% higher MAp44, G/G: 29% higher MAp44 |
32.1/25.0 |
| rs113938200 | Exon 09 | 3:g.18247374C>T | p.D368N | CCP1 | C/T: 39% lower MAp44 | 0.3/0.1 |
| rs698090 | Exon 09 | 3:g.187246512C>T | n.a. | 3′UTR MAp44 | C/C: 23% lower MASP-3, C/T: 13% higher MAp44, C/C: 23% higher MAp44 | 37.4/41.0 |
| rs72549154 | Exon 12 | 3:g.187236144G>T | p.R576 M | MASP-3 SP | G/T: 14% lower MASP-1 and 13% higher MASP-3 | 3.5/3.0 |
| rs850312 | Exon 12 | 3:g.187236020C>T | p.L617= | MASP-3 SP | C/C: lower on-admission MASP-3 levels in critically ill children | 34.0/24.0 |
| rs67143992 | Exon 12 | 3:g.187235533C>T | n.a. | 3′UTR MASP-3 |
C/T: 12% higher MASP-1, 9% higher MAp44 and 16% lower MASP-3, T/T: 31% lower MASP-3 and 19% higher MAp44 |
19.2/9.0 |
In bold: SNPs belonging to the upstream regulatory region haplotypes; n.a.: not applicable; CUB1: Complement C1r/C1s, Uegf, Bmp1; CCP: Complement Control Protein; dbSNP: Single Nucleotide Polymorphism Database; HGVS: Human Genome Variation Society (allele name available in http://www.ensembl.org); MAF: minor allele frequency; MASP: MBL-associated Serine Protease; MAp44: MBL-associated protein of 44 kDa; SP: Serine Protease; UTR: untranslated.
% Indicates the relative change compared to the homozygotes for the major allele.
Among the non synonymous variants described in the MASP1 gene, the p.G426E (rs38343199) is located in the CCP2 domain and occurs in around 3% allele frequency among Europeans. No association was found between this amino acid substitution and systemic lupus erythematosus, systemic inflammatory response syndrome (SIRS) and/or sepsis (Weiss et al., 2007).
3.2. MASP-1
MASP-1 was discovered in 1992 as a C1s-like protein associated to MBL (Matsushita and Fujita, 1992). MASP-1 protein contains 699 amino acid residues and a 19 amino acid leader peptide (Thiel, 2007). It is primarily expressed in the liver, being up-regulated by IL-6 during the acute phase response (Knittel et al., 1997). MASP-1 is considered a promiscuous protease because its substrate binding groove is wide and resembles that of trypsin rather than early complement proteases (Dobó et al., 2009). Normal MASP-1 concentration in serum/plasma is around 11 μg/mL (range 4–30 μg/mL), 20-fold higher than of MASP-2 (Thiel et al., 2012). Among patients with different cardio- and cerebrovascular diseases, MASP-1 levels were highest in patients with subacute myocardial infarction (median 12 μg/mL) and lowest in those with acute ischemic stroke (median 8.6 μg/mL) (Frauenknecht et al., 2013). In addition, the expression of MASP-1 was observed to be up-regulated in primary uterine leiomyosarcoma (Davidson et al., 2014) and in HCV infected-hepatocyte cell lines (Saeed et al., 2013). Having much higher propensity for autoactivation than MASP-2 (Ambrus et al., 2003), MASP-1 activates MASP-2 in heterocomplexes of large oligomeric MBL and produces 60% of C2a responsible for C3 convertase formation (Degn et al., 2012, Héja et al., 2012b). In fact, MASP-1 autoactivation seems to control the initiation of the lectin pathway (Megyeri et al., 2013). Nevertheless, MASP-1 does not cleave C4 and therefore is not capable of generating C3 convertase alone (Ambrus et al., 2003). Direct activation of C3 by MASP-1 can occur but is physiologically irrelevant (Ambrus et al., 2003). MASP-1 was reported to be essential for the development of autoimmune-associated inflammatory tissue injury by activating the alternative pathway of complement in an experimental model of inflammatory arthritis (Banda and Takahashi, 2010). Nevertheless, the reported role of MASP-1 on activation of the alternative pathway was not confirmed in humans (Degn et al., 2012). MASP-1 was shown to play a role in the coagulation, cleaving factor XIII and fibrinogen and mediating the formation of cross-linked fibrin, although with a lower catalytic efficiency compared with thrombin (Krarup et al., 2008). In fact, antithrombin in the presence of heparin is a more potent inhibitor of MASP-1 than C1 inhibitor (Presanis et al., 2004). Furthermore, MASP-1 caused obstructive thrombosis in a mouse model of arterial injury (Bonte et al., 2012).
Proteolytic activity of MASP-1 induces Ca2+ signaling, NF-kappaB and p38 MAPK pathways in endothelial cells through protease-activated receptor 4 (PAR4) (Megyeri et al., 2009). This activity leads to the release of IL-6 and IL-8, activating the chemotaxis of neutrophil granulocytes (Jani et al., 2014). MASP-1 is also able to modulate the immune response by the release of proinflammatory bradykinin from high-molecular weight kininogen (Dobó et al., 2011).
3.3. MASP-3
MASP-3 is ubiquitously expressed with mean serum concentration of 5.2–6.4 mg/L (ranging from 2 to 12.9 mg/L) (Degn et al., 2010, Skjoedt et al., 2010b) and evolutionarily highly conserved (Stover et al., 2003). MASP-3 occurs in serum mainly associated with Ficolin-3, and in lower amounts with Ficolin-2 and MBL (Skjoedt et al., 2010b). So far, the biological role of MASP-3 remains unclear. MASP-3 does not cleave any complement components and it is not inhibited by C1-inhibitor (Zundel et al., 2004; Yongqing et al., 2012). However, MASP-3 may reduce the lectin pathway activity due to competition with MASP's binding sites on the recognition molecules (Degn et al., 2009). Recently, MASP-3 was proposed to have a role in the activation of the alternative pathway in mice (Iwaki et al., 2011), however, in humans MASP-3 seems not to be required for activating the alternative pathway (Degn et al., 2012).
Together with CL-K1, MASP-3 seems to have an important role in early embryonic development, as shown by the effect of five rare MASP-3 exon 12 mutations (p.H497Y, p.C630R, p.G666E, p.G687R and p.W290X) in four independent families with autosomal recessive 3MC syndrome (Mingarelli, Malpuech, Michels and Carnevale), characterized by various developmental disorders. All the implicated mutations are predicted to damage the serine protease domain, abolishing MASP-3 enzymatic activity (Rooryck et al., 2011, Sirmaci et al., 2010).
MASP-3 may play a role in defense against acquired infections in intensive care unit (ICU). Diagnosed hospital-acquired-infection is a major problem occurring in approximately 30% of children admitted to ICU. Patients who acquired an infection at ICU had on-admission lower MASP-3 levels (P < 0.00). These results remained independently associated with acquisition of infection after correcting for baseline characteristics, risk factors, and the other lectin pathway proteins in multivariate logistic regression analysis. Low MASP-3 levels were also associated with prolonged need for intensive care irrespective of infection (Ingels et al., 2014).
3.4. MAp44
MAp44 (also called MAP-1 or MBL-associated protein 1) regulates negatively the complement activation by competing with MASP-1, 2 and 3 for MBL and ficolin binding sites (Degn et al., 2009, Skjoedt et al., 2010a, Pavlov et al., 2012, Degn et al., 2013). Similarly to MASP-3, MAp44 is largely expressed not only in the liver, but also in bladder, brain, cervix, colon and prostate (Degn et al., 2009), and does not behave as an acute phase protein. In contrast to MASP-3, its levels are highest in heart (Degn et al., 2009) and also decrease slightly in serum during the first 6 months after birth (Degn et al., 2010). Normal MAp44 serum levels are around 1.5 μg/mL (range 0.3–3.2 μg/mL) (Degn et al., 2010, Degn et al., 2009). MAp44 has been associated with cardioprotective effects, preserving cardiac function, decreasing infarct size and preventing thrombogenesis in murine models of ischaemia/reperfusion injury and arterial thrombosis by inhibiting MBL and C3 deposition (Bonte et al., 2012, Pavlov et al., 2012). The inhibitory effect on MBL deposition however, was only evident at pharmacological levels (10 μg/mL) on N-acetyl-d-glucosamine, bovine serum albumine (GlcNAc-BSA) – coated, but not mannan-coated plates, which was interpreted as an attenuating mechanism for MBL recognition of endogenous ligands (Pavlov et al., 2012). Yet the precise mechanism enabling MAp44 to inhibit MBL deposition awaits further elucidation. Nevertheless, MAp44 levels were not related to cardio- and cerebrovascular diseases in humans but associated with cardiovascular risk factors such as dyslipidaemia, obesity and hypertension (Frauenknecht et al., 2013).
Based on these findings, MAp44 has been recently suggested as a new therapeutic approach to be considered for the treatment of patients with myocardial ischemia/reperfusion injury and thrombogenesis (Pavlov et al., 2012), as well as with inflammatory arthritis (Banda et al., 2014).
3.5. MASP2 gene
The MASP2 gene is located on chromosome 1p36.23–31 (Stover et al., 1999a, Stover et al., 1999b), has 12 exons covering more than 20 Kb and encodes two proteins, MASP-2 and MAp19 (Fig. 3 ). Exon 1 is not translated, exon 2 encodes the signal peptide, and together with exon 3, the CUB1 domain. Exon 4 encodes the EGF-like domain, exons 6 and 7 encode the second CUB1 domain, exons 8–9 and 10–11 the two CCP domains, and exon 12 encodes the serine protease domain and the 3′ UTR region (Stover et al., 2001). MAp19 is a truncated protein produced by alternative splicing and polyadenylation of the primary transcript of MASP2 gene. Exon 5 is exclusive to the MAp19 transcript and contains an in-frame stop codon which leads to the premature termination of the translation process, and a polyadenylation signal, resulting in a truncated protein of 19 KDa (Stover et al., 1999a, Stover et al., 1999b, Takahashi et al., 1999).
Fig. 3.
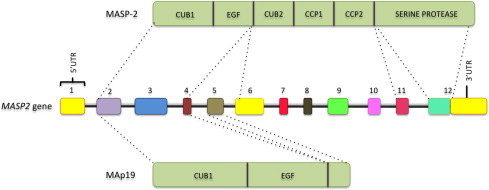
MASP2 gene and the mannose-binding lectin associated serine protease 2 (MASP-2) and MAp19 proteins. Exons are numbered and not depicted in scale. CUB: C1r/C1s, Uegf, and bone morphogenetic protein; EGF: epidermal growth factor; CCP: complement control protein.
Various polymorphisms encompassing the region from the MASP2 promoter to the last exon were associated with MASP-2 and MAp19 protein levels in serum (Table 2 ). Ten haplotypes with these polymorphisms were associated with low, intermediate and high producing haplotypes (Boldt et al., 2011a, Boldt et al., 2011b). MASP-2 deficiency may occur in homozygotes or compound heterozygotes for p.D120G, p.156_159dupCHNH and p.R439H variants, due to different reasons. The Asp-Gly substitution in residue 120 disrupts calcium ion binding of the first CUB1 domain in individuals with the p.D120G SNP. This turns MASP-2 and MAp19 unable to bind MBL and ficolins, abrogating complement activation and reducing MASP-2 concentration in up to 0.3% homozygote Europeans (Stengaard-Pedersen et al., 2003, Thiel et al., 2007, Valles et al., 2009). Yet the rare duplication of four aminoacids within the EGF domain (p.156_159dupCHNH) was found in Asians and probably results in MASP-2 misfolding (Thiel et al., 2009, Thiel et al., 2007). The p.R439H variant occurs with a predicted frequency of 2.6% among Africans and disrupts the activation peptide of the SP domain of a MASP-2 molecule still able to bind MBL. This probably reduces binding opportunities with full-working MASP-2 in heterozygotes and blocks complement activation in p.439H homozygotes (Thiel et al., 2009, Thiel et al., 2007). Beside these, three other non synonymous substitutions were associated with the modulation of MASP-2 levels, without obvious reasons: p.P126L (in the first CUB1 domain), p.V377A and p.Y371D (in the CCP2 domain) (Schadt et al., 2008, Thiel et al., 2009, Thiel et al., 2007). Interestingly, the g.21370G>T polymorphism responsible for p.Y371D is imbedded within a region with one of the strongest signal of selective sweep in the human genome (Ronen et al., 2013). Furthermore, polymorphisms in introns 4 and 9 were associated with opposite MASP-2 and MAp19 levels, probably due to an interference in mutually alternative mRNA splicing (Boldt et al., 2013a, Boldt et al., 2011a, Boldt et al., 2011b).
Table 2.
MASP2 single nucleotide polymorphisms associated with protein levels and function (Boldt et al., 2011a, Boldt et al., 2011b, Boldt et al., 2013a, Boldt et al., 2013b, Thiel et al., 2007, Thiel et al., 2009).
| dbSNP | Gene region | HGVS name | Reference: NP_006601.2 | Protein domain | Protein levelsa | Protein functiona | MAF (%) 1000 genomes project (all populations) |
|---|---|---|---|---|---|---|---|
| rs7548659 | Promoter | 1:g.11047382G>T | n.a. | n.a. | Variable | n.a. | 38 |
| rs61735600 | Exon 3 | 1:g.11046672C>T | p.R99Q | CUB1 | ≥600 ng/mL | Normal | 2 |
| rs72550870 | Exon 3 | 1:g.11046609T>C | p.D120G | CUB1 | ≤200 ng/mL | Cannot bind MBL, cannot activate C4 | 2 |
| rs56392418 | Exon 3 | 1:g.11046591G>A | p.P126L | CUB1 | ≤200 ng/mL | Normal | 3 |
| rs2273343 | Exon 4 | 1:g.11045488T>C | p. H155R | EGF | <200 ng/mL | Normal | 1 |
| – | Exon 4 | c.466_477dupTGCCACAACCAC @ | p. CHNHdup | EGF | <200 ng/mL | Cannot bind MBL, cannot induce C4 fragment deposition | 0.26@ |
| rs2273344 | Intron 4 | 1:g.11045065C>T | n.a. | n.a. | ≥600 ng/mL | n.a. | 15 |
| rs9430347 | Intron 5 | 1:g.11044788T>C | n.a. | n.a. | ≥600 ng/mL | n.a. | 15 |
| rs17409276 | Intron 9 | 1:g.11031148G>A | n.a. | n.a. | ≥600 ng/mL | n.a. | 15 |
| rs12711521 | Exon 10 | 1:g.11030859C>A | p.D371Y | CCP2 | Variable | Normal | 37 |
| rs2273346 | Exon 10 | 1:g.11030840A>G | p.V377A | CCP2 | ≤200 ng/mL | Normal | 9 |
| rs12085877 | Exon 12 | 1:g.11027630C>T | p.R439H | SP | ≤200 ng/mL | Binds MBL but does not autoactivate, cannot activate C4 | 2 |
| rs1782455 | Exon 12 | 1:g.11027467G>A | p.S493= | SP | Variable | n.a. (synonymous) | 29 |
n.a.: not applicable. CUB1: Complement C1r/C1s, Uegf, Bmp1; CCP: Complement Control Protein; EGF: Epidermal Growth Factor; dbSNP: Single Nucleotide Polymorphism Database; HGVS: Human Genome Variation Society (allele name available in http://www.ensembl.org); MAF: minor allele frequency; MBL: mannose (or mannan)-binding lectin; @ Reference: NM_006610, only in Chinese of Hong Kong (Thiel et al., 2007).
Associated with the minor allele.
3.6. MASP-2
MASP-2 was characterized by Thiel and others in the late 90s as an protein similar to MASP-1 (Thiel et al., 1997). It binds MBL and ficolins as a homodimer in a calcium-dependent manner through its epidermal growth factor (EGF)-like domain, assisted by the two surrounding CUB domains (Feinberg et al., 2003). Although found sufficient to autoactivate and initiate the lectin pathway of complement (Gál et al., 2005), MASP-2 is activated by MASP-1 at physiological conditions, afterwards cleaving the complement components C4 and C2, thus generating the C3 convertase C4bC2b. In fact, MASP-1 is required for efficient activation of the lectin pathway (Thiel et al., 1997, Degn et al., 2012, Héja et al., 2012a, Héja et al., 2012b, Kocsis et al., 2010, Megyeri et al., 2013). Compared to C1s, the serine protease of the classical pathway, MASP-2 is up to 1000 times more catalytic active and up to 50-fold more rapidly inhibited by C1-inhibitor (Kerr et al., 2008). MASP-2 also acts in the coagulation cascade, cleaving prothrombin in the same way as factor Xa, generating cross-linked fibrin covalently bound on bacterial surfaces (Gulla et al., 2010).
MASP-2 is predominantly expressed in hepatocytes and its promoter is regulated by STAT3, IL-1b and IL-6 (Unterberger et al., 2007). MASP-2 levels have been shown to be stable over time in healthy individuals, around 400–500 ng/mL in serum/plasma (range 70–1200 ng/mL) (Møller-Kristensen et al., 2003, Ytting et al., 2007). MASP-2 levels lower than 100 ng/mL are considered deficient (Thiel, 2007). MASP-2 levels were lower in myocardial infarction and acute stroke patients, compared with patients with stable coronary arterial disease and controls (Frauenknecht et al., 2013). This is in line with the observation that myocardial ischemia induces complement activation with MASP-2 consumption (Zhang et al., 2013). Among critically ill children, higher MASP-2 levels (around 360 ng/mL) were associated with malignancy, while lower MASP-2 levels (around 150 ng/mL) were associated with cardiac disease (Ingels et al., 2014). High MASP-2 levels were also associated with the development of severe infections in adult patients with haematological cancer undergoing chemotherapy (Ameye et al., 2012).
3.7. MAp19
MAp19 is a 19 kDa truncated protein produced by the alternative splicing of MASP2 gene, presenting only the first CUB domain and the EGF domain, with no serine protease activity. The exon 5 exclusive to MAp19 has an in-frame stop codon and a polyadenylation signal, coding four amino acid residues in the C-terminal tail and the 3′ UTR of the protein (Takahashi et al., 1999). It is secreted by the liver to the plasma, and expressed by Kupffer cells with similar median level of MASP-2 (217 ng/mL, ranging from 26 to 675 ng/mL). Nevertheless, only a minor fraction is associated with MBL and ficolins, and this binding occurs with ten times lower affinity compared with MASP-2 (Degn et al., 2011). MAp19 is excreted in human urine and may play a role in the inhibition of calcium oxalate renal stone formation (Degn et al., 2011, Kang et al., 1999). The nucleocapsid N protein of severe acute respiratory syndrome coronavirus interacts in vitro with MAp19, but the functional significance of it remains unknown (JL Liu et al., 2009a, Liu et al., 2009b).
4. MASPs and infectious diseases
Both insufficient and excessive activation of the lectin pathway may be harmful to the host. Whereas deficiency of any lectin pathway component may be clinically irrelevant in healthy individuals due to redundancy of the immune system, these deficiencies are frequently associated with increased susceptibility to secondary infections or autoimmune complications in immunocompromised patients (Degn and Thiel, 2013, Granell et al., 2006, Ingels et al., 2014, García-Laorden et al., 2006, Neth et al., 2000). Low MASP-2 levels, for example, were associated with an increased risk of severe chemotherapy-induced neutropenia in children with cancer (Schlapbach et al., 2007) and an acute decrease of MASP-2 levels in the early phase of septic shock correlated with in-hospital mortality (Charchaflieh et al., 2012). In contrast, increased levels of innate immunity proteins, which can in part be explained by polymorphisms in the corresponding genes, may contribute to exaggerated inflammatory responses (Boldt et al., 2012). High postoperative levels of MASP-2, for example, were associated with poor prognosis in colorectal cancer patients (Ytting et al., 2008).
Genetic integrity of the genes encoding lectin pathway components is essential for complement activation and regulation. SNPs in the MASP1 and MASP2 genes can reduce protein serum concentrations or affect its enzymatic activity, impairing complement activation and possibly coagulation and development protease cascades (Ammitzbøll et al., 2013, Rooryck et al., 2011, Thiel et al., 2009, Thiel et al., 2007). As expected, a growing number of infectious diseases have been associated with MASP1 and MASP2 SNPs since the first report on MASP deficiency in 2003, where a patient with persistent inflammatory disease, episodes of severe pneumococcal pneumonia and progressive lung fibrosis presented homozygosity for p.D120G in the MASP-2 CUB1 domain (Stengaard-Pedersen et al., 2003). Although the p.D120G variant was included in 2007 in the list of primary immunodeficiency diseases (Geha et al., 2007), it has low clinical penetrance (Garcia-laorden et al., 2008) and is relatively rare, reasons for which it was not found associated with any common disease investigated to date (Miller et al., 2010, Olesen et al., 2006, Ramasawmy et al., 2008, Schafranski et al., 2008, Segat et al., 2008, Ytting et al., 2011). In contrast, more frequent variants were actually found to play a role in susceptibility to infection and treatment response, as discussed below.
4.1. Viral diseases
MBL, one of the β-inhibitors identified more than six decades ago as able to inactivate influenza virus (Anders et al., 1990, Dommett et al., 2006), actually recognizes a range of different viruses (reviewed by (Messias-Reason and Boldt, 2008)). Although the inhibiting activity of PRMs as MBL does not necessarily depend on complement activation, and thus on the efficiency of MASP activation (Thielens et al., 2002), MASPs, as well as polymorphisms associated with MASP levels and function, were found to play a role in the immune response to different viral infections.
4.1.1. Human T-lymphotropic virus 1
The human T-lymphotropic virus 1 (HTLV-1) is a retrovirus endemic in many countries. The majority (95%) of individuals infected with HTLV-1 remain asymptomatic lifelong, whereas others develop severe diseases, such as the HTLV-1 associated myelopathy/tropical spastic paraparesis and adult T-cell leukemia (Izumo et al., 2000). In a study involving 96 patients of Northwestern Brazil, MASP2 p.Y371D (rs12711521) was associated with susceptibility to HTLV-1 infection. Specifically, aspartic acid at residue 371 was associated with a dominant higher risk for HTLV infection both in heterozygous and homozygous individuals (OR = 2.71 [95%CI = 1.56–4.74], P < 0.001) and was independent of genetically determined MBL deficiency, also associated with the disease. The possession of both MASP2*371D and MBL2 low-producing haplotypes increased the risk for HTLV-1 infection by approximately 60%, but no association with viral load was seen (Coelho et al., 2013). Although MASP-2 levels were not evaluated in the study, it is known that the p.371D variant is associated with a dominant effect on MASP-2 levels ≥600 ng/mL, and an additive effect on MAp19 levels ≤100 ng/mL (Boldt et al., 2013a). Thus, compound MBL deficiency and MASP-2 overproduction may play a role in the susceptibility to HTLV-1 infection. This association however, awaits replication in other cohorts.
4.1.2. Hepatitis C (HCV)
Approximately 170 million people worldwide are infected with hepatitis C virus (HCV) (Barth et al., 2006). Circa 60% of them progress to the chronic form of the disease, characterized by liver fibrosis, which may progress to cirrhosis and hepatocarcinoma. Liver injury in HCV patients has been suggested to be the result of persistent inflammatory response induced by immune mediators. Nonetheless, appropriate activation of the innate immune system is important for synthesis of antiviral peptides and inhibition of viral replication (Thimme et al., 2006).
In this regard, Ficolin-2 and MBL were found to bind HCV envelope glycoproteins E1 and E2, activating complement against HCV-infected hepatocytes (Brown et al., 2010, Liu et al., 2009a, Liu et al., 2009b), and MBL deficiency was repeatedly associated with HCV infection (Koutsounaki et al., 2008, Melo et al., 2009, Segat et al., 2007). Whereas early increased Ficolin-2 levels correlated with a better response to treatment (Hu et al., 2013), MBL deficiency was associated with no treatment response (Alves Pedroso et al., 2008, Eurich et al., 2012). Regarding MASP-2, Tulio et al. (2011) investigated 104 South-Brazilian patients with chronic hepatitis C and found an association of homozygotes for the p.371D variant with susceptibility to HCV (OR = 6.33 [95%CI = 1.85–21.7], P = 0.003). Confirming the findings in healthy populations (Boldt et al., 2013a, Boldt et al., 2011a, Boldt et al., 2011b), MASP-2 levels were higher in patients with p.371D, than in those without these variant. Genotypes causing partial MBL deficiency (YA/YO) were also positively associated with HCV infection in the same patient group (Alves Pedroso et al., 2008). Thus similarly to HTLV-1 infection, MBL deficiency and the MASP2*371D variant may exert a synergistic effect that increases susceptibility to HCV infection, but replication within other cohorts is needed to confirm the results.
Nevertheless there was no association of MASP2 polymorphisms or MASP-2 levels with treatment response and liver fibrosis, suggesting that MASP-2 does not have an immunomodulatory role in fibrosis progression and in the treatment with IFN and ribavirin (Tulio et al., 2011). In contrast, MBL deficiency was negatively associated with the severity of liver fibrosis (Alves Pedroso et al., 2008). Lower levels of functional MBL protein would be expected to impair the reported thrombin-like activity of the MBL/MASP-1 complex (Ambrus et al., 2003, Presanis et al., 2004), reducing fibrin cross-linking and the generation of fibrotic liver tissue. In fact, based on the premises that both MBL and MASP-1 are mainly produced in the liver and that MASP-1 has a reactivity profile similar to thrombin, a study was carried out to investigate MBL and the MBL/MASP-1 complex in the development of HCV-related liver fibrosis in English patients. Corroborating previous findings that MBL deficiency exerts a protective effect against liver fibrosis, median MBL serum levels were higher in patients with severe liver fibrosis than in those with mild disease (P = 0.002). Enzymatic activity of MBL/MASP-1 complexes correlated with MBL levels (Spearman's rho = 0.78, P = 0.01) and was also found to be raised in HCV patients with severe fibrosis (171.5 units/min) compared to those with mild fibrosis (80.3 units/min), non-HCV mild fibrosis (41.5 units/min) or controls (24.1 units/min). Moreover, MBL/MASP-1 complex activity remained associated after adjustment for MBL concentration in the multivariate model: OR = 7.26 per log10 units, [95%CI = 2.36–22.40], P = 0.001 (Brown et al., 2007). Very similar results were obtained by others in an Egyptian cohort, again presenting a strong association of HCV-induced liver fibrosis with the activity of the MBL/MASP-1 complex, adjusted for MBL levels (P = 0.003) (El Saadany et al., 2011). Furthermore, both MBL2 and MASP1 genes were up-regulated, presenting higher mRNA expression in hepatocyte cell lines infected with the genotype 2a JFH-1 HCV clone, as revealed by two different groups using microarray and real-time RT-PCR analysis (Blackham et al., 2010, Saeed et al., 2013). Activated recombinant MASP-1 was actually shown to directly cause hepatic stellate cells (HSC) to differentiate to a myofibroblast phenotype in vitro, as measured both by morphology and by the expression of alpha smooth muscle actin (α-SMA) and tissue inhibitor of metalloproteinase-1 (TIMP-1) (Saeed et al., 2013). This cell differentiation culminates in the deposition of excess fibrogenic extracellular matrix proteins and may occur through binding to a protein of the Protease-activated Receptor (PAR) family, as suggested for human vascular endothelial cells (Megyeri et al., 2009).
4.1.3. Human endogenous retrovirus
Human endogenous retroviruses (HERVs) and herpes viruses have been consistently associated with Multiple Sclerosis (MS), an inflammatory disease characterized by damage of nerve cells in the brain and spinal cord. Even though activated HERVs are rarely found in healthy individuals, MS patients have elevated levels of anti-HERV Gag and Env antibodies in both serum and cerebrospinal fluid directed towards distinct HERV epitopes (Christensen et al., 2007). In a study with 25 cases of sporadic MS and 42 MS patients with familial MS, including 32 unaffected relatives, the authors found a significant negative correlation between MASP-3 levels and seroreactivity to HERV peptides, but only in patients with active disease (Spearman's r = −0.631, P = 0.001). Low MASP-2 levels were also correlated with high MASP-3 levels, again only in MS patients with active disease, but with borderline significance (Spearman's r = −0.401, P = 0.06). Interestingly, higher MASP-2 levels were more common in MBL-deficient individuals, regardless of being affected or not (P = 0.0036). In this study, higher MASP-2 expression seemed to occur as a compensation mechanism for MBL deficiency, and higher MASP-3 levels may play a protective role against establishment of the disease (Christensen et al., 2007).
4.2. Bacterial infections
Composition and specific structural features of bacterial PAMPs may favour or prevent binding to PRMs of the lectin pathway of complement. This was initially shown in the early eighties by Kawakami et al. on the so called RaRf complex (later identified as MBL) and its interaction with Salmonella enterica serovar Typhimurium (Kawakami et al., 1982). The authors suggested a crucial role for lipopolysaccharide (LPS) in MBL binding, a feature later on found to be inhibited by lipooligosaccharide sialyation (Devyatyarova-Johnson et al., 2000, Jack et al., 1998) and capsulation (Krarup et al., 2005). Beside recognizing and binding different bacterial PAMPs (reviewed by (Messias-Reason and Boldt, 2008)), thereby initiating the complement cascade, PRMs of the lectin pathway were reported to opsonize bacteria (Kuhlman et al., 1989) and elicit cytokine release from phagocytes (Jack et al., 2001). Nevertheless PRM-mediated phagocytosis may potentially be inhibited by MASP-3 (Duus et al., 2010). Thus, the relative concentration and functional integrity of MASPs may balance the lectin pathway-mediated antibacterial response between complement activation/formation of the membrane-attack complex and opsonization/phagocytosis.
4.2.1. Leprosy
Leprosy, also known as Hansen's disease, is caused by Mycobacterium leprae, which invades macrophages and Schwann cells of the host. Although only a small group of infected individuals develop the disease, different immunologic responses lead to a wide spectrum of clinical manifestations, with progressive and irreversible disabilities (WHO, 2012). Molecular mechanisms involved in the infection and immune evasion by the bacillus are still not well understood due the challenges in cultivating M. leprae in vitro and in vivo (Tabouret et al., 2010).
M. leprae takes advantage of C3 opsonization to enter phagocytes, in order to establish intracellular infection (Schlesingert and Horwitz, 1994). For this reason, low levels of lectin pathway PRMs have long been proposed as beneficial against the infection, by disfavoring bacterial dissemination into host tissues (Garred et al., 1994). There is in fact evidence for a protective effect of low MBL and Ficolin-1 levels, as well as corresponding haplotypes/genotypes of these genes and FCN2, against the infection and development of lepromatous leprosy, the most severe form of the disease, which is characterized by a strong Th2 response (Boldt et al., 2013b, De Messias-Reason et al., 2009, De Messias-Reason et al., 2007, Dornelles et al., 2006, Garred et al., 1994, Sapkota et al., 2010). In the first study about a possible association between leprosy and MASP-2 levels generated by MASP2 polymorphisms, leprosy patients had lower MASP-2 serum levels compared with controls and a higher frequency of low MASP-2 producing alleles. The p.126L and p.439H polymorphisms, which are associated with MASP-2 deficiency, had a dominant effect on leprosy susceptibility (P = 0.007). Yet genotypes composed of intermediate producing MASP2 haplotypes were protective against the disease (P = 0.0014). There was also an association between the lepromatous form of the disease and low producing MASP2 polymorphisms. Thus in contrast to PRMs, low MASP-2 levels increased the susceptibility to leprosy per se and to the lepromatous leprosy form (Boldt et al., 2013a). Replication of these studies in other populations is needed in order to confirm these genetic associations.
4.2.2. Tuberculosis
Tuberculosis is caused by the Mycobacterium tuberculosis and affects mostly the lungs (pulmonary TB), but can also affect other organs (extrapulmonary TB). About one third of the world population is infected with this bacterium. Although only a small proportion of infected people develop the disease, which is curable, 3 million of the 9 million people that acquire the disease every year do not have access to treatment. Without treatment, the mortality rates are high (around 70% of patients die in 10 years), placing tuberculosis as the second cause of death due to a single infectious organism (the first is AIDS) (World Health Organization, 2014).
MBL is able to recognize different mycobacterial cell wall components containing mannose (mannosyl-lipoarabinomannan (ManLAM), arabinosyl-LAM, lipomannan and phosphatidylinositol mannoside) (Polotsky et al., 1997). Nevertheless, not all mycobacteria causing tuberculosis bind MBL equally well, e.g. Mycobacterium africanum binds recombinant human MBL more efficiently than M. tuberculosis. Furthermore, in a Ghanaian cohort, MBL deficiency was associated with protection against M. africanum-tuberculosis only (Thye et al., 2011).
MBL deficiency nevertheless was shown to increase the susceptibility to the infection in Northwestern Brazil (da Cruz et al., 2013). In addition, ficolin-2 has been found to recognize ManLAM and protect against a virulent M. tuberculosis strain both in vitro and in vivo (Luo et al., 2013). MASP-2 deficiency caused by homozygosity for the p.120G amino acid substitution was also recently reported in two persons with pulmonary tuberculosis (Sokolowska et al., 2015). MASP2 SNPs with a possible regulatory role in gene expression have been further associated with susceptibility to the disease, e.g. the heterozygous T/C genotype of a SNP located in intron 7 (rs6695096) increased the susceptibility to tuberculosis in Chinese individuals (OR = 1.67 [95%CI = 1.08–2.60], P < 0.05), especially in those cooking with solid fuel. The other investigated SNP – MASP2*Y371D–was not associated (Chen et al., 2014). The effect of other MASP2 SNPs/haplotypes and MASP1 polymorphisms have not yet been investigated. Gene-environment interactions as those investigated by Chen et al. (2014) may explain contrasting results of TB association studies with the other components of the lectin pathway, which are thus in need of replication in different cohorts.
4.2.3. Pseudomonas infection
The bacteria Pseudomonas aeruginosa causes various opportunistic infections, such as wound, pulmonary, urinary tract and blood stream infections, especially in immunocompromised individuals and cystic fibrosis patients (Chatzinikolaou et al., 2000, Santucci et al., 2003). Important characteristics of this pathogen are its resistance to many antibacterial agents, the capacity to survive in different environments and the expression of virulence factors (Trautmann et al., 2005).
A MASP-2 deficient mice model showed no significant association with survival or expression of cytokines in mice infected by P. aeruginosa. The authors concluded that the lectin pathway is not essential for complement activation in P. aeroginosa infection (Kenawy et al., 2012). In previous studies with cystic fibrosis patients, mutations in the MBL2 gene were associated with deterioration of lung function and a higher frequency of P. aeruginosa bacterial colonization (Gabolde et al., 1999, Garred et al., 1999, Haerynck et al., 2012). In contrast, no association between bacterial infections and MASP-2 levels and/or MASP2 polymorphisms has been found in two independent studies (Carlsson et al., 2005, Haerynck et al., 2012), despite the report of extremely severe lung disease in a cystic fibrosis patient homozygous for the p.120G variant (Olesen et al., 2006). In one of these studies, the authors nevertheless observed that MBL activation decreased significantly in MASP-2 deficient cystic fibrosis patients, but surprisingly not in healthy controls (Carlsson et al., 2005).
In contrast, a MASP1 synonymous SNP (rs850312) of exon 12, encoding the serine protease domain of MASP-3, was found associated with P. aeruginosa colonization in humans. Cystic fibrosis patients heterozygous or homozygous for the less frequent allele of this polymorphism were earlier colonized with P. aeruginosa than homozygous wild type patients (15 vs. 25 years of age, respectively, P = 0.0048). The association may in fact be due to a hitch-hiking effect with another MASP1 variant, since the authors investigated only three SNPs of this gene (Haerynck et al., 2012).
4.2.4. Streptococcal infection
Rheumatic fever (RF) and its most severe sequel chronic rheumatic heart disease (RHD) are chronic inflammations occurring in genetically predisposed children and teenagers, resulting from oropharynx infection by β-hemolytic Streptococcus group A. It affects the heart, joints, nervous system and skin and may progress to rheumatic heart disease, with stenosis of valve tissue and permanent heart damage (Chang, 2012). MBL and Ficolin-2 promote complement deposition on the streptococcal cell wall through binding to GlcNAc (Neth et al., 2000) and lipoteichoic acid (Lynch et al., 2004), respectively. Nevertheless, MBL levels were significantly higher in patients with RHD, than in controls, probably causing undesirable complement activation and tissue damage in RHD patients, whereas MBL2 variants leading to low MBL levels were found to protect against RF (Messias Reason et al., 2006, Schafranski et al., 2004). Instead, MASP-2 levels were found to be lower in 145 patients with history of RF from south Brazil (103 with RHD and 42 without cardiac lesion [RFo]), than in controls. Furthermore, the low MASP-2 producing p.377A and p.439H variants were negatively associated with RF (P = 0.02, OR = 0.36) and RHD (P = 0.01, OR = 0.25) and haplotypes producing higher MASP-2 concentrations, sharing the intron 9–exon 12g.1961795C, p.371D, p.377V and p.439R polymorphisms increased the susceptibility to RHD (P = 0.02, OR = 4.9). Thus, MASP2 gene polymorphisms and protein levels seem to play an important role in the development of RF and establishment of RHD (Catarino et al., 2014).
Post-streptococcal acute glomerulonephritis (PSAGN) also results from previous infection of the skin or throat caused by nephritogenic strains of group A beta-hemolytic streptococci. Antibodies produced to fight the infection may eventually settle in the glomeruli, causing inflammation (Hisano et al., 2007). In a study with kidney tissue obtained by percutaneous renal biopsy from 18 patients with PSAGN, the authors found glomerular co-localization of MBL and MASP-1. Glomerular deposits of fibrinogen were detected through immunohistochemical characterization, in five of seven patients with MBL/MASP-1 deposits but in only two of eleven patients without these complex deposits. Moreover in three of seven patients with MBL/MASP-1 deposits hematuria was prolonged, whereas in all patients without such deposits, urinalysis was normal (P < 0.025) (Hisano et al., 2007).
4.2.5. Pneumococcal infection
Streptococcus pneumoniae is the most common causative bacteria of respiratory infections from early infancy to old age (Krone et al., 2014). Pneumococcal otitis media is common among children, while pneumococcal pneumonia is a frequent cause of morbidity and mortality among elderly people. Also individuals with underlying special conditions, such as chronic diseases, those receiving immunosuppressive therapies, HIV positive or asplenic patients, are at risk of serious pneumococcal infection (Mészner, 2014).
Considering that complement activation protects against microorganism invasion through mechanisms either dependent or independent of antibodies, different studies investigated a role of complement pathways using complement deficient mice. The elimination of IgG and IgM opsonized pneumococci in a guinea pig model of pneumococcal bacteraemia was found in the early eighties to completely rely on complement activation (Brown et al., 1982). Although the classical pathway was initially proposed as mainly responsible for S. pneumoniae opsonization (Brown et al., 2002), neither the classical, nor the alternative activation pathway compensated for the absence of the lectin pathway in MASP-2 deficient mice, which were highly susceptible to pneumococcal infection and failed to opsonize S. pneumoniae (Ali et al., 2012). In fact, these mice presented similar infection survival times and mortality rates to those previously found for C1q and C4 deficient mice, employing the same strains of bacteria and mice, and the same dose and route of infection.
This study also demonstrated that mouse Ficolin-A and COLEC11, and not MBL-A or -C, are critical pattern recognition molecules that activate the complement via the lectin pathway on the surface of S. pneumonia (Brown et al., 2002). The authors also demonstrated that mouse Ficolin-A, which is closely related to human Ficolin-2, and COLEC11, but not MBL-A or -C, are critical pattern recognition molecules that activate the complement via the lectin pathway on the surface of S. pneumoniae (Ali et al., 2012). Another group concomitantly proved both Ficolins-1 and -2 to be essential for survival in ficolin-deficient mice infected with Streptococcus pneumonia (Endo et al., 2012). Taking into account the results in MASP-2 deficient mice, it is not surprising that the first patient described as homozygous for the p.D120G mutation presented recurrent infections, including severe pneumococcal pneumonia (Stengaard-Pedersen et al., 2003). Nevertheless no other investigation was conducted in humans with pneumococcal bacteremia, besides a case-control study with 54 HIV positive patients. In this particular study, the authors did not find any association with three polymorphisms associated with MASP-2 levels (p.R99Q, p.D120G and p.P126L). (Horcajada et al., 2009). In the light of the reported associations of MBL with respiratory infections, including a meta-analysis showing an association between MBL deficiency and death in patients with pneumococcal infection (reviewed by (Eisen, 2010)), further investigations on MASP levels and polymorphisms with susceptibility to pneumococcal disease should be warranted.
4.3. MASPs and parasitic diseases
Taking into account the capacity of PRMs of the lectin pathway in recognizing and binding different protozoan parasites (Ambrosio and De Messias-Reason, 2005, Kahn et al., 1996, Klabunde et al., 2002, Korir et al., 2014) and to modulate cytokine and chemokine levels during infection (Boldt et al., 2006), associations between dysfunctional MASP molecules, as well as MASP levels, are expected.
4.3.1. Malaria
Malaria, an infective disease caused by the parasite Plasmodium falciparum, is endemic in areas of sub-Saharan Africa, where between one and two million affected children under 5 years of age, die each year (Gupta et al., 1999). Various genetic variants modulate the immune response in malaria-affected populations and probably are maintained among them by balancing selection. With regard to lectin pathway components, MBL seems to play an important role during malaria infection by controlling parasitemia. Low protein levels and also low MBL producing polymorphisms were associated with malaria associated with severe anemia in Gabonese children (Boldt et al., 2006, Luty et al., 1998) and with positive parasite counts in asymptomatic P. falciparum infected Gabonese adults (Boldt et al., 2009) and Ghanaian children (Holmberga et al., 2008), as well as with placental malaria in primiparous Ghanaian women (Holmberg et al., 2012).
Despite of the relative importance of the lectin pathway in fighting P. falciparum infection, MASP1 and MASP2 polymorphisms have only been investigated in placental malaria. Among several variants genotyped in 269 primiparous Ghanaian women, of whom 54% had placental malaria, none of the investigated MASP1 variants were associated with the disease. Nevertheless, the p.439H variant encoded by MASP2 exon 12 showed a protective effect (OR = 0.55 [95%CI = 0.34–0.89], P = 0.014), especially against the presence of the hemozoin malarial pigment in the placenta, a feature suggestive of chronic placental infection (OR = 0.47 [95%CI = 0.26–0.83], P = 0.008). There was also a trend for a protective effect against low birth weight (OR = 0.51 [95%CI = 0.24–0.99], P = 0.057). Furthermore, the allele encoding the major p.126P variant, which occurs in absolute linkage disequilibrium with the allele encoding the major p.439R variant, was found to have the opposite effect, increasing susceptibility to the disease (P = 0.005). The p.439H mutation actually disrupts the activation peptide of MASP-2, which is still able to bind PRMs of the lectin pathway, but cannot activate complement (Thiel et al., 2009). The reasons why a dysfunctional MASP-2 protein protects against placental malaria nevertheless are not well understood, especially if taking into account that MBL deficiency presented the opposite effect, increasing susceptibility to placental malaria in the same patient group (Holmberg et al., 2012). Thus, these associations cannot solely be due to balanced/disturbed complement activation, a process dependent on intact MBL and MASP-2 molecules. Phagocytosis is expected to be hampered, since MASP-2 with the p.439H variant is able to bind PRMs of the lectin pathway and thus to block the interaction site with phagocytic receptors (Duus et al., 2010). In addition, p.439H-containing MASP-2 cannot generate opsonizing C3 fragments by complement activation (Thiel et al., 2009). This would mean less hemozoin uptake, explaining the strong negative association with hemozoin placental levels, and less consequent hemozoin-driven inflammation and tissue damage (Kalantari et al., 2013), explaining the negative association with chronic placental malaria. Functional studies on the p.439H MASP-2 variant will possibly clarify some of these issues.
4.3.2. Chagas disease
Chagas disease is one of the most important neglected tropical diseases worldwide, affecting 8–13 million people in Latin America, with an incidence of 40,000 new cases per year. The causal parasite Trypanosoma cruzi, a flagellated protozoan, is transmitted to humans through the sting of a blood-sucking triatomine bug (Lescure et al., 2010). After the infection, the lectin pathway becomes efficiently activated through the binding of MBL and Ficolins-2 and -3 to N- and O-glycosylated molecules on the parasite metacyclic trypomastigotes and amastigote surfaces (Fig. 4 ) (Kahn et al., 1996). The lysis of T. cruzi is dependent on MASP-2, which cleaves C2 and C4. In nonimmune serum depleted of MBL and ficolins, the classical pathway was inefficient to activate the complement response, and the alternative pathway was just slowly activated in the presence of the parasite. Therefore, the lectin pathway seems to play an important role by controling T. cruzi infection in humans (Cestari and Ramirez, 2010, Cestari et al., 2009). This issue is nevertheless controversial in T. cruzi-infected murine models (Gibson et al., 2012, Ribeiro et al., 2015). Susceptibility to T. cruzi infection was not increased in MASP-2-deficient mice, even though this mice lack functional activity of the lectin pathway (Ribeiro et al., 2015).
Fig. 4.
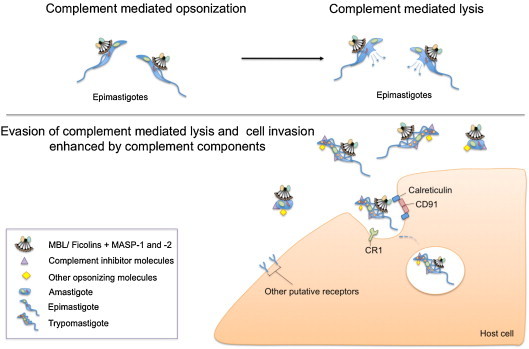
Dual role of complement proteins during Trypanosoma cruzi infection (Sánchez Valdéz et al., 2013). Epimastigotes, which are the flagellated forms in Triatominae vectors, are susceptible to complement-mediated lysis. Under natural conditions however, they are never confronted with it. On the other hand, the amastigote and trypomastigote forms infecting humans, resist complement-mediated lysis. Moreover, they use recognition by MBL/ficolins/MASP complexes to invade host cells. Several complement inhibitor molecules are expressed on T. cruzi surface, such as: Gp 58/68 (Fischer et al., 1988), Trypanosoma cruzi-decay accelerating factor (Norris et al., 1991), Complement regulator protein (Norris, 1998), Complement C2 receptor inhibitor trispanning (Inal et al., 2005) and Trypanosoma cruzi calreticulin (Ferreira et al., 2004). They are mostly glycoproteins and act destabilizing complexes formed by the proteins of the complement system.
T. cruzi becomes infective when it is able to evade the complement activation and thereby invading host cells. Several pathogens present molecules that stop the complement cascade and inhibit C3 convertase formation (Cestari et al., 2012). One of the molecules expressed in T. cruzi trypomastigotes (infectious state), is the complement C2 receptor inhibitor trispanning (CRIT). Derived peptides of this molecule inhibit both C2 binding and parasite killing. Biochemical studies have shown that the CRIT extracellular domain 1 inhibits MASP-2 cleavage of C2 and C3 convertase formation (Cestari et al., 2008).
Chagasic patients can present the indeterminate or symptomatic (cardiac, digestive or cardiodigestive) form of the disease. The persistence of the parasite could result in desialyation of myocardial and endothelial cells, with subsequent complement-driven tissue injury and onset of symptoms (Aiello et al., 2002). Higher levels/functional efficiency of lectin pathway components would thus be expected to increase these harmful effects and to be positively associated with symptomatic disease, which in fact was observed for MBL and for the FCN2*p.258S variant (Luz et al., 2013, Luz et al., 2010). The opposite was nevertheless observed regarding MASP2 polymorphisms in 208 chronic chagasic patients, of whom 59.1% were symptomatic. They actually had higher frequency of low producing genotypes (homozygotes for p.377A and heterozygotes for p.120G or for p.126L, excluding all those with the intron 9g.1961795*T allele) than controls (P = 0.011).
Comparing chronic symptomatic and indeterminate Chagas patients, a higher frequency of the MASP2 CD haplotype (1:g.11031148*C, p.371D) was observed among the first group (OR = 3.11 [95% CI = 1.46–6.61], P = 0.002), so as genotypes with CD, but without the 1:g.11047382*A in the promoter (OR = 11.11 [95%CI = 1.44–85.86], P = 0.0007). Cardiac symptomatic patients had an even stronger association with these genotypes (OR = 13.54 [95%CI = 1.7–107.63], P = 0.002). Again, high PRM levels and low MASP-2 levels were postulated by the authors to lead to increased MBL-driven phagocytosis and to higher inflammatory activity and injury in chronic Chagas disease.
Summarizing, there are strong evidences that complement activation by T. cruzi depends on MASP-2, which in association with MBL and ficolins, cleaves C2 and efficiently lysis the parasite. MASP2 polymorphisms might be useful prognostic biomarkers of Chagas disease, by allowing fast identification of patients with a higher risk to develop the chronic symptomatic form (Boldt et al., 2011a, Boldt et al., 2011b). This possibility however, is conditioned to the replication of the results of this genetic association.
5. Concluding remarks
Complement system has an important function in fighting infections by ultimately acting on pathogen lysis and infection clearance. The activation of MASPs through the lectin pathway of complement is an essential step in the establishment of an immune response against pathogen infection. MASP serum levels, sometimes driven by known genetic variants, have been related to a number of viral, bacterial and protozoan infections. These variants have been associated with different individual susceptibility to disease as well as disease outcome and complications. However, since it is unlikely to have prior disease protein measurements, it is usually difficult to differentiate between serum levels that were implicated in the cause of disease and those resulting from disease development or treatment response, if there is no associated polymorphism.
In general, low genetic expression, as well as non functional MASP proteins, lead to a compromised immune response against the pathogen, facilitating infection and disease progression. However, the complement system presents a dual role in disease susceptibility. It might be harmful if there is an excess of activation, promoting tissue damage, or by its lack of activity, predisposing to infection susceptibility. The observation of such dual behavior reinforces the importance of the fine balancing in complement regulation. MASP-1 and MASP-2 also participate in the coagulation system and MASP-1 can activate endothelial cells inducing pro-inflammatory responses (Gulla et al., 2010; Dobó et al., 2014), which can increase difficulty in understanding their roles in infectious diseases.
In this review, several pioneering studies were selected and discussed in order to pinpoint the importance of MASP proteins and gene polymorphisms in the susceptibility to infections and in the clinical progression to chronic infectious diseases. The results underline not only the expected importance of MASPs in the activation of the lectin pathway, but also in the generation of fibrin clots within the context of the coagulation cascade. Based on the proposed functions for MASP-3, MAp44 and MAp19, other immunomodulating functions are equally anticipated. Much work remains to be done, especially regarding the MASP1 polymorphism and functional studies on the role played by the truncated MAp19 and MAp44 proteins, as well as replication of unique disease association studies done with MASP2. Other diseases previously associated with polymorphisms and levels of MBL and ficolins, should be investigated as well. Then, first and foremost, MASP protein levels and genetic polymorphisms modulating MASP levels and function may be evaluated in immunocompromised and other at-risk patients to improve disease outcome using early therapeutic and preventive measures.
Acknowledgments
This work was supported by CNPq – Brazil (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and CAPES – Brazil (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).
References
- Abbas A.K., Lichtman A.H., Pillai S. 7 ed. EUA: Sauders Elsevier Inc.; 2012. Cellular and Molecular Immunology. [Google Scholar]
- Aiello V.D., Reis M.M., Benvenuti L.A., Higuchi M.D.L., Ramires J.A.F., Halperin J.a. A possible role for complement in the pathogenesis of chronic chagasic cardiomyopathy. J. Pathol. 2002;197:224–229. doi: 10.1002/path.1095. [DOI] [PubMed] [Google Scholar]
- Ali Y.M., Lynch N.J., Haleem K.S., Fujita T., Endo Y., Hansen S., Holmskov U., Takahashi K., Stahl G.L., Dudler T., Girija U.V., Wallis R., Kadioglu A., Stover C.M., Andrew P.W., Schwaeble W.J. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog. 2012;8:e1002793. doi: 10.1371/journal.ppat.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves Pedroso M.L., Boldt a.B.W., Pereira-Ferrari L., Steffensen R., Strauss E., Jensenius J.C., Ioshii S.O., Messias-Reason I. Mannan-binding lectin MBL2 gene polymorphism in chronic hepatitis C: association with the severity of liver fibrosis and response to interferon therapy. Clin. Exp. Immunol. 2008;152:258–264. doi: 10.1111/j.1365-2249.2008.03614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio A.R., De Messias-Reason I.J.T. Leishmania (Viannia) braziliensis: interaction of mannose-binding lectin with surface glycoconjugates and complement activation. An antibody-independent defence mechanism. Parasite Immunol. 2005;27:333–340. doi: 10.1111/j.1365-3024.2005.00782.x. [DOI] [PubMed] [Google Scholar]
- Ambrus G., Gál P., Kojima M., Szilágyi K., Balczer J., Antal J., Gráf L., Laich A., Moffatt B.E., Schwaeble W., Sim R.B., Závodszky P., Gal P., Kojima M., Szilagyi K., Balczer J., Antal J., Graf L., Laich A., Moffatt B.E., Schwaeble W., Sim R.B., Zavodszky P. Natural substrates and inhibitors of mannan-binding lectin-associated serine protease-1 and -2: a study on recombinant catalytic fragments. J. Immunol. 2003;170:1374–1382. doi: 10.4049/jimmunol.170.3.1374. [DOI] [PubMed] [Google Scholar]
- Ameye L., Paesmans M., Thiel S., Jensenius J.C., Aoun M. M-ficolin levels are associated with the occurrence of severe infections in patients with haematological cancer undergoing chemotherapy. Clin. Exp. Immunol. 2012;167:303–308. doi: 10.1111/j.1365-2249.2011.04512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammitzbøll C.G., Steffensen R., Nielsen H.J., Thiel S., Stengaard-Pedersen K., Bøgsted M., Jensenius J.C. Polymorphisms in the MASP1 gene are associated with serum levels of MASP-1, MASP-3, and MAp44. PLOS ONE. 2013;8:e73317. doi: 10.1371/journal.pone.0073317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders E.M., Hartley C.a., Jackson D.C. Bovine and mouse serum beta inhibitors of influenza A viruses are mannose-binding lectins. Proc. Natl. Acad. Sci. U. S. A. 1990;87:4485–4489. doi: 10.1073/pnas.87.12.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda N.K., Takahashi M. Essential role of complement mannose-binding lectin-associated serine proteases-1/3 in the murine collagen antibody-induced model of inflammatory arthritis. J. Immunol. 2010;185:5598–5606. doi: 10.4049/jimmunol.1001564.Essential. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda N.K., Mehta G., Kjaer T.R., Takahashi M., Schaack J., Morrison T.E., Thiel S., Arend W.P., Holers V.M. Essential role for the lectin pathway in collagen antibody-induced arthritis recealed through use of adenovirus programming complement inhibitor MAp44 expression. J. Immunol. 2014;193:2455–2468. doi: 10.4049/jimmunol.1400752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H., Liang T.J., Baumert T.F. Hepatitis C virus entry: molecular biology and clinical implications. Hepatology. 2006;44:527–535. doi: 10.1002/hep.21321. [DOI] [PubMed] [Google Scholar]
- Blackham S., Baillie A., Al-Hababi F., Remlinger K., You S., Hamatake R., McGarvey M.J. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 2010;84:5404–5414. doi: 10.1128/JVI.02529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt A.B., Goeldner I., Stahlke E.R.S., Thiel S., Jensenius J.C., de Messias-Reason I.J.T. Leprosy association with low MASP-2 levels generated by MASP2 haplotypes and polymorphisms flanking MAp19 Exon 5. PLOS ONE. 2013;8:e69054. doi: 10.1371/journal.pone.0069054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt A.B., Luty A., Grobusch M., Dietz K., Dzeing A., Kombila M., Kremsner P., Kun J. Association of a new mannose-binding lectin variant with severe malaria in Gabonese children. Genes Immun. 2006;7:393–400. doi: 10.1038/sj.gene.6364312. [DOI] [PubMed] [Google Scholar]
- Boldt A.B., Sanchez M.I.N., Stahlke E.R.S., Steffensen R., Thiel S., Jensenius J.C., Prevedello F.C., Mira M.T., Kun J.F.J., Messias-Reason I.J.T. Susceptibility to leprosy is associated with M-ficolin polymorphisms. J. Clin. Immunol. 2013;33:210–219. doi: 10.1007/s10875-012-9770-4. [DOI] [PubMed] [Google Scholar]
- Boldt A.B., Luz P.R., Messias-Reason I.J.T. MASP2 haplotypes are associated with high risk of cardiomyopathy in chronic Chagas disease. Clin. Immunol. 2011;140:63–70. doi: 10.1016/j.clim.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Boldt A.B., Messias-Reason I.J., Lell B., Issifou S., Pedroso M.L.A., Kremsner P.G., Kun J.F. Haplotype specific-sequencing reveals MBL2 association with asymptomatic Plasmodium falciparum infection. Malar. J. 2009;8:97. doi: 10.1186/1475-2875-8-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt A.B., Goeldner I., Messias-Reason I.J.T. Relevance of the lectin pathway of complement in rheumatic diseases. Adv. Clin. Chem. 2012;56 doi: 10.1016/B978-0-12-394317-0.00012-1. [DOI] [PubMed] [Google Scholar]
- Boldt A.B., Grisbach C., Steffensen R., Thiel S., Kun J., Jensenius J., Messias-Reason I. Multiplex sequence-specific polymerase chain reaction reveals new MASP2 haplotypes associated with MASP-2 and MAp19 serum levels. Hum. Immunol. 2011;72:753–760. doi: 10.1016/j.humimm.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Bonte L.R. La, Pavlov V.I., Tan Y.S., Takahashi K., Takahashi M., Banda N.K., Zou C., Fujita T., Stahl G.L. MBL-associated serine protease -1 (MASP-1) is a significant contributor to coagulation in a murine model of occusive thrombosis. J. Immunol. 2012;188:885–891. doi: 10.4049/jimmunol.1102916.MBL-Associated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E.J., Hosea S.W., Hammer C.H., Burch C.G. A quantitative analysis of the interactions of antipneumococcal antibody and complement in experimental pneumococcal bacteremia. J. Clin. Invest. 1982;69:85–98. doi: 10.1172/JCI110444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.S., Hussell T., Gilliland S.M., Holden D.W., Paton J.C., Ehrenstein M.R., Walport M.J., Botto M. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16969–16974. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.S., Keogh M.J., Owsianka A.M., Adair R., Patel A.H., Arnold J.N., Ball J.K., Sim R.B., Tarr A.W., Hickling T.P. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell. 2010;1:664–674. doi: 10.1007/s13238-010-0088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.S., Keogh M.J., Tagiuri N., Grainge M.J., Presanis J.S., Ryder S.D., Irving W.L., Ball J.K., Sim R.B., Hickling T.P. Severe fibrosis in hepatitis C virus-infected patients is associated with increased activity of the mannan-binding lectin (MBL)/MBL-associated serine protease 1 (MASP-1) complex. Clin. Exp. Immunol. 2007;147:90–98. doi: 10.1111/j.1365-2249.2006.03264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson M., Sjöholm a.G., Eriksson L., Thiel S., Jensenius J.C., Segelmark M., Truedsson L. Deficiency of the mannan-binding lectin pathway of complement and poor outcome in cystic fibrosis: bacterial colonization may be decisive for a relationship. Clin. Exp. Immunol. 2005;139:306–313. doi: 10.1111/j.1365-2249.2004.02690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino S.J.S., Boldt A.B., Beltrame M.H., Nisihara R.M., Schafranski M.D., de Messias-Reason I.J. Association of MASP2 polymorphisms and protein levels with rheumatic fever and rheumatic heart disease. Hum. Immunol. 2014;75:1197–1202. doi: 10.1016/j.humimm.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Cestari I.D.S., Ansa-Addo E., Deolindo P., Inal J.M., Ramirez M.I. Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J. Immunol. 2012;188:1942–1952. doi: 10.4049/jimmunol.1102053. [DOI] [PubMed] [Google Scholar]
- Cestari I.D.S., Ramirez M.I. Inefficient complement system clearance of Trypanosoma cruzi metacyclic trypomastigotes enables resistant strains to invade eukaryotic cells. PLoS ONE. 2010;5:e9721. doi: 10.1371/journal.pone.0009721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestari I.D.S., Evans-osses I., Freitas J.C., Inal J.M., Ramirez M.I. Complement C2 receptor inhibitor trispanning confers an increased ability to resist complement-mediated lysis in Trypanosoma cruzi. J. Infect. Dis. 2008;198:1276–1283. doi: 10.1086/592167. [DOI] [PubMed] [Google Scholar]
- Cestari I.D.S., Krarup A., Sim R.B., Inal J.M., Ramirez M.I. Role of early lectin pathway activation in the complement-mediated killing of Trypanosoma cruzi. Mol. Immunol. 2009;47:426–437. doi: 10.1016/j.molimm.2009.08.030. [DOI] [PubMed] [Google Scholar]
- Chang C. Cutting edge issues in rheumatic fever. Clin. Rev. Allergy Immunol. 2012;42:213–237. doi: 10.1007/s12016-011-8271-1. [DOI] [PubMed] [Google Scholar]
- Charchaflieh J., Wei J., Labaze G., Hou Y.J., Babarsh B., Stutz H., Lee H., Worah S., Zhang M. The role of complement system in septic shock. Clin. Dev. Immunol. 2012;2012:407324. doi: 10.1155/2012/407324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzinikolaou I., Abi-Said D., Bodey G.P., Rolston K.V.I., Tarrand J.J., Samonis G. Recent experience with pseudomonas aeruginosa bacteremia in patients with cancer. Arch. Intern. Med. 2000;160:501. doi: 10.1001/archinte.160.4.501. [DOI] [PubMed] [Google Scholar]
- Chen M., Daha M.R., Kallenberg C.G.M. The complement system in systemic autoimmune disease. J. Autoimmun. 2010;34:J276–J286. doi: 10.1016/j.jaut.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Chen M., Deng J., Su C., Li J., Wang M., Abuaku B.K., Hu S., Tan H., Wen S.W. Impact of passive smoking, cooking with solid fuel exposure, and MBL/MASP-2 gene polymorphism upon susceptibility to tuberculosis. Int. J. Infect. Dis. 2014;29:1–6. doi: 10.1016/j.ijid.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Christensen T., Petersen T., Thiel S., Brudek T., Ellermann-Eriksen S., Møller-Larsen A. Gene-environment interactions in multiple sclerosis: innate and adaptive immune responses to human endogenous retrovirus and herpesvirus antigens and the lectin complement activation pathway. J. Neuroimmunol. 2007;183:175–188. doi: 10.1016/j.jneuroim.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Coelho A., Brandão L., Guimarães R., Loureiro P., de Lima Filho J., de Alencar L., Crovellha S., Segat L. Mannose binding lectin and mannose binding lectin-associated serine protease-2 genes polymorphisms in human T-lymphotropic virus infection. J. Med. Virol. 2013;85:1829–1835. doi: 10.1002/jmv. [DOI] [PubMed] [Google Scholar]
- da Cruz H.L., da Silva R.C., Segat L., de Carvalho M.S., Brandao L.A., Guimarães R.L., Santos F.C., de Lira L.A., Montenegro L.M., Schindler H.C., Crovella S. MBL2 gene polymorphisms and susceptibility to tuberculosis in a northeastern Brazilian population. Infect. Genet. Evol. 2013;19:323–329. doi: 10.1016/j.meegid.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Dahl M.R., Thiel S., Matsushita M., Fujita T., Willis a.C., Christensen T., Vorup-Jensen T., Jensenius J.C. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15:127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- Davidson B., Abeler V.M., Førsund M., Holth A., Yang Y., Kobayashi Y., Chen L., Kristensen G.B., Shih I.-M., Wang T.-L. Gene expression signatures of primary and metastatic uterine leiomyosarcoma. Hum. Pathol. 2014;45:691–700. doi: 10.1016/j.humpath.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Messias-Reason I.J., Kremsner P.G., Kun J.F.J. Functional haplotypes that produce normal ficolin-2 levels protect against clinical leprosy. J. Infect. Dis. 2009;199:801–804. doi: 10.1086/597070. [DOI] [PubMed] [Google Scholar]
- De Messias-Reason I.J., Boldt A.B.W., Moraes Braga A.C., Von Rosen Seeling Stahlke E., Dornelles L., Pereira-Ferrari L., Kremsner P.G., Kun J.F.J. The association between mannan-binding lectin gene polymorphism and clinical leprosy: new insight into an old paradigm. J. Infect. Dis. 2007;196:1379–1385. doi: 10.1086/521627. [DOI] [PubMed] [Google Scholar]
- Degn S.E., Hansen A.G., Steffensen R., Jacobsen C., Jensenius J.C., Thiel S. MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J. Immunol. 2009;183:7371–7378. doi: 10.4049/jimmunol.0902388. [DOI] [PubMed] [Google Scholar]
- Degn S.E., Jensen L., Gál P., Dobó J., Holmvad S.H., Jensenius J.C., Thiel S. Biological variations of MASP-3 and MAp44, two splice products of the MASP1 gene involved in regulation of the complement system. J. Immunol. Methods. 2010;361:37–50. doi: 10.1016/j.jim.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Degn S.E., Jensen L., Olszowski T., Jensenius J.C., Thiel S. Co-complexes of MASP-1 and MASP-2 associated with the soluble pattern-recognition molecules drive lectin pathway activation in a manner inhibitable by MAp44. J. Immunol. 2013;191:1334–1345. doi: 10.4049/jimmunol.1300780. [DOI] [PubMed] [Google Scholar]
- Degn S.E., Kjaer T.R., Kidmose R.T., Jensen L., Hansen A.G., Tekin M., Jensenius J.C., Andersen G.R., Thiel S. Complement activation by ligand-driven juxtaposition of discrete pattern recognition complexes. Proc. Natl. Acad. Sci. U. S. A. 2014;111:13445–13450. doi: 10.1073/pnas.1406849111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn S.E., Thiel S. Humoral pattern recognition and the complement system. Scand. J. Immunol. 2013;78:181–193. doi: 10.1111/sji.12070. [DOI] [PubMed] [Google Scholar]
- Degn S.E., Thiel S., Nielsen O., Hansen A.G., Steffensen R., Jensenius J.C. MAp19, the alternative splice product of the MASP2 gene. J. Immunol. Methods. 2011;373:89–101. doi: 10.1016/j.jim.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn S.E., Jensen L., Hansen A.G., Duman D., Tekin M., Jensenius J.C., Thiel S. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J. Immunol. 2012;189:3957–3969. doi: 10.4049/jimmunol.1201736. [DOI] [PubMed] [Google Scholar]
- Devyatyarova-Johnson M., Rees I.H., Robertson B.D., Turner M.W., Klein N.J., Jack D.L. The lipopolysaccharide structures of Salmonella enterica serovar typhimurium and Neisseria gonorrhoeae determine the attachment of human mannose-binding lectin to intact organisms. Infect. Immun. 2000;68:3894–3899. doi: 10.1128/IAI.68.7.3894-3899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobó J., Harmat V., Beinrohr L., Sebestyén E., Závodszky P., Gál P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J. Immunol. 2009;183:1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
- Dobó J., Major B., Kékesi K.a., Szabó I., Megyeri M., Hajela K., Juhász G., Závodszky P., Gál P. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS ONE. 2011;6:e20036. doi: 10.1371/journal.pone.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobó J., Schroeder V., Jenny L., Cervenak L., Závodszky P., Gál P. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol. Immunol. 2014;61(2):69–78. doi: 10.1016/j.molimm.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Dommett R.M., Klein N., Turner M.W. Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens. 2006;68:193–209. doi: 10.1111/j.1399-0039.2006.00649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornelles L.N., Pereira-Ferrari L., Messias-Reason I. Mannan-binding lectin plasma levels in leprosy: deficiency confers protection against the lepromatous but not the tuberculoid forms. Clin. Exp. Immunol. 2006;145:463–468. doi: 10.1111/j.1365-2249.2006.03161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkelberger J.R., Song W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- Duus K., Thielens N.M., Lacroix M., Tacnet P., Frachet P., Holmskov U., Houen G. CD91 interacts with mannan-binding lectin (MBL) through the MBL-associated serine protease-binding site. FEBS J. 2010;277:4956–4964. doi: 10.1111/j.1742-4658.2010.07901.x. [DOI] [PubMed] [Google Scholar]
- Eisen D.P. Mannose-binding lectin deficiency and respiratory tract infection. J. Innate. Immun. 2010;2:114–122. doi: 10.1159/000228159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Saadany S.a., Ziada D.H., Farrag W., Hazaa S. Fibrosis severity and mannan-binding lectin (MBL)/MBL-associated serine protease 1 (MASP-1) complex in HCV-infected patients. Arab J. Gastroenterol. 2011;12:68–73. doi: 10.1016/j.ajg.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Endo Y., Takahashi M., Iwaki D., Ishida Y., Nakazawa N., Kodama T., Matsuzaka T., Kanno K., Liu Y., Tsuchiya K., Kawamura I., Ikawa M., Waguri S., Wada I., Matsushita M., Schwaeble W.W.J., Fujita T. Mice deficient in ficolin, a lectin complement pathway recognition molecule, are susceptible to Streptococcus pneumoniae infection. J. Immunol. 2012;189:5860–5866. doi: 10.4049/jimmunol.1200836. [DOI] [PubMed] [Google Scholar]
- Eurich D., Boas-Knoop S., Yahyazadeh A., Neuhaus R., Somasundaram R., Ruehl M., Puhl G., Neuhaus P., Neumann U.P., Bahra M. Role of mannose-binding lectin-2 polymorphism in the development of acute cellular rejection after transplantation for hepatitis C virus-induced liver disease. Transpl. Infect. Dis. 2012;14:488–495. doi: 10.1111/j.1399-3062.2012.00747.x. [DOI] [PubMed] [Google Scholar]
- Feinberg H., Uitdehaag J., Davies J. Crystal structure of the CUB1-EGF-CUB2 region of mannose-binding protein associated serine protease-2. EMBO J. 2003;22:2348–2359. doi: 10.1093/emboj/cdg236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira V., Valck C., Sanchez G., Gingras a., Tzima S., Molina M.C., Sim R., Schwaeble W., Ferreira a. The classical activation pathway of the human complement system is specifically inhibited by calreticulin from Trypanosoma cruzi. J. Immunol. 2004;172:3042–3050. doi: 10.4049/jimmunol.172.5.3042. [DOI] [PubMed] [Google Scholar]
- Fischer E., Ouaissi M.A., Velge P., Cornette J., Kazatchkine M.D. The trypomastigote form of T. cruzi from damage by the human alternative complement pathway. Immunology. 1988:299–303. [PMC free article] [PubMed] [Google Scholar]
- Frauenknecht V., Thiel S., Storm L., Meier N., Arnold M., Schmid J.-P., Saner H., Schroeder V. Plasma levels of mannan-binding lectin (MBL)-associated serine proteases (MASPs) and MBL-associated protein in cardio- and cerebrovascular diseases. Clin. Exp. Immunol. 2013;173:112–120. doi: 10.1111/cei.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabolde M., Feingold J., Besmond C. Association of variant alleles of mannose binding lectin with severity of pulmonary disease in cystic fibrosis: cohort study. Br. Med. J. 1999;319:1166–1167. doi: 10.1136/bmj.319.7218.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gál P., Harmat V., Kocsis A., Bián T., Barna L., Ambrus G., Végh B., Balczer J., Sim R.B., Náray-Szabó G., Závodszky P. A true autoactivating enzyme structural insight into mannose-binding lectin-associated serine protease-2 activations. J. Biol. Chem. 2005;280:33435–33444. doi: 10.1074/jbc.M506051200. [DOI] [PubMed] [Google Scholar]
- Garcia-laorden M.I., Sole-violan J., De Castro R., Aspa J. Mannose-binding lectin – associated serine protease 2 in susceptibility, severity, and outcome of pneumonia in adults. J. Allergy Clin. Immunol. 2008;122(2):368–374.e2. doi: 10.1016/j.jaci.2008.05.037. [DOI] [PubMed] [Google Scholar]
- Garred P., Harboe M., Oettinger T., Koch C., Svejgaard A. Dual role of mannan-binding protein in infections: another case of heterosis? Eur. J. Immunogenet. 1994;21:125–131. doi: 10.1111/j.1744-313X.1994.tb00183.x. [DOI] [PubMed] [Google Scholar]
- Garred P., Pressler T., Madsen H.O., Frederiksen B., Svejgaard a, Høiby N., Schwartz M., Koch C. Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J. Clin. Invest. 1999;104:431–437. doi: 10.1172/JCI6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha R.S., Notarangelo L.D., Casanova J.-L., Chapel H., Conley M.E., Fischer A., Hammarström L., Nonoyama S., Ochs H.D., Puck J.M., Roifman C., Seger R., Wedgwood J. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J. Allergy Clin. Immunol. 2007;120:776–794. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson A., Cheever A.W., Alan R., Rothfuchs A.G., Roffe E., Roffê E., Ezekowitz R.A.B., Takahashi K., Steindel M., Sher A., Báfica A. Mannose-binding lectin regulates host resistance and pathology during experimental infection with Trypanosoma cruzi. PLoS ONE. 2012;7:e47835. doi: 10.1371/journal.pone.0047835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.R., Girija U.V., Keeble A.H., Panchal R., Mitchell D.a., Moody P.C.E., Wallis R. Structural basis of mannan-binding lectin recognition by its associated serine protease MASP-1: implications for complement activation. Structure. 2011;19:1635–1643. doi: 10.1016/j.str.2011.08.014. [DOI] [PubMed] [Google Scholar]
- Granell M., Urbano-Ispizua A., Suarez B., Rovira M., Fernández-Avilés F., Martínez C., Ortega M., Uriburu C., Gaya A., Roncero J.M.A., Navarro A., Carreras E., Mensa J., Vives J., Rozman C., Montserrat E., Lozano F. Mannan-binding lectin pathway deficiencies and invasive fungal infections following allogeneic stem cell transplantation. Exp. Hematol. 2006;34:1435–1441. doi: 10.1016/j.exphem.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Gulla K.C., Gupta K., Krarup A., Gal P., Schwaeble W.J., Sim R.B., O’Connor C.D., Hajela K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology. 2010;129:482–495. doi: 10.1111/j.1365-2567.2009.03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Snow R., Donnelly C. Immunity to non-cerebral severe malaria is acquired after one or two infections. Nat. Med. 1999;5:3–6. doi: 10.1038/6560. [DOI] [PubMed] [Google Scholar]
- Haerynck F., Van Steen K., Cattaert T., Loeys B., Van Daele S., Schelstraete P., Claes K., Van Thielen M., De Canck I., Mahachie John J.M., De Baets F. Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum. Immunol. 2012;73:1175–1183. doi: 10.1016/j.humimm.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Héja D., Harmat V., Fodor K., Wilmanns M., Dobó J., Kékesi K.a., Závodszky P., Gál P., Pál G., Harmats V., Fodor K., Wilmanns M., Dobó J., Kékesi K.a., Závodszky P., Gál P., Pál G. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J. Biol. Chem. 2012;287:20290–20300. doi: 10.1074/jbc.M112.354332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Héja D., Kocsis A., Dobó J., Szilágyi K., Szász R., Závodszky P., Pál G., Gál P. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc. Natl. Acad. Sci. U. S. A. 2012;109:10498–10503. doi: 10.1073/pnas.1202588109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen M.L., Brandt J., Andrieu J.-P., Nielsen C., Jensen P.H., Holmskov U., Jorgensen T.J.D., Palarasah Y., Thielens N.M., Hansen S. Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J. Immunol. 2013;191:6117–6127. doi: 10.4049/jimmunol.1302121. [DOI] [PubMed] [Google Scholar]
- Hisano S., Matsushita M., Fujita T., Takeshita M., Iwasaki H. Activation of the lectin complement pathway in post-streptococcal acute glomerulonephritis. Pathol. Int. 2007;57:351–357. doi: 10.1111/j.1440-1827.2007.02107.x. [DOI] [PubMed] [Google Scholar]
- Holmberg V., Onkamo P., Lahtela E., Lahermo P., Bedu-Addo G., Mockenhaupt F.P., Meri S. Mutations of complement lectin pathway genes MBL2 and MASP2 associated with placental malaria. Malar. J. 2012;11:61. doi: 10.1186/1475-2875-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberga V., Schustera F., Ekkehart Dietzc J., Viscontia C.S., Anemanad S.D., Bienzlea U., Mockenhaupt F.P., Holmberg V., Schuster F., Dietz E., Sagarriga Visconti J.C., Anemana S.D., Bienzle U. Mannose-binding lectin variant associated with severe malaria in young African children. Microbes Infect. 2008;10:342–348. doi: 10.1016/j.micinf.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Holmskov U., Thiel S., Jensenius J.C. Collections and ficolins: humoral lectins of the innate immune defense. Annu. Rev. Immunol. 2003;21:547–578. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- Horcajada J.P., Lozano F., Muñoz A., Suarez B., Fariñas-Alvarez C., Almela M., Smithson A., Martínez E., Mallolas J., Mensa J., Gatell J.M. Polymorphic receptors of the innate immune system (MBL/MASP-2 and TLR2/4) and susceptibility to pneumococcal bacteremia in HIV-infected patients: a case-control study. Curr. HIV Res. 2009;7:218–223. doi: 10.2174/157016209787581382. [DOI] [PubMed] [Google Scholar]
- Hu Y.-L., Luo F.-L., Fu J.-L., Chen T.-L., Wu S.-M., Zhou Y.-D., Zhang X.-L. Early increased ficolin-2 concentrations are associated with severity of liver inflammation and efficacy of anti-viral therapy in chronic hepatitis C patients. Scand. J. Immunol. 2013;77:144–150. doi: 10.1111/sji.12014. [DOI] [PubMed] [Google Scholar]
- Inal J., Hui K., Miot S. Complement C2 receptor inhibitor trispanning: a novel human complement inhibitory receptor. J. Immunol. 2005;174:356–366. doi: 10.4049/jimmunol.174.1.356. [DOI] [PubMed] [Google Scholar]
- Ingels C., Vanhorebeek I., Steffensen R., Derese I., Jensen L., Wouters P.J., Hermans G., Thiel S., Van den Berghe G. Lectin pathway of complement activation and relation with clinical complications in critically ill children. Pediatr. Res. 2014;75:99–108. doi: 10.1038/pr.2013.180. [DOI] [PubMed] [Google Scholar]
- Iwaki D., Kanno K., Takahashi M., Endo Y., Matsushita M., Fujita T. The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. J. Immunol. 2011;187:3751–3758. doi: 10.4049/jimmunol.1100280. [DOI] [PubMed] [Google Scholar]
- Izumo S., Umehara F., Osame M. HTLV-I-associated myelopathy. Neuropathology. 2000;20:65–68. doi: 10.1046/j.1440-1789.2000.00320.x. [DOI] [PubMed] [Google Scholar]
- Jack D.L., Read R., Tenner A., Frosch M., Turner M., Klein N. Mannose-binding lectin regulates the inflammatory response of human professional phagocytes to Neisseria meningitidis serogroup B. J. Infect. Dis. 2001;184:1152–1162. doi: 10.1086/323803. [DOI] [PubMed] [Google Scholar]
- Jack D.L., Dodds a.W., Anwar N., Ison C.a., Law a., Frosch M., Turner M.W., Klein N.J. Activation of complement by mannose-binding lectin on isogenic mutants of Neisseria meningitidis serogroup B. J. Immunol. 1998;160:1346–1353. [PubMed] [Google Scholar]
- Jani P.K., Kajdácsi E., Megyeri M., Dobó J., Doleschall Z., Futosi K., Tímár C.I., Mócsai A., Makó V., Gál P., Cervenak L. MASP-1 induces a unique cytokine pattern in endothelial cells: a novel link between complement system and neutrophil granulocytes. PLoS ONE. 2014;9:e87104. doi: 10.1371/journal.pone.0087104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S., Wleklinski M., Ezekowitz R.A.B., Coder D., Aruffo A., Farr A. The major surface glycoprotein of Trypanosoma cruzi amastigotes are ligands of the human serum mannose-binding protein. Infect. Immun. 1996;64:2649–2656. doi: 10.1128/iai.64.7.2649-2656.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantari P., Deoliveira R.B., Chan J., Corbett Y., Rathinam V., Stutz A., Latz E., Gazzinelli R.T., Golenbock D.T., Fitzgerald K.A. Article dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium – derived hemozoin and DNA during malaria. Cell Rep. 2013;6:196–210. doi: 10.1016/j.celrep.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang I., Kim J.I., Chang S.G., Lee S.J., Choi S.L., Ha J., Kim S.S. Mannan-binding lectin (MBL)-associated plasma protein present in human urine inhibits calcium oxalate crystal growth. FEBS Lett. 1999;462:89–93. doi: 10.1016/S0014-5793(99)01509-4. [DOI] [PubMed] [Google Scholar]
- Kawakami M., Ihara I., Suzuki A., Harada Y. Properties of a new complement-dependent bactericidal factor specific for Ra chemotype salmonella in sera of conventional and germ-free mice. J. Immunol. 1982;129:2198–2201. [PubMed] [Google Scholar]
- Kenawy H.I., Ali Y.M., Rajakumar K., Lynch N.J., Kadioglu A., Stover C.M., Schwaeble W.J. Absence of the lectin activation pathway of complement does not increase susceptibility to Pseudomonas aeruginosa infections. Immunobiology. 2012;217:272–280. doi: 10.1016/j.imbio.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Kerr F.K.F., Thomas A.A.R., Wijeyewickrema L.C., Whisstock J.C., Boyd S.E., Kaiserman D., Matthews A.Y., Bird P.I., Thielens N.M., Rossi V., Pike R.N. Elucidation of the substrate specificity of the MASP-2 protease of the lectin complement pathway and identification of the enzyme as a major physiological target of the. Mol. Immunol. 2008;45:670–677. doi: 10.1016/j.molimm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Kimura A., Sakaguchi E., Nonaka M. Multi-component complement system of Cnidaria: C3, Bf, and MASP genes expressed in the endodermal tissues of a sea anemone, Nematostella vectensis. Immunobiology. 2009;214:165–178. doi: 10.1016/j.imbio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Kjaer T.R., Thiel S., Andersen G.R. Toward a structure-based comprehension of the lectin pathway of complement. Mol. Immunol. 2013;56:413–422. doi: 10.1016/j.molimm.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Kjaer T.R., Le le T.M., Pedersen J.S., Sander B., Golas M.M., Jensenius J.C., Andersen G.R., Thiel S. Structural insights into the initiating complex of the lectin pathway of complement activation. Structure. 2015;23:342–351. doi: 10.1016/j.str.2014.10.024. [DOI] [PubMed] [Google Scholar]
- Klabunde J., Uhlemann A.-C., Tebo A.E., Kimmel J., Schwarz R.T., Kremsner P.G., Kun J.F.J. Recognition of Plasmodium falciparum proteins by mannan-binding lectin, a component of the human innate immune system. Parasitol. Res. 2002;88:113–117. doi: 10.1007/s00436-001-0518-y. [DOI] [PubMed] [Google Scholar]
- Knittel T., Fellmer P., Neubauer K., Kawakami M., Grundmann A., Ramadori G. The complement-activating protease P100 is expressed by hepatocytes and is induced by IL-6 in vitro and during the acute phase reaction in vivo. Lab. Investig. 1997;77:221–230. [PubMed] [Google Scholar]
- Kocsis A., Kékesi K.a., Szász R., Végh B.M., Balczer J., Dobó J., Závodszky P., Gál P., Pál G. Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation. J. Immunol. 2010;185:4169–4178. doi: 10.4049/jimmunol.1001819. [DOI] [PubMed] [Google Scholar]
- García-Laorden M.I., García-Saavedra A., de Castro F.R., Violán J.S., Rajas O., Blanquer J., Borderías L., Rodríguez-Gallego C. Low clinical penetrance of mannose-binding lectin–associated serine protease 2 deficiency. J. Allergy Clin. Immunol. 2006;118(6):1383–1386. doi: 10.1016/j.jaci.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Korir J.C., Nyakoe N.K., Awinda G., Waitumbi J.N. Complement activation by merozoite antigens of Plasmodium falciparum. PLOS ONE. 2014;9 doi: 10.1371/journal.pone.0105093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsounaki E., Goulielmos G.N., Koulentaki M., Choulaki C., Kouroumalis E., Galanakis E. Mannose-binding lectin MBL2 gene polymorphisms and outcome of hepatitis C virus-infected patients. J. Clin. Immunol. 2008;28:495–500. doi: 10.1007/s10875-008-9201-8. [DOI] [PubMed] [Google Scholar]
- Krarup A., Gulla K.C., Gál P., Hajela K., Sim R.B. The action of MBL-associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Biochim. Biophys. Acta. 2008;1784:1294–1300. doi: 10.1016/j.bbapap.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Krarup A., Sørensen U.B.S., Matsushita M., Jensenius J.C., Thiel S. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules Mannan-binding lectin, L-ficolin, and H-ficolin. Infect. Immun. 2005;73:1052–1060. doi: 10.1128/IAI.73.2.1052-1060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krone C.L., van de Groep K., Trzciński K., Sanders E.A.M., Bogaert D. Immunosenescence and pneumococcal disease: an imbalance in host–pathogen interactions. Lancet. Respir. Med. 2014;2:141–153. doi: 10.1016/S2213-2600(13)70165-6. [DOI] [PubMed] [Google Scholar]
- Kuhlman M., Joiner K., Ezekowitz R.A.B. The human mannose-binding protein functions as an opsonin. J. Exp. Med. 1989;169:1733–1745. doi: 10.1084/jem.169.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen I.a., Thielens N.M., Christiansen M., Houen G. MASP interactions with plasma-derived MBL. Mol. Immunol. 2012;52:79–87. doi: 10.1016/j.molimm.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Lescure F.-X., Le Loup G., Freilij H., Develoux M., Paris L., Brutus L., Pialoux G. Chagas disease: changes in knowledge and management. Lancet. Infect. Dis. 2010;10:556–570. doi: 10.1016/S1473-3099(10)70098-0. [DOI] [PubMed] [Google Scholar]
- Liu J., Ali M.a.M., Shi Y., Zhao Y., Luo F., Yu J., Xiang T., Tang J., Li D., Hu Q., Ho W., Zhang X. Specifically binding of L-ficolin to N-glycans of HCV envelope glycoproteins E1 and E2 leads to complement activation. Cell. Mol. Immunol. 2009;6:235–244. doi: 10.1038/cmi.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Cao C., Ma Q. Study on interaction between SARS-CoV N and MAP19, Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi – Chinese. J. Cell. Mol. Immunol. 2009;25:777–779. [PubMed] [Google Scholar]
- Luo F., Sun X., Wang Y., Wang Q., Yanhong W., Pan Q., Zhang X.L. Ficolin-2 defends against virulent Mycobacteria tuberculosis infection in vivo, and its insufficiency is associated with infection in humans. PLOS ONE. 2013;8:e73859. doi: 10.1371/journal.pone.0073859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luty A.J.F., Kun F.J., Kremsner P.G. Mannose-binding lectin plasma levels and gene polymorphisms in Plasmodium falciparum. Malaria. 1998;178:1996–1999. doi: 10.1086/515690. [DOI] [PubMed] [Google Scholar]
- Luz P.R., Boldt A.B., Grisbach C., Kun J.F.J., Velavan T.P., Messias-Reason I.J.T. Association of L-ficolin levels and FCN2 genotypes with chronic Chagas disease. PLOS ONE. 2013;8:e60237. doi: 10.1371/journal.pone.0060237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz P.R., Miyazaki M.I., Neto N.C., Nisihara R.M., Messias-Reason I.J. High levels of mannose-binding lectin are associated with the risk of severe cardiomyopathy in chronic Chagas disease. Int. J. Cardiol. 2010;143:448–450. doi: 10.1016/j.ijcard.2009.09.467. [DOI] [PubMed] [Google Scholar]
- Lynch N.J., Roscher S., Hartung T., Morath S., Matsushita M., Maennel D.N., Kuraya M., Fujita T., Schwaeble W.J. L-ficolin specifically binds to lipoteichoic acid, a cell wall constituent of Gram-Positive bacteria, and activates the lectin pathway of complement. J. Immunol. 2004;172:1198–1202. doi: 10.4049/jimmunol.172.2.1198. [DOI] [PubMed] [Google Scholar]
- Ma Y.J., Skjoedt M.-O., Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway – the fifth lectin pathway initiation complex. J. Innate Immun. 2013;5:242–250. doi: 10.1159/000345356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M., Fujita T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 1992;176:1497–1502. doi: 10.1084/jem.176.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M., Thiel S., Jensenius J.C., Terai I., Fujita T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J. Immunol. 2000;165:2637–2642. doi: 10.4049/jimmunol.165.5.2637. [DOI] [PubMed] [Google Scholar]
- Megyeri M., Harmat V., Major B., Végh Á., Balczer J., Héja D., Szilágyi K., Datz D., Pál G., Závodszky P., Gál P., Dobó J. Quantitative characterization of the activation steps of mannan-binding lectin (MBL)-associated serine proteases (MASPs) points to the central role of MASP-1 in the initiation of the complement lectin pathway. J. Biol. Chem. 2013;288:8922–8934. doi: 10.1074/jbc.M112.446500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megyeri M., Makó V., Beinrohr L., Doleschall Z., Prohászka Z., Cervenak L., Závodszky P., Gál P. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J. Immunol. 2009;183:3409–3416. doi: 10.4049/jimmunol.0900879. [DOI] [PubMed] [Google Scholar]
- Melo F.M., Vasconcelos L.R.S., Silva B.S., Moura P., Cavalcanti M.S.M., Pereira L.M.M.B., Lacerda H.R. Structural polymorphism of the mannose-binding lectin 2 (MBL2) gene in HCV-infected patients with a serological marker for thyroid autoimmunity. Int. J. Immunogenet. 2009;36:377–381. doi: 10.1111/j.1744-313X.2009.00871.x. [DOI] [PubMed] [Google Scholar]
- Messias Reason I.J., Schafranski M.D., Jensenius J.C., Steffensen R. The association between mannose-binding lectin gene polymorphism and rheumatic heart disease. Hum. Immunol. 2006;67:991–998. doi: 10.1016/j.humimm.2006.08.296. [DOI] [PubMed] [Google Scholar]
- Messias-Reason I.J., Boldt A.B. Mannose-binding lectin, a multifunctional molecule of the innate immune system: biology, genetics and disease association. In: Torres S.L., Marin M.S., editors. Vol. 11788. Nova Science Publishers, Inc.; Hauppauge, NY: 2008. pp. 119–174. (Genetic Predispositon to Disease). [Google Scholar]
- Mészner Z. Pneumococcal disease prevention from early infancy to old age. Orv. Hetil. 2014;155:243–247. doi: 10.1556/OH.2014.29753. [DOI] [PubMed] [Google Scholar]
- Miller C., Wilgenbusch S., Michaels M., Chi D.S., Youngberg G., Krishnaswamy G. Molecular defects in the mannose binding lectin pathway in dermatological disease: case report and literature review. Clin. Mol. Allergy. 2010;8(6):1476–7961. doi: 10.1186/1476-7961-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller-Kristensen M., Jensenius J.C., Jensen L., Thielens N., Rossi V., Arlaud G., Thiel S. Levels of mannan-binding lectin-associated serine protease-2 in healthy individuals. J. Immunol. Methods. 2003;282:159–167. doi: 10.1016/j.jim.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Neth O., Jack D.L., Dodds a.W., Holzel H., Klein N.J., Turner M.W. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect. Immun. 2000;68:688–693. doi: 10.1128/IAI.68.2.688-693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka M., Miyazawa S. Evolution of the initiating enzymes of the complement system. Genome Biol. 2001;3(1) doi: 10.1186/gb-2001-3-1-reviews1001. reviews 1001.1–1001.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris K.A. Stable transfection of Trypanosoma cruzi epimastigotes with the trypomastigote-specific complement regulatory protein cDNA confers complement resistance. Infect. Immun. 1998;66:2460–2465. doi: 10.1128/iai.66.6.2460-2465.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris K.A., Bradt B., Cooper N.R., So M. Characterization of a Trypanosoma cruzi C3 binding protein with functional and genetic similarities to the human complement regulatory protein, decay-accelerating factor. J. Immunol. 1991;147:2240–2247. [PubMed] [Google Scholar]
- Olesen H.V., Jensenius J.C., Steffensen R., Thiel S., Schiøtz P.O. The mannan-binding lectin pathway and lung disease in cystic fibrosis – disfunction of mannan-binding lectin-associated serine protease 2 (MASP-2) may be a major modifier. Clin. Immunol. 2006;121:324–331. doi: 10.1016/j.clim.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Paréj K., Hermann A., Donáth N., Závodszky P., Gál P., Dobó J. Dissociation and re-association studies on the interaction domains of mannan-binding lectin (MBL)-associated serine proteases. MASP-1 and MASP-2, provide evidence for heterodimer formation. Mol. Immunol. 2014;59:1–9. doi: 10.1016/j.molimm.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Pavlov V.I., Skjoedt M.-O., Siow Tan Y., Rosbjerg A., Garred P., Stahl G.L. Endogenous and natural complement inhibitor attenuates myocardial injury and arterial thrombogenesis. Circulation. 2012;126:2227–2235. doi: 10.1161/CIRCULATIONAHA.112.123968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polotsky V.Y., Belisle J.T., Mikusova K., Ezekowitz R.A., Joiner K.A. Interaction of human mannose-binding protein with Mycobacterium avium. J. Infect. Dis. 1997;175:1159–1168. doi: 10.1086/520354. [DOI] [PubMed] [Google Scholar]
- Presanis J., Hajela K., Ambrus G., Gál P., Sim R.B. Differential substrate and inhibitor profiles for human MASP-1 and MASP-2. Mol. Immunol. 2004;40:921–929. doi: 10.1016/j.molimm.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Ramasawmy R., Spina G.S.G.G.S., Fae K.C., Pereira A.C., Nisihara R., Reason I.J.M., Grinberg M., Tarasoutchi F., Kalil J., Guilherme L., Kellen C., Pereira A.C., Nisihara R., Jose I., Reason M., Grinberg M., Tarasoutchi F., Guilherme L., Fae K.C., Kalil J. Association of mannose-binding lectin gene polymorphism but not of mannose-binding serine protease 2 with chronic severe aortic regurgitation of rheumatic etiology. Clin. Vaccine Immunol. 2008;15:932–936. doi: 10.1128/CVI.00324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Ding Q., Zhang X. Ficolins and infectious diseases. Virol. Sin. 2014;29:25–32. doi: 10.1007/s12250-014-3421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro C.H., Lynch N.J., Stover C.M., Ali Y.M., Valck C., Noya-leal F., Schwaeble W.J., Ferreira A. Deficiency in mannose-binding lectin-associated serine protease-2 does not increase susceptibility to Trypanosoma cruzi infection. Am. J. Trop. Med. Hyg. 2015;92(2):320–324. doi: 10.4269/ajtmh.14-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D., Hajishengallis G., Yang K., Lambris J.D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronen R., Udpa N., Halperin E., Bafna V. Learning natural selection from the site frequency spectrum. Genetics. 2013;195:181–193. doi: 10.1534/genetics.113.152587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooryck C., Diaz-Font A., Osborn D.P.S., Chabchoub E., Hernandez-Hernandez V., Shamseldin H., Kenny J., Waters A., Jenkins D., Kaissi A.A., Leal G.F., Dallapiccola B., Carnevale F., Bitner-Glindzicz M., Lees M., Hennekam R., Stanier P., Burns A.J., Peeters H., Alkuraya F.S., Beales P.L. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat. Genet. 2011;43:197–203. doi: 10.1038/ng.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed a, Baloch K., Brown R.J.P., Wallis R., Chen L., Dexter L., McClure C.P., Shakesheff K., Thomson B.J. Mannan binding lectin-associated serine protease 1 is induced by hepatitis C virus infection and activates human hepatic stellate cells. Clin. Exp. Immunol. 2013;174:265–273. doi: 10.1111/cei.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez Valdéz F.J., Pérez Brandán C., Zago M.P., Labriola C., Ferreira A., Basombrío M.Á. Trypanosoma cruzi carrying a monoallelic deletion of the calreticulin (TcCRT) gene are susceptible to complement mediated killing and defective in their metacyclogenesis. Mol. Immunol. 2013;53:198–205. doi: 10.1016/j.molimm.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Santucci S.G., Gobara S., Santos C.R., Fontana C., Levin A.S. Infections in a burn intensive care unit: experience of seven years. J. Hosp. Infect. 2003;53:6–13. doi: 10.1053/jhin.2002.1340. [DOI] [PubMed] [Google Scholar]
- Sapkota B.R., Macdonald M., Berrington W.R., Misch E.A., Ranjit C., Siddiqui M.R., Kaplan G., Hawn T.R. Association of TNF, MBL, and VDR polymorphisms with leprosy phenotypes. Hum. Immunol. 2010;71:992–998. doi: 10.1016/j.humimm.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Endo Y., Matsushita M., Fujita T. Molecular characterization of a novel serine protease involved in activation of the complement system by mannose-binding protein. Int. Immunol. 1994;6(4):665–669. doi: 10.1093/intimm/6.4.665. [DOI] [PubMed] [Google Scholar]
- Schadt E.E., Molony C., Chudin E., Hao K., Yang X., Lum P.Y., Kasarskis A., Zhang B., Wang S., Suver C., Zhu J., Millstein J., Sieberts S., Lamb J., GuhaThakurta D., Derry J., Storey J.D., Avila-Campillo I., Kruger M.J., Johnson J.M., Rohl C.a., Van Nas A., Mehrabian M., Drake T.a., Lusis A.J., Smith R.C., Guengerich F.P., Strom S.C., Schuetz E., Rushmore T.H., Ulrich R. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:1020–1032. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafranski M.D., Pereira Ferrari L., Scherner D., Torres R., de Messias-Reason I.J., Derbli M., Pereira L., De Messias-reason I.J. Functional MASP2 gene polymorphism in patients with history of rheumatic fever. Hum. Immunol. 2008;69:41–44. doi: 10.1016/j.humimm.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Schafranski M.D., Stier A., Nisihara R., Messias-Reason I.J.T. Significantly increased levels of mannose-binding lectin (MBL) in rheumatic heart disease: a beneficial role for MBL deficiency. Clin. Exp. Immunol. 2004;138:521–525. doi: 10.1111/j.1365-2249.2004.02645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlapbach L.J., Aebi C., Otth M., Leibundgut K., Hirt A., Ammann R.A. Deficiency of mannose-binding lectin-associated serine protease-2 associated with increased risk of fever and neutropenia in pediatric cancer patients. Pediatr. Infect. Dis. J. 2007;26:989–994. doi: 10.1097/INF.0b013e31811ffe6a. [DOI] [PubMed] [Google Scholar]
- Schlesingert L.S., Horwitz M.A. A role for natural antibody in the pathogenesis of leprosy: antibody in nonimmune serum mediates C3 Fixation to the Mycobacterium leprae surface and hence phagocytosis by human mononuclear phagocytes. Infect. Immun. 1994;62:280–289. doi: 10.1128/iai.62.1.280-289.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segat L., Fabris A., Padovan L., Milanese M., Pirulli D., Lupo F., Salizzoni M., Amoroso A., Crovella S. MBL2 and MASP2 gene polymorphisms in patients with hepatocellular carcinoma. J. Viral Hepat. 2008;15:387–391. doi: 10.1111/j.1365-2893.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- Segat L., Silva Vasconcelos L.R., Montenegro de Melo F., Santos Silva B., Arraes L.C., Moura P., Crovella S. Association of polymorphisms in the first exon of mannose binding lectin gene (MBL2) in Brazilian patients with HCV infection. Clin. Immunol. 2007;124:13–17. doi: 10.1016/j.clim.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Sirmaci A., Walsh T., Akay H., Spiliopoulos M., Sakalar Y.B., Hasanefendioğlu-Bayrak A., Duman D., Farooq A., King M.-C., Tekin M. MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndromes. Am. J. Hum. Genet. 2010;87:679–686. doi: 10.1016/j.ajhg.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjoedt M.-O., Hummelshoj T., Palarasah Y., Honore C., Koch C., Skjodt K., Garred P. A novel mannose-binding lectin/ficolin-associated protein is highly expressed in heart and skeletal muscle tissues and inhibits complement activation. J. Biol. Chem. 2010;285:8234–8243. doi: 10.1074/jbc.M109.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjoedt M.-O., Palarasah Y., Munthe-fog L., Jie Ma Y., Weiss G., Skjodt K., Koch C., Garred P. MBL-associated serine protease-3 circulates in high serum concentrations predominantly in complex with Ficolin-3 and regulates Ficolin-3 mediated complement activation. Immunobiology. 2010;215:921–931. doi: 10.1016/j.imbio.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Sokolowska A., Szala A., St Swierzko A., Kozinska M., Niemiec T., Blachnio M., Augustynowicz-Kopec E., Dziadek J., Cedzynski M. Mannan-binding lectin-associated serine protease-2 (MASP-2) deficiency in two patients with pulmonary tuberculosis and one healthy control. Cell. Mol. Immunol. 2015;12:119–121. doi: 10.1038/cmi.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengaard-Pedersen K., Thiel S., Gadjeva M., Møller-Kristensen M., Sørensen R., Jensen L.T., Sjøholm A.G., Fugger L., Jensenius J.C. Inherited deficiency of mannan-binding lectin-associated serine protease 2. N. Engl. J. Med. 2003;349:554–560. doi: 10.1056/NEJMoa022836. [DOI] [PubMed] [Google Scholar]
- Stover C.M., Endo Y., Takahashi M., Lynch N.J., Constantinescu C., Vorup-Jensen T., Thiel S., Friedl H., Hankeln T., Hall R., Gregory S., Fujita T., Schwaeble W. The human gene for mannan-binding lectin-associated serine protease-2 (MASP-2), the effector component of the lectin route of complement activation, is part of a tightly linked gene cluster on chromosome 1p36.2-3. Genes Immun. 2001;2:119–127. doi: 10.1038/sj.gene.6363745. [DOI] [PubMed] [Google Scholar]
- Stover C.M., Lynch N.J., Dahl M.R., Hanson S., Takahashi M., Frankenberger M., Ziegler-Heitbrock L., Eperon I., Thiel S., Schwaeble W.J. Murine serine proteases MASP-1 and MASP-3, components of the lectin pathway activation complex of complement, are encoded by a single structural gene. Genes Immun. 2003;4:374–384. doi: 10.1038/sj.gene.6363970. [DOI] [PubMed] [Google Scholar]
- Stover C.M., Thiel S., Thelen M., Lynch N.J., Vorup-Jensen T., Jensenius J.C., Schwaeble W.J. Two constituents of the initiation complex of the mannan-binding lectin activation pathway of complement are encoded by a single structural gene. J. Immunol. 1999;162:3481–3490. [PubMed] [Google Scholar]
- Stover C.M., Schwaeble W.J., Lynch N.J., Thiel S., Speicher M.R. Assignment of the gene encoding mannan-binding lectin-associated serine protease 2 (MASP2) to human chromosome 1p36.3→p36.2 by in situ hybridization and somatic cell hybrid analysis. Cytogenet. Cell Genet. 1999;84:148–149. doi: 10.1159/000015243. [DOI] [PubMed] [Google Scholar]
- Tabouret G., Astarie-dequeker C., Demangel C., Malaga W., Bello N.F., Perez E., Guilhot C. Mycobacterium leprae phenolglycolipid-1 expressed by engineered M. bovis BCG modulates early interaction with human phagocytes. PLoS Pathog. 2010;6(10):e1001159. doi: 10.1371/journal.ppat.1001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada F., Seki N., Matsuda Y., Takayama Y., Kawakami M. Localization of the genes for the 100-kDa complement-activating components of Ra-reactive factor (CRARF and Crarf) to human 3q27-q28 and mouse 16B2-B3. Genomics. 1995;25:757–759. doi: 10.1016/0888-7543(95)80027-J. [DOI] [PubMed] [Google Scholar]
- Takada F., Takayama Y., Hatsuse H., Kawakami M. A new member of the C1s family of complement proteins found in a bactericidal factor, Ra-reactive factor, in human serum. Biochem. Biophys. Res. Commun. 1993;196:1003–1009. doi: 10.1006/bbrc.1993.2349. [DOI] [PubMed] [Google Scholar]
- Takahashi M., Endo Y., Fujita T., Matsushita M. A truncated form of mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative polyadenylation is a component of the lectin complement pathway. Int. Immunol. 1999;11:859–863. doi: 10.1093/intimm/11.5.859. [DOI] [PubMed] [Google Scholar]
- Tarr A.W., Urbanowicz R.a., Ball J.K. The role of humoral innate immunity in hepatitis C virus infection. Viruses. 2012;4:1–27. doi: 10.3390/v4010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol. Immunol. 2007;44:3875–3888. doi: 10.1016/j.molimm.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Thiel S., Jensen L., Degn S.E., Nielsen H.J., Gál P., Dobó J., Jensenius J.C. Mannan-binding lectin (MBL)-associated serine protease-1 (MASP-1), a serine protease associated with humoral pattern-recognition molecules: normal and acute-phase levels in serum and stoichiometry of lectin pathway components. Clin. Exp. Immunol. 2012;169:38–48. doi: 10.1111/j.1365-2249.2012.04584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel S., Kolev M., Degn S., Steffensen R., Hansen A.G., Ruseva M., Jensenius J.C. Polymorphisms in mannan-binding lectin (MBL)-associated serine protease 2 affect stability, binding to MBL, and enzymatic activity. J. Immunol. 2009;182:2939–2947. doi: 10.4049/jimmunol.0802053. [DOI] [PubMed] [Google Scholar]
- Thiel S., Steffensen R., Christensen I., Ip W.K., Lau Y., Reason I., Eiberg H., Gadjeva M., Ruseva M., Jensenius J.C. Deficiency of mannan-binding lectin associated serine protease-2 due to missense polymorphisms. Genes Immun. 2007;8:154–163. doi: 10.1038/sj.gene.6364373. [DOI] [PubMed] [Google Scholar]
- Thiel S., Vorup-Jensen T., Stover C. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–510. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- Thielens N.M., Cseh S., Thiel S., Vorup-Jensen T., Rossi V., Jensenius J.C., Arlaud G.J. Interaction properties of human mannan-binding lectin (MBL)-associated serine proteases-1 and -2, MBL-associated protein 19, and MBL. J. Immunol. 2001;166:5068–5077. doi: 10.4049/jimmunol.166.8.5068. [DOI] [PubMed] [Google Scholar]
- Thielens N.M., Tacnet-Delorme P., Arlaud G.J. Interaction of C1q and mannan-binding lectin with viruses. Immunobiology. 2002;205:563–574. doi: 10.1078/0171-2985-00155. [DOI] [PubMed] [Google Scholar]
- Thimme R., Lohmann V., Weber F. A target on the move: innate and adaptive immune escape strategies of hepatitis C virus. Antiviral Res. 2006;69:129–141. doi: 10.1016/j.antiviral.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Trautmann M., Lepper P.M., Haller M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control. 2005;33:S41–S49. doi: 10.1016/j.ajic.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Tulio S., Faucz F., Werneck R. MASP2 gene polymorphism is associated with susceptibility to hepatitis C virus infection. Hum. Immunol. 2011;72:912–915. doi: 10.1016/j.humimm.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thye T., Niemann S., Walter K., Homolka S., Intermann D.D., Chinbua M.A., Enimil A., Gyapong J., Osei I., Owusu-Dabo E., Rusch-Gerdes S., Horstmann R.D., Ehlers S., Meyer C.G. Variant G57E of mannose binding lectin associated with protection against tuberculosis caused by Mycobacterium africanum but not by M. tuberculosis. PLoS ONE. 2011;6:e20908. doi: 10.1371/journal.pone.0020908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterberger C., Hanson S., Klingenhoff A., Oesterle D., Frankenberger M., Endo Y., Matsushita M., Fujita T., Schwaeble W., Weiss E.E.H., Ziegler-Heitbrock L., Stover C.M. Stat3 is involved in control of MASP2 gene expression. Biochem. Biophys. Res. Commun. 2007;364:1022–1025. doi: 10.1016/j.bbrc.2007.10.114. [DOI] [PubMed] [Google Scholar]
- Valles X., Sarrias M., Casals F., Farnós M., Piñer R., Suárez B., Morais L., Mandomando I., Sigaúque B., Roca A., Alonso P., Torres A., Thielens N., Lozano F. Genetic and structural analysis of MBL2 and MASP2 polymorphisms in south-eastern African children. Tissue Antigens. 2009;74:298–307. doi: 10.1111/j.1399-0039.2009.01328.x. [DOI] [PubMed] [Google Scholar]
- Wallis R., Dodd R.B. Interaction of mannose-binding protein with associated serine proteases: effects of naturally occurring mutations. J. Biol. Chem. 2000;275:30962–30969. doi: 10.1074/jbc.M004030200. [DOI] [PubMed] [Google Scholar]
- Walport M. Complement. First of two parts. N. Engl. J. Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- Weiss G., Madsen H.O., Garred P. A novel mannose-binding lectin-associated serine protease 1/3 gene variant. Scand. J. Immunol. 2007;65:430–434. doi: 10.1111/j.1365-3083.2007.01925.x. [DOI] [PubMed] [Google Scholar]
- World Health Organization Leprosy. 2012;101:1–3. [Google Scholar]
- World Health Organization . WHO Libr. Cat. Data; 2014. Global Tuberculosis Report. [Google Scholar]
- Yongqing T., Drentin N., Duncan R.C., Wijeyewickrema L.C., Pike R.N. Mannose-binding lectin serine proteases and associated proteins of the lectin pathway of complement: two genes, five proteins and many functions? Biochim. Biophys. Acta. 2012;1824(1):253–262. doi: 10.1016/j.bbapap.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Ytting H., Christensen I.J., Steffensen R., Alsner J., Thiel S., Jensenius J.C., Hansen U., Nielsen H.J. Mannan-binding lectin (MBL) and MBL-associated serine protease 2 (MASP-2) genotypes in colorectal cancer. Clin. Immunol. 2011;73:122–127. doi: 10.1111/j.1365-3083.2010.02480.x. [DOI] [PubMed] [Google Scholar]
- Ytting H., Christensen I.J., Thiel S., Jensenius J.C., Svendsen M.N., Nielsen L., Lottenburger T., Nielsen H.J. Biological variation in circulating levels of mannan-binding lectin (MBL) and MBL-associated serine protease-2 and the influence of age, gender and physical exercise. Scand. J. Immunol. 2007;66:458–464. doi: 10.1111/j.1365-3083.2007.01991.x. [DOI] [PubMed] [Google Scholar]
- Ytting H., Jarle I., Thiel S., Christian J., Jørgen H. Pre- and postoperative levels in serum of mannan-binding lectin associated serine protease-2 — a prognostic marker in colorectal cancer. Hum. Immunol. 2008;69:414–420. doi: 10.1016/j.humimm.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Zhang M., Hou Y.J.Y., Cavusoglu E., Lee D.C., Steffensen R., Yang L., Bashari D., Villamil J., Moussa M., Fernaine G., Jensenius J.C., Marmur J.D., Ko W., Shevde K. MASP-2 activation is involved in ischemia-related necrotic myocardial injury in humans. Int. J. Cardiol. 2013;166:499–504. doi: 10.1016/j.ijcard.2011.11.032.MASP-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Thangamani S., Ho B., Ding J.L. The ancient origin of the complement system. EMBO J. 2005;24:382–394. doi: 10.1038/sj.emboj.7600533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel S., Cseh S., Lacroix M., Dahl M.R., Matsushita M., Andrieu J.P., Schwaeble W.J., Jensenius J.C., Fujita T., Arlaud G.J., Thielens N.M. Characterization of recombinant mannan-binding lectin-associated serine protease (MASP)-3 suggests an activation mechanism different from that of MASP-1 and MASP-2. J. Immunol. 2004;172(7):4342–4350. doi: 10.4049/jimmunol.172.7.4342. [DOI] [PubMed] [Google Scholar]