

IL-27 signaling activates the production of antiviral proteins in the skin and inhibits Zika virus infection.
Abstract
In the skin, antiviral proteins and other immune molecules serve as the first line of innate antiviral defense. Here, we identify and characterize the induction of cutaneous innate antiviral proteins in response to IL-27 and its functional role during cutaneous defense against Zika virus infection. Transcriptional and phenotypic profiling of epidermal keratinocytes treated with IL-27 demonstrated activation of antiviral proteins OAS1, OAS2, OASL, and MX1 in the skin of both mice and humans. IL-27–mediated antiviral protein induction was found to occur in a STAT1- and IRF3-dependent but STAT2-independent manner. Moreover, using IL27ra mice, we demonstrate a significant role for IL-27 in inhibiting Zika virus morbidity and mortality following cutaneous, but not intravenous, inoculation. Together, our results demonstrate a critical and previously unrecognized role for IL-27 in cutaneous innate antiviral immunity against Zika virus.
INTRODUCTION
Zika virus (ZIKV) is an emerging pathogen that is transmitted to humans via a mosquito bite, and it can lead to congenital infection and neurodevelopmental defects if transmitted in utero, as well as Guillain-Barré syndrome in adults (1). Given that ZIKV inoculation begins with a mosquito bite, the skin serves as the first line of defense against systemic dissemination of ZIKV. One of the earliest innate immune defenses against viral infection is the production of antiviral proteins (AVPs), a specific subset of antimicrobial proteins (AMPs) that have activity against viruses (2–4). While the innate immune circuits that regulate antibacterial peptide and protein production are relatively well defined, little is known about the role and regulation of AVPs in the skin.
AVPs, including MX dynamin-like GTPase (guanosine triphosphatase) (MX) and the oligoadenylate synthase (OAS) family of molecules, are critical propagators of the cellular antiviral state. Previous studies have largely focused on induction of AVPs by interferons (IFNs), which is why these proteins are often referred to as IFN-stimulated genes (ISGs) (5). However, many viruses, including skin-tropic herpes simplex virus (HSV) and vaccinia virus, as well as mosquito-borne viruses such as dengue virus and ZIKV, have evolved escape mechanisms to block IFN and downstream signaling targets to escape host immunity (6, 7). Furthermore, while IFN-α is used to treat viral illnesses such as hepatitis B and cancers, including chronic myeloid leukemia (8), IFN-α is associated with an extensive adverse effect profile, including neurotoxicity, nephrotoxicity, and potentially fatal cytokine storm (9). Therefore, identification of factors to enhance innate antiviral immunity that are not dependent on IFN signaling may lead to the development of potential novel therapeutics against viral diseases.
Given the critical role of keratinocytes in skin antimicrobial host defense (3, 10–16), we sought to investigate the expression, regulation, and function of keratinocyte innate AVPs by the IFN-independent cytokine, interleukin-27 (IL-27), and the impact this signaling pathway has on in vitro and in vivo ZIKV infection.
IL-27 is a member of the IL-12 superfamily of cytokines (17, 18). IL-27 is a heterodimer composed of p28 and Epstein-Barr virus–induced 3 (EBI3), and it binds to the heterodimeric IL-27 receptor, composed of IL27RA and GP130 (19, 20). IL27RA is highly expressed on T cells; therefore, studies have largely focused on the biological function of IL-27 on T cells (21–28). For instance, IL-27 appears to have an inhibitory role on inflammation in the context of parasitic infections such as Toxoplasma gondii, Trypanosoma cruzi, Leishmania, or Plasmodium and Il27ra−/− mice phenocopy mice lacking IL-10 (19, 29, 30). However, IL-27 signaling is also capable of acting on naïve CD8+ T cells to enhance the generation of cytotoxic T lymphocytes (31), maintain plasmacytoid dendritic cells (32), and block HIV viral replication in CD4+ T cells (33).
We describe that IL-27 signaling leading to AVP production is IL27RA, STAT1 (signal transducers and activators of transcription 1), and IRF3 (interferon regulatory factor 3) dependent, but STAT2 independent. Functionally, IL-27 significantly suppressed ZIKV infection in human keratinocytes. Small interfering RNA (siRNA) and CRISPR-Cas9 gene editing, as well as mouse genetic knockout (KO) approaches, revealed that IL-27–mediated OAS2 signaling is STAT1 and IL27RA dependent, but STAT2, TYK2, and IFNAR1 independent. The in vivo relevance of IL-27 signaling was further demonstrated in murine infection models, where concurrent loss of Ifnar1 and Il27ra (Ifnar1−/−Il27ra−/−) resulted in significantly greater ZIKV-induced morbidity and mortality after subcutaneous virus inoculation compared with IL27ra. IL-27 conferred no protection against intravenous ZIKV infection, demonstrating the skin-specific activity of IL-27. These observations highlight IL-27 as a potential novel therapeutic agent against ZIKV by illustrating its abilities to enhance the skin’s own innate antiviral defenses.
RESULTS
IL-27 induces the expression of AVPs in keratinocytes to mediate antiviral function
Recently, our laboratory has demonstrated that IL-27 plays a key role in wound healing by inducing epidermal proliferation of keratinocytes and changing the expression of keratins and differentiation markers in these cells (17). Follow-up experiments where IL-27 was used to stimulate primary human keratinocytes revealed that keratinocytes respond to IL-27 by modulating expression of numerous genes, including many that encode AVPs. To evaluate IL-27’s ability to regulate gene expression, we treated human epidermal keratinocytes with IL-27 and analyzed the expression of multiple genes using a DNA microarray. We found that recombinant human IL-27 potently induces the expression of multiple keratinocyte AVPs, including OAS1, OAS2, OAS3, OASL, and MX1 (Fig. 1A), additional ISGs containing interferon gamma activated site (GAS) motifs and/or IFN-stimulated response element (ISRE) motifs, such as IFN regulatory factor 1 (IRF1), IFN-induced helicase C domain–containing protein 1 (IFIH1, MDA5), IFN-stimulated exonuclease gene 20 (ISG20), as well as IL-32, and Basic leucine zipper transcription factor, ATF-like 2 (BATF2) [suppressor of Activator Protein-1 (AP-1), regulated by IFN], and multiple genes from the guanylate-binding protein (GBP) family.
Fig. 1. IL-27 Induces the expression of AVPs in keratinocytes and inhibits ZIKV infection in vitro and in vivo.
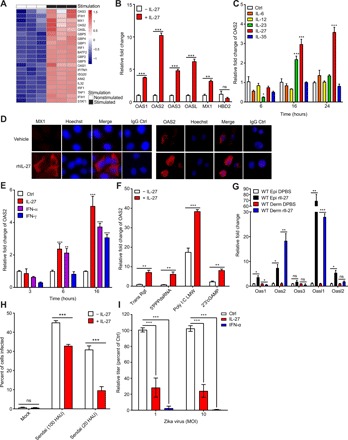
(A) DNA microarray of normal human epidermal keratinocytes treated for 24 hours with recombinant human IL-27 (100 ng/ml). (B) Quantitative PCR (qPCR) analysis examining the expression of multiple AVPs by primary human keratinocytes treated with IL-27 (100 ng/ml). The relative fold change for each condition was determined on the basis of OAS2 expression compared with nontreated cells. n = 6 biological replicates. (C) qPCR analysis examining the expression of OAS2 by primary human keratinocytes stimulated with 100 ng/ml of IL-6, IL-12, IL-23, IL-27, or IL-35 for 6, 16, and 24 hours. The relative fold change for each condition was determined on the basis of OAS2 expression compared with nontreated cells. n = 3 biological replicates. (D) Representative immunofluorescence staining showing MX1 and OAS2 (red) and Hoechst (blue) in NHEKs stimulated with rhIL-27 (100 ng/ml) for 48 hours. Original magnification, ×400 (50 μm). (E) qPCR analysis examining the expression of OAS2 by primary human keratinocytes stimulated with IL-27 (100 ng/ml), IFN-α (50 U/ml), or IFN-γ (50 U/ml) for 3, 6, or 16 hours. The relative fold change for each condition was determined on the basis of OAS2 expression compared with nontreated cells. n = 3 biological replicates. (F) qPCR analysis examining the expression of OAS2 by primary human keratinocytes transfected with 5′PPPdsRNA (100 ng/ml), poly I:C LMW, and 2′3′cGAMP and simultaneously stimulated with IL-27 (100 ng/ml) for 24 hours. The relative fold change for each condition was determined on the basis of OAS2 expression compared with non–IL-27–treated cells. n = 3 biological replicates. Trans Rgt, Transfection Reagent. (G) qPCR analysis examining the expression of indicated AVPs in response to subcutaneous injection of recombinant murine IL-27 (100 ng/ml) into the right ear of C57BL6/J mice. Tissue from both the treated right ear and nontreated left ear was harvested at 18 hours after injection. The relative fold change of each AVP in rIL-27–treated ears to DPBS-treated ears in both the epidermis and dermis. n = 3 mice. ns, not significant. (H) Quantification of Sendai virus envelope protein immunostaining of primary human keratinocytes pretreated for 24 hours with recombinant human IL-27 (100 ng/ml) and infected for 48 hours with Sendai virus at 100 and 20 HAU. Percentage of cells infected in IL-27–treated keratinocytes was compared with the percentage of cells infected in nontreated keratinocytes. n = 4 technical replicates in three separate experiments. (I) Focus-forming assay of supernatants from primary human keratinocytes pretreated with recombinant human IL-27 (100 ng/ml) or IFN-α (50 U/ml) for 4 hours and infected with Zika virus (Dakar strain) at 1 or 10 multiplicity of infection (MOI) for 48 hours. Titers were calculated as focus-forming units per milliliter and graphed as percent of Ctrl (Control, no cytokine treatment). To test for statistical significance, a two-tailed Student’s unpaired t test (B and E to H) or two-way analysis of variance (ANOVA) with Tukey’s multiple comparison test (C and D) was performed using GraphPad Prism software. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
To confirm that IL-27 induces the expression of AVPs, we stimulated human keratinocytes with IL-27 for 24 hours and found a significant increase in the mRNA expression of OAS1, OAS2, OAS3, OASL, and MX1 (Fig. 1, B and C). We also show confirmation of MX1 and OAS2 on the protein level (Fig. 1D). Notably, IL-27 failed to induce the expression of human β-defensin 2 (HBD2), an AMP whose effects are predominantly antibacterial, rather than antiviral (Fig. 1B) (34). Since IL-27 is a member of the IL-12 family of cytokines, we sought to determine whether other IL-12 family members could also induce AVPs in keratinocytes. Although we observed that both IL-27 and IL-23 significantly induced the production of OAS2, the induction of OAS2 by IL-23 was less than that of IL-27 at both 16 and 24 hours after stimulation and, in fact, decreased at 24 hours after stimulation (Fig. 1C). Other IL-12 family cytokines (IL-6, IL-12, and IL-35) failed to induce AVP expression. Because AVPs have classically been shown to be regulated by IFNs (5), we compared the ability of IL-27 to induce AVPs to that of a type I IFN (IFN-α) and type II IFN (IFN-γ). As expected, keratinocytes stimulated with IFN-α showed a significant increase in OAS2 expression at multiple time points, and IFN-γ induced a significant increase in OAS2 expression at 16 hours after stimulation (Fig. 1E). Notably, at a concentration of 50 U/ml, both IFN-α– and IFN-γ–mediated expression of OAS2 was similar in terms of fold change to IL-27–induced OAS2 expression. We next sought to determine whether IL-27 treatment of keratinocytes was associated with cytotoxicity. We found that neither IL-27 nor IFNs tested here were associated with any major cytotoxicity in primary human keratinocytes when delivered at concentrations that significantly increase AVP production (fig. S1, A and B).
To define the signaling pathway by which IL-27 drives AVP expression in human keratinocytes, we next evaluated the effect of IL-27 in the presence or absence of nucleic acid agonists of pattern recognition receptor (PRR) signaling, molecules that play a key role in activating pathways that produce IFNs and ISGs (35, 36). Since previous studies have shown that PRR signaling leads to AVP production, and our current research suggested that IL-27 also generates AVPs, we wanted to determine whether the presence of PRR agonists would lead to an increase in AVP production by IL-27. Intracellular (transfected) low–molecular weight poly I:C (poly I:C LMW) increased OAS2 expression in human keratinocytes, and the addition of IL-27 further enhanced OAS2 expression (Fig. 1F). Other Toll-like receptor (TLR) ligands, such as 5′PPPdsRNA (a RIG-I ligand), 2′3′cGAMP (a cGAS ligand), and imiquimod (IMQ) [a single-stranded RNA (ssRNA) analog that serves as a TLR 7 agonist], did not induce potent OAS2 expression in the absence of IL-27. However, IL-27 was still capable of inducing OAS2 expression in the presence of these ligands (Fig. 1F and fig. S1C). Together, our data suggest that IL-27 not only activates OAS2 transcription but also enhances other innate antiviral immune pathways, as seen by the additive effect that IL-27 exerts on poly I:C–mediated OAS2 expression.
We next sought to determine whether IL-27 would induce cutaneous AVP production in vivo. Injection of recombinant murine IL-27 into the skin of ears of C57BL6/J wild-type mice resulted in a potent induction of the AVPs Oas1, Oas2, Oasl1, and Oasl2 (Fig. 1G), a pattern of expression that mirrored the antiviral gene signature observed upon stimulation of human keratinocytes with IL-27 (Fig. 1B).
The ability of IL-27 to activate AVPs, including OAS family members known to play a role as antiviral effector proteins of RNA viruses (37) in keratinocytes, both independently and in conjunction with nucleic acid signaling, suggested to us that IL-27 may functionally mediate antiviral defenses against RNA viruses. Virus transmission and infection of the skin is frequently facilitated through microinjuries associated with skin barrier dysfunction, trauma, or mosquito bites (3). We found that human keratinocytes treated with IL-27 were less susceptible to infection by Sendai virus and ZIKV (Fig. 1, H and I). IL-27 inhibited infection of keratinocytes by Sendai virus, a negative sense ssRNA paramyxovirus, at both 100 hemagglutination units (HAUs) and 20 HAUs (Fig. 1H). For ZIKV, at a multiplicity of infection of 1 and 10, IL-27 also inhibited viral keratinocyte infection (Fig. 1I), while IFN-α was used as a positive control. Together, our data suggest that IL-27’s effect on human keratinocytes is broadly antiviral.
IL-27 induces OAS2 in an IL27RA/STAT1/IRF3-dependent and IFNAR1/TYK2/STAT2- and IFNGR1-independent manner
Given the similarities between IL-27–mediated AVPs and the classical IFN-mediated production of ISGs, we next determined whether IL-27 requires IFN signaling to induce OAS2. We used siRNA transfection to knock down specific genes in primary human keratinocytes and stimulated these cells with IL-27. Successful knockdown of target genes was confirmed using quantitative reverse transcription polymerase chain reaction (qRT-PCR) (fig. S2). Knockdown of IL27RA and STAT1 significantly inhibited IL-27–mediated OAS2 expression, whereas knockdown of IFNGR1, STAT2, and IFNAR1 did not inhibit OAS2 expression (Fig. 2A). Knockdown of STAT2 and IFNAR1 increased IL-27–mediated OAS2 expression. We next used siRNA to target numerous genes associated with viral sensing and response pathways, including the type I IFN and RIG-I–like receptor signaling pathways. Knockdown of JAK1, IRF1, IRF3, IRF7, TRAF6, TRIM25, BATF2, IFIH1 (MDA5), and MAP3K7 (also known as TAK1), significantly inhibited OAS2 transcription upon IL-27 stimulation, suggesting that IL-27–mediated OAS2 production requires these signaling molecules (Fig. 2B). In contrast, knockdown of JAK2, AZI2 (NAP1), TBK1, IKBKE, MAVS, and TLR3 did not suppress OAS2 transcription following IL-27 stimulation in keratinocytes.
Fig. 2. IL-27 enhances the expression of AVPs in a STAT1-/IL27RA-dependent and STAT2-/IFNAR1-/IFNGR1-independent manner.
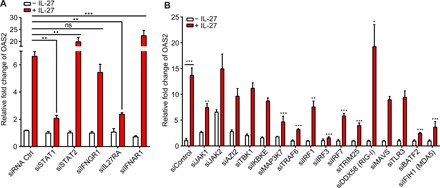
(A) qPCR analysis examining the expression of OAS2 by primary human keratinocytes transfected with siRNA of STAT1, STAT2, IFNGR1, IL27RA, and IFNAR1 for 5 hours and subsequently stimulated with IL-27 (100 ng/ml) for 24 hours. To determine statistical significance, the OAS2 expression of siRNA-transfected cells treated with IL-27 was compared with the OAS2 expression of IL-27–treated cells transfected with an siRNA control. n = 3 biological replicates. (B) qPCR analysis examining the expression of OAS2 by primary human keratinocytes transfected with siRNA of immune signaling molecules associated with type I IFN, RIG-I, or MDA5 signaling for 5 hours and subsequently stimulated with IL-27 (100 ng/ml) for 24 hours. To determine statistical significance, the OAS2 expression of siRNA-transfected cells treated with IL-27 was compared with the OAS2 expression of IL-27–treated cells transfected with an siRNA control. n = 3 biological replicates. To test for statistical significance, two-way ANOVA with Tukey’s multiple comparison test (A and B) was performed using GraphPad Prism software. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
To further investigate STAT1 signaling upon IL-27 stimulation, we treated primary human keratinocytes with IL-27 and analyzed these cells at different time points after stimulation and also stained for p-STAT1 and p-STAT2. p-STAT1 was found to translocate to the nucleus as early as 15 min after IL-27 treatment (Fig. 3A). Although absolute number of cells demonstrating nuclear p-STAT1 translocation remained relatively unchanged from 15 to 90 min after IL-27 stimulation, the intensity of staining decreased from 30 to 90 min (fig. S3A). Conversely, p-STAT2, a key component of the p-STAT1/p-STAT2 heterodimer required for classical type I IFN signaling, failed to translocate to the nucleus of human keratinocytes in response to IL-27 at all time points examined, whereas IFN-α used as a positive control did induce p-STAT2 translocation (Fig. 3B and fig. S3B). The rapid and transient nuclear translocation of p-STAT1 in response to IL-27 in the absence of p-STAT2 translocation supports the view that IL-27 activates a STAT1-dependent pathway distinct from the STAT1-STAT2 heterodimers activated by type I IFN signaling. To further confirm that the IL-27–AVP pathway is STAT1 dependent, but independent of type I IFN signaling, we next examined human keratinocytes using CRISPR-Cas9 KO of STAT1, STAT2, and TYK2, known mediators of the type I IFN pathway. Figure S3C shows the Sanger sequence for the STAT1, STAT2, and TYK2 KO keratinocytes, confirming specific CRIPSR-Cas9 targeting. We observed that IL-27–mediated OAS2 production was dependent on STAT1, but not STAT2 or TYK2, confirming that IL-27–mediated OAS2 production is STAT1 dependent, but STAT2 and TYK2 independent (Fig. 3C).
Fig. 3. Rapid and transient nuclear translocation of p-STAT1, but not p-STAT2, is characteristic of IL-27 signaling.
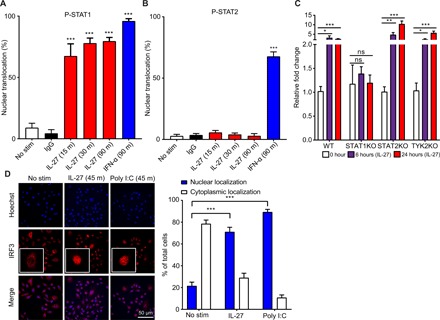
(A) Quantification of immunofluorescence staining for p-STAT1 (Cy3) in primary human keratinocytes treated with a time course of 100 ng/ml of IL-27 or 50 U/ml of IFN-α for the indicated times. Nuclear translocation was quantified by examining five different fields per slide. The nuclear translocation (%) at each treated time point was compared with the nuclear translocation (%) of the nontreated control. n = 3 biological replicates. IgG, immunoglobulin G. (B) Quantification of immunofluorescence staining for p-STAT2 (Cy3) in primary human keratinocytes treated with a time course of 100 ng/ml of IL-27 or 50 U/ml of IFN-α for the indicated times. Nuclear translocation was quantified by examining five different fields per slide. The nuclear translocation (%) at each treated time point was compared with the nuclear translocation (%) of the nontreated control. n = 3 biological replicates. No stim, No stimulation. (C) qPCR analysis examining the expression of OAS2 in immortalized human keratinocytes with CRISPR-Cas9 KO of STAT1, STAT2, and TYK2 treated for 6 and 24 hours with recombinant human IL-27 (100 ng/ml). Each experimental condition was compared with the non-CRISPR control. n = 3 biological replicates. (D) Immunofluorescence staining for IRF3 (Cy3) in primary human keratinocytes treated for 45 min with 100 ng/ml of IL-27 or poly I:C LMW. Nuclear translocation was quantified by examining five different fields per slide. The nuclear translocation (%) for both IL-27 and poly I:C was compared with the nuclear translocation (%) of the nontreated control. n = 3 biological replicates. To test for statistical significance, a two-tailed Student’s unpaired t test (A to C) or two-way ANOVA with Tukey’s multiple comparison test (D) was performed using GraphPad Prism software. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
IRF3 plays a key role in the MAVS and the STING/TBK1 signaling cascade downstream of TLR3 and additional TLRs, as well as additional RNA and DNA sensors (38–40). Since we demonstrated that IL-27 signaling is dependent on IRF3 and that IL-27 enhances poly I:C–mediated production of AVPs, we wanted to examine whether IRF3 was activated in response to IL-27 signaling. We found that IRF3 translocates to the nucleus within 45 min in primary human keratinocytes stimulated with IL-27 (Fig. 3D). The percentage of cells exhibiting IFRF3 nuclear translocation in response to IL-27 was similar to the percentage of cells exhibiting nuclear translocation in response to transfected dsRNA, which was used as positive control.
Since we found that STAT1 signaling is required for IL-27–mediated expression of OAS2 in keratinocytes, we next assessed whether STAT1 was required for the in vivo antiviral activity toward ZIKV. We found that Stat1KO mice succumbed to ZIKV infection through both intravenous (Fig. 4A) and subcutaneous inoculation routes (Fig. 4B) at 13 days after infection, whereas wild-type 129S6 mice experienced no mortality (Fig. 4, A and B). Genetic KO of Stat1 led to a gradual decline in weight before death in mice that were infected with ZIKV via either intravenous (Fig. 4C) or subcutaneous (Fig. 4D) injection. In addition, Stat1KO mice infected with ZIKV experienced similar clinical disease signs to infected Ifnar1KO and Ifnar1/Il27ra double KO (dKO) mice (fig. S4). Thus, our work is in agreement with previous data, which demonstrated that Stat1KO mice are susceptible to ZIKV infection (41, 42).
Fig. 4. STAT1 signaling plays a key role in antiviral defense against Zika virus in vivo.
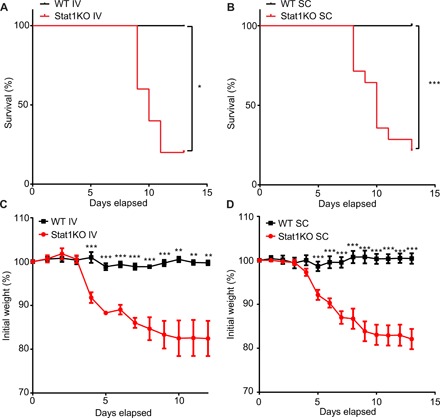
(A) Survival curve of 129S6 (WT) and Stat1KO mice injected intravenously (IV) with Zika virus (HPF2013 strain). n = 5 mice (129S6) and 5 mice (Stat1KO) per experimental group. (B) Survival curve of 129S6 (WT) and Stat1KO mice injected subcutaneously (SC) with Zika virus (HPF2013 strain). n = 9 mice (129S6) and 14 mice (Stat1KO) per experimental group. (C) Line graph detailing percentage of initial weight in 129S6 (WT) and Stat1KO mice injected intravenously with Zika virus (HPF2013 strain). The percentage of initial weight of wild-type (WT) mice was compared with the percentage of initial weight of Stat1KO at each time point. n = 5 mice (129S6) and 5 mice (Stat1KO) per experimental group. (D) Line graph detailing percentage of initial weight in 129S6 (WT) and Stat1KO mice injected SC with Zika virus (HPF2013 strain). The percentage of initial weight of WT mice was compared with the percentage of initial weight of Stat1KO at each time point. n = 9 mice (129S6) and 14 mice (Stat1KO) per experimental group. To test for statistical significance, a Mantel-Cox test (A and B) or a two-tailed Student’s unpaired t test (C and D) was performed using GraphPad Prism software. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
IL-27 signaling reduces mortality and weight loss due to ZIKV infection in the absence of type I IFN signaling
STAT1 is a key regulatory molecule in both IL-27 and IFN signaling. Having observed that IL-27 can inhibit viral infection in cell culture and that STAT1 signaling is critical to antiviral competence in mice, we sought to determine the role that IL-27 and type I IFNs play in inhibiting viral infection in vivo. C57BL6/J (B6), Il27raKO, Ifnar1KO, and Ifnar1/Il27ra dKO mice bred on a B6 background were infected with ZIKV subcutaneously, and mortality, weight, and disease signs were measured over 12 days after infection. Although wild-type B6 mice are uniformly resistant to ZIKV, Ifnar1KO mice are highly susceptible (42). No mortality or changes in weight were observed in either wild-type mice or Il27raKO mice, regardless of route of infection (Fig. 5, A to D). Notably, 100% of Ifnar1/Il27ra dKO mice succumbed to subcutaneous ZIKV inoculation, but only 50% of Ifnar1KO mice had died at 13 days after infection, identifying a protective role for IL-27 in controlling ZIKV infection (Fig. 5A). While we could not detect any differences in the viral titers in the serum from Ifnar1/Il27ra dKO compared with Ifnar1KO mice 2 days after infection (fig. S5), Ifnar1/Il27ra dKO mice significantly exhibited greater weight loss than Ifnar1KO starting at day 5 after injection (Fig. 5C). In contrast, intravenous injection of ZIKV led to 100% mortality by 10 days after infection and significant decreases in weight in both Ifnar1KO and Ifnar1/Il27ra dKO mice (Fig. 5, B and D). Significant changes in both weight and mortality in Ifnar1/Il27ra dKO mice compared with Ifnar1KO mice in response to subcutaneous, but not intravenous, ZIKV infection suggested that cutaneous IL-27 signaling specifically may play a protective role against ZIKV-associated mortality in the absence of IFN signaling.
Fig. 5. IL-27 signaling reduces in vivo mortality and weight loss due to Zika virus infection in the absence of type I IFN signaling.
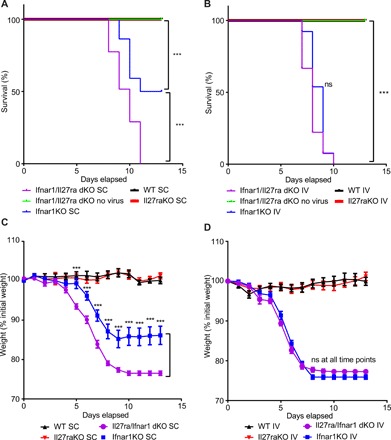
(A) Survival curve of B6 (WT), Ifnar1KO, Il27raKO, and Ifnar1/Il27ra dKO injected subcutaneously (SC) with 104 Zika virus (HPF2013 strain). n = 19 mice (B6), 23 mice (Il27raKO), 22 mice (Ifnar1KO), and 30 mice (Ifnar1/Il27ra dKO) per experimental group. (B) Survival curve of B6 (WT), Ifnar1KO, Il27raKO, and Ifnar1/Il27ra dKO injected intravenously (IV) with 104 Zika virus (HPF2013 strain). n = 7 mice (B6), 13 mice (Il27raKO), 13 mice (Ifnar1KO), and 27 mice (Ifnar1/Il27ra dKO) per experimental group. (C) Line graph detailing percentage of initial weight in B6 (WT), Ifnar1KO, Il27raKO, and Ifnar1/Il27ra dKO mice injected SC with Zika virus (HPF2013 strain). The percentage of initial weight of WT mice was compared with the percentage of initial weight of each genotype at each time point. n = 19 mice (B6), 23 mice (Il27raKO), 22 mice (Ifnar1KO), and 30 mice (Ifnar1/Il27ra dKO) per experimental group. (D) Line graph detailing percentage of initial weight in B6 (WT), Ifnar1KO, Il27raKO, and Ifnar1/Il27ra dKO mice injected intravenously with Zika virus (HPF2013 strain). The percentage of initial weight of WT mice was compared with the percentage of initial weight of each genotype at each time point. n = 7 mice (B6), 13 mice (Il27raKO), 13 mice (Ifnar1KO), and 27 mice (Ifnar1/Il27ra dKO) per experimental group. To test for statistical significance, a Mantel-Cox test (A and B) or a two-tailed Student’s unpaired t test for each time point (C and D) was performed using GraphPad Prism software. Data are means ± SEM. ***P < 0.001.
IL-27 signaling reduces the occurrence and severity of neurological disease signs due to ZIKV infection in the absence of type I IFN signaling
Having observed that cutaneous IL-27 signaling reduces mortality from ZIKV infection in the absence of IFN signaling, we next sought to determine whether IL-27 signaling was protective against the disease signs commonly associated with ZIKV infection in Ifnar1−/− mice. Constitutional symptoms such as scruffy fur and mild central nervous system weaknesses that herald subsequent substantial neurological signs appeared earlier in Ifnar1/Il27raKO compared with Ifnar1KO mice, manifesting as early as 5 days after infection (Fig. 6). Beginning as early as 7 days after infection, a greater percentage of Ifnar1/Il27ra dKO mice began to exhibit neurological signs compared with Ifnar1KO mice, with a larger number of Ifnar1/Il27raKO mice continuing to exhibit neurological signs until succumbing at day 10 after infection via the subcutaneous route (Fig. 6). Together, the increased morbidity and mortality seen in Ifnar1/Il27ra dKO mice following subcutaneous ZIKV infection compared with Ifnar1KO mice suggested that cutaneous IL-27 signaling may play a relevant and previously unknown role in inhibiting ZIKV infection in the skin and may affect neurological symptoms. Intact IL-27 signaling may therefore serve a protective antiviral role, even in the absence of type I IFN signaling.
Fig. 6. IL-27 signaling reduces the severity and occurrence of in vivo neurological symptoms due to Zika virus infection in the absence of type I IFN signaling.
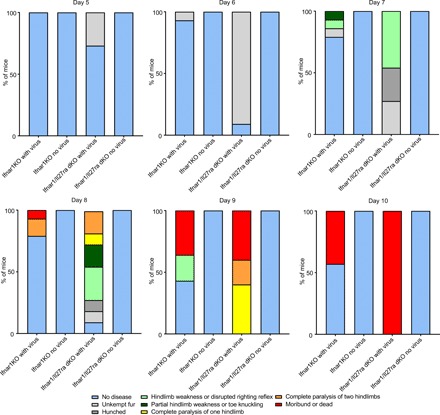
Bar graphs detailing morbidity and mortality of B6 (WT), Ifnar1KO, Il27raKO, and Ifnar1/Il27ra dKO injected subcutaneously with Zika virus (HPF2013 strain) at 5 to 10 days after infection. Symptoms were scored in a sequential fashion, where lesser symptoms (e.g., hunched, unkempt fur) were not counted if a mouse presented with more severe symptoms (e.g., hindleg weakness). n = 10 to 12 mice (B6), 12 to 14 mice (Il27raKO), 14 to 16 mice (Ifnar1KO), and 11 to 12 mice (Ifnar1/Il27ra dKO) per experimental group. Experiment was repeated three times.
DISCUSSION
There is an emerging body of evidence that suggests proinflammatory cytokines mediate the production of AMPs (10, 43). Here, we found that keratinocytes generate AVPs including OAS1, OAS2, OASL, and MX1 in response to IL-27. In vivo, IL-27 demonstrates a protective function against subcutaneous ZIKV infection in mice in the absence of type I IFN signaling.
Recent studies demonstrating that ZIKV has developed mechanisms to inhibit type I IFN signaling (44) highlight the need to investigate alternative pathways that may suppress ZIKV infection, which remains an infectious disease concern in the Americas and elsewhere (45, 46), with the potential to cause severe birth defects (47) and neurological symptoms (48), ZIKV is an emerging pathogen that is transmitted to humans via mosquito bites. ZIKV evades the IFN-mediated antiviral response in mice with the help of several nonstructural proteins, such as NS1 and NS4B. NS1 and NS4B have been shown to inhibit IFN-β signaling at the level of TANK-binding kinase 1 (49) and NS5, which inhibits STAT2 (44). Thus, in vivo mouse infections necessitate the use of Ifnar1−/− mice (42). However, type I IFN signaling has recently been associated with increased risk for fetal demise in mice infected with ZIKV (50). Therefore, identification of innate antiviral immune factors that are not dependent on IFN signaling may lead to the development of therapeutics against ZIKV and other viruses. When comparing Ifnar1/Il27ra dKO mice to Ilfnar1KO mice, the increased morbidity and mortality seen in Ifnar1/Il27ra dKO mice following subcutaneous ZIKV infection compared with Ilfnar1KO mice suggested that IL-27 signaling may play a relevant and previously unknown role in inhibiting ZIKV infection. Although the pathogenesis of ZIKV infection is not fully understood, it is currently thought that the skin is the first site of inoculation and viral replication, given that human primary fibroblasts, epidermal keratinocytes, and immature dendritic cells have been shown to be permissive to ZIKV infection and replication and that other flaviviruses are first inoculated into the skin (51, 52). Subsequently, the virus spreads to the draining lymph node, where it replicates, leading to viremia and hematogenous spread to other tissues in the body. Given that we observed that subcutaneous, but not intravenous, inoculation of ZIKV in dKO resulted in the amelioration of neurological disease signs, it is possible that inhibiting viral replication in the skin reduced the ability of ZIKV to spread to peripheral tissues, including neuronal tissues. However, we cannot exclude the possibility that IL-27 may also play a protective role directly in neuronal ZIKV infection or if unknown effects of IL-27 may contribute to preventing development of neurological injury during ZIKV infection. We speculate that intact IL-27 signaling may serve a pivotal protective antiviral role in the skin and potentially in the spread from the skin to the neuronal systems.
The AVPs OAS1, OAS2, OAS3, OASL, and MX1 have previously been shown to be induced primarily by type I IFNs (53). We found that IL-27 induces expression of these AVPs in a manner that is STAT1 and IL27RA dependent, but STAT2 and IFNAR1 independent. Together, these findings suggest that early induction of AVPs by IL-27 is independent of type I IFN signaling in skin cells, given the prominent role of STAT2 and IFNAR1 in classical type I IFN induction of ISGs (53). As STAT2 is known to be inhibited by ZIKV (44), IL-27 could play a novel host-defensive role to subvert viral inhibition of type I IFN regulatory molecules, such as STAT2. siRNA knockdown and CRISPR-Cas9 KO of STAT2 resulted in a significant increase in IL-27–mediated AVP expression over baseline, which may indicate the greater availability of free STAT1 to signal through the IL-27–AVP pathway. In further support of the role of IL-27 in specifically inducing STAT1 signaling, we found that p-STAT1, but not p-STAT2, was rapidly activated in response to IL-27, suggesting that IL-27 directly signals through STAT1 to generate AVPs, as opposed to indirectly producing AVPs via type I IFN signaling. Together, our data indicate that IL-27 is required to generate AVPs in the early cutaneous immune response to ZIKV infection via a STAT1- and IL27RA-dependent approach. Since IL-27 signaling does not appear to require STAT2, an IL-27–based approach would be resistant to the STAT2-antagonizing activity of ZIKV (44, 49).
Of note, IRF3 also appeared to play a role in IL-27 signaling. IL-27 stimulation of human keratinocytes led to rapid nuclear translocation of IRF3, and knockdown of IRF3 and MAP3K7 (also known as TAK1) led to significantly decreased IL-27–mediated OAS2 expression. A recent study (54) suggested that TAK1 signaling upstream of IRF3 leads to nuclear translocation of IRF3 and subsequent production of IFN-inducible genes. Although we do not discuss the direct interplay between STAT1 and IRF3 here, both genes appear to play important roles in IL-27–mediated AVP production.
Further research is needed to delineate the mechanisms behind why and how cutaneous IL-27 signaling protects the host from ZIKV infection, while systemic IL-27 signaling provides no additional protection when type I IFN signaling is ablated.
MATERIALS AND METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact A.S.M. (amanda.macleod@duke.edu).
Experimental model and subject details
Key resources can be found in the Supplementary Materials.
Mice
Male and female mice were bred and/or housed in a specific pathogen–free facility at the Duke University School of Medicine. For all studies, both male and female mice were used. All procedures with animals were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols were approved by the Duke University Animal Care and Use Committee (Animal Welfare Assurance). C57BL6/J, Balb/c, Ifnar1−/−, and Il27ra−/− mice were purchased from the Jackson laboratory (Bar Harbor, ME). Stat1−/− and 129S6 mice were purchased from Taconic Biosciences. Ifnar1−/− and Il27ra−/− were crossbred in Duke’s breeding facility to generate Ifnar1−/− Il27ra−/− dKO mice. All mice were used at 4 to 7 weeks of age and were age and gender matched for each experiment.
Viruses
Zika virus–Dakar (DAK; Zika virus/A.africanus-tc/SEN/1984/41525-DAK) (GenBank accession no. KU955591) was used for the in vitro experiments in human keratinocytes, and Zika virus–French Polynesia (PF; Zika virus strain H/PF/2013) (GenBank accession no. KJ776791.2) was used for in vivo infection studies. Strains were provided by S. Weaver at the University of Texas Medical Branch and H.L. (coauthor of this study), respectively. Sendai virus strain Cantell was obtained from Charles River Laboratory. Zika virus stocks were prepared as described (55). All viruses were titered by focus-forming assay in Vero cells. Vero cells (CCL-81) were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and were verified as mycoplasma free by the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich, St. Louis, MO, USA). In vitro Zika and Sendai virus infections were performed in serum-free media conditions for 1 hour, after which human keratinocyte medium with indicated treatment conditions was replenished.
Percentage of cells infected with Sendai virus quantification
Cells were immunostained for Sendai virus (anti-Sendai, Chicken, Abcam ab33988) and nuclei [4′,6-diamidino-2-phenylindole (DAPI)], and cells were identified as Sendai virus infected or uninfected by counting Sendai-positive cells using a Cellomics ArrayScan VTI High Content Screening Reader (Duke Functional Genomics Facility, Durham, NC, USA). Percentage of infected cells was calculated as the number of Sendai virus + cells/the number of total cells (Sendai/DAPI) per field. Values represent the means ± SEM (n = 4 fields) from three independent experiments, with >3000 cells counted per field.
Human keratinocytes
Human adult keratinocytes were purchased from Gibco and maintained for up to six passages in T-75 flasks. Cells were grown in 37°C serum-free EpiLife cell culture medium with EpiLife Defined Growth Supplement containing 0.05 mM Ca2+. Keratinocytes were used at approximately 75 to 80% confluence. For experiments, cells were plated at 200,000 cells per well in six-well plates, 75,000 cells per well in 12-well plates, or 75,000 cells per chamber in two-chamber chamber slides, respectively (Labtek, Grand Rapids, MI).
Method Details
In vivo Zika virus infection
B6, Il27raKO, Ifnar1KO, and Ifnar1/Il27ra dKO mice were injected subcutaneously with 103 to 105 Zika virus (HPF2013) once and observed for weight, survival, and neurological symptoms over the course of 13 to 14 days. For intravenous infections, ZIKV was injected intravenously into the tail vein.
Cytokine stimulation of human keratinocytes
Medium was removed, and new medium containing cytokines was used to stimulate keratinocytes for a variety of time points. Unless stated otherwise, IL-6, IL-23, IL-27, IL-35 (all from BioLegend), and IL-12 (Tonbo) were added to 1 to 2 ml of medium at a concentration of 100 ng/ml; IFN-α (BioLegend) or IFN-γ (PeproTech) was added to 1 to 2 ml of media at a concentration of 50 U/ml. For the cytotoxicity study, cells were suspended in CellTiter 96 AQueous One Solution Reagent and then incubated at 37°C for 1 to 4 hours in a humidified, 5% CO2 atmosphere. Absorbance was read at 490 nm using a 24-well plate reader.
TLR and PRR ligand stimulation of human keratinocytes
Medium was removed, and new medium containing cytokines was used to stimulate keratinocytes for a variety of time points. Poly I:C LMW, 5′PPPdsRNA, 5′PPPdsRNA control, cGAMP, and imiquimod were obtained from InvivoGen. Unless stated otherwise, TLR and PRR ligands were added to 1 to 2 ml of medium at a concentration of 100 ng/ml.
Immunofluorescence staining
Briefly, human primary keratinocytes were grown to subconfluency before IL-27 stimulation. For MX1 staining, cells were fixed with 100% ice-cold methanol and incubated in a blocking solution of 1×Dulbecco’s Phospate Buffered Saline (DPBS) containing 2.5% normal goat serum (Jackson ImmunoResearch), 2.5% normal donkey serum (Jackson ImmunoResearch), 1% bovine serum albumin (Calbiochem), 2% fish gelatin (Sigma), and 0.1% Triton X-100 (Sigma) for 1 hour at room temperature. Then, cells were incubated with anti-MX1 antibody (Abcam; final concentration 0.060 mg/ml) or appropriate immunoglobulin G (IgG) controls in a solution of fresh blocking solution and 0.1% Triton X-100 (mixed at a ratio 1:1) for 2 hours at room temperature. After the washing steps, cells were incubated with an Alexa Fluor 555–conjugated secondary antibody (Thermo Fisher Scientific Jackson). After subsequent washing, nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific), washed in 1×DPBS, and mounted with ProLong Gold antifade reagent (Thermo Fisher Scientific). For OAS2 staining, stimulated keratinocytes were paraformaldehyde (PFA) fixed (4% PFA) and incubated in a blocking solution of 1×DPBS containing 5% normal goat serum (Jackson ImmunoResearch) and 0.1% Triton X-100 for 1 hour at room temperature before incubation with anti-OAS2 antibody (Proteintech 0.33 mg/ml) or appropriate IgG control in 1% bovine serum albumin with 0.1% Triton X-100 for 2 hours at room temperature. Secondary antibody and Hoechst staining are described above. For staining with p-STAT1, p-STAT2, and IRF3, cells were fixed in 4% PFA followed by ice-cold methanol, washed, and stained with rabbit polyclonal anti-pSTAT1, anti-pSTAT2, or anti-IRF3 antibodies (all from Cell Signaling Technologies). After subsequent washes, cells were incubated with secondary Cy3-conjugated goat anti-rabbit antibody (Life Technologies) before Hoechst staining and mounting.
Focus-forming assay
Supernatants were collected at 48 hours postinfection or after from sera of infected mice and stored at −80°C until virus titration. Serial dilutions of thawed supernatants and sera were used to infect naïve Vero cells in triplicate wells of a 48-well plate. At 1 hour postinfection, Vero cells were overlaid with 2% methylcellulose and 2% fetal bovine serum in Opti-MEM. At 72 hours postinfection, cells were fixed in a 1:1 methanol/acetone solution and immunostained with mouse anti-4G2 antibody (1:500), generated from the D1-4G2-4-15 hybridoma cell line against flavivirus envelope protein (ATCC). Following binding of horseradish peroxidase–conjugated secondary antibody (Jackson ImmunoResearch, 1:500), infected foci were visualized with the VIP Peroxidase Substrate Kit (Vector Laboratories) and counted at ×40 magnification to determine the viral titer (focus-forming units per milliliter).
RNA isolation and qRT-PCR
For murine ear tissue, epidermal ear sheets were separated and floated on trypsin GNK for 15 min at 37°C. Then, two curved forceps were used to separate the epidermis from the dermis of each ear sheet, and both layers of skin were then homogenized using scissors and placed into TRIzol for quantitative PCR (qPCR) analysis. Total RNA was isolated using a Direct-zol Kit (Genesee Scientific, Research Triangle Park, NC) or using TRIzol reagent (Invitrogen). RNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA), and the resulting cDNA was amplified using Fast SYBR Green Master Mix (Applied Biosystems). Primers for amplification are listed in table S1 online. Fold induction of gene expression was normalized to the house keeping glyceraldehyde-3-phosphate dehydrogenase and calculated using the 2(−ΔΔCt) method.
siRNA knockdown
siRNA constructs were obtained from either Dharmacon or OriGene. siRNA transfection buffer and transfection reagent were obtained from OriGene. Cells were plated 1 day before transfection, and medium was changed to serum-free medium 2 hours before transfection. siRNA constructs were resuspended in 1× transfection buffer at a concentration of 0.02 nmol/μl. Each construct (1.7 μl) was added to a 200-μl master mix that contained both 1× transfection buffer and transfection reagent at volumes indicated by the manufacturer. siRNA master mixes were allowed to incubate at room temperature for 15 to 30 min before being added dropwise to wells. Five hours after siRNA master mixes were added, media were changed to serum-containing medium, and cells were stimulated with the indicated cytokines as described above.
Quantification and statistical analysis
Statistical analysis
Analyses of all in vitro and in vivo experiments were conducted using Prism software and depicts means ± SEM. Data are presented as means ± SEM. Group sizes were determined on the basis of the results of preliminary experiments without predetermination of sample size. Preliminary experiments were performed to determine requirements for sample size, taking into account resources available and ethical, reductionist animal use. Mice were randomly assigned to groups. Mouse studies were not performed in a blinded fashion. Generally, each mouse of the different experimental groups was reported. Statistical analysis was calculated using Prism software (GraphPad), with significance reported for P < 0.05 and is detailed in the figure legends.
Microarray data analysis and computation
Human Affymetrix microarray data were processed and normalized using the affy (56) and limma (57) Bioconductor (58) packages from the R statistical programming environment. Raw data were normalized to eliminate systematic differences across samples using the robust multiarray average approach, and differential expression between IL-27 stimulated and control samples was calculated using a linear model with an empirical Bayes’ estimation of the test statistic. The false discovery rate was used to correct for multiple hypothesis testing.
Data and software availability
Data showing the IL-27 stimulated human keratinocyte dataset (shown in Figure 1) can be found at GSE143228.
Supplementary Material
Acknowledgments
We would like to acknowledge and thank D. Erdmann, S. Degan, and J. Zhang for help in providing human skin tissue from plastic surgery procedures. We thank the Duke Functional Genomics Facility for use of the Cellomics ArrayScan. We also thank J. Coers for the helpful discussions on our manuscript and S. Kim for help with siRNA work. Funding: This work was supported by funds from NIH R01 AI139207 and R21AI139207 (to A.S.M.). A.S.M. is/was supported by a Dermatology Foundation Award, a Duke Physician-Scientist Strong Start Award, and a Silab company partnership grant. NIH grants R21NS100545 (to S.M.H.), R21AI129851 (to S.M.H.), T32-CA009111 (M.J.M.), and NIAMS P30-AR075043 (to J.E.G.) provided additional support. The Virology Unit of the Duke Regional Biocontainment Laboratory received partial support for construction from the NIH, National Institute of Allergy and Infectious Diseases (UC6-AI058607). J.T.K. and C.H. were supported by the Eugene A. Stead Student Research Fellowship. C.H. was also supported by the Poindexter Fellowship. S.M.H. has an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (1016810). Author contributions: J.T.K., C.H., and A.S.M. designed the studies and wrote the manuscript. J.K. and C.H. performed most of the experiments and analyzed and interpreted the data. J.Su., P.H., A.S.M., and L.F. participated in performing the experiments and helped to interpret the experimental results. J.Su. and J.Sh. helped to revise the manuscript and perform experiments for revision. M.J.M., K.F.L., and S.M.H. designed and performed in vitro Sendai and ZIKV studies and helped in editing the manuscript. G.S. and the core performed in vivo virus studies and provided intellectual expertise. J.E.G., R.U., and M.K.S. generated and supplied CRISPR-Cas9 KO keratinocytes. H.M.L. supplied ZIKV titers. D.L.C. performed biocomputational analyses. Competing interests: The MacLeod laboratory has previously received funds from Silab Company (to A.S.M.), and funding from this partnership was not directly used for this study. A.S.M. also consults for Silab. Silab did not have any influence on the content of this project. A.S.M. is also consulting for the LEO Foundation. The spouse of A.S.M. is employed at Precision Biosciences and has stock and stock options. All other authors declare that they have no competing interests. Data and materials availability: Data showing the IL-27–stimulated human keratinocyte dataset (shown in Fig. 1) are available and can be found at GSE143228. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay3245/DC1
REFERENCES AND NOTES
- 1.Pierson T. C., Diamond M. S., The emergence of Zika virus and its new clinical syndromes. Nature 560, 573–581 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Zhu J., Zhang Y., Ghosh A., Cuevas R. A., Forero A., Dhar J., Ibsen M. S., Schmid-Burgk J. L., Schmidt T., Ganapathiraju M. K., Fujita T., Hartmann R., Barik S., Hornung V., Coyne C. B., Sarkar S. N., Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40, 936–948 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Handfield C., Kwock J., MacLeod A. S., Innate antiviral immunity in the skin. Trends Immunol. 39, 328–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadler A. J., Williams B. R. G., Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider W. M., Chevillotte M. D., Rice C. M., Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 32, 513–545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann H.-H., Schneider W. M., Rice C. M., Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 36, 124–138 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith G. L., Benfield C. T. O., Maluquer de Motes C., Mazzon M., Ember S. W. J., Ferguson B. J., Sumner R. P., Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 94, 2367–2392 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Lazear H. M., Schoggins J. W., Diamond M. S., Shared and distinct functions of type I and type III interferons. Immunity 50, 907–923 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tisoncik J. R., Korth M. J., Simmons C. P., Farrar J., Martin T. R., Katze M. G., Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 76, 16–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai Y., Li D., Li C., Muehleisen B., Radek K. A., Park H. J., Jiang Z., Li Z., Lei H., Quan Y., Zhang T., Wu Y., Kotol P., Morizane S., Hata T. R., Iwatsuki K., Tang C., Gallo R. L., The antimicrobial protein REG3A regulates keratinocyte proliferation and differentiation after skin injury. Immunity 37, 74–84 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macleod A. S., Hemmers S., Garijo O., Chabod M., Mowen K., Witherden D. A., Havran W. L., Dendritic epidermal T cells regulate skin antimicrobial barrier function. J. Clin. Invest. 123, 4364–4374 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eyerich S., Eyerich K., Pennino D., Carbone T., Nasorri F., Pallotta S., Cianfarani F., Odorisio T., Traidl-Hoffmann C., Behrendt H., Durham S. R., Schmidt-Weber C. B., Cavani A., Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 119, 3573–3585 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacLeod A. S., Mansbridge J. N., The innate immune system in acute and chronic wounds. Adv. Wound Care (New Rochelle) 5, 65–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacLeod D. T., Nakatsuji T., Wang Z., di Nardo A., Gallo R. L., Vaccinia virus binds to the scavenger receptor MARCO on the surface of keratinocytes. J. Invest. Dermatol. 135, 142–150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernard E., Hamel R., Neyret A., Ekchariyawat P., Molès J.-P., Simmons G., Chazal N., Desprès P., Missé D., Briant L., Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 476, 1–10 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Duangkhae P., Erdos G., Ryman K. D., Watkins S. C., Falo L. D. Jr., Marques E. T. A. Jr., Barratt-Boyes S. M., Interplay between keratinocytes and myeloid cells drives dengue virus spread in human skin. J. Invest. Dermatol. 138, 618–626 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Yang B., Suwanpradid J., Sanchez-Lagunes R., Choi H. W., Hoang P., Wang D., Abraham S. N., MacLeod A. S., IL-27 facilitates skin wound healing through induction of epidermal proliferation and host defense. J. Invest. Dermatol. 137, 1166–1175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tait Wojno E. D., Hunter C. A., Stumhofer J. S., The immunobiology of the interleukin-12 family: Room for discovery. Immunity 50, 851–870 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida H., Hunter C. A., The immunobiology of interleukin-27. Annu. Rev. Immunol. 33, 417–443 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Cao Y., Doodes P. D., Glant T. T., Finnegan A., IL-27 induces a Th1 immune response and susceptibility to experimental arthritis. J. Immunol. 180, 922–930 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Chihara N., Madi A., Kondo T., Zhang H., Acharya N., Singer M., Nyman J., Marjanovic N. D., Kowalczyk M. S., Wang C., Kurtulus S., Law T., Etminan Y., Nevin J., Buckley C. D., Burkett P. R., Buenrostro J. D., Rozenblatt-Rosen O., Anderson A. C., Regev A., Kuchroo V. K., Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar N. P., Moideen K., Banurekha V. V., Nair D., Sridhar R., Nutman T. B., Babu S., IL-27 and TGFβ mediated expansion of Th1 and adaptive regulatory T cells expressing IL-10 correlates with bacterial burden and disease severity in pulmonary tuberculosis. Immun. Inflamm. Dis. 3, 289–299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z., Liu J.-Q., Talebian F., Wu L.-C., Li S., Bai X.-F., IL-27 enhances the survival of tumor antigen-specific CD8+ T cells and programs them into IL-10-producing, memory precursor-like effector cells. Eur. J. Immunol. 43, 468–479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas S., Ghilardi N., Li J., de Sauvage F. J., IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 100, 15047–15052 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mittal A., Murugaiyan G., Beynon V., Hu D., Weiner H. L., IL-27 induction of IL-21 from human CD8+ T cells induces granzyme B in an autocrine manner. Immunol. Cell Biol. 90, 831–835 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pennock N. D., Gapin L., Kedl R. M., IL-27 is required for shaping the magnitude, affinity distribution, and memory of T cells responding to subunit immunization. Proc. Natl. Acad. Sci. U.S.A. 111, 16472–16477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters A., Fowler K. D., Chalmin F., Merkler D., Kuchroo V. K., Pot C., IL-27 induces Th17 differentiation in the absence of STAT1 signaling. J. Immunol. 195, 4144–4153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider R., Yaneva T., Beauseigle D., El-Khoury L., Arbour N., IL-27 increases the proliferation and effector functions of human naïve CD8+ T lymphocytes and promotes their development into Tc1 cells. Eur. J. Immunol. 41, 47–59 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Villarino A., Hibbert L., Lieberman L., Wilson E., Mak T., Yoshida H., Kastelein R. A., Saris C., Hunter C. A., The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity 19, 645–655 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Hamano S., Himeno K., Miyazaki Y., Ishii K., Yamanaka A., Takeda A., Zhang M., Hisaeda H., Mak T. W., Yoshimura A., Yoshida H., WSX-1 is required for resistance to Trypanosoma cruzi infection by regulation of proinflammatory cytokine production. Immunity 19, 657–667 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Morishima N., Owaki T., Asakawa M., Kamiya S., Mizuguchi J., Yoshimoto T., Augmentation of effector CD8+ T cell generation with enhanced granzyme B expression by IL-27. J. Immunol. 175, 1686–1693 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Harker J. A., Wong K. A., Dallari S., Bao P., Dolgoter A., Jo Y., Wehrens E. J., Macal M., Zuniga E. I., Interleukin-27R signaling mediates early viral containment and impacts innate and adaptive immunity after chronic lymphocytic choriomeningitis virus infection. J. Virol. 92, e02196-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fakruddin J. M., Lempicki R. A., Gorelick R. J., Yang J., Adelsberger J. W., Garcia-Pineres A. J., Pinto L. A., Lane H. C., Imamichi T., Noninfectious papilloma virus-like particles inhibit HIV-1 replication: Implications for immune control of HIV-1 infection by IL-27. Blood 109, 1841–1849 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sørensen O. E., Thapa D. R., Rosenthal A., Liu L., Roberts A. A., Ganz T., Differential regulation of β-Defensin expression in human skin by microbial stimuli. J. Immunol. 174, 4870–4879 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T., The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Sun L., Wu J., Du F., Chen X., Chen Z. J., Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type-I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drappier M., Michiels T., Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 15, 19–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu W., Jain A., Gao Y., Dozmorov I. M., Mandraju R., Wakeland E. K., Pasare C., Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc. Natl. Acad. Sci. U.S.A. 112, 13994–13999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vercammen E., Staal J., Beyaert R., Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 21, 13–25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazear H. M., Lancaster A., Wilkins C., Suthar M. S., Huang A., Vick S. C., Clepper L., Thackray L., Brassil M. M., Virgin H. W., Nikolich-Zugich J., Moses A. V., Gale M. Jr., Früh K., Diamond M. S., IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLOS Pathog. 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo Y.-P., Tsai K.-N., Luo Y.-C., Chung P.-J., Su Y.-W., Teng Y., Wu M.-S., Lin Y.-F., Lai C.-Y., Chuang T.-H., Dai S.-S., Tseng F.-C., Hsieh C.-H., Tsai D.-J., Tsai W.-T., Chen C.-H., Yu G.-Y., Establishment of a mouse model for the complete mosquito-mediated transmission cycle of Zika virus. PLOS Negl. Trop. Dis. 12, e0006417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazear H. M., Govero J., Smith A. M., Platt D. J., Fernandez E., Miner J. J., Diamond M. S., A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang S. C., Tan X.-Y., Luxenberg D. P., Karim R., Dunussi-Joannopoulos K., Collins M., Fouser L. A., Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203, 2271–2279 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grant A., Ponia S. S., Tripathi S., Balasubramaniam V., Miorin L., Sourisseau M., Schwarz M. C., Sánchez-Seco M. P., Evans M. J., Best S. M., García-Sastre A., Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song B.-H., Yun S.-I., Woolley M., Lee Y.-M., Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 308, 50–64 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Fauci A. S., Morens D. M., Zika virus in the Americas—Yet another arbovirus threat. N. Engl. J. Med. 374, 601–604 (2016). [DOI] [PubMed] [Google Scholar]
- 47.de Magalhães-Barbosa M. C., Prata-Barbosa A., Robaina J. R., Raymundo C. E., Lima-Setta F., da Cunha A. J. L. A., Trends of the microcephaly and Zika virus outbreak in Brazil, January-July 2016. Travel Med. Infect. Dis. 14, 458–463 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Cao-Lormeau V.-M., Blake A., Mons S., Lastère S., Roche C., Vanhomwegen J., Dub T., Baudouin L., Teissier A., Larre P., Vial A.-L., Decam C., Choumet V., Halstead S. K., Willison H. J., Musset L., Manuguerra J.-C., Despres P., Fournier E., Mallet H.-P., Musso D., Fontanet A., Neil J., Ghawché F., Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 387, 1531–1539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y., Liu Q., Zhou J., Xie W., Chen C., Wang Z., Yang H., Cui J., Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 3, 17006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yockey L. J., Jurado K. A., Arora N., Millet A., Rakib T., Milano K. M., Hastings A. K., Fikrig E., Kong Y., Horvath T. L., Weatherbee S., Kliman H. J., Coyne C. B., Iwasaki A., Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 3, eaao1680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barzon L., Trevisan M., Sinigaglia A., Lavezzo E., Palù G., Zika virus: From pathogenesis to disease control. FEMS Microbiol. Lett. 363, fnw202 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Hamel R., Dejarnac O., Wichit S., Ekchariyawat P., Neyret A., Luplertlop N., Perera-Lecoin M., Surasombatpattana P., Talignani L., Thomas F., Cao-Lormeau V.-M., Choumet V., Briant L., Desprès P., Amara A., Yssel H., Missé D., Biology of Zika virus infection in human skin cells. J. Virol. 89, 8880–8896 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Der S. D., Zhou A., Williams B. R. G., Silverman R. H., Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 95, 15623–15628 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Refaat A., Owis M., Abdelhamed S., Saiki I., Sakurai H., Retrospective screening of microarray data to identify candidate IFN-inducible genes in a HTLV-1 transformed model. Oncol. Lett. 15, 4753–4758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quicke K. M., Bowen J. R., Johnson E. L., McDonald C. E., Ma H., O’Neal J. T., Rajakumar A., Wrammert J., Rimawi B. H., Pulendran B., Schinazi R. F., Chakraborty R., Suthar M. S., Zika virus infects human placental macrophages. Cell Host Microbe 20, 83–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gautier L., Cope L., Bolstad B. M., Irizarry R. A., affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huber W., Carey V. J., Gentleman R., Anders S., Carlson M., Carvalho B. S., Bravo H. C., Davis S., Gatto L., Girke T., Gottardo R., Hahne F., Hansen K. D., Irizarry R. A., Lawrence M., Love M. I., MacDonald J., Obenchain V., Oleś A. K., Pagès H., Reyes A., Shannon P., Smyth G. K., Tenenbaum D., Waldron L., Morgan M., Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay3245/DC1
Data Availability Statement
Data showing the IL-27 stimulated human keratinocyte dataset (shown in Figure 1) can be found at GSE143228.