

MAP6 enters the lumen and induces coiling in a previously unidentified class of neuronal microtubules.
Abstract
Neuronal activities depend heavily on microtubules, which shape neuronal processes and transport myriad molecules within them. Although constantly remodeled through growth and shrinkage events, neuronal microtubules must be sufficiently stable to maintain nervous system wiring. This stability is somehow maintained by various microtubule-associated proteins (MAPs), but little is known about how these proteins work. Here, we show that MAP6, previously known to confer cold stability to microtubules, promotes growth. More unexpectedly, MAP6 localizes in the lumen of microtubules, induces the microtubules to coil into a left-handed helix, and forms apertures in the lattice, likely to relieve mechanical stress. These features have not been seen in microtubules before and could play roles in maintaining axonal width or providing flexibility in the face of compressive forces during development.
INTRODUCTION
There is perhaps no cell type that must meet more extreme demands than neurons. From their birth and migration within the tight quarters of the developing neural crest to the precise navigation of axons tips extending toward targets that may be meters away, neurons must both maintain a proper shape and be sufficiently flexible to adapt their morphology to new functional demands. This dual capacity for persistence and plasticity derives largely from microtubules, which themselves can be stable, labile, or a combination of both (1).
Microtubules are hollow cylinders that are constantly being made and disassembled according to the cell’s needs to grow, change shape, and move organelles or cargo within the intracellular space. They are composed of tubulin heterodimers, consisting of α-tubulin and β-tubulin, which join end to end to form protofilaments that associate laterally around a 25-nm-wide core. The β subunit is key to the microtubule’s renowned dynamic instability: Guanosine triphosphate (GTP) bound to the β subunit is quickly hydrolyzed to guanosine diphosphate (GDP) soon after assembly, leading to a conformational change of tubulin that makes the microtubule more prone to depolymerization (2, 3). In purified tubulin preparations, microtubules alternate between phases of growth and shrinkage, known rather colorfully as rescues and catastrophes. In terminally differentiated neurons, however, dynamic microtubules coexist with long-lived microtubules characterized by slow turnover. These stable microtubules are thought to be crucial for the maintenance of axonal and dendritic architecture. Recent studies indicate that there is actually a whole repertoire of stable microtubule subtypes, but the specific roles of these subpopulations in neurons are not yet known (4). Further complicating this picture is the fact that different regions of the same microtubule can have different degrees of lability or stability (1). A number of proteins and posttranslational modifications are thought to influence microtubule perdurance, particularly the microtubule-associated proteins (MAPs) (1), but their mechanisms remain elusive.
Our laboratory has been studying MAP6 (also known as stable tubule-only peptide), a structural MAP that has the unusual property of protecting microtubules against drug- and cold-induced depolymerization (5–8). This extreme stability appears to be a property unique to MAP6, and it serves a physiological purpose: MAP6-deficient mice suffer severe deficits in synaptic plasticity similar to those seen in schizophrenia, along with concomitant cognitive deficits (9). They also display axonal tract growth defects (10, 11), possibly due to microtubule lability that can be alleviated by microtubule-stabilizing drugs (12, 13). To better understand the mechanism by which neuronal microtubules acquire stability, we decided to study MAP6 activity at the level of individual microtubules.
RESULTS AND DISCUSSION
MAP6 forms microtubule intraluminal particles
We first analyzed microtubules extracted from primary cultured neurons of either wild-type or MAP6-deficient mouse embryos by cryo–electron microscopy. We observed the coexistence of two populations of microtubules in the wild-type condition (Fig. 1A): 64% contained intraluminal particles, and 36% did not. By contrast, the majority (73%) of MAP6-KO (knockout) neurons contained no visible intraluminal particles at all. This observation caught our attention for three reasons. First, MAP6 has been thought to be a structural MAP and thus to bind to the exterior of the microtubule lattice. Second, although intraluminal particles have been previously reported in microtubules observed in certain neuronal compartments (14), their identity has been completely unknown. Third, there is evidence that microtubules are regulated, at least in part, through the lumen: Tubulin acetylation, which occurs on long-lived microtubules, occurs within the lumen, and taxol, the major stabilizing agent, binds to the microtubule lumen (15); furthermore, so-called microtubule inner proteins (MIPs) within cilia and flagella have been found to be responsible for doublet microtubule stabilization (16). Could MAP6 be an inner protein for neuronal microtubules?
Fig. 1. MAP6 localizes inside microtubules.

(A) Cryo–electron microscopy images showing the two types of microtubules extracted from primary cultured neurons of mouse embryos. Black and white stars indicate microtubules with and without intraluminal particles, respectively. Inset shows a high-magnification image of the area denoted by the dashed rectangle (a band-pass filter was applied and added to the original image to improve contrast). The percentages of microtubules with or without these particles in wild-type (WT) versus MAP6-KO neurons are indicated on the right. The total length of measured microtubules was 278 and 242 μm for wild-type and MAP6-KO conditions, respectively. Scale bars, 50 nm (horizontal) and 25 nm (vertical). (B) Main panels: Cryo–electron tomography of microtubules copolymerized in vitro with purified tubulin and MAP6 in the presence of GMPCPP. Arrowheads, microtubule inner particles. Bottom: (a to d) Transverse sections along the lines indicated in top panels showing localization of dots inside hollow tubes (a and b) or at the inner side of open protofilament sheets (c and d). (C) Cryo–electron microscopy images of in vitro pre-polymerized and taxol-stabilized microtubules (MTs), incubated with or without MAP6. Scale bars, 50 nm.
Because it would be difficult to answer this question in cells, we used in vitro reconstitution experiments to polymerize microtubules from purified tubulin in the presence of recombinant neuronal MAP6 (fig. S1A). Cryo–electron tomography revealed regularly spaced particles within microtubules copolymerized with MAP6 (Fig. 1B). Cross sections of the microtubules show that these particles are localized within the lumen and on the inner face of unclosed protofilament sheets at microtubule’s growing end (Fig. 1B, a to d, and movie S1). These intraluminal particles have a mean diameter of 9 nm [9.27 ± 1.74 (SD), n = 85] and are spaced at ~31-nm intervals [31.1 ± 6.7 (SD), n = 189]. When preformed taxol-stabilized microtubules were incubated with MAP6, no particles appeared within the microtubules (Fig. 1C), but MAP6–green fluorescent protein (GFP) did decorate the lattice exterior (fig. S1, B and C). This indicates that the intraluminal particles form only when MAP6 is present during co-polymerization.
MAP6 modulates microtubule dynamics
We next used total internal reflection fluorescence (TIRF) microscopy to analyze the influence of low concentrations of MAP6 on microtubule growth from stable seeds (Fig. 2A and fig. S2). MAP6 strongly inhibited microtubule depolymerization by reducing the shrinking rate and promoting rescue events (Fig. 2B). Unexpectedly, MAP6 also increased the frequency of catastrophes (Fig. 2B). This is unusual for a structural stabilizing MAP but would be expected for end-binding proteins that accumulate at the microtubule tips such as end-binding proteins (EBs) and members of the kinesin family (KIF2A, KIF18A, and KIF3C) (17–19). Thus, MAP6 exerts a strong stabilizing effect that likely involves binding along the lattice; at the same time, it destabilizes the microtubule ends. The combination of these two activities appears to produce slow-growing, largely stable microtubules with highly dynamic extremities.
Fig. 2. MAP6 promotes the growth of stable, curved microtubules.
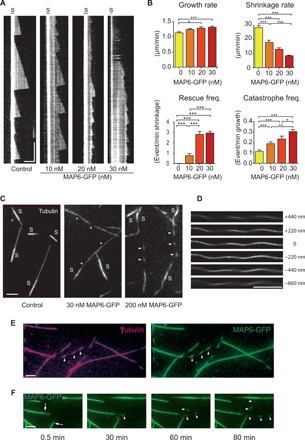
(A) Kymographs show microtubules grown from GMPCPP seeds (S) in the presence of 10 μM tubulin and 0 (control), 10, 20, or 30 nM MAP6-GFP (n = 93, 41, 38, and 40 microtubules, respectively). (B) Graphs show rates of growth and shrinkage and frequencies of rescue and catastrophe, extracted from kymographs presented in (A). (C) Pictures of microtubules grown as in (A) from GMPCPP seeds in the presence of 0 (control), 30, or 200 nM MAP6-GFP for 90 min. Note the modification of microtubule appearance, with increasing concentrations of MAP6, from a straight, uniform conformation (control, left), to a slightly curved shape with portions showing less fluorescence (*, middle), and finally to a dashed line appearance (arrowheads, right). (D) Microtubules grown in the presence of 200 nM MAP6-GFP and imaged using confocal Airyscan. Distance variations in the z axis are indicated to the right. (E) Microtubules first grown with 15 μM tubulin and then incubated with 10 μM tubulin and 150 nM MAP6-GFP for an additional 90 min (arrowheads point to areas of less fluorescence that create a dashed appearance). Magenta, tubulin; green, MAP6-GFP. (F) Time course of microtubule polymerization after addition of 150 nM MAP6-GFP. Preexisting microtubules are polymerized without MAP6-GFP (arrows), and dash-shaped microtubules are polymerized in the presence of MAP6-GFP (arrowheads). Note the rapid binding of MAP6-GFP to preexisting microtubules. Scale bars, 5 μm (horizontal) and 5 min (vertical). Bars, means; error bars, SEM. *P < 0.05; **P < 0.01; ***P < 0.001, Kruskal-Wallis analysis of variance (ANOVA) followed by post hoc Dunn’s multiple comparisons test.
MAP6 induces microtubule helical deformation
Besides influencing microtubule dynamics, MAP6 also altered the conformation of the growing microtubule. This effect, already visible at 30 nM MAP6, was quite notable at 150 to 200 nM (Fig. 2C). When observed by TIRF microscopy, microtubules grown in the presence of MAP6 took on the appearance of a dashed line instead of the continuous line visible in the control condition, because parts of the strand curve away from the illuminated field (~100 nm) at the bottom of the perfusion chamber (fig. S3A) (wide-field illumination is able to show the continuity of the strand). The orientation and the periodicity of the “dashes” compared to the overall direction of the microtubule indicate a left-handed, long-range helicity with a pitch of 5.5 ± 0.8 μm (SD, n = 478), which we confirmed using confocal microscopy with deconvolution (Airy scanning microscopy) at several focal illuminations along the z axis (Fig. 2D and movie S2). These curvatures cannot be explained by an increase in microtubule flexibility, because the overall growth of these three-dimensional (3D) helical microtubules remained linear for the duration of the experiment (movie S3). When MAP6 was added to preformed microtubules, only the newly growing sections were helical, while the preexisting ones remained linear (Fig. 2, E and F). These results indicate that, as for the formation of intraluminal particles, MAP6-dependent changes in microtubule structure require MAP6 to be present at the time of polymerization. This suggests that MAP6 acts at the tip of growing microtubules.
Intraluminal localization of MAP6 is required for microtubule helical deformation
The co-occurrence of tip destabilization, intraluminal particle formation, and microtubule conformational changes suggests that these events are linked. To see whether we could parse these events, we tested several MAP6 mutants with deletion of domains conserved among the neuronal isoforms of MAP6 family: the microtubule stabilizing Mn and Mc domains (MAP6ΔMn and MAP6ΔMc; fig. S4) and the N-terminal domain (MAP6Δ4-35; Fig. 3A) (20, 21). These domains are all required for microtubule curvature and microtubule inner particle formation (Fig. 3, B and D, and fig. S4). MAP6Δ4-35 was of particular interest, because it retains the known microtubule-binding domains (Fig. 3A) and accordingly bound to and stabilized microtubules to a similar degree as the full-length MAP6 by reducing shrinkage rates and increasing rescue events but failed to stimulate catastrophes (Fig. 3C). Overall, these data imply that the presence of MAP6 inside the lumen of growing microtubules is necessary for it to induce microtubule helicity.
Fig. 3. MAP6Δ4-35 mutant stabilizes microtubules but does not lead to catastrophes, curvature, apertures, or particle densities within the lumen.
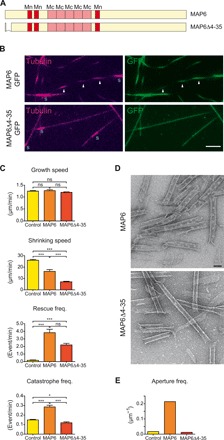
(A) Scheme of MAP6 and MAP6Δ4-35 showing the temperature-sensitive (Mc) and temperature-insensitive (Mn) microtubule stabilization domains and the deletion present in the MAP6Δ4-35 mutant. (B) TIRF images of microtubules grown for 90 min from GMPCPP seeds in the presence of either 200 nM MAP6-GFP or MAP6Δ4-35-GFP. Arrowheads point to the dashed appearance of microtubules. Scale bar, 5 μm. (C) Dynamical parameters of microtubules polymerized in the absence or presence of either MAP6-GFP or MAP6Δ4-35-GFP (n = 93, 29, and 43 microtubules, respectively). (D) Electron microscopy images of microtubules copolymerized with either MAP6-GFP or MAP6Δ4-35-GFP in the presence of GMPCPP and processed for negative staining. Scale bar, 50 nm. (E) Aperture frequency in microtubules copolymerized in the absence or presence of either MAP6-GFP or MAP6Δ4-35-GFP. *P < 0.05; ***P < 0.001, Kruskal-Wallis ANOVA followed by post hoc Dunn’s multiple comparison. ns, not significant.
MAP6 induces the formation of stable apertures in growing microtubules
Another consequence of the presence of MAP6 during microtubule polymerization is the apparition of numerous apertures in the lattice (Fig. 4, A and B). The fact that aperture frequency (~0.2 μm−1) and size (~100 nm) did not change significantly over time (15 to 90 min; Fig. 4, C and D) indicates that MAP6-induced holes are not prone to efficient repair, such as that described for enzymatically or mechanically damaged microtubules (22, 23). MAP6 did not induce aperture formation in preformed taxol-stabilized microtubules (Fig. 4E), confirming that MAP6 must be present during microtubule polymerization to induce the mechanical stress that leads to aperture formation. Accordingly, the MAP6Δ4-35 mutant, which neither formed particles inside the lumen nor induced curvature, showed no more tendency to form apertures than control (Fig. 3E). It is worth noting that a cellular cryo–electron microscopy study recently reported similarly sized apertures within neuronal microtubules (24), which suggests that our results are relevant to the in vivo condition.
Fig. 4. MAP6 produces apertured microtubules.
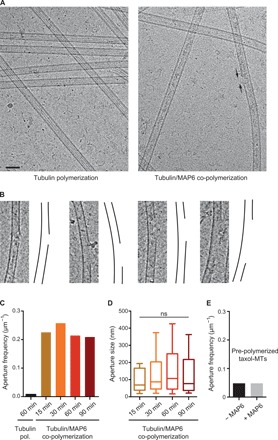
(A) Cryo–electron microscopy images of microtubules polymerized with tubulin alone (left) or in the presence of MAP6 (right). Arrows point to one aperture in the microtubule lattice. Scale bar, 50 nm. (B) Examples of apertures observed in microtubules polymerized with MAP6. In each case, the micrograph is on the left, and a drawing of the microtubule wall is on the right. To improve contrast, a band-pass filter was applied and added to original images. (C) Aperture frequency on microtubules polymerized with tubulin alone or in the presence of MAP6 at several polymerization times. The total length of measured microtubules was 370, 164, 291, 569, and 248 μm in the control (no MAP6) and at 15, 30, 60, and 90 min, respectively. (D) Distribution of aperture size in the presence of MAP6 at indicated polymerization times. Whiskers represent the 10th and 90th percentiles of the data. Bars within boxes represent the median. Kruskal-Wallis ANOVA followed by post hoc Dunn’s multiple comparisons test; n = 38, 75, 122, and 52 for assembly at 15, 30, 60, and 90 min, respectively. (E) Aperture frequency in preformed taxol-stabilized microtubules incubated without or with MAP6. The total length of measured microtubules was 319 and 237 μm without and with MAP6, respectively.
MAP6 induces microtubule bending: The frustrated core-shell model
How can we link the presence of MAP6 particles inside the lumen of growing microtubules with the formation of helical microtubules containing stable apertures? The induction of microtubule curvature by MAP6 can be explained by a frustrated core-shell model built on our previous tubulin bistability model (25, 26) based on two assumptions: (i) After GTP hydrolysis, the GDP-tubulin can exist as long or short tubulin states (Fig. 5A; see also Supplementary Text), and (ii) a cooperative (allosteric) interaction within a protofilament favors the switching of adjacent tubulins. In the absence of MAPs or external forces, following GTP hydrolysis, GDP-tubulins will predominantly switch to their short state and the microtubule is straight [Fig. 5B and (26)]. In the presence of MAP6, this picture changes drastically as large-scale coherent superhelices are formed. We propose that when tubulin and MAP6 are copolymerized, MAP6 forms an elastic network inside the microtubule lumen by binding to GTP-tubulin. The GDP-tubulin resulting from GTP hydrolysis tries to shorten, but the intraluminal MAP6 elastic network exerts an opposite effect favoring the long state (Fig. 5C). To solve this conflict and decrease the stored energy to a minimum, the lattice might adopt a frustrated conformation where, on one side of the microtubule lattice, a block of adjacent tubulin dimers adopts the short state conformation, while, on the opposite side, tubulin dimers stay in the long state conformation. This leads to a local bending in the direction of the short tubulin block. Once the curvature is established, the conformational switching of GDP-tubulin to the short state propagates via allosteric interaction along the protofilament axis and induces a continuous curvature of the microtubule, resulting in a global helical reshaping, a process reminiscent of the formation of polymorphic, helically coiled states of bacterial flagella (27, 28). As this spontaneous symmetry breaking mechanism is due to the coherent conformational switch of an entire block of protofilaments, the superhelical pitch is imposed by the intrinsic, lattice-imposed helicity of the protofilament along the microtubule axis (29, 30). This is in agreement with our data (Figs. 2 and 5D). Microtubules nucleated from guanylyl-(alpha, beta)-methylene-diphosphonate (GMPCPP) microtubule seeds are likely to be composed of 14 protofilaments both because of the seeds (31) and the fact that MAP6 promotes the growth of 14-protofilament microtubules (fig. S5). The observation confirms that the superhelical shape of microtubules grown in the presence of MAP6 is left-handed, with a pitch of around 6 μm, in agreement with the intrinsic left-handed twisted protofilament structure observed in the 14-protofilament microtubules (without MAP) (32). This fact alone strongly supports the cooperativity assumption of our model.
Fig. 5. MAP6 induces microtubule bending: The frustrated core-shell model.
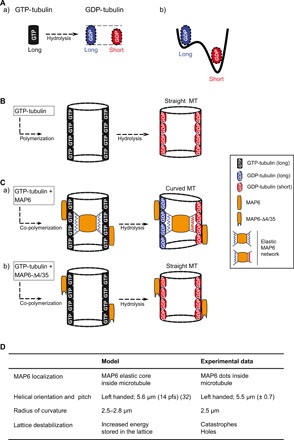
(A) After GTP hydrolysis, the GDP-bound tubulin protofilaments can exist in two distinct states (a), with slightly differing elongations (see Supplementary Text): a long tubulin state (blue) and a short tubulin state (red). (b) The short state of the GDP-tubulin is energetically favored with respect to the long one. (B) For microtubules polymerized with tubulin alone, GTP-tubulin is incorporated into the lattice in its long state. After GTP hydrolysis, the short GDP-tubulin state is energetically dominant. The microtubule shortens but stays straight. To allow visualization of the lumen, only two protofilaments, one on each side of the microtubule, are shown. (C) For microtubules copolymerized with tubulin and MAP6 (a), MAP6 binds to the nonhydrolyzed, long GTP-tubulin state and forms an elastic network inside the microtubule, stabilizing the initial (long) state at the time point of binding. After hydrolysis, the GDP-tubulin tends to shorten but enters into conflict with the force exerted by the intraluminal MAP6 network to maintain the tubulin long state. The resulting frustration is minimized by a symmetry-breaking mechanism [i.e., compact tubulin dimers (red) on one side of the cylinder and elongated tubulin dimers (blue) on the opposite side], leading to a curved ground state and in which areas of high local stress form holes or apertures (not shown in the scheme; Fig. 4). The observed radius of curvature of the superhelix, about 2.5 μm, is theoretically realized for a length difference of about 0.6% between the short and long state of GDP-tubulin (see Supplementary Text for more details). (b) MAP6Δ4-35 mutant only binds outside the microtubule where it stabilizes the lattice without inducing microtubule curvature. Note that MAP6 also binds on the microtubule external surface (a). (D) Model validation: The table shows the agreements between data predicted from the model with those experimentally obtained.
We also measured the approximate width of the helix [around 700 nm (fig. S3B)] and calculated the average radius of curvature of helical microtubules (see Materials and Methods). The obtained value of 2.5 μm allows us to predict from the model a length difference of about 0.6% between the short and long state of GDP-tubulin (see Fig. 5D and Supplementary Text). MAP6Δ4-35 mutant that does not localize inside the microtubule wall fails to induce microtubule curvature (Fig. 5C).
Another consequence of the model is that the increased energy stored in the microtubule wall must be somehow released. It is possible that this extra stored energy destabilizes the microtubule lattice locally, leading to loss of specific dimers to relieve the mechanical stress; such a scenario could account for the increase in catastrophe frequency despite MAP6’s potent stabilizing effect (Fig. 2) and the formation of apertures (Fig. 4).
What might be the functional relevance of helical microtubules with persistent holes and intraluminal densities in neurons? The heterogeneous distribution of neuronal MIPs that we and others find (14, 24) indicates that there are specialized functions for different kinds of stable microtubules. One possibility is that proteins inside microtubules and apertures in the microtubule lattice modify the accessibility of acetylation sites to αTAT1 (15, 33). By occupying greater width, a helical structure might influence the spatial organization of the microtubule network (e.g., by inhibiting microtubule co-alignment and bundling), help determine neurite or axonal width, or confer resistance to compressive loads such as might be encountered during development or regeneration (e.g., moving through a glial scar) (34). Given that MAP6 belongs to a family of four genes (MAP6, MAP6D1, FAM154A, and FAM154B) (21, 35), understanding the functions of these other proteins could help clarify the special roles of various microtubule types in neurons.
Luminal material has been known to exist within cellular microtubules for decades, but the identity and function of the particles have remained largely unknown (14, 24, 36). To our knowledge, the tubulin acetyl transferase αTAT1, which catalyzes tubulin acetylation in the lumen of microtubules, is the only protein so far thought to function inside microtubules—yet the evidence for αTAT1 localization within microtubules remains indirect (37). MAP6 would therefore be the first identified MIP in neurons. Further work will be needed to tease out the specific neuronal functions that require such a class of stable, helical microtubules.
MATERIALS AND METHODS
Preparation of tubulin
Tubulin was purified from bovine brain and labeled with ATTO-565 or ATTO-496 [ATTO-TEC GmbH, Germany (38)].
Recombinant protein expression
MAP6 with an N-terminal histidine-Tag was cloned either directly in pFastbac-Htb or in fusion with GFP and an additional histidine-Tag at the C termini. Bacmids and baculovirus were produced using DH10BAC and sf9 insect cells as indicated by the manufacturer (Thermo Fisher Scientific). Protein expression was carried out in High Five cells in suspension at 106 cells/ml for 48 hours at 27°C.
Purification of His-MAP6
Each population of 108 High Five cells was resuspended in 5 ml of lysis buffer [50 mM Hepes (pH 7.4), 400 mM NaCl, 0.5% Triton X-100, and 1 protease inhibitor tablet mini (Roche)] and centrifuged at 200,000g for 30 min at 4°C. Heat-stable proteins present in solution were recovered in the supernatant after incubation at 90°C for 15 min and centrifugation (200,000g for 30 min at 4°C). The soluble fraction was diluted three times with the dilution buffer [50 mM Hepes (pH 7.4) and 0.5% Triton X-100], passed through a 4-ml Q-Sepharose column, and loaded on a 5-ml SP Sepharose column. After washing with Q/SP column buffer [50 mM Hepes (pH 7.4), 150 mM NaCl, and 0.5% Triton X-100], bound proteins were eluted with five column volumes of elution buffer [50 mM Hepes (pH 7.4), 400 mM NaCl, and 0.05% Triton X-100]. Imidazole (2 mM) was then added, and the solution was loaded on a cobalt column (1 ml; Thermo Fisher Scientific). After washing with 10 mM imidazole in 50 mM Hepes (pH 7.4) and 400 mM NaCl, bound proteins were eluted by increasing the imidazole concentration up to 200 mM. The eluate volume was reduced to 200 μl by concentration on Amicon Ultra 30K (Merck Millipore) and injected on a gel filtration column (Superdex 200 10/300; Thermo Fisher Scientific) in 20 mM Hepes (pH7.4) and KCl 100 mM. Collected fractions containing the protein were pooled, concentrated as above on Amicon Ultra 30K, centrifuged at 200,000g for 10 min at 4°C, aliquoted, quick-frozen in liquid nitrogen, and stored at −80°C. Protein concentration was quantified by SDS–polyacrylamide gel electrophoresis (PAGE) and Coomassie blue staining against a bovine serum albumin (BSA) concentration ladder using ChemiDoc (Bio-Rad) scanning and ImageJ software.
Purification of His-MAP6-GFP-His, His-MAP6ΔMn-GFP-His, His-MAP6ΔMc-GFP-His, or His-MAP6-Δ4-35-GFP-His protein
Each 108 High Five cell pellet was resuspended in 5 ml of lysis buffer A [50 mM Hepes (pH 7.4), 500 mM arginine, 1 mM EGTA/EDTA, and 1 protease inhibitor tablet mini (Roche)]. After centrifugation (150,000g for 30 min at 4°C), the supernatant was diluted 10 times in dilution buffer [50 mM Hepes (pH 7.4), 100 mM NaCl, 0.1% Triton X-100, and 1 mM EGTA/EDTA] and centrifuged again at 150,000g for 30 min at 4°C. The supernatant was passed through a 4-ml Q-Sepharose and loaded on a 5-ml SP Sepharose. After washing with Q/SP column buffer [50 mM Hepes (pH 7.4), 150 mM NaCl, and 0.05% Triton X-100], bound proteins were eluted with a five-column volume of SP elution buffer [50 mM Hepes (pH 7.4), 400 mM NaCl, and 0.05% Triton]. Imidazole (6 mM) was then added, and the solution was loaded on a cobalt column (1 ml; Thermo Fisher Scientific). The cobalt column was first washed with 50 mM Hepes (pH 7.4) and 400 mM NaCl in the presence of 20 mM imidazole, and the bound proteins were further eluted in the presence of 200 mM imidazole. The proteins were further purified using Superdex-200 columns as described above for His-MAP6.
TIRF microscopy
Perfusion chambers were prepared with functionalized polyethylene glycol (PEG)–silane glass coverslips. Coverslips were first sonicated in acetone for 30 min, washed in acetone (30 min) and 96% ethanol (EtOH) (15 min), and rinsed with water. Coverslips were next treated with a 2% Hellmanex solution (Hellma) for 2 hours and rinsed in water. They were next sonicated in 1 M NaOH for 15 min, rinsed with water, further sonicated in 96% EtOH, and rinsed again with water. Coverslips were dried under clean air stream, treated with plasma, and incubated overnight at room temperature under gentle agitation in a 1 mg/ml solution of PEG-silane (30 kDa, Creative PEGWorks) or PEG-silane-biotin (3.5 kDa, Laysan Bio) in EtOH containing 0.02% HCl. Coverslips were next rinsed in 96% EtOH and water, air-dried, and stored at 4°C. Perfusion chambers were assembled between PEG-silane–coated and PEG-silane-biotin–coated coverslip using double adhesive tape face to form a flow chamber of around 5 μl in volume.
Preparation of microtubule seeds
Microtubule seeds were obtained by polymerizing 10 μM tubulin (50% of biotinylated tubulin and 50% of ATTO-565–labeled tubulin) in the presence of 1 mM GMPCPP in BRB80 buffer at 37°C for 1 hour. Microtubule seeds were then centrifuged for 5 min at 100,000g, resuspended in BRB80 with 1 mM GMPCPP, aliquoted, and stored in liquid nitrogen. For each experiment, one aliquot was thawed at 35°C and diluted (1:300 to 1:500) in BRB80 + 1% BSA.
Microtubule polymerization assay in TIRF microscopy
The flow chamber was successively perfused with neutravidin (25 μg/ml; Pierce) in 1% BSA, Poly(l-lysine) (PLL)–g-PEG (2-kDa solution; 0.1 mg/ml; JenKem) in 10 mM Hepes (pH 7.4), 1% pluronic F127 in BRB80, and 1% BSA in BRB80 buffer and finally with the microtubule seed solution. Microtubule polymerization was initiated with 10 μM tubulin (containing 10% of ATTO-565–labeled tubulin and 90% unlabeled tubulin) with the indicated concentration of MAP6 proteins in TIRF buffer {BRB80 plus 50 mM KCl, 4 mM dithiothreitol (DTT), 1% BSA, glucose (1 mg/ml), catalase (70 μg/ml), glucose oxidase (600 μg/ml), and 0.05% methylcellulose [1500 centipoise (cP)]}. Chambers were sealed with dental paste and visualized on an inverted microscope (Eclipse Ti, Nikon) equipped with an iLas TIRF system (Roper Scientific), a cooled charge-coupled device camera (EMCCD Evolve 512, Photometrics) with 512 × 512–pixel imaging array (16 × 16 μm pixels), and a warm stage controller (LINKAM MC60), all under the control of MetaMorph software (version 7.7.5, Molecular Devices). Samples were excited with 491- and 561-nm lasers and observed using an Apochromat 60× or 100× oil-immersion objective (numerical aperture, 1.49). Time-lapse imaging was performed at one frame per 5 s with a 100-ms exposure time during 45 to 90 min at 32°C.
Analysis of microtubule dynamics
Image analysis was performed in ImageJ software, with an in-house plugin for extracting the four parameters determining microtubule dynamics from the kymograph. Growth and shrinkage rates were calculated from the slopes corresponding to the microtubules’ growing and shrinking phases, respectively. The frequencies of catastrophe and rescue events were calculated by dividing the number of events by the time spent in the growing or the shrinking phase, respectively. Graphs and statistical analysis were made using GraphPad Prism.
Wide-field TIRF and Airyscan imaging of microtubules
Microtubules were allowed to polymerize as described above, except that the perfusion chamber was not sealed but kept under wet atmosphere for 45 min. Free tubulin was removed by gentle perfusion with BRB containing 0.05% methylcellulose (1500 cP), and microtubules were fixed with 0.5% of glutaraldehyde in the same buffer for 5 min. Imaging buffer [BRB80, 0.1% methylcellulose (1500 cP), catalase (82 μg/ml), glucose oxidase (0.58 mg/ml), and glucose (1 mg/ml)] was then perfused, and flow chambers were sealed with paste. Imaging was done using TIRF microscopy as described above in TIRF or wide-field illumination or, for higher-resolution analysis, with a confocal LSM710/Airyscan (Zeiss) microscope.
Determination of the radius of curvature of helical microtubule
The radius of curvature (Rc) for the helical microtubule was calculated using the formula Rc = r + p2/4π2r, where r is the radius and p is the pitch of the helix. It yields a value of ~2.5 μm for a radius of 0.35 μm and a pitch of 5.5 μm.
MAP6-GFP interaction with taxol-stabilized microtubules
Microtubules were polymerized from 70 μM tubulin in BRB buffer plus 1 mM GTP for 30 min at 35°C. Taxol (100 μM) was then added, and the incubation was continued for additional 15 min. The polymerization mix was diluted to 15 μM tubulin and centrifuged at 200,000g for 10 min at 35°C. Microtubules were resuspended in one volume of binding buffer (BRB80 plus 50 mM KCl, 5 mM DTT, 0.05% Tween 20, and 10 μM taxol), and their concentration was quantified using a tubulin concentration ladder and analysis of the scanned Coomassie blue-stained SDS-PAGE gels with ImageJ software. Microtubules (200 nM) were incubated with increasing concentrations of MAP6-N-GFP (5 to 500 nM in triplicates) for 10 min at 35°C in 10 μl of binding buffer and centrifuged at 200,000g for 10 min at 35°C. The supernatants were discarded, the pellets were resuspended with 200 μl of 3 M urea in 100 mM tris-HCl (pH 8.0), and the quantities of MAP6-GFP co-sedimented with tubulin were quantified against a MAP6-GFP calibration curve using PHERAstar FS (BMG Labtech). Concentrations of bound MAP6-GFP were plotted as a function of the concentration of MAP6-GFP input. GraphPad Prism was used for curve fitting (one-site binding equation) and Kdapp determination.
Extraction of neuronal microtubules for cryo–electron microscopy analysis
Mouse hippocampal neurons were prepared as previously described (21). In accordance with the policy of the Institut des Neurosciences of Grenoble (GIN) and the French legislation, experiments were done in compliance with the European Community Council Directive of 24th November 1986 (86/609/EEC). The research involving animals was authorized by the Direction Départementale de la protection des populations—Préfecture de l’Isère-France and by the ethics committee of GIN number 004 accredited by the French Ministry for of Research. At 8 days of culture (8 days in vitro), they were rinsed with PEM buffer (80 mM Pipes, 2 mM EGTA, and 1 mM MgCl2), permeabilized for 3 min at 37°C in PEM buffer supplemented with 10 μM taxol and 0.5% Triton X-100, and washed three times with PEM-taxol. Neurons were then scraped in PEM-taxol and centrifuged for 3 min at 40g, and the supernatants were immediately frozen.
Transmission electron microscopy and negative staining
Tubulin was polymerized at 3 μM in BRB80 buffer supplemented with 0.5 mM GMPCPP and 10% dimethyl sulfoxide (DMSO) in the absence or presence of 1 μM MAP6 or MAP-GFP for 40 min at 32°C. A 4-μl sample drop was applied to a glow-discharge copper carbon–coated grid, blotted, and stained with 2% uranyl acetate for 30 s. Grids were observed using a FEI Tecnai F20 200 kV FEG equipped with a 4K × 4K FEI Ceta CMOS (complementary metal-oxide semiconductor) camera (FEI Company Ltd.).
Cryo–electron microscopy and tomography
Vitreous ice-embedded microtubules were prepared under controlled temperature and humidity using the vitrification robot EM-GP2 (Leica). Tubulin (6 μM) was polymerized at 30°C in the presence of MAP6 (0.3 μM) in BRB80 buffer supplemented with 50 mM KCl and 1 mM GTP. Sample (4 μl) was applied onto holey carbon grids in the EM-GP2 chamber at various incubation times (15, 30, 60, and 90 min) followed by blotting and vitrification in liquid ethane. For control experiments, microtubules were polymerized from 60 μM tubulin in BRB80 buffer supplemented with 50 mM KCl and 1 mM GTP at 32°C for 60 min. For experiments using preformed microtubules, taxol-stabilized microtubules (1.5 μM) were incubated with MAP6 (0.3 μM) for 30 min at 32°C. For experiments using extracts, extracts containing microtubules were rapidly thawed before vitrification. Specimens were observed under low-dose conditions using a FEI Tecnai F20 equipped with a 4K × 4K FEI Ceta CMOS camera and operated at 200 kV. Image analysis was performed in ImageJ software.
For cryo–electron tomography, 10-nm gold nanoparticles coated with cationic BSA (Aurion Gold Tracers, 210111) were prepared in BRB80 as described previously (39) and used as fiducial markers for 3D reconstructions. Tubulin (6 μM) was polymerized in the presence of MAP6 (2 μM) in BRB80 buffer supplemented with 0.5 mM GMPCPP at 32°C during 40 min. Sample (4 μl) was mixed with 1 μl of fiducial markers and deposited onto a holey carbon–coated grid (Quantifoil, R 3.5/1) in the humid and temperature-controlled EM-GP2 chamber before blotting and vitrification. Specimens were observed on a FEI Tecnai F20 equipped with a 4K × 4K FEI Ceta CMOS camera and operated at 200 kV. Tomographic tilt series were acquired at a magnification of ×29,000 between ±60° (2° increments) under low electron dose (1 to 2 e/Å2) and at a defocus of −4 to −5 μm, using FEI tomography software. Tomograms were reconstructed using IMOD and binned by a factor of 2 (final pixel size of 0.70 nm).
Supplementary Material
Acknowledgments
We thank L. Demacedo and A. Vinit for technical support in cell culture and protein biochemistry, E. Neumann for help with the electron microscope, and the zootechnicians of the Grenoble Institute Neuroscience (GIN). Funding: This work was supported by INSERM, CEA, CNRS, Université Grenoble Alpes and by awards from the French Agence Nationale de la Recherche to A.A., I.A., and G.S. (2017-CE11-0026 MAMAs). This work was supported by grants from Agence Nationale de la Recherche ANR-15-IDEX-02 NeuroCoG in the framework of the “Investissements d’avenir” program. GIN is a member of the Grenoble Center of Excellence in Neurodegeneration (GREEN). The Photonic Imaging Center of Grenoble Institute Neuroscience (Univ. Grenoble Alpes—Inserm U1216) is part of the ISdV core facility and certified by the IBiSA label. This work used the EM facilitie at the Grenoble Instruct-ERIC Center (ISBG; UMS 3518 CNRS CEA-UGA-EMBL) with support from the French Infrastructure for Integrated Structural Biology (FRISBI; ANR-10-INSB-05-02) and GRAL, a project of the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003) within the Grenoble Partnership for Structural Biology. The IBS Electron Microscope facility is supported by the Auvergne Rhône-Alpes Region, the Fonds Feder, the Fondation pour la Recherche Médicale, and GIS-IBiSA. Author contributions: C.D. prepared recombinant proteins. C.D., C.C., and M.S. performed in vitro reconstitution experiments and analyzed data. J.D., I.A., and C.D. performed and analyzed cryo–electron microscopy, cryo–electron tomography, and negative staining experiments. J.D., M.B., and G.S. set up cryo–electron tomography acquisition. S.G.-F. performed neuronal cultures. E.D. performed image analysis. C.B. performed molecular biology. I.K. and H.M. performed and wrote theoretical modeling. C.D., I.A., A.A., and C.B. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Any additional datasets, analysis details, and material recipes are available upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz4344/DC1
REFERENCES AND NOTES
- 1.Baas P. W., Rao A. N., Matamoros A. J., Leo L., Stability properties of neuronal microtubules. Cytoskeleton 73, 442–460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alushin G. M., Lander G. C., Kellogg E. H., Zhang R., Baker D., Nogales E., High resolution microtubule structures reveal the structural transitions in αβ–tubulin upon GTP hydrolysis. Cell 157, 1117–1129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchison T., Kirschner M., Dynamic instability of microtubule growth. Nature 312, 237–242 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Tas R. P., Kapitein L. C., Exploring cytoskeletal diversity in neurons. Science 361, 231–232 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Delphin C., Bouvier D., Seggio M., Couriol E., Saoudi Y., Denarier E., Bosc C., Valiron O., Bisbal M., Arnal I., Andrieux A., MAP6-F is a temperature sensor that directly binds to and protects microtubules from cold-induced depolymerization. J. Biol. Chem. 287, 35127–35138 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Job D., Rauch C. T., Margolis R. L., High concentrations of STOP protein induce a microtubule super-stable state. Biochem. Biophys. Res. Commun. 148, 429–434 (1987). [DOI] [PubMed] [Google Scholar]
- 7.Baas P. W., Heidemann S. R., Microtubule reassembly from nucleating fragments during the regrowth of amputated neurites. J. Cell Biol. 103, 917–927 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillaud L., Bosc C., Fourest-Lieuvin A., Denarier E., Pirollet F., Lafanechère L., Job D., STOP proteins are responsible for the high degree of microtubule stabilization observed in neuronal cells. J. Cell Biol. 142, 167–179 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrieux A., Salin P. A., Vernet M., Kujala P., Baratier J., Gory-Faureé S., Bosc C., Pointu H., Proietto D., Schweitzer A., Denarier E., Klumperman J., Job D., The suppression of brain cold-stable microtubules in mice induces synaptic defects associated with neuroleptic-sensitive behavioral disorders. Genes Dev. 16, 2350–2364 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deloulme J.-C., Gory-Fauré S., Mauconduit F., Chauvet S., Jonckheere J., Boulan B., Mire E., Xue J., Jany M., Maucler C., Deparis A. A., Montigon O., Daoust A., Barbier E. L., Bosc C., Deglon N., Brocard J., Denarier E., Le Brun I., Pernet-Gallay K., Vilgrain I., Robinson P. J., Lahrech H., Mann F., Andrieux A., Microtubule-associated protein 6 mediates neuronal connectivity through Semaphorin 3E-dependent signalling for axonal growth. Nat. Commun. 6, 7246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimenez U., Boulan B., Mauconduit F., Taurel F., Leclercq M., Denarier E., Brocard J., Gory-Fauré S., Andrieux A., Lahrech H., Deloulme J. C., 3D imaging of the brain morphology and connectivity defects in a model of psychiatric disorders: MAP6-KO mice. Sci. Rep. 7, 10308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrieux A., Salin P., Schweitzer A., Bégou M., Pachoud B., Brun P., Gory-Fauré S., Kujala P., Suaud-Chagny M. F., Höfle G., Job D., Microtubule stabilizer ameliorates synaptic function and behavior in a mouse model for schizophrenia. Biol. Psychiatry 60, 1224–1230 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Daoust A., Bohic S., Saoudi Y., Debacker C., Gory-Fauré S., Andrieux A., Barbier E. L., Deloulme J.-C., Neuronal transport defects of the MAP6 KO mouse—a model of schizophrenia—and alleviation by Epothilone D treatment, as observed using MEMRI. Neuroimage 96, 133–142 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Garvalov B. K., Zuber B., Bouchet-Marquis C., Kudryashev M., Gruska M., Beck M., Leis A., Frischknecht F., Bradke F., Baumeister W., Dubochet J., Cyrklaff M., Luminal particles within cellular microtubules. J. Cell Biol. 174, 759–765 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nogales E., Whittaker M., Milligan R. A., Downing K. H., High-resolution model of the microtubule. Cell 96, 79–88 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Ichikawa M., Bui K. H., Microtubule inner proteins: A meshwork of luminal proteins stabilizing the doublet microtubule. Bioessays 40, 1700209 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Gardner M. K., Zanic M., Gell C., Bormuth V., Howard J., Depolymerizing kinesins Kip3 and MCAK shape cellular microtubule architecture by differential control of catastrophe. Cell 147, 1092–1103 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Mohan R., Katrukha E. A., Doodhi H., Smal I., Meijering E., Kapitein L. C., Steinmetz M. O., Akhmanova A., End-binding proteins sensitize microtubules to the action of microtubule-targeting agents. Proc. Natl. Acad. Sci. U.S.A. 110, 8900–8905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzik-Lendrum S., Rayment I., Gilbert S. P., Homodimeric kinesin-2 KIF3CC promotes microtubule dynamics. Biophys. J. 113, 1845–1857 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosc C., Frank R., Denarier E., Ronjat M., Schweitzer A., Wehland J., Job D., Identification of novel bifunctional calmodulin-binding and microtubule-stabilizing motifs in STOP proteins. J. Biol. Chem. 276, 30904–30913 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Gory-Fauré S., Windscheid V., Bosc C., Peris L., Proietto D., Franck R., Denarier E., Job D., Andrieux A., STOP-like protein 21 is a novel member of the STOP family, revealing a Golgi localization of STOP proteins. J. Biol. Chem. 281, 28387–28396 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Schaedel L., John K., Gaillard J., Nachury M. V., Blanchoin L., Théry M., Microtubules self-repair in response to mechanical stress. Nat. Mater. 14, 1156–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vemu A., Szczesna E., Zehr E. A., Spector J. O., Grigorieff N., Deaconescu A. M., Roll-Mecak A., Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 361, eaau1504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atherton J., Stouffer M., Francis F., Moores C. A., Microtubule architecture in vitro and in cells revealed by cryo-electron tomography. Acta Crystallogr. D Struct. Biol. 74, 572–584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohrbach H., Johner A., Kulić I. M., Cooperative lattice dynamics and anomalous fluctuations of microtubules. Eur. Biophys. J. 41, 217–239 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Ziebert F., Mohrbach H., Kulić I. M., Why microtubules run in circles: Mechanical hysteresis of the tubulin lattice. Phys. Rev. Lett. 114, 148101 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Asakura S., Polymerization of flagellin and polymorphism of flagella. Adv. Biophys. 1, 99–155 (1970). [PubMed] [Google Scholar]
- 28.Calladine C. R., Construction of bacterial flagella. Nature 255, 121–124 (1975). [DOI] [PubMed] [Google Scholar]
- 29.Chrétien D., Wade R. H., New data on the microtubule surface lattice. Biol. Cell 71, 161–174 (1991). [DOI] [PubMed] [Google Scholar]
- 30.Ray S., Meyhöfer E., Milligan R. A., Howard J., Kinesin follows the microtubule’s protofilament axis. J. Cell Biol. 121, 1083–1093 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hyman A. A., Chrétien D., Arnal I., Wade R. H., Structural changes accompanying GTP hydrolysis in microtubules: Information from a slowly hydrolyzable analogue guanylyl-(alpha,beta)-methylene-diphosphonate. J. Cell Biol. 128, 117–125 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wade R. H., Chrétien D., Cryoelectron microscopy of microtubules. J. Struct. Biol. 110, 1–27 (1993). [DOI] [PubMed] [Google Scholar]
- 33.Soppina V., Herbstman J. F., Skiniotis G., Verhey K. J., Luminal localization of α-tubulin K40 acetylation by cryo-EM analysis of fab-labeled microtubules. PLOS ONE 7, e48204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazarus C., Soheilypour M., Mofrad M. R., Torsional behavior of axonal microtubule bundles. Biophys. J. 109, 231–239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dacheux D., Roger B., Bosc C., Landrein N., Roche E., Chansel L., Trian T., Andrieux A., Papaxanthos-Roche A., Marthan R., Robinson D. R., Bonhivers M., Human FAM154A (SAXO1) is a microtubule-stabilizing protein specific to cilia and related structures. J. Cell Sci. 128, 1294–1307 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Burton P. R., Luminal material in microtubules of frog olfactory axons: Structure and distribution. J. Cell Biol. 99, 520–528 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szyk A., Deaconescu A. M., Spector J., Goodman B., Valenstein M. L., Ziolkowska N. E., Kormendi V., Grigorieff N., Roll-Mecak A., Molecular basis for age-dependent microtubule acetylation by tubulin acetyltransferase. Cell 157, 1405–1415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyman A., Drechsel D., Kellogg D., Salser S., Sawin K., Steffen P., Wordeman L., Mitchison T., Preparation of modified tubulins. Methods Enzymol. 196, 478–485 (1991). [DOI] [PubMed] [Google Scholar]
- 39.Coquelle F. M., Blestel S., Heichette C., Arnal I., Kervrann C., Chrétien D., Cryo-electron tomography of microtubules assembled in vitro from purified components. Methods Mol. Biol. 777, 193–208 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Nawrotek A., Knossow M., Gigant B., The determinants that govern microtubule assembly from the atomic structure of GTP-tubulin. J. Mol. Biol. 412, 35–42 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Igaev M., Grubmüller H., Microtubule assembly governed by tubulin allosteric gain in flexibility and lattice induced fit. eLife 7, e34353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krebs A., Goldie K. N., Hoenger A., Structural rearrangements in tubulin following microtubule formation. EMBO Rep. 6, 227–232 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venier P., Maggs A. C., Carlier M.-F., Pantaloni D., Analysis of microtubule rigidity using hydrodynamic flow and thermal fluctuations. J. Biol. Chem. 269, 13353–13360 (1994). [PubMed] [Google Scholar]
- 44.Arnal I., Wade R. H., How does taxol stabilize microtubules? Curr. Biol. 5, 900–908 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Kellogg E. H., Hejab N. M. A., Howes S., Northcote P., Miller J. H., Díaz J. F., Downing K. H., Nogales E., Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from cryo-EM structures. J. Mol. Biol. 429, 633–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elie-Caille C., Severin F., Helenius J., Howard J., Muller D. J., Hyman A. A., Straight GDP-tubulin protofilaments form in the presence of taxol. Curr. Biol. 17, 1765–1770 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Amos L. A., Amos W. B., The bending of sliding microtubules imaged by confocal light microscopy and negative stain electron microscopy. J. Cell Sci. Suppl. 14, 95–101 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang R., LaFrance B., Nogales E., Separating the effects of nucleotide and EB binding on microtubule structure. Proc. Natl. Acad. Sci. U.S.A. 115, E6191–E6200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shima T., Morikawa M., Kaneshiro J., Kambara T., Kamimura S., Yagi T., Iwamoto H., Uemura S., Shigematsu H., Shirouzu M., Ichimura T., Watanabe T. M., Nitta R., Okada Y., Hirokawa N., Kinesin-binding–triggered conformation switching of microtubules contributes to polarized transport. J. Cell Biol. 217, 4164–4183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouzar L., Müller M. M., Messina R., Nöding B., Köster S., Mohrbach H., Kulić I. M., Helical superstructure of intermediate filaments. Phys. Rev. Lett. 122, 098101 (2019). [DOI] [PubMed] [Google Scholar]
- 51.J. Howard, Applied Mechanics Reviews (Sinauer-Associates, 2001), vol. 55, chap. 3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz4344/DC1