

This study identifies an oligonucleotide that activates a growth factor receptor and exerts its antiapoptotic activity in vivo.
Abstract
Although growth factors have great therapeutic potential because of their regenerative functions, they often have intrinsic drawbacks, such as low thermal stability and high production cost. Oligonucleotides have recently emerged as promising chemical entities for designing synthetic alternatives to growth factors. However, their applications in vivo have been recognized as a challenge because of their susceptibility to nucleases and limited distribution to a target tissue. Here, we present the first example of oligonucleotide-based growth factor mimetics that exerts therapeutic effects at a target tissue after systemic injection. The aptamer was designed to dimerize a growth factor receptor for its activation and mitigated the progression of Fas-induced fulminant hepatitis in a mouse model. This unprecedented functionality of the aptamer can be reasonably explained by its high nuclease stability and migration to the liver parenchyma. These mechanistic analyses provided insights for the successful application of aptamer-based receptor agonists.
INTRODUCTION
The hepatocyte growth factor (HGF) is a natural protein that stimulates fundamental cellular functions, such as proliferation, differentiation, and migration (1, 2). HGF was first detected in the serum of hepatectomized rats and was identified as a mitogen that supports the unique regenerative capacity of the liver tissue (3, 4). Upon liver damage, the plasma concentration of the HGF rises and the secreted HGF activates its cognate receptor, Met, which is expressed in hepatocytes to stimulate DNA synthesis during liver regeneration (5). Because of its regenerative activity, the administration of recombinant HGF (rHGF) has attracted much attention as a therapeutic option for the treatment of liver diseases, such as acute liver failure and liver cirrhosis (6, 7). Although promising, such growth factor–based therapeutics often suffer from intrinsic drawbacks, including low thermal stability and batch-to-batch variation (8, 9), which impose a strict quality control. The cost of recombinant growth factors also represents an obstacle to their practical applications (8, 9). In these contexts, synthetic alternatives to rHGF have been highly sought after.
Recently, DNA aptamers have emerged as promising chemical entities for designing growth factor mimetics (10–13). DNA aptamers can be synthesized chemically via well-established solid-phase synthesis methods, which have several advantages over biological production of recombinant proteins in terms of synthetic scalability and cost effectiveness (14, 15). DNA aptamers are highly thermally stable compared to recombinant proteins, and their thermal denaturation is reversible (14, 15). In addition, one can rationally design an assembly of DNA aptamers that works as functional mimetics of growth factors because of the structural programmability of DNA (10–13). Growth factors bind to their cognate receptors, receptor tyrosine kinases (RTKs), thus inducing their dimerization and subsequent activation. The activated RTKs can trigger intracellular signal transduction events and transcriptional changes to regulate various cellular functions. Analogous approaches have been reported for the design of agonistic aptamers for the insulin receptor (16) and immune costimulatory receptors (17–21).
Despite the potential of aptamer-based growth factor mimetics as synthetic alternatives to recombinant growth factors (10–13), no previous study has succeeded in delivering them to a target tissue in quantities that are sufficient to exert agonistic functions. Susceptibility to nuclease degradation, which hinders the delivery of the oligonucleotides to the site of action, is one of the most critical issues in this field of research. Here, we report an HGF-mimetic DNA aptamer that activated Met in the liver parenchyma and exerted its antiapoptotic activity in vivo. This single-stranded agonistic aptamer exhibited a high serum nuclease stability even in a chemically unmodified form, thus allowing sufficient delivery to the liver parenchyma of mice after systemic injection, as demonstrated by a series of biodistribution assays (including real-time confocal imaging of liver tissues from living mice). Mechanistic analyses provided a structural basis for the design of a nuclease-resistant agonistic DNA aptamer, even without chemical modification. In addition, we observed in vivo behavior and clearance profile of the aptamer in detail to obtain insight into future design of therapeutic DNA aptamer. Notably, the aptamer alleviated liver damage in a mouse model of fulminant hepatitis, which demonstrated its in vivo therapeutic potential. This aptamer is the first example of oligonucleotide-based growth factor mimetics that exerts therapeutic effects at the target tissue after systemic injection.
RESULTS
Nuclease stability of Met-binding DNA aptamers
First, we focused on the nuclease stability of an agonistic DNA aptamer that reproduces the activation mechanism of HGF (Fig. 1A). Previously, we reported that a 50-mer DNA aptamer (“Apt-mono” in Fig. 1, B and C) functions as a Met antagonist by competing with HGF for Met binding (22). This aptamer was identified as the minimal binding motif of an original Met-binding aptamer named CLN0003 (Fig. 1D) (23). Later, we revealed that a tandem dimer of the Met-binding aptamer (“Apt-dimer” in Fig. 1, B and C) functions as a Met agonist, as it was able to induce Met dimerization and subsequent phosphorylation (11). A series of in vitro experiments using cultured cells demonstrated that the Apt-dimer reproduces the biological activities of the HGF, such as stimulation of cell migration and proliferation (11).
Fig. 1. Met-binding DNA aptamers and their nuclease stability.
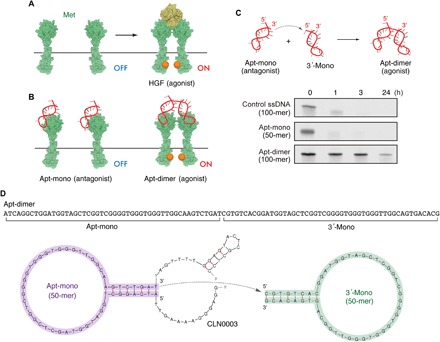
(A and B) Schematic representation of HGF-induced Met activation and the Met activation potential of the Apt-mono and the Apt-dimer. (C) (Top) Schematic representation of the oligonucleotides used in the present work. (Bottom) Nuclease stability of the oligonucleotides in serum. Each oligonucleotide (2 μM) was incubated in PBS containing 50% FBS at 37°C. (D) Sequence and predicted secondary structure of the aptamers used in the present work. The sequence of the Apt-mono (purple) was identified as the minimal binding motif of a Met-binding aptamer (CLN0003) reported previously (22). The Apt-dimer [termed “ss-0” in a previous work (11)] was designed as a tandem dimer of the Met-binding aptamer, as depicted in the figure. The stem sequence of the 3′-mono (green) was replaced with an alternative complimentary sequence to prevent the misfolding of the aptamer.
To study in vivo applicability of these aptamers, we evaluated the nuclease stability of these aptamers by incubating them in a 50% fetal bovine serum (FBS) solution. Unexpectedly, only the Apt-dimer exhibited a high nuclease stability, as the bands corresponding to the full-length DNA could be observed in polyacrylamide gel electrophoresis (PAGE) even after 24 hours of incubation with 50% FBS (Fig. 1C). In contrast, the bands corresponding to the Apt-mono and control DNA, a 100-mer single-stranded DNA (ssDNA) that does not form a specific secondary structure, had almost disappeared after 1 hour of incubation. We hypothesized that the 3′ half of the Apt-dimer (3′-mono in Fig. 1D) served as a protective motif against 3′ exonuclease activity, which is a major factor in the degradation of oligonucleotides in serum (24). This notion was supported by the following facts: A secondary structure prediction suggested that 3′-mono folded into a stem-loop structure in a physiological condition (Fig. 1D), and previous reports demonstrated that the introduction of a stable hairpin structure at the 3′ terminus of the oligonucleotides enhanced their stability against 3′ exonuclease activity (25, 26). Consistently, the aptamer was readily digested in a 50% FBS solution when the 3′-terminal sequence of 3′-mono was substituted with a poly-T sequence to prevent duplex formation at the stem (fig. S1). In addition, a mutant of Apt-dimer whose 3′-mono sequence was replaced with a hairpin-forming, non-aptamer sequence also exhibited high nuclease stability (fig. S1). These results indicate that 3′-mono plays a critical role in improving the serum stability of the aptamer, as well as in converting the Met-binding aptamer into a potent Met agonist (11).
To test the generality of this design principle, we tested the serum stability of a recently reported agonistic DNA aptamer for the fibroblast growth factor receptor 1 (FGFR1) (13), which is synthesized as the tandem dimer form of stem-loop–forming FGFR1-binding aptamers (fig. S2). As expected, the aptamer dimer exhibited a higher nuclease stability compared with its monomeric counterpart, and inhibition of duplex formation at the 3′ stem deteriorated the stabilizing effect of the dimerization. It is noteworthy that the original monomeric aptamer was designed to adopt a thermally stable stem-loop structure by increasing the GC content in the stem sequence to improve the serum stability according to the literature (27). While the strategy of increasing GC content in the stem sequence was effective in a typical cell culture condition (10% FBS solution), the aptamer was immediately digested in the 50% FBS condition (13). In sharp contrast, the tandem dimer aptamer exhibited a higher nuclease stability compared with the aptamer monomer, which indicated the critical role of the 3′ half of the aptamer in improving the nuclease stability. These results suggest that the tandem dimer of a stem-loop–forming DNA aptamer represents a general platform for the design of oligonucleotide-based RTK agonists with high nuclease resistance based on a chemically unmodified DNA.
Liver accumulation of the HGF-mimetic aptamer
Having developed a potent HGF mimic aptamer with high nuclease stability, we next evaluated whether the aptamer localizes to liver parenchyma after intravenous injection in mice. While oligonucleotides tend to accumulate in liver tissues after systemic injection, microscopic distribution of the aptamer in the liver remains unclear (28). In addition to observing the distribution of the oligonucleotides, we performed functional analyses using the Apt-dimer and a mutant of the Apt-dimer named “G4 mutant” as an unfunctional control. In the mutant aptamer, four G-to-A mutations were introduced to inhibit G-quadruplex formation, which works as essential binding motifs for Met (Fig. 2, A and B) (22). In circular dichroism (CD) measurements, the Apt-dimer showed a positive peak around 265 nm and a negative peak around 245 nm in the presence of the K+ cation, indicating the formation of a parallel G-quadruplex structure, whereas the G4 mutant did not show such characteristic peaks (fig. S3A). Competition assay using fluorescently labeled HGF indicated that the Apt-dimer competed with HGF for binding to the receptor, while the G4 mutant did not (fig. S3B). Therefore, the Apt-dimer forms a G4 structure, which is essential for receptor binding. The binding site of the aptamer on the receptor overlaps at least partly with that of HGF. Introduction of four G-to-A mutations inhibits the formation of the G4 structure, thereby decreasing the affinity to the receptor. These results are consistent with the observation reported in the previous study on the Met-binding aptamer monomer (22). We also confirmed that the introduction of these mutations substantially decreased the agonistic activity of the aptamer (Fig. 2C) while maintaining its high nuclease stability in serum (Fig. 2B).
Fig. 2. Design of an unfunctional mutant of the Apt-dimer (G4 mutant) and microdistribution of the oligonucleotides in liver tissues.
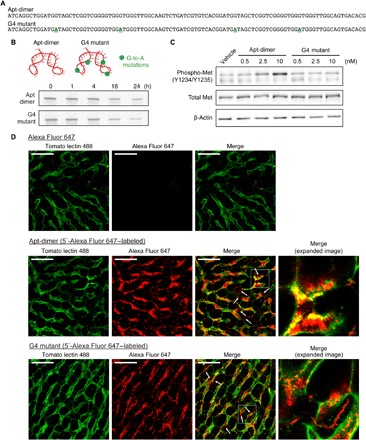
(A) Sequences of the oligonucleotides used in the experiments depicted in Fig. 2. The four G-to-A mutations of the G4 mutant are highlighted. (B) Nuclease stability of the Apt-dimer and G4 mutant. Each oligonucleotide (2 μM) was incubated in PBS containing 50% FBS at 37°C. After incubation, the samples were immediately analyzed using denaturing 15% PAGE. (C) Western blotting analysis of the phosphorylation level of Met in SCCVII cells after 15 min of incubation with oligonucleotides. (D) Confocal imaging of liver tissues. Alexa Fluor 647 carboxylic acid (0.5 nmol) or a 5′-Alexa Fluor 647–labeled oligonucleotide (0.5 nmol) was intravenously injected (red). Blood vessels were visualized by injecting 25 μg of DyLight 488–conjugated tomato lectin (green). Ten minutes after the oligonucleotide injection, the liver was excised and observed directly using a confocal laser scanning microscope. Scale bars, 50 μm.
Regarding the distribution of the oligonucleotides, first we assessed the microdistribution of the Apt-dimer in a liver tissue specimen excised 10 min after the injection of 5′-Alexa Fluor 647–labeled oligonucleotides. These oligonucleotides were co-injected with DyLight 488–labeled tomato lectin to clarify the position of endothelial cells (29). As shown in Fig. 2D, the fluorescence signal from the Apt-dimer showed substantial colocalization with the signal from tomato lectin, indicating the accumulation of the injected oligonucleotides in the liver endothelium. Notably, further observations revealed the presence of red fluorescence on the abluminal side of sinusoidal endothelial cells (white arrows in Fig. 2D), which was indicative of the leakage of the aptamer from the endothelial layers to the liver parenchyma. A similar distribution profile was observed for G4 mutant–treated mice; thus, the binding affinity of the aptamer to Met had little effect on its distribution profile. Met expression was mainly observed in liver parenchymal cells, rather than endothelial cells (fig. S4), with a distribution pattern that was different from that of the Apt-dimer. The distribution of Apt-dimer may be explained by its nonselective deposition onto the sinusoidal wall, as was also observed after the systemic injection of other types of oligonucleotide therapeutics (30).
To visualize the accumulation of Apt-dimer in the liver tissue in real time, we performed liver tissue imaging using intravital real-time confocal laser scanning microscopy (IVRTCLSM; fig. S5) (31). Immediately after the injection of the aptamer, the fluorescence signal in the blood vessels of the liver increased; however, it started to decay a few minutes later, which indicates the translocation of the oligonucleotides from the blood circulation to the tissues. Within several minutes after the injection, the fluorescent signal was aligned with the sinusoidal walls (Fig. 3A and movie S1), which is consistent with the observations performed in the liver tissue sections (Fig. 2D). Notably, a closer inspection revealed the presence of an obvious increase in the fluorescence signals in the liver parenchyma 5 to 20 min after the injection compared with that detected before the injection (dotted circles in Fig. 3A), which was confirmed by the fluorescence intensity quantification of liver parenchyma (Fig. 3A, right). A comparable profile was observed in the liver of G4 mutant (5′-Alexa 647–modified)–treated mice (Fig. 3B and movie S2). In sharp contrast, the free Alexa 647 dye was immediately cleared from the blood, with no accumulation in the liver after systemic injection (Fig. 3C and movie S3), indicating that the fluorescence distribution profiles shown in Fig. 3 (A and B) properly reflect the distribution of the Apt-dimer and G4 mutant, respectively, rather than that of the free dye liberated from digested oligonucleotides. Clearance profile of the Apt-dimer was then observed by continuous real-time imaging of the liver. The fluorescence intensity of the liver parenchyma gradually decreased 30 min or later after the injection, eventually to the background level within 2 hours (fig. S5C and movie S4).
Fig. 3. Intravital real-time imaging of liver tissues after systemic injection of the oligonucleotides.
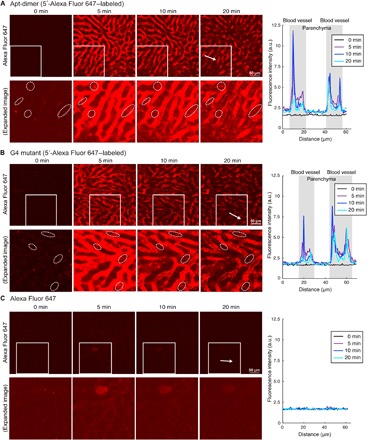
(A) 5′-Alexa Fluor 647–labeled Apt-dimer (0.5 nmol), (B) 5′-Alexa Fluor 647–labeled G4 mutant (0.5 nmol), or (C) Alexa Fluor 647 (0.5 nmol) was intravenously injected. (Left) Confocal images. Lower panels show enlarged and brighter images of the boxed region in the upper panels. The dotted circles highlight the leakage of Apt-dimer to the liver parenchyma. (Right) Intensity profile of Alexa Fluor 647 in the region indicated by the white arrows in the left figures. Scale bars, 50 μm. a.u., arbitrary units.
In addition to the evaluation of the microdistribution of the oligonucleotides described above, we assessed their biodistribution at the organ level, as it is another determinant of aptamer functionality. The fluorescence intensity from each tissue (Fig. 4, left) or tissue homogenate (Fig. 4, right) was measured 10 min after injection of the 5′-Alexa Fluor 647–modified oligonucleotides. The results indicated that both oligonucleotides were highly accumulated in the liver and kidney. Of particular interest, the Apt-dimer exhibited enhanced accumulation in some organs, including the liver and spleen, compared with the G4 mutant. Although the actual mechanism underlying this observation remains unclear, the compact folding of the G4 structures in the Apt-dimer sequence (22) might change the biodistribution or in vivo circulation time of the aptamers, as suggested in a previous report (32). Of note, most of the Apt-dimer was cleared from the body within 4 hours after the injection (fig. S6). These results and those of the biodistribution assays revealed that the Apt-dimer features enhanced accumulation in liver tissue after systemic administration and subsequent translocation to the liver parenchyma, which suggests its potential use as an antiapoptotic agent for liver diseases.
Fig. 4. Tissue distribution of the aptamer.
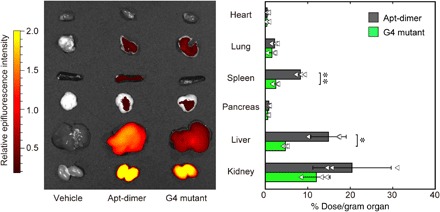
Tissue distribution of the 5′-Alexa Fluor 647–labeled oligonucleotides. (Left) Representative ex vivo fluorescence image of the organs 10 min after intravenous injection of Alexa Fluor 647–labeled oligonucleotides (0.5 nmol). The organs were excised after perfusion with PBS and imaged using IVIS (excitation/emission = 640/680 nm). (Right) Biodistribution of oligonucleotides quantified by fluorescence intensity in the supernatant of the organ homogenates. The results are expressed as the mean ± SD (n = 3). Statistical significance was examined by two-sided Student’s t test (**P < 0.01; *P < 0.05).
Agonistic functions and therapeutic potential of the Met agonist aptamer
To address whether the aptamer can activate Met signaling in the liver, we evaluated the phosphorylation level of Met by Western blotting 10 min after intravenous injection of the aptamer (Fig. 5B). The administration of the Apt-dimer substantially increased the phosphorylation level of Met and downstream signaling, such as extracellular signal–regulated kinase (ERK) and Akt. These results suggest that a portion of Apt-dimer is delivered to the liver in a functional form by avoiding nuclease digestion. Indeed, 3% dose per gram of tissue of the Apt-dimer was detected in the liver by quantitative real-time polymerase chain reaction (PCR)–based analysis (fig. S7). Because Met was expressed mainly in hepatocytes in the liver parenchyma (fig. S4), these data strongly suggest the functionality of the Apt-dimer to activate the HGF/Met-signaling cascade in hepatocytes. Immunohistochemical analysis performed using a phospho-Met–specific antibody also revealed the aptamer-induced Met activation throughout the liver tissues, including hepatocytes (Fig. 5C and fig. S8). These results are consistent with the direct observations of Apt-dimer distribution, which showed Apt-dimer leakage to the liver parenchyma (Figs. 2D and 3A). In contrast, the G4 mutant did not induce Met phosphorylation, as assessed using Western blotting and immunohistochemical analysis, supporting the contention that Met phosphorylation was induced in a manner that was specific to the Apt-dimer.
Fig. 5. Agonistic functions and therapeutic potential of the HGF-mimic aptamer.
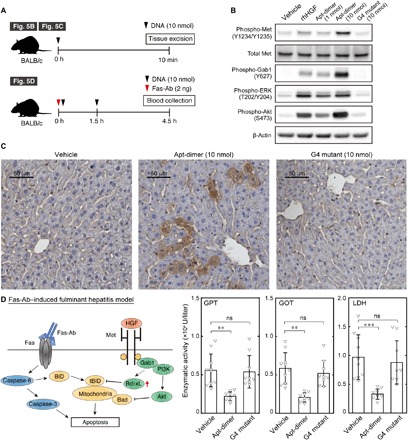
(A) Schematic representation of the experimental outlines. (B) Immunoblotting analysis of Met activation in mouse livers after intravenous injection of recombinant human HGF (rhHGF) or oligonucleotides. The liver was excised 10 min after the intravenous injection of rhHGF (1 μg) or oligonucleotides (1 to 10 nmol). (C) Immunohistochemistry of Met activation in the mouse liver after intravenous injection of the oligonucleotides. The liver was excised 10 min after the intravenous injection of oligonucleotides (10 nmol). (D) (Left) Schematic representation of the apoptotic signal induced by Fas-Ab and the antiapoptotic signal induced by Met activation. (Right) Effect of Apt-dimer administration on a fulminant hepatitis mouse model. An anti–Fas-Ab (2 ng) was co-injected with vehicle control (n = 10), Apt-dimer (10 nmol, n = 9), or G4 mutant (10 nmol, n = 9) intravenously. The oligonucleotide (10 nmol) was administered 1.5 hours later. The average activity of glutamate pyruvate transaminase (GPT), glutamate oxaloacetate transaminase (GOT), and lactate dehydrogenase (LDH) is shown with error bars (SD). Statistical significance was examined by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test (***P < 0.001; **P < 0.01; ns, not significant).
Last, we evaluated the therapeutic potential of the Apt-dimer using a fulminant hepatitis mouse model. In the present study, an agonistic Fas antibody (Fas-Ab) with proapoptotic function was systemically injected into mice to induce massive apoptosis of hepatocytes, which leads to liver failure (Fig. 5D, left) (33–35). Although its mechanism of action has been thought to be multidimensional, rHGF exerts its therapeutic effects via its antiapoptotic function, which is stimulated by activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (6, 36). However, its high production cost and poor pharmacokinetics have been major obstacles for its clinical application. In this experiment, the oligonucleotide sample was co-injected with the Fas-Ab and was further injected 1.5 hours after the initial injection. Blood was collected 4.5 hours after the initial injection to evaluate liver tissue damage by measuring the leakage of liver enzymes to the plasma. As shown in Fig. 5D, the administration of the Apt-dimer mitigated the progression of fulminant hepatitis, as the levels of liver enzymes in the plasma decreased significantly compared with the control groups. In this fulminant hepatitis model, Fas-Ab was reported to cause liver damage mainly by inducing hepatocyte apoptosis, which suggests that the therapeutic effect of the Apt-dimer can be attributed to its antiapoptotic effect in hepatocytes (37, 38). This contention was supported by immunohistochemical analysis showing the aptamer-induced Met activation throughout liver tissues, including hepatocytes (Fig. 5C and fig. S6), which is consistent with our observations that the Apt-dimer leaked to the liver parenchyma (Figs. 2D and 3A). These results demonstrate the therapeutic potential of the Apt-dimer to modulate the function of hepatocytes to treat liver diseases as a synthetic alternative to rHGF.
Safety analyses
We also performed comprehensive safety analyses to assist with the future clinical translation of the Apt-dimer system. The acute and chronic toxicity of the Apt-dimer was assessed by gross morphology observation, histopathological examination, and a blood test performed on normal mice, 24 hours and 7 days after the injection (refer to additional supplementary file). We found that the Apt-dimer induced no detectable change in any of the tissues that were tested, including those from the liver, kidneys, spleen, and lungs, at both the macroscopic and microscopic levels, 24 hours and 7 days after the injection was administered. The blood test revealed no detectable increase in markers for liver damage (aspartate aminotransferase and alanine aminotransferase), kidney function (blood urea nitrogen and creatinine), general tissue damage (lactate dehydrogenase), damage to pancreas (amylase) and biliary function (total bilirubin), and electrolytes (Na, K, Cl) at both the acute and chronic stages. Another issue regarding the DNA aptamer is the potential of immunogenicity due to its recognition by innate immune receptors, such as Toll-like receptors. To address this issue, immunogenicity of the Apt-dimer was evaluated by measuring the serum levels of inflammatory molecules 4 hours after administering the injection. In the enzyme-linked immunosorbent assay (ELISA) performed, the serum concentrations of interleukin-6 (IL-6) and interferon-β (IFN-β) were below the detection limit, demonstrating that the immunogenicity of the Apt-dimer is low. Therefore, the therapeutic dose of the aptamer was safely administered without any detectable acute and chronic toxicity at the dose that provided a therapeutic outcome in the model of fulminant hepatitis (Fig. 5D).
DISCUSSION
The present work demonstrated that the Apt-dimer, a chemically unmodified 100-mer aptamer, works as a functional mimetic of HGF in vivo. This aptamer activated Met in mouse hepatocytes (Fig. 5C) and mitigated the progression of antibody-induced fulminant hepatitis in a mouse model at early phase after the injection (Fig. 5D), while prolonged functional analyses of this aptamer are needed for its clinical translation in a future study. Comprehensive analyses of acute and chronic toxicity demonstrated that the Apt-dimer is highly safe at the dose that provided therapeutic outcome in the model of fulminant hepatitis. Notably, this study provided the first example of unmodified agonistic aptamer that exhibits therapeutic effect after systemic delivery. This bivalent agonistic aptamer featured an unexpectedly high nuclease stability in serum compared with an unstructured control DNA and its monomeric counterpart (Fig. 1C), in addition to the remarkable activity to evoke Met signaling (11). Our data suggest that the aptamer sequence of the 3′ half of the Apt-dimer served as a protective motif against the 3′ exonuclease activity by adopting a stem-loop structure. As stem-loop structures are a common structural motif of aptamers, this “self-contained” strategy would be a useful approach for the design of an agonistic aptamer with high nuclease stability. Although some chemical modifications can be implemented into aptamers, to render them nuclease resistant (14, 15), this strategy may deteriorate the affinity of the original aptamer and raise the production costs substantially. Therefore, the use of an unmodified aptamer-based agonist with sufficient nuclease stability for successful delivery to the site of action would be the most practical and straightforward approach. Notably, the current aptamer could be delivered to the liver parenchyma without the need of local injection or conjugation with delivery agents (39).
In addition to nuclease stability, the in vivo distribution profile of aptamers is a determinant of their function as therapeutics. To the best of our knowledge, this study presented the first intravital real-time fluorescence imaging of aptamer accumulation at histological resolution, which enabled the direct evaluation of how the aptamer behaves in the target tissue. Our observations revealed that a large fraction of the injected aptamer was deposited in the liver sinusoidal endothelium, whereas a portion of the aptamer was translocated to the liver parenchyma. The functional analyses strongly suggested the activation of the HGF/Met-signaling cascade in hepatocytes in normal and disease model mice (Fig. 5), further supporting the translocation of the aptamer to the liver parenchyma. The relatively small size of the aptamer might allow such translocation, indicating a potential advantage of aptamers as an alternative to recombinant proteins. The initial accumulation of the aptamers in the liver sinusoidal endothelium may be explained by the capture of the oligonucleotide by scavenger receptors expressed on endothelial cells, as some of these receptors were implicated in the nonspecific clearance of anionic biopolymers, such as heparin, hyaluronic acid, and oligonucleotides (40). Modifying such interactions with the scavenger receptors would be worthy of further investigation aimed at improving the performance of the aptamer. Together, our intravital imaging analyses provided valuable mechanistic insights into aptamer behavior in the target tissue, as well as into the processes that hamper the optimal functioning of the aptamers. Further development based on these findings and a functional comparison of the aptamer with existing therapeutics, including recombinant proteins, should be performed in the future for the clinical translation of the aptamer. Although the current work focused on the treatment of liver disease, the Apt-dimer holds promise as a therapeutic option for various diseases and situations of tissue damage that have been recognized as potential targets of rHGF, including amyotrophic lateral sclerosis and spinal cord injury (6). By identifying the DNA structural basis that is ideal for obtaining high nuclease stability and assessing the in vivo behavior of the DNA aptamer, this study provides a rationale for the design of growth factor mimetics for therapeutic applications.
MATERIALS AND METHODS
General information
Reagents were purchased from standard suppliers and used without further purification. All DNA samples were purchased from Fasmac, Eurofins Genomics, and GeneDesign. The DNA annealing was performed with a thermal cycler (95°C for 5 min and then cooled at 0.1°C/s to 25°C or cooled to room temperature). Dulbecco’s phosphate-buffered saline (DPBS) was obtained from Fujifilm Wako Pure Chemical Corporation (048-29805). Recombinant human HGF was purchased from PeproTech (100-39). BALB/c mice (female, 5 to 7 weeks old) were purchased from CLEA Japan and Charles River Laboratories.
Serum nuclease stability of oligonucleotides
Oligonucleotide samples (2 μM) were incubated in PBS containing 50% non–heat-inactivated FBS at 37°C. After incubation, the samples were immediately analyzed using denaturing 15% PAGE.
Cell culture
SNU-5 cells were cultured in Iscove's modified Dulbecco's medium supplemented with 20% FBS and 1% antibiotic-antimycotic (Life Technologies, 15240). SCCVII cells were cultured in RPMI 1640 supplemented with 10% FBS and 1% antibiotic-antimycotic. The cells were cultured in a humidified incubator (37°C, 5% CO2 humidified atmosphere).
CD measurements
CD measurement was performed using a CD spectrometer (J-1500, JASCO) over a wavelength of 210 to 320 nm using a quartz cuvette at 37°C. DNA samples were prepared in 20 mM tris-HCl buffer (pH 7.6) supplemented with or without 100 mM KCl. DNA refolding was performed using a thermal cycler (95°C for 5 min and then cooled at 0.1°C/s to 25°C) before measurements. The scanning speed was set to 100 nm/min.
Flow cytometry
SNU-5 cells were incubated with HiLyte Fluor 647–labeled HGF (5 nM) in the presence of oligonucleotides (0 to 1000 nM) in DPBS supplemented with 0.5% bovine serum albumin (BSA) for 15 min at 37°C. After incubation, the cells were washed with DPBS two times and analyzed by flow cytometry. The fluorescent signal from each cell was measured by flow cytometry (Guava eacyCyte, Merck Millipore). HiLyte Fluor 647–labeled HGF was prepared using the Ab-10 Rapid HiLyte Fluor 647 Labeling Kit (LK36, Dojindo) according to the manufacturer’s protocol.
Western blotting of cell lysate
SCCVII cells were seeded at 35-mm dishes and cultured in RPMI 1640 supplemented with 10% FBS overnight. Subsequently, the cells were cultured in the starving medium (RPMI 1640 supplemented with 0.1% BSA) for 24 hours. After the starvation, the medium was replaced and then the cells were stimulated with an appropriate oligonucleotide sample for 15 min at 37°C. The cells were washed with DPBS twice and lysed with lysis buffer [20 mM tris-HCl (pH 8.0), 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 10% glycerol] supplemented with inhibitor cocktail (Nacalai Tesque, 25955-11) and phosphatase inhibitor cocktail (Nacalai Tesque, 07575-51). The cell lysates were centrifuged at 10,000g for 20 min at 4°C, and then the supernatants were recovered. The total protein concentration of each cell lysate was adjusted to the same value according to the result obtained by bicinchoninic acid (BCA) assay. The cell lysates were separated by SDS-PAGE and then transferred to the polyvinylidene difluoride (PVDF) membrane. The membrane was reacted with the following primary antibodies at 4°C overnight: anti–phospho-Met Y1234/Y1235 (1:2000; 3077, Cell Signaling Technology), anti-Met (1:2000; 3127, Cell Signaling Technology), and anti–β-actin (1:2000; 4967, Cell Signaling Technology). Subsequently, the membrane was reacted with the following secondary antibodies at room temperature for 1 hour: anti-mouse immunoglobulins/horseradish peroxidase (HRP) (1:3000; P0447, Dako) and anti-rabbit immunoglobulins/HRP (1:3000; P0488, Dako). The membranes were probed by using ImmunoStar LD (Fujifilm Wako Pure Chemical Corporation, 296-69901).
Distribution of Apt-dimer in liver tissue
To observe the distribution of the aptamers in liver tissue, mice were intravenously injected with Alexa Fluor 647 carboxylic acid (0.5 nmol) or 5′-Alexa Fluor 647–labeled oligonucleotides (0.5 nmol). Five minutes after intravenous injection of the aptamers, the mice were additionally injected with 25 μg of DyLight 488–conjugated tomato lectin (Vector Laboratories, DL-1174) to visualize the blood vessels. After another 5 min, the mice were sacrificed and the liver samples were gently excised, washed with PBS, and subjected to direct observation using a confocal laser scanning microscope (Leica TCS SP8, Leica Microsystems). Alexa Fluor 647 and DyLight 488 were excited using laser light at 638 and 488 nm, respectively.
IVRTCLSM imaging
Under anesthesia using 2% isoflurane, mouse liver was exposed, and then a coverslip holder was placed onto the liver to flatten the tissue surface. The tissue was observed using a Nikon A1R confocal laser scanning microscope system attached to an upright Eclipse FN1 and equipped with a CFI Apo LWD 40× objective (Nikon). After injection of Alexa Fluor 647 carboxylic acid (0.5 nmol) or 5′-Alexa Fluor 647–labeled oligonucleotide (0.5 nmol) from the tail vein, observation was performed using 640-nm laser and 700/75-nm emission filter.
Biodistribution of aptamers
The 5′-Alexa Fluor 647–labeled Apt-dimer or G4 mutant (0.5 nmol) was injected to BALB/c mice via the tail vein. After injection, the mice were perfused using PBS and the organs were excised at an appropriate time. The excised organs were imaged using an IVIS imaging system (PerkinElmer) (excitation/emission = 640/680 nm). To quantify the biodistribution of the aptamers, the excised organs were weighed and homogenized in passive lysis buffer (Promega, E1941) using a Multi-beads Shocker (Yasui Kikai Corporation). The fluorescence intensity of Alexa Fluor 647 (excitation/emission = 640/680 nm) in the supernatant was measured using a microplate reader (Infinite M200 Pro, Tecan Japan).
Western blotting of mouse liver lysate
The Apt-dimer or G4 mutant (10 nmol) was injected to BALB/c mice via the tail vein. After 10 min, the liver samples were excised and homogenized with a lysis buffer by using disposable homogenizer (Nippi, 320152). The lysis buffer [20 mM tris-HCl (pH 8.0), 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 10% glycerol] contains a protease inhibitor cocktail (Nacalai Tesque, 25955-11) and a phosphatase inhibitor cocktail (Nacalai Tesque, 07575-51). The homogenates were centrifuged at 10,000 to 12,000g for 20 min, and then the supernatants were recovered. The total protein concentration of each cell lysate was measured with the standard BCA assay after the dilution. The total protein concentration of each cell lysate was adjusted to the same value before the subsequent assays. The homogenates were separated by SDS-PAGE and then transferred to a PVDF membrane. The membrane was reacted with the following primary antibodies at 4°C overnight: anti–phospho-Met (Y1234/Y1235) (1:1000; 3077, Cell Signaling Technology), anti-Met (1:2000; 3127, Cell Signaling Technology), anti–phospho-Gab1 (Y627) (1:1000; 3233, Cell Signaling Technology), anti–phospho-ERK1/2 (T202/Y204) (1:2000; 4370, Cell Signaling Technology), anti–phospho-Akt (S473) (1:2000; 4060, Cell Signaling Technology), and anti–β-actin (1:1000; 4967, Cell Signaling Technology). Subsequently, the membrane was reacted with the following secondary antibodies at room temperature for 1 hour: anti-mouse immunoglobulins/HRP (1:3000; P0447, Dako) and anti-rabbit immunoglobulins/HRP (1:3000; P0488, Dako). The membranes were probed by using ImmunoStar LD (Fujifilm Wako Pure Chemical Corporation, 296-69901).
Quantitative PCR analysis
The assay was conducted according to the reference with a slight modification (22). The Apt-dimer (0.5 nmol) was injected into BALB/c mice via the tail vein, and liver samples were excised after 10 min after the injection. The aptamer was extracted from the excised tissue using a standard plasmid DNA purification kit. Frozen tissue samples (<100 mg) were weighed and homogenized in 300 μl of buffer P1 (19051, Qiagen) using a multi-bead shocker. Buffer P2 (300 μl; 19052, Qiagen) was added, and samples were mixed gently by inversion. After 5 min of incubation at room temperature, 300 μl of buffer P3 (19053, Qiagen) was added. Samples were gently mixed by inversion and incubated on ice for 5 min. After centrifugation at 10,000g for 5 min, the supernatant was incubated with 30 μl of proteinase K (9034, Takara) at 55°C overnight. The oligonucleotide was extracted with phenol:chloroform:isoamyl alcohol (25:24:1) extraction followed by isopropanol precipitation. The obtained DNA pellet was resuspended in 500 μl of nuclease-free water. The spike-in DNA samples were prepared to construct standard curve by adding an appropriate amount of DNA to the homogenate of liver tissues excised from nontreated mice just before the addition of P2 buffer. Quantitative PCR was performed by using PikoReal 96 (Thermo Scientific) and KOD SYBR qPCR mix (QKD-201, TOYOBO) according to the manufacturer’s instructions. The extracted DNA sample was used after diluting it with nuclease-free water (1:100).
Immunohistochemistry
For the observation of Met activation in liver by systemic administration of oligonucleotides, Apt-dimer or G4 mutant (10 nmol) was injected to BALB/c mice via the tail vein. After 10 min, the mice were perfused by using PBS, followed by fixation using 4% paraformaldehyde. Liver samples were excised, embedded in paraffin, and sectioned at 4-mm thickness. After deparaffinization and rehydration, endogenous peroxidase was blocked by incubating with 3% H2O2 in methanol for 10 min at ambient temperature. Subsequently, the sections were incubated with 2% skim milk in PBS for 30 min at ambient temperature, followed by incubation with rabbit anti–phospho-Met polyclonal antibody (1:100 in PBS containing 2% skim milk, ab5662, Abcam) for 2 hours at ambient temperature. The sections were then washed with PBS three times and incubated with Histofine Simple Stain Mouse MAX PO (Nichirei Bioscience) for 45 min at ambient temperature. The chromogen diaminobenzidine (DAB) was applied to visualize the antigen-antibody reaction. The nuclei were counterstained with hematoxylin. The obtained tissue sections were observed by using a microscope (BZ-X710, Keyence). For the observation of c-Met location in liver, BALB/c mice were perfused by using PBS and fixed by using 4% paraformaldehyde. Excised liver samples were further immersion-fixed in 4% paraformaldehyde at 4°C overnight, followed by cryoprotection with 30% sucrose in PBS. Tissues were frozen in Tissue-Tek O.C.T. Compound (Sakura Finetek Japan) and sectioned at 20-μm thickness. The sections were blocked with 1% BSA in PBS at ambient temperature for 15 min and incubated with rabbit anti-Met monoclonal antibody (1:100 in PBS containing 1% BSA, ab51067, Abcam) at 4°C overnight. Then, the sections were incubated with an Alexa Fluor 647–conjugated secondary antibody (1:250 in PBS containing 1% BSA, ab150075, Abcam) at ambient temperature for 1 hour. Blood vessels were visualized by incubating with DyLight 488–labeled tomato lectin (1:250 dilution in PBS) at ambient temperature for 1 hour. The nuclei were stained by using Hoechst 33342. The obtained sections were mounted in Vectashield mounting media (Vector Laboratories) and observed by using a confocal laser scanning microscope (Leica TCS SP8). Alexa Fluor 647, DyLight 488, and Hoechst 33342 were excited by using laser light at 638, 488, and 405 nm, respectively.
Treatment of fulminant hepatitis mouse model
The Apt-dimer or G4 mutant (10 nmol) was co-injected with 2 ng of anti-CD95 antibody (554254, BD Biosciences) to BALB/c mice via the tail vein. After 1.5 hours, a second injection of Apt-dimer or G4 mutant (10 nmol) was performed. After 4.5 hours of initial injection, serum glutamate pyruvate transaminase, glutamate oxaloacetate transaminase, and lactate dehydrogenase concentrations were quantified by using the Dri-Chem 7000IZ System (Fujifilm).
Blood chemistry test
The Apt-dimer (10 nmol) was injected into BALB/c mice via the tail vein. Whole blood was collected after 24 hours or 7 days from the injection, and serum samples were prepared by centrifugation at 1700g for 30 min. The serum samples were frozen and sent to Oriental Yeast Co. Ltd. for blood chemical test service.
Quantification of IL-6 and IFN-β
The Apt-dimer (10 nmol) was injected into BALB/c mice via the tail vein. Whole blood was collected 4 hours after administering the injection, and serum samples were prepared by centrifugation at 1700g for 30 min. Concentrations of IL-6 and IFN-β in the serum samples were determined by using the Mouse IL-6 Quantikine ELISA Kit (M6000B, R&D Systems) and Mouse IFN-beta Quantikine ELISA Kit (MIFNB0, R&D Systems), respectively. The assay was performed according to the manufacture’s protocols. Serum samples prepared after 1.5 hours (for IFN-β) or 4 hours (for IL-6) from the single intraperitoneal injection (100 μg) of lipopolysaccharide (128-05171, Fujifilm) were used as positive controls.
Gross morphology and H&E staining of organs
The Apt-dimer (10 nmol) was injected into BALB/c mice via the tail vein. After 24 hours or 7 days after the injection was administered, the tissues were excised and washed with PBS. The images were obtained using a EOS Kiss X10 camera equipped with S18-55 IS STM lens (Canon). For hematoxylin and eosin (H&E) staining, the mice were perfused by using PBS, followed by fixation using 4% paraformaldehyde. The tissues were embedded in paraffin, cut into sections (4 μm thickness), and stained with H&E. The obtained tissue sections were observed by using a microscope (BZ-X710, Keyence).
Ethical compliance
All animal experiments were approved by the Animal Care and Use Committee of the University of Tokyo and Animal Care and Use Committee of the Innovation Center of NanoMedicine and performed in accordance with the guidelines for the care and use of laboratory animals as stated by these institutions.
Supplementary Material
Acknowledgments
We thank N. Nishiyama and M. Matsui (Tokyo Institute of Technology) for giving the opportunity for immunohistochemistry experiments. We thank K. Kataoka (Innovation Center of NanoMedicine) for the use of an intravital real-time confocal laser scanning microscopy. Funding: This work was supported by Core Research for Evolutional Science and Technology (CREST) of Molecular Technology (no. JPMJCR13L4), Japan Science and Technology Agency, grants from the Uehara Memorial Foundation (to S.S.) and KAKENHI (no. 17 K14512), and Japan Society for the Promotion of Science (to R.U.). Author contributions: R.U. and S.S. conceived the project. R.U., N.Y., and S.U. designed the experiments. R.U., S.U., N.K., N.Y., A.U., M.A., and K.T. performed the experiments. H.C. and S.S. supervised the project. R.U., S.U., N.Y., H.C., and S.S. wrote the manuscript. Competing interests: K.T. is hired by Center of Open Innovation Network for Smart Health (COINS), Center of Innovation (COI) Program from MEXT. R.U. and S.S. have filed a patent application (International Application No. PCT/JP2015/075581, international filing date: September 9, 2015; Publication No. WO/2016/039371, publication date: March 17, 2016). All other authors declare no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data are available from the authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay2801/DC1
REFERENCES AND NOTES
- 1.Birchmeier C., Birchmeier W., Gherardi E., Vande Woude G. F., Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4, 915–925 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Trusolino L., Bertotti A., Comoglio P. M., MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 11, 834–848 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Nakamura T., Nawa K., Ichihara A., Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 122, 1450–1459 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Michalopoulos G., Houck K. A., Dolan M. L., Luetteke N. C., Control of hepatocyte replication by two serum factors. Cancer Res. 44, 4414–4419 (1984). [PubMed] [Google Scholar]
- 5.Michalopoulos G. K., DeFrances M. C., Liver regeneration. Science 276, 60–66 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto K., Funakoshi H., Takahashi H., Sakai K., HGF–Met pathway in regeneration and drug discovery. Biomedicine 2, 275–300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ido A., Moriuchi A., Numata M., Murayama T., Teramukai S., Marusawa H., Yamaji N., Setoyama H., Kim I.-D., Chiba T., Higuchi S., Yokode M., Fukushima M., Shimizu A., Tsubouchi H., Safety and pharmacokinetics of recombinant human hepatocyte growth factor (rh-HGF) in patients with fulminant hepatitis: A phase I/II clinical trial, following preclinical studies to ensure safety. J. Transl. Med. 9, 55 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell A. C., Briquez P. S., Hubbell J. A., Cochran J. R., Engineering growth factors for regenerative medicine applications. Acta Biomater. 30, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z., Wang Z., Lu W. W., Zhen W., Yang D., Peng S., Novel biomaterial strategies for controlled growth factor delivery for biomedical applications. NPG Asia Mater. 9, e435 (2017). [Google Scholar]
- 10.Ramaswary V., Monsalve A., Sautina L., Segal M. S., Dobson J., Allen J. B., DNA aptamer assembly as a vascular endothelial growth factor receptor agonist. Nucleic Acid Ther. 25, 227–234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ueki R., Ueki A., Kanda N., Sando S., Oligonucleotide-based mimetics of hepatocyte growth factor. Angew. Chem. Int. Ed. 55, 579–582 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Ueki R., Atsuta S., Ueki A., Sando S., Nongenetic reprogramming of the ligand specificity of growth factor receptors by bispecific DNA aptamers. J. Am. Chem. Soc. 139, 6554–6557 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Ueki R., Atsuta S., Ueki A., Hoshiyama J., Li J., Hayashi Y., Sando S., DNA aptamer assemblies as fibroblast growth factor mimics and their application in stem cell culture. Chem. Commun. 55, 2672–2675 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Keefe A. D., Pai S., Ellington A., Aptamers as therapeutics. Nat. Rev. Drug Discov. 9, 537–550 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J., Rossi J., Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 16, 181–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yunn N.-O., Koh A., Han S., Lim J. H., Park S., Lee J., Kim E., Jang S. K., Berggren P. O., Ryu S. H., Agonistic aptamer to the insulin receptor leads to biased signaling and functional selectivity through allosteric modulation. Nucleic Acids Res. 43, 7688–7701 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNamara J. O. II, Kolonias D., Pastor F., Mittler R. S., Chen L., Giangrande P. H., Sullenger B., Gilboa E., Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 118, 376–386 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dollins C. M., Nair S., Boczkowski D., Lee J., Layzer J. M., Gilboa E., Sullenger B. A., Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 15, 675–682 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pratico E. D., Sullenger B. A., Nair S. K., Identification and characterization of an agonistic aptamer against the T cell costimulatory receptor, OX40. Nucleic Acid Ther. 23, 35–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastor F., Soldevilla M. M., Villanueva H., Kolonias D., Inoges S., de Cerio A. L., Kandzia R., Klimyuk V., Gleba Y., Gilboa E., Bendandi M., CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids 2, e98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soldevilla M. M., Villanueva H., Bendandi M., Inoges S., López-Díaz de Cerio A., Pastor F., 2-fluoro-RNA oligonucleotide CD40 targeted aptamers for the control of B lymphoma and bone-marrow aplasia. Biomaterials 67, 274–285 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Ueki R., Sando S., A DNA aptamer to c-Met inhibits cancer cell migration. Chem. Commun. 50, 13131–13134 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Boltz A., Piater B., Toleikis L., Guenther R., Kolmar H., Hock B., Bi-specific aptamers mediating tumor cell lysis. J. Biol. Chem. 286, 21896–21905 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw J.-P., Kent K., Bird J., Fishback J., Froehler B., Modified deoxyoligonucleotides stable to exonuclease degradation in serum. Nucleic Acids Res. 19, 747–750 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang J. Y., Termsamani J., Agrawal S., Self-stabilized antisense oligodeoxynucleotide phosphorothioates: Properties and anti-HIV activity. Nucleic Acids Res. 21, 2729–2735 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshizawa S., Ueda T., Ishido Y., Miura K., Watanabe K., Hirao I., Nuclease resistance of an extraordinarily thermostable mini-hairpin DNA fragment, d(GCGAAGC) and its application to in vitro protein synthesis. Nucleic Acids Res. 22, 2217–2221 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kahn I. M., Coulson J. M., A novel method to stabilise antisense oligonucleotides against exonuclease degradation. Nucleic Acids Res. 21, 2957–2958 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juliano R. L., The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson R. T., Levine S. T., Haynes S. M., Gutierrez P., Baratta J. L., Tan Z., Longmuir K. J., Use of labeled tomato lectin for imaging vasculature structures. Histochem. Cell Biol. 143, 225–234 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Rifai A., Brysch W., Fadden K., Clark J., Schlingensiepen K. H., Clearance kinetics, biodistribution, and organ saturability of phosphorothioate oligodeoxynucleotides in mice. Am. J. Pathol. 149, 717–725 (1996). [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumoto Y., Nomoto T., Cabral H., Matsumoto Y., Watanabe S., Christie R. J., Miyata K., Oba M., Ogura T., Yamasaki Y., Nishiyama N., Yamasoba T., Kataoka K., Direct and instantaneous observation of intravenously injected substances using intravital confocal micro-videography. Biomed. Opt. Express 1, 1209–1216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perschbacher K., Smestad J. A., Peters J. P., Standiford M. M., Denic A., Wootla B., Warrington A. E., Rodriguez M., Maher L. J. III, Quantitative PCR analysis of DNA aptamer pharmacokinetics in mice. Nucleic Acid Ther. 25, 11–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosai K., Matsumoto K., Nagata S., Tsujimoto Y., Nakamura T., Abrogation of fas-induced fulminant hepatic failure in mice by hepatocyte growth factor. Biochem. Biophys. Res. Commun. 244, 683–690 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Igney F. H., Krammer P. H., Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2, 277–288 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Ichim G., Tait S. W. G., A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 16, 539–548 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Wang X., DeFrances M. C., Dai Y., Pediaditakis P., Johnson C., Bell A., Michalopoulos G. K., Zarnegar R., A mechanism of cell survival: Sequestration of Fas by the HGF receptor met. Mol. Cell 9, 411–421 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Lacronique V., Mignon A., Fabre M., Viollet B., Rouquet N., Molina T., Porteu A., Henrion A., Bouscary D., Varlet P., Joulin V., Kahn A., Bcl–2 protects from lethal hepatic apoptosis induced by an anti–Fas antibody in mice. Nat. Med. 2, 80–86 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez I., Matsuura K., Khatib K., Reed J. C., Nagata S., Vassalli P., A bcl-2 transgene expressed in hepatocytes protects mice from fulminant liver destruction but not from rapid death induced by anti-Fas antibody injection. J. Exp. Med. 183, 1031–1036 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M., He F., Li H., Yang S., Zhang J., Ghosh P., Wang H.-H., Nie Z., Near-infrared light-activated DNA-agonist nanodevice for nongenetically and remotely controlled cellular signaling and behaviors in live animals. Nano Lett. 19, 2603–2613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sørensen K. K., Simon-Santamaria J., McCuskey R. S., Smedsrød B., Liver sinusoidal endothelial cells. Compr. Physiol. 20, 1751–1774 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay2801/DC1