

MHC restriction determines whether S. aureus infection elicits protection, but vaccine efficacy is unconstrained by MHC haplotype.
Abstract
Recurrent Staphylococcus aureus infections are common, despite robust immune responses. S. aureus infection elicited protective antibody and T cell responses in mice that expressed the Major Histocompatibility Complex (MHC) of the H-2d haplotype, but not H-2b, demonstrating that host genetics drives individual variability. Vaccination with a-toxin or leukotoxin E (LukE) elicited similar antibody and T cell responses in mice expressing H-2d or H-2b, but vaccine-elicited responses were inhibited by concomitant infection in H-2d–expressing mice. These findings suggested that competitive binding of microbial peptides to host MHC proteins determines the specificity of the immunodominant response, which was confirmed using LukE-derived peptide-MHC tetramers. A vaccine that elicited T cell and antibody responses protected mice that expressed H-2d or H-2b, demonstrating that vaccination can overcome MHC-restricted immunodominance. Together, these results define how host genetics determine whether immunity elicted by S. aureus is protective and provide a mechanistic roadmap for future vaccine design.
INTRODUCTION
Staphylococcus aureus is a frequent cause of infections ranging from mild skin and soft tissue infections (SSTI) to severe, invasive infections such as bacteremia, osteoarticular infections, pneumonia, and septic shock (1). Long recognized as an important cause of health care–associated infections, methicillin-resistant S. aureus has emerged in recent decades as a leading cause of community-associated infection in otherwise healthy children and adults (1, 2). Recurrent S. aureus infections are common, with recurrence rates within a year as high as 50% in children and adults following SSTI (3, 4). This suggests that naturally acquired immunity against S. aureus is incomplete; however, the high rate of colonization [up to 70%; (5)] coupled with the relatively low rate of symptomatic infection suggests that some level of immunity exists in the general population.
Antistaphylococcal immunity is detectable in most adults, regardless of prior infection or colonization status (6, 7). However, the determinants of protective immunity against S. aureus infections in humans are unclear. Efforts to vaccinate high-risk populations, such as hemodialysis recipients or adults undergoing cardiovascular surgery, have failed despite seemingly adequate immunogenicity in vaccine recipients (8, 9). These failures highlight major limitations in the field, including a lack of understanding of the precise mechanisms by which highly immunogenic S. aureus antigens drive protective (or nonprotective) responses, the impact of the host genetic background on the elicited protective responses, and few identified serologic correlates of protection (4). In addition, the recent elucidation of the role of staphylococcal protein A (SpA) in evading infection-elicited immunity highlights the ability of the pathogen to divert the immune response away from a protective phenotype (10). These observations underscore the complexity of immune evasion strategies used by S. aureus, and several animal models of recurrent S. aureus infection have emerged as critical tools for developing a better understanding of the molecular mechanisms of protective immunity (11–16).
We previously reported a mouse model of recurrent S. aureus SSTI, in which primary infection elicits strong polyclonal antibody responses in both BALB/c and C57BL/6 mice (11). However, only the BALB/c response, which is characterized by T helper 17 (TH17) polarization and an antibody response that is enriched against antigens controlled by the saeRS regulatory operon, is protective against secondary SSTI (17). In this study, we aimed to identify the mechanisms by which S. aureus SSTI elicits protective versus nonprotective adaptive immune phenotypes in BALB/c and C57BL/6 mice, respectively. In doing so, we identified the murine major histocompatibility complex (MHC) haplotype as the genetic basis for divergent protective versus nonprotective antibody and T cell responses following infection and demonstrated that the effects of MHC restriction can be overcome by a multivalent vaccine based on a subset of sae-regulated antigens.
RESULTS
Genetically determined variability in infection-elicited protective antibody and T cell responses
The staphylococcal α-hemolysin (Hla) is a critical virulence determinant in mouse models of infection, and vaccination of mice with Hla elicits antibody-mediated protection against SSTI and pneumonia (18–20). We previously demonstrated that primary SSTI elicits strong anti-Hla antibody responses and subsequent protection against secondary SSTI in BALB/c mice, but not C57BL/6 mice, suggesting a genetic basis for protective responses (11). We therefore hypothesized that Hla-specific antibody levels would also be highly variable in humans, reflecting the population-level genetic heterogeneity. To test this, we quantified anti-Hla antibody levels by enzyme-linked immunosorbent assay (ELISA) in a cohort of 120 healthy adults, for which the systemic T cell responses have previously been reported (21). There was a marked heterogeneity in anti-Hla immunoglobulin G (IgG) levels, with a normal distribution (Fig. 1A). There was also heterogeneity in antibody levels against four other saeRS-regulated antigens (leukotoxin E, LukE; leukocidin S-PV, LukS-PV; a serine protease, SplB; and a cysteine protease, SspB), although only anti-LukE IgG levels were normally distributed (fig. S1). In contrast, antibody levels against LukS-PV, SplB, and SspB were skewed toward lower values, suggesting that responses against these antigens are subdominant, compared with Hla and LukE, although the weaker responses against LukS-PV could also be explained by the fact that the lukSF-PV genes are not as widely distributed among sequenced S. aureus isolates.
Fig. 1. Host genetics determined the strength of protective antibody and T cell responses.
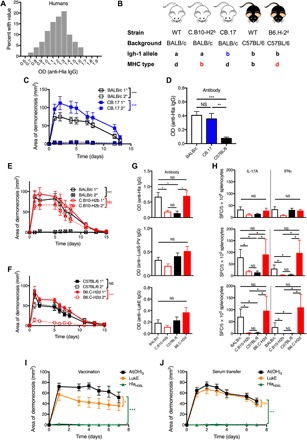
(A) Anti-Hla IgG levels demonstrated significant variability among 120 healthy adults, with a normal distribution (D’Agostino and Pearson normality test, P = 0.6). (B) Mouse strains used in this study. (C and D) Primary SSTI protected BALB/c and CB.17 mice against secondary SSTI and elicited higher anti-Hla IgG levels, compared with C57BL/6 mice (eight mice per group, pooled from two experiments). (E and F) Primary SSTI protected BALB/c and B6.C-H2d mice, but not C57BL/6 or C.B10-H2b mice, against secondary SSTI (eight mice per group, pooled from two experiments). (G) Protection in mice of the H-2d haplotype correlated with higher levels of anti-Hla IgG (top), but not anti–LukS-PV IgG (middle) or anti-LukE (bottom) (seven to eight mice per group, pooled from two experiments). (H) Top: There was a trend toward stronger IL-17A responses against Hla in mice of the H-2d haplotype, but the differences were not significant. Anti-Hla IFNγ responses did not correlate with the H-2d haplotype (seven to eight mice per group, pooled from two experiments). In contrast, IL-17A and IFNγ responses against LukS-PV (middle) and LukE (bottom) were strongest in mice of the H-2d haplotype (four mice per group from one representative experiment). (I) HlaH35L vaccination of BALB/c mice elicited strong protection against dermonecrosis, compared with weaker protection afforded by LukE vaccination (eight mice per group from one representative experiment). (J) Adoptive transfer of serum from HlaH35L-vaccinated mice, but not LukE-vaccinated mice, protected against dermonecrosis (13 mice per group, pooled from two experiments). For the lesion size experiments, data were compared using two-way analysis of variance (ANOVA) with repeated measures and Tukey’s posttest. For all other experiments, data were compared using one-way ANOVA with Tukey’s posttest. OD, optical density; SFC, spot-forming colonies. All data are plotted as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. NS, not significant.
The variability of these antibody levels suggested that individual genetic variability may drive the strength of the antibody response. Therefore, we sought to define the genetic basis for the divergent anti-Hla antibody responses in BALB/c and C57BL/6 mice. We first tested whether differences in the antibody repertoire were responsible by infection of BALB/c, C57BL/6, and CB.17 mice, which are congenic BALB/c mice bearing the C57BL/6 immunoglobulin (Ig) heavy chain allele (Fig. 1B and table S1). Notably, both BALB/c and CB.17 mice were protected against secondary infection, whereas C57BL/6 mice were not (Fig. 1C; C57BL/6 data omitted for clarity). Consistent with these findings, there were comparably high anti-Hla IgG levels before secondary SSTI in BALB/c and CB.17 mice, but lower levels in C57BL/6 mice (Fig. 1D). These results demonstrated that the inability of SSTI to elicit anti-Hla antibodies in C57BL/6 mice was not due to a “hole” in the antibody repertoire and suggested a different genetic determinant of infection-elicited responses.
A primary determinant of genetic variability in immune responses is the MHC. MHC haplotypes differ in BALB/c and C57BL/6 mice; BALB/c mice are of the H-2d haplotype and express I-Ad and I-Ed, and C57BL/6 mice are of the H-2b haplotype and express class II I-Ab and no I-E (table S1). To test the hypothesis that the MHC haplotype determines the strength of the protective response, we infected with SSTI BALB/c, C57BL/6, C.B10-H2b (BALB/c background, congenic for H-2b), and B6.C-H2d (C57BL/6 background, congenic for H-2d) mice (Fig. 1B and table S1). There was strong protection against secondary infection in BALB/c (Fig. 1E) and B6.C-H2d mice (Fig. 1F), consistent with both of these strains sharing the H-2d MHC haplotype. In contrast, there was minimal or no protection against secondary infection in C57BL/6 (Fig. 1F) or C.B10-H2b (Fig. 1E) mice, suggesting that the MHC haplotype determined protective immunity following SSTI. There were also higher anti-Hla IgG levels in mice that expressed the MHC H-2d haplotype and were protected against secondary SSTI, compared with those that expressed H-2b and were not protected (Fig. 1G, top). These findings suggested that the failure of SSTI to elicit protective anti-Hla antibody responses in C57BL/6 mice was due to the specificity of the MHC haplotype. We previously demonstrated that protective immune responses against S. aureus SSTI in BALB/c mice were directed against sae-regulated antigens, including Hla, LukE, and LukS-PV (11, 17). However, while Hla-specific antibody responses correlated with protection, the LukS-PV– and LukE-specific antibody levels did not (Fig. 1G, middle and bottom).
We previously showed that TH17-polarized immune responses, independent of antibody, also contributed to protection in BALB/c mice (11, 17). Therefore, we quantified antigen-specific effector T cell responses by interleukin-17A (IL-17A) and interferon γ (IFNγ) ELISpots in the BALB/c, C57BL/6, C.B10-H2b, and B6.C-H2d mice. There was a trend toward decreased anti-Hla IL-17A responses in C.B10-H2b and C57BL/6 mice, compared with BALB/c and B6.C-H2d mice; however, the differences were not significant nor were there significant differences in anti-Hla IFNγ responses among the mouse strains (Fig. 1H, top). In contrast, there were significantly stronger IL-17A and IFNγ responses against LukS-PV (Fig. 1H, middle) and LukE (Fig. 1H, bottom) in the mice expressing H-2d, compared with those that express H-2b. Therefore, the Hla-specific antibody responses, but not effector T cell responses, tracked with the H-2d haplotype. In contrast, the LukE- and LukS-PV–specific effector T cell responses, but not antibody responses, similarly tracked with the H-2d haplotype. We also noted that the preferential skewing toward IL-17A over IFNγ responses observed in BALB/c mice was not recapitulated in the B6.C-H2d mice (Fig. 1H, middle and bottom), consistent with a role for non-MHC background genes in the skewing of the effector T cell responses. Nevertheless, the strength of the IL-17A response and the protection against reinfection were comparable in BALB/c and B6.C-H2d mice, underscoring the importance of the antigen specificity of the immunodominant T cell response.
Together, these findings suggested a hypothesis that protective immunity against recurrent infection was dependent on Hla-specific antibody responses and LukE- and LukS-PV–specific effector T cell responses. To test this, BALB/c mice were vaccinated with HlaH35L or LukE, followed by S. aureus SSTI. As reported by others (19), vaccination with HlaH35L strongly protected mice against dermonecrosis (Fig. 1I). Vaccination with LukE, on the other hand, elicited moderately strong protection against dermonecrosis, as the lesion size was reduced by approximately 40%, compared with control mice. To test the hypothesis that Hla-specific protection, but not that afforded by LukE, was mediated by antibody, serum from vaccinated mice was adoptively transferred into naïve BALB/c mice before infection. Consistent with this hypothesis, serum from HlaH35L-vaccinated mice strongly protected against dermonecrosis, but serum from LukE-vaccinated mice did not (Fig. 1J). Together, these findings demonstrate that infection-mediated protective immunity is determined by the MHC haplotype and that protection is characterized by Hla-specific antibody responses and, to a lesser extent, LukE/LukS-PV–specific effector T cell responses.
A conserved leukotoxin epitope drives T cell responses against LukE and LukS-PV
In addition to Hla, LukE, and LukS-PV, we previously reported that responses against two other sae-regulated antigens, SplB and SspB, were associated with protection in BALB/c mice (17). Therefore, we compared effector T cell responses against each of these antigens following infection of BALB/c and C57BL/6 mice by ELISpot for IL-17A and IFNγ. In BALB/c mice, LukE and LukS-PV stimulated the strongest IL-17A responses, compared with Hla, SplB, and SspB (Fig. 2A), suggesting that TH17 responses are targeted toward these two antigens. Compared with the IL-17A responses, there were weaker IFNγ against each of these antigens in BALB/c mice (Fig. 2B). In contrast to the responses in BALB/c mice, there were minimal IL-17A or IFNγ responses against LukE, LukS-PV, or SplB in C57BL/6 mice (Fig. 2, A and B). S. aureus–specific T cell responses (following stimulation with heat-killed S. aureus) were comparably elicited in BALB/c and C57BL/6 mice, but were biased toward TH17 in BALB/c mice and toward TH1 in C57BL/6 mice (Fig. 2, A and B), consistent with our previous observations (11). These findings confirm that infection elicits strong antigen-specific T cell responses in BALB/c mice (targeted primarily against LukE and LukS-PV), but, although there are S. aureus–specific T cell responses following infection of C57BL/6 mice, they are not targeted toward the five sae-regulated antigens.
Fig. 2. Distinct leukotoxin epitopes drove protective T cell responses in BALB/c and C57BL/6 mice.
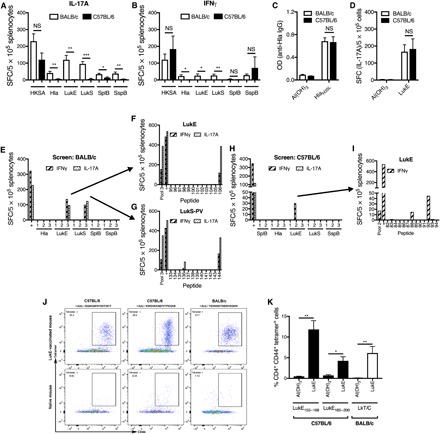
(A) Infection of BALB/c mice resulted in stronger IL-17A responses by ELISpot against LukE and LukS-PV, compared with C57BL/6 mice. There were also stronger responses against Hla, SplB, and SspB in BALB/c mice, but the magnitude of the differences was less. (B) Infection also resulted in stronger IFNγ responses in BALB/c mice for Hla, LukE, and LukS-PV, although the overall responses were weak (six mice per group, pooled from two experiments; HKSA, heat-killed S. aureus). Following vaccination with HlaH35L or LukE, there were comparably strong anti-Hla antibody (C) and anti-LukE IL-17A (D) responses, respectively, in BALB/c and C57BL/6 mice (four mice per group from one representative experiment). (E) Screening of two to three peptide pools from each of the five sae-regulated antigens revealed one pool each from LukE and LukS-PV that elicited strong responses by ELISpot (pool no. 3 for each). (F and G) Deconvolution of these pools identified a conserved immunogenic epitope (designated “LkT/C”) in LukE [(C) no. 106] and LukS-PV [(D) no. 144] (three mice per group from one representative experiment). (H) Screening of peptide pools in LukE-vaccinated C57BL/6 mice revealed one pool that elicited strong responses (pool no. 2). (I) Deconvolution of this pool identified two distinct immunogenic epitopes (no. 88 and no. 92) (three mice per group from one representative experiment). “+” indicates concanavalin A, a positive control. (J and K) Validation of class II pMHC tetramers. Splenocytes and draining lymph nodes from LukE-vaccinated C57BL/6 or BALB/c mice were isolated and stained with one of three PE-conjugated tetramers, followed by enrichment using anti-PE microbeads and magnetic columns. (K) Compared with control mice, LukE-vaccinated mice had large populations of tetramer+ CD44+ CD4+ T cells (three to five mice per group pooled from two experiments). Results were compared with Student’s t test. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Because infection-elicited protective antibody and T cell responses were determined by the MHC haplotype, we hypothesized that the strength of the protective response to vaccination would also depend on the genetic background. On the basis of the above findings, we focused on Hla-specific antibody responses and LukE-specific effector T cell responses. In contrast to responses following infection, vaccination with HlaH35L elicited comparably strong Hla-specific antibody responses in BALB/c and C57BL/6 mice (Fig. 2C). Similarly, there were no differences in LukE-specific IL-17A responses following LukE vaccination of BALB/c or C57BL/6 mice (Fig. 2D). These findings demonstrated that C57BL/6 mice were able to mount antibody and T cell responses to protective antigens and suggested that host genetics are only critical for determining the strength of infection-elicited immune responses, but not those elicited by vaccination.
One potential explanation for why LukE-specific T cells are not detectable by IFNγ and IL-17 ELISpot assays in C57BL/6 mice, but infection elicited comparable anti-LukE IgG responses, is that T cells might respond by release of a cytokine other than IFNγ or IL-17. To address this possibility, we sought to quantify the number of LukE-specific CD4+ T cells during infection and/or vaccination, irrespective of cytokine production, by use of epitope-specific peptide-MHC (pMHC) tetramers. To generate pMHC tetramers, we aimed to identify immunogenic epitopes in each of the five antigens. To accomplish this, overlapping peptides covering the sequence of each of the antigens were synthesized. As a screening step, splenocytes from previously infected BALB/c mice were stimulated with two to three pools per antigen of these peptides, followed by IL-17A and IFNγ ELISpot (Fig. 2E). Consistent with the relatively weak Hla-, SplB-, and SspB-specific TH1 and TH17 responses, no peptide pools from these antigens stimulated responses following infection of BALB/c mice (Fig. 2E). In contrast, one pool each from LukE and LukS-PV stimulated strong IL-17A and IFNγ responses (Fig. 2E; pool no. 3 for each), within which we identified two peptides that stimulated strong responses, LukE296–311 (Fig. 2F; peptide no. 106) and LukS-PV297–312 (Fig. 2G; peptide no. 144). The amino acid sequences of these peptides were identical, and each is located directly at the C terminus of the respective protein (Table 1). We designated this peptide LkT/C (leukotoxin/leukocidin). To assess whether this peptide sequence is conserved among staphylococcal leukocidins, a Basic local alignment search tool (BLAST) search was performed and revealed homologous peptide sequences at the C terminus of γ-hemolysin A (HlgA), γ-hemolysin C (HlgC), and leukocidin A (LukA), with varying degrees of sequence similarity, and there was no homology identified in Hla (Table 1). We were unable to identify any immunogenic epitopes within Hla, LukE, LukS-PV, or LukE in previously infected C57BL/6 mice (fig. S2A), but we did identify one peptide within SspB that stimulated IFNγ responses, SspB297–312, (fig. S2A, pool no. 3, and S2B, peptide no. 182).
Table 1. Sequence homology at the C terminus of staphylococcal pore-forming toxins.
| Protein | amino acid position | amino acid sequence |
| LukE | 296–311 | YEVNWKTHEIKVKGHN |
| LukS-PV | 297–312 | YEVNWKTHEIKVKGHN |
| HlgC | 300–315 | YEVNWKTHEIKVKGQN |
| HlgA | 292–309 | YEVNWKTHEVKIKSIT |
| LukA (LukG) | 326–341 | YEVDWKNKTVKVVDKY |
| Hla | 308–319 | YKIDWEKEEMTN |
To validate that LkT/C drives T cell responses against both LukE and LukS-PV during skin infection, we quantified LukE-, LukS-PV–, or LkT/C-specific effector T cell responses in previously infected BALB/c and C57BL/6 mice. As expected, there were strong IL-17A responses and moderate IFNg responses against LkT/C, LukE, and LukS-PV in splenocytes from infected BALB/c mice, but no detectable responses in C57BL/6 mice (fig. S2C). Therefore, we hypothesized that LkT/C was presented by either I-Ad or I-Ed but not by I-Ab. We found that LkT/C bound I-Ed with high affinity and with lower affinity to I-Ad and I-Ab (Table 2). Together, these findings demonstrate that strong effector T cell responses were elicited only in infected BALB/c mice because LkT/C was successfully presented in the context of I-Ed, which is absent in C57BL/6 mice.
Table 2. MHC binding of identified epitopes.
N/D, not done. IC50, half-maximal inhibitory concentration.
| Protein | Position | Sequence | IC50 (nM) | ||
| I-Ab | I-Ad | I-Ed | |||
| LukS-PV | 297–312 | YEVNWKTHEIKVKGHN | 17293 | 3211 | 2.2 |
| LukE | 296–311 | YEVNWKTHEIKVKGHN | 17293 | 3211 | 2.2 |
| LukE | 153–168 | IGGNGSFNYSKTISYT | 586 | N/D | N/D |
| LukE | 185–200 | KWGVKANEFVTPDGKK | 4050 | N/D | N/D |
Because vaccination of C57BL/6 mice with LukE elicited T cell responses, but LkT/C specifically binds I-Ed, we hypothesized that LukE vaccine-specific responses in C57BL/6 mice would be driven by a different immunogenic epitope. Consistent with this notion, the anti-LukE T cell responses following vaccination in C57BL/6 mice were not recapitulated following stimulation with LkT/C, confirming that this peptide is not presented by I-Ab (fig. S2D). Therefore, we sought to identify the immunogenic epitope(s) in LukE-vaccinated C57BL/6 mice and found one peptide pool for LukE that stimulated a moderately strong IFNγ response (Fig. 2H, pool no. 2). Within this pool, we identified two immunogenic epitopes, LukE153–168 (no. 88) and LukE185–200 (no. 92), each of which was subsequently found to bind I-Ab with moderate affinity (Fig. 2I and Table 2). These results suggested that vaccine-elicited LukE T cell responses in C57BL/6 mice are driven by distinct epitopes that preferentially bind MHC I-Ab with lower affinity than LkT/C binding to I-Ed.
pMHC tetramers track LukE-specific T cell populations in BALB/c and C57BL/6 mice
Our findings that antibody responses were dissociated from effector T cell responses suggested that follicular helper T (TFH) cell responses may more accurately predict the magnitude of the antibody response. However, antigen-specific TFH cells are best characterized by the expression of CXCR5, programmed cell death protein1 (PD-1), inducible T cell costimulator (ICOS) or Bcl6, and pMHC tetramers, as there is no readily accepted functional assay comparable to the IFNγ and IL-17 ELISpot assay for TH1 and TH17, respectively (22). Using the immunogenic peptides identified from BALB/c and C57BL/6 mice, pMHC LkT/C:I-Ed, LukE153–168:I-Ab, and LukE185–200:I-Ab tetramers were synthesized by the National Institutes of Health (NIH) Tetramer Core Facility. To validate the tetramers, BALB/c and C57BL/6 mice were vaccinated with LukE. Following vaccination, phycoerythrin (PE)–labeled tetramer+ cells were enriched using anti-PE magnetic beads and columns, and activated epitope-specific T cells were quantified by flow cytometry as tetramer+ CD44hi CD4+ T cells (23). Using this approach, we validated high frequencies of LukE153–168- and LukE185–200-specific and LkT/C-specific CD44hi CD4+ T cells following LukE vaccination in C57BL/6 and BALB/c mice, respectively (Fig. 2, J and K).
Immunodominant non-LukE peptides prevent presentation of LukE-specific epitopes
Our observations that infection-elicited immune responses were absent in C57BL/6 mice, but vaccine-specific responses were preserved, suggested the hypothesis that other immunodominant epitopes presented during infection interfered with the presentation of Hla and LukE epitopes and, subsequently, anti-Hla antibody and anti-LukE T cell responses. To test this, we developed a model of concomitant infection and vaccination in which mice were vaccinated with HlaH35L (or LukE) and/or infected with S. aureus, followed by quantification of anti-Hla IgG levels or LukE-specific T cell responses (Fig. 3A). In both BALB/c and C57BL/6 mice, vaccination with HlaH35L resulted in stronger responses, compared with infection (Fig. 3B). In BALB/c mice, there was a trend toward higher anti-Hla IgG levels in the mice that received concomitant infection and vaccination, compared with vaccination alone, but the differences were not significant. In contrast, C57BL/6 mice that received concomitant SSTI and HlaH35L vaccination had significantly lower anti-Hla IgG levels, compared with those that received HlaH35L vaccination alone (Fig. 3B). Consistent with the lower anti-Hla IgG levels, concomitant SSTI inhibited the efficacy of HlaH35L vaccination against SSTI (fig. S3A).
Fig. 3. Inhibition of vaccine-elicited antibody and T cell responses by concomitant infection.
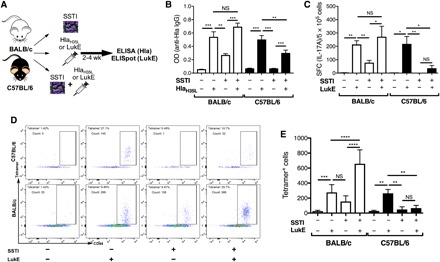
(A) Experimental design for concomitant infection/vaccination. BALB/c or C57BL/6 mice were infected with S. aureus, vaccinated with HlaH35L or LukE, or received concomitant infection and HlaH35L/LukE vaccination, followed by quantification of anti-Hla antibody or anti-LukE IL-17A responses. (B) In BALB/c mice, concomitant SSTI/Hla vaccination resulted in a trend toward higher anti-Hla IgG levels, compared with vaccination alone, but the differences were not significant. In contrast, concomitant SSTI/Hla vaccination in C57BL/6 mice resulted in lower anti-Hla IgG levels, compared with vaccination alone (seven to nine mice per group pooled from two experiments). (C) In BALB/c mice, concomitant SSTI/LukE vaccination resulted in a trend toward higher anti-LukE IL-17A responses, compared with vaccination alone, but the differences were not significant. In contrast, concomitant vaccination and infection resulted in strongly decreased anti-LukE responses in C57BL/6 mice, compared with vaccination alone (four to five mice per group from one representative experiment). (D and E) Immunodominance of LukE-specific T cell responses were confirmed by quantifying tetramer+ CD44hi CD4+ T cells following concomitant LukE vaccination and infection. (D) Representative flow plots of tetramer+ CD4+ CD44hi T cells following concomitant LukE vaccination and infection (five mice per group pooled from two experiments). Data were compared using one-way ANOVA with Tukey’s posttest. All data are plotted as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Vaccination with LukE resulted in stronger IL-17A and IFNγ responses, compared with SSTI, in both BALB/c and C57BL/6 mice (Fig. 3C and fig. S3C). In BALB/c mice, the combination of SSTI and LukE vaccination appeared comparable to LukE alone, suggesting neither interference nor synergism between these treatments. In contrast, C57BL/6 mice that received concomitant SSTI and LukE vaccination had significantly weaker anti-LukE responses, compared with LukE alone. These observations confirm that infection can indeed interfere with the induction of LukE-specific T cell responses. Neither the anti-LukE antibody (fig. S3D) nor the anti-Hla TH17 (fig. S3B) responses reflected these differences, suggesting that immunodominance by nonprotective S. aureus antigens may preferentially inhibit either antibody or effector T cell responses, depending on the antigen.
To confirm that other immunodominant epitopes presented during SSTI interfere with LukE vaccine-elicited responses in C57BL/6 mice, but not BALB/c mice, the concomitant SSTI/LukE vaccination experiments were repeated, followed by tetramer-based quantification of specific T cell responses. As we observed by ELISpot, vaccination with LukE resulted in strong LukE-specific T cell responses in the spleen and draining lymph nodes of BALB/c and C57BL/6 mice (Fig. 3, D and E). In contrast to the IL-17A and IFNγ ELISpot assays, concomitant SSTI and LukE vaccination of BALB/c mice resulted in significantly more LukE-specific T cells, compared with LukE alone. This suggests that LukE-specific T cells with different cytokine profiles are elicited by concurrent infection and vaccination. In contrast to BALB/c mice, and consistent with the ELISpot assays, concomitant SSTI and LukE vaccination resulted in strongly attenuated LukE-specific responses in C57BL/6 mice, compared with LukE vaccination alone (Fig. 3, D and E). Last, to confirm that the immunodominant antigens presented during S. aureus SSTI specifically inhibited vaccination with S. aureus–specific antigens, we determined whether SSTI would inhibit responses to an irrelevant antigen, 2W-ovalbumin (2W-OVA); the 2W peptide is a variant of the peptide 52–68 from the I-E α chain (24, 25) and has been used to track endogenous populations of 2W-specific CD4+ T cells (26). Naïve C57BL/6 mice were immunized subcutaneously with splenocytes from 2W-OVA transgenic C57BL/6 mice [donor-specific transfer (DST)], in the presence or absence of concomitant SSTI. 2W-specific T cells were identified using 2W:I-Ab pMHC tetramers. Consistent with the notion that this immunodominance is specific to S. aureus antigens, there were no significant differences in the percentage or numbers of 2W-specific T cells or 2W-specific conventional T (Tconv) cells, (defined as Fox3p−), regardless of whether mice received DST alone or DST and concomitant SSTI (fig. S4). Together, these results demonstrate that S. aureus SSTI inhibits LukE-specific T cell responses elicited by vaccination in C57BL/6 mice, whereas SSTI boosts vaccine-elicited responses in BALB/c mice.
TFH cell responses following infection and vaccination
The ability of mice that express MHC H-2b to generate strong LukE and LukS-PV antibody responses following infection, compared with weak effector T cell responses, raised the possibility of differential expansion of antigen-specific effector TH1/TH17 cells, compared with TFH cells, which are critical for B cell help and subsequent antibody generation. To test this possibility, we quantified TFH cell responses following SSTI or LukE vaccination of BALB/c and C57BL/6 mice. To quantify LukE-specific TFH cells, we gated on the CD4+ CD44+ tetramer+ population and then quantified cells within this population that were PD-1hi BCL6hi (Fig. 4A). As expected, both infection and LukE vaccination expanded a population of LukE-specific TFH cells in BALB/c mice (Fig. 4C), consistent with strong anti-LukE antibody responses in these mice. However, in C57BL/6 mice, only LukE vaccination resulted in an expansion of LukE-specific TFH cells (Fig. 4C). In contrast, despite strong LukE antibody responses, no LukE-specific TFH cells were detected in C57BL/6 mice following SSTI. This could be explained by the ability of T cells with other specificities to provide cognate help to LukE- or LukS-PV–specific B cells, as long as those antigens are linked during B cell receptor–mediated internalization (27, 28). Therefore, we also quantified bulk TFH cell responses in BALB/c and C57BL/6 mice following infection or LukE vaccination by quantification of CD4+ CD44+ PD-1hi BCL6hi T cells (Fig. 4B). Consistent with our hypothesis, infection resulted in an expansion of bulk TFH cells in both BALB/c and C57BL/6 mice (Fig. 4D). Therefore, it is possible that expanded TFH cell populations with other epitope specificities may result in generation of anti-LukE antibodies in C57BL/6 mice following SSTI. This is the subject on ongoing investigation.
Fig. 4. TFH cell responses following infection and vaccination.
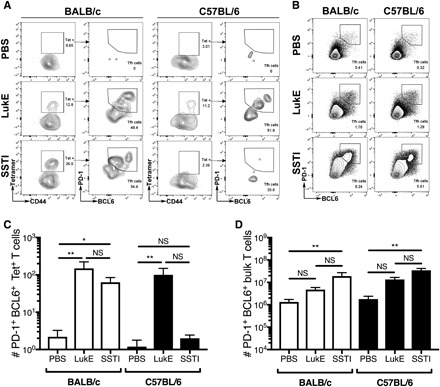
LukE-specific (A and C) and bulk (B and D) TFH cell responses were quantified in BALB/c and C57BL/6 mice following SSTI or vaccination with LukE. [(A) and (C)] LukE-specific TFH cells were quantified as CD4+ CD44+ pMHC tetramer+ BCL6hi PD-1hi cells. (A) Representative flow plots. (C) There were strong LukE-specific TFH cell responses following infection and vaccination of BALB/c mice; in contrast, only vaccination resulted in expansion of LukE-specific TFH in C57BL/6 mice, whereas infection did not. [(B) and (D)] Bulk TFH cells were quantified as CD4+ CD44+ PD-1hi BCL6hi cells. (B) Representative flow plots. (D) There were strong bulk TFH cell responses following infection of both BALB/c and C57BL/6 mice. For both sets of experiments, there were five mice per group from one representative experiment. Data were compared using one-way ANOVA with Tukey’s posttest. Data are presented as means ± SEM. *P < 0.05; **P < 0.01.
A multivalent vaccine elicits protection and circumvents MHC restriction and immunodominance
There is consensus in the field that a multivalent vaccine will be necessary to prevent S. aureus infection. The immunodominance toward or away from protective immune responses that we observed during infection raises the possibility that individual components of a multivalent vaccine may act to inhibit one another in unanticipated ways, depending on the MHC alleles of the recipient. Therefore, we developed an investigational quadrivalent vaccine (“4S”) based on the four sae-regulated antigens that elicited TH17 responses during S. aureus SSTI and tested whether the 4S vaccine would elicit T cell–mediated protection in BALB/c and C57BL/6 mice. Although the T cell responses against LukE and LukS-PV were directed against a conserved epitope in BALB/c mice, we elected to include both in the vaccine because there may be distinct epitopes that elicit responses in C57BL/6 mice. There were no significant differences in the strength of the IL-17A, IFNγ, or antibody responses against any of the antigens between vaccinated BALB/c and C57BL/6 mice (Fig. 5, A to C). These results suggested that, despite the importance of the MHC haplotype in infection-elicited responses, MHC restriction can be overcome by vaccination with the four sae-regulated antigens that did not appear to inhibit antigen-specific T cell responses against one another. Consistent with this conclusion, both 4S-vaccinated C57BL/6 (Fig. 5D) and BALB/c (Fig. 5F) mice had significantly smaller lesions following SSTI with S. aureus, compared with control mice. Vaccination also resulted in superior enhancement of bacterial clearance from the lesions in C57BL/6 mice 7 days after infection, compared with BALB/c mice (Fig. 5, E and G). We found that protection was not antibody dependent, since adoptive transfer of serum from 4S-vaccinated mice into naïve C57BL/6 mice before skin infection did not result in smaller skin lesions or enhanced bacterial clearance from the lesions (Fig. 5, H and I). In contrast, adoptive transfer of serum from Hla-immunized mice was protective. Last, adoptive transfer of T cells from 4S-vaccinated mice provided modest protection against SSTI, resulting in a 25% reduction in lesion size (Fig. 5J). Although transfer of T cells from Hla-vaccinated mice resulted in a trend toward smaller lesions, the differences were not significant. Reflecting the modest protection afforded by 4S-specific T cells, there were no significant differences in bacterial clearance from the lesions among the groups (Fig. 5K). It should be noted that transfer of 4S-specific T cells resulted in more modest protection than vaccination alone, suggesting that other mechanisms may also contribute to 4S vaccine–mediated protection. Together, these results suggest that the modest protection afforded by the 4S vaccine is mediated by T cells.
Fig. 5. Vaccine-elicited T cell–mediated protection circumvented MHC-restricted immunodominance.
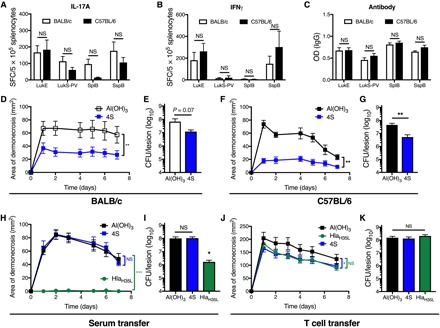
(A to C) Following vaccination with the 4S vaccine (LukE, LukS-PV, SplB, and SspB), there were no significant differences in antigen-specific IL-17A (A), IFNγ (B), or antibody (C) responses against any of the antigens in BALB/c and C57BL/6 mice, although there were trends toward stronger IL-17A responses in BALB/c mice (three mice per group from one representative experiment). (D to G) Vaccination with the 4S vaccine resulted in significantly smaller skin lesions in BALB/c (D) and C57BL/6 (F) mice, compared with controls. Vaccination resulted in a trend toward fewer bacteria recovered from the lesions of BALB/c mice (E) and significantly fewer bacteria from the lesions of C57BL/6 mice (G) 7 days after infection (six to eight mice per group from one representative experiment). (H and I) 4S-mediated protection was independent of antibody, as there were no significant differences in lesion size (H) or number of bacteria recovered from the lesions (I) between mice that received serum from 4S-vaccinated or Al(OH)3 control C57BL/6 mice. In contrast, C57BL/6 mice that received serum from HlaH35L-vaccinated BALB/c mice were protected (eight mice per group from one representative experiment). (J) Transfer of T cells from 4S-vaccinated mice resulted in moderately smaller lesions following SSTI, compared with controls. Although there was a trend toward smaller lesions in mice that received T cells from HlaH35L-vaccinated mice, the differences were not significant (P = 0.08). (K) Consistent with the modest reduction in lesion size, there were no significant differences in the number of bacteria recovered from the lesions 7 days after infection (10 to 14 mice per group pooled from two separate experiments). Data were compared using Student’s t test [(A), (B), (C), (E), and (G)], two-way ANOVA with repeated measures and Tukey’s posttest [(D), (F), (H), and (J)] or one-way ANOVA with Tukey’s posttest [(I) and (K)]. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
An optimized vaccine protects mice that express either MHC H-2d or H-2b
We hypothesized that a vaccine that elicits both T cell responses against LukE, LukS-PV, SplB, and SspB, and antibody responses against Hla would override MHC restriction and optimally protect against S. aureus SSTI. To test this, we vaccinated BALB/c and C57BL/6 mice with an optimized vaccine (“5S,” 4S + HlaH35L) and compared protection with this vaccine with the antibody-based HlaH35L vaccine. Vaccination with the optimized 5S vaccine resulted in comparable IL-17A, IFNγ, and antibody responses against each of the antigens in BALB/c and C57BL/6 mice, with the exception of higher anti–LukS-PV IgG levels in vaccinated C57BL/6 mice (Fig. 6, A to C). Vaccination resulted in nearly complete protection against dermonecrosis in both BALB/c and C57BL/6 mice (Fig. 6, D and F), as well as fewer bacteria recovered from the lesions, with a nearly 3 log reduction in bacterial colony-forming units (CFU) in both BALB/c and C57BL/6 mice (Fig. 6, E and G). Although the Hla-based vaccine also protected against dermonecrosis, the 5S vaccine resulted in modestly increased clearance of bacteria from the skin lesions in both BALB/c and C57BL/6 mice (Fig. 6, E and G). Together, these results demonstrate that an optimized vaccine consisting of Hla and the four additional sae-regulated antigens results in strong protection against S. aureus SSTI in mice that express MHC H-2b or H-2d. These observations bode well for multivalent vaccine efforts, suggesting that a carefully curated multicomponent vaccine may be effective, independent of host genetic background.
Fig. 6. An optimized vaccine overcame MHC restriction and immunodominance.
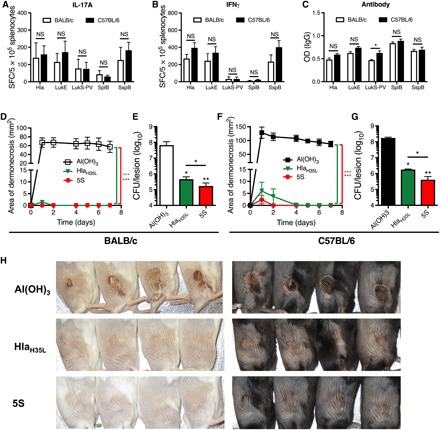
BALB/c and C57BL/6 mice were vaccinated and boosted with one of two vaccines: HlaH35L or 5S (HlaH35L, LukE, LukS-PV, SplB, and SspB). (A to C) Vaccination with the 5S vaccine resulted in comparably strong IL-17A (A), IFNγ (B), and antibody (C) responses against the five antigens in BALB/c and C57BL/6 mice, with the exception of stronger LukS-PV–specific antibody responses in C57BL/6 mice (three mice per group from one representative experiment). (D to G) Vaccination with 5S or HlaH35L protected against dermonecrosis in BALB/c (D) and C57BL/6 (F) mice, and vaccination with 5S resulted in superior clearance of bacteria 7 days after infection from the skin lesions of BALB/c (E) and C57BL/6 mice (G), compared with controls or HlaH35L-vaccinated mice (six to eight mice per group from one representative experiment). (H) Representative skin lesions from vaccinated BALB/c and C57BL/6 mice 7 days after infection. Data were compared using Student’s t test [(A) to (C)], two-way ANOVA with repeated measures and Tukey’s posttest [(D) and (F)] or one-way ANOVA with Tukey’s posttest [(E) and (G)]. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. (Photo credit: Fan Zhao, the University of Chicago.)
DISCUSSION
We identified notable heterogeneity in the anti-Hla IgG levels in healthy adults, which suggests that host genetics, in the context of a lifetime of exposure to S. aureus, is the primary driver of anti-Hla antibody responses in humans. While the complex genetics of human antistaphylococcal immunity cannot be fully recapitulated in mice, understanding the mechanistic bases in mouse models may advance our understanding of individual susceptibility to, and adaptive immune responses elicited by, S. aureus infection in the human population. To this end, we identified the murine MHC as the genetic basis for divergent adaptive immune phenotypes elicited by S. aureus SSTI. The protective phenotype requires the MHC class II H-2d haplotype and is characterized by strong antibody responses against Hla and TH17 responses against LukE and LukS-PV. In contrast, the nonprotective phenotype, in the context of MHC class II H-2b, is characterized by polyclonal antibody responses against other S. aureus antigens and T cell responses not directed against LukE or LukS-PV. Only infection-elicited antibody and T cell responses were dependent on the MHC haplotype, whereas subunit vaccination circumvented the impact of the MHC haplotype and elicited similarly strong responses in mice that expressed either H-2d (BALB/c) or H-2b (C57BL/6). This suggests that the differences observed were not due to inherent deficiencies in T or B cell repertories but due to inhibition of antigen presentation by immunodominant antigens that more strongly bind MHC class II.
We investigated the determinants of infection-elicited antibody responses against Hla in mice and excluded the possibility that there was a hole in the C57BL/6 antibody repertoire, because SSTI elicited high anti-Hla IgG levels in CB.17 mice (BALB/c background expressing the C57BL/6 antibody repertoire) and protected against secondary SSTI. Instead, we found that the MHC haplotype determined whether primary infection elicited protective anti-Hla antibody responses against recurrent SSTI, because both protection and anti-Hla IgG levels were only elicited in mice that have the H-2d haplotype, but not H-2b. Neither Hla-specific TH17/IL-17A nor TH1/IFNγ responses were dependent on the MHC haplotype. In contrast to infection-elicited responses, vaccination with HlaH35L resulted in equally strong antibody responses in BALB/c and C57BL/6 mice. Concomitant infection competitively inhibited, albeit modestly, the anti-Hla antibody responses, but there was no inhibition of Hla-specific effector TH17 responses. Although the mechanism for this inhibition is not clear, we hypothesize that competition for epitope binding to MHC class II inhibits only the Hla-specific TFH cell response, whereas the effector TH1 and TH17 responses are not affected. Unfortunately, we were not able to generate tetramers to track Hla-specific TFH cell responses; however, we were able to track the protective T cell responses against LukE and LukS-PV.
The immunogenicity of an epitope is determined by processing of the native antigen by unfolding and cleavage, ability of the epitope to bind MHC, and recognition of the pMHC complex by the T cell receptor (29). We found that, similar to anti-Hla antibody responses, effector TH1 and TH17 responses against LukE and LukS-PV were determined by the MHC haplotype. Infection-elicited responses against each of these antigens in mice of the H-2d haplotype were driven by a conserved C-terminal epitope, called LkT/C. Moreover, anti-LukE and LukS-PV effector T cell responses, like the anti-Hla antibody responses, were also competitively inhibited by concomitant infection. The demonstrated strong binding of LkT/C to class II I-Ed suggests that differences in binding affinity for MHC are most likely to explain the phenotypes that we observed, so-called “determinant selection” by MHC (30). In contrast, because LukE153–168 and LukE185–200 bind I-Ab with moderate affinity, our findings suggest that epitopes from other nonprotective antigens outcompete these epitopes during S. aureus infection of mice that express class II H-2b. However, the lack of antigenic competition during vaccination with LukE alone allows for strong anti-LukE responses. We cannot rule out, however, an additional contribution of other genetic differences between C57BL/6 and BALB/c mice to the differences that we observed. Together, we speculate that the immunodominance that we observed during SSTI may be specific to either TFH or effector T cells, depending on the antigen.
Genes within the MHC class II loci involved in antigen presentation are among the most polymorphic loci in vertebrates (31) and, together with T cell receptor diversity, determine the rate of disease clearance. Extensive MHC polymorphism has long been theorized to be maintained by pathogen-driven selection, by buffering populations against widespread epidemics even at the expense of increased susceptibility to some types of infections in select individuals. Consistent with this theory, we show that in a BALB/c background, one of the two functional MHC class II genes (I-Ed) is able to bind with high affinity to the LkT/C epitope, whereas in C57BL/6 mice, competition for the only functional MHC class II genes (I-Ab) resulted in the dominant presentation of epitopes from nonprotective antigens. In this context, there is reduced presentation of epitopes from protective antigens. This resulted in S. aureus SSTI eliciting a protective response in BALB/c mice and an equally robust but nonprotective immune response in C57BL/6 mice. Despite the considerable complexity of the human leukocyte antigen (HLA) gene locus, we speculate that a similar variation in immunodominance may, at least in part, explain why recurrent S. aureus infections are common in some patients, while most healthy individuals remain free of repeated and serious infections.
We hypothesized that one strategy to avoid the effects of immunodominance of nonprotective antigens would be to vaccinate with a selected combination of protective antigens. We demonstrated that vaccination with four of the sae-regulated antigens (LukE, LukS-PV, SplB, and SspB) resulted in antibody-independent protection against dermonecrosis in C57BL/6 mice, providing evidence for the hypothesis that a multivalent vaccine could override MHC-restricted T cell responses. However, the protection was relatively modest, compared with previous reports of anti–Hla antibody–mediated protection (12, 19). We found that addition of HlaH35L to the quadrivalent 4S vaccine enhanced efficacy, and the resulting 5S vaccine was superior to either HlaH35L alone (albeit modestly) or the 4S vaccine. This vaccine protected equally well in BALB/c and C57BL/6 mice. In this context, the anti-Hla antibody responses are complemented by T cell responses against the other four sae-regulated antigens for improved protection. Thus, our findings suggest a potential multiantigen immune correlate of protection in which strong TH17 responses against a subset of sae-regulated antigens and antibody responses against Hla would predict/generate stronger protective immunity across broad MHC backgrounds.
Mouse models have obvious advantages to define mechanisms of protective immunity against S. aureus; however, the species specificity of staphylococcal toxins suggests that results from mouse models need to be interpreted with caution. For example, rodent neutrophils are resistant to lysis by Panton-Valentine leukocidin (PVL) (32); this can be explained by the recent demonstration that PVL binds the complement receptors C5aR1 and C5aR2 in humans and rabbits, but not mice (33). Other pore-forming toxins with identified cellular receptors that may mediate species-specific effects include Hla (34), LukED (35, 36), HlgAB (37), HlgCB (37), and LukAB (38). Notably, mice are also resistant to staphylococcal superantigens (39). There are also several mouse models of recurrent S. aureus infection that draw seemingly discrepant conclusions regarding the mechanisms of protective immunity. In our model, antibody- and TH17-mediated protection was observed in BALB/c mice (11). However, in a mouse model of S. aureus peritonitis, memory TH1 cells were protective (14). A recent study confirmed that C57BL/6 mice were not protected against secondary SSTI (16); however, IL-1β−/− C57BL/6 mice were partially protected against secondary infection in an antibody-independent manner due to clonal expansion of memory γδ T cells. Together, integration of each of the distinct mouse models of recurrent infection suggests that, depending on the site of infection and the genetic background of the mice studied, protective immunity depends on a combination of TH1-, TH17-, and γδ T cell–mediated mechanisms in concert with protective antibody responses.
This work has several potentially important implications when choosing the optimal target population for a S. aureus vaccine. The finding that concomitant infection inhibits anti-Hla antibody and anti-LukE effector T cell responses is intriguing, but the impact of this immunodominant mechanism on vaccine responses is likely to be limited. For example, it would not be anticipated that many patients would have clinically apparent infections at the time of vaccination, but given the high rate of S. aureus colonization at the population level, it will be critical to determine whether colonization itself might interfere with vaccine efficacy. Second, the nearly ubiquitous presence of S. aureus raises the question of whether preexisting antibody and T cell immunity from prior infection and/or colonization might similarly inhibit vaccine efficacy. If this is the case, then it would provide justification for targeting the pediatric population with a S. aureus vaccine, before the onset of potentially deleterious immune responses. Work is ongoing in our group to address these issues.
Our findings also have the potential to affect vaccine design. First, they raise the possibility that genetic heterogeneity may affect vaccine-elicited responses. Fortunately, our findings that vaccination with a pentavalent sae-based vaccine protected both BALB/c and C57BL/6 mice should bode well for vaccine design. Second, identification of the immunogenic epitopes that mediate protection in multiple mouse strains could guide vaccine design. For example, one could envision a vaccine incorporating LkT/C as part of a multiepitope vaccine, which excludes irrelevant nonprotective but highly immunogenic regions of the individual full-length proteins. In this case, one epitope may also confer protective responses against multiple antigens. Third, identification of immunogenic epitopes and development of pMHC tetramers will enable detailed mechanistic investigation of protective (and nonprotective) CD4+ T cell responses. Given the seemingly adequate immunogenicity of the IsdB and capsule-based candidate vaccines but lack of protection (8, 9), it is critical to learn from past failures and better predict vaccine efficacy in the future.
There are several unanswered questions raised by this work. First, we have not yet identified the immunodominant epitopes in C57BL/6 mice that outcompete protective responses. We hypothesize that these will come from one or more antigens that elicited strong antibody or T cell responses in C57BL/6 mice, but not BALB/c mice (17). Second, future studies should compare the protection afforded by full-length LukE and LukS-PV with LkT/C or LukE153–168- and LukE185–200-specific T cell responses. Although our sae-based vaccine strongly protected mice against SSTI, the importance of each of the individual antigens has not yet been established, and the benefit of adding the four additional antigens to Hla is not yet clear. In particular, it remains to be determined whether the LukE- and LukS-PV–specific responses are conserved or whether each antigen protects independently. Third, our findings do not fully explain the discrepancy between antigen-specific antibody and effector T cell responses. The demonstration that there is expansion of bulk, but not LukE-specific, TFH cells following infection of C57BL/6 mice suggests that nonspecific TFH cells are providing cognate help to LukE-specific B cells, but how this occurs is not yet clear. Similarly, identification of immunogenic epitopes in Hla will be necessary to develop pMHC tetramers to track Hla-specific TFH cell responses. Last, these findings will need to be translated from mice to the human population. This is particularly important given the diversity of HLA haplotypes in the human population and the failure of previous vaccines to prevent infections in clinical practice despite success in animal models. We propose that a detailed mechanistic understanding in the mouse models is a critical foundation on which to base translational studies.
MATERIALS AND METHODS
Patients and samples
All human studies were approved by the Institutional Review Board at the University of Chicago, and informed consent was obtained from all subjects before enrollment. The population and sample collection procedures have been previously reported (21). Briefly, as part of a study of skin infection in otherwise healthy adults, uninfected controls were adults (matched for age, gender, and race with infected patients) presenting to the Emergency Department at the University of Chicago between September 2010 and September 2012 with minor, noninfectious complaints; blood was drawn on enrollment. Serum was prepared from 3 ml of nonheparinized blood.
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Chicago (protocol no. 72405) or the Research Institute at Nationwide Children’s Hospital (protocol no. AR17-00072) and adhered to the standards of the NIH Guide for the Care and Use of Laboratory Animals. For most experiments, female BALB/c and C57BL/6 mice were purchased from Taconic. For the MHC experiments, female BALB/c, C57BL/6, C.B10-H2b, and B6.C-H2d mice were purchased from Jackson. For the experiments using CB.17 mice, female CB.17, BALB/c, and C57BL/6 mice were purchased from Charles River Laboratories. 2W-OVA transgenic C57BL/6 mice were a gift from J.J. Moon (Harvard University) and were bred at the University of Chicago. Primary infection was performed when the mice were 7 to 8 weeks old.
Mouse model of S. aureus infection
Our mouse model of S. aureus skin infection using strain 923, a USA300 isolate, has been reported (11, 40). Briefly, an overnight culture of S. aureus was diluted 1:100 into fresh tryptic soy broth and grown at 37° (250 rpm) until the exponential phase [OD600 (optical density at 600 nm), 1.8 for 3 hours], at which point the bacteria were pelleted by centrifugation, washed in phosphate-buffered saline (PBS), and resuspended in fresh PBS at a concentration of 1.5 × 107 CFU/50 μl. Mice were sedated with ketamine and xylazine, and their flanks were shaved and cleansed, following which S. aureus (or PBS control) was injected subcutaneously. In the recurrent SSTI model, secondary infection was performed on the opposite flank 6 to 8 weeks following primary infection. To assess the severity of skin infection, lesions were photographed daily, and the size was measured. To quantify the number of bacteria in the skin lesions, mice were euthanized 7 days after infection, following which the lesions were removed aseptically and homogenized in PBS, and serial dilutions were plated on mannitol salt agar for colony enumeration.
Vaccination
The truncated open reading frames of LukE, SplB, and SspB were polymerase chain reaction–amplified, restriction-digested, and cloned in frame with a His-tag in pET28a (Novagen). The resulting plasmid was expressed in Escherichia coli (DE3, BL21; Invitrogen). The proteins were chromatography-purified from E. coli using the His-Bind Kit (Novagen). The constructs for purification of HlaH35L and LukS-PV were provided by J. B. Wardenburg (Washington University, St. Louis). HlaH35L, cloned in pET24b with a His-tag, was purified from E. coli (BL21), as described (20). LukS-PV, cloned in pGEX with a glutathione S-transferase (GST) tag, was purified from E. coli (BL21), and the GST removed, as described (20). Endotoxin was removed using an endotoxin removal kit (Pierce). Mice were vaccinated with one of three vaccines: HlaH35L, 4S (LukE, LukS-PV, SplB, and SspB), or 5S (4S + HlaH35L). Vaccines were prepared by adding 10 μg of each antigen, adjuvanted with Al(OH)3 (Alhydrogel; Brenntag) at a final concentration of 0.1% in a total volume of 200 μl. Vaccinations were administered subcutaneously 5 and 2 weeks before infection. For the serum adoptive transfer experiments, naïve mice received 150 μl of serum from Al(OH)3, LukE, 4S, or HlaH35L-vaccinated mice via retro-orbital injection 1 day before infection. For the T cell adoptive transfer experiments, T lymphocytes were isolated by negative selection using the Pan T cell Isolation Kit II or the CD8+ T cell Isolation Kit II (Miltenyi Biotec). One day before infection, each recipient mouse received 8 × 106 T cells or PBS in a volume of 200 μl by retro-orbital injection.
Quantification of T lymphocyte responses
To quantify antigen-specific T cell responses, 96-well plates were coated with anti–IL-17 or anti-IFNγ antibody (Becton Dickinson Biosciences), and 5 × 105 splenocytes from infected or vaccinated mice were added to each well. The splenocytes were incubated with purified antigen or peptide (1 μg/ml) or heat-killed S. aureus (5 × 105 CFU per well) for 24 hours at 37°C. Following washing, biotin-labeled detection antibodies were added to the wells, followed by avidin–horseradish peroxidase substrate (eBioscience). Spots were counted following addition of the substrate solution (BD Biosciences) using an Immunospot series 1 analyzer (Cellular Technology).
Quantification of antibody responses
To quantify antigen-specific antibodies in the mouse samples by ELISA, 96-well plates (Costar, Corning Inc.) were coated with the purified antigens (5 μg/ml; purified Hla was purchased from Sigma-Aldrich). Mouse serum was diluted 1:200 in PBS and added to the antigen-containing wells. Detection of antigen-specific IgG was performed using alkaline phosphatase (AP)–conjugated goat anti-mouse IgG (1:5000; AffiniPure, Jackson ImmunoResearch) and AP substrate p-nitrophenyl phosphate (Sigma-Aldrich) following the manufacturer’s recommendations. Absorbance was measured using a GENios spectrophotometer (Tecan). For the human samples, serum was also diluted 1:200, and the samples were processed in the same way as the mouse samples, with the secondary antibody being goat anti-human IgG. To control for plate-to-plate variability in the human studies, a set of standards was included on each plate, and the sample values were corrected on the basis of the values of the standards.
Identification of immunodominant epitopes
A library of 16 AA peptides, each overlapping by 8 AA, spanning the entire sequence of LukE, LukS-PV, Hla, SplB, and SspB was synthesized (A&A Labs, San Diego). ELISpot assays for quantification of IL-17A or IFNγ responses were performed as described above, with the following modifications. Splenocytes from convalescent BALB/c or C57BL/6 mice (8 weeks after infection) were incubated with 1 of 14 peptide pools (12 to 16 peptides each, each peptide at 10 μg/ml), with a response considered positive if the value was greater than double the mean negative control wells and was above the threshold of 10 spot-forming colonies/5 × 105 splenocytes. For each positive pool, splenocytes were incubated with each of the peptides from the pool to determine which of the peptides was immunogenic. The same procedures were used to identify immunogenic epitopes in C57BL/6 mice following immunization with LukE.
MHC purification and binding assays
Purification of class II MHC molecules by affinity chromatography and the performance of assays, based on the inhibition of binding of a high-affinity radiolabeled peptide to quantitatively measure peptide binding, were performed as detailed elsewhere (41). Briefly, the mouse B cell lymphoma LB27.4 was used as a source of MHC molecules. A high-affinity radiolabeled peptide (0.1 to 1 nM; peptide ROIV, sequence YAHAAHAAHAAHAAHAA) was coincubated at room temperature (RT) with purified MHC in the presence of a cocktail of protease inhibitors and an inhibitor peptide. Following a 2-day incubation, MHC-bound radioactivity was determined by capturing MHC/peptide complexes on monoclonal antibody (Y3JP)–coated Lumitrac 600 plates (Greiner Bio-One), and measuring bound counts per minute using the TopCount (Packard Instrument Co.) microscintillation counter. The concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide was calculated. Under the conditions used, where [label] < [MHC] and half-maximal inhibitory concentration (IC50) ≥ [MHC], the measured IC50 values are reasonable approximations of the true Kd (dissociation constant) values (42, 43). Each competitor peptide was tested at six different concentrations covering a 100,000-fold range and in three or more independent experiments. As a positive control, the unlabeled version of the radiolabeled probe was also tested in each experiment.
Quantification of T cell responses by flow cytometry
For the quantification of epitope-specific T cells, PE-conjugated I-Ed LukE296–311 and I-Ab LukE153–168 and LukE185–200 class II pMHC tetramers were synthesized by the NIH Tetramer Core Facility. Lymphocytes were isolated from spleens and draining lymph nodes and processed into single-cell suspensions and then blocked with anti-CD16/32 antibody (clone 2.4G2, Bio X Cell), followed by incubation for 60 min with tetramers at RT. Tetramer enrichment procedures were performed as described previously (23). Briefly, following tetramer staining, tetramer-bound cells were enriched using anti-PE magnetic beads and columns (Miltenyi Biotec). The enriched fractions were collected and used for flow cytometric analysis. Cells were stained for flow cytometry using AquaFluor LiveDead (Life Technologies) solution to exclude dead cells, and a cocktail of dump antibodies was used to exclude unwanted cells: DX5, CD11b (M1/70, BioLegend), F4/80 (BM8), CD19 (1D3), and TER119. Additional antibodies against CD3 (145-2C11), CD4 (RM4-5), CD8 (53-6.7), and CD44 (IM7) were used to stain T cells.
For quantification of TFH cells, draining lymph nodes were collected 2 weeks following a second vaccination with LukE, and tetramer staining was performed as described above. For surface staining, cells were incubated with anti-CD49b (DX5), anti-TER119, anti-CD19, anti-CD45 (30-F11), anti-CD3 (17A2), anti-CD4, anti-CD8, anti-CD44, and anti–PD-1 (29F.1A12). For transcription factor staining, cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for 30 min in the dark at RT following two subsequent washes with stain buffer [PBS and 2% fetal calf serum (FCS)] after surface staining. Cells were then washed twice with Permeabilization Wash Buffer and incubated with anti-Foxp3 (FJK-16s) and anti–Bcl-6 (7D1) overnight in the dark at 4°C. Antibodies were purchased from eBioscience, BioLegend, or Becton Dickinson. Flow cytometry was performed on an LSRII or Fortessa (BD Biosciences) cytometer and analyzed with FlowJo software.
To quantify OVA-specific responses in the context of S. aureus SSTI, splenocytes were prepared as single-cell suspensions from 2W-OVA transgenic C57BL/6 mice and lysed with ACK (ammonium chloride potassium) buffer. The cells were washed twice in PBS and were subcutaneously injected (10 × 106 cells per mouse) into naïve C57BL/6 mice, along with concomitant S. aureus SSTI (or PBS). To quantify 2W-specific T cells, mice were euthanized 8 days later, and lymphocytes from draining lymph nodes (dLNs) and splenocytes were stained with a fixable LIVE/DEAD stain (Aqua, Invitrogen) and blocked with anti-CD16/32 antibody (2.4G2), followed by staining with 2W-specific I-Ab pMHC tetramers PE-2W(EAWGALANWAVDSA) and allophycocyanin (APC)–2W(EAWGALANWAVDSA) for 1 hour at RT (tetramers were obtained from the NIH tetramer facility). Cells were then stained with anti-CD49b (DX5), anti-TER119, anti-CD19 (1D3), anti-CD3 (145-2C11), anti-CD4 (RM4-5), anti-CD8 (53-6.7), and anti-CD44 (IM7). Surface-stained cells were then fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for 30 min at RT, followed by two washes with staining buffer (PBS and 2% FCS). Cells were then washed twice with Permeabilization Wash Buffer and incubated with anti-Foxp3 (FJK-16s) overnight at 4°C. Flow cytometry was performed on a BD LSRII and analyzed with FlowJo software. 2W-specific T cells were identified as PE-2W:I-Ab tetramer and APC-2W:I-Ab tetramer double-positive cells. Tconv cells were identified as Foxp3− cells.
Data analysis
Data were compared using Student’s t test, one-way analysis of variance (ANOVA) with the Tukey posttest, or two-way ANOVA with repeated measures and the Tukey posttest, where appropriate. Differences were considered significant when P < 0.05. All data were analyzed using GraphPad Prism.
Supplementary Material
Acknowledgments
We thank J. B. Wardenburg (Washington University, St. Louis) for the gift of plasmids for purification of LukS-PV and HlaH35L, R. S. Daum (University of Maryland) for guidance, and the NIH Tetramer Core Facility (https://tetramer.yerkes.emory.edu) for the synthesis of class II tetramers. Funding: This study was funded by the National Institute of Allergy and Infectious Diseases (AI076596 and AI125489 to C.P.M. and AI118182 to A.S.C.), the National Institute of Arthritis and Musculoskeletal Diseases (AR059414 to M.-L.A.), and the Immune Epitope Database and Analysis Program, contracts no. HHSN272201200010C and HHSN27220140045C (to A.S.). Author contributions: Y.S., F.Z., P.B., Z.L., D.W., M.-L.A., A.S., A.S.C., and C.P.M. conceived and designed the experiments. Y.S., F.Z., P.B., Z.L., Q.T., D.W., and C.P.M. performed the experiments. Y.S., F.Z., P.B., Z.L., Q.T., D.W., M.-L.A., A.S., A.S.C., and C.P.M. analyzed the data. Y.S., F.Z., D.W., M.-L.A., A.S., A.S.C., and C.P.M. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaw7713/DC1
REFERENCES AND NOTES
- 1.David M. Z., Daum R. S., Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23, 616–687 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herold B. C., Immergluck L. C., Maranan M. C., Lauderdale D. S., Gaskin R. E., Boyle-Vavra S., Leitch C. D., Daum R. S., Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA 279, 593–598 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Miller L. G., Eells S. J., David M. Z., Ortiz N., Taylor A. R., Kumar N., Cruz D., Boyle-Vavra S., Daum R. S., Staphylococcus aureus skin infection recurrences among household members: An examination of host, behavioral, and pathogen-level predictors. Clin. Infect. Dis. 60, 753–763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritz S. A., Tiemann K. M., Hogan P. G., Epplin E. K., Rodriguez M., Al-Zubeidi D. N., Bubeck Wardenburg J., Hunstad D. A., A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin. Infect. Dis. 56, 1554–1561 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar N., David M. Z., Boyle-Vavra S., Sieth J., Daum R. S., High Staphylococcus aureus colonization prevalence among patients with skin and soft tissue infections and controls in an urban emergency department. J. Clin. Microbiol. 53, 810–815 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verkaik N. J., de Vogel C. P., Boelens H. A., Grumann D., Hoogenboezem T., Vink C., Hooijkaas H., Foster T. J., Verbrugh H. A., van Belkum A., van Wamel W. J. B., Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J. Infect. Dis. 199, 625–632 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Kolata J. B., Kühbandner I., Link C., Normann N., Vu C. H., Steil L., Weidenmaier C., Bröker B. M., The fall of a dogma? Unexpected high T-cell memory response to Staphylococcus aureus in humans. J. Infect. Dis. 212, 830–838 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Fowler V. G. Jr., Allen K. B., Moreira E. D., Moustafa M., Isgro F., Boucher H. W., Corey G. R., Carmeli Y., Betts R., Hartzel J. S., Chan I. S. F., McNeely T. B., Kartsonis N. A., Guris D., Onorato M. T., Smugar S. S., DiNubile M. J., Sobanjo-ter Meulen A., Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: A randomized trial. JAMA 309, 1368–1378 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Fattom A., Matalon A., Buerkert J., Taylor K., Damaso S., Boutriau D., Efficacy profile of a bivalent Staphylococcus aureus glycoconjugated vaccine in adults on hemodialysis: Phase III randomized study. Hum. Vaccin. Immunother. 11, 632–641 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pauli N. T., Kim H. K., Falugi F., Huang M., Dulac J., Dunand C. H., Zheng N.-Y., Kaur K., Andrews S. F., Huang Y., DeDent A., Frank K. M., Charnot-Katsikas A., Schneewind O., Wilson P. C., Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J. Exp. Med. 211, 2331–2339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomery C. P., Daniels M., Zhao F., Alegre M.-L., Chong A. S., Daum R. S., Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infect. Immun. 82, 2125–2134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampedro G. R., DeDent A. C., Becker R. E. N., Berube B. J., Gebhardt M. J., Cao H., Bubeck Wardenburg J., Targeting Staphylococcus aureus α-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J. Infect. Dis. 210, 1012–1018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H. K., Kim H.-Y., Schneewind O., Missiakas D., Identifying protective antigens of Staphylococcus aureus, a pathogen that suppresses host immune responses. FASEB J. 25, 3605–3612 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown A. F., Murphy A. G., Lalor S. J., Leech J. M., O'Keeffe K. M., Mac Aogáin M., O'Halloran D. P., Lacey K. A., Tavakol M., Hearnden C. H., Fitzgerald-Hughes D., Humphreys H., Fennell J. P., van Wamel W. J., Foster T. J., Geoghegan J. A., Lavelle E. C., Rogers T. R., McLoughlin R. M., Memory Th1 cells Are protective in invasive Staphylococcus aureus infection. PLOS Pathog. 11, e1005226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez M., Kolar S. L., Müller S., Reyes C. N., Wolf A. J., Ogawa C., Singhania R., De Carvalho D. D., Arditi M., Underhill D. M., Martins G. A., Liu G. Y., O-acetylation of peptidoglycan limits helper T cell priming and permits Staphylococcus aureus reinfection. Cell Host Microbe 22, 543–551.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dillen C. A., Pinsker B. L., Marusina A. I., Merleev A. A., Farber O. N., Liu H., Archer N. K., Lee D. B., Wang Y., Ortines R. V., Lee S. K., Marchitto M. C., Cai S. S., Ashbaugh A. G., May L. S., Holland S. M., Freeman A. F., Miller L. G., Yeaman M. R., Simon S. I., Milner J. D., Maverakis E., Miller L. S., Clonally expanded γδ T cells protect against Staphylococcus aureus skin reinfection. J. Clin. Invest. 128, 1026–1042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao F., Cheng B. L., Boyle-Vavra S., Alegre M.-L., Daum R. S., Chong A. S., Montgomery C. P., Proteomic identification of saeRS-dependent targets critical for protective humoral immunity against Staphylococcus aureus skin infection. Infect. Immun. 83, 3712–3721 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bubeck Wardenburg J., Schneewind O., Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205, 287–294 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy A. D., Bubeck Wardenburg J., Gardner D. J., Long D., Whitney A. R., Braughton K. R., Schneewind O., DeLeo F. R., Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202, 1050–1058 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bubeck Wardenburg J., Bae T., Otto M., Deleo F. R., Schneewind O., Poring over pores: α-hemolysin and panton-valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13, 1405–1406 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Alegre M.-L., Chen L., David M. Z., Bartman C., Boyle-Vavra S., Kumar N., Chong A. S., Daum R. S., Impact of Staphylococcus aureus USA300 colonization and skin infections on systemic immune responses in humans. J. Immunol. 197, 1118–1126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hale J. S., Ahmed R., Memory T follicular helper CD4 T cells. Front. Immunol. 6, 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moon J. J., Chu H. H., Hataye J., Pagán A. J., Pepper M., McLachlan J. B., Zell T., Jenkins M. K., Tracking epitope-specific T cells. Nat. Protoc. 4, 565–581 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rees W., Bender J., Teague T. K., Kedl R. M., Crawford F., Marrack P., Kappler J., An inverse relationship between T cell receptor affinity and antigen dose during CD4+ T cell responses in vivo and in vitro. Proc. Natl. Acad. Sci. U.S.A. 96, 9781–9786 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudensky A. Y., Rath S., Preston-Hurlburt P., Murphy D. B., Janeway C. A. Jr., On the complexity of self. Nature 353, 660–662 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Moon J. J., Chu H. H., Pepper M., McSorley S. J., Jameson S. C., Kedl R. M., Jenkins M. K., Naive CD4+ T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanzavecchia A., Antigen-specific interaction between T and B cells. Nature 314, 537–539 (1985). [DOI] [PubMed] [Google Scholar]
- 28.Gefen T., Vaya J., Khatib S., Rapoport I., Lupo M., Barnea E., Admon A., Heller E. D., Aizenshtein E., Pitcovski J., The effect of haptens on protein-carrier immunogenicity. Immunology 144, 116–126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berzofsky J. A., Immunodominance in T lymphocyte recognition. Immunol. Lett. 18, 83–92 (1988). [DOI] [PubMed] [Google Scholar]
- 30.Rosenthal A. S., Determinant selection and macrophage function in genetic control of the immune response. Immunol. Rev. 40, 136–152 (1978). [DOI] [PubMed] [Google Scholar]
- 31.Sommer S., The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2, 16 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Löffler B., Hussain M., Grundmeier M., Brück M., Holzinger D., Varga G., Roth J., Kahl B. C., Proctor R. A., Peters G., Staphylococcus aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLOS Pathog. 6, e1000715 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spaan A. N., Henry T., van Rooijen W. J. M., Perret M., Badiou C., Aerts P. C., Kemmink J., de Haas C. J. C., van Kessel K. P. M., Vandenesch F., Lina G., van Strijp J. A. G., The staphylococcal toxin panton-valentine leukocidin targets human C5a receptors. Cell Host Microbe 13, 584–594 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Inoshima N., Wang Y., Bubeck Wardenburg J., Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J. Invest. Dermatol. 132, 1513–1516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alonzo F. III, Kozhaya L., Rawlings S. A., Reyes-Robles T., DuMont A. L., Myszka D. G., Landau N. R., Unutmaz D., Torres V. J., CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493, 51–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reyes-Robles T., Alonzo F. III, Kozhaya L., Lacy D. B., Unutmaz D., Torres V. J., Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14, 453–459 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spaan A. N., Vrieling M., Wallet P., Badiou C., Reyes-Robles T., Ohneck E. A., Benito Y., de Haas C. J. C., Day C. J., Jennings M. P., Lina G., Vandenesch F., van Kessel K. P. M., Torres V. J., van Strijp J. A. G., Henry T., The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 5, 5438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DuMont A. L., Yoong P., Day C. J., Alonzo F. III, McDonald W. H., Jennings M. P., Torres V. J., Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. U.S.A. 110, 10794–10799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salgado-Pabón W., Schlievert P. M., Models matter: The search for an effective Staphylococcus aureus vaccine. Nat. Rev. Microbiol. 12, 585–591 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Montgomery C. P., Boyle-Vavra S., Daum R. S., Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLOS ONE 5, e15177 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sidney J., Southwood S., Moore C., Oseroff C., Pinilla C., Grey H. M., Sette A., Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr. Protoc. Immunol. Chapter 18, Unit 18.13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Y., Prusoff W. H., Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973). [DOI] [PubMed] [Google Scholar]
- 43.Gulukota K., Sidney J., Sette A., DeLisi C., Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J. Mol. Biol. 267, 1258–1267 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaw7713/DC1