

Stem cell identity is maintained by diverse Polycomb complexes cooperating to ensure robust silencing of key developmental genes.
Abstract
The transcriptional repressors Polycomb repressive complex 1 (PRC1) and PRC2 are required to maintain cell fate during embryonic development. PRC1 and PRC2 catalyze distinct histone modifications, establishing repressive chromatin at shared targets. How PRC1, which consists of canonical PRC1 (cPRC1) and variant PRC1 (vPRC1) complexes, and PRC2 cooperate to silence genes and support mouse embryonic stem cell (mESC) self-renewal is unclear. Using combinatorial genetic perturbations, we show that independent pathways of cPRC1 and vPRC1 are responsible for maintenance of H2A monoubiquitylation and silencing of shared target genes. Individual loss of PRC2-dependent cPRC1 or PRC2-independent vPRC1 disrupts only one pathway and does not impair mESC self-renewal capacity. However, loss of both pathways leads to mESC differentiation and activation of a subset of lineage-specific genes co-occupied by relatively high levels of PRC1/PRC2. Thus, parallel pathways explain the differential requirements for PRC1 and PRC2 and provide robust silencing of lineage-specific genes.
INTRODUCTION
Embryonic stem cells (ESCs) have the potential to self-renew and differentiate into any cell type of the embryo. Self-renewal and pluripotency are sustained by an autoregulatory network of transcription factors cooperating with diverse chromatin modifiers to establish and maintain ESC-specific gene expression (1). Silencing of lineage-specific genes, critical to prevent both spontaneous differentiation and exit from pluripotency, is thought to involve epigenetic chromatin modifications by Polycomb group (PcG) proteins (2, 3).
In vertebrates, PcG proteins assemble into diverse chromatin-modifying complexes, represented mainly by Polycomb repressive complex 1 (PRC1) and PRC2 (4, 5). PRC2 contains Suz12, Eed, and the histone methyltransferases Ezh1/2, which mono-, di-, and trimethylate lysine 27 of histone H3 (H3K27me1/2/3) (6–9). PRC1 is defined by an E3 ligase activity that catalyzes monoubiquitination of histone H2A at lysine 119 (H2AK119ub1) (10–12). The E3 ligase activity of mammalian PRC1 resides in the Ring1/Rnf2 subunit (also known as Ring1A/Ring1B) when paired with a Pcgf protein (13–15). Ring1/Rnf2 combines with one of six mammalian Pcgf proteins to promote the assembly of biochemically distinct multisubunit PRC1 complexes (16, 17).
PRC1 complexes can be broadly subdivided into canonical and variant complexes (cPRC1 and vPRC1, respectively), based on distinct modes of genomic targeting. cPRC1 localizes at PRC2 targets by binding to H3K27me3 via a chromobox domain–containing (CBX) protein (9, 18, 19). In mouse ESCs (mESCs), Cbx7 is the predominant CBX protein responsible for cPRC1 targeting (16, 20, 21). H3K27me3 binding by CBX proteins prompted an initial model for PcG protein recruitment as stepwise, hierarchical targeting and coordinated gene silencing by PRC2 and cPRC1 (PRC2/cPRC1). cPRC1 complexes can support chromatin compaction and long-range interactions between large Polycomb chromatin domains, which has been proposed to facilitate transcriptional gene repression (22, 23).
vPRC1 incorporates Rybp/Yaf2 in place of CBX proteins (13, 15–17). Hence, variant complexes are recruited to target genes independently of H3K27me3 and PRC2. Sequence-specific recruitment of vPRC1, which at least partly involves recognition of hypomethylated CpG islands (CGIs) (24, 25), has been proposed to subsequently promote PRC2 targeting. PRC2 can recognize H2AK119ub1 (14, 26), leading to a revised pathway in which vPRC1 acts most upstream by signaling for PRC2 targeting via H2AK119ub1 and, by extension, cPRC1 recruitment. Thus, through “writing” and “reading” of histone modifications, PRC1 and PRC2 engage in a feedback mechanism that promotes the formation of a repressive chromatin domain. This model is supported by recent evidence demonstrating that PRC2 recruitment is severely reduced upon complete catalytic inactivation of PRC1 (27, 28). However, there is also mounting evidence for sequence-specific binding of PRC2 subunits (29–31). Hence, a multitude of different, parallel signals may direct PRC1 and PRC2 recruitment in mammals.
PRC1 subunit composition affects not only targeting but also catalytic activity. Pcgf-Ring1 heterodimers associated with vPRC1 have intrinsically higher enzymatic activity in vitro compared to their cPRC1 counterparts (13, 32, 33). In addition, the vPRC1 subunit Rybp enhances monoubiquitination by the catalytic core complex (33). Hence, in vivo vPRC1 complexes are responsible for catalyzing the majority of H2AK119ub1, targeting its deposition at distinct genomic regions (34, 35). In contrast, the contribution of cPRC1 to the pool of H2AK119ub1 remains controversial.
Beyond serving as targeting signal, the relevance of H2AK119ub1 to silencing is unclear since the capacity of PRC1 to condense chromatin structure appears to be independent of its catalytic activity (22, 23, 36). Furthermore, vPRC1 complexes have been implicated in both gene repression and transcriptional activation (34, 35, 37). Together, these observations argue for distinct modes of regulation by vPRC1 and PRC2/cPRC1.
Whereas PRC1 and PRC2 have been shown to be essential in early embryonic development (38–41), their relevance for ESC pluripotency and self-renewal remains controversial (3). Accumulating evidence suggests that PRC2 subunits are required for differentiation in vitro, but loss of Suz12 or Eed does not substantially perturb gene expression and self-renewal capacity in mESCs (42–44). Although EZH1/2 were recently shown to be required for self-renewal of human ESCs (hESCs) (45, 46), they were dispensable for proliferation in hESCs cultured with MEK and GSK3 inhibitors, which maintain a naïve ground state mimicking mESCs (46). In contrast, loss of both PRC1 catalytic core subunits Ring1 and Rnf2 resulted in derepression of lineage-specific genes and spontaneous differentiation, indicating an essential role for maintaining mESC identity (47). This phenotypic discrepancy between PRC1 and PRC2 could reflect distinct roles in stem cell fate maintenance. Alternatively, PRC2-dependent and PRC2-independent PRC1 complexes might play redundant roles in mESC self-renewal. To this extent, loss of PRC2-dependent cPRC1 at shared target genes might be compensated by vPRC1 and vice versa, whereas loss of all PRC1 complexes upon Ring1/Rnf2 loss would eliminate compensation.
To test this hypothesis, we used chemical genetics to evaluate the impact of disrupting PRC2/cPRC1 and vPRC1 on H2AK119ub1 deposition, lineage-specific gene silencing, and maintenance of mESC identity. We show that simultaneous, but not individual, depletion of Rybp (vPRC1) and Eed or Suz12 (both PRC2) diminishes H2AK119ub1 at shared Polycomb target loci and triggers spontaneous differentiation and loss of self-renewal, similar to Ring1/Rnf2 deletion. Specifically, maintenance of repressive chromatin at shared targets involves cooperation between cPRC1 and Pcgf1-containing vPRC1 complexes. Rapid and synchronous PRC1 displacement before differentiation onset reveals that aberrant activation is limited to promoters co-occupied by particularly high levels of cPRC1 and vPRC1. Together, these findings reconcile observations previously interpreted as differential requirements for PRC1 and PRC2 and demonstrate that PRC2/cPRC1 and vPRC1 act redundantly to silence lineage-specific genes and ensure robust maintenance of mESC self-renewal.
RESULTS
vPRC1, cPRC1, and PRC2 target lineage-specific genes
To investigate the roles of PRC2, cPRC1, and vPRC1 in gene repression (Fig. 1A), we analyzed their genomic distribution and histone modifications by chromatin immunoprecipitation sequencing (ChIP-seq). We profiled endogenous Rnf2 (PRC1), Eed (PRC2), and Cbx7 (cPRC1), as well as Rybp, Pcgf1, and Pcgf6 (vPRC1) in mESCs cultivated in serum conditions. We determined PRC1-dependent H2AK119ub1 and PRC2-dependent H3K27me3. In addition, we used available BioCap data to identify unmethylated CGIs (48).
Fig. 1. cPRC1 and vPRC1 partially overlap with PRC2 at silent CGI TSSs in mESCs.
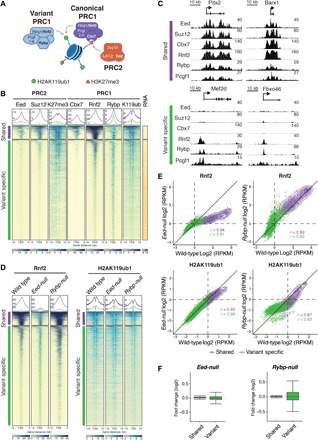
(A) Schematic of core PRC1 and PRC2 complexes and their catalytic activities. Targeting of Cbx7-containing cPRC1 relies on PRC2-mediated H3K27me3. Rybp-containing vPRC1 targeting is H3K27me3 independent. (B) Meta plots and heat maps of Eed, Suz12, H3K27me3 (K27me3), Cbx7, Rnf2, Rybp, and H2AK119ub1 (K119ub) centered around the TSS of CGI promoters (±5 or ±10 kb) show relative enrichment in wild-type mESCs. TSSs are classified by k-means clustering into “shared” (965 TSSs, violet) and “variant-specific” (7333 TSSs, green) and “noBioCap” (19,712 TSSs, not shown). TSSs were grouped by enrichment of cPRC1 (Rnf2, Cbx7), vPRC1 (Rnf2, Rybp), and PRC2 (Eed) subunits, and unmethylated CGIs (BioCap). RNA shows heat map of gene expression measured by QuantSeq. Colored scales (bottom) show dynamic range of ChIP-seq and QuantSeq signals. (C) Genomic screenshots of PcG proteins at representative “shared” and “variant-specific” Polycomb target genes in wild-type mESCs. (D) Meta plots and heat maps compare Rnf2 and H2AK119ub1 enrichments at “shared” and “variant-specific” genes in wild-type, Eed-null, and Rybp-null mESCs. (E) Density scatterplots compare ChIP-seq signals of Rnf2 (±5 kb around TSS) and H2AK119ub1 (±10 kb around TSS) in wild-type and Eed-null mESCs (left) and wild-type and Rybp-null mESCs (right). r, Pearson’s correlation coefficient; “shared” TSSs, violet; “variant-specific” TSSs, green. RPKM, reads per kilobase of transcript per million mapped reads. (F) Box plots show gene expression changes relative to wild type in Eed-null mESCs and Rybp-null mESCs at “shared” (violet) and “variant-specific” (green) genes.
We classified PRC1 and PRC2 target genes via k-means clustering (k = 3) of transcription start sites (TSSs), yielding three distinct sets of genes. Cluster 1 contained 965 inactive CGI TSSs, co-occupied by Cbx7, Rybp, Pcgf1, Pcgf6, Rnf2, and Eed (“shared targets”; Fig. 1, B and C, and fig. S1, A and B). Cluster 2 contained 7333 transcriptionally active CGI TSSs, enriched for Rybp and Rnf2 but not Cbx7 (“variant-specific targets”; Fig. 1, B and C and fig. S1, A and B). Occupancy of PRC2 subunits Eed and Suz12 was also strongly reduced relative to cluster 1. Similar to Rybp, Pcgf1 and Pcgf6 were enriched in cluster 2. Thus, the distribution of Pcgf proteins substantially overlaps in mESCs (fig. S1A), distinct from a human cancer cell line (13) and consistent with another recent report (35). The colocalization of Rybp, Pcgf1, Pcgf6, and Rnf2 at both inactive and active CGI promoters is consistent with diverse vPRC1 complexes contributing to gene repression and activation (34, 35, 37, 49). Last, cluster 3 contained 19,712 TSSs that lacked enrichment of PcG proteins and BioCap signal, supporting the critical role of CGI in recruiting mammalian Polycomb complexes (24, 25) (“no BioCap”; fig. S1A). “Shared target genes” bound by cPRC1, vPRC1, and PRC2 subunits were enriched for key regulators of lineage specification and differentiation (fig. S1, C and D) (50).
PRC2/cPRC1 and vPRC1 silence shared target genes independently
First, we evaluated the role of PRC2 in transcriptional silencing and self-renewal by examining Eed-null haploid mESCs from the Haplobank repository (51). Haploid mESCs are indistinguishable from their diploid counterparts with regard to growth and developmental potential but facilitate reverse genetics via gene-trap insertion (51). Gene-trap disruption is reversible, restoring normal gene expression and allowing the exclusion of unspecific secondary mutations (fig. S1, E to G). Notably, over time, haploid mESCs tend to spontaneously convert into diploid mESCs (52). All analyses were performed on haploid-derived diploid mESCs.
For relative comparison of PcG protein occupancy and histone modifications between wild-type and mutant mESCs, we performed calibrated ChIP-seq experiments. For vPRC1, we limited our analyses to profiling of Rybp and Pcgf1. Calibration was achieved by ligation of barcoded adapters to immunoprecipitated genomic DNA from different genotypes and subsequent pooled multiplex library amplification to avoid skewed results due to differential ChIP enrichment.
Haploid-derived Eed-null mESCs lacked Eed protein and displayed substantially reduced levels of genomic H3K27me3 (fig. S2, A and B), as expected given that Eed is essential for PRC2 assembly and function (44). Cbx7 binding was strongly reduced in Eed-null mESCs, consistent with PRC2-dependent recruitment of cPRC1 (fig. S2, A and B). The genomic distribution of Rnf2 was also substantially reduced upon loss of Eed, suggesting that cPRC1 represents most of the chromatin-bound PRC1 in mESCs, in agreement with previous observations (50) (Fig. 1, D and E, and fig. S2B). In contrast, and consistent with H3K27me3-independent targeting, binding of Rybp and Pcgf1 was unchanged or even increased in mESCs relative to wild type (fig. S2, A and B). Higher abundance of these variant-specific subunits was particularly evident at shared TSSs, suggesting that vPRC1 competes with PRC2/cPRC1 to bind these targets in wild-type mESCs. Moreover, despite the marked loss of the PRC1 catalytic subunit, H2AK119ub1 levels were only marginally reduced at shared target genes in Eed-null mESCs (Fig. 1, D and E, and fig. S2B). These data suggest that vPRC1 targeting is sufficient to maintain this mark in the absence of cPRC1, consistent with other in vitro and in vivo analyses (13, 33, 50). Transcriptome profiling by QuantSeq, which provides quantitative 3′-end mRNA analysis (53), revealed that shared and variant-specific genes were mostly unaffected in Eed-null mESCs relative to wild type (Fig. 1F). Together, these data suggest that PRC2 and, by extension, cPRC1 (PRC2/cPRC1) are not required to maintain lineage-specific gene repression, in agreement with previous reports (44).
Similar Pcgf1 and Pcgf6 distribution could reflect redundant functions of distinct vPRC1 complexes at PcG target genes. Thus, to probe the physiological relevance of vPRC1 and avoid potential compensatory mechanisms, we disrupted all variant complexes by deleting the common subunit Rybp. We used CRISPR-Cas9 to generate Rybp loss-of-function (LOF) mESCs lacking Rybp expression (fig. S1G). ChIP-seq profiling in these mutant cells revealed a marked reduction in Pcgf1 binding at both shared and variant-specific targets, indicating efficient disruption of Pcgf1-containing vPRC1 recruitment upon Rybp loss (fig. S2, A and C).
In addition, H2AK119ub1 was substantially reduced at shared and variant-specific TSSs (Fig. 1, D and E, and fig. S2C), consistent with Rybp stimulating vPRC1-dependent catalytic activity (33). The reduction was less pronounced at shared targets with the highest H2AK119ub1 levels. Eed and Cbx7 protein expression was elevated, and enrichment of H3K27me3 and Cbx7 was increased at shared target genes in Rybp-null mESCs (figs. S1G and S2, A and C), suggesting that PRC2/cPRC1 may compensate for vPRC1 displacement. Alternatively, the Rybp paralog Yaf2 is expressed in Rybp-null mESCs and may partially compensate at these targets (33). Notably, Rose et al. (33) reported that conditional Rybp deletion leads to reduced H3K27me3 genome-wide. To reconcile these contradictory results, we propose that the up-regulation of Eed expression that we observe enhances PRC2 activity at steady state to counteract the erosion of this mark immediately after Rybp depletion.
Transcriptome analyses of Rybp-null mESCs revealed significant up- and down-regulation of variant-specific targets, whereas Polycomb-dependent silencing of shared target genes was unaffected by disruption of vPRC1 (Fig. 1F). Overall, our results suggest that PRC2/cPRC1 and vPRC1 are each sufficient to maintain robust silencing of shared lineage-specific genes and mESC self-renewal.
PRC2/cPRC1 and vPRC1 act redundantly in mESCs
To test whether PRC2/cPRC1 and vPRC1 complexes act redundantly to maintain H2AK119ub1 and silencing at shared target genes, we aimed to establish Rybp LOF in Eed-null mESCs. We found that mESC colonies harboring constitutive Eed and Rybp LOF mutations were not viable so we used the Auxin-based degron system to generate conditional Rybp LOF in Eed-null mESCs. We established two independent Eed-null mESC clones expressing the F-box protein osTir1, and both alleles of endogenous Rybp were fused to an Auxin-inducible degron (AID)–3×FLAG–green fluorescent protein (GFP) tag (dRybp Eed-null) (Fig. 2A).
Fig. 2. Combined depletion of Rybp and Eed or Suz12 results in loss of mESC self-renewal.
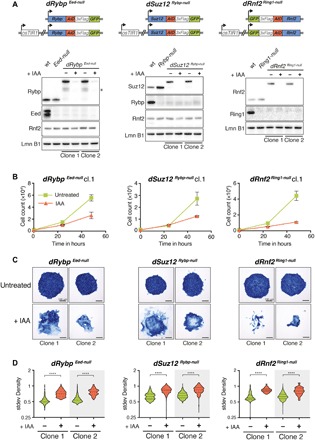
(A) Schematic representation of degron mESC lines. In Eed-null mESCs, AID-3×FLAG-GFP was inserted biallelic at the 3′ end of the endogenous Rybp ORF (left). In Rybp-null mESCs, AID-3×FLAG-GFP was inserted biallelic at the 3′ end of the endogenous Suz12 ORF (middle). In Ring1-null mESCs, AID-3×FLAG-GFP was inserted biallelic at the 5′ end of the endogenous Rnf2 ORF (right). All degron mESC lines constitutively express osTir1. Western blot of PcG proteins and histone modifications in wild-type, Eed-null mESCs, and two independent clones of dRybp Eed-null mESCs (left). Western blot of PcG proteins and histone modifications in wild-type mESCs, Rybp-null mESCs, and two independent clones of dSuz12 Rybp-null mESCs (middle). Western blot of PcG proteins and histone modifications in wild-type mESCs, Ring1-null mESCs, and two independent clones of dRnf2 Ring1-null mESCs (right). IAA treatment (250 μM) for 24 hours leads to degradation of AID-fusion protein. * indicates unspecific signal. Lamin B1 (Lmn B1) serves as loading control. (B) Proliferation assays of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs grown under serum conditions for 72 hours without (untreated) or with 250 μM IAA. Live cells were quantified by flow cytometry every 24 hours. Displayed are the median and SD of four replicate measurements starting at 24 hours IAA (time point, 0 hours). (C) Representative images of alkaline phosphatase (AP) staining of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESC colonies cultured in the absence (untreated) or presence of 250 μM IAA for 5 days. (D) Box plots display SD of density quantifying the degree of mESC colony heterogeneity in response to 5 days of IAA treatment. Statistical significance calculated using unpaired t test (****P < 0.0001).
In addition, we sought to generate the reciprocal combination in Rybp-null mESCs. However, C-terminal modification disrupts Eed function, and N-terminal fusion was hampered by alternative translation start sites (54) (fig. S1, F and G). Instead, we chose to target the other essential PRC2 core subunit by engineering two independent Rybp-null mESC lines with conditional Suz12 LOF (dSuz12 Rybp-null). As positive controls, we also established Ring1-null mESCs with conditional Rnf2 LOF (dRnf2 Ring1-null) to eliminate all PRC1.
All three degron mESC lines expressed their respective AID-3×FLAG-GFP fusion proteins in the absence of Auxin (IAA) (lanes 3 and 5) but not upon IAA treatment (lanes 4 and 6), as expected (Fig. 2A). Moreover, we observed the expected protein interactions and bulk histone modifications in degron mESCs, suggesting that assembly and function of PcG complexes were not affected by protein fusions in the absence of IAA (fig. S3, A and B).
Fluorescence-activated cell sorting (FACS) analysis revealed rapid and uniform reduction of GFP to background levels within 2 hours of IAA treatment (fig. S3C), indicative of synchronous depletion of the AID-3×FLAG-GFP fusion proteins. IAA-treated dRnf2 Ring1-null mESCs displayed strongly diminished cell proliferation compared to untreated controls (Fig. 2B and fig. S3D), consistent with impaired self-renewal upon loss of both Rnf2 and Ring1 (47). IAA treatment also reduced cell proliferation of dRybp Eed-null and dSuz12 Rybp-null mESC lines (Fig. 2B and fig. S3D). In addition, neither dRybp Eed-null, dSuz12 Rybp-null, nor dRnf2 Ring1-null mESCs could be cultured for prolonged periods in the presence of IAA. In contrast, proliferation of wild-type, Ring1-null, Eed-null, and Rybp-null mESCs was unaltered by IAA treatment (fig. S3E). Thus, unlike loss of Eed or Rybp alone, combined disruption of Rybp and Eed or Rybp and Suz12 significantly compromised mESC self-renewal.
To assess whether the observed proliferation defects coincided with spontaneous differentiation and exit from pluripotency, we performed alkaline phosphatase (AP) staining. To quantify the morphological phenotype, we determined the heterogeneity of AP staining across all colonies for each genotype as a standard deviation of optical density (stdev Density) (fig. S4, A and B). IAA treatment of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs yielded sparse, partially fragmented colonies with significantly whiter, unstained cells compared to control conditions, consistent with spontaneous differentiation and the loss of self-renewal capacity (Fig. 2, C and D). By comparison, IAA treatment had no impact on AP staining of Eed-null, Rybp-null, and Ring1-null mESC colonies (fig. S3, F and G).
To further validate that the cellular phenotype stems from combined deregulation of PRC2/cPRC1 and vPRC1, we engineered two additional independent dRybp mESC lines: one with constitutive Suz12 LOF (dRybp Suz12-null) and another one with both Suz12 and Eed LOF (dRybp Suz12-null, Eed-null) (fig. S5, A and B). IAA treatment resulted in significantly reduced proliferation of dRybp Suz12-null and dRybp Suz12-null, Eed-null mESCs and a higher fraction of colonies with partial AP staining (fig. S5, C to F). Thus, PRC2/cPRC1 and vPRC1 are both required to prevent spontaneous differentiation of mESCs.
Unlike serum-grown mESCs, which exhibit varied expression of pluripotency factors, mESC cultivation in defined medium with inhibitors of two kinases (MEK and GSK3), known as “2i” medium, enforces a uniform naïve ground state with little propensity for spontaneous differentiation (55). To evaluate the requirement of PRC2/cPRC1 and vPRC1 for self-renewal in a homogeneous population of naïve mESCs, we examined cell proliferation of dRybp Eed-null and dSuz12 Rybp-null mESCs in 2i conditions. Upon IAA treatment, all degron mESC lines grown in defined 2i medium showed severe proliferation defects similar to those grown under serum conditions (fig. S5, G and H). IAA-treated cells detached from tissue culture plates, thus preventing the quantification of differentiation levels by AP staining. Together, these data suggest that PRC2/cPRC1 and vPRC1 act redundantly to promote self-renewal and maintain mESC identity.
Loss of mESC self-renewal is linked to reduced PRC1-mediated H2AK119ub1 at shared Polycomb target genes
To investigate whether aberrant mESC self-renewal upon loss of both PRC2/cPRC1 and vPRC1 is related to changes in PRC1 occupancy and H2AK119ub1, we performed calibrated ChIP-seq. Degradation of Rnf2 upon IAA treatment of dRnf2 Ring1-null mESCs resulted in a marked loss of chromatin-bound Rnf2 at Polycomb target TSSs, as expected, concomitant with a significant reduction of H2AK119ub1 (Fig. 3, A to D, and fig. S6, A and B). Similarly, IAA treatment of dRybp Eed-null mESCs led to diminished H2AK119ub1 at shared and variant-specific Polycomb target TSSs, consistent with the proposed broad role of vPRC1 in depositing this repressive histone modification (33, 34) (Fig. 3, A to D, and fig. S6, A and B). Moreover, in the absence of PRC2-dependent cPRC1, Rybp degradation further reduced Rnf2 binding at target genes, suggesting displacement of vPRC1 from chromatin in dRybp Eed-null mESCs. IAA-induced degradation of Suz12 in dSuz12 Rybp-null mESCs also decreased Rnf2 binding, in agreement with efficient cPRC1 displacement (Fig. 3, A to D, and fig. S6, A and B). Notably, loss of cPRC1 coincided with substantially reduced H2AK119ub1, particularly at shared target genes, arguing that cPRC1 is critical for the maintenance of this repressive histone mark in the absence of vPRC1. Thus, in contrast to single LOF mutations in Rybp or Eed/Suz12, combined depletion severely reduces total PRC1 targeting and activity similar to Ring1/Rnf2 (Fig. 3A and fig. S6C). While vPRC1 complexes shape H2AK119ub1 broadly throughout the genome (34), deposition of this mark at shared TSSs depends on the catalytic activities of both vPRC1 and cPRC1 complexes. Together, these results suggest that cPRC1 and vPRC1 cooperate to maintain repression of shared target genes and support the link between the cellular phenotype and loss of PRC1-dependent chromatin modifications.
Fig. 3. cPRC1 and vPRC1 maintain H2AK119ub1 at “shared” Polycomb target genes.
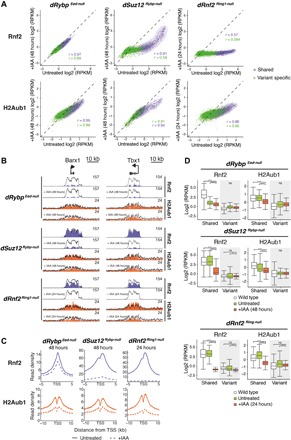
(A) Density scatterplots compare ChIP-seq signals of Rnf2 (top, ±5 kb around TSS) and H2AK119ub1 (H2Aub1) (bottom, ±10 kb around TSS) in untreated and IAA-treated dRybp Eed-null (48 hours, left), dSuz12 Rybp-null (48 hours, middle), and dRnf2 Ring1-null (24 hours, right) mESCs. r, Pearson’s correlation coefficient; “shared” TSS signal, violet; “variant-specific” TSS signal, green. (B) Genomic screenshots of Rnf2 (violet) and H2AK119ub1 (H2Aub1, orange) enrichments at two representative “shared” Polycomb target genes in untreated and IAA-treated dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs. Superimposed are levels in wild-type mESCs (black line). (C) Meta plots of Rnf2 (top) and H2AK119ub1 (bottom) enrichments at “shared” Polycomb target genes in untreated and IAA-treated dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs as indicated. (D) Box plots compare ChIP-seq signal of Rnf2 (left, ±5 kb around TSS) and H2AK119ub1 (H2Aub1) (right, ±10 kb around TSS) at “shared” and “variant-specific” Polycomb target genes in wild-type mESCs (white), untreated (green), and IAA-treated (red) dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs. Significance (P value) was calculated using Wilcoxon rank test (****P < 2.2 × 10−16; ns, not significant).
PRC2/cPRC1 and vPRC1 cooperate to maintain silencing of lineage-specific genes
Our data suggest that PRC2/cPRC1 and vPRC1 act redundantly in mESCs to repress chromatin at lineage-specific developmental genes and support self-renewal. Disruption of PRC1 via codepletion of Ring1 and Rnf2 causes derepression of Polycomb target genes and down-regulation of pluripotent stem cell markers (47); therefore, we expected similar gene expression changes in dRybp Eed-null and dSuz12 Rybp-null mESCs treated with IAA. To test this hypothesis, we quantified mRNA expression by QuantSeq, which requires 10 times fewer reads than conventional mRNA profiling methods and thus facilitated a direct comparison of multiple conditions in the same sequencing experiment.
Consistent with the observed phenotypes, complete loss of PRC1 activities in dRnf2 Ring1-null mESCs resulted in marked changes in gene expression (Padj < 0.1; Fig. 4A): 2613 genes were significantly deregulated 24 hours after IAA treatment, and most remained deregulated within the 4581 genes that were altered 48 hours after treatment. More genes were up-regulated than down-regulated upon loss of PRC1, in line with its critical role in maintenance of gene silencing.
Fig. 4. Combined cPRC1 and vPRC1 disruption triggers activation of lineage-specific genes.
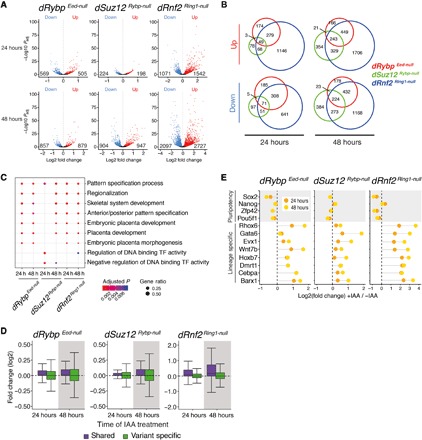
(A) Volcano plots show gene expression changes of IAA-treated relative to untreated dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs at 24 hours (top) and 48 hours (bottom). Down (blue), number of repressed genes; up (red), number of up-regulated genes. Differential gene expression (Padj < 0.1) was calculated by taking into account two independent clones per genotype, each sequenced in triplicate. (B) Venn diagrams show overlap of differentially expressed genes in dRybp Eed-null (red circles), dSuz12 Rybp-null (green circles), and dRnf2 Ring1-null (blue circles) mESCs at 24 and 48 hours of IAA treatment. (C) Analysis of Gene Ontology (GO) categories of up-regulated genes in response to IAA treatment of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs at 24 and 48 hours. The color indicates the significance, and the size represents the fraction of genes in each category. (D) Box plots of gene expression changes at “shared” (violet) and “variant-specific” (green) Polycomb target genes in dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs after 24 and 48 hours of IAA addition. (E) Dot charts display dynamic expression changes after 24 and 48 hours of IAA treatment at selected mESC pluripotency markers (gray) or lineage-specific PcG target genes.
Despite comparable cellular phenotypes, fewer gene expression changes were observed upon IAA treatment of dRybp Eed-null and dSuz12 Rybp-null mESCs (Fig. 4A). Relative to untreated controls, vPRC1 disruption in dRybp Eed-null mESCs led to the deregulation of 1074 genes after 24 hours and 1736 genes after 48 hours, while Suz12 degradation in dSuz12 Rybp-null mESCs led to the deregulation of 422 genes after 24 hours and 1851 genes after 48 hours. Nevertheless, the genes that were altered in dRybp Eed-null and dSuz12 Rybp-null mESCs significantly correlated and overlapped with those altered upon loss of Rnf2 in dRnf2 Ring1-null mESCs (Fig. 4B and fig. S7A). Thus, the transcriptome changes due to sequential displacement of cPRC1 and vPRC1 showed a high degree of concordance with those arising from complete loss of PRC1.
Gene Ontology (GO) analyses revealed that the genes up-regulated upon IAA treatment of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs were enriched for transcription factors linked to developmental processes, including pattern specification, embryonic organ development, and skeletal systems development (Fig. 4C). Shared Polycomb target genes were preferentially up-regulated in response to IAA treatment, consistent with the aberrant activation of lineage-specific transcriptional regulators (Fig. 4D). In contrast, variant-specific genes displayed both up-regulation and down-regulation (Fig. 4D). GO term analysis revealed that down-regulated genes were enriched for genes associated with RNA processing and metabolism, consistent with the observed proliferation defects (fig. S7B). In addition, unlike lineage-specific regulators, pluripotency transcription factors including Pou5f1 (Oct4), Sox2, and Zfp42 (Rex1) were down-regulated in dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs upon IAA treatment (Fig. 4E), consistent with the observed exit from pluripotency. Similar changes in gene expression were detected upon IAA treatment of degron mESCs cultured in 2i conditions (fig. S7, C and D).
The observed gene expression changes are in agreement with the loss of self-renewal and the spontaneous differentiation of IAA-treated dRybp Eed-null mESCs and dSuz12 Rybp-null mESCs, resembling IAA-treated dRnf2 Ring1-null mESCs. We conclude that the disruption of both vPRC1 and PRC2/cPRC1 in mESCs leads to loss of PRC1-mediated H2AK119ub1 and aberrant activation of genes encoding key developmental regulators. vPRC1 and PRC2/cPRC1 act redundantly to maintain repression of these target genes, providing robust control to safeguard mESC self-renewal and pluripotency.
PRC1/2 enrichment levels and chromatin marks predict Polycomb-dependent repression
Next, we examined how the gene expression changes were related to PRC1 occupancy and histone modifications. On average, genes that were up-regulated upon IAA treatment showed reductions in both Rnf2 occupancy and H2AK119ub1 (Fig. 5, A and B). However, many genes without transcriptional changes (“unchanged”) also showed diminished Rnf2 and H2AK119ub1 levels in IAA-treated cells (Fig. 5, A and B). Only a small fraction of Polycomb target genes were derepressed upon 48 hours of IAA treatment of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs, revealing a distinct requirement for PRC1 activity.
Fig. 5. Polycomb-repressed genes are marked high PRC2, cPRC1, and vPRC1 occupancy.
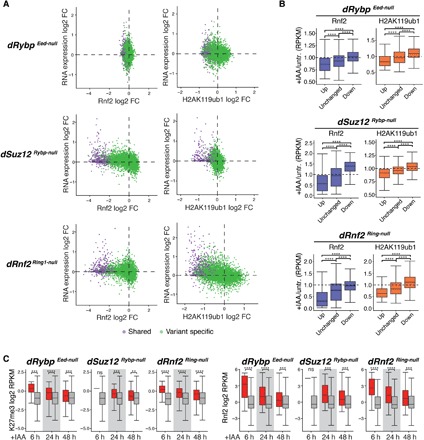
(A) Scatterplots show changes in Rnf2 (top, ±5 kb around TSS) and in H2AK119ub1 (bottom, ±10 kb around TSS) signals relative to gene expression changes in IAA-treated dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs. “Shared” genes, violet; “variant-specific” genes, green. (B) Box plots show changes in Rnf2 (±5 kb around TSS) and H2AK119ub1 (±10 kb around TSS) signals of up-regulated (up), unchanged, and down-regulated (down) genes after 48 hours of IAA treatment of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs. Significance (P value) calculated using Wilcoxon test (****P < 2.2 × 10−16). (C) Box plots display wild-type levels of H3K27me3 (±10 kb around TSS) and Rnf2 (±5 kb around TSS) of genes up-regulated (red) or unchanged (gray) at 6, 24, and 48 hours of IAA treatment of dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs. Significance (P value) calculated using Wilcoxon test (**P < 0.01; ***P < 0.001; ****P < 1 × 10−4; ns, not significant).
First, to distinguish direct changes due to loss of PRC1 activity from indirect changes due to differentiation, we analyzed gene expression within 6 hours of IAA treatment. In dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs, we observed altered expression of 63, 1, and 555 genes, respectively (fig. S8A). Among the up-regulated genes, we noticed a clear bias for shared Polycomb-bound TSSs at the early time points compared to 48 hours of IAA treatment (fig. S8B). While shared genes account for only 12% of all PcG target genes, they were significantly overrepresented in the fraction of up-regulated genes at 6 and 24 hours of IAA treatment in dRybp Eed-null, dSuz12 Rybp-null, and dRnf2 Ring1-null mESCs, suggesting that shared genes are particularly and directly vulnerable to loss of PRC1 activity (fig. S8B).
Next, we sought to determine whether the subset of shared Polycomb target genes that require PRC1 activity to maintain silencing exhibits any unifying features in wild-type mESCs, which may predict their derepression upon PRC1 loss. CGI promoters of shared Polycomb target genes are also bound by Trithorax group (TrxG) proteins, which mediate active chromatin modifications, including H3K4me3. Consequently, colocalization of PcG proteins and TrxG proteins is associated with “bivalent” nucleosomes bearing H3K27me3 and H3K4me3 (56). Bivalent chromatin modifications have been proposed to confer a “poised” promoter state at lineage-specific genes, which is repressed but favors rapid activation in response developmental cues (56). We hypothesized that shared target genes with elevated H3K4me3 would require PRC1 to maintain silencing; however, H3K4me3 enrichment was unrelated or correlated negatively to PRC1 dependence (fig. S8C).
Next, we hypothesized that rapid aberrant activation of shared PcG target genes due to PRC1 loss might be associated with relatively low levels of repressive histone modifications and PcG protein occupancy. Unexpectedly, H3K27me3 and H2AK119ub1 were higher at up-regulated shared genes compared to unchanged shared genes at 6 and 24 hours of IAA treatment (Fig. 5C and fig. S8D). Similarly, PRC1 (Rnf2) and PRC2 (Suz12) levels were also significantly higher at shared TSSs that require PRC1 to maintain silencing at these early time points (Fig. 5C and fig. S8D). Elevated levels of total PRC1 reflect higher Cbx7 and Rybp occupancy, in agreement with observations that both cPRC1 and vPRC1 are required for robust transcriptional repression (fig. S8D).
Together, we conclude that only a subset of silent lineage-specific genes relies on PRC1 for transcriptional repression in mESCs. These PRC1-dependent TSSs are controlled by cPRC1/PRC2 and vPRC1, which establish large domains of repressive Polycomb modifications.
Pcgf1-containing PRC1 and cPRC1 are required to maintain mESC self-renewal
Last, we sought to identify which vPRC1 complex cooperates with cPRC1 to maintain H2AK119ub1 at shared Polycomb TSSs. Pcgf1, Pcgf3, Pcgf5, and Pcgf6 nucleate highly heterogeneous vPRC1 complexes, display distinct modes genomic targeting, and have variable contributions to genomic H2AK119ub1 in vivo (34, 35). Pcgf1-containing PRC1 is broadly responsible for H2AK119ub1 at most Polycomb-bound TSSs (34), and its genomic localization substantially overlaps with Eed, Cbx7, and Rybp at shared TSSs (fig. S1A) (35). Hence, we decided to focus our investigation on the interaction between cPRC1/PRC2 and Pcgf1-containing vPRC1. First, we engineered dSuz12 mESCs with IAA-dependent expression of endogenous Suz12 fused to AID-3×FLAG-GFP (Fig. 6A and fig. S9A, lanes 1 and 2). Next, we used CRISPR-Cas9 gene editing in dSuz12 mESCs to obtain two independent clones with Pcgf1 LOF (dSuz12 Pcgf1-null) (Fig. 6A and fig. S9A, lanes 3 to 6). Notably, compared to untreated controls, IAA addition to dSuz12 Pcgf1-null mESCs severely impaired cell proliferation and impeded prolonged culturing (Fig. 6B and fig. S9B). In contrast, proliferation was not significantly affected in dSuz12 mESCs, which could be long-term cultured in the presence of IAA (fig. S9B). The cellular phenotype in dSuz12 Pcgf1-null mESCs closely resembled the effects observed in IAA-treated dRybp Eed-null and dSuz12 Rybp-null mESCs, suggesting defects in self-renewal capacity. AP staining of IAA-treated colonies of dSuz12 Pcgf1-null mESCs, but not dSuz12 mESCs, revealed sparse, partially fragmented colonies with whiter, unstained cells compared to control conditions, consistent with spontaneous differentiation and the loss of self-renewal capacity (Fig. 6C and fig. S9, C and D). Last, we interrogated the impact of cPRC1 displacement on H2AK119ub1 and expression of a panel of shared Polycomb target genes in dSuz12 Pcgf1-null mESCs. In agreement with functional redundancy between cPRC1 and Pcgf1-containing vPRC1, Suz12-dependent displacement of Rnf2 led to H2AK119ub1 reduction in dSuz12 Pcgf1-null mESCs but not in dSuz12 mESCs (Fig. 6, D and E, and fig. S9, E to H). Notably, the drop in H2AK119ub1 at shared Polycomb-bound TSSs in dSuz12 Pcgf1-null mESCs was less pronounced after 48 hours of IAA treatment compared to dSuz12 Rybp-null mESCs (Fig. 3A). This suggests that PRC2/cPRC1 might cooperate with multiple vPRC1 complexes to regulate H2AK119ub1 at shared targets. Nevertheless, combined depletion of Suz12 and Pcgf1 was sufficient to abrogate repression resulting in strong transcriptional up-regulation of repressed lineage-specific genes in dSuz12 Pcgf1-null mESCs (Fig. 6F). Given the substantial genomic overlap at shared Polycomb-bound TSSs, we conclude that Pcgf1-containing vPRC1 and cPRC1 cooperate to maintain H2AK119ub1 and transcriptional repression at TSSs of shared Polycomb target genes.
Fig. 6. Combined Pcgf1 and Suz12 depletion reduces H2AK119ub1, triggers activation of lineage-specific genes and results in loss of mESC self-renewal.
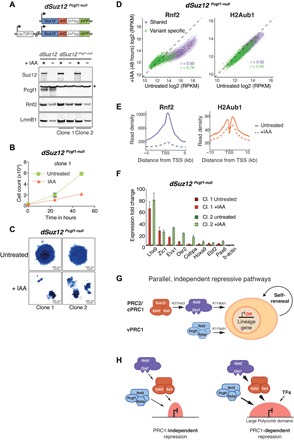
(A) Schematic representation of dSuz12 Pcgf1-null mESCs. Pcgf1-null mutation in mESCs expressing osTIR1 and endogenous Suz12 fused to AID-3×FLAG-GFP. Western blot of dSuz12 mESCs and two independent clones of dSuz12 Pcgf1-null mESCs show IAA-dependent fusion protein degradation and confirm Pcgf1 LOF mutation. * indicates unspecific band. Lamin B1 (Lmn B1) serves as loading control. (B) Proliferation assays of two independent clones of dSuz12 Pcgf1-null mESCs grown under serum conditions for 72 hours without (untreated) or with 250 μM IAA. Live cells were quantified by flow cytometry every 24 hours. Displayed is the median and SD of four replicate measurements starting at 24 hours IAA (time point, 0 hours). (C) Representative image of AP staining of dSuz12 Pcgf1-null mESC colonies cultured in the absence (untreated) or presence or 250 μM IAA for 5 days (+IAA). (D) Density scatterplots compare ChIP-seq signals of Rnf2 (left, ±5 kb around TSS) and H2AK119ub1 (H2Aub1) (right, ±10 kb around TSS) in dSuz12 Pcgf1-null mESCs without and with IAA treatment for 48 hours. r, Pearson’s correlation coefficient; “shared” TSS signal, violet; “variant-specific” TSS signal, green. (E) Meta plots of Rnf2 (left) and H2AK119ub1 (right) enrichments at “shared” Polycomb target genes in untreated and IAA-treated dSuz12 Pcgf1-null mESCs for 48 hours. (F) Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis compares expression of panel of “shared” Polycomb target genes in two independent clones of dSuz12 Pcgf1-null mESCs without and with IAA treatment for 48 hours. Gene expression is normalized to GAPDH and is shown relative to the average expression in untreated cells for each individual cell line. Error bars show SD (n = 2). (G) Cartoon illustrates model of parallel, independent mechanisms of PRC2/cPRC1 and vPRC1 targeting to ensure robust repression of lineage-specific genes and safeguard against exit from mESC pluripotency and spontaneous differentiation. (H) Cartoon displays shared PcG target genes embedded in small (left) or large (right) Polycomb repressive chromatin domains (red). PRC1 is critical for repression of PcG target genes located within large Polycomb domains marked by relatively high levels of PRC2/cPRC1 and vPRC1, as well as repressive histone modifications. Silencing of TSSs with smaller Polycomb domains is PRC1 independent and may be buffered by additional repressive chromatin modifications or the lack of activating transcription factors (TFs).
DISCUSSION
Our work reveals that, in mESCs, PRC2/cPRC1 and vPRC1 act in parallel, redundant pathways. At least one pathway must be engaged to suppress transcription at a subset of lineage-specific promoters that would otherwise trigger spontaneous differentiation (Fig. 6G). We show that in mESCs lacking Eed/Suz12 (PRC2/cPRC1) and Rybp (vPRC1), transcriptional derepression and exit from pluripotency closely resemble the consequences of Ring1/Rnf2 double depletion (total PRC1), arguing that cPRC1 and vPRC1 separately orchestrate PRC1 activity at co-occupied target genes to facilitate silencing. At the same time, our data do not exclude a cPRC1-independent contribution of PRC2 to gene silencing. In contrast and consistent with previous reports, individual disruption of vPRC1, or precluding cPRC1 targeting via loss of H3K27me3, did not affect normal proliferation and gene expression (33, 44), suggesting that one PRC1 complex family can compensate for the lack of the other.
A study by Morey et al. (50) used single and double knockdowns of Cbx7 and Rybp to distinguish cPRC1 and vPRC1 functions in mESCs. Consistent with our results, the authors reported synthetic reduction of H2AK119ub1 and derepression of selective target genes upon double depletion. However, they did not observe loss of mESC self-renewal. We speculate that phenotypic differences might arise from limited knockdown efficiency or from redundancy by alternative canonical complexes including Cbx2-containing cPRC1. Another study reported that unlike upon codepletion of Eed and Rybp, mESCs lacking Eed together with Rnf2 continue to proliferate and maintain self-renewal capacity (57). Rnf2 and its closely related homolog Ring1 are shared among all PRC1 complexes (16, 17). Since up-regulation of Ring1 can compensate for Rnf2 loss (11), vPRC1 function is likely preserved in these cells.
In an effort to identify which vPRC1 complexes synergize with cPRC1, we codepleted Pcgf1 together with Suz12. vPRC1 complexes harboring Pcgf1 are broadly responsible for deposition of H2AK119ub1 at Polycomb-bound TSSs (34, 35), and their genomic distribution overlaps with PRC2/cPRC1 at shared Polycomb target TSSs (fig. S1A) (35). Consistent with redundant functions at shared targets, induced Suz12 degradation in dSuz12 Pcgf1-null mESCs reduced H2AK119ub1, which led to transcriptional up-regulation and triggered loss of self-renewal. Since similar molecular and cellular effects were observed upon IAA treatment of dSuz12 Rybp-null mESCs, we conclude that Pcgf1-containing vPRC1 cooperates with cPRC1 to maintain H2AK119ub1 at shared Polycomb target genes. In addition, the synthetic lethality of Pcgf1 with Suz12 excludes the possibility that PRC1-independent functions of Rybp account for the loss of self-renewal observed in IAA-treated dSuz12 Rybp-null and dRybp Eed-null mESCs (58).
While this manuscript was under review, two independent studies reported the consequences of combinatorial deletions of Pcgf genes on Polycomb silencing (34, 35). To investigate the contributions of cPRC1 and vPRC1 complexes to H2AK119ub1 and gene silencing in mESCs, the two groups created LOF mutations in cPRC1-associated Pcgf2/Pcgf4 alone or in combination with deletions of vPRC1-associated Pcgf genes. In agreement with our findings of functional redundancy between cPRC1 and Pcgf1-containing vPRC1 complexes, Scelfo et al. (35) reported that Pcgf1/Pcgf2/Pcgf4 are required for maintenance of H2AK119ub1 at common targets. To determine the contribution of cPRC1, Fursova et al. (34) compared conditional Pcgf1/Pcgf3/Pcgf5 triple knockout (TKO) mESCs with conditional quadruple deletion of Pcgf1/Pcgf3/Pcgf5/Pcgf2. H2AK119ub1 levels were strongly reduced in Pcgf1/3/5 TKO mESCs, and no further reduction was observed upon additional Pcgf2 deletion. While the authors conclude that this suggests minimal contribution of cPRC1, it may be difficult to discern any further reduction from the existing low H2AK119ub1 levels in Pcgf1/3/5 TKO mESCs. Neither study reported spontaneous differentiation and loss of mESC self-renewal capacity in response to codepletion of cPRC1- and vPRC1-associated Pcgf proteins. Notably, unlike in mESCs with complete loss of PRC2-dependent H3K27me3, Cbx7 targeting was partially maintained in Pcgf2/Pcgf4 LOF mESCs (34), suggesting that vPRC1-associated Pcgf proteins can contribute to cPRC1 assembly. Hence, cPRC1 function may be partly preserved upon Pcgf2/Pcgf4 deletion, accounting for the differences between the studies.
Our results reconcile previous observations leading to the interpretation that PRC1, but not PRC2, is required for mESC self-renewal (3). We demonstrate that PRC2 is critical for directing cPRC1 activity to maintain repression of lineage-specific target genes when vPRC1 function is compromised. Thus, PRC2/cPRC1 and vPRC1 coordinate redundant mechanisms that ensure robust repression of key lineage-specific genes not only for differentiation but also for maintaining mESC identity (Fig. 6G).
Recent studies demonstrated that PRC2 is required for maintenance of self-renewal and pluripotency in primed hESCs (45, 46). However, this phenotype was rescued upon addition of 2i, converting PRC2 mutant hESCs into a naïve state that resembles the developmental stage of mESCs (46). We predict that similar to its role in mESCs, vPRC1 can compensate for the lack of PRC2/cPRC1 in naïve hESCs. Conversely, redundant control of lineage-specific gene repression by cPRC1 and vPRC1 may be lost or weakened in primed mESCs and hESCs.
Even though approximately 1000 silent genes have high occupancy of PRC2/cPRC1 and vPRC1, only a fraction of these were derepressed upon cPRC1 and vPRC1 displacement in IAA-treated dRybp Eed-null and dSuz12 Rybp-null mESCs or loss of total PRC1 activity in IAA-treated dRnf2 Ring1-null mESCs. Unexpectedly, aberrantly up-regulated shared genes are embedded in large Polycomb domains with particularly high levels of PRC1 and PRC2 occupancy and histone modifications in mESCs (Fig. 6H). While some PcG target promoters may lack activating transcription factors in mESCs, our results argue that transcription of other target genes is repressed by Polycomb-dependent mechanisms. Large Polycomb domains may exert their repressive activity by reducing nucleosome mobility and displacement, thereby hampering accessibility for transcription factors or impeding productive elongation by RNA polymerase II. In another vein, PRC1-independent silencing of shared genes may involve additional chromatin modifications that maintain repression in the absence of PRC1 and PRC2. Binding of other chromatin modifiers to shared PcG target loci might even contribute to limiting the formation of larger repressive Polycomb chromatin domains.
In mammals, cPRC1 and vPRC1 complexes have radiated into diverse PcG protein assemblies with distinct modes of targeting and mechanisms of chromatin regulation (4, 5, 59). Localization of cPRC1 complexes to PRC2 target loci is predominantly mediated by binding to H3K27me3-modified nucleosomes (fig. S2A) (21, 50). In contrast, PRC2-independent targeting of vPRC1 to silent and active TSSs relies mostly on sequence specificity for CGIs (24, 25). Nevertheless, PRC2/cPRC1 and vPRC1 binding patterns also show substantial overlap at silent lineage-specific genes (33, 50) (this study). In extension, genomic profiling of Pcgf proteins revealed significant colocalization at shared and variant-specific target genes arguing for redundancy even among different vPRC1 complexes (35). Independent parallel modes of PRC1 targeting may provide fail-safe repression of key lineage-specific genes to maintain mESC pluripotency and self-renewal (Fig. 6G). In addition, distinct noncatalytic and catalytic activities of cPRC1 and vPRC1 might further promote robustness of repression in the face of developmental transitions. To this extent, we have recently shown that ectopic recruitment of cPRC1 or vPRC1 can initiate transcriptional repression, yet only cPRC1 can engage in a feedback mechanism with PRC2 to promote long-term epigenetic silencing (60). Hence, sequence-specific binding of vPRC1 might facilitate formation of repressive chromatin, which is subsequently propagated through replication and cell division by cPRC1.
MATERIALS AND METHODS
Cell culture conditions of mESCs
All mESCs used in this study were derived from haploid HMSc2 termed AN3-12 (51). mESCs grown under serum conditions were cultivated feeder free in base medium composed of high-glucose Dulbecco’s Modified Eagle Medium supplemented with 13.5% fetal bovine serum (FBS; Sigma), 10 mM Hepes (pH 7.4), 2 mM GlutaMAX (Gibco), 1 mM sodium pyruvate (Sigma), penicillin (100 U/ml) (Sigma), streptomycin (0.1 mg/ml) (Sigma), 1× nonessential amino acids (Sigma), 50 mM β-mercaptoethanol (Gibco) supplemented with 10% FBS (Sigma), and recombinant leukemia inhibitory factor (LIF; produced in-house). mESCs grown in 2i conditions were cultivated feeder free in base medium supplemented with 1× N2 Supplement (Thermo Fisher Scientific), 1× B-27 Supplement minus Vitamin A (Thermo Fisher Scientific), GSK3i (CHIR-99021), MEKi (PD0325901), and recombinant LIF.
Handling of haploid mESCs
Haploid mESCs spontaneously turn diploid in culture. We used FACS to isolate haploid mESCs facilitating homozygous genetic manipulation. Genomic DNA was stained for FACS with Hoechst 33342 (20 μg/ml; Thermo Fisher Scientific) for 30 min at 37°C and 5% CO2. For all biochemical and molecular assays, diploid mESCs were used.
Gene-trap cell lines
Eed-null mESCs, generated by gene-trap insertion, were obtained from the Haplobank repository (51). Briefly, translation of full-length Eed was terminated by integration of a retrovirus-based enhanced gene-trap cassette between exons 2 and 3.
Generation of AID-tagged mESCs
All genetic manipulations were performed on haploid mESCs to ensure biallelic genetic modification. Ring1-null, Rybp-null, and Pcgf1-null mESCs were generated with CRISPR-Cas9 gene editing (table S2), and LOF was confirmed by western blotting. Next, wild-type and mutant LOF mESCs were transduced with F-box protein osTir1, and a clone with intermediate osTir1 was isolated from each genotype for further genetic manipulation. Using CRISPR-Cas9–mediated homology-directed repair in Eed-null mESCs, we modified the endogenous Rybp gene by introducing a C-terminal fusion with AID followed by 3×FLAG and a GFP CDS (dRybp Eed-null). Eed expression was restored in these cells by transduction with FlpO recombinase to reverse the disruptive orientation of the gene-trap cassette and obtain dRybp mESCs. In dRybp and dRybp Eed-null mESCs, we used CRISPR gene editing of Suz12 to obtain dRybp Suz12-null and dRybp Suz12-null, Eed-null, respectively. In wild-type, Pcgf1-null, and Rybp-null mESCs, we genetically modified endogenous Suz12 to generate a C-terminal fusion with GFP-3xFLAG-AID (dSuz12, dSuz12 Rybp-null, and dSuz12 Pcgf1-null). Ring1-null mESCs were modified by CRISPR-Cas9–mediated homology-directed repair to generate an N-terminal fusion of GFP-3×FLAG-AID with endogenous Rnf2 (dRnf2 Ring1-null). Two independent clones were isolated per genotype.
Western blot
Nuclear extract from 10 × 106 mESCs was obtained by lysis in Buffer A [final: 25 mM Hepes (pH 7.6), 5 mM MgCl2, 25 mM KCl, 0.05 mM EDTA, 10% glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 1× cOmplete Mini protease inhibitor] followed by collection in radioimmunoprecipitation assay buffer [final: 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate (DOC), 0.1% SDS, and 50 mM tris (pH 8.0)]. Nuclear extracts were homogenized in a Diagenode Bioruptor, and concentration was determined by Bradford assay (Bio-Rad). Total protein (20 μg/lane) was run on Novex Life Technology NuPAGE 4 to 12% bis-tris gels in the Invitrogen NuPAGE MES SDS Running Buffer and transferred on a Merck Chemicals Immobilon-P membrane (polyvinylidene difluoride, 45 μm). The membrane was blocked [5% nonfat dry milk in 1× phosphate-buffered saline (PBS) and 0.1% Tween 20] and incubated with primary antibodies. Last, the membrane was incubated with corresponding secondary horseradish peroxidase–coupled antibodies (5% nonfat dry milk in 1× PBS and 0.1% Tween 20), developed using Clarity Western ECL Substrate (Bio-Rad) and imaged by a ChemiDoc XRS+ Imaging system (Bio-Rad).
AP staining
One thousand cells were grown on gelatin-coated glass coverslips for 5 days to form colonies at low density. The cells were seeded in regular mESC medium and allowed to adhere to the coverslips, and then after 12 to 16 hours, the medium was exchanged with either regular mESC medium or with medium containing 250 μM IAA. At day 5, coverslips were washed with 100 mM tris (pH 8) before AP activity assay, which was performed using the VECTOR Blue AP Substrate Kit (Vector Laboratories, VECSK-5300) according to the manufacturer’s instructions. After AP staining, colonies were fixed by incubating with Histofix solution (4% formaldehyde) overnight. The next day, coverslips were washed with 1× PBS and mounted onto glass slides using Fluorescence Mounting Medium (Dako, S3023) and left to dry for at least 16 hours. Images from representative colonies were acquired with a bright-field microscope (Axioplan 2) using a 10× objective. The Pannoramic FLASH 250 II (3DHISTECH) wide-field microscope was used to scan whole slides to obtain sufficient images for quantification of staining intensity and morphology parameters. The images obtained from scanning whole slides were processed with a custom workflow created by T. Lendl (IMBA/IMP BioOptics Core) using Definiens Developer image analysis software.
Growth curves
A total of 3300 cells per well were seeded in regular mESC medium on flat-bottom 96-well plates in four replicates. The cells were allowed to adhere to the wells for 4 to 6 hours, and then the medium was exchanged either with regular mESC medium or with medium containing 250 μM IAA. Cell count was measured after 24, 48, and 72 hours by flow cytometry on LSR Fortessa (BD Biosciences).
Chromatin immunoprecipitation
mES cells (25 × 106) were collected, washed once in 1× PBS, and cross-linked with formaldehyde at a final concentration of 1% for 7 min. The cross-linking was stopped on ice and with glycine at a final concentration of 0.125 M. The cross-linked cells were pelleted by centrifugation for 5 min at 1200g at 4°C. Nuclei were prepared by washes with NP-Rinse buffer 1 [final: 10 mM tris (pH 8.0), 10 mM EDTA (pH 8.0), 0.5 mM EGTA, and 0.25% Triton X-100] followed by NP-Rinse buffer 2 [final: 10 mM tris (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, and 200 mM NaCl]. Afterward, the cells were prepared for shearing by sonication by two washes with Covaris shearing buffer [final: 1 mM EDTA (pH 8.0), 10 mM tris-HCl (pH 8.0), and 0.1% SDS] and resuspension of the nuclei in 0.9 ml of Covaris shearing buffer (with 1× protease inhibitors cOmplete Mini; Roche). The nuclei were sonicated for 15 min (duty factor, 5.0; Peak Incident Power, 140.0; cycles per burst, 200; at 4°C) in 1-ml Covaris glass cap tubes using a Covaris E220 High-Performance Focused Ultrasonicator. Lysates were incubated in 1× IP buffer [final: 50 mM Hepes/KOH (pH 7.5), 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, and 0.1% SDS], with the following antibodies at 4°C on a rotating wheel: H3K27me3 (Diagenode, C15410195), Rnf2 (Cell Signaling Technology, D22F2), Suz12 (Cell Signaling Technology, D39F6), Pcgf1 (Abcam, ab202395), Pcgf6 (Abcam, ab200038), H3K4me3 (Millipore, 05-745R), Cbx7 (Abcam, ab21873), Rybp (Sigma-Aldrich, PRS2227), and H2AK119ub (Cell Signaling Technology, D27C4). ChIPs were washed 5× with 1× IP buffer [final: 50 mM Hepes/KOH (pH 7.5), 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, and 0.1% SDS] or 1.5× IP buffer for H3K27me3 and H2AK119ub1, followed by 3× with DOC buffer [10 mM tris (pH 8), 0.25 mM LiCl, 1 mM EDTA, 0.5% NP40, and 0.5% DOC] and 1× with Tris-EDTA (TE) buffer (+50 mM NaCl).
ChIP-seq library preparation
Libraries were prepared using the NEXTFLEX ChIP-Seq kit (Bioo Scientific) following the “No size-selection cleanup” protocol and doubling the incubation times for all enzymatic steps. Each sample of ChIPed DNA was end-repaired and ligated to unique barcoded adapters to produce individual libraries. Libraries corresponding to samples to be directly compared to each other (e.g., +IAA versus −IAA) were pooled together and purified using 1 volume of Agencourt AMPure XP (Beckman Coulter). The pooled libraries were eluted with 25 μl of elution buffer (NEXTFLEX ChIP-Seq kit) and amplified using the KAPA Real-Time Library Amplification Kit (PEQLAB) following the kit instructions. Last, the amplified libraries were size-selected to fragments of 200 to 800 base pairs by running them on 1.5% agarose gel and staining with 1× SYBR Gold (Thermo Fisher Scientific) to visualize the DNA on a blue light light-emitting diode screen and cut the appropriate fragments. The size-selected libraries were gel-purified with the Monarch DNA Gel extraction kit (New England Biolabs).
Transcriptional profiling of 3′ end mRNA
A total of 75,000 cells per well were seeded on 24-well plates in regular mESC medium in triplicate. After 4 to 6 hours, fresh medium was added to cultivate mESC for 48 hours. Subsequently, mESCs were changed into medium with or without 250 μM IAA for a duration of 6, 24, or 48 hours before collecting all samples at the same time (reverse time course). Then, the wells were washed 1× with PBS, and cells were immediately lysed with RLT buffer (RNeasy kit), and total RNA was isolated using the RNeasy kit (QIAGEN) following the on-column deoxyribonuclease digestion protocol.
Supplementary Material
Acknowledgments
We are grateful to all members of the Bell laboratory and to M. Leeb, C. Buecker, V. A. Herzog, N. Fasching, M. Muhar, and P. Voigt for experimental advice and helpful discussions. We thank Life Science Editors for editorial assistance. We thank M. Brand, G. Winter, and U. Elling for sharing reagents and experimental advice. We thank T. Lendl and G. Petri at the IMP/IMBA BioOptics Facility for mESC colony imaging and quantitative analysis and the VBCF-NGS team (www.vbcf.ac.at) for deep sequencing services. We apologize to colleagues whose work could not be cited due to space limitations. Funding: This work was supported by the Austrian Academy of Sciences, the New Frontiers Group of the Austrian Academy of Sciences (NFG-05), the Human Frontiers Science Programme Career Development Award (CDA00036/2014-C), and start-up funding from the Norris Comprehensive Cancer Center at Keck School of Medicine of USC. J.A.Z.-M. was supported by Boehringer Ingelheim Fonds PhD Fellowship. This work was also supported by the following grant from the National Institutes of Health: NIH K01CA229995 to S.K.R. Author contributions: J.A.Z.-M. and O.B. initiated and designed the study. J.A.Z.-M., C.P., S.G., Q.Z., and H.F.M. performed the experiments. J.A.Z.-M., J.W., T.R.B., B.R., D.B., and S.K.R. performed bioinformatic analyses. J.J. and J.Z. provided DNA constructs and experimental advice. O.B. supervised all aspects of the project. The manuscript was prepared by J.A.Z.-M. and O.B. All authors discussed results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: QuantSeq and ChIP-seq data are deposited at the Gene Expression Omnibus (GEO) database under accession no. GSE136584. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaax5692/DC1
REFERENCES AND NOTES
- 1.Orkin S. H., Hochedlinger K., Chromatin connections to pluripotency and cellular reprogramming. Cell 145, 835–850 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bracken A. P., Helin K., Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 9, 773–784 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Aloia L., Di Stefano B., Di Croce L., Polycomb complexes in stem cells and embryonic development. Development 140, 2525–2534 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Aranda S., Mas G., Di Croce L., Regulation of gene transcription by Polycomb proteins. Sci. Adv. 1, e1500737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuettengruber B., Bourbon H. M., Di Croce L., Cavalli G., Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V., Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Müller J., Hart C. M., Francis N. J., Vargas M. L., Sengupta A., Wild B., Miller E. L., O’Connor M. B., Kingston R. E., Simon J. A., Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Kuzmichev A., Jenuwein T., Tempst P., Reinberg D., Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell 14, 183–193 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y., Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Cao R., Tsukada Y. I., Zhang Y., Role of Bmi-1 and Ring1A in H2A ubiquitylation and hox gene silencing. Mol. Cell 20, 845–854 (2005). [DOI] [PubMed] [Google Scholar]
- 11.de Napoles M., Mermoud J. E., Wakao R., Tang Y. A., Endoh M., Appanah R., Nesterova T. B., Silva J., Otte A. P., Vidal M., Koseki H., Brockdorff N., Polycomb group proteins ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7, 663–676 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R. S., Zhang Y., Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., Reinberg D., PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blackledge N. P., Farcas A. M., Kondo T., King H. W., McGouran J. F., Hanssen L. L. P., Ito S., Cooper S., Kondo K., Koseki Y., Ishikura T., Long H. K., Sheahan T. W., Brockdorff N., Kessler B. M., Koseki H., Klose R. J., Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157, 1445–1459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tavares L., Dimitrova E., Oxley D., Webster J., Poot R., Demmers J., Bezstarosti K., Taylor S., Ura H., Koide H., Wutz A., Vidal M., Elderkin S., Brockdorff N., RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148, 664–678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kloet S. L., Makowski M. M., Baymaz H. I., Van Voorthuijsen L., Karemaker I. D., Santanach A., Jansen P. W. T. C., Di Croce L., Vermeulen M., The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 23, 682–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauri S., Comoglio F., Seimiya M., Gerstung M., Glatter T., Hansen K., Aebersold R., Paro R., Gstaiger M., Beisel C., A high-density map for navigating the human Polycomb complexome. Cell Rep. 17, 583–595 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Wang L., Brown J. L., Cao R., Zhang Y., Kassis J. A., Jones R. S., Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell 14, 637–646 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Min J., Zhang Y., Xu R.-M., Structural basis for specific binding of polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 17, 1823–1828 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morey L., Pascual G., Cozzuto L., Roma G., Wutz A., Benitah S. A., Di Croce L., Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 10, 47–62 (2012). [DOI] [PubMed] [Google Scholar]
- 21.O’Loghlen A., Muñoz-Cabello A. M., Gaspar-Maia A., Wu H. A., Banito A., Kunowska N., Racek T., Pemberton H. N., Beolchi P., Lavial F., Masui O., Vermeulen M., Carroll T., Graumann J., Heard E., Dillon N., Azuara V., Snijders A. P., Peters G., Bernstein E., Gil J., MicroRNA regulation of Cbx7 mediates a switch of polycomb orthologs during ESC differentiation. Cell Stem Cell 10, 33–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eskeland R., Leeb M., Grimes G. R., Kress C., Boyle S., Sproul D., Gilbert N., Fan Y., Skoultchi A. I., Wutz A., Bickmore W. A., Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francis N. J., Kingston R. E., Woodcock C. L., Chromatin compaction by a polycomb group protein complex. Science 306, 1574–1577 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Farcas A. M., Blackledge N. P., Sudbery I., Long H. K., McGouran J. F., Rose N. R., Lee S., Sims D., Cerase A., Sheahan T. W., Koseki H., Brockdorff N., Ponting C. P., Kessler B. M., Klose R. J., KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. eLife 2012, e00205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendenhall E. M., Koche R. P., Truong T., Zhou V. W., Issac B., Chi A. S., Ku M., Bernstein B. E., GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLOS Genet. 6, e1001244 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalb R., Latwiel S., Baymaz H. I., Jansen P. W. T. C., Müller C. W., Vermeulen M., Müller J., Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 21, 569–571 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Blackledge N. P., Fursova N. A., Kelley J. R., Huseyin M. K., Feldmann A., Klose R. J., PRC1 catalytic activity is central to Polycomb system function. bioRxiv 27, 667667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamburri S., Lavarone E., Fernández-Pérez D., Zanotti M., Manganaro D., Conway E., Pasini D., Histone H2AK119 mono-ubiquitination is essential for Polycomb-mediated transcriptional repression. bioRxiv 8, 690461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H., Liefke R., Jiang J., Kurland J. V., Tian W., Deng P., Zhang W., He Q., Patel D. J., Bulyk M. L., Shi Y., Wang Z., Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549, 287–291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Højfeldt J. W., Laugesen A., Willumsen B. M., Damhofer H., Hedehus L., Tvardovskiy A., Mohammad F., Jensen O. N., Helin K., Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 25, 225–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perino M., Van Mierlo G., Karemaker I. D., Van Genesen S., Vermeulen M., Marks H., Van Heeringen S. J., Veenstra G. J. C., MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat. Genet. 50, 1002–1010 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Taherbhoy A. M., Huang O. W., Cochran A. G., BMI1-RING1B is an autoinhibited RING E3 ubiquitin ligase. Nat. Commun. 6, 7621 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Rose N. R., King H. W., Blackledge N. P., Fursova N. A., Ember K. J., Fischer R., Kessler B. M., Klose R. J., RYBP stimulates PRC1 to shape chromatin-based communication between Polycomb repressive complexes. eLife 5, e18591 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fursova N. A., Blackledge N. P., Nakayama M., Ito S., Koseki Y., Farcas A. M., King H. W., Koseki H., Klose R. J., Synergy between variant PRC1 complexes defines Polycomb-mediated gene repression. Mol. Cell 74, 1020–1036.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scelfo A., Fernández-Pérez D., Tamburri S., Zanotti M., Lavarone E., Soldi M., Bonaldi T., Ferrari K. J., Pasini D., Functional landscape of PCGF proteins reveals both RING1A/B-dependent-and RING1A/B-independent-specific activities. Mol. Cell 74, 1037–1052.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pengelly A. R., Kalb R., Finkl K., Müller J., Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev. 29, 1487–1492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao Z., Lee P., Stafford J. M., Von Schimmelmann M., Schaefer A., Reinberg D., An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voncken J. W., Roelen B. A. J., Roefs M., De Vries S., Verhoeven E., Marino S., Deschamps J., Van Lohuizen M., Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. U.S.A. 100, 2468–2473 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Carroll D., Erhardt S., Pagani M., Barton S. C., Surani M. A., Jenuwein T., The Polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 21, 4330–4336 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J., Mager J., Schnedier E., Magnuson T., The mouse PcG gene eed is required for Hox gene repression and extraembryonic development. Mamm. Genome 13, 493–503 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Pasini D., Bracken A. P., Jensen M. R., Denchi E. L., Helin K., Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23, 4061–4071 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riising E. M., Comet I., Leblanc B., Wu X., Johansen J. V., Helin K., Gene silencing triggers Polycomb repressive complex 2 recruitment to CpG Islands genome wide. Mol. Cell 55, 347–360 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Shen X., Liu Y., Hsu Y. J., Fujiwara Y., Kim J., Mao X., Yuan G. C., Orkin S. H., EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 32, 491–502 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chamberlain S. J., Yee D., Magnuson T., Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 26, 1496–1505 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collinson A., Collier A. J., Morgan N. P., Sienerth A. R., Chandra T., Andrews S., Rugg-Gunn P. J., Deletion of the Polycomb-group protein EZH2 leads to compromised self-renewal and differentiation defects in human embryonic stem cells. Cell Rep. 17, 2700–2714 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shan Y., Liang Z., Xing Q., Zhang T., Wang B., Tian S., Huang W., Zhang Y., Yao J., Zhu Y., Huang K., Liu Y., Wang X., Chen Q., Zhang J., Shang B., Li S., Shi X., Liao B., Zhang C., Lai K., Zhong X., Shu X., Wang J., Yao H., Chen J., Pei D., Pan G., PRC2 specifies ectoderm lineages and maintains pluripotency in primed but not naïve ESCs. Nat. Commun. 8, 672 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Endoh M., Endo T. A., Endoh T., Fujimura Y.-i., Ohara O., Toyoda T., Otte A. P., Okano M., Brockdorff N., Vidal M., Koseki H., Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development 135, 1513–1524 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Long H. K., Sims D., Heger A., Blackledge N. P., Kutter C., Wright M. L., Grützner F., Odom D. T., Patient R., Ponting C. P., Klose R. J., Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. eLife 2013, e00348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hisada K., Sánchez C., Endo T. A., Endoh M., Román-Trufero M., Sharif J., Koseki H., Vidal M., RYBP represses endogenous retroviruses and preimplantation- and germ line-specific genes in mouse embryonic stem cells. Mol. Cell. Biol. 32, 1139–1149 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morey L., Aloia L., Cozzuto L., Benitah S. A., Di Croce L., RYBP and Cbx7 define specific biological functions of Polycomb complexes in mouse embryonic stem cells. Cell Rep. 3, 60–69 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Elling U., Wimmer R. A., Leibbrandt A., Burkard T., Michlits G., Leopoldi A., Micheler T., Abdeen D., Zhuk S., Aspalter I. M., Handl C., Liebergesell J., Hubmann M., Husa A.-M., Kinzer M., Schuller N., Wetzel E., Van De Loo N., Martinez J. A. Z., Estoppey D., Riedl R., Yang F., Fu B., Dechat T., Ivics Z., Agu C. A., Bell O., Blaas D., Gerhardt H., Hoepfner D., Stark A., Penninger J. M., A reversible haploid mouse embryonic stem cell biobank resource for functional genomics. Nature 550, 114–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi S., Lee J., Kohda T., Matsuzawa A., Kawasumi M., Kanai-Azuma M., Kaneko-Ishino T., Ishino F., Induction of the G2/M transition stabilizes haploid embryonic stem cells. Development 141, 3842–3847 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Herzog V. A., Reichholf B., Neumann T., Rescheneder P., Bhat P., Burkard T. R., Wlotzka W., Von Haeseler A., Zuber J., Ameres S. L., Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montgomery N. D., Yee D., Montgomery S. A., Magnuson T., Molecular and functional mapping of EED motifs required for PRC2-dependent histone methylation. J. Mol. Biol. 374, 1145–1157 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wray J., Kalkan T., Gomez-Lopez S., Eckardt D., Cook A., Kemler R., Smith A., Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat. Cell Biol. 13, 838–845 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voigt P., Tee W.-W., Reinberg D., A double take on bivalent promoters. Genes Dev. 27, 1318–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leeb M., Pasini D., Novatchkova M., Jaritz M., Helin K., Wutz A., Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 24, 265–276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Lai P., Jia J., Song Y., Xia Q., Huang K., He N., Ping W., Chen J., Yang Z., Li J., Yao M., Dong X., Zhao J., Hou C., Esteban M. A., Gao S., Pei D., Hutchins A. P., Yao H., RNA helicase DDX5 inhibits reprogramming to pluripotency by miRNA-based repression of RYBP and its PRC1-dependent and -independent functions. Cell Stem Cell 20, 462–477.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Simon J. A., Kingston R. E., Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moussa H. F., Bsteh D., Yelagandula R., Pribitzer C., Stecher K., Bartalska K., Michetti L., Wang J., Zepeda-Martinez J. A., Elling U., Stuckey J. I., James L. I., Frye S. V., Bell O., Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat. Commun. 10, 1931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaax5692/DC1