

Abstract
DNA aptamers specifically recognizing microbial cells and viruses have a range of analytical and therapeutic applications. This article describes recent advances in the development of aptamers targeting specific pathogens (e.g., live bacteria, whole viral particles, and virally-infected mammalian cells). Specific aptamers against pathogens have been used as affinity reagents to develop sandwich assays, to label and to image cells, to bind with cells for flow-cytometry analysis, and to act as probes for development of whole-cell biosensors. Future applications of aptamers to pathogens will benefit from recent advances in improved selection and new aptamers containing modified nucleotides, particularly slow off-rate modified aptamers (SOMAmers).
Keywords: Aptamer, Bacteria, Bioanalytical assay, Biosensor, DNA, In-vitro selection, Pathogen, Protein, SELEX, SOMAmer
1. Introduction
Infectious diseases are the leading cause of death worldwide [1], and rank as the third highest cause of death in Canada and the USA after malignant neoplasms and cardiovascular disease [2], [3]. The rapidly growing global population, the rise of antimicrobial resistance, and recent resurgence in the incidence of infectious disease, most notably tuberculosis and smallpox, highlight the severity of the threat [4].
Pathogen surveillance is critical to diagnosis, control, and prevention of infectious diseases. Technology in the area of routine pathogen detection in the laboratory and the clinic has been slow to develop. Common diagnostic and surveillance methods include culture, biochemical tests, and antibody-based methods (e.g., agglutination and enzyme-linked assays) [5]. Definitive identification involves the amplification-based methods of regular and real-time PCR (quantitative PCR, or qPCR) [6]. Although fast and sensitive, PCR and qPCR are too complex to carry out in a basic clinical laboratory setting lacking highly-trained personnel. There is continued need for point-of-care assays that are simple, sensitive, and economical.
Aptamers are a class of flexible reagents with the potential to play a role in point-of-care assays. This group of synthetic nucleic-acid molecules can be generated in vitro against virtually any target molecule. The affinity of aptamers for their targets is comparable to, or even higher than, most monoclonal antibodies. Typical dissociation constants for aptamer-target complexes are found in the picomolar to low micromolar range [7]. Aptamers possess key advantages over antibodies due to their increased flexibility and the nature of in vitro evolution. They have low variability between batches, do not rely on animals for their production, can be created against toxic or non-immunogenic targets, are economical and fast to produce, and can be synthesized against a complex target without prior knowledge of a specific target molecule or without purifying a known target molecule.
Aptamers can be chemically modified and incorporated into a variety of simple assays for pathogen detection, as well as more complex assay formats, including flow cytometry, cell imaging, and aptamer-based biosensors. Aptamers have long shelf-lives and they can be used to coat surfaces and devices for point-of-care testing applications. These devices could potentially be re-useable after heating due to the thermal stability of aptamers.
This article discusses aptamer technology pertinent to pathogenic targets. The objective is two-fold:
-
(1)
to summarize and to evaluate recent developments in the selection of aptamers against pathogens; and,
-
(2)
to evaluate the analytical applications of these aptamers.
2. Selection of aptamers against pathogenic targets
2.1. Selection of aptamers against purified molecular targets
Aptamers can be evolved via an iterative process called Systematic Evolution of Ligands by Exponential Enrichment (SELEX). Different SELEX methodologies have been previously reviewed [8]. Briefly, a single-stranded random DNA or RNA library containing 1014–1016 unique sequences is incubated with a target of interest. The target-bound and unbound nucleic-acid sequences are separated, and the target-bound sequences are amplified for use as inputs in the next round of selection. The random nucleic-acid library typically contains 40–100 nucleotide (nt) single-stranded sequences with a randomized stretch of nts in the center and fixed primer sequences on either end. As many as 20 rounds of selection are carried out until a pool of aptamer sequences with high target affinity is obtained. These aptamers can then be cloned and sequenced.
Most aptamers selected against pathogenic targets have been evolved using purified target molecules, using the common SELEX procedures (Fig. 1 ). It is easier to obtain high-affinity aptamers in fewer rounds of SELEX with a purified target. Various bacterial or viral molecules have been used, mostly proteins. Carbohydrates have also been targeted including the lipopolysaccharide (LPS) of E. coli O111:H4 and cell-surface extracts of C. jejuni [9], [10], [11]. Table 1 summarizes molecules purified from pathogens against which aptamers have been selected. The use of amino-acid side-chain mimics in nt structure is a recent breakthrough that has allowed aptamers to be selected against previously impossible protein targets [12]. In the future, aptamers incorporating such chemical modifications will probably expand the SELEX target repertoire even further.
Figure 1.
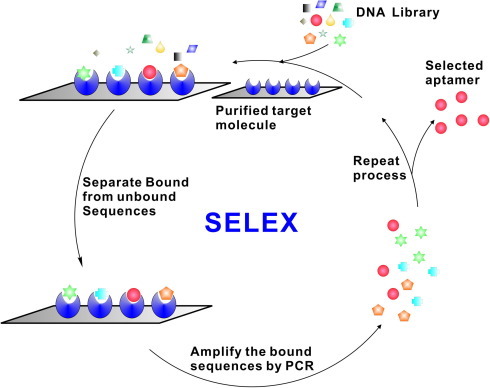
Conventional SELEX for generating DNA aptamers [8]. A random DNA library, typically containing 1014–1016 unique sequences of single-stranded oligonucleotides, is incubated with the target molecule of interest. The target-bound and unbound sequences are separated, and the target-bound sequences are amplified by PCR for use as inputs in the next round of selection. The reiterated rounds of selection are carried out to generate a pool of aptamer sequences that have high affinity for the target molecule. These aptamers are then cloned and sequenced.
Table 1.
Purified pathogen molecules that have been used in selection of aptamers by SELEX
| Target molecules | Ref. |
|---|---|
| HIV-1 | |
| Integrase | [14] |
| Reverse transcriptase | [15], [16] |
| Nucleocapsid protein | [17], [18] |
| Tat protein | [19] |
| R5 SV glycoprotein (gp120) | [20], [21] |
| Drug-resistant reverse transcriptase | [22] |
| Hepatitis C virus | |
| RdRp | [23] |
| NS3 | [24] |
| NS3 helicase | [25], [26] |
| 3′X tail | [27] |
| NS3 protease | [26], [28] |
| NS5B RNA polymerase | [29] |
| IRES (internal ribosome entry site) | [27], [30] |
| Hepatitis B virus | |
| HBsAg surface antigen | [31] |
| Influenza virus | |
| H5N1 HA protein | [32] |
| SARS coronavirus | |
| NTPase, Helicase | [33], [34] |
| Apple stem pitting virus | |
| Coat proteins | [35] |
| Foot and mouth disease virus | |
| VP1 protein | [36] |
| Prion proteins | |
| PrPsc | [37] |
| PrPsc fibrils | [38] |
| rPrPsc | [39] |
| rPrPc | [40] |
| Mammalian prion proteins | [41] |
| Escherichia coli | |
| Release factor 1 | [42] |
| Core RNA Polymerase | [43] |
| Lipopolysaccharide O111: B4 | [10], [11] |
| Mycobacterium | |
| M. avium sub. paratuberculosis MAP0105c gene product | [44] |
| M. tuberculosis MPT64 protein | [45] |
| M. tuberculosis polyphosphate kinase 2 | [46] |
| Francisella tularensis | |
| Protein lysate | [47] |
| Campylobacter jejuni | |
| Surface extract | [48] |
| Protein lysate | [49] |
| Salmonella enterica | |
| serovar Typhi Type IVB pilus | [50] |
| Outer membrane proteins | [51] |
| Listeria monocytogenes | |
| Internalin A | [52] |
| Leishmania infantum | |
| H2 Antigen | [53] |
| Burkholderia pseudomallei | |
| BipD/BopE/BPSL2748 | [54] |
| Ustilago maydis [corn pathogen] | |
| RNA-binding protein Rrm4 | [55] |
| Venezuelan equine encephalitis virus | |
| Capsid protein | [56] |
| Bacterial toxins | |
| Staphylococcal enterotoxin B, Cholera toxin | [57] |
| Botulinum neurotoxin | [58], [59] |
| Shiga toxin | [59] |
2.2. Selection of aptamers against live cells
Although aptamers can be selected using purified target molecules, their applications to target whole live cells may sometimes be limited, because the binding sites of cell-surface molecules can be different from their isolated forms. As a result, whole live cells have become increasingly common SELEX targets. SELEX using whole cells allows molecules to be targeted in their native conformations on the cell surface. It also negates the need for complex purification and target-partitioning steps. Cell-SELEX against mammalian cells has been extensively reviewed [13].
Bacterial cells present unique challenges over mammalian cells due to their cell-wall structure. Gram-negative bacteria possess a negatively-charged outer membrane that can repel nucleic-acid molecules. Capsules covering the surface of many pathogenic bacteria can also obscure target proteins. Both of these factors make it difficult to generate aptamers that bind to the surface molecules of bacterial cells. In addition, bacteria often grow much faster than culture cells of mammalian tissue. Short generation times lead to rapid changes in protein expression and high surface variation between cultures and colonies. Such variation can impede consistent measurement of aptamer binding.
The bacterial cell-SELEX procedure developed by our group is described in Fig. 2 [76]. Briefly, pathogenic bacterial cells are incubated in solution with a randomized nucleic-acid library. Cell-bound nucleic acids are separated from unbound nucleic acids via centrifugation. Cells are then washed, and cell-bound nucleic acids are eluted using low salt concentrations at high temperature. Cell-bound aptamers can also be amplified directly from the cell surface. Aptamer pools obtained after each round are screened for increased target-cell-binding affinity (e.g., by flow-cytometry analysis). Rounds of counter-selection against non-target cells, in which the sequences bound to the nontarget cells are removed, can be introduced after the first two rounds of positive selection. Alternatively, positive selection and counter-selection rounds can be alternated. These studies demonstrated the feasibility of selecting aptamers for both Gram-positive and Gram-negative bacteria. A summary of pathogenic whole cell and whole virus targets is presented in Table 2 .
Figure 2.
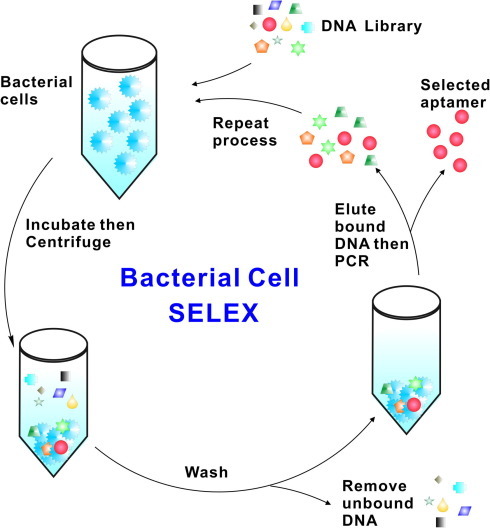
Bacterial cell SELEX [75]. A random DNA library, typically containing 1014–1016 unique sequences of single-stranded oligonucleotides, is incubated with the target bacterial cells. The mixture is centrifuged and the cells are washed to remove the unbound sequences. The cell-bound DNA sequences are eluted off, amplified by PCR, and used as a new DNA pool for the next round of selection. The reiterated rounds of selection are carried out to generate a pool of aptamer sequences that have high affinity for the target cells. These aptamers are then cloned and sequenced.
Table 2.
Whole bacterial cells and viral particles used in selection of aptamers by SELEX
| Pathogens | Ref. |
|---|---|
| Rous Sarcoma Virus particles | [60] |
| Bacillus anthracis spores | [59], [61], [62] |
| Live African Trypanosomes | [63], [64] |
| Trypanosoma cruzi | [65] |
| Bacillus thurigensis spores | [59], [66] |
| Human Influenza A virus particles | [67] |
| Vaccinia virus particles | [68] |
| Mycobacterium tuberculosis | [69] |
| Lactobacillus acidophilus | [70] |
| Escherichia coli DH5α | [71] |
| MS-2 Bacteriophage particles | [59] |
| Mammalian cells expressing Hepatitis C E2 envelope glycoprotein | [72] |
| Vaccinia-infected mammalian cells | [73], [74] |
| Staphylococcus aureus | [75] |
| Streptococcus pyogenes | [76] |
| Pseudomonas aeruginosa | [77] |
| Campylobacter jejuni | [78] |
As another important feature of whole cell-SELEX, specific target molecules do not need to be identified or purified ahead of time. Aptamers binding to the cell surface can be used to purify and to identify their respective target molecules post-SELEX. For example, aptamers against cell-surface molecules conjugated to magnetic beads can be used to pull the target out of a cell-lysate or crude-membrane preparation via either affinity interaction or photocrosslinking. Once the aptamer-protein complexes are eluted from the magnetic beads, they can be electrophoresed and identified via mass spectrometry.
While the majori of work in post-SELEX target elucidation has been done with mammalian cells, the principles also apply to whole-cell pathogens. Aptamers derived against the surfaces of whole African Trypanosomes [63] were used to identify continuously expressed proteins in the variant surface coat of Trypanosoma brucei including a 42-kDa protein in the flagellar attachment zone [64] and a variant surface glycoprotein [79]. Our group has identified a putative target of a Lactobacillus acidophilus specific DNA aptamer as the S-layer protein [70]. We have also identified putative target molecules of several aptamers selected against Group A Streptococcus [80].
3. Analytical applications of selected aptamers
Aptamers have proved useful in a variety of assays that conventionally use antibodies, including ELISA-like assays, western blotting, flow cytometry, microscopy, affinity chromatography, and capillary electrophoresis. Also, many novel technologies (e.g., aptamer-based biosensors and nano-devices, and ligation and amplification assays) are developed by taking advantage of aptamers. Today the applications of aptamers derived via whole cell-SELEX are limited, mainly because the number of aptamers derived against viral or bacterial targets is limited. Following is a summary of whole-cell aptamer technologies that have potential applications against pathogens, or have already been applied for pathogen detection.
3.1. Sandwich assays
In a traditional sandwich assay, an antibody is immobilized on a solid surface. Upon target binding, a secondary antibody attached to an enzyme (ELISA) or fluorophore is added. The secondary antibody binds to a different epitope on the target molecule than the primary antibody. Several sandwich assays using aptamers in place of antibodies have been developed to measure bacterial antigens.
An aptamer-linked immunosorbent assay was developed for detection of Francisella tularensis antigen [47]. A mixture of 25 different aptamers was used as both a capture and detection reagent. An aptamer cocktail-coated micro-titer plate was incubated with bacterial antigen, after which a biotin-labeled aptamer cocktail was added and the bound antigen detected via streptavidin-horseradish peroxidase. The aptamer assay demonstrated specificity and affinity comparable to that of an antibody-based ELISA.
The sandwich format can also be used to screen an aptamer pool for candidates with high affinity and selectivity to pathogenic antigens. A selected ssDNA aptamer population specific for Leishmania infantum histone H2A protein was screened using purified recombinant H2A-coated microtiter-plate wells [53]. Recovered aptamers were found to be specific for H2A after only three SELEX rounds.
3.2. Flow cytometry
Flow cytometry is a common method for cell sorting, counting, and detection. Cells are passed through a flow cell individually, and forward and side-scattered light intensities are measured. A laser beam is also used to excite fluorescent molecules, and fluorescence intensity is measured. Hence, cells bound to a fluorescent probe or dye can be identified and sorted from the population.
Antibodies are often used as probes, but aptamers have been found to be equally efficient. In addition to target identification and purification, aptamers are useful probes for identification and collection of cells expressing a target molecule of interest. Flow cytometry has been used both to evaluate target recognition by an aptamer pool and to isolate cell populations expressing a target. However, there are difficulties in using aptamers for cell typing in this fashion, as discovered in a standardized flow-cytometry assay by the Ellington group [81]. This problem is also faced when using antibodies in cell typing. There is not always a correlation between binding to the cell and biomarker expression on the cell surface; sometimes, the cell-SELEX procedure selects for sequences with high affinity but poor specificity.
Specific aptamer sequences can be fluorescently-labeled and used in tandem to sort bacterial cells or virus-infected cells. Cells are identified based on a distinct pattern of multiple aptamer binding. Closely-related cells give their own distinct patterns of binding to aptamers, and aptamers evolved against different cells can identify specific cells in a population. For example, SELEX carried out on vaccinia-infected cells generated a panel of aptamers with high affinity to cells expressing viral proteins [74]. When used together in flow-cytometric analysis, these aptamers could identify vaccinia infection in a variety of cell lines. The aptamers also generated distinct intensities and patterns of binding dependent on the strain of vaccinia.
Another study selected aptamers against whole Staphylococcus aureus cells and used a fluorescently-labeled panel of five high-affinity aptamers to differentiate different S. aureus strains [75]. The aptamers were shown to bind different cell-surface targets via a competitive flow-cytometry experiment; using five aptamers combined, rather than individually, was superior at detecting bacteria in pyogenic fluid.
Another useful way to employ flow cytometry is in determination of binding affinity between the target and the aptamer pool obtained from each round of selection. Bacterial cell-SELEX targets against which this technique has been applied include L. acidophilus [70], M. tuberculosis [69], C. jejuni [78], S. aureus [75], P. aeruginosa [77], and S. pyogenes [76] . Recently, flow cytometry was used to screen aptamers that are specific for vaccinia-virus infected A549 cells [73].
Conjugation of aptamers to fluorescent nanoparticles or nanorods allows for increased fluorescence intensity over regular fluorescent labels when cells are analyzed via flow cytometry, and hence increased sensitivity of detection. Using fluorescent nanorods as aptamer scaffolds for flow cytometry can potentially render useful medium-affinity aptamers derived from cell-SELEX as assembly of aptamers on the nanorod surface increases their binding affinity [83].
3.3. Aptamer biosensors
All biosensors comprise a biological molecular-recognition element (MRE] and a signal-transduction element (STE). A classical biosensor functions to transduce target binding by the MRE into a readable output. The term aptasensor applies to biosensors in which an aptamer is used as one or both of these elements. Aptasensors have seen increasing use for detection of pathogens in environmental and clinical samples. An excellent review of some of these methods has been published [84].
A variety of technologies have been applied to the development of aptasensors for pathogen detection. The earliest aptasensors for bacterial detection were based on an electrochemi-luminescence (ECL) sandwich format [59], [61], [62]. SELEX was carried out using heat-killed anthrax spores as a separation matrix. Resultant ssDNA aptamers with high affinity for the spores were conjugated to magnetic beads or to biotin. The magnetic bead-aptamer conjugates acted as capture reagents and the biotinylated aptamers as reporters for detection of as few as 10 spores with a linear dynamic range of 10–6x106.
Aptasensors with fluorescent labels were also used for pathogen detection. The same group that created the ECL anthrax sensor designed another sensor by conjugating aptamers selected against Bacillus thurigensis spores to CdSe quantum dots [66]. This assay could detect as few as 1000 CFU/mL of spores.
Quantum dots were also integrated into a sandwich assay with aptamer-conjugated magnetic beads for detection of Campylobacter jejuni [48]. Surface proteins extracted from C. jejuni were used as SELEX targets and the two highest affinity resultant aptamers were used as capture and detection ligands. The capture aptamer-magnetic bead conjugates could be adhered to the surface of a cuvette via a magnet, for uniform, reproducible laser excitation. Unlike the previous sensor for anthrax, this sensor detected both live and heat-killed cells at levels as low as 2.5 CFU in complex food matrices. The assay was also portable; fluorescence could be measured via bench-top or hand-held fluorometers.
A FRET-based assay was developed for detection of a peptide from foot and mouth disease (FMD) virus VP1 structural protein [36]. A polyclonal mixture of aptamers was labeled with Alexa Fluor 546-14-dUTP and incubated with quencher-bound FMD peptide. Binding of the FMD peptide to the aptamers resulted in a “lights-off” response that could measure levels of FMD peptide as low as 25 ng/mL. The peptide-bound aptamers were also fixed to a solid support, and a competitive assay with introduced analyte resulted in a “lights-on” response.
A similar approach was used to detect Escherichia coli using aptamers evolved against E. coli outer membrane proteins (OMPs) [10]. Aptamers were labeled with Alexa Fluor, and E. coli cells were heavily labeled with Black Hole Quencher (BHQ). Fluorescent aptamer/BHQ-labeled bacteria complexes were attached to a microtiter plate; upon introduction of unlabeled E. coli cells, the quencher-labeled cells were competed away and a measurable signal was generated. This method could detect as few as 30 unlabeled live cells.
A DNA capture-element (DCE) sensing system based on fluorescence quenching was also developed by this group for both aptamer selection and sensing of B. thurigensis, B. anthracis spores, botulinum neurotoxin and MS-2 bacteriophage [84]. Magnetic beads were attached to a dual fluorescently-labeled and quencher-labeled DNA aptamer; upon interaction of the aptamer with a target cell, the dsDNA is denatured into ssDNA. This dissociates the quencher and generates a fluorescent signal. The magnetic bead-aptamer-target-cell complexes can then be purified out of solution and re-detected with a complementary DNA probe attached to a quencher.
Recently, a fiber-optic sandwich biosensor was created using a fluorescently-labeled detection aptamer and an antibody-coated optical waveguide surface for detection of Listeria monocytogenes in food [52]. Fiber-optic biosensors measure the interaction between cells and aptamers by detecting waves emitted via excitation of a fluorescent reporter, usually an antibody. However, aptamers are ideal reagents for inclusion in such sensors. Due to their small size, aptamers exhibit high binding density on the cell surface, yielding enhanced signal and sensitivity [52]. The DNA aptamer was fluorescently-labeled and found to be specific for Internalin A, an invasion protein and pathogenicity marker. This sensor could detect as few as 103 CFU/mL of L. monocytogenes, and was successful at detecting cells spiked into chicken, beef, and turkey.
Several novel aptasensors relying on fluorescently-labeled aptamers have been developed for unlabeled Hepatitis C virus (HCV). One is a chip-based diagnostic sensor for HCV made from sol-gel immobilized anti-HCV core protein aptamers on a 96-well plate [89]. A secondary fluorescently-labeled anti-core antibody was used for detection. HCV particles in infected patient sera could be detected; normal patient samples did not generate a signal.
Aptamers do not need to be fluorescently-labeled for use in pathogen sensing. Their presence can be also detected via PCR amplification. Aptamers were selected against Salmonella typhimurium OMPs and conjugated to magnetic beads for collection of S. typhimurium from chicken-carcass rinse samples and subsequent quantitative PCR detection [51]. The assay had a limit of detection (LOD) of 10–100 CFU/mL of rinsate. A recirculation format of the assay was created by holding the aptamer-coated magnetic beads in place on a tube with a capture magnet, and running diluted chicken rinse through. The LOD of the recirculation assay was 4–40 CFU/mL of rinsate; both assays allow for sample processing and work on complex chicken-litter samples.
Similarly, antibody-coated magnetic beads were used to collect E. coli from complex samples with detection via PCR amplification of an E. coli-specific aptamer [86]. The assay could detect as few as 10 cells/mL of sample with a linear dynamic range of the order of 10–107 E. coli/mL, and was specific for the target cells.
Electrochemical detection has recently been applied to bacterial detection, specifically in the design of a probe for Salmonella Typhi detection in complex food samples [87]. This probe method is rapid, portable, and label-free. The electrodes are also reusable after reconstitution in buffer. A single-walled carbon nanotube (SWCNT) layer is deposited on a glass-rod electrode. The layer is coated with an aptamer specific for S. Typhi type IV pili. Conformational changes induced by target cell binding to the aptamers result in a change of charge on the SWCNTs and a subsequent change in potential recorded by the electrode. This system could detect below 1 CFU/mL of sample in less than 1 min with a linear dynamic range of 0.2–103 CFU/mL.
Another group developed a resonance sensor that could detect HCV helicase at levels as low as 100 pg/mL [88]. The principal components of this sensor are nanomechanical microcantilevers. RNA aptamers specific to HCV helicase are attached to the cantilever surface; aptamer-target binding induces stress on the cantilever surface and a dynamic response change that can be measured.
3.4. Cell labeling and imaging
Aptamers and aptamer-nanoparticle conjugates can be labeled and used in microscopy for detection of target cells. Fluorescence microscopy is recommended as a confirmatory method to be used in addition to flow cytometry or fluorescence measurement for aptamer-based cell typing [90]. Microscopy can be carried out with fluorescently-labeled aptamers and aptamer-nanoparticle conjugates.
Aptamers specific for Trypanosoma cruzi were used as probes to label the parasite surface fluorescently [65]. Fluorescence in-situ hybridization (FISH) with fluorescently-labeled DNA aptamer probes has been successful at imaging Pseudomonas aeruginosa [77]. Optical microscopy has also been used to visualize the density of cells captured by aptamers on a microfluidic device [82], [83].
However, there are instances where the target cell/aptamer interaction cannot be microscopically visualized. For example, a modified avidin method could detect spores binding to aptamer-coated magnetic beads; this interaction could not be visualized under the microscope [59], [85] – most probably due to the hindrance from the spacing of the aptamers on the beads and the size of both the beads and the spores.
While it is easier for aptamers to label molecules on the cell surface, they have been used for intracellular imaging. Imaging of bacterial RNA in live cells (E. coli) has been accomplished using protein complementation regulated by the interaction of an RNA-binding protein and an aptamer [91]. The RNA-binding protein is engineered to express GFP only in the aptamer-bound state; mRNA and rRNA can be tagged with aptamer and localized.
4. Therapeutic potential of aptamers
The inhibitory activity of certain aptamers on their target molecules has therapeutic ramifications, particularly for cell-surface molecules and receptors. Most research on therapeutic aptamers has focused on cancer therapy.
A number of inhibitory aptamers have been isolated for pathogens, including Trypanosoma cruzi [63], [64], [65], Hepatitis C Virus [72], SARS coronavirus [33], [34], E. coli lipopolysaccharide (LPS) [9], [10], and M. tuberculosis polyphosphate kinase 2 [46].
Also, many of the aptamer-based technologies developed for chemotherapeutic drug delivery to a host cell surface could be modified for use with antimicrobials.
5. SOMAmer arrays
The advent of slow off-rate modified aptamer (SOMAmer) technology [12] has the potential to extend the applicability of aptamer-based pathogen surveillance, detection and sensing formats. Gold and colleagues [12], [92] developed a new generation of aptamers, termed SOMAmers, with two key innovations, utilizing chemically modified nts to generate SOMAmers with protein-like properties and employing kinetic manipulations to improve the specificity of SOMAmers.
Fig. 3 shows a SOMAmer-selection process. The process begins with incorporation of modified nts (four dUTP analogs) into the DNA library by polymerase extension of a primer annealed to a biotinylated template. After separation from biotinylated templates, the DNA library containing modified nts is incubated with the target molecule. The slow off-rate enrichment and partition process is subsequently performed with the partition step of high stringency, including the use of extensive washing with large volumes. After separation of the bound DNA from the unbound DNA, the enriched DNA sequences are amplified by PCR to prepare the DNA pool for the next round of selection. Because the chemically-modified dUTP analogs in the modified DNA library and the enriched DNA pools contain functional groups that mimic amino-acid side-chains, the SOMAmers generated are thought to have some “protein-like” properties [12]. The expanded chemical diversity due to the incorporation of the modified nts extends SELEX to selection of aptamers against more difficult targets (e.g., carbohydrates on microbial cell surface and wide range of protein targets). Indeed, huge potential has been demonstrated from the success of obtaining aptamers (with pool Kd values below 30 nM) against 1200 human proteins [12].
Figure 3.
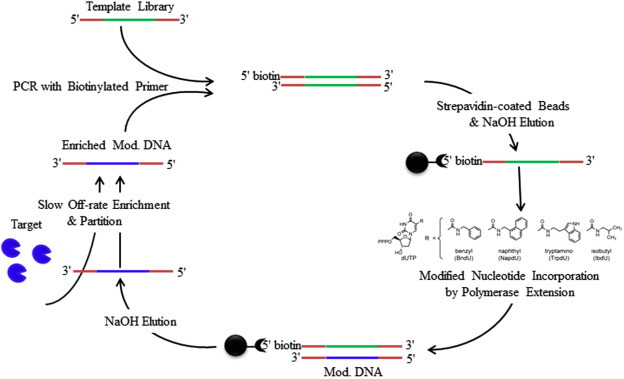
SOMAmer selection technique [12]. Four dUTP analogs were modified with chemical groups that mimic amino-acid side-chains. These modified nucleotides (nts) were incorporated into DNA library by polymerase extension of a primer annealed to a biotinylated template. After separation from biotinylated templates, the single-stranded DNA library containing the modified nts was incubated with the target molecule. The slow off-rate enrichment and partition process involving high stringency was used to separate and to enrich the DNA sequences that were bound to the target molecule. The bound DNA sequences were amplified by PCR and were used as new DNA pool for the next round of selection. The reiterated rounds of selection were carried out to generate a pool of SOMAmers that have high affinity for the target molecule.
SOMAmers were applied to develop a new proteomic array technology by taking advantage of both protein-binding and complementary base-pairing features of aptamers. Biotin-conjugated SOMAmers were first incubated with samples and SOMAmer-protein complexes were then captured onto streptavidin-coated beads. After washing the beads, target proteins in complexes were then modified with a NHS-biotin tag. The complexes were photocleaved from the first set of beads and challenged with a polyanionic competitor (dextran sulfate) for selective disruption of non-specific binding interactions. The complexes were further attached via the target protein biotin to a second set of avidin-coated beads. After the free SOMAmers were washed away, the bound SOMAmers were released from their targets and quantified. The number of SOMAmers was directly proportional to the number of target molecules (i.e. one SOMAmer per analyte). The utility of the SOMAmer assay was demonstrated by measuring 813 different proteins with pM levels of LODs and 7 logs of overall dynamic range. SOMAmer arrays have been successfully applied to identify novel biomarkers and proteome profiles in chronic kidney disease and non-small-cell lung cancer.
Both SOMAmer selection and array technology could potentially be developed for pathogen surveillance, typing of pathogen species and strain, and detection of microbial infection. They could enable comparison of proteome profiles between pathogens or infected and healthy individuals. Another exciting application could be high-throughput, multiplex antimicrobial resistance screening or comparison of antibiotic resistance profiles of different bacterial species or strains. Also, comparison of proteome profiles between patients could be used for diagnosis of infection and identification of potential treatment options.
6. Concluding remarks
Aptamers are an emerging class of reagents exciting due to their flexibility. They can be integrated into a myriad of existing and novel assay formats, and can be selected to have affinity and selectivity for virtually any target. The recent development of SOMAmer technology has opened up new targets to SELEX [12].
Although initially slow to develop, selection of aptamers against bacterial and viral targets has grown steadily in recent years. Such targets include purified cell-surface components and whole cells. Aptamers have shown promise in pathogen detection, collection, and therapy. Existing diagnostic and detection technologies have been modified to include aptamers targeting bacteria and viruses. In addition, a variety of novel aptamer-based assays have been developed for pathogen detection. Many existing aptamer-based assays initially designed for cancer cells could also be used for bacterially or virally-infected cells. The assays simply need to be modified to include aptamers against pathogenic targets in lieu of cancer-related targets.
SOMAmer technology has also demonstrated the feasibility of selecting aptamers against more than 1000 targets and of aptamer arrays that can simultaneously detect hundreds of targets [12]. Such aptamer-based assays for pathogenic detection can fill the existing need for rapid, reliable point-of-care testing in both the laboratory and the clinic. Also, aptamers can be extended towards much needed antimicrobial and antiviral applications.
Acknowledgments
The authors acknowledge the financial support from the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, the Canada Research Chairs Program, Alberta Health and Wellness, the Alberta Water Research Institute, and the Canadian Water Network.
Contributor Information
X. Chris Le, Email: xc.le@ualberta.ca.
Xing-Fang Li, Email: xingfang.li@ualberta.ca.
References
- 1.The World Health Organization (WHO), Report No. 310: Fact sheet, WHO, Geneva, Switzerland, 2008.
- 2.Statistics Canada (SC), Leading causes of death in Canada-2007, Report No. 84-215-x2010001, SC, Ottawa, Ontario, Canada, 2007.
- 3.Xu J., Kochanek K.D., Murphy S.L., Tejada-Vera B. National Vital Statistics Reports. 2010;58:1. [PubMed] [Google Scholar]
- 4.Noji E.K. J. Contingen. Crisis Manage. 2001;9:223. [Google Scholar]
- 5.Gerber M.A., Shulman S.T. Clin. Microbiol. Rev. 2004;17:571. doi: 10.1128/CMR.17.3.571-580.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lantz P., Abu Al-Soud W., Knutsson R., Hahn-Hägerdal B., Rådström P. Abu Al-Soud, R. Knutsson, B. Hahn-Hägerdal, P. Rådström, Biotechnol. Annu. Rev. 2000;5:87. doi: 10.1016/s1387-2656(00)05033-x. [DOI] [PubMed] [Google Scholar]
- 7.White R.R., Sullenger B.A., Rusconi C.P. J. Clin. Invest. 2000;106:929. doi: 10.1172/JCI11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamula C.L.A., Guthrie J.W., Zhang H., Li X.-F., Le X.C. Trends Anal. Chem. 2006;25:681. [Google Scholar]
- 9.Bruno J.G. In Vitro Cell. Dev. Biol. Anim. 2010;46:107. doi: 10.1007/s11626-009-9257-7. [DOI] [PubMed] [Google Scholar]
- 10.Bruno J.G., Carrillo M.P., Phillips T. Folia Microbiol. (Prague) 2008;53:295. doi: 10.1007/s12223-008-0046-6. [DOI] [PubMed] [Google Scholar]
- 11.Dwarakanath S., Bruno J.G., Shastry A., Phillips T., John A.A., Kumar A., Stephenson L.D. Biochem. Biophys. Res. Commun. 2004;325:739. doi: 10.1016/j.bbrc.2004.10.099. [DOI] [PubMed] [Google Scholar]
- 12.Gold L. PLoS ONE. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sefah K., Shangguan D., Xiong X., O’Donoghue M.B., Tan W. Nat. Protoc. 2010;5:1169. doi: 10.1038/nprot.2010.66. [DOI] [PubMed] [Google Scholar]
- 14.Allen P., Collins B., Brown D., Hostomsky Z., Gold L. Virology. 1996;225:306. doi: 10.1006/viro.1996.0605. [DOI] [PubMed] [Google Scholar]
- 15.Tuerk C., Gold L. Science (Washington, DC) 1990;249:505. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 16.Tuerk C., MacDougal S., Gold L. Proc. Natl. Acad. Sci. USA. 1992;89:6988. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S.J., Kim M.Y., Lee J.H., You J.C., Jeong S. Biochem. Biophys. Res. Commun. 2002;291:925. doi: 10.1006/bbrc.2002.6521. [DOI] [PubMed] [Google Scholar]
- 18.Jeong Y.Y., Kim S.H., Jang S.I., You J.C. BMB Rep. 2008;41:511. doi: 10.5483/bmbrep.2008.41.7.511. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto R., Baba T., Kumar P.K. Genes Cells. 2000;5:389. doi: 10.1046/j.1365-2443.2000.00331.x. [DOI] [PubMed] [Google Scholar]
- 20.Khati M., Schuman M., Ibrahim J., Sattentau Q., Gordon S., James W. J. Virol. 2003;77:12692. doi: 10.1128/JVI.77.23.12692-12698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen C., Forzan M., Sproat B., Pantophlet R., McGowan I., Burton D., James W. Virology. 2008;381:46. doi: 10.1016/j.virol.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li N., Wang Y., Pothukuchy A., Syrett A., Husain N., Gopalakrisha S., Kosaraju P., Ellington A.D. Nucleic Acids Res. 2008;36:6739. doi: 10.1093/nar/gkn775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones L.A., Clancy L.E., Rawlinson W.D., White P.A. Antimicrob. Agents Chemother. 2006;50:3019. doi: 10.1128/AAC.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar P.K., Machida K., Urvil P.T., Kakiuchi N., Vishnuvardhan D., Shimotohno K., Taira K., Nishikawa S. Virology. 1997;237:270. doi: 10.1006/viro.1997.8773. [DOI] [PubMed] [Google Scholar]
- 25.Hwang B., Cho J.S., Yeo H.J., Kim J.H., Chung K.M., Han K., Jang S.K., Lee S.W. RNA. 2004;10:1277. doi: 10.1261/rna.7100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuda K., Umehara T., Sekiya S., Kunio K., Hasegawa T., Nishikawa S. Biochem. Biophys. Res. Commun. 2004;325:670. doi: 10.1016/j.bbrc.2004.10.089. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda K., Toyokawa Y., Kikuchi K., Konno K., Ishihara R., Fukazawa C., Nishikawa S., Hasegawa T. Nucleic Acids Symp. Ser. (Oxford) 2008;52:205. doi: 10.1093/nass/nrn104. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda K., Vishnuvardhan D., Sekiya S., Hwang J., Kakiuchi N., Taira K., Shimotohno K., Kumar P.K., Nishikawa S. Eur. J. Biochem. 2000;267:3685. doi: 10.1046/j.1432-1327.2000.01400.x. [DOI] [PubMed] [Google Scholar]
- 29.Biroccio A., Hamm J., Incitti I., De Francesco R., Tomei L. J. Virol. 2002;76:3688. doi: 10.1128/JVI.76.8.3688-3696.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kikuchi K., Umehara T., Nishikawa F., Fukuda K., Hasegawa T., Nishikawa S. Biochem. Biophys. Res. Commun. 2009;386:118. doi: 10.1016/j.bbrc.2009.05.135. [DOI] [PubMed] [Google Scholar]
- 31.Liu J., Yang Y., Hu B., Ma Z., Huang H., Yu Y., Liu S., Lu M., Yang D. Virol. Sinica. 2010;25:27. doi: 10.1007/s12250-010-3091-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng C., Dong J., Yao L., Chen A., Jia R., Huan L., Guo J., Shu Y., Zhang Z. Biochem. Biophys. Res. Commun. 2008;366:670. doi: 10.1016/j.bbrc.2007.11.183. [DOI] [PubMed] [Google Scholar]
- 33.Jang K.J., Lee N.R., Yeo W.S., Jeong Y.J., Kim D.E. Biochem. Biophys. Res. Commun. 2008;366:738. doi: 10.1016/j.bbrc.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahn D.G., Jeon I.J., Kim J.D., Song M.S., Han S.R., Lee S.W., Jung H., Oh J.W. Analyst (Cambridge, UK) 2009;134:1896. doi: 10.1039/b906788d. [DOI] [PubMed] [Google Scholar]
- 35.Lautner G., Balogh Z., Bardoczy V., Meszaros T., Gyurcsanyi R.E. Analyst (Cambridge, UK) 2010;135:918. doi: 10.1039/b922829b. [DOI] [PubMed] [Google Scholar]
- 36.Bruno J.G., Carrillo M.P., Phillips T. J. Biomol. Tech. 2008;19:109. [PMC free article] [PubMed] [Google Scholar]
- 37.Proske D., Gilch S., Wopfner F., Schatzl H.M., Winnacker E.L., Famulok M. Chembiochem. 2002;3:717. doi: 10.1002/1439-7633(20020802)3:8<717::AID-CBIC717>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 38.Rhie A., Kirby L., Sayer N., Wellesley R., Disterer P., Sylvester I., Gill A., Hope J., James W., Tahiri-Alaoui A. J. Biol. Chem. 2003;278:39697. doi: 10.1074/jbc.M305297200. [DOI] [PubMed] [Google Scholar]
- 39.Weiss S., Proske D., Neumann M., Groschup M.H., Kretzschmar H.A., Famulok M., Winnacker E.L. J. Virol. 1997;71:8790. doi: 10.1128/jvi.71.11.8790-8797.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takemura K., Wang P., Vorberg I., Surewicz W., Priola S.A., Kanthasamy A., Pottahil R., Chen S.G., Sreevatsan S. Exp. Biol. Med. (Maywood) 2006;231:204. doi: 10.1177/153537020623100211. [DOI] [PubMed] [Google Scholar]
- 41.Bibby D.F., Gill A.C., Kirby L., Farquhar C.F., Bruce M.E., Garson J.A. J. Virol. Methods. 2008;151:107. doi: 10.1016/j.jviromet.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Sando S., Ogawa A., Nishi T., Hayami M., Aoyama Y. Bioorg. Med. Chem. Lett. 2007;17:1216. doi: 10.1016/j.bmcl.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Kulbachinskiy A., Feklistov A., Krasheninnikov I., Goldfarb A., Nikiforov V. Eur. J. Biochem. 2004;271:4921. doi: 10.1111/j.1432-1033.2004.04461.x. [DOI] [PubMed] [Google Scholar]
- 44.Bannantine J.P., Radosevich T.J., Stabel J.R., Sreevatsan S., Kapur V., Paustian M.L. Clin. Vaccine Immunol. 2007;14:518. doi: 10.1128/CVI.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin L., Zheng R., Ma Z., Feng Y., Liu Z., Yang H., Wang J., Jin R., Lu J., Ding Y., Hu Z. Clin. Chem. Lab. Med. 2009;47:405. doi: 10.1515/CCLM.2009.097. [DOI] [PubMed] [Google Scholar]
- 46.Shum K.T., Lui E.L., Wong S.C., Yeung P., Sam L., Wang Y., Watt R.W., Tanner J.A. Biochemistry. 2011;50:3261. doi: 10.1021/bi2001455. [DOI] [PubMed] [Google Scholar]
- 47.Vivekananda J., Kiel J.L. Lab. Invest. 2006;86:610. doi: 10.1038/labinvest.3700417. [DOI] [PubMed] [Google Scholar]
- 48.Bruno J.G., Phillips T., Carrillo M.P., Crowell R. J. Fluoresc. 2009;19:427. doi: 10.1007/s10895-008-0429-8. [DOI] [PubMed] [Google Scholar]
- 49.S. McMasters, D. Stratis-Cullum, Evaluation of aptamers as molecular recognition elements for pathogens using capillary electrophoretic analysis, in: B.M. Cullum, J.C. Carter (Editors), Smart Medical and Biomedical Sensor Technology IV, SPIE Proc., Vol. 6380, 2006.
- 50.Pan Q., Zhang X.L., Wu H.Y., He P.W., Wang F., Zhang M.S., Hu J.M., Xia B., Wu J. Antimicrob. Agents Chemother. 2005;49:4052. doi: 10.1128/AAC.49.10.4052-4060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joshi R., Janagama H., Dwivedi H.P., Senthil Kumar T.M., Jaykus L.A., Schefers J., Sreevatsan S. Mol. Cell. Probes. 2009;23:20. doi: 10.1016/j.mcp.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 52.Ohk S., Koo O., Sen T., Yamamoto C., Bhunia A. J. Appl. Microbiol. 2010;109:808. doi: 10.1111/j.1365-2672.2010.04709.x. [DOI] [PubMed] [Google Scholar]
- 53.Ramos E., Pineiro D., Soto M., Abanades D.R., Martin M.E., Salinas M., Gonzalez V.M. Lab. Invest. 2007;87:409. doi: 10.1038/labinvest.3700535. [DOI] [PubMed] [Google Scholar]
- 54.Gnanam A.J., Hall B., Shen X., Piasecki S., Vernados A., Galyov E.E., Smither S.J., Kitto G.B., Titball R.W., Ellington A.D., Brown K.A. Trans. Royal Soc. Trop. Med. Hyg. 2008;102:S55. doi: 10.1016/S0035-9203(08)70015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konig J., Julius C., Baumann S., Homann M., Goringer H.U., Feldbrugge M. RNA. 2007;13:614. doi: 10.1261/rna.334307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang J., Lee M.S., Watowich S.J., Gorenstein D.G. FEBS Lett. 2007;581:2497. doi: 10.1016/j.febslet.2007.04.072. [DOI] [PubMed] [Google Scholar]
- 57.Bruno J.G., Kiel J.L. BioTechniques. 2002;32:178. doi: 10.2144/02321dd04. [DOI] [PubMed] [Google Scholar]
- 58.Tok J.B., Fischer N.O. Chem. Commun. (Cambridge). 2008;16:1883. doi: 10.1039/b717936g. [DOI] [PubMed] [Google Scholar]
- 59.Fan M., McBurnett S.R., Andrews C.J., Allman A.M., Bruno J.G., Kiel J.L. J. Biomol. Tech. 2008;19:311. [PMC free article] [PubMed] [Google Scholar]
- 60.Pan W., Craven R.C., Qiu Q., Wilson C.B., Wills J.W., Golovine S., Wang J.F. Proc. Natl. Acad. Sci. USA. 1995;25:11509. doi: 10.1073/pnas.92.25.11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruno J.G., Kiel J.L. Biosens. Bioelectron. 1999;14:457. doi: 10.1016/s0956-5663(99)00028-7. [DOI] [PubMed] [Google Scholar]
- 62.B. Zhen, Y.J. Song, Z.B. Guo, J. Wang, M.L. Zhang, S.Y. Yu, R.F. Yang, Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 34 (2002) 635. [PubMed]
- 63.Homann M., Goringer H.U. Nucleic Acids Res. 1999;27:2006. doi: 10.1093/nar/27.9.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Homann M., Lorger M., Engstler M., Zacharias M., Goringer H.U. Comb. Chem. High Throughput Screen. 2006;9:491. doi: 10.2174/138620706777935324. [DOI] [PubMed] [Google Scholar]
- 65.Ulrich H., Magdesian M.H., Alves M.J., Colli W. J. Biol. Chem. 2002;277:20756. doi: 10.1074/jbc.M111859200. [DOI] [PubMed] [Google Scholar]
- 66.Ikanovic M., Rudzinski W.E., Bruno J.G., Allman A., Carrillo M.P., Dwarakanath S., Bhahdigadi S., Rao P., Kiel J.L., Andrews C.J. J. Fluoresc. 2007;17:193. doi: 10.1007/s10895-007-0158-4. [DOI] [PubMed] [Google Scholar]
- 67.Gopinath S.C., Misono T.S., Kawasaki K., Mizuno T., Imai M., Odagiri T., Kumar P.K. J. Gen. Virol. 2006;87:479. doi: 10.1099/vir.0.81508-0. [DOI] [PubMed] [Google Scholar]
- 68.Nitsche A., Kurth A., Dunkhorst A., Panke O., Sielaff H., Junge W., Muth D., Scheller F., Stocklein W., Dahmen C., Pauli G., Kage A. BMC Biotechnol. 2007;15:48. doi: 10.1186/1472-6750-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen F., Zhou J., Luo F., Mohammed A.B., Zhang X.L. Biochem. Biophys. Res. Commun. 2007;357:743. doi: 10.1016/j.bbrc.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 70.Hamula C.L.A., Zhang H., Guan L., Li X.-F., Le X.C. Anal. Chem. 2008;80:7812. doi: 10.1021/ac801272s. [DOI] [PubMed] [Google Scholar]
- 71.So H.M., Park D.W., Jeon E.K., Kim Y.H., Kim B.S., Lee C.K., Choi S.Y., Kim S.C., Chang H., Lee J.O. Small. 2008;4:197. doi: 10.1002/smll.200700664. [DOI] [PubMed] [Google Scholar]
- 72.Chen F., Hu Y., Li D., Chen H., Zhang X.L. PLoS One. 2009;4:e8142. doi: 10.1371/journal.pone.0008142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang Z., Parekh P., Turner P., Moyer R.W., Tan W. Clin. Chem. 2009;55:813. doi: 10.1373/clinchem.2008.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parekh P., Tang Z., Turner P.C., Moyer R.W., Tan W. Anal. Chem. 2010;82:8642. doi: 10.1021/ac101801j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cao X., Li S., Chen L., Ding H., Xu H., Huang Y., Li J., Liu N., Cao W., Zhu Y., Shen B., Shao N. Nucleic Acids Res. 2009;37:4621. doi: 10.1093/nar/gkp489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamula C.L.A., Le X.C., Li X.-F. Anal. Chem. 2011;83:3640. doi: 10.1021/ac200575e. [DOI] [PubMed] [Google Scholar]
- 77.Wang K.Y., Zeng Z.L., Yang X.Y., Li W.B., Lan X.P. Eur. J. Clin. Microbiol. Infect. Dis. 2011;30:273. doi: 10.1007/s10096-010-1074-0. [DOI] [PubMed] [Google Scholar]
- 78.Dwivedi H.P., Smiley R.D., Jaykus L.A. Appl. Microbiol. Biotechnol. 2010;87:2323. doi: 10.1007/s00253-010-2728-7. [DOI] [PubMed] [Google Scholar]
- 79.Lorger M., Engstler M., Homann M., Goringer H.U. Eukaryot. Cell. 2003;2:84. doi: 10.1128/EC.2.1.84-94.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.C.L.A. Hamula, PhD Thesis, Selection of DNA aptamers binding to live bacterial cells, University of Alberta, Canada, Fall 2010.
- 81.Li N., Ebright J.N., Stovall G.M., Chen X., Nguyen H.H., Singh A., Ellington A.D. J. Proteome Res. 2009;8:2438. doi: 10.1021/pr801048z. [DOI] [PubMed] [Google Scholar]
- 82.Herr J.K., Smith J.E., Medley C.D., Shangguan D., Tan W. Anal. Chem. 2006;78:2918. doi: 10.1021/ac052015r. [DOI] [PubMed] [Google Scholar]
- 83.Huang Y.F., Sefah K., Bamrungsap S., Chang H.T., Tan W. Langmuir. 2008;24:11860. doi: 10.1021/la801969c. [DOI] [PubMed] [Google Scholar]
- 84.Torres-Chavolla E., Alocilja E.C. Biosens. Bioelectron. 2009;24:3175. doi: 10.1016/j.bios.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bruno J.G., Carrillo M.P., Phillips T., Andrews C.J. J. Fluoresc. 2010;20:1211. doi: 10.1007/s10895-010-0670-9. [DOI] [PubMed] [Google Scholar]
- 86.Lee H.J., Kim B.C., Kim K.W., Kim Y.K., Kim J., Oh M.K. Biosens. Bioelectron. 2009;24:3550. doi: 10.1016/j.bios.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 87.Zelada-Guillen G.A., Riu J., Duzgun A., Rius F.X. Angew. Chem. Int. Ed. Engl. 2009;48:7334. doi: 10.1002/anie.200902090. [DOI] [PubMed] [Google Scholar]
- 88.Hwang K.S., Lee S.M., Eom K., Lee J.H., Lee Y.S., Park J.H., Yoon D.S., Kim T.S. Biosens. Bioelectron. 2007;23:459. doi: 10.1016/j.bios.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 89.Lee S., Kim Y.S., Jo M., Jin M., Lee D.K., Kim S. Biochem. Biophys. Res. Commun. 2007;358:47. doi: 10.1016/j.bbrc.2007.04.057. [DOI] [PubMed] [Google Scholar]
- 90.Liu G., Mao X., Phillips J.A., Xu H., Tan W., Zeng L. Anal. Chem. 2009;81:10013. doi: 10.1021/ac901889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Valencia-Burton M., McCullough R.M., Cantor C.R., Broude N.E. Nat. Methods. 2007;4:421. doi: 10.1038/nmeth1023. [DOI] [PubMed] [Google Scholar]
- 92.Vaught J.D., Bock C., Carter J., Fitzwater T., Otis M., Schneider D., Rolando J., Waugh S., Wilcox S.K., Eaton B.E. J. Am. Chem. Soc. 2010;132:4141. doi: 10.1021/ja908035g. [DOI] [PubMed] [Google Scholar]