

Abstract
A rapid, convenient and reliable pseudorabies virus (PRV) detection system was developed by using the loop-mediated isothermal amplification (LAMP) method. Six special primers were designed successfully based on the PRV DNA-binding protein (DBP) gene. The assay was optimized to amplify PRV DNA by incubation at 63 °C for 1 h. The LAMP products had a ladder-like pattern of bands from 188 bp when electrophoresed on an agarose gel and its specificity was confirmed by digestion with Hinc II enzyme. Two naked-eye detection methods were developed for use in the field. The detection limit of the LAMP assay was found to be 10 fg DNA sample which was 100–1000-fold higher than that of PCR. By using DNA (or cDNA) samples extracted from three different PRV strains and six other viruses known to be related genetically to PRV or to cause similar clinical signals in pig, the system was identified to amplify only the PRV DNA. A comparison between the LAMP and PCR assay using five clinical samples showed good correlation.
Keywords: LAMP, PRV, Detection
1. Introduction
Pseudorabies virus (PRV) is a member of the herpesviridae family, which infects pigs with high mortality. This virus disease causes serious reduction to the pig industry and leads to severe financial loss, which requires a better detection system for improving the virus surveillance (Pejsak and Truszczyński, 2006). Traditionally, PRV detection is based on direct virus isolation (Pensaert and Kluge, 1989) or detection of antigens by an immunohistological method (Ducatelle et al., 1982). However, both methods are time-consuming, requiring at least 2–3 days. PCR has allowed detection of the virus rapidly, but requiring precise instruments and an elaborate method for detection the amplified product (Osorio, 1991, Balasch et al., 1998, Lee et al., 2007).
Loop-mediated isothermal amplification (LAMP) is a nucleic acid amplification method, which amplifies DNA with high specificity, sensitivity and rapidity under isothermal condition using a set of six specially designed primers and a DNA polymerase with strand displacement activity (Notomi et al., 2000). It has been used widely for infectious diseases diagnosis, such as detection of H5N1 avian influenza virus (Imai et al., 2007), severe acute respiratory syndrome coronavirus (Hong et al., 2004), hepatitis B virus (Li et al., 2005), foot-and-mouth disease virus (Dukes et al., 2006), Taura syndrome virus (Kiatpathomchai et al., 2007) and swine vesicular disease virus (Blomström et al., 2007). In this study, LAMP was applied in PRV detection and its specificity and sensitivity were assessed.
2. Materials and methods
2.1. Samples
PRV (strain Fa) was obtained from China Institute of Veterinary Drug Control (CIVDC); PRV (strain SA215, Bartha), porcine circovirus-2 (PCV-2), porcine epidemic diarrhea virus (PEDV), porcine reproductive and respiratory syndrome virus (PRRSV), swine transmissible gastroenteritis coronavirus (TGEV), swine vesicular disease virus (SVDV) and swine fever virus (SFV) were all derived from their passage in cell culture which were provided by Shanghai Entry-Exit Inspection and Quarantine Bureau (SHCIQ) (Table 1 ). The clinical samples of porcine brain tissue were collected from suspect cases of pseudorabies.
Table 1.
Sample list
| Virus | Strain/isolate | Country of origin | Provider |
|---|---|---|---|
| PRV | Strain SA 215, strain Bartha | China | SHCIQ |
| PRV | Strain Fa | China | CIVDC |
| PCV-2 | Isolate ZZ | China | SHCIQ |
| PEDV | Strain DX | China | SHCIQ |
| PRRSV | Strain SD1 | China | SHCIQ |
| TGEV | Strain SC-Y | China | SHCIQ |
| SVDV | Strain HK/70 | China | SHCIQ |
| SFV | Strain LJL12 | China | SHCIQ |
2.2. DNA extraction
Total genomic DNA was extracted using the QlAamp® DNA Blood Mini Kit (Qiagen GmbH, Germany). After elution in 20 μl nuclease-free H2O, DNA samples were stored at −70 °C until used.
2.3. LAMP primers designing
The DNA-binding protein (DBP) gene (GenBank Accession number U80909) was chosen as the suitable target for designing the LAMP primers, as it provides a relatively conserved nucleic acid fragment of 188 bp. Primers were designed using Primer Explorer V3 software (http://primerexplorer.jp/e/).
2.4. LAMP-based detection
The basic LAMP reaction was carried out in a volume of 25 μl containing 1× ThermoPol buffer (New England Biolabs Inc., USA), 6.0 mM MgSO4, 1.0 M betaine (Sigma, Germany), 1.2 mM dNTPs, 0.2 μM each of outer primer, 1.6 μM each of inner primer and 0.4 μM each of loop primer, 8 U of Bst polymerase (Large Fragment; New England Biolabs Inc., USA) with 1 μl total DNA as template. The amplification was performed at 65 °C in a laboratory water bath (Kangle, China, 25–99 °C, ±0.3 °C) for 1 h. Initial experiments were performed to optimize the assay by varying the concentration of MgSO4, dNTPs, primers, amplification temperature and reaction time.
The amplified products were separated on a 1.5% agarose gel in 0.5× TBE buffer with the voltage of 120 V for 1 h and visualized by staining with ethidium bromide. The result could also be visualized directly with the naked eye according to the white precipitate of magnesium pyrophosphate generated in the reaction or the green color produced by the intercalating dye Picogreen® (Invitrogen, USA). To confirm the specificity of amplification products, 2 μl of the reaction mixture was digested with Hinc II at 37 °C for 4 h.
2.5. Sensitivity and specificity of LAMP for PRV detection
The sensitivity of LAMP was demonstrated and compared with PCR (Balasch et al., 1998, Chen et al., 2002, Lee et al., 2007) using various PRV DNA dilutions (10−1 to 10−9) as templates (original concentration of DNA sample was 10 ng/μl).
The specificity of LAMP was examined by the use of DNA (or cDNA) extracted from three different PRV strains and six other viruses known to be related genetically to PRV or to cause similar clinical signals in pig.
2.6. Evaluation of the LAMP assay using clinical samples
The evaluation of the LAMP assay was carried out using DNA extracted from clinical samples with PCR running in parallel.
3. Results
3.1. Designing the LAMP primers
LAMP primers were designed using the Primer Explorer V3 software based on a conserved fragment of the DBP gene (Fig. 1 ). After screening according to the end stability of the primers (the free energy of the 3′ ends of F2/B2, F3/B3, LF/LB and the 5′-end of F1c/B1c should be −4 kcal/mol or less), one set of primers was chosen (Table 2 ). The outer primers (F3 and B3) help to displace the primary strand. The inner primers (FIP and BIP) each have two distinct sequences corresponding to the sense and anti-sense sequence of the target. FIP comprises F1c and F2 while BIP comprises B1c and B2. The forward loop primer (LF) is designed using the complementary strand corresponding to the region between F1 and F2 while reverse loop primer (LB) was designed using the complementary strand corresponding to the region between B1 and B2.
Fig. 1.
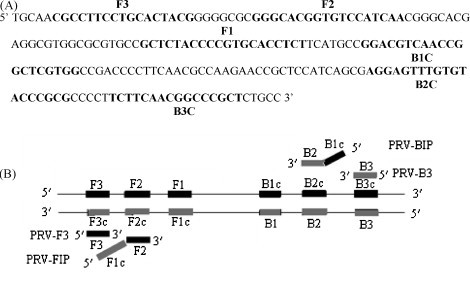
The conserved fragment of the DBP gene used to design LAMP primers (A) and the schematic diagram of LAMP primers (B).
Table 2.
Details of LAMP primers used for LAMP amplification of PRV
| Primer name | Type | Length (bp) | Sequence (5′–3′) |
|---|---|---|---|
| F3 | Forward outer | 17 | CGCCTTCCTGCACTACG |
| B3 | Reverse outer | 16 | AGCGGGCCGTTGAAGA |
| FIP (F1c + F2) | Forward inner | 38 | AGAGGTGCACGGGGTAGAGCGGGCACGGTGTCCATCAA |
| BIP (B1c + B2) | Reverse inner | 39 | GGACGTCAACCGGCTCGTGGCGCGGGTACACAAACTCCT |
| LF | Forward loop | 18 | ACGCGCCACGCCTCGTGC |
| LB | Reverse loop | 18 | CGACCCCTTCAACGCCAA |
3.2. Optimizing LAMP assay conditions for PRV detection
The LAMP reaction conditions were optimized by varying the concentration of MgSO4, dNTPs, primers, amplification temperature and reaction time. The results indicated that the reaction could be carried out when the Mg2+ concentration is higher than 4 mM and the optimal amplification was got at 8 mM (Fig. 2(A)). As shown in Fig. 2(B), the ladder-like bands could be obtained with dNTPs ranging from 0.4 to 1.2 mM while 1.0 mM is the best. Fig. 2(C) demonstrates that the target gene was amplified in the primer ratios ranging from 1:1 to 1:10 while the ratio at 1:8 gave the best amplification. As shown in Fig. 2(D and E), the amplification could be detected initially at 0.5 h and reached maximal at 1 h while the most clear pattern could be obtained at 63 °C.
Fig. 2.

Optimization of the LAMP reaction for PRV detection. (A) Effect of Mg2+ on the LAMP reaction: 1–6, Mg2+ was 2, 4, 6, 8, 10 and 12 mM, respectively. (B) Effect of dNTPs on the LAMP reaction: 1–7, dNTPs was 0, 0.2, 0.4, 0.6, 0.8, 1.0 and 1.2 mM, respectively. (C) The effect of ratio of outer and inner primers on the LAMP reaction: 1–5, ratio was 1:1, 1:2, 1:4, 1:8 and 1:10, respectively. (D) The effect of temperature on the LAMP reaction: 1–3, temperature located at 60, 63 and 65 °C, respectively. (E) The effect of reaction time on the LAMP reaction: 1–4, the amplification time was 15, 30, 45 and 60 min, respectively.
On the basis of the above results, the LAMP assay conditions were optimized in a 25 μl reaction volume as follows: 1× ThermoPol buffer, 8.0 mM MgSO4, 1.0 M betaine, 1.0 mM dNTPs, 0.2 μM each of outer primer, 1.6 μM each of inner primer and 0.4 μM each of loop primer, 8 U of Bst polymerase with 1 μl total DNA as template. The amplification was carried out at 63 °C for 1 h.
3.3. Interpretation of the LAMP products
Amplification products of LAMP were detected by agarose gel electrophoresis and visual inspection. As shown in Fig. 3(A), the target DNA sequence was amplified at 63 °C with a ladder-like pattern on the gel. The positive reaction was also indicated clearly by the white precipitate of magnesium pyrophosphate in Fig. 3(B) and the green color produced by the intercalating dye Picogreen® shown in Fig. 3(C). The specificity of the amplification product was confirmed by Hinc II digestion as indicated in Fig. 3(A, lane 2). Products of predictable sizes of the 80 and 108-bp motifs were resolved on the gel.
Fig. 3.
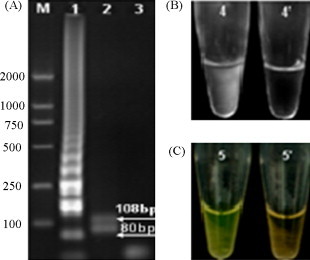
Agarose gel electrophoresis (A) and visual inspection of LAMP (B and C). 1, LAMP products of PRV; 2, LAMP products digested with Hinc II; 3, negative control; 4, positive LAMP reaction visualized by turbidity; 4′, negative LAMP reaction visualized by turbidity; 5, positive LAMP reaction visualized by adding Picogreen®; 5′, negative LAMP reaction with Picogreen®.
3.4. Sensitivity of LAMP for PRV detection
The sensitivity of LAMP was demonstrated and compared with PCR tests by using various PRV DNA dilutions (10−1 to 10−9) as templates. As shown in Fig. 4 , LAMP was able to detect template at 10−6 dilution (about 10 fg DNA), whereas PCR could only amplify the 10−3 to 10−4 dilution DNA sample. Therefore, the detection sensitivity of LAMP was 100–1000-fold higher than the PCR.
Fig. 4.
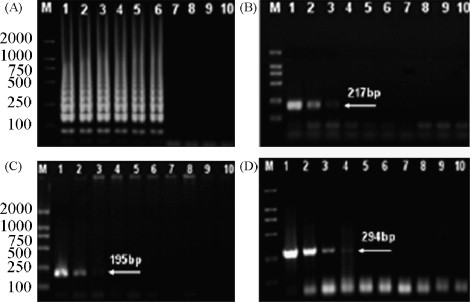
Sensitivity of LAMP for PRV detection. (A) The result obtained by LAMP. (B–D) The results obtained according to PCR tests by Chen et al. (2002), Balasch et al. (1998) and Lee et al. (2007), respectively. 1–9, DNA sample located at 10−1, 10−2, …, 10−9 dilutions, respectively; 10, negative control.
3.5. Specificity of LAMP for PRV detection
Three different PRV strains and six other viruses were investigated to confirm the specificity of the LAMP for PRV detection. The results showed that only the PRV is amplified while no amplification is performed in all other tested viruses (Fig. 5 ).
Fig. 5.
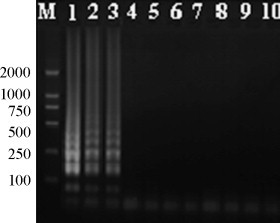
Specificity of LAMP for PRV detection. 1–9, PRV strain Fa, SA215, Bartha, porcine circovirus-2, porcine epidemic diarrhea virus, porcine reproductive and respiratory syndrome virus, swine transmissible gastroenteritis coronavirus, swine vesicular disease virus and swine fever virus, respectively; 10, negative control.
3.6. Evaluation of the LAMP assay using clinical samples
The evaluation of the LAMP assay was carried out using DNA extracted from five clinical samples with PCR running in parallel. The results of LAMP assay correlated well with the PCR, meanwhile, sample 2 was positive by LMAP, but not amplified by PCR (Table 3 ).
Table 3.
The LAMP assay and PCR with clinical samples
| Clinical samples | PCR | LAMP |
|---|---|---|
| 1 | + | + |
| 2 | − | + |
| 3 | − | − |
| 4 | + | + |
| 5 | + | + |
Note: “+”: positive; “−”: negative.
4. Discussion
To the best of our knowledge, this is the first report of LAMP applied successfully to establish the PRV detection system. Initial experiments were performed to optimize the assay conditions by using different concentration of MgSO4, dNTPs, primers, amplification temperature and reaction time. Among all factors analyzed, Mg2+ had the greatest affect on LAMP; it is possible that Mg2+ affects DNA polymerase activity and primer annealing (Saiki et al., 1998). The LAMP system developed in this study worked well during 60–65 °C, which would be useful for PRV detection in field practice when using a regular laboratory water bath. The results demonstrated that LAMP amplification products could be detected in 0.5 h, faster than PCR, so that diagnosis, including DNA extraction, amplification and product detection could be completed within 1 h after arrival of the samples. It could make the technique far more suitable for practice in the field.
It was observed that a large amount of pyrophosphate ion was produced when nucleic acid was amplified by LAMP, yielding white precipitate of magnesium pyrophosphate in the reaction mixture which allowed naked-eye detection (Mori et al., 2001). Alternatively, LAMP products could also be detected by adding intercalating dye Picogreen®. Positive amplification exhibited a green color while a negative reaction corresponded to an orange color (Dukes et al., 2006). The two visual inspection of LAMP product (Fig. 3(B and C)) are better detection methods as there is no need for gel electrophoresis and staining with ethidium bromide. Furthermore, it was found that all samples which tested positive/negative by visual inspection were also positive/negative when analyzed by electrophoresis. It may facilitate the application of LAMP, especially as a field test.
As a test for virus, the sensitivity is essential in cases where low concentration of virus is expected. The detection limit of the LAMP system established was about 10 fg DNA, much more sensitive than the PCR for PRV detection. Six other viruses were used in the study to confirm the specificity of LAMP; the results showed no DNA amplification in all viruses used, which makes LAMP more attractive for PRV detection in the field. The high specificity of LAMP is most probably attributable to recognition of the target sequence by six independent sequences in the initial stage and by four independent sequences during the second reaction stage (Notomi et al., 2000). A good correlation was obtained between the LAMP and PCR results when using five clinical samples collected from suspect cases of pseudorabies, which demonstrated the value in practice of LAMP. The LAMP assay could detect all the samples shown positive by PCR as well as sample 2.
References
- Balasch M., Pujols J., Segalés J., Pumarola M. Aujeszky's disease (pseudorabies) virus detection in cerebrospinal fluid in experimentally infected pigs. Vet. Microbiol. 1998;60:99–106. doi: 10.1016/s0378-1135(97)00156-9. [DOI] [PubMed] [Google Scholar]
- Blomström A.L., Hakhverdyan M., Reid S.M., Dukes J.P., King D.P., Belák S., Berg M. A one-step reverse transcriptase loop-mediated isothermal amplification assay for simple and rapid detection of swine vesicular disease virus. J. Virol. Methods. 2007;147:188–193. doi: 10.1016/j.jviromet.2007.08.023. [DOI] [PubMed] [Google Scholar]
- Chen H.C.H., He Q.G., Li X.C.H., Wu B., Fang L.R., Jin M.L., Qiu D.X., Wu M.Z.H. Standard Press; Beijing, China: 2002. Diagnostic Techniques for Aujeszky's Disease. [Google Scholar]
- Ducatelle R., Coussement W., Hoorens J. Immunoperoxidase study of Aujeszky's disease virus in pigs. Res. Vet. Sci. 1982;32:294–302. [PubMed] [Google Scholar]
- Dukes J.P., King D.P., Alexandersen S. Novel reverse transcription loop-mediated isothermal amplification for rapid detection of foot-and-mouth disease virus. Arch. Virol. 2006;151:1093–1106. doi: 10.1007/s00705-005-0708-5. [DOI] [PubMed] [Google Scholar]
- Hong T.C., Mai Q.L., Cuong D.V., Parida M., Minekawa H., Notomi T., Hasebe F., Morita K. Development and evaluation of a novel loop-mediated isothermal amplification method for rapid detection of severe acute respiratory syndrome coronavirus. J. Clin. Microbiol. 2004;42:1956–1961. doi: 10.1128/JCM.42.5.1956-1961.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai M., Ninomiya A., Minekawa H., Notomi T., Ishizaki T., Tu P.V., Tien N.T.K., Tashiro M., Odagiri T. Rapid diagnosis of H5N1 avian influenza virus infection by newly developed influenza H5 hemagglutinin gene-specific loop-mediated isothermal amplification method. J. Virol. Methods. 2007;141:173–180. doi: 10.1016/j.jviromet.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Kiatpathomchai W., Jareonram W., Jitrapakdee S., Flegel T.W. Rapid and sensitive detection of Taura syndrome virus by reverse transcription loop-mediated isothermal amplification. J. Virol. Methods. 2007;146:125–128. doi: 10.1016/j.jviromet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Li Q.Y., Xu Q.Y., Liu N., Zhang F.X. Loop-mediated isothermal amplification for rapid detection of hepatitis B virus. Lett. Biotech. 2005;16:647–648. [Google Scholar]
- Lee C.S., Moon H.J., Yang J.S., Park S.J., Song D.S., Kang B.K., Park B.K. Multiplex PCR for the simultaneous detection of pseudorabies virus, porcine cytomegalovirus, and porcine circovirus in pigs. J. Virol. Methods. 2007;139:39–43. doi: 10.1016/j.jviromet.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Mori Y., Nagamine K., Tomita N., Notomi T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001;289:150–154. doi: 10.1006/bbrc.2001.5921. [DOI] [PubMed] [Google Scholar]
- Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe k., Amino N., Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:e63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio F.A. Diagnosis of pseudorabies (Aujeszky's disease) virus infections. In: Morrison R.B., editor. Proceedings of the First International Symposium on the Eradication of Pseudorabies (Aujeszky's disease) Virus; St. Paul, USA; 1991. pp. 17–32. [Google Scholar]
- Pensaert M.B., Kluge J.P. Pseudorabies virus (Aujeszky's disease) In: Pensaert M.B., editor. Virus Infections of Porcines. Elsevier; Amsterdam: 1989. pp. 39–65. [Google Scholar]
- Pejsak Z.K., Truszczyński M.J. Truszczynsk: Aujeszky's disease (pseudorabies) In: Straw B.E., Zimmerman J.J., D’Allaire S., Taylor D.J., editors. Diseases of Swine. 9th ed. Blackwell Publishing Ltd.; Ames: 2006. pp. 419–433. [Google Scholar]
- Saiki R.K., Gelfand D.H., Stoffel S., Scharf S.J., Higuchi R., Horn G.T., Mullis K.B., Erlich H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1998;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]