

Highlights
-
•
Antiviral agents function as either viral targets or host factors.
-
•
Virus-targeting antivirals (VTAs) function through a direct (DVTAs) or an indirect (InDVTAs) method in the viral life cycle.
-
•
Host-targeting antivirals (HTAs) include reagents that target the host proteins that are involved in the viral life cycle.
Keywords: direct virus-targeting antiviral, indirect virus-targeting antiviral, host-targeting antiviral
Abstract
The prevalence of chronic viral infectious diseases, such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), and influenza virus; the emergence and re-emergence of new viral infections, such as picornaviruses and coronaviruses; and, particularly, resistance to currently used antiviral drugs have led to increased demand for new antiviral strategies and reagents. Increased understanding of the molecular mechanisms of viral infection has provided great potential for the discovery of new antiviral agents that target viral proteins or host factors. Virus-targeting antivirals can function directly or indirectly to inhibit the biological functions of viral proteins, mostly enzymatic activities, or to block viral replication machinery. Host-targeting antivirals target the host proteins that are involved in the viral life cycle, regulating the function of the immune system or other cellular processes in host cells. Here we review key targets and considerations for the development of both antiviral strategies.
Current antiviral strategies
Viruses comprise a large group of pathogens that are responsible for causing severe infectious diseases. Over the past 30 years, antiviral agents that target viral proteins or host factors have been successfully developed. Based on their inhibitory mechanisms, antiviral reagents can be divided into two groups: (i) inhibitors that target the viruses themselves or (ii) inhibitors that target host cell factors. Virus-targeting antivirals (VTAs) can function directly (DVTAs) or indirectly (InDVTAs) to inhibit biological functions of viral proteins, mostly enzymatic activities, or they block the correct formation of the viral replication machinery (Table 1 ). Host-targeting antivirals (HTAs) include reagents that target the host proteins that are involved in the viral life cycle (Figure 1 ), regulating the function of the immune system or other cellular processes in host cells. With increased knowledge of viral protein and host factors, the scientific community has achieved great progress in mechanism-based antiviral discovery against chronic viral infectious diseases, and in understanding of the emergence of new viral diseases and of the resistance to traditional antivirals. This review will highlight recent achievements in antiviral development and discuss various strategies for preventing virus attachment and entry into the host cell, as well as strategies for preventing virus replication and transcription within the host cell.
Table 1.
A summary of the antivirals described in this review
| Group | Subgroup | Name | Structure formula | Target and mechanism |
|---|---|---|---|---|
| Direct virus-targeting antivirals (DVTAs) | Attachment inhibitors | BMS488043 | 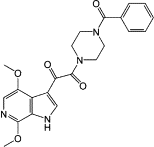 |
Block HIV-1 gp120–CD4 interaction |
| BMS663068 | 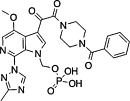 |
|||
| ICAM-1 | Block HRV–receptor interaction | |||
| Oseltamivir | 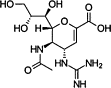 |
Influenza NAI | ||
| Zanamivir | 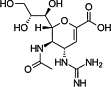 |
|||
| Laninamivir | 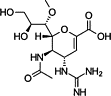 |
|||
| Peramivir | 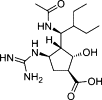 |
|||
| Entry inhibitors | T20 peptide (Enfuvirtide) | YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF | Block the conformational changes of HIV-1 gp41 | |
| Cp32M | VEWNEMTWMEWEREIENYTKLIYKILESSQEQ | |||
| Sifuvirtide | SWETWEREIENYTRQIYRILEESQEQQDRNERDLLE | |||
| T2635 | TTWEAWDRAIAEYAARIEALIRAAQEQQEKNEAALREL | |||
| Pleconaril | 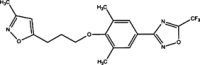 |
Replace the natural pocket factor and inhibit picornaviral uncoating | ||
| BTA798 | 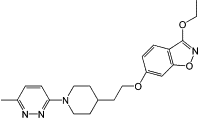 |
|||
| Protease inhibitors | Amprenavir | 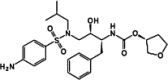 |
HIV-1 PIs | |
| Atazanavir | 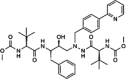 |
|||
| Darunavir | 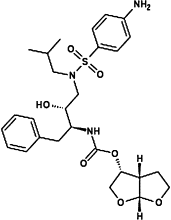 |
|||
| Fosamprenavir | 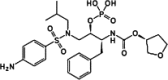 |
|||
| Indinavir | 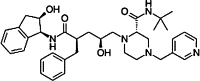 |
|||
| Lopinavir | 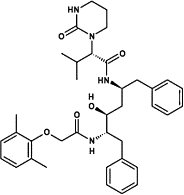 |
|||
| Nelfinavir | 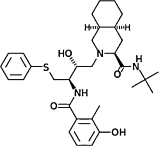 |
|||
| Ritonavir | 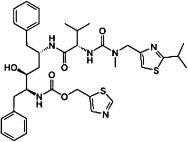 |
|||
| Saquinavir |  |
|||
| Tipranavir |  |
|||
| Polymerase inhibitors | Zidovudine (AZT) |  |
HIV-1 NRTI | |
| Didanosine (ddi) |  |
|||
| Zalcitabine (ddC) | 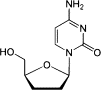 |
|||
| Stavudine (d4T) | 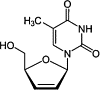 |
|||
| Lamivudine (3TC) | 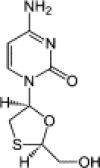 |
|||
| Nevirapine | 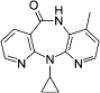 |
HIV-1 NNRTI | ||
| Delavirdine | 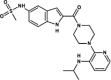 |
|||
| Efavirenz | 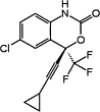 |
|||
| Etravirine | 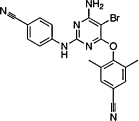 |
|||
| Rilpivirine | 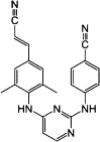 |
|||
| Integrase inhibitors | Raltegravir | 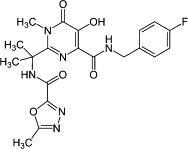 |
Integrase strand transfer inhibitor (INSTI) | |
| Dolutegravir | 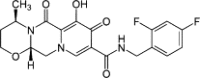 |
|||
| Elvitegravir (Stribild) | 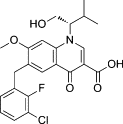 |
|||
| Methyltransferase inhibitors | Aurintricarboxylic acid | 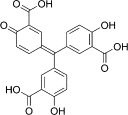 |
Inhibit the 2′-O activity of DENV MTase | |
| Sinefungin | 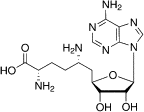 |
An analog of SAM that inhibits the activity of flavivirus MTase | ||
| BG323 | 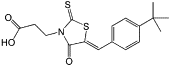 |
Inhibit the guanylyltransferase activity of DENV MTase | ||
| Helicase inhibitors | Biphenyls | 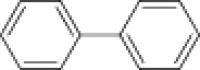 |
Inhibit HPV E1 helicase activity | |
| Biphenysulfonacetic acid | ||||
| Triclocarban (CID 7547) | 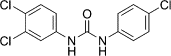 |
Inhibit SV40 Tag helicase activity | ||
| Bisphenol A (BPA; CID 6623) | 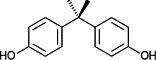 |
|||
| Triphenylmethanes (CID 42618092) | 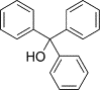 |
Inhibit HCV NS3 helicase activity | ||
| Aurintricarboxylic acid (ATA) | 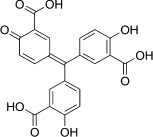 |
|||
| Indirect virus-targeting antivirals (InDVTAs) | RTC blockers | BMS790052 | 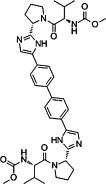 |
Inhibit the hyperphosphorylation of NS5A |
| RNP blockers | Nucleozin | 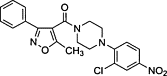 |
Inhibit the nuclear accumulation of influenza NP | |
| The first 25 amino acids of PB1 | GPLGSMDVNPTLLFLKVPAQNAISTTFPYT | Inhibit the interaction of PA-PB1 and influenza polymerase activity | ||
| Suramin | 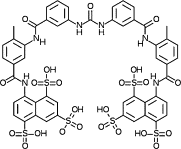 |
Bind to the RNA-binding cavity and inhibit SFTSV replication | ||
| Others | CHEMBL1207308 | 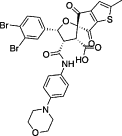 |
Inhibit the interaction of HPV E1–E2 | |
| Host-targeting antivirals (HTAs) | Cyclophilin inhibitors | Alisporivir (Debio-025) | 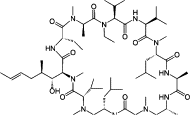 |
Inhibit the function of cyclophilins |
| NIM811 | 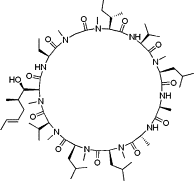 |
|||
| SCY635 | 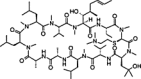 |
|||
| HIV-1 co-receptor antagonists | Aplaviroc | 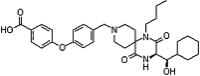 |
CCR5 antagonist | |
| Maraviroc | 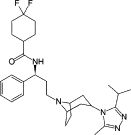 |
|||
| Vicriviroc | 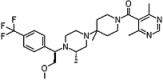 |
|||
| Cenicriviroc | 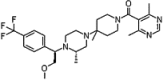 |
Figure 1.
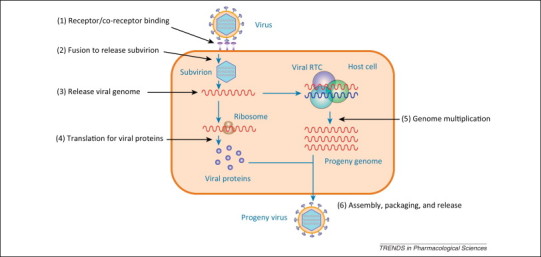
A schematic representation of the viral life cycle. The life cycle of a virus can be divided into six basic steps: 1. receptor or co-receptor binding; 2. fusion; 3. release of the viral genome; 4. translation of viral proteins; 5. genome multiplication; and 6. assembly, packaging, and release of the progeny virus. Because viral life cycles have extremely large discrepancies according to the type of virus, we refer the readers to find more detail in [143].
Direct virus-targeting antivirals
Attachment inhibitors
The first step in viral invasion is the attachment to host cells via an interaction with functional receptor(s). For enveloped viruses, the viral proteins located on the outer envelope of the virion are responsible for the recognition of receptors and the attachment to host cells. HIV (a member of the Retroviridae family) is a typical enveloped virus, and its invasion is mediated by the envelope proteins gp120 and gp41, which are arranged on the viral membrane as a trimer of three trans-membrane gp41 and three noncovalently attached gp120 surface subunits [1] (Figure 2 ). gp120 recognizes the CD4 receptor and launches the conformational changes that expose the binding sites for the binding of a co-receptor, (i.e., CCR5 and CXCR4 [2]). Antagonists that block the interactions between HIV and its receptor and co-receptors have therefore been developed as anti-HIV therapeutics. Attempts to find specific inhibitors that block the interaction between HIV-1 and CD4 were initiated using a soluble extracellular domain of CD4 protein that retained the ability to bind gp120. Although the preliminary results revealed that either soluble CD4 protein or a CD4–immunoglobulin fusion protein showed good in vitro anti-HIV activity, all failed in clinical trials due to poor pharmacokinetic features (e.g., the half-life of CD4–immunoglobulin fusion protein in mice is only 2.4 h) 3, 4, 5. Small molecule inhibitors that occupy a specific region within the CD4-binding pocket of gp120 were subsequently developed to block the gp120–CD4 interaction (Figure 2A). For example, BMS488043 [6] and BMS663068 [7] were found to significantly reduce HIV-1 proliferation and have good pharmaceutical characteristics.
Figure 2.
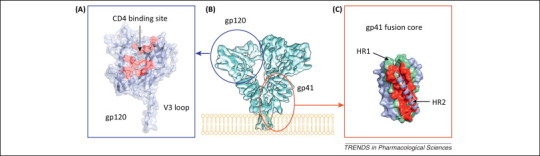
Mechanisms for antivirals targeting HIV-1 entry. (A) The crystal structure of HIV-1 gp120 with a complete V3 loop (PDB code: 2B4C[144]) is shown as a colored cartoon diagram covered by a transparent molecular surface. The gp120 molecule is colored in blue, and the CD4-binding site is highlighted in red. (B) The architecture of the HIV-1 Env (gp120–gp41) trimer presented as a cryo-EM map shown from a perspective parallel to the viral membrane [1]. The relative positions of gp120 and gp41 are circled. (C) The crystal structure of the HIV-1 gp41 fusion core (PDB code: 1DLB). HR1 and HR2 are colored green and blue, respectively. The core formed by the three HR1 and two HR2 fragments is covered with a molecular surface, whereas the remaining HR2 fragment is shown as a cartoon diagram. The binding site for the HR2 region is highlighted in red. (B) Reproduced and modified, with permission, from [1]. Abbreviations: HIV, human immunodeficiency virus; PDB, Protein Data Bank; cryo-EM, cryo-electron microscopy; HR, heptad repeat domain.
Another success is influenza neuraminidase (NA) inhibitor (NAI). Influenza NA is a surface glycoprotein and functions at two steps of the viral life cycle: (i) cleaves the cell receptor sialic acid residues, which bind to influenza hemagglutinin (HA), and allows the release of the progeny virus; and (ii) cleaves the sialic acid moieties on the mucin that bathes the airway epithelial cells or co-binds the receptor with HA [8]. In line with the structure of NA 9, 10, several NAIs have been successfully developed to competitively occupy the sialic acid-binding pocket of NA. Among these NAIs, oseltamivir and zanamivir were first used clinically as an anti-flu therapy [11]. Oseltamivir is a prodrug that is readily absorbed by the gastrointestinal tract and is converted by hepatic esterases to the active compound (oseltamivir carboxylate). Zanamivir has poor oral bioavailability and is currently available as a dry powder mixed with lactose. Moreover, laninamivir and peramivir were also approved in North Asia recently. Laninamivir has excellent in vitro activity against wild type, as well as oseltamivir-resistant, influenza viruses currently circulating [12]. Additionally, peramivir is another NAI that differs structurally from other inhibitors through novel substitutions that result in multiple binding interactions with the active site and allows the antiviral to be active against NAI-resistant viruses [13].
Non-enveloped viruses, such as the picornavirus (Picornaviridae family) and human papillomavirus (HPV) (Papillomaviridae family), interact with their functional receptors through viral capsid proteins. Picornaviruses are typical non-enveloped viruses, and some members, including enterovirus 71 (EV71) and human rhinoviruses (HRVs), are responsible for causing severe human infection diseases. The non-enveloped capsids of picornaviruses are icosahedral structures comprising 60 copies of viral structural proteins VP1–4 14, 15. VP1–3 each adopt a β-barrel configuration and are arranged with icosahedral symmetry such that VP1 surrounds the 5-fold axes and VP2 and VP3 alternate around the 2- and 3-fold axes [16]. Although the receptor-binding sites on the surface of picornavirus capsids are not conserved [17], these sites have been used to discover inhibitors that block virus–receptor interactions. For example, the canyon structure on the surface of the HRV capsid serves to bind to the HRV receptor, and the soluble portion of the intercellular adhesion molecule-1 (ICAM-1) [18] and numerous compounds that compete with the putative HRV receptor binding site have been shown to bind in a nearby hydrophobic pocket to inhibit virus attachment to the receptor [19]. However, none of these compounds have been clinical successes to date.
Entry inhibitors
After attaching to host cells, a virus will release its genome into the cytoplasm through endocytosis or direct membrane fusion. Because this viral entry is one of the key early steps in the viral life cycle (Figure 1), entry inhibitors have been successfully developed for antiviral therapies. As an enveloped virus, HIV-1 uses gp41 to facilitate its entry process after gp41 is activated by the binding of gp120 to the receptor. The extracellular portion of gp41 contains two heptad repeat domains (HR1 and HR2) separated by a loop region and a hydrophobic fusion peptide (FP) at the N terminus. During the fusion process, the FP of gp41 is inserted into the host cell membrane, and HR1 adopts a triple-stranded coiled-coil structure, forming a meta-stable prefusion intermediate. HR2 subsequently folds into the hydrophobic grooves of the HR1 coiled-coil to form a stable six-helix bundle that juxtaposes the viral and cellular membranes for fusion (Figure 2C).
Fusion inhibitors are designed to block the conformational changes that are required for membrane fusion. The T20 peptide (Enfuvirtide), which is a peptidic mimic of HR2 and acts by competitively binding to HR1, is the first and the only clinically approved fusion inhibitor 20, 21. T20 can inhibit a broad range of HIV strains at the nanomolar level, but the poor bioavailability of the drug (it has a plasma half-life of ∼4 h) make the clinical application of this drug difficult 22, 23. To overcome this pharmacokinetic disadvantage, a series of modified peptides including Cp32M [24], Sifuvirtide [25], and T2635 [26] have been generated to stabilize the helical structure of the HR2-like peptide by incorporating intrahelical salt bridges between the helix turns. In particular, the plasma half-life of sifuvirtide is 5-fold greater than that of T20 [27]. Moreover, chemical modifications including the PEGylation [28], glycosylation [29], and more have also been introduced to improve the pharmacokinetics of HR2-based fusion inhibitors.
Alternatively, a subset of bioavailable small molecule fusion inhibitors have been developed targeting the HR2-binding pocket on the HR1 trimer 30, 31, 32 or other undefined regions [33]. A similar strategy has been successfully used against many viruses that use a class I fusion mechanism, including influenza virus, Ebola virus (Filoviridae family), and severe acute respiratory syndrome coronavirus (SARS-CoV) (Coronaviridae family) [34], but this approach is rare in antiviral development targeting viruses with a class II/III fusion mechanism. However, the difficulty in production and delivery means that none of these small molecule fusion inhibitors can be approved for clinical use.
Non-enveloped viruses use a different strategy for virus entry. After attaching to their receptors, a non-enveloped virus releases its genome into the host cell through a conformational shift of its capsid protein(s). For example, the capsid of EV71 harbors 60 copies of a hydrophobic ‘pocket factor’, a natural lipid (sphingosine), at the base of the canyon in the capsid protein VP1 [35] (Figure 3 ). The expulsion of sphingosine following the binding of the virus to its receptor triggers the opening of the capsid to release the viral genome. Pleconaril [36] and BTA798 are two examples of hydrophobic compounds that can replace the natural pocket factor and inhibit picornaviral uncoating by generating resistance to the expulsion of pocket factor 37, 38, 39. Using the skeletons of pleconaril and related molecules, a novel class of imidazolidinones has been further synthesized with significant anti-EV71 activity (IC50 in the range of 0.001–25 μM) [40].
Figure 3.
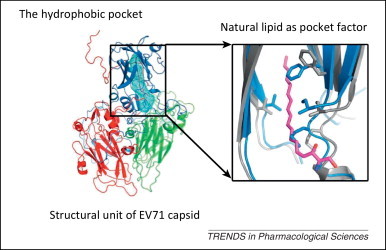
Pocket factor-binding site as a target for antivirals against picornaviral uncoating. The crystal structure of one structural unit, viral structural proteins 1 to 4 (VP1–4), of the enterovirus 71 (EV71) virus is shown as a cartoon diagram with VP1–4 depicted in blue, red, green, and yellow, respectively. The hydrophobic pocket (blue mesh) in VP1 EV71 is occupied by a natural lipid (magenta). The inset shows an enlarged structural comparison of the hydrophobic pockets of the mature virus (gray, with pocket factor shown in magenta) and empty particles (blue). Reproduced and modified, with permission, from [145].
Protease inhibitors
Most viruses encode one or several proteases that play crucial roles in the viral life cycle. The viral proteases carry out the proteolysis of a polyprotein precursor and release functional viral proteins, allowing them to function correctly and individually in replication/transcription and maturation 41, 42. Viral proteases also effectively protect viral proteins by modulating host cell pathways, including ubiquitination and ISGylation 43, 44, 45, 46, 47, 48. In contrast to the diversity of viral protease functions and structures, the catalytic active site of viral proteases generates stringent substrate specificity in protein cleavage. Synthetic substrate peptides, which can be designed according to the natural substrates of individual viral proteases, usually generate high-affinity binding and thus provide potent candidates for further drug discovery. One of the great successes is the HIV-1 protease inhibitors (PIs). There are ten PIs currently approved to treat HIV-1 infection: amprenavir (APV), atazanavir (ATZ), darunavir (TMC114), fosamprenavir (Lexiva), indinavir (IDV), lopinavir (LPV), nelfinavir (NFV), ritonavir (RTV), saquinavir (SQV), and tipranavir (TPV) [49]. All HIV-1 PIs share relatively similar chemical structures derived from its natural peptidic substrate and, therefore, the cross-resistance to PIs occurs at the active site of HIV-1 protease [50]. As a result, PIs are commonly used as the combination with other anti-HIV drugs to avoid drug resistance. Because most of the pharmaceutical disadvantages have been overcome, PIs have become the most potent types of antiviral drugs. Current progress in generating antiviral PIs has been systematically reviewed elsewhere 51, 52.
Polymerase inhibitors
Almost all viruses encode polymerases in the central steps of replication and transcription. Thus, polymerases are becoming the most attractive and suitable targets for antiviral development. Based on the function and structure of viral polymerases, there are two major types of polymerase inhibitors: (i) nucleoside and nucleotide substrate analogs and (ii) allosteric inhibitors. Nucleoside/nucleotide analogs play a dominant role in antiviral drugs targeting viral polymerases. Nucleoside analogs are first triphosphated by the host cell to produce the active inhibitor and then act as an inhibitor by competing with the natural nucleoside triphosphates and terminating the growing viral nucleic acids. The disadvantage of nucleoside analogs is that the initial phosphorylation step, that is, production of the monophosphorylated form, required for activation to a triphosphate may not correctly occur in the host cell [53]. Therefore, monophosphate nucleotide analogs were developed as polymerase inhibitors to avoid this problem. To date, most of the approved antiviral drugs for anti-HIV therapy utilize this mechanism, including Zidovudine (AZT, 3′-azido-2′,3′-dideoxythymidine), Didanosine (ddi, 2′,3′-dideoxyinosine), Zalcitabine (ddC, 2′,3′-dideoxycytidine), Stavudine (d4T, 2′,3′-dideoxy-2′,3′-didehydrothymidine), Lamivudine (3TC, (–)-β-l-3′-thia-2′,3′-dideoxycytidine), and others. The same strategy was also successfully used in the development of antivirals against a wide range of viruses, including cytomegalovirus (CMV) [54] and herpes simplex virus (HSV) [55] (Herpesviridae family), hepatitis B virus (HBV) (Hepadnaviridae family) [56], and HCV [57].
During the course of a polymerase cycle, the relative orientation of the polymerase domains undergoes a slight shift and this shift causes the conformational change of a specific site, the allosteric site, in the viral polymerase. Therefore, compounds that bind to the allosteric site could conceivably block the structural movement of polymerase domains and thus inhibit the function of viral polymerases. Antiviral inhibitors that work by this mechanism are known as ‘allosteric inhibitors’. The allosteric inhibitors of HIV reverse transcriptase (RT) are also known as non-nucleoside reverse transcriptase inhibitors (NNRTIs). NNRTIs and the hydrophobic binding site on HIV RT were first identified by screening compound libraries against HIV-1 RT combined with structural biological analysis 58, 59, 60, 61. The binding of NNRTI to HIV-1 RT prevents the flexibility and movement of RT required for the elongation of the nucleic acid. To date, there are five NNRTIs, that is, nevirapine, delavirdine, efavirenz, etravirine, and rilpivirine, that have been approved by the FDA for clinical use to treat HIV-1 infection. Furthermore, the development of NNRTIs against other viral polymerases, such as that of HCV, is ongoing, and other polymerases are likely to have suitable sites for allosteric inhibitors [62].
Integrase inhibitors
Integrase is an enzyme that helps the retrovirus to facilitate the incorporation of proviral DNA into the host cell genome and catalyzes a vital function in viral replication. Inhibitors of integrase represent the newest class of antiretroviral drugs (Figure 4 ). Raltegravir, an integrase strand transfer inhibitor (INSTI), was the first generation drug of this class to be approved and is a potent and well-tolerated antiviral agent [63]. Dolutegravir is the most advanced second generation INSTI, and it possess good tolerability, once-daily dosing with no need for a pharmacological enhancer, and relatively little cross-resistance with raltegravir [64]. Another INSTI, elvitegravir, was recently approved for the treatment of HIV infection as part of a fixed dose combination known as Stribild [65]. Furthermore, inhibitors of the lens epithelium-derived growth factor (LEDGF)/p75 binding site of integrase (LEDGINs) have also been developed, but these are still in a very early stage. Each of these drugs contributes a new benefit to the class and will extend the treatment options for patients with HIV-1 infection.
Figure 4.
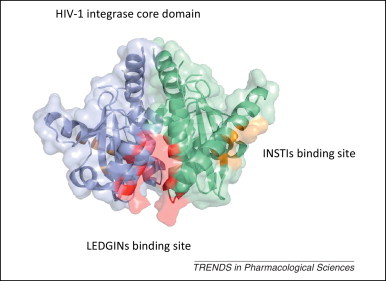
The sites in HIV-1 integrase targeted by antivirals. The crystal structure of the two dimeric HIV-1 integrase core domains are shown as a cartoon diagram covered with a transparent molecular surface and colored as blue and green (PDB code: 3ZSQ). The binding sites for INSTIs and LEDGINs are colored gold and red, respectively. Abbreviations: HIV, human immunodeficiency virus; PDB, Protein Data Bank; INSTI, integrase strand transfer inhibitor; LEDGIN, lens epithelium-derived growth factor (LEDGF)/p75 binding site of integrase.
Methyltransferase inhibitors
The 5′ terminus of the genomic RNA in a subset of RNA viruses requires a cap structure, which can be directly taken from the mRNA of host cells, as in influenza virus, or synthesized by viral enzymes, as in flavivirus and coronavirus. In the latter cases, a cap structure is generated by a series of enzymes and attached to the first nucleotide of the genome RNA via a 5′-5′ triphosphate linker. For example, the genome of flavivirus is capped by a type 1 cap structure (m7GpppAm), which is methylated at the N-7 position of the guanine (m7G) and on the 2′ OH of the ribose of the first nucleotide (Am) of the genome RNA [66]. Three enzymatic steps are required to form the cap structure, including an RNA triphosphatase (NS3), a guanylyl transferase (GTase), and a methyltransferase (MTase), provided by the N terminus of NS5 protein. Based on the structure of MTase and its complex with different substrates, three key ligand-binding sites were identified (Figure 5 ). The binding site for S-adenosyl methionine (SAM), which acts as the methyl donor, is located at the core domain of MTase, whereas the binding site for the receiver GTP molecule and the guanine moiety of the RNA cap is formed by the core domain and the N-terminal extension. Moreover, there is another site that binds RNA between the SAM and GTP pockets; this site is known to be a second, lower affinity binding site that is formed by the core domain and by a channel between two helices of the N-terminal extension.
Figure 5.
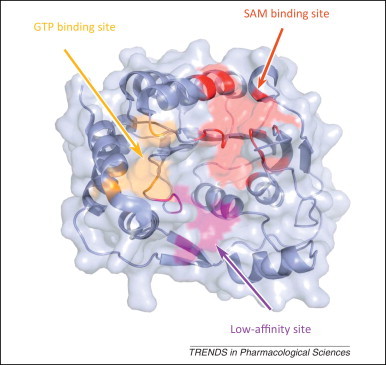
The sites in DENV MTase targeted by antivirals. The crystal structure of DENV MTase (PDB code: 3P8Z) is shown as a blue cartoon covered with transparent molecular surface. The SAM binding site, GTP binding site, and low-affinity site are highlighted in red, gold, and magenta, respectively. Abbreviations: DENV, dengue virus; PDB, Protein Data Bank; SAM, S-adenosyl methionine; MTase, methyltransferase.
Because these sites are crucial for MTase activity and virus replication, all are valid sites for the design of DVTAs (Figure 4). Ribavirin was first shown to inhibit the binding of the RNA cap to dengue virus (DENV) MTase by competitively binding to the GTP/RNA cap pocket [67]. Through structure-based drug design, aurintricarboxylic acid was found to inhibit the 2′-O activity of DENV MTase with an IC50 of 2 μM by binding to the lower affinity site [68]. Another inhibitor, sinefungin, which is an analog of SAM with the methylated sulfur of SAM replaced by a carbon and amine, can inhibit flavivirus activity with an IC50 as low as 0.7 μM [69]. Recently, the compound BG323 has been discovered using high-throughput screening (HTS) methods as an in vitro inhibitor of the guanylyltransferase activity of DENV MTase (and thus DENV replication) [70]. However, these inhibitors must be used at a high concentration to outcompete the high-affinity endogenous ligand, and it is necessary to solve this problem before this mechanism can be clinically exploited.
Helicase inhibitors
Some viruses utilize a viral helicase to separate one strand of DNA or RNA from the complementary strand in a process driven by adenosine triphosphate (ATP) hydrolysis. The most widely studied viral helicase is the flaviviral helicase, which is the helicase domain of nonstructural protein 3 (NS3) encoded by HCV. SARS-CoV (nsp13 protein), Simian virus 40 (SV40) (Polyomaviridae family) (TAg protein), and HPV (E1 protein) are also known to encode helicases for their replication [71].
The inhibitors of helicase-catalyzed ATP hydrolysis are the most straightforward class, and several nucleotide and nucleobase analogs have been discovered targeting this mechanism. Biphenyls and triphenylmethanes have been studied as inhibitors of SV40 TAg [72], HPV E1 [73], and HCV NS3 helicase [74]. Biphenyls and biphenysulfonacetic acid were found to inhibit HPV growth, an inhibitor of HPV E1-catalyzed ATP hydrolysis with an IC50 value >2 μM [75]. Compounds similar to triclocarban (CID7547) and Bisphenol A (BPA; CID6623) were found to inhibit the helicase activity of SV40 TAg. Based on the complex structure of the HCV NS3 helicase domain and soluble blue HT (PDB code 2ZJO), triphenylmethanes, a more potent triphenylmethane [76], and aurintricarboxylic acid (ATA) [77] were developed to inhibit the helicase activity of HCV NS3.
The nucleic acid binding site of helicase is also a promising site for the discovery of antiviral compounds that directly inhibit helicase-catalyzed unwinding [78]. For example, many DNA-binding pharmacophores, such as anthracyclines, acridones, tropolones, and amidinoanthracyclines, have been optimized as HCV helicase inhibitors [78]. Although there are numerous efforts that have been launched to find antivirals targeting viral helicase, little progress had been made to push them into clinical usage, and these projects have met great challenges on cytotoxicity, bioavailability, and pharmacokinetic properties [79].
Indirect virus-targeting antivirals
Replication and transcription complex blockers
Following viral entry, viral proteins, together with a number of host cell factors, assemble a viral replication and transcription complex (RTC) that is responsible for the production of the viral genome or other nucleic acids [80]. Therefore, reagents that can efficiently block the formation of the viral RTC could conceivably inhibit viral proliferation.
For example, once HCV enters the host cell, the genome of HCV will work as mRNAs to produce viral proteins and form an RTC in which the host factors account for viral replication and transcription. NS5A is a membrane-associated nonstructural phosphoprotein, and it is believed that NS5A has no enzymatic activity but plays a critical role in regulating the formation of the HCV RTC. Gao et al. identified a potent HCV inhibitor, BMS790052, that targets NS5A and produced few side effects in a Phase I clinical study [81]. Although the exact mechanism by which BMS790052 exerts its effects is yet to be defined, the resistance profile reveals that inhibitor sensitivity maps to the N terminus of domain 1 of NS5A. This inhibitor is further shown to block the hyperphosphorylation of NS5A, which is believed to play an essential role in the viral life cycle. This work, for the first time, proved the concept that small molecules targeting a non-traditional viral protein without any known enzymatic activity can also have profound antiviral effects with considerable promise for the treatment of HCV infection.
During the replication of EV71 and other picornaviruses, polymerase (also named 3Dpol), 3B (also named VPg), and protease (also named 3Cpro) participate in the formation of viral RTC, together with host polyA-binding protein 1 (PABP-1) and polyC-binding protein 2 (PCBP-2) [15]. Unlike many other viruses, the replication of the picornavirus genome is initiated by the 5′ end of genome covalently linked to VPg through a so-called VPg uridylylation process [82]. Although VPg-binding sites vary in picornaviruses 14, 15, 83, 84, the binding of VPg to 3Dpol is known to be critical for VPg uridylylation and virus replication. In our recent study, we demonstrated that a ten amino acid peptide of VPg can effectively inhibit the VPg uridylylation process with an EC50 as low as 50 nM (Z. Lou et al., unpublished). These results shed light on discovering future InDVTAs.
Ribonucleoprotein complex inhibitors
Throughout the life cycle of a negative sense single-stranded RNA (ssRNA) virus, the genome length RNA is encapsidated by a virally encoded nucleoprotein (NP), instead of a naked RNA, and associated with RdRp (polymerase complex) to form a stable ribonucleoprotein (RNP) complex, which is responsible for virus replication, transcription, and assembly [85]. During this process, NP can protect the RNA against exogenous nucleases or the innate immune system in the host cell.
Based on the crystallographic achievements regarding the ssRNA virus RNP complex 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96 over the past few years, great progress has been made in this new putative antiviral strategy. Kao et al. first identified a compound, nucleozin, that triggers the aggregation of and inhibits the nuclear accumulation of NP; this compound can inhibit the replication of influenza virus with a nanomolar median effective concentration (EC50) [97]. In a parallel effort, Gerritz et al. discovered a series of influenza replication inhibitors and showed that they interfere with NP-dependent processes via the formation of higher order NP oligomers with an EC50 of 60 nM [98]. Notably, the structure of the NP in complex with a representative compound from this class of inhibitors revealed that two molecules of an inhibitor in an antiparallel orientation lock two adjacent NP protomers. This unexpected quaternary complex explained the viral inhibition via ligand-induced formation of stable NP oligomers [98]. In addition to inhibiting NP, the disruption of the polymerase complex of influenza virus has been proposed for antiviral strategies. Based on the complex structure of the PA C-terminal domain (PAC) and the first 25 amino acids of PB1 [99], a subset of modifications on N-terminal peptide of PB1 was shown to diminish the binding affinity of PA and PB1, inhibit polymerase activity, and attenuate the replication of influenza virus 100, 101, 102. Moreover, the structure of SFTSV NP in complex with Suramin, an antiviral inhibitor, revealed that the blockers bind to the RNA-binding cavity and can attenuate SFTSV replication; this indicated that targeting RNP formation may be a new therapeutic antiviral approach 103, 104. Because both the polymerase complex and NP show significant conservation between different influenza viruses, these results demonstrated that targeting the formation of viral RNP is a valid approach to the development of small molecule therapies against serious antiviral resistance to currently available drugs, such as adamantanes or neuraminidase inhibitors.
Other key interaction blockers during the viral life cycle
Other key interactions between viral proteins and/or host factors play essential roles in virus entry, replication, and maturation. A very interesting example is the interaction between HPV E1 and E2 protein, helping E1 helicase to tether to the HPV origin of replication. This interaction can therefore be used to guide the development of antivirals treating HPV infection. The disruption of the HPV E1–E2 interaction was first facilitated by a very potent inhibitor, CHEMBL1207308. This inhibitor can effectively diminish the interaction between HPV E1 and E2 and thus inhibit HPV proliferation with an EC50 of 6 nM [105].
Although a few InDVTAs have been discovered for antiviral therapy, their mechanisms of working are still required to be further investigated. We cannot conclude the possibility that InDVTAs may have additional functions (or may be the major function) and the category of antivirals will be refined according to further knowledge.
Host-targeting antivirals
Viruses utilize many host factors for their efficient proliferation. The correct functions of these host factors are crucial for virus replication and, therefore, compounds that regulate the function of host factors can be introduced as antiviral agents. Next we discuss two host factors that are involved in broad spectrum virus infection.
Interferon
Apart from the acquired immune response, host cells mount a number of defenses, such as the innate immune response, to virus infection. Interferon (IFN) is one of the most crucial molecules in the innate immune response and acts as the primary switch for initiating antiviral immunity in vertebrates. Upon being infected by a virus, host cells produce and secrete type I (mainly IFN-α and IFN-β) and type III (IFN-λ) IFNs. These secreted IFNs interact with the membrane-anchored IFN receptors (IFNARs), mainly IFNAR1 and INFAR2, and subsequently stimulate and upregulate the expression of hundreds of IFN-stimulated genes (ISGs) to inhibit the replication of viruses [106]. Among these ISGs, IFN-inducible transmembrane (IFITM) proteins restrict the entry of influenza virus, West Nile virus (WNV), and DENV. Additionally, Mx (myxovirus resistance) GTPases can inhibit the correct function of viral nucleocapsids and polymerases, such as influenza virus [107].
Due to the significance of IFN in the host cells to restrict viral infections, IFN is used in certain instances as a primary antiviral therapy, particularly in the absence of an effective antiviral or vaccination strategy. The use of IFN for antiviral therapy was first developed for treating HBV infection [108] and subsequently applied to treat HCV in combination with ribavirin [109]. IFN-β [110] and IFN-γ [111] have also been evaluated for use in anti-HCV treatment. To further improve the therapeutic efficacy of IFN-α therapy, several options are being investigated. Some clinical benefits were observed in pilot studies with ofloxacin 112, 113 and an immunomodulatory peptide, α1-thymosin [114]. In particular, PEG-modified IFN-α [115] combined with ribavirin is now standard treatment for HCV infection.
Cyclophilin inhibitors
Cyclophilins (Cyps) are key cellular factors that function in numerous cellular processes, including transcriptional regulation, the immune response, protein secretion, and mitochondrial function 116, 117. Cyclophilin A (CypA) is a key member of the Cyp family and was first identified as a mediator of the immunosuppressive function of cyclosporin A (CsA) through the formation of a CsA–CypA complex. This complex binds to and inhibits the function of the protein phosphatase calcineurin [118], which normally functions to dephosphorylate NF-AT, a transcription factor important for T cell activation.
CypA is also known to play critical roles in the proliferation of viruses, including HIV-1, influenza virus, HCV, vesicular stomatitis virus (VSV), vaccinia virus, SARS-CoV, rotavirus (RV), and HPV 117, 119, by interacting with viral proteins or facilitating IFN-β production 14, 120, 121, 122, 123. CypA was first revealed to be incorporated into HIV-1 virions by interacting with the capsid protein (CA), and an interaction between newly synthesized HIV-1 CA and CypA is required for HIV-1 to induce dendritic cell maturation 114, 124. CypA also interacts with other HIV-1 proteins, such as Vpr and p6, to regulate HIV infection 88, 91, 125, 126, 127, 128. Several lines of evidence indicate that CypA and cyclophilin B (CypB) [129] function in the replication of HCV by either increasing the affinity of HCV polymerase NS5B for viral RNA to enhance HCV replication [130] or binding to the HCV NS5A protein to aid viral replication [131].
The discovery of CypA inhibitors as antiviral agents started with the immunosuppressive drug CsA that inhibits HCV [129] and HIV [132]. On the basis of these results, CypA antagonists, which are often derived from CsA, have been developed, including alisporivir (Debio-025), NIM811, and SCY635; these compounds lack immunosuppressive effects but retain high-affinity CypA binding and show very good antiviral effects against HIV or HCV infection 133, 134, 135.
HIV-1 co-receptors antagonists
HIV-1 co-receptor antagonists that block the interactions between HIV-1 and CCR5 and/or CXCR4 have also been introduced to the anti-HIV efforts, and a few of these have been successful. For instance, CCR5 antagonists that potently inhibit HIV-1 replication and have good pharmaceutical properties, including aplaviroc [136], maraviroc (the only clinically used anti-HIV drug as a co-receptor antagonist) [137], vicriviroc [138], and cenicriviroc 139, 140, have been successfully advanced to Phase II/III clinical trials or approved for clinical usage in the past 5 years. However, the precise mechanisms of these co-receptor antagonists are still not clear. Recently, the crystal structures of CCR5 [141] and CXCR4 [142] have been reported. These two structures provided the atomic information of the ligand-binding pocket and the sites for co-receptor oligomerization. Further structural study on the complex of CCR5 and CXCR4 with their antagonists will promote the investigation into the inhibitory mechanisms of these inhibitors and help us to increase their anti-HIV efficacy.
Concluding remarks
At present, diverse antiviral drugs are clinically approved or in the later stages of clinical trials. Most are based on the conserved mechanisms described in this review, in particular through targeting of viral polymerases and proteases. However, the majority of drug-resistant infection cases have been reported with the usage of antivirals based on this strategy. The requirement for new antiviral drugs to treat chronic infectious diseases and the emergence of more efficient new viruses serve as catalysts for research to find additional targets and mechanisms for antiviral development.
A few novel strategies have been introduced for antiviral research, including inhibitors of viral MTase and helicase, blockages of viral RTC formation (e.g., nucleozin to influenza), and host factor antagonists or agonists. However, protease and polymerase inhibitors still occupy a dominant place among antivirals. This is because we still do not have very precise knowledge on the mechanisms underlying alternative strategies, and this requires further investigations on their mechanism before these strategies can be widely used for antiviral development, in particular those targeting drug-resistant viral infections. In our opinion, protease and polymerase inhibitors will still be the first, and probably the major, choice in the development of therapies against emerging novel viruses.
In addition, we must consider another important aspect for antiviral development. For some viruses, currently available drugs can effectively eliminate the virus in the host, but genetic components are left behind. Thus, the host still suffers from the infection because the virus can integrate its genetics into the host cell. For example, treatment with the combination of INF and adefovir (or other antivirals) can effectively, if not completely, reduce HBV titer in human liver. However, the covalently closed circular DNA (cccDNA) cannot be removed and still exists in the nuclei of infected liver cells, where it continuously coordinates the expression of HBV antigens. Therefore, new mechanisms and strategies to completely remove viral components integrated in host cells, as well as to kill the virus itself, are required in future antiviral development.
Acknowledgments
This work was supported by the Ministry of Science and Technology of China ‘973’ Project (grant numbers 2013CB911103 and 2014CB542800) and the National Natural Science Foundation of China (grant numbers 81322023, 31000332, 31100208, 31170678, and 31370733).
Contributor Information
Zhiyong Lou, Email: louzy@xtal.tsinghua.edu.cn.
Zihe Rao, Email: raozh@xtal.tsinghua.edu.cn.
References
- 1.Mao Y. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc. Natl. Acad. Sci. U.S.A. 2013;110:12438–12443. doi: 10.1073/pnas.1307382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgiev I.S. Elicitation of HIV-1-neutralizing antibodies against the CD4-binding site. Curr. Opin. HIV AIDS. 2013;8:382–391. doi: 10.1097/COH.0b013e328363a90e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyczak J.B., Morrison S.L. Biological and pharmacokinetic properties of a novel immunoglobulin–CD4 fusion protein. Arch. Virol. 1994;139:189–196. doi: 10.1007/BF01309464. [DOI] [PubMed] [Google Scholar]
- 4.Schooley R.T. Recombinant soluble CD4 therapy in patients with the acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. A Phase I–II escalating dosage trial. Ann. Intern. Med. 1990;112:247–253. doi: 10.7326/0003-4819-112-4-247. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson J.M. Treatment of advanced human immunodeficiency virus type 1 disease with the viral entry inhibitor PRO 542. Antimicrob. Agents Chemother. 2004;48:423–429. doi: 10.1128/AAC.48.2.423-429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanna G.J. Antiviral activity, pharmacokinetics, and safety of BMS-488043, a novel oral small-molecule HIV-1 attachment inhibitor, in HIV-1-infected subjects. Antimicrob. Agents Chemother. 2012;55:722–728. doi: 10.1128/AAC.00759-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowicka-Sans B. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. 2012;56:3498–3507. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hooper K.A., Bloom J.D. A mutant influenza virus that uses an n1 neuraminidase as the receptor-binding protein. J. Virol. 2013;87:12531–12540. doi: 10.1128/JVI.01889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colman P.M. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature. 1983;303:41–44. doi: 10.1038/303041a0. [DOI] [PubMed] [Google Scholar]
- 10.Varghese J.N. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 A resolution. Nature. 1983;303:35–40. doi: 10.1038/303035a0. [DOI] [PubMed] [Google Scholar]
- 11.Ison M.G. Antivirals and resistance: influenza virus. Curr. Opin. Virol. 2011;1:563–573. doi: 10.1016/j.coviro.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Yamashita M. Laninamivir and its prodrug, CS-8958: long-acting neuraminidase inhibitors for the treatment of influenza. Antivir. Chem. Chemother. 2010;21:71–84. doi: 10.3851/IMP1688. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez J.E. Clinical experience in adults and children treated with intravenous peramivir for 2009 influenza A (H1N1) under an Emergency IND program in the United States. Clin. Infect. Dis. 2011;52:695–706. doi: 10.1093/cid/cir001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C. Crystal structure of enterovirus 71 RNA-dependent RNA polymerase complexed with its protein primer VPg: implication for a trans mechanism of VPg uridylylation. J. Virol. 2013;87:5755–5768. doi: 10.1128/JVI.02733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Y. Enterovirus 71 VPg uridylation uses a two-molecular mechanism of 3D polymerase. J. Virol. 2012;86:13662–13671. doi: 10.1128/JVI.01712-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porta C. Rational engineering of recombinant picornavirus capsids to produce safe, protective vaccine antigen. PLoS Pathog. 2013;9:e1003255. doi: 10.1371/journal.ppat.1003255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fry E.E. Crystal structure of equine rhinitis A virus in complex with its sialic acid receptor. J. Gen. Virol. 2010;91:1971–1977. doi: 10.1099/vir.0.020420-0. [DOI] [PubMed] [Google Scholar]
- 18.Marlin S.D. A soluble form of intercellular adhesion molecule-1 inhibits rhinovirus infection. Nature. 1990;344:70–72. doi: 10.1038/344070a0. [DOI] [PubMed] [Google Scholar]
- 19.Murray M.A., Babe L.M. Inhibitory effect of dibenzofuran and dibenzosuberol derivatives on rhinovirus replication in vitro; effective prevention of viral entry by dibenzosuberenone. Antiviral Res. 1999;44:123–131. doi: 10.1016/s0166-3542(99)00061-3. [DOI] [PubMed] [Google Scholar]
- 20.Wild C.T. Peptides corresponding to a predictive α-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilby J.M. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 22.Kilby J.M. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retroviruses. 2002;18:685–693. doi: 10.1089/088922202760072294. [DOI] [PubMed] [Google Scholar]
- 23.Lalezari J.P. A Phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy. AIDS. 2003;17:691–698. doi: 10.1097/00002030-200303280-00007. [DOI] [PubMed] [Google Scholar]
- 24.He Y. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16332–16337. doi: 10.1073/pnas.0807335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Y. Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr. Pharm. Des. 2013;19:1800–1809. doi: 10.2174/1381612811319100004. [DOI] [PubMed] [Google Scholar]
- 26.Dwyer J.J. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Y. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008;283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 28.Veronese F.M., Pasut G. PEGylation, successful approach to drug delivery. Drug Discov. Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 29.Sinclair A.M., Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J. Pharm. Sci. 2005;94:1626–1635. doi: 10.1002/jps.20319. [DOI] [PubMed] [Google Scholar]
- 30.Wang H. ADS-J1 inhibits human immunodeficiency virus type 1 entry by interacting with the gp41 pocket region and blocking fusion-active gp41 core formation. Antimicrob. Agents Chemother. 2009;53:4987–4998. doi: 10.1128/AAC.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang S. Design, synthesis, and biological activity of novel 5-((arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl)phenyl)thi azolidin-4-ones as HIV-1 fusion inhibitors targeting gp41. J. Med. Chem. 2011;54:572–579. doi: 10.1021/jm101014v. [DOI] [PubMed] [Google Scholar]
- 32.Katritzky A.R. Design, synthesis, and structure–activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009;52:7631–7639. doi: 10.1021/jm900450n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray E.J. A low-molecular-weight entry inhibitor of both CCR5- and CXCR4-tropic strains of human immunodeficiency virus type 1 targets a novel site on gp41. J. Virol. 2010;84:7288–7299. doi: 10.1128/JVI.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrison S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren J. Picornavirus uncoating intermediate captured in atomic detail. Nat. Commun. 2013;4:1929. doi: 10.1038/ncomms2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang G. In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch. Virol. 2011;157:669–679. doi: 10.1007/s00705-011-1222-6. [DOI] [PubMed] [Google Scholar]
- 37.Rotbart H.A. Treatment of picornavirus infections. Antiviral Res. 2002;53:83–98. doi: 10.1016/s0166-3542(01)00206-6. [DOI] [PubMed] [Google Scholar]
- 38.Phelps D.K., Post C.B. A novel basis of capsid stabilization by antiviral compounds. J. Mol. Biol. 1995;254:544–551. doi: 10.1006/jmbi.1995.0637. [DOI] [PubMed] [Google Scholar]
- 39.Tsang S.K. Stabilization of poliovirus by capsid-binding antiviral drugs is due to entropic effects. J. Mol. Biol. 2000;296:335–340. doi: 10.1006/jmbi.1999.3483. [DOI] [PubMed] [Google Scholar]
- 40.Shia K.S. Design, synthesis, and structure–activity relationship of pyridyl imidazolidinones: a novel class of potent and selective human enterovirus 71 inhibitors. J. Med. Chem. 2002;45:1644–1655. doi: 10.1021/jm010536a. [DOI] [PubMed] [Google Scholar]
- 41.Summers D.F., Maizel J.V., Jr Evidence for large precursor proteins in poliovirus synthesis. Proc. Natl. Acad. Sci. U.S.A. 1968;59:966–971. doi: 10.1073/pnas.59.3.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pelham H.R. Translation of encephalomyocarditis virus RNA in vitro yields an active proteolytic processing enzyme. Eur. J. Biochem. 1978;85:457–462. doi: 10.1111/j.1432-1033.1978.tb12260.x. [DOI] [PubMed] [Google Scholar]
- 43.Thiel V. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 44.Harcourt B.H. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratia K. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlieker C. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell. 2007;25:677–687. doi: 10.1016/j.molcel.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balakirev M.Y. Deubiquitinating function of adenovirus proteinase. J. Virol. 2002;76:6323–6331. doi: 10.1128/JVI.76.12.6323-6331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Z. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism function. J. Virol. 2010;84:7832–7846. doi: 10.1128/JVI.00217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llibre J.M. From TMC114 to darunavir: five years of data on efficacy. AIDS Rev. 2013;15:112–121. [PubMed] [Google Scholar]
- 50.Nijhuis M. Implications of antiretroviral resistance on viral fitness. Curr. Opin. Infect. Dis. 2001;14:23–28. doi: 10.1097/00001432-200102000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Pearlman B.L. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect. Dis. 2012;12:717–728. doi: 10.1016/S1473-3099(12)70060-9. [DOI] [PubMed] [Google Scholar]
- 52.Andany N., Loutfy M.R. HIV protease inhibitors in pregnancy: pharmacology and clinical use. Drugs. 2013;73:229–247. doi: 10.1007/s40265-013-0017-3. [DOI] [PubMed] [Google Scholar]
- 53.Oberg B. Rational design of polymerase inhibitors as antiviral drugs. Antiviral Res. 2006;71:90–95. doi: 10.1016/j.antiviral.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 54.Andrei G. Drug targets in cytomegalovirus infection. Infect. Disord. Drug Targets. 2009;9:201–222. doi: 10.2174/187152609787847758. [DOI] [PubMed] [Google Scholar]
- 55.Billich A. Entecavir (Bristol-Myers Squibb) Curr. Opin. Investig. Drugs. 2001;2:617–621. [PubMed] [Google Scholar]
- 56.Palumbo E. New drugs for chronic hepatitis B: a review. Am. J. Ther. 2008;15:167–172. doi: 10.1097/MJT.0b013e318155a191. [DOI] [PubMed] [Google Scholar]
- 57.Wegzyn C.M., Wyles D.L. Antiviral drug advances in the treatment of human immunodeficiency virus (HIV) and chronic hepatitis C virus (HCV) Curr. Opin. Pharmacol. 2012;12:556–561. doi: 10.1016/j.coph.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 58.Miyasaka T. A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J. Med. Chem. 1989;32:2507–2509. doi: 10.1021/jm00132a002. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka H. A new class of HIV-1-specific 6-substituted acyclouridine derivatives: synthesis and anti-HIV-1 activity of 5- or 6-substituted analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) J. Med. Chem. 1991;34:349–357. doi: 10.1021/jm00105a055. [DOI] [PubMed] [Google Scholar]
- 60.Goldman M.E. Pyridinone derivatives: specific human immunodeficiency virus type 1 reverse transcriptase inhibitors with antiviral activity. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6863–6867. doi: 10.1073/pnas.88.15.6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rawal R.K. Structure–activity relationship studies on clinically relevant HIV-1 NNRTIs. Curr. Med. Chem. 2012;19:5364–5380. doi: 10.2174/092986712803833326. [DOI] [PubMed] [Google Scholar]
- 62.Beaulieu P.L. Non-nucleoside inhibitors of the HCV NS5B polymerase: progress in the discovery and development of novel agents for the treatment of HCV infections. Curr. Opin. Investig. Drugs. 2007;8:614–634. [PubMed] [Google Scholar]
- 63.Karmon S.L., Markowitz M. Next-generation integrase inhibitors: where to after raltegravir? Drugs. 2013;73:213–228. doi: 10.1007/s40265-013-0015-5. [DOI] [PubMed] [Google Scholar]
- 64.Messiaen P. Clinical use of HIV integrase inhibitors: a systematic review and meta-analysis. PLoS ONE. 2013;8:e52562. doi: 10.1371/journal.pone.0052562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sax P.E. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379:2439–2448. doi: 10.1016/S0140-6736(12)60917-9. [DOI] [PubMed] [Google Scholar]
- 66.Noble C.G., Shi P.Y. Structural biology of dengue virus enzymes: towards rational design of therapeutics. Antiviral Res. 2013;96:115–126. doi: 10.1016/j.antiviral.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 67.Benarroch D. A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 2004;279:35638–35643. doi: 10.1074/jbc.M400460200. [DOI] [PubMed] [Google Scholar]
- 68.Milani M. Flaviviral methyltransferase/RNA interaction: structural basis for enzyme inhibition. Antiviral Res. 2009;83:28–34. doi: 10.1016/j.antiviral.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong H. Structural and functional analyses of a conserved hydrophobic pocket of flavivirus methyltransferase. J. Biol. Chem. 2010;285:32586–32595. doi: 10.1074/jbc.M110.129197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stahla-Beek H.J. Identification of a novel antiviral inhibitor of the flavivirus guanylyltransferase enzyme. J. Virol. 2012;86:8730–8739. doi: 10.1128/JVI.00384-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keum Y.S., Jeong Y.J. Development of chemical inhibitors of the SARS coronavirus: viral helicase as a potential target. Biochem. Pharmacol. 2012;84:1351–1358. doi: 10.1016/j.bcp.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seguin S.P. A screen for modulators of large T antigen's ATPase activity uncovers novel inhibitors of Simian Virus 40 and BK virus replication. Antiviral Res. 2012;96:70–81. doi: 10.1016/j.antiviral.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White P.W. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486. Antimicrob. Agents Chemother. 2005;49:4834–4842. doi: 10.1128/AAC.49.12.4834-4842.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shadrick W.R. Discovering new medicines targeting helicases: challenges and recent progress. J. Biomol. Screen. 2012;18:761–781. doi: 10.1177/1087057113482586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Faucher A.M. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase. J. Med. Chem. 2004;47:18–21. doi: 10.1021/jm034206x. [DOI] [PubMed] [Google Scholar]
- 76.Chen C.S. Structure-based discovery of triphenylmethane derivatives as inhibitors of hepatitis C virus helicase. J. Med. Chem. 2009;52:2716–2723. doi: 10.1021/jm8011905. [DOI] [PubMed] [Google Scholar]
- 77.Chen Y. Characterization of aurintricarboxylic acid as a potent hepatitis C virus replicase inhibitor. Antivir. Chem. Chemother. 2009;20:19–36. doi: 10.3851/IMP1286. [DOI] [PubMed] [Google Scholar]
- 78.Belon C.A., Frick D.N. NS3 helicase inhibitors. In: He Y., Tan S.L., editors. Hepatitis C: Antiviral Drug Discovery and Development. Caister Academic; 2011. pp. 237–256. [Google Scholar]
- 79.Kwong A.D. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discov. 2005;4:845–853. doi: 10.1038/nrd1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagy P.D., Pogany J. The dependence of viral RNA replication on co-opted host factors. Nat. Rev. Microbiol. 2012;10:137–149. doi: 10.1038/nrmicro2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao M. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ambros V., Baltimore D. Protein is linked to the 5′ end of poliovirus RNA by a phosphodiester linkage to tyrosine. J. Biol. Chem. 1978;253:5263–5266. [PubMed] [Google Scholar]
- 83.Ferrer-Orta C. The structure of a protein primer–polymerase complex in the initiation of genome replication. EMBO J. 2006;25:880–888. doi: 10.1038/sj.emboj.7600971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gruez A. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J. Virol. 2008;82:9577–9590. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kranzusch P., Whelan S. Architecture and regulation of negative-strand viral enzymatic machinery. RNA Biol. 2012;9:941–948. doi: 10.4161/rna.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ariza A. Nucleocapsid protein structures from orthobunyaviruses reveal insight into ribonucleoprotein architecture and RNA polymerization. Nucleic Acids Res. 2013;41:5912–5926. doi: 10.1093/nar/gkt268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arranz R. The structure of native influenza virion ribonucleoproteins. Science. 2013;338:1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 88.Guo Y. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5046–5051. doi: 10.1073/pnas.1200808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hastie K.M. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U.S.A. 2011;108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hastie K.M. Crystal structure of the Lassa virus nucleoprotein–RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. U.S.A. 2011;108:19365–19370. doi: 10.1073/pnas.1108515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li B. Bunyamwera virus possesses a distinct nucleocapsid protein to facilitate genome encapsidation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:9048–9053. doi: 10.1073/pnas.1222552110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moeller A. Organization of the influenza virus replication machinery. Science. 2013;338:1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qi X. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature. 2011;468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raymond D.D. Phleboviruses encapsidate their genomes by sequestering RNA bases. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19208–19213. doi: 10.1073/pnas.1213553109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Raymond D.D. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11769–11774. doi: 10.1073/pnas.1001760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reguera J. Structural basis for encapsidation of genomic RNA by La Crosse Orthobunyavirus nucleoprotein. Proc. Natl. Acad. Sci. U.S.A. 2013;110:7246–7251. doi: 10.1073/pnas.1302298110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kao R.Y. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 2010;28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gerritz S.W. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl. Acad. Sci. U.S.A. 2011;108:15366–15371. doi: 10.1073/pnas.1107906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.He X. Crystal structure of the polymerase PA(C)–PB1(N) complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 100.Wunderlich K. Identification of high-affinity PB1-derived peptides with enhanced affinity to the PA protein of influenza A virus polymerase. Antimicrob. Agents Chemother. 2011;55:696–702. doi: 10.1128/AAC.01419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wunderlich K. Identification of a PA-binding peptide with inhibitory activity against influenza A and B virus replication. PLoS ONE. 2009;4:e7517. doi: 10.1371/journal.pone.0007517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Manz B. Disruption of the viral polymerase complex assembly as a novel approach to attenuate influenza A virus. J. Biol. Chem. 2011;286:8414–8424. doi: 10.1074/jbc.M110.205534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou H. The nucleoprotein of severe fever with thrombocytopenia syndrome virus processes a stable hexameric ring to facilitate RNA encapsidation. Protein Cell. 2013;4:445–455. doi: 10.1007/s13238-013-3901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y. Structure of Crimean-Congo haemorraghic fever virus nucleoprotein: superhelical homo-oligomers and the role of caspase-3 cleavage. J. Virol. 2012;86:12294–12303. doi: 10.1128/JVI.01627-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goudreau N. Optimization and determination of the absolute configuration of a series of potent inhibitors of human papillomavirus type-11 E1-E2 protein–protein interaction: a combined medicinal chemistry, NMR and computational chemistry approach. Bioorg. Med. Chem. 2007;15:2690–2700. doi: 10.1016/j.bmc.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 106.Thomas C. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2012;146:621–632. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yan N., Chen Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012;13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hotho D.M. Natural killer cell activity and function in chronic HCV-infected patients during peg interferon and ribavirin: early effects of active substance use. Antiviral Res. 2013;97:347–355. doi: 10.1016/j.antiviral.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 109.Scott L.J., Perry C.M. Interferon-α-2b plus ribavirin: a review of its use in the management of chronic hepatitis C. Drugs. 2002;62:507–556. doi: 10.2165/00003495-200262030-00009. [DOI] [PubMed] [Google Scholar]
- 110.Enomoto M. Dynamics of hepatitis C virus monitored by real-time quantitative polymerase chain reaction during first 2 weeks of IFN-β treatment are predictive of long-term therapeutic response. J. Interferon Cytokine Res. 2002;22:389–395. doi: 10.1089/107999002753675820. [DOI] [PubMed] [Google Scholar]
- 111.Kumashiro R. Interferon-γ brings additive anti-viral environment when combined with interferon-α in patients with chronic hepatitis C. Hepatol. Res. 2002;22:20–26. doi: 10.1016/s1386-6346(01)00113-9. [DOI] [PubMed] [Google Scholar]
- 112.Negro F. Treatment of chronic hepatitis C with α-interferon plus ofloxacin in patients not responding to α-interferon alone. J. Hepatol. 1998;29:369–374. doi: 10.1016/s0168-8278(98)80053-6. [DOI] [PubMed] [Google Scholar]
- 113.Tsutsumi M. Effects of combination therapy with interferon and ofloxacin on chronic type C hepatitis: a pilot study. J. Gastroenterol. Hepatol. 1996;11:1006–1011. doi: 10.1111/j.1440-1746.1996.tb00022.x. [DOI] [PubMed] [Google Scholar]
- 114.Moscarella S. Interferon and thymosin combination therapy in naive patients with chronic hepatitis C: preliminary results. Liver. 1998;18:366–369. doi: 10.1111/j.1600-0676.1998.tb00819.x. [DOI] [PubMed] [Google Scholar]
- 115.Kozlowski A. Development of pegylated interferons for the treatment of chronic hepatitis C. BioDrugs. 2001;15:419–429. doi: 10.2165/00063030-200115070-00001. [DOI] [PubMed] [Google Scholar]
- 116.Handschumacher R.E. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 117.Liu X. Insights into the roles of cyclophilin A during influenza virus infection. Viruses. 2013;5:182–191. doi: 10.3390/v5010182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu J. Calcineurin is a common target of cyclophilin–cyclosporin A and FKBP–FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 119.Bienkowska-Haba M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009;5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tian X. Hepatitis B virus (HBV) surface antigen interacts with and promotes cyclophilin A secretion: possible link to pathogenesis of HBV infection. J. Virol. 2010;84:3373–3381. doi: 10.1128/JVI.02555-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bose S. Requirement for cyclophilin A for the replication of vesicular stomatitis virus New Jersey serotype. J. Gen. Virol. 2003;84:1687–1699. doi: 10.1099/vir.0.19074-0. [DOI] [PubMed] [Google Scholar]
- 122.Luo C. Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A. Biochem. Biophys. Res. Commun. 2004;321:557–565. doi: 10.1016/j.bbrc.2004.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.He H. Cyclophilin A inhibits rotavirus replication by facilitating host IFN-I production. Biochem. Biophys. Res. Commun. 2012;422:664–669. doi: 10.1016/j.bbrc.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 124.Colgan J. Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerization. J. Virol. 1996;70:4299–4310. doi: 10.1128/jvi.70.7.4299-4310.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zander K. Cyclophilin A interacts with HIV-1 Vpr and is required for its functional expression. J. Biol. Chem. 2003;278:43202–43213. doi: 10.1074/jbc.M305414200. [DOI] [PubMed] [Google Scholar]
- 126.Bruns K. Structural characterization of the HIV-1 Vpr N terminus: evidence of cis/trans-proline isomerism. J. Biol. Chem. 2003;278:43188–43201. doi: 10.1074/jbc.M305413200. [DOI] [PubMed] [Google Scholar]
- 127.Solbak S.M. HIV-1 p6-Another viral interaction partner to the host cellular protein cyclophilin A. Biochim. Biophys. Acta. 2012;1824:667–678. doi: 10.1016/j.bbapap.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 128.Solbak S.M. The host–pathogen interaction of human cyclophilin A and HIV-1 Vpr requires specific N-terminal and novel C-terminal domains. BMC Struct. Biol. 2011;11:49. doi: 10.1186/1472-6807-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Watashi K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 130.Liu Z. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 2009;83:6554–6565. doi: 10.1128/JVI.02550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tellinghuisen T.L. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 2008;82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wainberg M.A. The effect of cyclosporine A on infection of susceptible cells by human immunodeficiency virus type 1. Blood. 1988;72:1904–1910. [PubMed] [Google Scholar]
- 133.Coelmont L. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitor. Antimicrob. Agents Chemother. 2009;53:967–976. doi: 10.1128/AAC.00939-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hopkins S. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 2010;54:660–672. doi: 10.1128/AAC.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ptak R.G. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob. Agents Chemother. 2008;52:1302–1317. doi: 10.1128/AAC.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lalezari J. Antiviral activity and safety of 873140, a novel CCR5 antagonist, during short-term monotherapy in HIV-infected adults. AIDS. 2005;19:1443–1448. doi: 10.1097/01.aids.0000183633.06580.8a. [DOI] [PubMed] [Google Scholar]
- 137.Cooper D.A. Maraviroc versus efavirenz, both in combination with zidovudine–lamivudine, for the treatment of antiretroviral-naive subjects with CCR5-tropic HIV-1 infection. J. Infect. Dis. 2010;201:803–813. doi: 10.1086/650697. [DOI] [PubMed] [Google Scholar]
- 138.Caseiro M.M. Vicriviroc plus optimized background therapy for treatment-experienced subjects with CCR5 HIV-1 infection: final results of two randomized phase III trials. J. Infect. 2012;65:326–335. doi: 10.1016/j.jinf.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 139.Marier J.F. Pharmacokinetics and pharmacodynamics of TBR-652, a novel CCR5 antagonist, in HIV-1-infected, antiretroviral treatment-experienced, CCR5 antagonist-naive patients. Antimicrob. Agents Chemother. 2011;55:2768–2774. doi: 10.1128/AAC.00713-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lalezari J. Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects. J. Acquir. Immune Defic. Syndr. 2011;57:118–125. doi: 10.1097/QAI.0b013e318213c2c0. [DOI] [PubMed] [Google Scholar]
- 141.Tan Q. Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu B. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 2006;4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang C.C. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang X. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 2011;19:424–429. doi: 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]