

Abstract
Recent epidemics of West Nile virus (WNV) around the world have been associated with significant rates of mortality and morbidity in humans. To develop standard WNV diagnostic tools that can differentiate WNV from Japanese encephalitis virus (JEV), four monoclonal antibodies (MAbs) specific to WNV envelope (E) protein were produced and characterized by isotyping, reactivity with denatured and native antigens, affinity assay, immunofluorescence assay (IFA), and epitope competition, as well as cross-reactivity with JEV. Two of the MAbs (6A11 and 4B3) showed stronger reactivity with E protein than the others (2F5 and 6H7) in Western blot analysis. 4B3 could bind with denatured antigen, as well as native antigens in indirect ELISA, flow cytometry analysis, and IFA; whereas 2F5 showed highest affinity with native antigen. 4B3 and 2F5 were therefore used to establish an antigen capture-ELISA (AC-ELISA) detection system. The sensitivity of this AC-ELISA was 3.95 TCID50/0.1 ml for WNV-infected cell culture supernatant. Notably, these MAbs showed no cross-reactivity with JEV, which suggests that they are useful for further development of highly sensitive, easy handling, and less time-consuming detection kits/tools in WNV surveillance in areas where JEV is epidemic.
Keywords: West Nile virus, Monoclonal antibody, Envelope protein domain III, Antigen capture-ELISA
1. Introduction
West Nile virus (WNV) is a member of the Japanese encephalitis virus (JEV) serocomplex of the genus Flavivirus, family Flaviviridae. Recent epidemics of WNV around the world have been associated with significant rates of mortality and morbidity in humans (Lanciotti et al., 1999, Gea-Banacloche et al., 2004, Gubler, 2007, Murgue et al., 2002). However, neither a specific treatment for WNV infection nor a preventive vaccine is available at present. In nature, WNV exists in an enzootic cycle between mosquitoes and birds, with birds being the principal amplifying host (Glaser, 2004). The rapid spread of WNV is most likely caused by the migration of infected wild birds after contact with pools of Culex mosquitoes (Malkinson et al., 2002, Rappole et al., 2000). As the clinical symptoms of WNV infection are non-specific compared to those of other encephalitis viruses, diagnosis relies mainly on laboratory tests.
Serological testing is the primary method of diagnosing WNV infection. The plaque reduction neutralization tests for type-specific diagnosis are laborious, expensive, and require live virus, which limits their application in large-scale surveillance. ELISA-based detection for IgM, IgG or IgA has been developed, and some of these assays are commercially available (Hogrefe et al., 2004, Levett et al., 2005, Martin et al., 2000, Prince and Lape-Nixon, 2005). However, the serological cross-reactions and cross-neutralizations found in the JEV serocomplex viruses limit the specificity of serological tests (Hogrefe et al., 2004, Martin et al., 2000, Niedrig et al., 2007).
WNV viremia can serve as a clear indicator of recent infection and is suitable for early detection because it begins within a few days after infection and is short-lived. WNV-infected mosquitoes can be easily detected by various virus-detection methods (Hunt et al., 2002, Marfin and Gubler, 2001). Viral isolation depends heavily on the quality of samples and requires the use of cell culture and a BSL-3 laboratory, with 6-day delay. Reverse-transcriptase polymerase chain reaction (RT-PCR) is expensive and prone to contamination. Indirect immunofluorescence assay (IFA) with well-identified specific monoclonal antibodies (MAbs) can confirm virus infection. WNV antigen detection tests with specific MAbs have been used for dead birds and mosquito surveillance programs in North America (Dauphin and Zientara, 2007). An MAb-based antigen capture-ELISA (AC-ELISA) that can differentiate WNV from St Louis encephalitis virus has also been developed (Hunt et al., 2002).
As a result of the antigenic cross-reaction in the JEV serocomplex flaviviruses, it is critical to distinguish between WNV and JEV in areas such as China and Japan where JEV is endemic. Molecular diagnostic methods that simultaneously discriminate between WNV and JEV using RT-PCR analyses have previously been reported (Shirato et al., 2003, Shirato et al., 2005). MAb is the most attractive option for the development of standardized viral diagnostic assays. In this study, four MAbs against WNV envelope protein domain III (EDIII) were characterized by isotyping, affinity assay, reactivity with denatured and native antigens, and epitope competition, as well as cross-reactivity with JEV. The results suggest the applicability of the MAbs to various analytical methods, such as immunoblotting, IFA, and AC-ELISA, for detection and pathogenic study of WNV.
2. Materials and methods
2.1. Preparation of recombinant WNV EDIII protein
The EDIII (residues 298–415) of WNV bird 5810 strain was expressed, purified and refolded as described previously (Yuan et al., 2005). Briefly, the recombinant protein was expressed in Escherichia coli as an inclusion body and refolded in an appropriate buffer. The refolded protein was purified by gel-filtration chromatography.
2.2. Production and purification of MAbs
Six BALB/c mice (from National Institute for the Control of Pharmaceutical and Biological Products, Beijing, China), aged 8 weeks, were primed intraperitoneally with 50 μg recombinant EDIII protein, mixed with complete Freund's adjuvant (Sigma–Aldrich). Two boosts were given at days 14 and 28 with 50 μg EDIII mixed with incomplete adjuvant (Sigma–Aldrich). Three days after the last boost, the titer of polyclonal antiserum was assessed using indirect ELISA (described below) with EDIII as antigen. The mouse with the highest titer was chosen to harvest splenocytes. Separated splenocytes were fused with SP2/0 myeloma cells at a ratio of 5:1 using 50% (w/v) polyethylene glycol, according to a previously described protocol (Kohler and Milstein, 1975). The hybridoma cells were obtained and subsequently cloned by limiting dilution. The cell lines that produced specific antibodies were subcloned successively 3–5 times by limiting dilution to ensure monoclonality and stability. Positive clones that secreted high-titer EDIII-specific antibodies in indirect ELISA were further identified. The immunoglobulin subclass was determined using SBA Clonotyping System/AP kit (Southern Biotechnology Associates). Four positive cell lines (6A11, 4B3, 2F5 and 6H7) were used to generate ascites in BALB/c mice and MAbs were purified by protein A or protein G chromatography, according to manufacturer's protocols (Pharmacia). The concentration of purified MAb was determined by bicinchoninic acid protein assay (Pierce Biotechnology).
2.3. Western blot analysis
To examine whether the ascites MAbs recognized the linear epitope of EDIII protein, Western blot analysis was performed under denaturing conditions. EDIII protein was run on 12% SDS-PAGE, then electrotransferred onto a nitrocellulose membrane (Amersham Biosciences UK) and blocked with 5% non-fat dry milk in Tris-buffered saline (TBS). Membranes were incubated for 2 h at room temperature with four ascites MAbs (1:2000), respectively, and then washed 3 times with 0.05% Tween-20 in TBS (TBST), and incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:5000 dilution; Santa Cruz) for 1 h at room temperature, and detected by SuperSignal West Pico Chemiluminescent substrate solution (Pierce Biotechnology). In the control experiment, EDIII protein was incubated with an irrelevant MAb H5, which is an anti-influenza antibody (1:2000).
The specificity of purified MAbs for WNV E protein was also evaluated by Western blot analysis. The recombinant E proteins of WNV (bird 5810 strain) and JEV (Beijing-1 strain) with a His tag were expressed on the membrane of 293T cells, by transiently transfecting pcDNA4-WNV E or pcDNA4-JEV E plasmids into 293T cells. The cell lysate and inactivated WNV (Chin01 strain) or JEV (Beijing-1 strain) were separated by 10% SDS-PAGE, and were then electrotransferred onto a nitrocellulose membrane and blocked. Membrane was incubated for 2 h with purified MAb (1 μg/ml) or anti-His MAb (0.5 μg/ml; Santa Cruz), as a positive control for the expression of JEV E protein. The membrane was washed 3 times with TBST, and incubated with HRP-conjugated goat anti-mouse IgG secondary antibody (1:5000) for 1 h, and detected by substrate solution.
2.4. Indirect ELISA
All ELISAs were carried out in 96-well microtiter ELISA Plates (Greiner Bio-One). Titers of hybridoma-cell-secreted MAbs were detected by indirect ELISA. Briefly, the wells were coated overnight at 4 °C with 20 ng/well of purified EDIII, or an equal amount of bovine serum albumin (BSA; Sigma–Aldrich), as a negative control, and diluted in 50 mM carbonate saline (pH 9.6). After blocked for 1 h at 37 °C with PBS containing 3% BSA (PBSA), the wells were washed 4 times with PBS containing 0.05% Tween-20 (PBST). Serially diluted MAbs in PBSA (100 μl) were added to each well in triplicate and incubated for 1 h at 37 °C. After wells were washed 4 times with PBST, HRP-conjugated anti-mouse IgG (1:2000) was added to each well and incubated at 37 °C for 40 min, then washed again. Antibody binding was visualized by addition of the mixture of H2O2 and 3,3′,5,5′-tetramethyl-benzidene substrate (TMB; Sigma–Aldrich). After incubation for 15 min at 37 °C, the reaction was stopped by addition of 0.1 M H2SO4, and absorbance was read at 450 nm with a reference wavelength of 595 nm on a model Sunrise plate reader (Tecan). The endpoint titers of purified MAbs were also determined by 10-fold serial dilution with indirect ELISA. In all ELISAs, the irrelevant MAb H5 was used as an antibody control. The positive cutoff ratio was set at 2 (ratio of OD value coated with EDIII/OD value coated with BSA). This value is comparable to “positive to negative” cutoff ratios used in other WNV diagnostic assays (Davidson et al., 2005, Estrada-Franco et al., 2003).
2.5. Cell surface staining detection by flow cytometry analysis
The percentage of 293T cells expressing WNV E protein was determined by cell surface staining with MAbs. A FACSCalibur flow cytometer (BD Biosciences) was used for flow cytometry analysis. WNV E protein was expressed on the membrane of 293T cells by transfection of pcDNA4-WNV E plasmids into 293T cells and cultured for 48 h. Single-cell suspensions were prepared and incubated with ascites (1:2000) at 4 °C for 1 h in 100 μl PBSA buffer, then washed 3 times with PBS buffer. Cells were adsorbed with FITC-conjugated anti-mouse IgG (1:500; Santa Cruz) at 4 °C for 1 h, and washed again. Fluorescent signals on the cell surface were detected and the percentage of positive cells was counted among 3 × 104 cells. Controls included cells without addition of primary MAb, cells with H5 MAb and normal mouse IgG.
2.6. Affinity analysis by surface plasmon resonance (SPR)
The affinity between MAb and purified EDIII was determined by SPR on a Biacore 3000 (Biacore, Inc). Firstly, EDIII was immobilized on the surface of a CM5 chip by amine coupling and then used to capture purified MAb. Analysis was performed at 25 °C at a constant flow rate of 30 μl/min, using HBS-EP buffer [10 mM HEPES (pH 7.4), 150 mM NaCl, 3.4 mM EDTA, 0.005% surfactant P20] as a running buffer. To determine the association rate, dissociation rate and affinity constant (K D), a concentration series from 0.4 to 400 nM of purified MAb was injected (240 μl, associated for 8 min and then dissociated over 10 min). The EDIII surface was regenerated by injection of 50 mM NaOH before each EDIII injection. Binding curves and kinetic parameters were analyzed with a global fit 1:1 binding algorithm with drifting baseline by BIAevaluation software version 3.2 (Biacore). The affinity constant K D was determined as k off/k on, using data from three independent experiments.
2.7. Immunofluorescence assay
Binding of mouse ascites MAbs with WNV- or JEV-infected cells was determined by IFA. Sub-confluent BHK-21 cells, which were grown in 24-well microplates with slides, were infected with WNV (Chin-01 strain) or JEV (Beijing-1 strain) at a multiplicity of infection of 0.1. After incubation for 3 days, serially diluted MAbs were added to virus-infected BHK-21 cells. After incubation at room temperature for 2 h, slides were washed 3 times with PBST, and FITC-labeled anti-mouse IgG was added at dilution of 1:1000. Slides were washed again after 1 h incubation, stained with Evans blue, and observed under fluorescence microscope at 200× magnification. Cells showing strong green fluorescence were recorded as positive. The highest dilution of mouse ascites MAb that showed a strong positive fluorescence signal was recorded as the IFA titer. The uninfected cells were used as a negative control at each dilution, and JEV-infected BHK-21 cells were used to evaluate the cross-reactivity of MAbs with JEV. The experiments that involved the use of WNV were performed in a BSL-3 laboratory.
2.8. Competitive-binding ELISA and AC-ELISA
The detector MAb was labeled with biotin using an EZ-link Sulfo-NHS-LC- Biotinylation kit (Pierce Biotechnology) according to the manufacturer's instructions. Experiments on epitope competition of the three purified MAbs (6A11, 4B3 and 2F5) were carried out using competitive-binding ELISA. The wells were coated and blocked as described in Section 2.4. After 100 μl unlabeled MAbs (5 μg/ml) were added and incubated for 1 h at 37 °C, wells were washed 3 times, followed by incubation with an equal amount of another biotin-labeled MAb for 1 h at 37 °C. Plates were washed again and incubated with HRP-conjugated streptavidin (diluted 1:2000 in PBS; Zhongshan Goldenbridge Biotechnology). After washing, the color was developed with the addition of 100 μl freshly prepared substrate solution (1:1 mixture of TMB and H2O2 solution) for 15 min at 37 °C. The color reaction was stopped by 100 μl 0.1 M H2SO4, and absorbance was read at 450 nm, with a reference wavelength of 595 nm. Wells with addition of the irrelevantly unlabeled MAb (H5) were used as a negative control, and wells with addition of unlabeled MAb (the same as the biotin-labeled MAb) were a positive control. Each pair of MAbs was assayed in triplicate. Results were expressed as a percentage of inhibition and derived by the following formula: percentage of inhibition (PI) = [(negative control OD − test MAb OD)/(negative control OD − positive control OD)] × 100%.
For the AC-ELISA, the purified MAb (1 μg/well), diluted in 50 mM carbonate saline (pH 9.6), was coated on wells overnight at 4 °C. After blocking for 3 h at 37 °C with PBS containing 5% non-fat dry milk, wells were washed 3 times with PBST. All the following washing procedures were the same as described above. Virus culture supernatant (103.5 TCID50/ml) or recombinant EDIII protein, serially diluted in PBS containing 1% non-fat dry milk (PBSM) was added to the wells (100 μl/well) and incubated for 3 h. Cell culture supernatant or BSA was used as a negative control. After washing, 100 μl per well biotin-labeled detector MAb (2 μg/well, diluted in PBSM) was added and incubated for 1 h at 37 °C. After washing, the wells were incubated for 30 min at 37 °C with 100 μl per well HRP-conjugated streptavidin and detected as above. In this ELISA test, the positive cutoff was also set at 2 (ratio of positive/negative).
3. Results
3.1. Generation and purification of MAbs against WNV EDIII protein
The positive-fused cell clones were screened using indirect ELISA with recombinant EDIII as antigen. The hybridomas with higher ELISA titers were selected for screening, and four MAbs (6A11, 4B3, 2F5 and 6H7) were finally isolated and cloned. Ascites was produced in BALB/c mouse by hybridomas. The heavy chain subclasses of MAbs were determined as IgG2a (6A11) and IgG1 (4B3, 2F5 and 6H7), and the light chains of all of these were kappa isotype. 6A11 was efficiently purified by protein A chromatography, and 4B3, 2F5 and 6H7 by protein G chromatography. The concentrations of purified MAbs were determined as 10–18 mg/ml.
3.2. Western blot analysis
The binding specificity and cross-reactivity of the MAbs against denatured EDIII protein and E protein were determined by Western blot analysis. Four MAbs reacted with both EDIII and E proteins. Two of the ascites MAbs (6A11 and 4B3) showed stronger reactivity with recombinant EDIII protein (12.5 kDa) than the others (2F5 and 6H7), and the irrelevant MAb (H5) against influenza virus did not bind to EDIII (Fig. 1A). WNV E protein, which was expressed on the surface of 293T cells, as well as that from inactivated WNV, showed specific binding to 2F5, and there was no cross-reactivity with recombinant E protein or inactivated JEV (Fig. 1B). MAb 2F5 did not show any non-specific binding to negative controls, which were 293T cells transfected with pcDNA4 vector and cultured BHK-21 cells. The binding analysis with other MAbs (6A11, 4B3 and 6H7) yielded similar results (data not shown). The JEV E protein was fused with a His tag at its C terminus, and expression of JEV E on 293T cells was confirmed by anti-His antibody (Fig. 1B). This indicated that the four MAbs recognized denatured WNV EDIII and E protein with different binding affinity and did not show cross-reactivity with E protein from JEV.
Fig. 1.
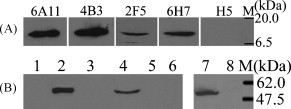
Western blot analysis of anti-EDIII MAbs with denatured antigen. (A) Reactivity of four MAbs with recombinant EDIII protein, using irrelevant MAb against influenza virus (H5) as a negative control. (B) Reactivity of MAb 2F5 with E proteins from WNV and JEV. Lanes 1–3: lysates of 293T cells transfected with pcDNA4, pcDNA4-WNV E and pcDNA4-JEV E plasmids, respectively; lane 4: inactivated WNV; lane 5: inactivated JEV; lane 6: cell culture supernatant of BHK-21 cells; lanes 7 and 8: the same as lanes 3 and 1, respectively. M: protein molecular weight markers. Left panel (lanes 1–6) was detected with MAb 2F5 and right panel (lanes 7 and 8) was detected with anti-His antibody.
3.3. Reactivity of MAbs with WNV EDIII protein in indirect ELISA
To examine the reactivity of the MAbs with EDIII protein under non-denaturing conditions, the indirect ELISA was performed with folded EDIII protein. The titers of four unpurified ascites MAbs were higher than 107 in indirect ELISA (data not shown). The reactivity of purified MAbs with EDIII is shown in Fig. 2 . Three MAbs (6A11, 4B3 and 2F5) showed strong positive binding with EDIII at concentrations of 0.02–20 μg/ml (absorbance ratio >10), compared with BSA-coated wells. Meanwhile, they showed obvious positive signals at the concentration of 0.002 μg/ml (absorbance ratio >2). Among these, MAb 2F5 showed highest sensitivity. The low absorbance ratios at the highest concentration tested (200 μg/ml) were due to the high non-specific binding of MAbs with BSA at this concentration. The irrelevant MAb H5 did not show specific binding to EDIII. It was surprising that MAb 6H7 showed no specific reactivity with EDIII after purification, so it was excluded from the following ELISAs.
Fig. 2.
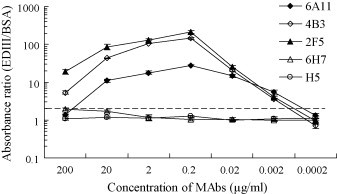
Absorbance ratio of MAbs binding to EDIII and BSA. The broken line indicates the absorbance ratio cutoff value, which was set at 2.
3.4. Flow cytometry analysis of MAbs
Cell surface expression of WNV E protein was detected by MAb staining and determined by flow cytometry. When stained with different MAbs, the percentage of fluorescent cells varied greatly. Representative profiles are shown in Fig. 3 . The percentage of fluorescent positive cells was 4.5 ± 2.6%, 26.9 ± 8.6%, 44.6 ± 8.0%, and 8.9 ± 1.2%, when stained with MAb 6A11, 4B3, 2F5 and 6H7, respectively. Controls, which included cells without MAb, and cells stained with the normal mouse serum or H5, were all negative. Among the four MAbs against EDIII, 2F5 showed the strongest binding to native E protein.
Fig. 3.
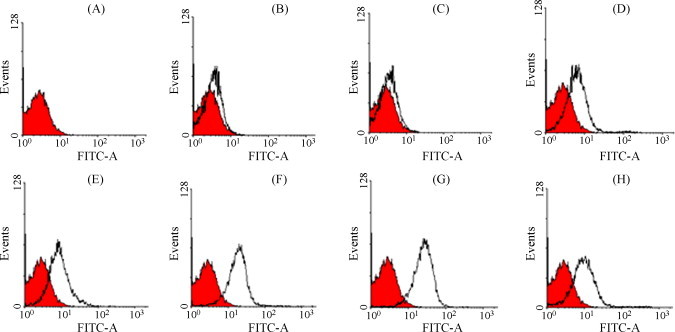
Flow cytometry analysis of cells that expressed WNV E proteins. The profile of transfected 293T cells without antibody staining was used to define the background of fluorescent intensity (A–H, red). The white profiles in (B–H) were compared with reference to the red profile. (B) Stained directly with FITC-conjugated anti-mouse IgG, without addition of primary antibody; (C) stained with normal mouse serum; (D–H) stained with H5, 6A11, 4B3, 2F5 and 6H7, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.5. Binding affinity between purified MAbs and recombinant EDIII protein
The binding affinity between recombinant EDIII protein and purified MAbs (6A11, 4B3 and 2F5) was analyzed by SPR in the solid phase. MAb 6H7 did not bind to immobilized EDIII protein under experimental conditions, so the binding affinity between 6H7 and EDIII was undetectable. MAb 2F5 bound to EDIII with an affinity of 1.8 ± 0.3 nM, which was the highest among the three MAbs. The affinity of 4B3 and 6A11 was similar, with a K D range from 407.1 ± 96.3 to 692.5 ± 112.1 nM (Fig. 4 ).
Fig. 4.
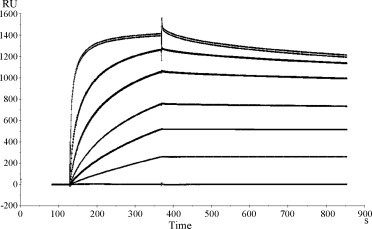
Biacore binding curves of purified MAbs 4B3 with immobilized EDIII protein. A concentration series from 0.4 to 400 nM of purified 4B3 was injected (240 μl, associated for 8 min and then dissociated over 10 min). The affinity constant KD was determined as koff/kon.
3.6. IFA
IFA was performed to further analyze whether the MAbs recognized the endogenously produced E protein in WNV-infected BHK-21 cells. Both normal mouse serum and three MAbs did not show non-specific binding to uninfected cells (data not shown). 6A11, 4B3 and 2F5 showed strong reactivity with WNV-infected cells, whereas normal mouse serum did not bind to infected cells (Fig. 5 ). The IFA titer of each MAb was determined, based on the highest dilution of ascites that gave a strong signal on WNV-infected BHK-21 cells. When BHK-21 cells were infected with WNV Chin-01 strain, the IFA titers of 6A11, 4B3, 2F5 and 6H7 were 2560, 7680, 5120 and 40, respectively. Notably, these MAbs showed no cross-reactivity with cells infected with JEV (Fig. 5). Validity of the MAbs used in IFA was confirmed in an independent laboratory using BHK-21 cells infected with WNV strain NY99 (data not shown).
Fig. 5.
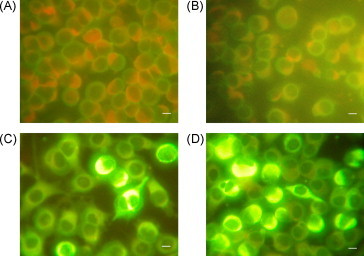
Detection of WNV-infected BHK-21 cells by immunofluorescence assay. The slides were stained with Evans blue and then observed by fluorescence microscopy. Scale bar: 20 μm. (A) WNV-infected cells stained with normal mouse serum. (B) JEV-infected cells stained with MAb 2F5. (C) WNV-infected cells stained with MAb 4B3. (D) WNV-infected cells stained with MAb 2F5.
3.7. Epitope competitions of purified MAbs and AC-ELISA
After biotinylation, the epitope competitions of three MAbs (6A11, 4B3 and 2F5) were assayed by competitive-binding indirect ELISA. The biotin-labeled 6A11 inhibited the binding of 4B3 to EDIII (57.2 ± 6.1%) and vice versa, which indicated that 6A11 and 4B3 recognized overlapping epitopes. 2F5 showed no competitive binding with 6A11 or 4B3, which meant that 2F5 recognized a different epitope from 6A11 or 4B3.
In order to establish a sensitive AC-ELISA for WNV detection, each pair of the three MAbs was evaluated. The highest sensitivity was obtained by using 2F5 as capture antibody, and biotin-conjugated 4B3 as detector antibody. To determine the detection limit of AC-ELISA, a serial dilution of EDIII protein and WNV culture supernatant (103.5 TCID50/ml) were used to construct the binding curve (Fig. 6 ). Cell culture supernatant without WNV infection was used as a negative control. According to the cutoff threshold (2, which is the ratio of positive/negative), it was deduced that as little as 10 ng/0.1 ml of recombinant EDIII protein and 3.95 TCID50/0.1 ml of virus culture supernatant could be detected (Fig. 6). This result revealed that MAbs 4B3 and 2F5 could be used to detect WNV in cell culture supernatant.
Fig. 6.
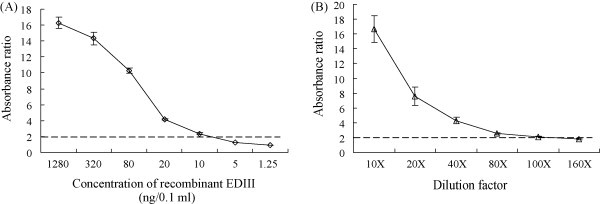
Sensitivity of AC-ELISA using MAbs. (A) Quantitative analysis using recombinant EDIII protein. 10 ng/0.1 ml was the detection limit. (B) Quantitative analysis using WNV cell culture supernatant. 80-fold dilution of cell culture supernatant was the detection limit, which was 3.95 TCID50/0.1 ml. The broken line indicates the absorbance ratio cutoff value, which was set at 2.
3.8. Properties of MAbs against WNV EDIII protein
Four MAbs against WNV EDIII protein were identified by isotyping, reactivity with denatured and native antigens, affinity assay and epitope competition ELISA, and IFA, and the results of these analyses were used to design the AC-ELISA. The properties of these MAbs are summarized in Table 1 .
Table 1.
Properties of MAbs against WNV EDIII protein in different assays.
| Assay | MAbs |
|||
|---|---|---|---|---|
| 6A11 | 4B3 | 2F5 | 6H7 | |
| Indirect ELISA | ++a | +++ | ++++ | –b |
| Western blot | +++ | ++++ | + | ++ |
| Flow cytometry | + | ++ | ++++ | + |
| Binding affinity | ++ | ++ | ++++ | – |
| Immunofluorescence | + | +++ | ++ | – |
| Antigen capture-ELISA | + | +++ | +++ | – |
+: Weak positive; ++++: strong positive.
–: Not detectable.
4. Discussion
WNV with greater epidemic potential and virulence emerged in the early 1990s and has spread rapidly across many countries (Lanciotti et al., 1999, Gea-Banacloche et al., 2004, Gubler, 2007, Murgue et al., 2002). Many methods have been developed for WNV diagnosis, in which serological testing is the primary method (Dauphin and Zientara, 2007). However, the cross-reactivity of the antibody response against flaviviruses limits the specificity of these tests (Calisher et al., 1989, Niedrig et al., 2007). As JEV is endemic in Southeast Asia, the cross-reactivity between WNV and JEV should be considered in WNV surveillance. MAbs with strong and specific reactivity to WNV antigens are the most attractive option for the development of standardized diagnostic tools. WNV EDIII-reactive MAbs have been reported by several groups (Beasley and Barrett, 2002, Oliphant et al., 2005, Sanchez et al., 2005, Throsby et al., 2006). Beasley and Barrett (2002) have described four MAbs that reacted with recombinant EDIII protein in a non-reducing Western blot assay and one of them bound to JEV. However, there is no further investigation of these MAbs expect for the neutralization activity. Furthermore, a panel of EDIII-specific MAbs has been identified by ELISA and neutralization assay, but the cross-reactivity of these MAbs with JEV has not been examined (Throsby et al., 2006).
Immunization with different forms of flavivirus antigens can produce antibodies with different properties (Aberle et al., 1999, Raviprakash et al., 2000, Sanchez et al., 2005). WNV-specific and neutralizing Abs have been mapped to EDIII (Oliphant et al., 2005, Throsby et al., 2006), and EDIII can induce specific immune responses and protection against WNV infection (Chu et al., 2007). To increase the possibility of obtaining EDIII-specific antibodies in the present study, mice were immunized with refolded EDIII protein, which was shown to be in native conformation by crystal structure analysis (PDB:2P5P).
Mouse ascites or purified MAbs were used in different assays in this study. The results of 6A11, 4B3 and 2F5 were similar in indirect ELISA for ascites or purified MAbs. The reason that 6H7 lost its specific reactivity with EDIII protein in indirect ELISA after purification may have been caused by the low pH of the elution buffer during purification, which probable destroyed the antigen binding site of this antibody. The binding affinity of MAb 2F5 (1.8 ± 0.3 nM) was comparable to that of MAb E16 (3.4 nM) (Oliphant et al., 2005) and CR4353 (6.5 nM) (Throsby et al., 2006), however, 4B3 and 6A11 showed a much lower K D (407.1 nM for 4B3 and 692.5 nM for 6A11). One reason is that the SPR analysis was performed by using refolded EDIII protein. The binding activity of 2F5 to refolded EDIII was higher than 4B3 and 6A11 in other assays, so the lower K D of 4B3 and 6A11 is reasonable. 4B3 and 6A11 seemed to bind with overlapping epitopes in competitive ELISA, but they showed different binding ability with the same antigen for denatured EDIII or E protein, as well as for native E protein or intact WNV particles. This may be explained by the different binding affinity constants of 4B3 and 6A11 with EDIII. It is interesting that 4B3 could bind to both denatured and native antigens. Crystal structural analysis of EDIII protein (Mukhopadhyay et al., 2003, Nybakken et al., 2005, Volk et al., 2004) has shown that EDIII is an Ig-like domain with seven β-strands, which constitute its main structure. Single β-strand is linear in native as well as in denatured protein. If 4B3 happens to recognize one of the seven β-strands, it is highly likely that 4B3 shows reactivity with both denatured and native antigens. This needs further investigation. In the present study, none of the MAbs showed protective activity in microtiter-based neutralization assays (data not shown). One possible reason is the natural scarcity of EDIII-specific neutralization antibodies. This is comparable with other study. In the 119 WNV-specific MAbs that have been cloned from three WNV-infected patients, only 10 exhibited neutralizing activity, and as few as two targeted domain III (Throsby et al., 2006).
Notably, none of the four MAbs showed cross-reactivity with JEV in any of our assays. WNV and JEV are genetically closely related, and the serological cross-reactions are usually found with these viruses (Martin et al., 2000). Considering that WNV and JEV are maintained in similar transmission cycles, and have overlapping geographic distributions in Southeast Asia, MAbs which can differentiate WNV from JEV are very useful for the surveillance system in these areas.
Of the many techniques developed for the rapid diagnosis of viral infections, the AC-ELISA is a sensitive and specific method that is capable of large-scale screening in surveillance programs. It also offers advantages over more traditional antigen detection methods, such as isolation in cell culture and plaque titration, which rely on the inoculation of samples into cells, or RT-PCR, which is expensive and prone to contamination. The availability of MAbs with strong reactivity to the target antigen is a crucial component for AC-ELISA development. Because of the possible presence of both conformational and linear antigens from live viruses or denatured samples, the recognition of the two forms of antigens by MAbs is extremely important for successful detection (He et al., 2007). To obtain the strongest signal for WNV detection by the AC-ELISA, different combination of MAbs were evaluated. It was found that a combination of MAbs 4B3 and 2F5 gave the highest signal. This is reasonable because, among the four MAbs, 4B3 showed the strongest reactivity with denatured antigen, and 2F5 had the highest affinity with native antigen. Therefore, the combination of MAbs 4B3 and 2F5 might contribute greatly to the sensitivity of the AC-ELISA. The detection limit was 3.95 TCID50/0.1 ml for WNV-infected cell culture supernatant in the present study. This sensitivity is comparable with that for severe acute respiratory syndrome coronavirus (Che et al., 2004, Shin et al., 2006), and higher than some AC-ELISAs for other arboviruses (Hall et al., 1987, Hildreth and Beaty, 1984). Sensitivity comparisons with other previously published AC-ELISAs for flaviviruses are difficult to make, because previous studies have used plaque forming units as endpoints to assess infectivity level (Hunt et al., 2002, Tsai et al., 1987).
In conclusion, four MAbs specific to WNV EDIII protein were produced and characterized. These MAbs could be used in immunoblot assay, flow cytometry analysis, IFA, and studies in WNV pathology. As they showed no cross-reactivity with JEV, these MAbs could be used to discriminate WNV from JEV in areas where JEV is epidemic. The detection limit of the established AC-ELISA re-emphasizes the sensitivity of specific MAbs for viral antigen detection, which suggests that these MAbs will be useful for further development of highly sensitive, easy handling, and less time-consuming detection kits/tools in WNV surveillance.
Acknowledgements
We thank Dr. David Cushley, who is an editor of International Science Editing service, for his assistance in language edit. We are grateful to Prof. Shi-Bo Jiang for comments and discussion of this manuscript (Dr. Jiang's stay in IMCAS is supported by CAS Overseas Outstanding Scientist Program). We also thank Dr. Zheng Fan, Ms. Zhen-Ying Liu for their technical supports.
This work was supported by National Basic Research Program 973 (grant no. 2005CB523001), National Key Technologies R&D Programs (grant no.2006BAF07B00), Chinese Academy of Sciences Knowledge Innovation Project (grant no. GFCX-YJ-23). The China–Japan Joint Laboratory of Molecular Immunology and Molecular Microbiology is, in part, supported by Japan MEXT (Ministry of Education, Culture, Sports, Science and Technology). G.F. Gao is a distinguished young investigator of National Natural Science Foundation of China (NSFC) (grant no. 30525010).
References
- Aberle J.H., Aberle S.W., Allison S.L., Stiasny K., Ecker M., Mandl C.W., Berger R., Heinz F.X. A DNA immunization model study with constructs expressing the tick-borne encephalitis virus envelope protein E in different physical forms. J. Immunol. 1999;163:6756–6761. [PubMed] [Google Scholar]
- Beasley D.W., Barrett A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002;76:13097–13100. doi: 10.1128/JVI.76.24.13097-13100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calisher C.H., Karabatsos N., Dalrymple J.M., Shope R.E., Porterfield J.S., Westaway E.G., Brandt W.E. Antigenic relationships between flaviviruses as determined by cross-neutralization tests with polyclonal antisera. J. Gen. Virol. 1989;70(Pt 1):37–43. doi: 10.1099/0022-1317-70-1-37. [DOI] [PubMed] [Google Scholar]
- Che X.Y., Hao W., Wang Y., Di B., Yin K., Xu Y.C., Feng C.S., Wan Z.Y., Cheng V.C., Yuen K.Y. Nucleocapsid protein as early diagnostic marker for SARS. Emerg. Infect. Dis. 2004;10:1947–1949. doi: 10.3201/eid1011.040516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.H., Chiang C.C., Ng M.L. Immunization of flavivirus West Nile recombinant envelope domain III protein induced specific immune response and protection against West Nile virus infection. J. Immunol. 2007;178:2699–2705. doi: 10.4049/jimmunol.178.5.2699. [DOI] [PubMed] [Google Scholar]
- Dauphin G., Zientara S. West Nile virus: recent trends in diagnosis and vaccine development. Vaccine. 2007;25:5563–5576. doi: 10.1016/j.vaccine.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Davidson A.H., Traub-Dargatz J.L., Rodeheaver R.M., Ostlund E.N., Pedersen D.D., Moorhead R.G., Stricklin J.B., Dewell R.D., Roach S.D., Long R.E., Albers S.J., Callan R.J., Salman M.D. Immunologic responses to West Nile virus in vaccinated and clinically affected horses. J. Am. Vet. Med. Assoc. 2005;226:240–245. doi: 10.2460/javma.2005.226.240. [DOI] [PubMed] [Google Scholar]
- Estrada-Franco J.G., Navarro-Lopez R., Beasley D.W., Coffey L., Carrara A.S., Travassos da Rosa A., Clements T., Wang E., Ludwig G.V., Cortes A.C., Ramirez P.P., Tesh R.B., Barrett A.D., Weaver S.C. West Nile virus in Mexico: evidence of widespread circulation since July 2002. Emerg. Infect. Dis. 2003;9:1604–1607. doi: 10.3201/eid0912.030564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gea-Banacloche J., Johnson R.T., Bagic A., Butman J.A., Murray P.R., Agrawal A.G. West Nile virus: pathogenesis and therapeutic options. Ann. Intern. Med. 2004;140:545–553. doi: 10.7326/0003-4819-140-7-200404060-00015. [DOI] [PubMed] [Google Scholar]
- Glaser A. West Nile virus and North America: an unfolding story. Rev. Sci. Tech. 2004;23:557–568. doi: 10.20506/rst.23.2.1504. [DOI] [PubMed] [Google Scholar]
- Gubler D.J. The continuing spread of West Nile virus in the western hemisphere. Clin. Infect. Dis. 2007;45:1039–1046. doi: 10.1086/521911. [DOI] [PubMed] [Google Scholar]
- Hall R.A., Kay B.H., Burgess G.W. An enzyme immunoassay to detect Australian flaviviruses and identify the encephalitic subgroup using monoclonal antibodies. Immunol. Cell Biol. 1987;65(Pt 1):103–110. doi: 10.1038/icb.1987.12. [DOI] [PubMed] [Google Scholar]
- He Q., Velumani S., Du Q., Lim C.W., Ng F.K., Donis R., Kwang J. Detection of H5 avian influenza viruses by antigen-capture enzyme-linked immunosorbent assay using H5-specific monoclonal antibody. Clin. Vacc. Immunol. 2007;14:617–623. doi: 10.1128/CVI.00444-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildreth S.W., Beaty B.J. Detection of eastern equine encephalomyelitis virus and Highlands J virus antigens within mosquito pools by enzyme immunoassay (EIA). I. A laboratory study. Am. J. Trop. Med. Hyg. 1984;33:965–972. doi: 10.4269/ajtmh.1984.33.965. [DOI] [PubMed] [Google Scholar]
- Hogrefe W.R., Moore R., Lape-Nixon M., Wagner M., Prince H.E. Performance of immunoglobulin G (IgG) and IgM enzyme-linked immunosorbent assays using a West Nile virus recombinant antigen (preM/E) for detection of West Nile virus- and other flavivirus-specific antibodies. J. Clin. Microbiol. 2004;42:4641–4648. doi: 10.1128/JCM.42.10.4641-4648.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A.R., Hall R.A., Kerst A.J., Nasci R.S., Savage H.M., Panella N.A., Gottfried K.L., Burkhalter K.L., Roehrig J.T. Detection of West Nile virus antigen in mosquitoes and avian tissues by a monoclonal antibody-based capture enzyme immunoassay. J. Clin. Microbiol. 2002;40:2023–2030. doi: 10.1128/JCM.40.6.2023-2030.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler G., Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Lanciotti R.S., Roehrig J.T., Deubel V., Smith J., Parker M., Steele K., Crise B., Volpe K.E., Crabtree M.B., Scherret J.H., Hall R.A., MacKenzie J.S., Cropp C.B., Panigrahy B., Ostlund E., Schmitt B., Malkinson M., Banet C., Weissman J., Komar N., Savage H.M., Stone W., McNamara T., Gubler D.J. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333–2337. doi: 10.1126/science.286.5448.2333. [DOI] [PubMed] [Google Scholar]
- Levett P.N., Sonnenberg K., Sidaway F., Shead S., Niedrig M., Steinhagen K., Horsman G.B., Drebot M.A. Use of immunoglobulin G avidity assays for differentiation of primary from previous infections with West Nile virus. J. Clin. Microbiol. 2005;43:5873–5875. doi: 10.1128/JCM.43.12.5873-5875.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkinson M., Banet C., Weisman Y., Pokamunski S., King R., Drouet M.T., Deubel V. Introduction of West Nile virus in the Middle East by migrating white storks. Emerg. Infect. Dis. 2002;8:392–397. doi: 10.3201/eid0804.010217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfin A.A., Gubler D.J. West Nile encephalitis: an emerging disease in the United States. Clin. Infect. Dis. 2001;33:1713–1719. doi: 10.1086/322700. [DOI] [PubMed] [Google Scholar]
- Martin D.A., Muth D.A., Brown T., Johnson A.J., Karabatsos N., Roehrig J.T. Standardization of immunoglobulin M capture enzyme-linked immunosorbent assays for routine diagnosis of arboviral infections. J. Clin. Microbiol. 2000;38:1823–1826. doi: 10.1128/jcm.38.5.1823-1826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Kim B.S., Chipman P.R., Rossmann M.G., Kuhn R.J. Structure of West Nile virus. Science. 2003;302:248. doi: 10.1126/science.1089316. [DOI] [PubMed] [Google Scholar]
- Murgue B., Zeller H., Deubel V. The ecology and epidemiology of West Nile virus in Africa, Europe and Asia. Curr. Top. Microbiol. Immunol. 2002;267:195–221. doi: 10.1007/978-3-642-59403-8_10. [DOI] [PubMed] [Google Scholar]
- Niedrig M., Sonnenberg K., Steinhagen K., Paweska J.T. Comparison of ELISA and immunoassays for measurement of IgG and IgM antibody to West Nile virus in human sera against virus neutralisation. J. Virol. Methods. 2007;139:103–105. doi: 10.1016/j.jviromet.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Nybakken G.E., Oliphant T., Johnson S., Burke S., Diamond M.S., Fremont D.H. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature. 2005;437:764–769. doi: 10.1038/nature03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant T., Engle M., Nybakken G.E., Doane C., Johnson S., Huang L., Gorlatov S., Mehlhop E., Marri A., Chung K.M., Ebel G.D., Kramer L.D., Fremont D.H., Diamond M.S. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince H.E., Lape-Nixon M. Evaluation of a West Nile virus immunoglobulin A capture enzyme-linked immunosorbent assay. Clin. Diagn. Lab Immunol. 2005;12:231–233. doi: 10.1128/CDLI.12.1.231-233.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappole J.H., Derrickson S.R., Hubalek Z. Migratory birds and spread of West Nile virus in the Western Hemisphere. Emerg. Infect. Dis. 2000;6:319–328. doi: 10.3201/eid0604.000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviprakash K., Kochel T.J., Ewing D., Simmons M., Phillips I., Hayes C.G., Porter K.R. Immunogenicity of dengue virus type 1 DNA vaccines expressing truncated and full length envelope protein. Vaccine. 2000;18:2426–2434. doi: 10.1016/s0264-410x(99)00570-8. [DOI] [PubMed] [Google Scholar]
- Sanchez M.D., Pierson T.C., McAllister D., Hanna S.L., Puffer B.A., Valentine L.E., Murtadha M.M., Hoxie J.A., Doms R.W. Characterization of neutralizing antibodies to West Nile virus. Virology. 2005;336:70–82. doi: 10.1016/j.virol.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Shin G.C., Chung Y.S., Kim I.S., Cho H.W., Kang C. Preparation and characterization of a novel monoclonal antibody specific to severe acute respiratory syndrome-coronavirus nucleocapsid protein. Virus Res. 2006;122:109–118. doi: 10.1016/j.virusres.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K., Miyoshi H., Kariwa H., Takashima I. Detection of West Nile virus and Japanese encephalitis virus using real-time PCR with a probe common to both viruses. J. Virol. Methods. 2005;126:119–125. doi: 10.1016/j.jviromet.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Shirato K., Mizutani T., Kariwa H., Takashima I. Discrimination of West Nile virus and Japanese encephalitis virus strains using RT-PCR RFLP analysis. Microbiol. Immunol. 2003;47:439–445. doi: 10.1111/j.1348-0421.2003.tb03381.x. [DOI] [PubMed] [Google Scholar]
- Throsby M., Geuijen C., Goudsmit J., Bakker A.Q., Korimbocus J., Kramer R.A., Clijsters-van der Horst M., de Jong M., Jongeneelen M., Thijsse S., Smit R., Visser T.J., Bijl N., Marissen W.E., Loeb M., Kelvin D.J., Preiser W., ter Meulen J., de Kruif J. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J. Virol. 2006;80:6982–6992. doi: 10.1128/JVI.00551-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai T.F., Bolin R.A., Montoya M., Bailey R.E., Francy D.B., Jozan M., Roehrig J.T. Detection of St. Louis encephalitis virus antigen in mosquitoes by capture enzyme immunoassay. J. Clin. Microbiol. 1987;25:370–376. doi: 10.1128/jcm.25.2.370-376.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk D.E., Beasley D.W., Kallick D.A., Holbrook M.R., Barrett A.D., Gorenstein D.G. Solution structure and antibody binding studies of the envelope protein domain III from the New York strain of West Nile virus. J. Biol. Chem. 2004;279:38755–38761. doi: 10.1074/jbc.M402385200. [DOI] [PubMed] [Google Scholar]
- Yuan F., Lou Z., Li X., Chen Y.W., Bell J.I., Rao Z., Gao G.F. Refolding, crystallization and preliminary X-ray structural studies of the West Nile virus envelope (E) protein domain III. Acta Crystallograph. Sect. F. Struct. Biol. Cryst. Commun. 2005;61:421–423. doi: 10.1107/S1744309105008195. [DOI] [PMC free article] [PubMed] [Google Scholar]