

Abstract
A TaqMan real-time RT-PCR assay was developed for detection of RNA transcripts produced by replicating CPV-2. A pair of primers and a TaqMan probe targeting the spliced NS2 mRNA were designed. A synthetic DNA fragment was constructed to mimic the spliced NS2 mRNA by PCR-based gene assembly and was used for generation of standard RNAs. The detection limit of the assay was 1 × 102 RNA copies and standard curve displayed a linear range from 1 × 102 to 1 × 109 copies and a good reproducibility. The assay was then applied to determine the mRNA loads in the tissues of dogs naturally infected by CPV-2. mRNA was detected in a variety of tissues, including the central nervous system.
Keywords: CPV-2, mRNA, Real-time RT-PCR, Infectious virus
1. Introduction
Canine parvovirus type 2 (CPV-2) emerged as causative agent of severe hemorrhagic gastroenteritis in dogs in 1978 (Kelly, 1978, Appel et al., 1979) and spread rapidly all over the world. Since 1979 the original type 2 underwent extensive antigenic changes and was fully replaced by the antigenic variants CPV-2a and CPV-2b (Mochizuki et al., 1993a, De Ybanez et al., 1995, Greenwood et al., 1996, Truyen et al., 1996, Truyen et al., 2000, Sagazio et al., 1998, Buonavoglia et al., 2000, Pereira et al., 2000). The variants -2a and -2b differ from the original type CPV-2 by a few amino acid changes in the VP2 protein, that account for an extended host range in vivo ed in vitro and for increased affinity to canine transferrin receptors (Parker et al., 2001, Hueffer et al., 2003). A new antigenic variant, type 2c, has been reported in dogs in Europe and Southern Asia (Buonavoglia et al., 2001, Nakamura et al., 2004, Decaro et al., 2006c).
CPV-2 is a member of the Parvoviridae family and is closely related to feline panleukopenia virus (FPV) and mink enteritis virus (MEV) (Parrish et al., 1982). Parvoviruses possess a non-enveloped capsid surrounding a single-stranded DNA genome of about 5 kb in length. The genome has two open reading frames (ORF). ORF2 encodes the two capsid proteins, VP1 and VP2 while ORF1 encodes two nonstructural (NS) proteins, NS1 and NS2, that are translated by alternative splicing of the transcribed RNA (mRNA). As a result, NS2 contains 87 amino-terminal amino acids in common with NS1 joined to 78 amino acids from an alternative open reading frame at the 3′-end of the ORF1 (Wang et al., 1998).
Several assays are available for detection of CPV-2 in the faeces of infected dogs. Hemagglutination or virus isolation may fail to detect small amounts of virus or the virus neutralized by secretory antibodies. CPV shedding in the faeces reaches high loads in the first days after infection, with a rapid decrease at 10–11 days post-infection (Decaro et al., 2005a, Elia et al., 2005). Also, in the late stage of infection the antibodies in the gut lumen may sequestrate most virus particles (Decaro et al., 2005c; Desario et al., 2005). Furthermore, as CPV-2-induced cytopathic effect in cell cultures is often not evident, staining by immunofluorescence is required, with additional laboratory work.
More recently, molecular methods such as PCR (Mochizuki et al., 1993b, Hirasawa et al., 1994, Truyen et al., 1994, Schunck et al., 1995, Senda et al., 1995, Uwatoko et al., 1995, Tempesta et al., 1998, Pereira et al., 2000, Buonavoglia et al., 2001) and real-time PCR (Decaro et al., 2005c) proved to be rapid, sensitive and specific assays for detection of CPV in clinical samples. However, detection of parvovirus DNA by PCR does not provide any information on virus infectivity.
Parvovirus DNA appears to be very stable and low levels of DNA can persist in serum and a range of tissues for months following acute infection (Soderlund-Venermo et al., 2002). Accordingly, detection of viral DNA does not equate necessarily with active viral infection and diagnostic assay targeting parvovirus B19 RNA transcripts have been developed (Bostic et al., 1999).
In this study a TaqMan RT-PCR method was developed for detection of RNA transcripts produced by replicating CPV-2. The load and distribution of CPV-2 mRNA in samples from infected dogs were estimated in comparison with the load of virus DNA, as evaluated by real-time PCR.
2. Materials and methods
2.1. Construction of synthetic RNA transcript and generation of standard RNA
A synthetic DNA fragment was constructed to mimic the spliced parvovirus mRNA encoding for the NS2 protein. A 200 bp fragment within the ORF1 sequence was chosen, that spans the RNA splicing region (Wang et al., 1998).
To generate synthetic DNA, overlapping primers were designed and used in repeated PCRs for the assembly and the amplification steps. The assembly was carried out in a reaction mix of 50 μl consisting of one unit of Kod DNA polymerase (Novagen, Madison, WI, USA), 5 μl of 10× buffer supplied by the manufacturer, 250 μM dNTP and 1 μM of each primer (Table 1 , A + B + C). The thermal conditions were as follows: 30 s at 94 °C, 1 min at 55 °C and 1 min at 72 °C repeated for 55 cycles with a final extention at 72 °C for 10 min. Because single-stranded ends of complementary oligonucleotide DNA fragments are filled in by PCR, cycling with DNA polymerase results in increasingly larger DNA fragments until the full-length sequence is obtained.
Table 1.
Sequence of primers and TaqMan probe used in the study
| Name | Polarity | Sequence 5′–3′ |
|---|---|---|
| Aa | Sense | GGATGTTCGCTGGAACAACTATACCAAACCAATTCAAAATGAAGAGCTAACATCTTTAATTAGAGGAGCACAAACAGCAATGGAT |
| Ba | Antisense | GCCGCTTGTGTCTCTAAGTCTTTGCAACTTTTTGGCGAGACTATCAACTTCCGATTCCCAGTCCATTTCTTCTTCTTCGGTTTGATGCATTGCTGTTTGTGCT |
| Ca | Antisense | TAGAACTTGGTCTTGACTCTGAGGATTGCTTGCCGCTTGTGTCTCTAAG |
| S1a | Sense | CGACTGGATGTTCGCTGGAACAA |
| AS1a | Antisense | CAGGATAGAACTTGGTCTTGACTC |
| R2forb | Sense | ACTAACATCTTTAATTAGAGGAG |
| R2revb | Antisense | CTCTAAGTCTTTGCAACTTT |
| R2Pbb | Antisense | FAM-TGGCGAGACTATCAACTTCCGAT-TAMRA |
Primers used for assembly and amplification steps of standard template.
Primers used in real-time RT-PCR.
In the amplification step an aliquot of annealed oligonucleotide mixture was used as template for a second PCR reaction containing two outside primers, S1/AS1 (Table 1) in order to amplify the full-length sequence. Amplification was performed as follows: 35 cycles at 94 °C for 30 s, 55 °C for 1 min, 72 °C for 1 min with a final extension at 72 °C for 10 min.
The PCR product was cloned into a linearized plasmid vector with overhanging 3′-deoxythymidine (T) residues (TOPO TA cloning kit, Invitrogen, Milan, Italy). RNA transcripts were generated in vitro with RiboMAXTM Large Scale RNA Production System-T7 (Promega Italia, Milan, Italy) from the T7 promoter, according to the manufacturer's guidelines. Residual DNA was eliminated by DNAse treatment and the transcript was purified with RNeasy columns (Qiagen S.p.A., Milan, Italy) and quantified by spectrophotometrical analysis. RNA copy numbers were then calculated and 10-fold serial dilutions were prepared in TE (Tris–HCl, EDTA, pH 8.0) buffer containing 30 μg carrier RNA (tRNA from Escherichia coli, Sigma–Aldrich Srl, Milan, Italy) per millilitre. Aliquots of each dilution were frozen at −70 °C and used only once.
2.2. Design of primers and probe
Primers and TaqMan probe were designed using Beacon Design software version 2.0 (Premier Biosoft International, Palo Alto, CA, USA) to amplify a 113 bp fragment of the spliced mRNA. Primers and probe were synthesized by MWG Biotech AG (Ebersberg, Germany). In order to avoid the amplification of genomic DNA, the TaqMan probe was designed such that the probe binding region encompassed the RNA splicing site (Fig. 1 ). The probe was labelled with 6-carboxyfluorescein (FAM) at the 5′-end and with 6-carboxytetramethylrhodamine (TAMRA) at the 3′-end. The position and sequence of the primers and probe used for TaqMan RT-PCR amplification are reported in Table 1.
Fig. 1.
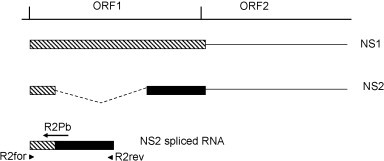
Schematic depicts splice junction spanned by primer pair R2for/R2rev and probe R2Pb for TaqMan RT-PCR strategy.
2.3. Reverse transcription
Duplicate of the standard dilutions and RNA templates were subjected simultaneously to reverse transcription (RT). One microliter of each duplicate of standard dilutions or template RNA was reverse transcribed in a reaction volume of 20 μl containing PCR buffer 1× (KCl 50 mM, Tris–HCl 10 mM, pH 8.3), MgCl2 5 mM, 1 mM of each deoxynucleotide (dATP, dCTP, dGTP and dTTP), RNase. Inhibitor 1 U, MuLV reverse transcriptase 2.5 U, random hexamers 2.5 U. Synthesis of c-DNA was carried out at 42 °C for 30 min, followed by a denaturation step at 99 °C for 5 min.
2.4. Real-time assay
For the real-time assay, 20 μl of c-DNA was added to 30 μl of reaction master mix. The master mix consisted of 25 μl of IQTM Supermix (Bio-Rad Laboratories Srl, Milan, Italy), 600 nM of each primer (R2for and R2rev), 200 nM of probe R2Pb. Fluorogenic PCR was carried out in a i-Cycler instrument (iQTM Real-Time Detection, Bio-Rad Laboratories Srl) with the following steps: activation of iTaq DNA polymerase at 95 °C for 10 min and 40 cycles consisting of denaturation at 95 °C for 15 s, primer annealing extension at 60 °C for 1 min.
Standard RNA (5000 copies/ml of faecal suspension) used in a real-time RT-PCR assay for avian influenza virus (Di Trani et al., 2006) was employed as an internal control (IC) in order to confirm the successful extraction of RNA, conversion to c-DNA and the TaqMan PCR reaction, as previously demonstrated (Elia et al., 2006).
2.5. Validation of the assay
The analytic specificity of spliced CPV-2 mRNA detection by real-time RT-PCR was assessed by testing RNA and DNA preparations of other canine pathogens, including canine coronavirus types I and II (CCoVI, CCoVII) (Decaro et al., 2005d), canine distemper virus (CDV) (Elia et al., 2006), canine adenovirus (CAdV) (Decaro et al., 2006a), reoviruses (Decaro et al., 2005b) and rotaviruses (Martella et al., 2001).
Within- and between-run precision of the quantitative real-time RT-PCR assay was assessed by multiple measurements of samples containing different amounts of spliced mRNA (1.43 × 106, 2.69 × 105, 2.45 × 104 and 2.76 × 102). The samples were evaluated in five replicates (within-run precision) and in four separate experiments (between-run precision). For all measurements, mean value, standard deviation and coefficients of variations (CVs) were calculated for the threshold cycle (C T) values.
2.6. Samples
Tissue samples including brain, cerebellum, spleen, lungs, liver, kidneys, bladder and mesenteric lymphnodes from nine CPV-2-positive dogs were collected at necroscopy. Infection by CPV-2 in the dogs had been diagnosed by real-time PCR (Decaro et al., 2005c) and viral characterization (CPV-2, -2a, -2b and -2c) was achieved by minor groove binder (MGB) probe assays (Decaro et al., 2006b). Twenty-five milligrams of each tissue sample were homogenized and used for RNA extraction (QIAampViral RNA Mini kit, Qiagen S.p.A., Milan, Italy), according to manufacturer's instructions.
To asses the kinetic of mRNA production during CPV infection, the CPV-2b strain 29/97 (Buonavoglia et al., 1998, Buonavoglia et al., 2000) was inoculated onto canine A-72 cell line maintained in Dulbecco Minimal Essential Medium with 5% FCS. The titre of the viral stock was 104.50TCID50. Each well of 24-well plates was seeded with 1 ml of cell suspension containing about 3 × 105 cells. The monolayers were infected shortly after trypsinization with 100 μl of a viral dilution containing 6.91 × 102 DNA copies/10 μl of template (m.o.i. of 0.001). Madin Darby Bovine Kidney (MDBK) cells, that are non-permissive to CPV-2 infection, were inoculated in parallel to compare CPV-2 replication in permissive and non-permissive cultural systems.
Supernatants and criolysates were taken separately at 24, 48, 72 and 96 h post-infection (p.i.) from the plates and used to evaluate the load of CPV-2 mRNA and DNA. Viral RNA was isolated using a commercial RNA isolation kit (RNeasy Total RNA Kit, Qiagen S.p.A., Milan, Italy) according to manufacturer's instructions. The RNA was eluted in 50 μl of nuclease free water and stored at −80 °C. Viral DNA was extracted using QIAamp viral kit, following the standard protocol (Qiagen S.p.A., Milan, Italy).
RNA extracted from mock-infected A-72 cells and three tissue samples (brain, spleen, liver) from an uninfected dog were included as negative controls.
2.7. TaqMan assay for quantitation of CPV-2 DNA
To compare the amount of viral DNA in the tissue samples and in the infected cell cultures, a real-time PCR assay was used as described previously (Decaro et al., 2005c).
3. Results
3.1. Analytical specificity and linear range of amplification of real-time RT-PCR assay
The specificity of the system for CPV-2 mRNA was assessed by running in the assay samples known to be positive for other infectious agents. No cross-reactivity with CCoVI and II, CDV, CAdV, reoviruses and rotaviruses was observed in this assay. Similarly, no detectable fluorescence signal was obtained in the negative controls (mock-infected cells and virus-negative tissue samples), confirming the assay was highly specific for the detection of CPV-2 spliced mRNA.
The linear range of quantitation of the real-time RT-PCR assay for CPV-2 mRNA was determined by using 10-fold serial dilutions of the standard RNA ranging from 1 × 100 to 1 × 109 copies. Standard RNA was first reverse transcribed and c-DNA used to determine the detectability and the linearity of the assay (Fig. 2 ). Threshold cycle (C T) values were measured in duplicate and were plotted against the known copy number of the standard sample. The generated standard curve covered a linear range of eight orders of magnitude and showed linearity over the entire quantitation range (slope = −3.585). The coefficient of linear regression (R 2) was equal to 0.999.
Fig. 2.
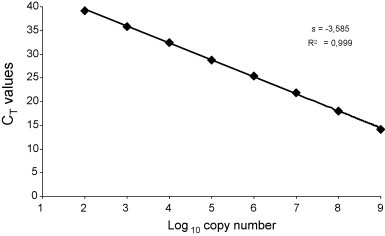
Standard curve of the real-time RT-PCR assay for CPV-2 mRNA. Tenfold dilutions of standard RNA prior to amplification were used, as indicated in the x-axis, whereas the corresponding cycle threshold (CT) values are presented on the y-axis. Each dot represents the result of duplicate amplification of each dilution. The coefficient of determination (R2) and the slope value (s) of the regression curve were calculated and are indicated.
3.2. Precision of the quantitative real-time RT-PCR
To assess the within- and between-run precision, samples containing different amounts of CPV-2 mRNA were assayed in replicate on the same plate or in multiple experiments (Fig. 3 ). The intra-assay CVs were in the range of 10.5% (samples containing 1.51 × 106 mRNA copies/μl of template) to 53.84% (samples containing 2.85 × 102 mRNA copies/μl of template), whereas the inter-assay CVs were comprised between 15.01% (samples containing 1.22 × 106 mRNA copies/μl of template) and 40.78% (samples containing 2.48 × 102 mRNA copies/μl of template).
Fig. 3.
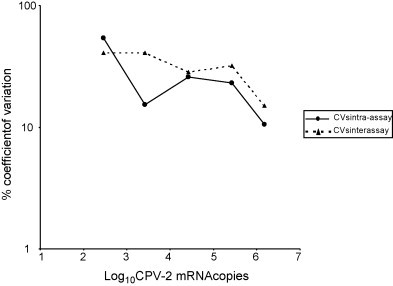
Coefficient of variation intra-assay and inter-assay over the dynamic range of the real-time RT-PCR for CPV-2 mRNA.
3.3. Detection kinetics of spliced CPV mRNA at different times p.i.
Table 2 shows the loads of mRNA at different hours p.i. in the cells inoculated with CPV-2b. Intracellular CPV-2 mRNA was detected as early as 24 h p.i., with significant increase from 1.09 × 104 to 2.95 × 106 copies/μl of template after 96 h p.i. A parallel increase in the number of viral DNA copies was detected both in the criolysates and in the supernatants.
Table 2.
CPV-2 DNA and mRNA detection in infected culture of A-72 cells
| Hours p.i. | Criolysate |
Supernatants |
||
|---|---|---|---|---|
| DNAa | NS2 mRNAb | DNAa | NS2 mRNAb | |
| 0 (inoculum) | 6.91 × 102 | Neg | – | – |
| 24 | 3.27 × 103 | 1.09 × 104 | 1.55 × 102 | Neg |
| 48 | 7.32 × 103 | 1.38 × 104 | 3.26 × 103 | 4.97 × 101 |
| 72 | 6.89 × 104 | 6.38 × 105 | 2.12 × 104 | 6.59 × 101 |
| 96 | 4.88 × 105 | 2.95 × 106 | 1.39 × 105 | 5.98 × 102 |
Copy number/10 μl of template.
Copy number/μl of template.
As expected, in the A-72 cells a few copies of mRNA were detected in the supernatants collected at 48 h p.i. and the mRNA load did not increase significantly at 72 and 96 h p.i.
In the non-permissive MDBK cells, CPV-2b infection did not result in virus replication, as viral RNA and DNA were not detected at all (data not shown).
3.4. Samples
The newly developed real-time RT-PCR assay was applied to determine the viral mRNA loads in different tissues of CPV-2 naturally infected dogs. The results were therefore compared with the real-time PCR assay specific for viral DNA (Table 3 ). Spliced mRNA was demonstrated in a variety of tissues, showing a wide bodily distribution of CPV-2, as previously demonstrated (Decaro et al., 2007). The mRNA load in the tissues markedly varied. In the tissues with the highest virus DNA loads (up to 7.46 × 108 DNA copies/10 μl of template) such as spleen, liver, lungs and mesenteric lymphnodes, the mRNA loads ranged from undetectable levels to 6.18 × 103 mRNA copies/μl of template.
Table 3.
Analysis of samples collected at necroscopy from CPV-2-positive dogs by real-time assays for DNA and mRNA
| Dog | Tissue sample | DNAa | mRNAb | Viral characterization |
|---|---|---|---|---|
| 242/05-A | Brain | 1.86 × 105 | 6.24 × 102 | CPV-2c |
| Heart | 4.21 × 106 | 4.96 × 102 | ||
| Lungs | 1.99 × 106 | Neg | ||
| Liver | 2.41 × 107 | Neg | ||
| Spleen | 2.61 × 107 | Neg | ||
| Kidney | 7.97 × 105 | Neg | ||
| Mesenteric lymphnode | 7.30 × 107 | Neg | ||
| Bladder | 7.93 × 104 | Neg | ||
| 280/05-A | Brain | 1.11 × 106 | 2.82 × 103 | CPV-2a |
| Lungs | 1.68 × 105 | 1.15 × 103 | ||
| Liver | 4.58 × 106 | 3.49 × 103 | ||
| Spleen | 9.14 × 106 | 6.18 × 103 | ||
| Mesenteric lymphnode | 7.27 × 106 | 3.18 × 103 | ||
| 315/06-D | Brain | 3.37 × 103 | Neg | CPV-2b |
| Lungs | Neg | Neg | ||
| Liver | 1.93 × 103 | Neg | ||
| Spleen | Neg | Neg | ||
| 436/06-A | Brain | 2.46 × 108 | 1.22 × 103 | CPV-2a |
| Cerebellum | 1.38 × 108 | 3.24 × 105 | ||
| Thymus | 1.45 × 107 | 4.83 × 103 | ||
| Spleen | 7.06 × 106 | 9.14 × 102 | ||
| Heart | 1.06 × 107 | 8.41 × 102 | ||
| Lungs | 1.86 × 08 | Neg | ||
| Liver | 7.46 × 108 | 1.07 × 103 | ||
| Kidney | 1.18 × 108 | 3.76 × 102 | ||
| Mesenteric lymphnode | 1.78 × 107 | 1.23 × 103 | ||
| 446/06 | Brain | 2.54 × 102 | Neg | CPV-2a |
| Cerebellum | 1.72 × 103 | 3.43 × 102 | ||
| 1/07 | Brain | 1.16 × 102 | Neg | CPV-2a |
| Cerebellum | 4.25 × 104 | 1.41 × 103 | ||
| 12/07-A | Brain | 5.36 × 105 | 2.63 × 104 | CPV-2a |
| Cerebellum | 2.92 × 103 | 2.18 × 106 | ||
| 12/07-B | Brain | 7.18 × 105 | 1.43 × 104 | CPV-2a |
| Cerebellum | 6.86 × 105 | 2.16 × 105 | ||
| 91/07-B | Brain | 3.26 × 107 | 6.28 × 103 | CPV-2a |
| Cerebellum | 1.08 × 107 | 5.55 × 103 | ||
Real-time titre are expressed as number of CPV-2 DNA copies/10 μl of template.
Real-time titre are expressed as number of CPV-2 mRNA copies/μl of template.
The highest viral mRNA load was found in the central nervous system (CNS), including brain and cerebellum, with the titres ranging from 3.43 × 102 to 2.18 × 106 mRNA copies/μl of template. In these tissues a correlation was found between the viral DNA and mRNA loads, as the mRNA was detected in most DNA-positive tissue samples.
4. Discussion
In this study, a real-time RT-PCR assay was designed to provide a complementary molecular tool for the study of CPV-2 pathogenesis. Thus far, a number of nucleic acid amplification techniques for the detection of CPV-2 have been described. However, all such techniques are targeted to CPV-2 DNA and a specific molecular method for detection of infectious CPV-2 particles was not available. Parvovirus DNA appears to be stable and low levels of DNA of human parvovirus B19 have been detected in serum and tissues for months after acute infection (Soderlund-Venermo et al., 2002). High titres of CPV-2 DNA can been detected for several weeks in blood in dogs even after the virus has disappeared from the intestinal content (Decaro et al., unpublished data). Also, during parvovirus replication in vitro, defective genomes may be generated and the real infectious virus titres may be overrated (Holland, 1991, Tempesta et al., 1998). The synthesis of parvovirus mRNAs is considered a marker of virus infection in cells (Bostic et al., 1999) and therefore accurate estimate of the viral RNA transcripts may be exploited as a good proxy for evaluation of virus replication.
To achieve selective amplification of CPV-2 mRNA, a real-time RT-PCR system was designed to target the spliced RNA transcript specific for the NS2 protein. The real-time RT-PCR assay proved to be highly specific, as no cross-amplification of other canine viruses was observed. The assay was highly reproducible and linear over a range of eight orders of magnitude, from 102 to 109 copies, allowing a precise calculation of CPV-2 mRNA loads. The reproducibility of the assay was high with relatively small intra- and inter-assay variability.
The TaqMan assay was used to investigate the viral mRNA loads in tissue samples from naturally infected dogs. CPV-2 mRNA was demonstrated in a variety of tissues, showing a wide distribution of the infectious virus in the organism. However, viral mRNA loads were not strictly correlated with the counterpart DNA titres, since in some tissues there were high viral DNA loads, but low RNA titres or no mRNA at all. The reasons for these inconsistencies may be threefold: (i) some tissues are not permissive to CPV-2 replication and the detection of viral DNA could be a consequence of the viraemia; (ii) the quantity of mRNA may depend on the stage of cell infection, decreasing as the infection course proceeds; (iii) viral RNA in some tissues may undergo quick degradation by the tissue RNases and therefore improper conservation after repeated cycles of freezing and thawing or even delayed storage of samples may result in the loss of virus mRNA (Peirson and Butler, 2007).
An intriguing finding of our study was the detection of CPV-2 mRNA in CNS of infected dogs. Involvement of the CNS tissues during parvovirus infection has been described in cats and virus replication in the nervous tissues may result in the onset of neurological signs (Csiza et al., 1972, Wilcox et al., 1984, Url et al., 2003). By converse, in dogs CPV antigen has never been detected in neurons, despite the presence of neurodegeneration (Agungpriyono et al., 1999, Url and Schmidt, 2005). In a recent study on CPV-2 distribution in tissues (Decaro et al., 2007), high titres of viral genome were detected in the CNS with median titres above 106 DNA copies/10 μl of template. In agreement with these results, we could observe the presence of CPV-2 mRNA in the brain and cerebellum tissues. This finding does not necessarily support the conclusion that neurons are permissive to CPV-2 replication as the presence of a full set of viral mRNAs may not be followed by the production of a full set of viral proteins (Gallinella et al., 2000). In addition, in our study virus mRNA was not extracted from individual cell types and therefore the precise source of the CPV-2 mRNA is not certain. Whether inflammatory cells rather than neurons were the actual source of the detected mRNA remains to be elucidated in future trials.
In conclusion, the method described in this study may represent an important tool for quantitation of CPV-2 mRNA load. Measurement of CPV-2 DNA and mRNA during infection and investigation of the relationship between these loads might provide a new perspective for further research in CPV-2 pathogenesis. In addition, considering the new interests in the research on antiviral drugs, the assay described here may also be applied to the study of new compounds able to inhibit parvovirus replication.
References
- Agungpriyono D.R., Uchida K., Tabaru H., Yamaguchi R., Tateyama S. Subacute massive necrotizing myocarditis by canine parvovirus type 2 infection with diffuse leukoencephalomalacia in a puppy. Vet. Pathol. 1999;36:77–80. doi: 10.1354/vp.36-1-77. [DOI] [PubMed] [Google Scholar]
- Appel M.J.G., Scott W.F., Carmichael L.E. Isolation and immunization studies of canine parvo-like virus from dogs with haemorrhagic enteritis. Vet. Rec. 1979;105:156–159. doi: 10.1136/vr.105.8.156. [DOI] [PubMed] [Google Scholar]
- Bostic J.R., Brown K.E., Young N.S., Koenig S. Quantitative analysis of neutralizing immune responses to human parvovirus B19 using a novel reverse transcriptase-polymerase chain reaction-based assay. J. Infect. Dis. 1999;179:619–626. doi: 10.1086/314648. [DOI] [PubMed] [Google Scholar]
- Buonavoglia C., Martella V., Fratelli A., Tempesta M., Cavalli A., Buonavoglia D., Bozzo G., Elia G., Decaro N., Carmichael L.E. Evidence for evolution of canine parvovirus type-2 in Italy. J. Gen. Virol. 2001;82:1555–1560. doi: 10.1099/0022-1317-82-12-3021. [DOI] [PubMed] [Google Scholar]
- Buonavoglia C., Pratelli A., Tempesta M., Martella V., Normanno G. Valutazione delle caratteristiche di innocuità e immunogenicità di una variante 2b di parvovirus del cane (CPV-2b) Vet. Anno. 1998;12(6):55–58. [Google Scholar]
- Buonavoglia D., Cavalli A., Fratelli A., Martella V., Greco G., Tempesta M., Buonavoglia C. Antigenic analysis of canine parvovirus strains isolated in Italy. N. Microbiol. 2000;23:93–96. [PubMed] [Google Scholar]
- Csiza C.K., Scott F.W., De Lahunta A., Gillespie J.H. Respiratory signs and central nervous system lesions in cats infected with panleukopenia virus. A case report. Cornell Vet. 1972;62:192–195. [PubMed] [Google Scholar]
- De Ybanez R.R., Vela C., Cortes E., Simarro I., Casal J.I. Identification of types of canine parvovirus circulating in Spain. Vet. Rec. 1995;136:174–175. doi: 10.1136/vr.136.7.174. [DOI] [PubMed] [Google Scholar]
- Decaro N., Campolo M., Desario C., Elia G., Martella V., Lorusso E., Buonavoglia C. Maternally derived antibodies in pups and protection from canine parvovirus infection. Biologicals. 2005;33:261–267. doi: 10.1016/j.biologicals.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Decaro N., Campolo M., Desario C., Ricci D., Camero M., Lorusso E., Elia G., Gavazza A., Martella V., Buonavoglia C. Virological and molecular characterization of a type 3 mammalian reovirus strain isolated from a dog with diarrhea in Italy. Vet. Microbiol. 2005;109:19–27. doi: 10.1016/j.vetmic.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Elia G., Martella V., Desario C., Campolo M., Di Trani L., Tarsitano E., Tempesta M., Buonavoglia C. A real-time PCR assay for rapid detection and quantitation of canine parvovirus type 2 DNA in the feces of dogs. Vet. Microbiol. 2005;105:19–28. doi: 10.1016/j.vetmic.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Decaro N., Martella V., Ricci D., Elia G., Desario C., Campolo M., Cavaliere N., Di Trani L., Tempesta M., Buonavoglia C. Genotyping-specific fluorogenic RT-PCR assays for the detection and quantitation of canine coronavirus type I and II RNA in faecal samples of dogs. J. Virol. Methods. 2005;130:72–78. doi: 10.1016/j.jviromet.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Campolo M., Elia G., Buonavoglia D., Colaianni M.L., Lorusso A., Mari V., Buonavoglia C. Infectious canine hepatitis: an “old” disease reemerging in Italy. Res. Vet. Sci. 2006 doi: 10.1016/j.rvsc.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Elia G., Martella V., Campolo M., Desario C., Camero M., Cirone F., Lo russo E., Lucente M.S., Narcisi D., Scalia P., Buonavoglia C. Characterisation of the canine parvovirus type 2 variants using minor groove binder probe technology. J. Virol. Methods. 2006;133:92–99. doi: 10.1016/j.jviromet.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Decaro N., Martella V., Desario C., Bellacicco A.L., Camero M., Manna L., Buonavoglia C. First detection of canine parvovirus type 2c in pups with haemorrhagic enteritis in Spain. J. Vet. Med. B: Infect. Dis. Vet. Public Health. 2006;53:468–472. doi: 10.1111/j.1439-0450.2006.00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Martella V., Elia G., Desario C., Campolo M., Lorusso E., Colaianni M.L., Lorusso A., Buonavoglia C. Tissue distribution of the antigenic variants of canine parvovirus type 2 in dogs. Vet. Microbiol. 2007;121:39–44. doi: 10.1016/j.vetmic.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desario C., Decaro N., Campolo M., Cavalli A., Cirone F., Elia G., Martella V., Lorusso E., Camero M., Buonavoglia C. Canine parvovirus infection: which diagnostic test for virus? J. Virol. Methods. 2005;126:179–185. doi: 10.1016/j.jviromet.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Di Trani L., Bedini B., Donatelli I., Campitelli L., Chiappini B., De Marco M.A., Delogu M., Buonavoglia C., Vaccari G. A sensitive one-step real-time PCR for detection of avian influenza viruses using a MGB probe and an internal positive control. BMC Infect. Dis. 2006;6:87. doi: 10.1186/1471-2334-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia G., Cavalli A., Cirone F., Lorusso E., Camero M., Buonavoglia D., Tempesta M. Antibody levels and protection to canine parvovirus type 2. J. Vet. Med. B. 2005;52:320–322. doi: 10.1111/j.1439-0450.2005.00870.x. [DOI] [PubMed] [Google Scholar]
- Elia G., Decaro N., Martella V., Cirone F., Lucente M.S., Lo russo E., Di Trani L., Buonavoglia C. Detection of canine distemper virus in dogs by real-time RT-PCR. J. Virol. Methods. 2006;136:171–176. doi: 10.1016/j.jviromet.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Gallinella G., Manaresi E., Zuffi E., Venturosi S., Bonsi L., Bagnara G.P., Musini M., Zerbini M. Different patterns of restriction to B19 parvovirus replication in human blast cell lines. Virology. 2000;278:361–367. doi: 10.1006/viro.2000.0673. [DOI] [PubMed] [Google Scholar]
- Greenwood N.M., Chalmers W.S.K., Baxendale W., Thompson H. Comparison of isolates of canine parvovirus by monoclonal antibody and restriction-enzyme analysis. Vet. Rec. 1996;138:495–496. doi: 10.1136/vr.138.20.495. [DOI] [PubMed] [Google Scholar]
- Hirasawa T., Kaneshige T., Mikazuki K. Sensitive detection of canine parvovirus DNA by the nested polymerase chain reaction. Vet. Microbiol. 1994;41:135–145. doi: 10.1016/0378-1135(94)90143-0. [DOI] [PubMed] [Google Scholar]
- Holland J.J. Defective viral genomes. In: Fields B.N., Knipe D.M., editors. Fundamental Virology. 2nd ed. Raven Press Ltd.; New York: 1991. [Google Scholar]
- Hueffer K., Parker J.S., Weichert W.S., Geisel R.E., Sgro J.Y., Parrish C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferring receptor. J. Virol. 2003;77:1718–1726. doi: 10.1128/JVI.77.3.1718-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly W.R. An enteric disease of dogs resembling feline panleukopenia. Aust. Vet. J. 1978;54:593. doi: 10.1111/j.1751-0813.1978.tb02426.x. [DOI] [PubMed] [Google Scholar]
- Martella V., Pratelli A., Elia G., Decaro N., Tempesta M., Buonavoglia C. Isolation and genetic characterization of two G3P5A[3] canine rotavirus strains in Italy. J. Virol. Methods. 2001;96:43–49. doi: 10.1016/s0166-0934(01)00312-3. [DOI] [PubMed] [Google Scholar]
- Mochizuki M., Harasawa R., Nakatami H. Antigenic and genomic variabilities among recently prevalent parvoviruses of canine and feline origin in Japan. Vet. Microbiol. 1993;38:1–10. doi: 10.1016/0378-1135(93)90070-n. [DOI] [PubMed] [Google Scholar]
- Mochizuki M., San Gabriel M.C., Nakatani H., Yoshida M., Harasawa R. Comparison of polymerase chain reaction with virus isolation and haemagglutination assays for the detection of canine parvoviruses in faecal specimens. Res. Vet. Sci. 1993;55:60–63. doi: 10.1016/0034-5288(93)90035-e. [DOI] [PubMed] [Google Scholar]
- Nakamura M., Tohya Y., Miyazawa T., Mochizuki M., Phung H.T., Nguyen N.H., Huynh L.M., Nguyen L.T., Nguyen P.N., Nguyen P.V., Nguyen N.P., Akashi H. A novel antigenic variant of canine parvovirus from a Vietnamese dog. Arch. Virol. 2004;149:2261–2269. doi: 10.1007/s00705-004-0367-y. [DOI] [PubMed] [Google Scholar]
- Parker J.S.L., Murphy W.J., Wang D., O’Brien S.J., Parrish C.R. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 2001;75:3896–3902. doi: 10.1128/JVI.75.8.3896-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish C.R., Carmichael L.E., Antczak D.F. Antigenic relationships between canine parvovirus type 2, feline panleukopenia virus and mink enteritis virus using conventional antisera and monoclonal antibodies. Arch. Virol. 1982;72:267–278. doi: 10.1007/BF01315223. [DOI] [PubMed] [Google Scholar]
- Peirson S.N., Butler J.N. RNA extraction from mammalian tissues. Methods Mol. Biol. 2007;362:315–327. doi: 10.1007/978-1-59745-257-1_22. [DOI] [PubMed] [Google Scholar]
- Pereira C.A., Monezi T.A., Mehnert D.U., D’Angelo M., Durigon E.L. Molecular characterisation of canine parvovirus in Brazil by polymerase chain reaction assay. Vet. Microbiol. 2000;75:127–133. doi: 10.1016/s0378-1135(00)00214-5. [DOI] [PubMed] [Google Scholar]
- Sagazio P., Tempesta M., Buonavoglia D., Cirone F., Buonavoglia C. Antigenic characterization of canine parvovirus strains isolated in Italy. J. Virol. Methods. 1998;73:197–200. doi: 10.1016/s0166-0934(98)00055-x. [DOI] [PubMed] [Google Scholar]
- Schunck B., Kraft W., Truyen U. A simple touch-down polymerase chain reaction for the detection of canine parvovirus and feline panleukopenia virus in feces. J. Virol. Methods. 1995;55:427–433. doi: 10.1016/0166-0934(95)00069-3. [DOI] [PubMed] [Google Scholar]
- Senda M., Parrish C.R., Harasawa R., Gamoh K., Muramatsu M., Hirayama N., Itoh O. Detection by PCR of wild-type canine parvovirus which contaminates dog vaccines. J. Clin. Microbiol. 1995;33:110–113. doi: 10.1128/jcm.33.1.110-113.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund-Venermo M., Hokynar K., Nieminen J., Rautakorpi H., Hedman K. Persistence of human parvovirus B19 in human tissues. Pathol. Biol. 2002;50:307–316. doi: 10.1016/s0369-8114(02)00307-3. [DOI] [PubMed] [Google Scholar]
- Tempesta M., Pratelli A., Buonavoglia D., Normanno G., Otranto D., Buonavoglia C. The polymerase chain reaction for the detection of defective interfering canine parvovirus particles. N. Microbiol. 1998;21:353–357. [PubMed] [Google Scholar]
- Truyen U., Platzer G., Parrish C.R. Antigenic type distribution among canine parvoviruses in dogs and cats in Germany. Vet. Rec. 1996;138:365–366. doi: 10.1136/vr.138.15.365. [DOI] [PubMed] [Google Scholar]
- Truyen U., Platzer G., Parrish C.R., Hanichen T., Hermanns W., Kaaden O.R. Detection of canine parvovirus DNA in paraffin-embedded tissues by polymerase chain reaction. Zentralbl Veterinarmed B. 1994;41:148–152. doi: 10.1111/j.1439-0450.1994.tb00218.x. [DOI] [PubMed] [Google Scholar]
- Truyen U., Steinel A., Bruckner L., Lutz H., Mostl K. Distribution of antigenic types of canine parvovirus in Switzerland, Austria and Germany. Schweiz. Arch. Tierheilkd. 2000;142:115–119. [PubMed] [Google Scholar]
- Url A., Schmidt P. Do canine parvoviruses affect canine neurons? An immunohistochemical study. Res. Vet. Sci. 2005;79:57–59. doi: 10.1016/j.rvsc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Url A., Truyen U., Rebel-Bauder B., Weissenbock H., Schmidt P. Evidence of parvovirus replication in cerebral neurons of cats. J. Clin. Microbiol. 2003;41:3801–3805. doi: 10.1128/JCM.41.8.3801-3805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwatoko K., Sunairi M., Nakajima M., Yamaura K. Rapid method utilizing the polymerase chain reaction for detection of canine parvovirus in feces of diarrheic dogs. Vet. Microbiol. 1995;43:315–323. doi: 10.1016/0378-1135(94)00102-3. [DOI] [PubMed] [Google Scholar]
- Wang D., Yuan W., Davis I., Parrish C.R. Nonstructural protein-2 and the replication of canine parvovirus. Virology. 1998;240:273–281. doi: 10.1006/viro.1997.8946. [DOI] [PubMed] [Google Scholar]
- Wilcox G.E., Flower R.L., Cook R.D. Recovery of viral agents from the central nervous system of cats. Vet. Microbiol. 1984;9:355–366. doi: 10.1016/0378-1135(84)90004-x. [DOI] [PubMed] [Google Scholar]