

Abstract
Multiple sclerosis (MS) is an autoimmune disease associated with environmental factors, possibly including several viruses such as the coronaviruses. Indeed, murine coronavirus (MHV) infection provides a well-known experimental model for MS studies. Intracerebral infection of C57BL/6 mice with MHV-A59 revealed that viral replication was efficient and that clearance of infectious virus occurred as soon as 7 days post-infection. Using cDNA arrays, analysis of gene expression profile in the brain revealed a modulation of 80 different genes following infection, with at least 27 of these genes having previously been directly related to innate or acquired immune responses. Concordingly, an important activation of auto-reactive T cells specific to myelin basic protein was demonstrated. Altogether, these results indicate that an MHV infection of the central nervous system (CNS) leads to an important host genomic response implicating immunity-related genes and to the activation of myelin-reactive autoimmune T cells.
Keywords: Autoimmunity, Coronavirus, Inflammation, Multiple sclerosis, T cells, Transcriptome
1. Introduction
Despite on-going efforts, the etiology of autoimmune diseases remains unknown. Genetic and environmental factors, such as viral infections, have often been associated with these pathologies. Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) and coronaviruses appear on the list of viruses that have been associated with this pathology. More specifically, human coronaviruses (HCoV) and virus-specific antibodies have been detected respectively in the brain and cerebrospinal fluids of MS patients (Burks et al., 1980, Murray et al., 1992, Stewart et al., 1992, Arbour et al., 2000, Salmi et al., 1982). Furthermore, acute infection of human microglia and astrocytes in primary cultures (Bonavia et al., 1997) and persistent infection of CNS cell lines (Arbour et al., 1999a, Arbour et al., 1999b) both demonstrate the neurotropism of human coronavirus. Moreover, infection of mice with the HCoV murine counterpart, murine hepatitis virus (MHV), provides an excellent experimental animal model for MS studies (Lane and Buchmeier, 1997). Indeed, this model has already been used to demonstrate the activation of autoreactive T cells in mice following infection (Kyuwa et al., 1991) and the activation of myelin-specific T cells in infected rats (Watanabe et al., 1983). In addition, naive animals that received T cells from infected rats restimulated by MBP, developed an EAE-like lesions (Watanabe et al., 1983). Finally, we have demonstrated the presence of T lymphocytes that recognize both HCoV and MBP in MS patients (Talbot et al., 1996).
The present study investigates the mouse CNS genomic response following intracerebral (i.c.) infection by MHV-A59. Viral replication was demonstrated by detection of infectious virus in both the brain and the liver but clearance took place as early as 7 days post-infection (p.i.) in both organs. In order to investigate the host response during CNS infection, global gene expression studies were performed. The gene expression profile revealed a modulation during infection with a special emphasis on immune response-related genes at 7 days p.i. Therefore, we next investigated the effects on immune cells and found the activation of myelin-specific T cells 7 days following MHV infection. Therefore, while an efficient clearance of infectious MHV-A59 virus takes place within the CNS and in periphery, the i.c. infection of genetically susceptible C57BL/6 mice induced both an important modulation of the gene expression profile within the CNS and the activation of self-reactive T cells. Moreover, these auto-reactive cells appear to be tissue-specific, recognizing myelin basic protein (MBP), as no such cells are detected against either ovalbumin or a mouse lung homogenate.
2. Materials and methods
2.1. Virus, cells, and animals
The A59 strain of MHV was initially obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA), plaque-purified twice, and passaged four times at a multiplicity of infection (MOI) of 0.01 on DBT cells as described previously (Daniel and Talbot, 1987). DBT astrocytoma cells induced as described previously (Kumanishi, 1967) were a kind gift of Dr. Michael J. Buchmeier (The Scripps Research Institute, La Jolla, CA, USA). Stock virus was constituted of a supernatant of DBT cells infected with MHV-A59 at a MOI of 0.001 and contained 3×107 plaque forming units (PFU)/mL. Four-week-old MHV-seronegative male C57BL/6 mice were purchased from Charles River Laboratories (St-Constant, Québec, Canada). Control mice were housed separately from infected mice. Mice were held for 72–96 h, then anesthetized intra-peritoneally with a ketamine–xylazine solution (200 mg/kg ketamine, 10 mg/kg xylazine) before intracerebral (i.c.) infection with 50 μL of virus diluted to 2×103 PFU/mL in phosphate buffered saline (PBS), which represents 100 PFU. Control mice were inoculated with the same dilution of a supernatant from non-infected DBT cells.
2.2. Viral load
Mice were sacrificed each day post-infection (p.i.) from day 1 to day 10 to harvest brain and liver. Tissues were homogenized in ice-cold PBS at a 1/10 (w/v) dilution. Mixtures were centrifuged at 4 °C for 20 min at 514×g. Viral load from supernatants was subsequently evaluated by plaque assays on DBT cells as previously described (Daniel and Talbot, 1987).
2.3. RNA preparation
Brains from three infected and three control mice were removed at 1, 2, 4 and 7 days p.i. Total RNA was extracted with Trizol LS (Invitrogen, Carlsbad, CA, USA) prior to the enrichment of polyA+ RNA using Atlas™ Pure Total RNA Labeling System (BD Biosciences, Palo Alto, CA, USA), according to the manufacturer's instructions. Aliquots of each DNase-treated total RNA were conserved for RT-PCR.
2.4. Atlas™ mouse 1.2 II expression array
All procedures for labeling and purifying the probes were carried out according to the manufacturer's instructions for the Atlas™ mouse 1.2 II cDNA array (cat. no. 7857-1 from Clontech; Palo Alto, CA, USA). Briefly, complex [α-33P]-dATP-labeled cDNA probes were generated by reverse transcription (RT) of mRNA from infected and control mouse brain. Total RNA from 3 mouse brains were pooled together at each time p.i. The probes were purified by column chromatography (ChromaSpin) and met the manufacturer's recommendation for specific activity (a total of 12×106 cpm for each probe). Membranes were pre-hybridized for 1 h in ExpressHyb Solution (Clontech) supplemented with 0.5 mg/mL of heat-denatured sheared salmon testes DNA (Sigma, Oakville, ON, Canada). Hybridization proceeded overnight at 68 °C in a roller bottle. Membranes were stringently washed with agitation for 30 min of prewarmed (68 °C) solution 1 (2× standard saline citrate (SSC), 1% (w/v) sodium dodecyl sulfate (SDS) four times and solution 2 (0.1× SSC, 0.5% (w/v) SDS) once. Subsequently, membranes were rinsed in 2× SSC at room temperature, exposed to a phosphor screen for 21 days and scanned (pixel size, 100 μm; PMT voltage, 688 V; local average background correction) using a PhosphoImager SI (Molecular Dynamics, Inc., Sunnyvale, CA, USA). Autoradiographic intensity was measured using ImageQuant software (version 5.0; Molecular Dynamics). To eliminate potential variations in RNA quantification between the uninfected and MHV-infected samples that could result from difference in the hybridization of the probe on the nylon membranes, the filters were normalized using the average nine housekeeping genes expression (ubiquitin, phospholipase A2, hypoxantine–guanine phosphoribosyltransferase, glyceraldehyde-3-phosphate dehydrogenase, myosin I, ornithine decarboxylase, β-actin, Ca2+ binding protein and ribosomal protein S29) and according to our own experience with this type of membrane, the value of 5000 arbitrary densitometric units was chosen as the minimum value that could be read by the PhosphoImager SI for each single spot (gene), in order to get reproducible data between the different hybridizations of the membranes. Moreover, as the cDNA array method is mainly for screening purposes and only semi-quantitative, the level of expression of any given gene had to be modulated by at least threefold to be considered significant, as suggested by others (Chapoval et al., 2001).
2.5. Reverse transcription (RT)-PCR
Some of the variations of gene expression observed with cDNA arrays were confirmed by RT-PCR. First-strand mouse cDNAs was prepared from 1 μg total RNA in a 20 μl reaction volume by random priming using Moloney Murine Leukemia Virus (M-MLV) (Invitrogen). A volume of 0.5–4 μl of the first-strand reaction were used for each PCR reaction with 2.5 U of rTaq DNA polymerase (Amersham Pharmacia Biotech, Piscataway, NL, USA). All PCR reactions began with a 3 min incubation at 94 °C and ended by a final extension step at 72 °C for 10 min. For each PCR cycle performed, denaturation was performed by incubation at 95 °C for 30 s, annealing was allowed to proceed for 45 s and the elongation temperature was 72 °C. Other PCR conditions and primer designs were optimized for semi-quantitative analysis specifically for each genes studied (described in Table 1 ). Ten or 20 μl of the reaction mixture (depending on the gene tested) was size-separated on a 1% (w/v) agarose gel and specifically amplified products were detected by ethidium bromide staining and UV transillumination. Semi-quantitative analysis was carried out using a computerized densitometric imager (ImageJ, NIH, Bethesda, MA, USA).
Table 1.
Primers and specific conditions for RT-PCR amplification of different genes expressed in mouse brain
| Gene | Primer | Primer sequence (5′→3′) | Location on gene | Fragment size (bp) | Number of cycles | Annealing temperature (°C) | Elongation length (s) |
|---|---|---|---|---|---|---|---|
| GAPDH | Forward | CGGAGTCAACGGATTTGGTCGTAT | 58–81 | 307 | 25 | 55 | 45 |
| Reverse | AGCCTTCTCCATGGTGGTGAAGAC | 364–341 | |||||
| CXCL-10 | Forward | GGATGGCTGTCCTAGCTCTG | 612–631 | 206 | 35 | 55 | 45 |
| Reverse | CCTTGGGAAGATGGTGGTTA | 817–798 | |||||
| Galectin-3 | Forward | AGCACTAATCAGGTGAGC | 1–18 | 841 | 40 | 52 | 60 |
| Reverse | GGCTTAGATCATGGCGTGG | 841–823 | |||||
| CD3γ | Forward | CCTTCTATCCAGCACCCAGA | 714–733 | 199 | 30 | 55 | 45 |
| Reverse | CAAGGTTGACAAGTGCTCCA | 912–893 | |||||
| Ii | Forward | TCACTGTATCCCCAACCACA | 3174–3193 | 200 | 35 | 55 | 45 |
| Reverse | CCCCTTACCTTGACCCAGTT | 3373–3354 |
2.6. Proliferation assay
Spleens were removed aseptically at 7 days p.i. Splenocytes were cultivated upon different antigenic stimulations. Cells were cultured at a concentration of 2×106 cells/mL in 200 μL in 96 wells plate with RPMI 1640 (Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS), 100 units/mL penicillin G (Invitrogen), 10 μg/mL streptomycin (Invitrogen), 1.25 μg/mL fungizone (Invitrogen), 0.01 M HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) and 55 μM 2-mercaptoethanol for 72 h at 37 °C in a humidified atmosphere containing 5% (v/v) carbon dioxide. Before incubation, 10 μL of the following antigens was added to each well: a dilution 1/20 (v/v) of a lysate of non-infected or MHV-A59 infected DBT cells, 0.2 mg/mL of bovine MBP, ovalbumin or amouse lung homogenate. [3H]thymidine (0.5 μCi) was added to each well 18 h before harvesting. Proliferations were considered positive in wells where it was at least twice superior compared to control antigen (lysate of DBT cells for MHV, no antigen for MBP, ovalbumin and mouse lung homogenate).
3. Results
3.1. Viral load and pathological symptoms
After i.c. inoculation, infectious virus titers in brain and liver were evaluated daily for 10 days (Fig. 1 ). MHV replicated rapidly in brain as shown by the detection of infectious virus as soon as day 1 p.i. and reached a maximal viral load at 2 days p.i. Afterwards, the viral load declined until day 7 p.i. where it was undetectable. Moreover, virus also replicated in liver after i.c. injection, with a maximal viral load at 4 days p.i. Infectious virus was undetectable in the liver at 7 days p.i. No infectious virus was detected in mice at 18, 21, 22 and 26 days p.i (n=3) (data not shown). Most of the mice did not present any pathological symptoms at all. However, some mice had a hunched back and tremors and very rarely presented hemiplegia. Furthermore, we made sure that all the mice were positively infected by detecting the viral RNA by RT-PCR.
Fig. 1.
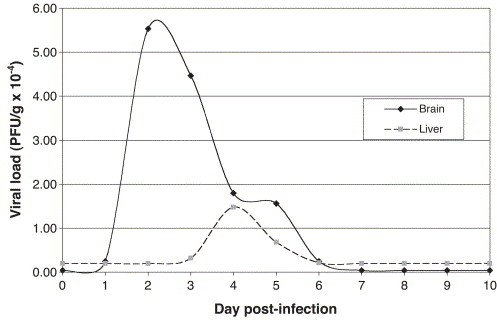
Virus titers following i.c. infection of C57BL/6 with 100 PFU of MHV-A59. Infectious titers were determined by plaque assay on DBT cells (day 1, 4, 7: n=10; day 2, 3, 5, 6, 8, 9, 10: n=7).
3.2. Gene expression
In order to better understand the mouse CNS response following infection, gene expression profiles in the brain of MHV-infected and control mice were analyzed at 1, 2, 4 and 7 days p.i. This was performed using cDNA arrays containing PCR-generated probes specific for murine genes. To limit inter-animal variations, RNA from 3 mouse brains was pooled for each condition tested. To ascertain that the results were reproducible and that the modulation observed between MHV-infected and mock-infected brains were accurate and not only due to random modulation, the threshold was set at 3-fold differences in the level of expression as suggested by others (Chapoval et al., 2001) and, according to our own experience with this type of nylon membrane, this expression level was to be above 5000 arbitrary densitometric units. According to these parameters, 80 of the 1176 genes tested showed significant changes in their level of expression and most of the variations were up-regulations observed on day 7 p.i. This global survey of gene expression indicates that several different types of genes appeared to be differentially expressed in the brains of MHV-infected mice compared to mock infected animals (Table 2 ) and a classification adapted from Ibrahim et al. (2001) is suggested. Nevertheless, we wished to focus on genes that are somehow associated to the immune response of the host, leaving the other categories of differentially expressed genes for future studies. Amongst these changes, several modulations of well-characterized immune-related genes were observed. The up-regulation of Ii (Ia-associated invariant chain), CD3γ, galectin-3, CXCL10 (IP-10) expression were then corroborated by semi-quantitative RT-PCR by using the RNA pool from 3 mouse brains used previously for the cDNA arrays analysis (Fig. 2 ). These up-regulations were also confirmed using RNA from an individual mouse (data not shown).
Table 2.
Identification of differentially expressed genes in mouse brain at 1, 2, 4 and 7 days following MHV infection
| Gene/protein name | Fold modulation |
|||
|---|---|---|---|---|
| Day 1 | Day 2 | Day 4 | Day 7 | |
| Immune response related | ||||
| Immune-related | ||||
| Complement component 1 (q subcomponent binding protein) | 2.2 ↓ | 6668.1 ↓ | 1.2 ↓ | – |
| Complement component 1 (q subcomponent alpha polypeptide) | 1.2 ↑ | 1.7 ↑ | 1.2 ↓ | 7.0 ↑ |
| Complement component 1 (q subcomponent beta polypeptide) | 1.4 ↑ | 1.3 ↑ | 1.3 ↑ | 6.7 ↑ |
| Interferon-induced protein with tetracopeptide repeats 3 (IFIT3) | – | 20.6 ↑ | 6.5 ↑ | 16.5 ↑ |
| Interferon dependent positive acting transcription factor 3 gamma (ISGF3 gamma) | 1.3 ↑ | 3.7 ↑ | 3.2 ↑ | 5.1↑ |
| Interferon gamma inducible protein 47 kDa | – | – | – | 30.7 ↑ |
| Interferon activated gene 204 | – | – | – | 5714.1 ↑ |
| Fc receptor, IgE, high affinity 1 (gamma polypeptide) | – | – | – | 19.9 ↑ |
| Fc receptor, IgG, high affinity 1 | – | – | – | 5187.0↑ |
| Fc receptor, IgG, low affinity III | – | – | – | 6.9 ↑ |
| CXCL 9 | – | – | – | 135.5 ↑ |
| CXCL 10 | 65.2 ↑ | 10658.4 ↑ | – | 10813.5 ↑ |
| CD68 Ag | – | – | 1.1 ↑ | 7.7 ↑ |
| CD8 Ag, alpha chain | – | – | – | 8.6 ↑ |
| CD3 antigen gamma polypeptide | – | – | – | 5095.0 ↑ |
| Lymphocyte antigen 86 | 2.1 ↑ | 1.8 ↑ | 2.1 ↑ | 8.8 ↑ |
| High mobility group 1 protein (HMGB1) | 3.7 ↑ | 1.1 ↑ | 1.0 | 17.0 ↑ |
| P lysozyme structural | – | – | – | 4.7 ↑ |
| Cytotoxic granule-associated RNA-Binding protein 1 (TIA) | 1.2 ↓ | 1.4 ↓ | 1.2 ↓ | 5.3 ↑ |
| CCL2/MCP-1 | – | – | – | 5.9 ↑ |
| Antigen processing and presentation | ||||
| Ia-associated invariant chain (Ii) | 1.2 ↑ | 20.6 ↑ | 4.1 ↑ | 123.6 ↑ |
| ATP-binding cassette, sub-family B (MDR/TAP) member 2 | – | – | – | 8166.1 ↑ |
| Cathepsin S | – | – | – | 7.8 ↑ |
| Cystatin F | – | – | – | 25.4 ↑ |
| CD83 Antigen | 3.6 ↑ | 1.2 ↓ | 1.1 ↓ | 7.8 ↑ |
| Extracellular matrix, cell adhesion | ||||
| Galectin-3 | – | – | – | 12.8 ↑ |
| CD18 (integrin beta 2) | – | – | – | 19.3 ↑ |
| Others | ||||
| Transcription | ||||
| Interferon regulatory factor 1 | – | – | – | 13.6 ↑ |
| Heat shock protein, 84 kDa | 20214.2 ↓ | 1.3 ↓ | 1.6 ↓ | 11167 ↑ |
| High mobility group protein 2 | – | – | – | 29.5 ↑ |
| Ribosomal protein S5 | 1.9 ↓ | 1.1 ↓ | 10.2 ↓ | 1.8 ↑ |
| Cellular nucleic acid binding protein | – | 1.8 ↑ | 1.8 ↑ | 4.9 ↑ |
| Signal transduction | ||||
| Protein tyrosine phosphatase receptor type C | – | – | – | 7.3 ↑ |
| PKC isoform iota (PKCI) | 1.1 ↑ | 1.1 ↑ | 5.8 ↓ | 1.1 ↑ |
| Annexin A2 | 1.3 ↓ | – | – | 3.6 ↑ |
| Cell structure, movement and secretion | ||||
| Adaptor protein, complex AP-1, sigma 1 | – | 1.5 ↑ | 16.7 ↓ | – |
| Septin 7/cdc10 | 1.5 ↑ | 3.2 ↑ | 2.0 ↑ | 1.4 ↑ |
| Importin beta | – | 5.4 ↓ | – | – |
| Lysosomal membrane glycoprotein 1 | 1.1 ↓ | 1.1 ↓ | 3.5 ↓ | 1.0 |
| Myelin-Associated glycoprotein (MAG) | 1.2 ↑ | 1.2 ↑ | 3.0 ↓ | 2.2 ↑ |
| GFAP | 1.1 ↓ | 1.2 ↑ | 1.5 ↓ | 4.1 ↑ |
| Vimentin | 1.2 ↑ | 1.2 ↑ | 1.2 ↑ | 4.9 ↑ |
| S100 calcium-binding protein A4 | – | 1.7 ↓ | 2.2 ↑ | 6.8 ↑ |
| Cell division and death | ||||
| Serine protease inhibitor (2–2) | – | – | – | 19.7 ↑ |
| Protein folding | ||||
| FK506 binding protein 6 (65 kDa) | 6951.0 ↓ | – | – | – |
| Chaperonin subunit 6b (zeta) | – | 6137.7 ↓ | – | – |
| Chaperonin subunit 3 (gamma) | 1.5 ↓ | 1.8 ↓ | 3.5 ↓ | – |
| Tubulin cofactor a | – | – | 10.0 ↓ | – |
| Peroxiredoxin 3 | 1.0 | 1.1 ↓ | 1.5 ↓ | 3.2 ↑ |
| Miscellaneous | ||||
| Growth hormone | 7.1 ↓ | 2.6 ↓ | 116.5 ↓ | 9.3 ↓ |
| Prolactin | 6.1 ↓ | 2.5 ↓ | 7.0 ↓ | – |
| Adenylate cyclase activating polypeptide 1 receptor 1 | 1.1 ↓ | 1.3 ↓ | 5680.7 ↓ | – |
| Alcohol dehydrogenase family 3 | – | 5207.7 ↓ | – | – |
| ATP-binding cassette subfamily A (ABC 1) member 1 | – | 5099.0 ↓ | – | – |
| Phosphodiesterase 6D CGMP-specific, rod, delta | – | – | 25.1↓ | – |
| Prosaposin | 1.2 ↓ | 1.8 ↑ | 8.4 ↓ | 1.8 ↑ |
| Lipocalin 2 | – | – | – | 15.3 ↑ |
| Malate dehydrogenase, mitochondrial | 1.2 ↓ | 1.9 ↑ | 2.3 ↓ | 415.0 ↑ |
| Adaptor-related protein complex AP-3 sigma subunit | 1.2 ↓ | 1.2 ↓ | 1.2 ↓ | 9095.1 ↑ |
| Nuclear receptor subfamily 4 group A member 1 | 1.8 ↑ | 1.8 ↑ | – | 5446.9 ↑ |
| LanC/GRP69A/p40 | 1.3 ↓ | 3.1↑ | 1.6 ↓ | 1.6 ↓ |
| Preproacrosin | 1.5 ↑ | 5.1↑ | 5.3 ↑ | 1.5 ↑ |
| Amyloid beta (A4)-like protein 1 | 3.0 ↓ | 1.8 ↓ | 3.4 ↓ | 1.3 ↓ |
| Glutamine fructose-6-phosphate transaminase 1 | – | 6954.9↓ | – | – |
| Mm3 muscarinic acetylcholine Receptor (MACHR) | – | 4.9 ↓ | – | – |
| Glycine receptor beta subunit | 1.1 ↑ | 1.2 ↑ | 5.4 ↑ | – |
| Cytochrome C oxidase Subunit Via, polypeptide 2 | 1.0 | 4.0 ↓ | 1.3 ↑ | 1.1 ↓ |
| Cytochrome b-245, alpha polypeptide | – | – | – | 6.1 ↑ |
| Carbonic anhydrase 11 | 1.8 ↓ | 3.2 ↓ | 1.4 ↓ | 2.0 ↓ |
| Carbonic anhydrase 2 | 1.2 ↓ | 1.6 ↓ | 3.2 ↓ | 1.0 |
| Acetyl-Coenzyme A Dehydrogenase, long chain | – | 3.1 ↓ | – | – |
| Solute carrier family 4 (anion exchanger), member 2 | – | 1.4 ↑ | 3.2 ↓ | – |
| Apolipoprotein CII | – | – | 6.3 ↑ | 2.8 ↑ |
| Apolipoprotein D | 1.7 ↑ | 1.2 ↑ | 1.2 ↓ | 5.7 ↑ |
| Sepiapterin reductase | – | – | 5.7 ↓ | – |
| NADH dehydrogenase Flavoprotein 1 | 1.5 ↓ | 2.0 ↓ | 5.5 ↓ | 1.9 ↑ |
| Glutamate oxaloacetate transaminase 2, mitochondrial | 1.1 ↑ | 1.1 ↑ | 4.3 ↓ | 1.5 ↓ |
| Glucosidase alpha acid | 1.5 ↓ | 1.1 ↓ | 3.5 ↓ | 1.2 ↓ |
| Amyloid beta (A4) precursor-like protein 1 | 3.0 ↓ | 1.8 ↓ | 3.4 ↓ | 1.3 ↓ |
| Heterogeneous nuclear ribonucleoprotein A1 | 1.1 ↓ | 1.5 ↓ | 3.3 ↓ | 2.0 ↑ |
Genes were considered as significantly modulated if the variation between the expression of control and infected mice was at least 3-fold and if the expression level was at least 5000 densitometric units. Values in bold represent significant modulation. The dash symbol (–) means that the densitometric unit was lower than 5000, therefore the modulation could not be addressed according to our criteria.
Fig. 2.
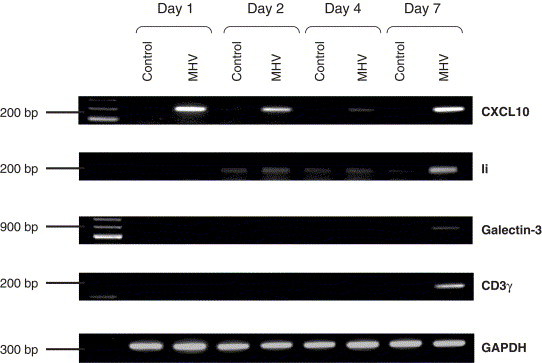
Expression of IP-10, Ii, galectin-3 and CD3γ in mouse brain. The level of expression was measured by RT-PCR of total RNA extracted from MHV-infected and control mouse brains. RT-PCR products were migrated on a 1% (w/v) agarose gel and revealed by ethidium bromide staining.
3.3. Proliferation assays
Knowing that these modulations in the expression of several immune response-related genes could have an impact on immune cells, we chose to conduct a proliferation assay ex vivo on splenocytes from mice at 7 days p.i. Amongst the 31 infected mice tested, all showed antibodies specific for MHV and 28 demonstrated a cellular immune response to MBP (Table 3 ). Moreover, as indicated in details in Table 3, there was a large variation in the number of proliferation positive wells between the infected mice. Nevertheless, our results clearly indicated that the vast majority of the mice had a cellular immune response to MHV and a significant increase in activated autoreactive T cells against MBP.
Table 3.
Proliferation assay associated with the activation of MBP-reactive T cells in spleens of mice at 7 days p.i. with MHV-A59
| Mouse number | Positive wells MHV (average cpm) | S.I. (mean) | Positive wells MBP (average cpm) | S.I. (mean) |
|---|---|---|---|---|
| 1 | 0/24 | – | 19/26 (3454) | 3.1 |
| 2 | 0/25 | – | 6/28 (3641) | 2.6 |
| 3 | 8/26 (5948) | 2.2 | 10/28 (2055) | 2.2 |
| 4 | 12/32 (4487) | 2.5 | 7/32 (2596) | 2.1 |
| 5 | 6/20 (4226) | 2.4 | 0/24 | – |
| 6 | 0/16 | 5/23 (3212) | 2.3 | |
| 7 | 23/26 (5561) | 7.1 | 2/28 (960) | 2.1 |
| 8 | 4/24 (1400) | 2.3 | 5/24 (4027) | 3.8 |
| 9 | 0/24 | – | 7/25 (3256) | 2.9 |
| 10 | 0/27 | – | 7/28 (5278) | 2.1 |
| 11 | 0/24 | – | 21/24 (3500) | 3.1 |
| 12 | 20/21 (6968) | 3.4 | 1/21 (2265) | 2.1 |
| 13 | 2/24 (7835) | 2.2 | 19/24 (8178) | 3.6 |
| 14 | 22/24 (7247) | 4.5 | 22/24 (2166) | 2.9 |
| 15 | 23/24 (5406) | 3.8 | 19/24 (2883) | 3.4 |
| 16 | 10/24 (2677) | 2.3 | 19/24 (3250) | 3.0 |
| 17 | 24/24 (7521) | 9.1 | 2/24 (968) | 2.1 |
| 18 | 24/24 (2965) | 3.4 | 20/24 (2216) | 3.1 |
| 19 | 24/24 (4234) | 4.8 | 14/24 (1568) | 2.5 |
| 20 | 21/23 (3086) | 3.6 | 21/23 (2331) | 3.3 |
| 21 | 3/24 (753) | 2.4 | 24/24 (1007) | 3.5 |
| 22 | 10/15 (1108) | 3.1 | 0/15 | – |
| 23 | 4/24 (2022) | 2.3 | 21/24 (3139) | 3.5 |
| 24 | 15/18 (3707) | 2.8 | 18/18 (3571) | 3.5 |
| 25 | 19/23 (3204) | 2.5 | 24/24 (3727) | 3.6 |
| 26 | 16/16 (3595) | 3.4 | 15/16 (2113) | 3.0 |
| 27 | 12/12 (7606) | 7.7 | 1/13 (968) | 2.1 |
| 28 | 2/10 (1320) | 2.4 | 0/10 | – |
| 29 | 14/16 (2666) | 2.7 | 16/16 (3294) | 3.8 |
| 30 | 3/16 (2198) | 2.3 | 12/24 (2077) | 3.6 |
| 31 | 16/16 (8150) | 8.6 | 7/16 (1030) | 2.3 |
| Total of wells | 339/670 | 364/702 |
Thirty mice were infected with 100 PFU and splenocytes were cultivated in 96-well plates as indicated in Section 2.6. The proliferation data (cpm) were considered positive in wells where the count was at least 1000 (before substraction of the background count) and where it was more than twofold superior compared to control antigen (DBT cell lysate for MHV and RPMI culture medium for MBP). Splenocytes from 30 uninfected mice were cultivated the same way. S.I.: Stimulation Index.
Specific activation to virus and to myelin basic protein (MBP) was observed in 48% of the wells containing splenocytes from MHV-infected mice (Fig. 3 ), whereas this activation was not significant in control mice (p<0.0001). The absence of antigen-specific proliferation measured upon stimulation by ovalbumin or a mouse lung homogenate demonstrated the specificity of the response to viral and myelin antigens.
Fig. 3.
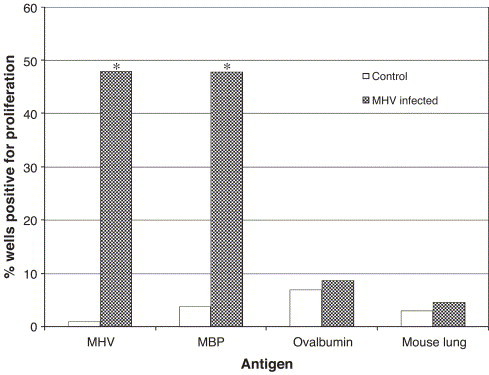
Specific activation of myelin-reactive T cells following MHV infection. Mice were injected i.c. with 100 PFU of MHV-A59. Seven days p.i., splenocytes were cultured with viral, myelin or control antigens for 48 h. Cells were pulsed with [3H]thymidine and harvested. Statistical significance was determined using the Student t-test for unpaired data. *p<0.0001. The proliferation data were considered positive in wells where it was at least twice superior compared to control antigen (lysate of DBT cells for MHV, no antigen for MBP, ovalbumin and mouse lung homogenate).
4. Discussion
MHV infection provides an excellent animal model for MS (Lane and Buchmeier, 1997), an autoimmune neurological disease which remains of unknown etiology even though several viruses have been related to the illness over the years, including coronaviruses. It has been reported that C57BL/6 mice are susceptible to MHV infection and show demyelination following infection (Lavi et al., 1984). MHV is neuroinvasive and can reach the brain through the olfactory nerve (Barthold, 1988, Lavi et al., 1988, Perlman et al., 1989, Barnett and Perlman, 1993). Intra-cerebral inoculation bypasses this natural way and allows the administration of the virus directly to the brain. The A59 strain leads to demyelination while being less neurovirulent than JHM (Robb et al., 1979). In order to get new insights into the mechanisms underlying the neuroimmunopathology induced by MHV, mice were infected with MHV-A59 and modulation of the transcriptome profile within the CNS was observed, as well as its possible consequences on immune cell responses.
In order to better characterize the model in the conditions studied, a viral load kinetics was first performed and illustrated that the virus was already replicating at the injection site at 1 day p.i. Maximal viral load in brain was detected 1 day later (2 days p.i.) and began to decline afterwards. Moreover, virus was detected in the periphery, as indicated by detection of infectious virus in liver. In both brain and liver, infectious virus became undetectable at 7 days p.i. Thus, experiments using cDNA arrays were performed at 1 day p.i., an early time after infection, at 2 days p.i., when the brain viral load was maximal, at 4 days p.i., when virus clearance was an ongoing process and at 7 days p.i., when infectious virus was undetectable.
According to our criteria described in Section 3.2, more than half (44 out of 80) of the significant changes in gene expression induced by MHV infection were up-regulation observed at 7 days p.i. (Table 2) and, as previously mentioned, most of these genes (26 out of 44) have been previously related to immune responses. Genes like Tap-1, Ii or cathepsin S encode proteins involved in antigen presentation. As part of a normal immune response, an increase in antigen presentation can contribute to the clearance on the infectious agent but has the potential to increase the presentation of self antigens, such as myelin components, which can be exposed following lysis of the infected cells or damage caused by local CNS inflammation. An up-regulation in the expression of some chemokines (CXCL9, CXCL10) was also observed. These chemokines bind CXCR3 on the surface of T and NK cells (Farber, 1997, Loetscher et al., 1996). CXCL9 and CXCL10 are involved in T-cell recruitment in the acute response following MHV infection of mouse (Liu et al., 2000, Liu et al., 2001a). CXCL10/IP-10 also contribute to demyelination in chronic response (Liu et al., 2001b). Moreover, IP-10 (−/−) mice infected with MHV recruited less T lymphocytes in the brain, had a delayed viral clearance from the brain and showed less demyelination in the CNS (Dufour et al., 2002). Moreover, the early increase in the level of expression of CXCL-10, as early as 1 and 2 days p.i. (Table 2), is of particular interest as this chemokine is known to be essential for the activation and recruitment of NK cells as early as 2 days p.i. to the CNS of MHV-A59-infected mice, helping to initiate virus clearance even before the T cells start to significantly infiltrate the CNS (Trifilo et al., 2004). Furthermore, it was also shown that these MHV-A59-activated NK cells produced IFN-γ within the CNS (Trifilo et al., 2004). The kinetics of CXCL-10 expression is in exact concordance with the literature as an increased expression in ependymal cells as early as 1 day p.i. was shown before (Lane et al., 1998) and its on-going sustained increase in expression correlates perfectly well with these results as this increase become concomitant with the increase of the gene coding for IFIT3, one of the IFN-γ-induced gene present on the ATLAS 1.2 II cDNA array. This latter result may indicate that IFN-γ is already being produced by infiltrating NK cells after the first burst of CXCL-10 increase. However, if not controlled adequately, the expression of chemokines such as CXCL-9 and CXCL-10 at a high level (Table 2) later in the process, when no more infectious virus is detected within the brain, could contribute to the recruitment of more and more immune cells, a situation that could lead to an autoimmune response partly related to the presence of MBP-autoreactive T cells. The HMGB1 gene is also slightly increased (3.7-fold) at 1 day p.i. This gene has a role in local inflammation and is expressed by microglia (Takata et al., 2003) and macrophage (Wang et al., 2004) and was recently shown to stimulate the production of IFN-γ by NK cells (Demarco et al., 2005). This early increase (1 day p.i.) in the level of expression may be associated with the early steps in the process of microglial cell activation. Moreover, as the expression of HMGB1 is increased 17-fold at 7 days p.i., this can be correlated with the infiltration of macrophages from the periphery. The early (1 day p.i.) and late increase (7 days p.i.) in the expression level of the HMGB 1 gene may also be associated with the concomitant elevation of the CD83 antigen, which is usually associated with dendritic cell (DC) maturation (Messmer et al., 2004). Moreover, even though they appear to be absent from the brain parenchyma, dendritic cells are one of the first leukocytes to be recruited within the CNS during the early phase of development of EAE, probably coming from a subpopulation of immature DC that reside in the meninges and the choroid plexus (Karman et al., 2004). Moreover, as they mature and participate to CNS inflammation, they are likely to also play a role against CNS infection (Pashenkov et al., 2003). Another IFN-induced gene with a significant increased level of expression is the transcription factor ISGF3-gamma. The expression of this particular gene is mainly controlled by type I IFN, especially IFNβ (Suhara et al., 1996), and the expression of the gene coding for this cytokine was previously shown to be increased as early as 3 days p.i. after a CNS infection by MHV-A59 (Rempel et al., 2004). As MHV-A59 replication is also affected by IFNβ (Garlinghouse et al., 1984), a local production of this cytokine by astrocytes following MHV infection (Wang et al., 1998) could contribute to the first steps of viral clearance.
Two recent reports (Rempel et al., 2004, Li et al., 2004) have also reported an increase in the level of expression of a set of proinflammatory cytokines early after infection by MHV-A59. Indeed, using a very precise method of RNAse Protection Assay, Rempel and colleagues observed a slight but significant increase in the expression of the genes coding for IL-12 p40 and IL-1β, IL-6 and TNF-α as early as 3 days p.i. Using the system of small cytokine array (GEArray), Li and colleagues were also able to observe an increase in the level of expression of these 4 proinflammatory cytokines as well as in the level of expression of IL-15 at 5 days p.i. However, by using the cDNA macroarray system, which allows to screen for the modulation of expression of 1176 different genes, we were not able to measure any significant modulation in the expression of the genes coding for those cytokines. The genes coding for IL-6 and TNF-α, not being present on the nylon membranes ATLAS mouse 1.2 II, could not be studied directly and there was no measurable modulation of any genes, present on the membrane, that are known to be regulated by TNF-α. However, by applying our criteria of 3-fold modulation and 5000 densitometric units (described in Section 2.4), no significant modulation in expression of the gene coding for IL-15, IL-1 β and IL-12 p40 could be observed. As the gene coding for IL-1 β and IL-12 p40 were shown to be significantly modulated during an MHV-A59 infection of the mouse CNS in two different reports, our result may be surprising. Nevertheless, our criteria for screening the significant modulations of the mouse transcriptome following infection appear to be appropriate as many of the significant modulations we observed are in perfect concordance with other studies on the subject.
Other genes that were up-regulated at 7 days p.i. are characteristic of leukocytes (CD68, CD45, CD18, galectin-3, Fc receptor IgG, CD3γ, CD8α), suggesting an infiltration of immune cells within the infected brain when no more infectious virus is detected (Fig. 1). Moreover, recent reports (Rempel et al., 2004, Phillips et al., 2002) have directly demonstrated that intracranial infection with either MHV-A59 or MHV-JHM induces an infiltration of T cells within the CNS at 7 days p.i. CD18 is involved in leukocyte extravasation (Gahmberg et al., 1998). Galectin-3 and Fc receptors are found at macrophage and microglia cell surface and are involved in binding and phagocytosis of myelin (Smith, 2001). Moreover, in a model of EAE, macrophage and microglial cells overexpress galectin-3 (Reichert and Rotshenker, 1999). An increase in the expression of CD3γ and CD8α point towards a T-cell infiltration into the CNS. The presence of immune cells in the brain could lead to direct or indirect damage. These modulations suggest that viral infection of the brain is followed by an increase in antigen presentation and cytokines and chemokines expression and by extravasation of leukocytes at the site of infection at 7 days p.i., when the infectious virus has been eliminated from the brain.
It has previously been demonstrated that MHV-JHM infection of mouse can lead to the activation of self-reactive T cells, which produce IL-2 in the presence of antigen-presenting cells and in the absence of viral antigen (Kyuwa et al., 1991). Furthermore, it was also reported several years ago that rats infected with MHV-JHM showed T-cell proliferation upon stimulation with myelin basic protein (MBP), and adoptive transfer of these cells to naive rats led to an experimental allergic encephalomyelitis (EAE)-like disease. However, prior to the current study, there had been no reports of T cell responses specific to a particular myelin antigen in MHV-infected mice. Therefore, as the robust genomic response observed (Table 2) indicates that several of the genes that were modulated in their expression are immune response-related, it led us to directly investigate the immune cell response, with a special emphasis on T cell, following MHV infection of the CNS.
Thus, we decided to evaluate the cellular immune response against the virus and a myelin protein, MBP, which appears to be a target of self-reactive T lymphocytes in MS and can induce an MS-like disease, EAE, in experimental animals through MBP-specific T cells. Bovine MBP was presented to infected mouse T cells. MBP represents 25–30% of total myelin proteins in the CNS (Deber and Reynolds, 1991). This protein is much conserved between species (Martenson, 1984) and has 79% identity with mouse MBP (alignment with SIM, using an algorithm described by Huang and Miller, 1991). In our model, the maximal T-cell response to MHV-A59 was detected with mouse splenocytes harvested at 7 days p.i. At this moment, proliferation upon stimulation with MBP was observed in T cells from MHV-infected but not from control mice (Fig. 3). It is worth noting that this activation was specific as indicated by the absence of proliferation when cells were stimulated by control antigens (ovalbumin or mouse lung homogenate). Not all the infected mice showed a specific immune response for MBP, nor for MHV-A59. Despite this observed variability between immune responses at 7 days p.i., the difference in proliferative responses to viral and myelin antigens in infected and control groups was highly significant (p<0.0001).
Thus, our results demonstrate for the first time the activation by murine coronavirus infection of self-reactive T cells that recognize a myelin antigen present within the CNS. These results are in agreement with the hypothesis associating viral infections and autoimmune diseases and are in correlation with the transcriptome profile modulation we observed within the CNS following MHV infection. Altogether, these results suggest a role for the inflammatory response in the autoimmune process, which may involve underlying mechanisms such as molecular mimicry or epitope spreading, or a combination (Talbot et al., 1996, Miller et al., 2001). However, at the moment, we do not have any direct proof that these autoreactive T cells can have any incidence related to the outcome of an autoimmune pathology.
In conclusion, following i.c. injection, MHV-A59 replicated in the CNS and in the periphery but complete clearance of infectious virus took place rapidly. Furthermore, this disappearance of infectious virus was associated with a robust host genomic response including upregulation of several genes encoding proteins related to different levels of the innate and acquired immune response, as indicated by results obtained with a powerful platform for parallel analysis on a genome-wide scale. Our observations open up future studies on several immune-response-related genes but also on other genes which role during response to MHV infection has not been anticipated. Using an MS animal model, cDNA arrays allowed us to further describe that an MHV infection can induce the activation of self-reactive T cells specific for a myelin protein that has been implicated in the neuropathology of EAE and MS.
The essential role of autoimmunity has been well characterized in the induction of demyelination within the CNS following an infection by MHV-JHM (for review see Matthews et al., 2002a, Haring and Perlman, 2001). On the other hand, to our knowledge, no clear demonstration of the importance of an autoimmune response following an MHV-A59 infection has ever been reported. Indeed, even though different reports (Matthews et al., 2002b, Sutherland et al., 1997) have strongly suggested that autoimmunity does not play an essential role in demyelination following an infection by this virus, it cannot be ruled out that the induction of the process implicates autoimmunity. Therefore, the detection of autoreactive T cells specific to a myelin antigen is most interesting as it could implicate a role for these cells in the induction of the demyelination process.
Acknowledgments
We gratefully thank Francine Lambert for excellent technical assistance. This work was supported by the Institute of Infection and Immunity, Canadian Institutes of Health Research (CIHR) funds to Pierre J. Talbot. Édith Gruslin was supported by a studentship from Natural Science and Engineering Research Council of Canada (NSERC).
References
- Arbour N., Côté G., Lachance C., Tardieu M., Cashman N.R., Talbot P.J. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J. Virol. 1999;73:3338–3350. doi: 10.1128/jvi.73.4.3338-3350.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N., Ekandé S., Côté G., Lachance C., Chagnon F., Cashman N.R., Talbot P.J. Persistent infection of human oligodendrocytic and neuronal cell lines by human coronavirus 229E. J. Virol. 1999;73:3326–3337. doi: 10.1128/jvi.73.4.3326-3337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N., Day R., Newcombe J., Talbot P.J. Neuroinvasion by human respiratory coronaviruses. J. Virol. 2000;74:8913–8971. doi: 10.1128/jvi.74.19.8913-8921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett E.M., Perlman S. The olfactory nerve and not the trigeminal nerve is the major site of CNS entry for mouse hepatitis virus, strain JHM. Virology. 1993;194:185–191. doi: 10.1006/viro.1993.1248. [DOI] [PubMed] [Google Scholar]
- Barthold S.W. Olfactory neural pathway in mouse hepatitis virus nasoencephalitis. Acta Neuropathol. 1988;76:502–506. doi: 10.1007/BF00686390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonavia A., Arbour N., Yong W.V., Talbot P.J. Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J. Virol. 1997;71:800–806. doi: 10.1128/jvi.71.1.800-806.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burks J.S., DeVald B.L., Jankovsky L.D., Gerdes J.C. Two coronaviruses isolated from central nervous system tissue of two multiple sclerosis patients. Science. 1980;209:933–934. doi: 10.1126/science.7403860. [DOI] [PubMed] [Google Scholar]
- Chapoval A.I., Ni J., Lau J.S., Wilcox R.A., Flies D.B., Liu D., Dong H., Sica G.L., Zhu G., Tamada K., Chen L. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001;2:269–274. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- Daniel C., Talbot P.J. Physico-chemical properties of murine hepatitis virus, strain A59. Brief report. Arch. Virol. 1987;96:241–248. doi: 10.1007/BF01320963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deber C.M., Reynolds J.S. Central nervous system myelin: structure, function and pathology. Clin. Biochem. 1991;24:113–134. doi: 10.1016/0009-9120(91)90421-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarco R.A., Fink M.P., Lotze M.T. Monocytes promote natural killer cell interferon gamma production in response to the endogenous danger signal HMGB1. Mol. Immunol. 2005;42:433–444. doi: 10.1016/j.molimm.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Dufour J.H., Dziejman M., Liu M.T., Leung J.H., Lane T.E., Luster A.D. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- Farber J.M. Mig and IP10: CXC chemokines that target lymphocytes. J. Leukoc. Biol. 1997;61:246–257. [PubMed] [Google Scholar]
- Gahmberg C.G., Valmu L., Fagerholm S., Kotovuori P., Ihanus E., Tian L., Pessa-Morikawa T. Leukocyte integrins and inflammation. Cell. Mol. Life Sci. 1998;54:549–555. doi: 10.1007/s000180050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlinghouse L.E., Jr., Smith A.L., Holford T. The biological relationship of mouse hepatitis virus (MHV) strains and interferon: in vitro induction and sensitivities. Arch. Virol. 1984;82:19–29. doi: 10.1007/BF01309365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring J., Perlman S. Mouse hepatitis virus. Curr. Opin. Microbiol. 2001;4:462–466. doi: 10.1016/S1369-5274(00)00236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Miller W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 1991;12:337–357. [Google Scholar]
- Ibrahim S.M., Mix E., Böttcher T., Koczan D., Gold R., Rolfs A., Thiesen H.-J. Gene expression profiling of the nervous system in murine experimental autoimmune encephalomyelitis. Brain. 2001;124:1927–1938. doi: 10.1093/brain/124.10.1927. [DOI] [PubMed] [Google Scholar]
- Karman J., Ling C., Sandor M., Fabry Z. Dendritic cells in the initiation of immune responses against central nervous system-derived antigens. Immunol. Lett. 2004;92:107–115. doi: 10.1016/j.imlet.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Kumanishi T. Brain tumors induced with Rous Sarcoma virus, Schmidt-Ruppin strain. I. Induction of brain tumors in adult mice with Rous chicken sarcoma cells. Jpn. J. Exp. Med. 1967;37:461–474. [PubMed] [Google Scholar]
- Kyuwa S., Yamaguchi K., Toyoda Y., Fujiwara K. Induction of self-reactive T cells after murine coronavirus infection. J. Virol. 1991;65:1789–1795. doi: 10.1128/jvi.65.4.1789-1795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane T.E., Buchmeier M.J. Murine coronavirus infection: a paradigm for virus-induced demyelinating disease. Trends Microbiol. 1997;5:9–14. doi: 10.1016/S0966-842X(97)81768-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane T.E., Asensio V.C., Yu N., Paoletti A.D., Campbell I.L., Buchmeier M.J. Dynamic regulation of alpha and beta chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 1998;160:970–978. [PubMed] [Google Scholar]
- Lavi E., Gilden D.H., Highkin M.K., Weiss S.R. Persistence of mouse hepatitis virus A59 RNA in a slow demyelinating infection in mice as detected by in situ hybridization. J. Virol. 1984;51:563–566. doi: 10.1128/jvi.51.2.563-566.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi E., Fishman P.S., Highkin M.K., Weiss S.R. Limbic encephalitis after inhalation of a murine coronavirus. Lab. Invest. 1988;58:31–36. [PubMed] [Google Scholar]
- Li Y., Fu L., Gonzales D.M., Lavi E. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J. Virol. 2004;78:3398–3406. doi: 10.1128/JVI.78.7.3398-3406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.T., Chen B.P., Oertel P., Buchmeier M.J., Armstrong D., Hamilton T.A., Lane T.E. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J. Immunol. 2000;165:2327–2330. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- Liu M.T., Armstrong D., Hamilton T.A., Lane T.E. Expression of Mig (monokine induced by interferon-gamma) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J. Immunol. 2001;166:1790–1795. doi: 10.4049/jimmunol.166.3.1790. [DOI] [PubMed] [Google Scholar]
- Liu M.T., Keirstead H.S., Lane T.E. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J. Immunol. 2001;167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- Loetscher M., Gerber B., Loetscher P., Jones S.A., Piali L., Lewis I.C., Baggiolini M., Moser B. Chemokine receptor specific for IP-10 and Mig: structure, function, and expression in activated lymphocytes. J. Exp. Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martenson R. Myelin basic protein speciation. Prog. Clin. Biol. Res. 1984;146:511–521. [PubMed] [Google Scholar]
- Matthews A.E., Weiss S.R., Paterson Y. Murine hepatitis virus—a model for virus-induced CNS demyelination. J. Neurovirology. 2002;8:76–85. doi: 10.1080/13550280290049534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews A.E., Lavi E., Weiss S.R., Paterson Y. Neither B cells nor T cells are required for CNS demyelination in mice persistently infected with MHV-A59. J. Neurovirology. 2002;8:257–264. doi: 10.1080/13550280290049697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messmer D., Yang H., Telusma G., Knoll F., Li J., Messmer B., Tracey K.J., Chiorazzi N. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 2004;173:307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- Miller S.D., Olson J.K., Croxford J.L. Multiple pathways to induction of virus-induced autoimmune demyelination: lessons from Theiler's virus infection. J. Autoimmun. 2001;16:219–227. doi: 10.1006/jaut.2000.0489. [DOI] [PubMed] [Google Scholar]
- Murray R.S., Brown B., Brian D., Cabirac G.F. Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann. Neurol. 1992;31:525–533. doi: 10.1002/ana.410310511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashenkov M., Teleshova N., Link H. Inflammation in the central nervous system: the role for dendritic cells. Brain Pathol. 2003;13:23–33. doi: 10.1111/j.1750-3639.2003.tb00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Jacobsen G., Afifi A. Spread of a neurotropic murine coronavirus into the CNS via the trigeminal and olfactory nerves. Virology. 1989;170:556–560. doi: 10.1016/0042-6822(89)90446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J., Chua M.M., Rall G.F., Weiss S.R. Murine coronavirus spike glycoprotein mediates degree of viral spread, inflammation and virus-induced immunopathology in the central nervous system. Virology. 2002;301:109–120. doi: 10.1006/viro.2002.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert F., Rotshenker S. Galectin-3/Mac-2 in experimental allergic encephalomyelitis. Exp. Neurol. 1999;160:508–514. doi: 10.1006/exnr.1999.7229. [DOI] [PubMed] [Google Scholar]
- Rempel J.D., Murray S.J., Meisner J., Buchmeier M.J. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology. 2004;318:381–392. doi: 10.1016/j.virol.2003.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb J.A., Bond C.W., Leibowitz J.L. Pathogenic murine coronaviruses. III. Biological and biochemical characterization of temperature-sensitive mutants of JHMV. Virology. 1979;94:385–399. doi: 10.1016/0042-6822(79)90469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmi A., Ziola B., Hovi T., Reunanen M. Antibodies to coronaviruses OC43 and 229E in multiple sclerosis patients. Neurology. 1982;32:292–295. doi: 10.1212/wnl.32.3.292. [DOI] [PubMed] [Google Scholar]
- Smith M.E. Phagocytic properties of microglia in vitro: implications for a role in multiple sclerosis and EAE. Microsc. Res. Tech. 2001;54:81–94. doi: 10.1002/jemt.1123. [DOI] [PubMed] [Google Scholar]
- Stewart J.N., Mounir S., Talbot P.J. Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology. 1992;191:502–505. doi: 10.1016/0042-6822(92)90220-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhara W., Yoneyama M., Yonekawa H., Fujita T. Structure of mouse interferon stimulated gene factor 3 gamma (ISGF3 gamma/p48) cDNA and chromosomal localization of the gene. J. Biochem. (Tokyo) 1996;119:231–234. doi: 10.1093/oxfordjournals.jbchem.a021227. [DOI] [PubMed] [Google Scholar]
- Sutherland R.M., Chua M.-M., Lavi E., Weiss S.R., Paterson Y. CD4+ and CD8+ T cells are not major effectors of mouse hepatitis virus A59-induced demyelinating disease. J. Neurovirol. 1997;3:225–228. doi: 10.3109/13550289709018297. [DOI] [PubMed] [Google Scholar]
- Takata K., Kitamura Y., Tsuchiya D., Kawasaki T., Taniguchi T., Shimohama S. Role of high mobility group protein-1 (HMG1) in amyloid-beta homeostasis. Biochem. Biophys. Res. Commun. 2003;301:699–703. doi: 10.1016/s0006-291x(03)00024-x. [DOI] [PubMed] [Google Scholar]
- Talbot P.J., Paquette J.-S., Ciurli C., Antel J.P., Ouellet F. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann. Neurol. 1996;39:233–240. doi: 10.1002/ana.410390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilo M.J., Montalto-Morrison C., Stiles L.N., Hurst K.R., Hardison J.L., Manning J.E., Masters P.S., Lane T.E. CXC chemokine ligand 10controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J. Virol. 2004;78:585–594. doi: 10.1128/JVI.78.2.585-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Haluskey J.A., Lavi E. Coronavirus MHV-A59 causes upregulation of interferon-β RNA in primary glial cell cultures. Adv. Exp. Med. Biol. 1998;440:451–454. doi: 10.1007/978-1-4615-5331-1_57. [DOI] [PubMed] [Google Scholar]
- Wang H., Yang H., Tracey K.J. Extracellular role of HMGB1 in inflammation and sepsis. J. Intern. Med. 2004;255:320–331. doi: 10.1111/j.1365-2796.2003.01302.x. [DOI] [PubMed] [Google Scholar]
- Watanabe R., Wege H., ter Meulen V. Adoptive transfer of EAE-like lesions from rats with coronavirus-induced demyelinating encephalomyelitis. Nature. 1983;305:150–153. doi: 10.1038/305150a0. [DOI] [PMC free article] [PubMed] [Google Scholar]