

Abstract
A novel two-step real-time RT-PCR assay using SYBR® Green I was developed for the detection of acute Bovine Viral Diarrhoea virus (BVDV) infection in whole blood from cattle. During infection animals experience a characteristic transient leucopenia and the number of cells per volume of blood changes over time; so quantitation of viral load by reference to a cellular housekeeping gene is not ideal as this may hide significant animal to animal variation.
Therefore, to facilitate comparison of different samples, an external RNA reference was used for normalisation whereby each sample was spiked with the RNA virus, Canine Enteric Coronavirus (CECov), prior to RNA extraction, for comparative purposes. Real-time RT-PCR was carried out with two primer sets designed to amplify either a 156 bp region of the BVDV 5′-UTR or a 280 bp region of the CECov nucleocapsid protein gene. Linearity and efficiency of the assay was established and the method assessed using samples from BVDV-challenged calves.
Viral RNA was quantified on days 6 and 14 post-challenge by real-time RT-PCR. Infectious virus isolation by traditional cell culture was negative after day 7. This study demonstrates encouraging results for rapid, sensitive and reliable detection of acute BVDV infection and provides an alternative real-time RT-PCR method for use on whole blood samples or samples where suitable housekeeping genes are not available.
Keywords: Real-time RT-PCR, Bovine Viral Diarrhoea virus, Pestivirus, Canine Enteric Coronavirus
Bovine Viral Diarrhoea virus (BVDV) is a pathogen of worldwide importance for cattle, causing significant economic losses (Brownlie, 1991). BVDV is classified within the Pestivirus genus of the Flaviviridae (Heinz et al., 2000). Pestiviruses are small, enveloped viruses that contain a single-strand positive sense RNA molecule of approximately 12.5 kb. The genome is transcribed as a single open reading frame, flanked by 5′- and 3′-untranslated regions (UTRs). The BVDV polyprotein is co- and post-translationally cleaved to give rise to a number of viral structural and non-structural proteins (Collett et al., 1988). Acute BVDV infection is often clinically unapparent or mild; clinical signs including transient pyrexia, leucopenia, oculonasal discharge and depression. Although frequently unnoticed, acute infection can contribute to or cause a variety of problems including diseases of the reproductive, respiratory and enteric systems (Baker, 1995). BVDV infection during pregnancy may result in foetal infection. Foetuses infected before the onset of immunocompetence can become persistently infected. Persistently infected (PI) animals continuously shed virus and act as a major reservoir of infection for other cattle (Paton et al., 1999).
Current laboratory-based BVDV detection methods include serological assays, virus isolation, antigen detection and detection of viral RNA by reverse transcription (RT) PCR (Sandvik, 2005). Real-time PCR is now being used to identify, classify and quantify many viral pathogens as it is a highly sensitive and rapid method for detecting viral nucleic acid sequences in clinical specimens. Furthermore, real-time PCR is a quantitative technique and as such may be used to assess viral RNA levels. Real-time RT-PCR methods for genotyping BVDV have been described previously (Bhudevi and Weinstock, 2001, Bhudevi and Weinstock, 2003, Lettellier and Kerkhofs, 2003). These papers have focused on the detection and classification of BVDV into either of the two recognised genotypes (BVDV-1 and BVDV-2) rather than monitoring the changes in viral load during the course of an acute or persistent infection. However, as real-time PCR is a quantitative technique it may also be used to assess viral RNA levels during the course of infection and recovery.
Using real-time PCR to quantify the viral RNA levels in BVDV-infected animals presents particular technical and biological challenges. Collection and processing of samples from different animals over a number of different days inevitably results in some degree of sample to sample variation in the technical detection of the target nucleic acids. This arises from differences in efficiency of RNA extraction, reverse transcription or PCR reactions which is made particularly significant by the sensitivity of this technique. These technical variations are often “controlled” for by normalisation of the signal to an internal standard, typically a “housekeeping” gene, facilitating the comparison of different data sets. This presents two further problems: firstly, whilst this is common practice, such housekeeping genes are rarely truly static and may alter under experimental conditions (Thellin et al., 1999, Whelan et al., 2003). To take account of this, best practice would recommend assessing the validity of a number of different housekeeping gene targets and comparing them under the test conditions used. This laborious task is the only way to objectively identify more stable mRNA sets. However, this process is not relevant to the BVDV situation during infection as, secondly, during acute infection with BVDV animals experience a measurable leucopenia and the number of cells per volume of blood changes over time. Traditional methods of virus isolation quantitate the amount of virus recovered from a fixed volume of blood irrespective of the cell numbers in this sample, which during acute BVDV infection will fluctuate (Polak and Zmudzinski, 2000). By standardising the viral RNA measured by real-time PCR to a housekeeping gene set the quantitation is essentially done on a per cell basis rather than a per volume basis rendering the comparison of samples from leucopenic calves more difficult and precluding a simple comparison with more traditional virological methods.
To overcome these difficulties a novel two-step real-time RT-PCR method using an external RNA reference standard was developed, which has been evaluated on samples from experimentally infected cattle. Each sample was spiked with a known amount of a second RNA virus, Canine Enteric Coronavirus (CECov), prior to RNA extraction. While the external standard is not an exact mimic of the true target it is subject to the same experimental procedures and provides an external standard for correction of differences in the efficiency of RNA extraction or RT-PCR. Furthermore, to facilitate ease of sample handling the method was based on quantification of BVDV RNA per volume of frozen whole blood.
Five Holstein–Friesian calves ranging in age from 2 to 4 months were obtained from a BVDV free-supplier and tested to confirm BVDV-free status by antigen and antibody ELISA. The calves were housed in a purpose-built barn with appropriate bio-security measures in place to avoid exposure to adventitious pathogens. After an initial acclimatisation period the calves were challenged intranasally with 2 × 106 TCID50 of virus 456497 (early passage BVDV-1; kindly donated by VLA, Weybridge, UK). EDTA blood samples were taken at appropriate days post-challenge to process for virus isolation and real-time PCR. All EDTA blood samples taken for virology were prepared by lysis of the erythrocytes using ammonium chloride lysis buffer. The samples were subsequently pelleted and washed, as described in Nobiron et al. (2003). To detect BVDV, the samples were diluted 1:5 in maintenance medium and then seeded, in duplicate, onto freshly prepared foetal bovine lung (FBL) cells and incubated for 5 days at 37 °C, 5% CO2. After one round of freeze thaw and dilution of material, the inoculum was passed once more onto FBLs, which had been previously seeded onto coverslips, and incubated for a further 5 days. Presence of virus was detected via immuno-fluorescence staining using a BVDV hyperimmune serum and a Cy-3-conjugated anti-bovine antibody. Scoring was performed using a fluorescence microscope and appropriate filter sets.
RNA was extracted from frozen whole blood (allowing samples from several different time points to be processed on the same day) using the QIAamp DNA blood mini kit (Qiagen). The DNA Blood Mini Kit was used with several modifications (made in discussion with the supplier) as it enabled purification of RNA from frozen whole blood while the Viral RNA Kit was not able to process frozen blood samples. First 400 μl blood was thawed and 50 μl CECov (1 × 105 TCID50/ml) added as an external RNA reference. This was added to 400 μl protease solution followed by 400 μl buffer AL. After incubation at 56 °C for 10 min, the sample was cooled and 460 μl 100% ethanol added before applying to the spin column. After two washes, RNA was eluted with 50 μl tissue culture grade dH2O (Invitrogen). RNA was reverse-transcribed using Superscript II (Invitrogen) and random primers (Amersham Pharmacia Biotech).
SYBR® Green I dye was used in this real-time RT-PCR method, which binds to any double stranded DNA produced in the reaction. Each reaction contained 10 μl SYBR® Green JumpStart Taq ReadyMix for QPCR (Sigma), 2.5 μl forward primer (10 pmol/μl), 2.5 μl reverse primer (10 pmol/μl) and 5 μl template cDNA. Samples were analysed in triplicate. The PCR conditions were 94 °C for 10 min followed by 35 cycles of 94 °C for 10 s; 53 °C for 20 s; 72 °C for 20 s; 80 °C for 10 s after which a plate read was taken. The reactions were carried out on a DNA Engine Opticon 2 (MJ Research) and data analysed using the Opticon Monitor Analysis Software version 2.02. The primers were designed to amplify a 156 bp region of BVDV 5′-UTR (sense 5′-TAGTCGTCAGTGGTTCACGCC; antisense 5′-CCTCTGCAGCACCCTATCAG), or a 280 bp region of CECov nucleocapsid gene (sense 5′-CTCGTGGYCGGAAGAGTAAT; antisense 5′-GCAACCCAGAMRACTCCATC) and were tested to confirm sensitivity and efficiency over a range of DNA concentrations. Primer efficiency was confirmed for each experiment using the formula:
Primer efficiency = 10−1/slope (Pfaffl, 2001), whereby a value of 2.0 indicates 100% efficiency. This ensured PCR products were amplified at an efficient rate and experiments were comparable. The standard samples used were plasmid DNA containing BVDV or CECov PCR products cloned into pGemT (Promega). As cDNA is a potentially more complex sample than the purified plasmid DNA used for the standard curve a test was carried out to confirm the components of a viral cDNA preparation would not be detrimental to the PCR reaction whereby standard samples were spiked with cDNA from uninfected bovine blood and the cycle threshold (CT) values compared to non-spiked samples. Melting curve analysis of the PCR products was also carried out for each experiment and confirmed that both primer–dimers and non-specific products were absent and fluorescence was measured at temperatures where only BVDV or CECov specific amplicons were detected (data not shown). The mean CT value for CECov was determined after multiple extractions of CECov spiked blood (mean CT = 12.457, S.D. = 0.484; n = 30). Samples that fell within the range of mean CECov CT ± 1 S.D. were corrected as described in Niesters (2001); the procedure was repeated for any samples that were outside of this range.
After BVDV challenge, leucopenia was evident in all calves (Fig. 1 ). The decrease in white blood cell counts ranged from 7% to 52% compared to the mean pre-challenge levels for all animals 3 days post-challenge. The mean reduction was statistically significant on day 3 (P = 0.009), day 5 (P = 0.025) and day 6 (P = 0.017). This demonstrates the characteristic leucopenia observed following acute BVDV infection and highlights the complexities faced when undertaking real-time PCR on acutely infected cattle if normalizing to cellular housekeeping genes.
Fig. 1.
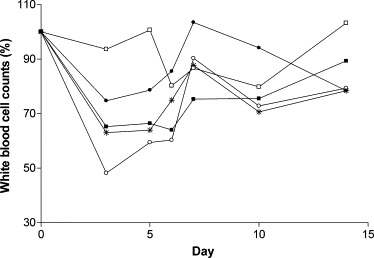
White blood cell counts following challenge. Counts were calculated as a percentage of the pre-challenge baseline values for calf 1 (●), calf 3 (○), calf 10 (□), calf 15 (*) and calf 20 (■).
Primers were designed to amplify a 156 bp region of the highly conserved BVDV 5′-UTR. The linearity of the 5′-UTR assay was confirmed using cDNA obtained from BVDV spiked blood (Fig. 2A) within the range of 7000 ng to 7 ng, from samples containing 2.1 × 104 TCID50 to 2.1 × 101 TCID50 BVDV, indicating the sensitivity of the technique. The PCR efficiency was 2.3 and the regression coefficient was 0.993. It was not possible to efficiently detect virus in samples containing less than 2.1 × 101 TCID50 BVDV. Linearity of the primers was also confirmed using plasmid DNA containing the 5′-UTR PCR product (Fig. 2B) as this was used for generation of 5′-UTR standard curve in each experiment. The linear range of the PCR was from 1 × 108 to 1 × 101 DNA copies/μl, with an efficiency of 2.02 and regression coefficient of 0.995. Spiking the plasmid DNA standards with cDNA obtained from a BVDV negative animal was not detrimental to the efficiency of the reaction (Fig. 2C) and there was no statistically significant difference between CT values obtained from plasmid compared to cDNA spiked plasmid (P = 0.997). The linear range of the CECov primers was also assessed using cDNA obtained from samples containing 1 × 102 to 5 × 103 TCID50 CECov with an efficiency of 1.87 and regression coefficient of 0.990 (data not shown).
Fig. 2.
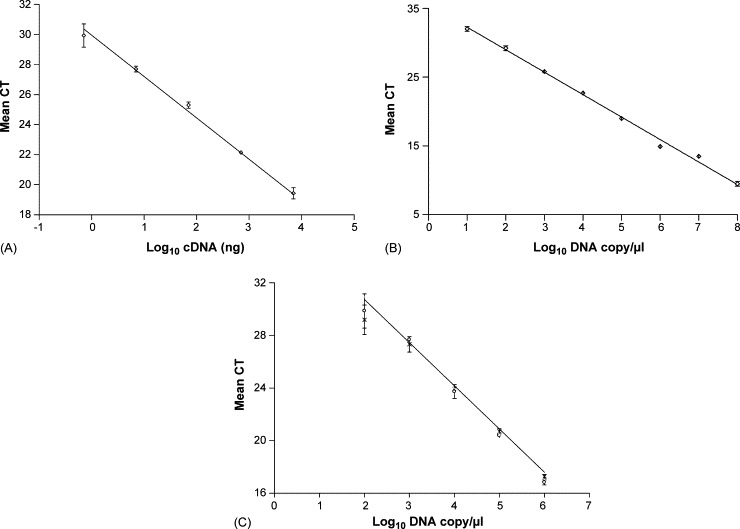
Optimisation of BVDV 5′-UTR real-time PCR primers. Mean CT values of (A) serially diluted cDNA from BVDV spiked blood, y = −2.76x + 30.06, r2 = 0.993, (B) plasmid DNA standard curve, y = −3.42x + 35.83, r2 = 0.995, and (C) plasmid DNA (○) and cDNA spiked plasmid DNA (×), y = −3.55x + 38.11, r2 = 0.997. Error bars show standard deviation.
A BVDV plasmid standard curve ranging from 1 × 108 to 1 × 101 copies/μl was used to assess 5′-UTR copy number in each sample. The PCR efficiency for each experiment was between 1.96 and 2.07 and the regression coefficient was greater than 0.992 (data not shown). The 5′-UTR CT value was adjusted accordingly based on the CT value of the CECov reaction.
By standard virus isolation one animal, calf 20, was virus negative throughout the study period, while virus could be isolated from the remaining calves for 1, 2 or 3 days between days 5 and 7 post-challenge. No virus was isolated from any animal on day 10 or 14 post-infection (Table 1 ).
Table 1.
Virus isolation following challenge
| Calf | Day post-challenge |
|||||
|---|---|---|---|---|---|---|
| 3 | 5 | 6 | 7 | 10 | 14 | |
| 1 | − | − | + | + | − | − |
| 3 | − | − | + | − | − | − |
| 10 | − | + | + | + | − | − |
| 15 | − | − | + | − | − | − |
| 20 | − | − | − | − | − | − |
Virus isolation from buffy coat was performed by immunofluorescence assay after two passages in FBL cells.
To validate this new methodology, two time points were chosen for analysis: day 6 post-infection as this represents the peak of the viraemia and also day 14 as this represents a stage when virus isolation indicates all animals to be virus free. BVDV viral RNA was detected in whole blood taken at days 6 and 14 post-challenge in every calf (Fig. 3 ). The estimates for viral genome copy number ranged from 1.95 × 105 to 5.88 × 105, and 1.92 × 105 to 3.22 × 105 5′-UTR copies/ml blood on days 6 and 14, respectively.
Fig. 3.
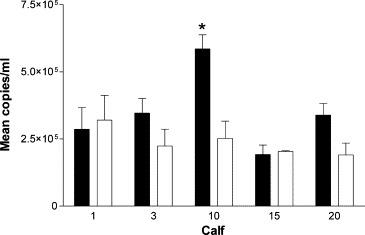
Quantification of BVDV RNA. Mean BVDV RNA copies/ml blood from calves on day 6 (■) and day 14 (□) post-challenge. Error bars show standard error of the mean “*” indicates significant values.
Viral RNA copies per ml of blood were higher on day 6 than on day 14 in three of the five calves although this difference was only statistically significant in calf 10 (P = 0.018). The remaining two calves had very similar levels of viral RNA on both days 6 and 14 post-infection. Calf 20 which had been consistently negative for infectious virus isolation was positive for viral RNA at both time points tested.
It was to be expected that there would be some discrepancy between the estimation of the BVDV 5′-UTR copy number of the viral RNA and the infectious viral load in this study and results confirm that RNA copies do not correlate with the number of infectious genomes present and therefore the potential for infectivity and virus transmission. Lettellier and Kerkhofs (2003) demonstrated using a BVDV type 1 virus stock that there were 2400 copies of virus genome per infectious particle. This presumably reflects a ratio of infectious to defective viral genomes. Similar ratios have been observed for Yellow Fever Virus (Bae et al., 2003). The results from the current study would support these observations and, in vivo at least, it is proposed that although BVDV RNA is still being detected, infectious virus has been complexed with antibodies thereby leading to cessation of viraemia. In the case of calf 20, virus neutralising antibody titres of 1:564 were measured on day 14 (data not shown), supporting this premise.
In order to compare samples it is necessary to use some form of standardisation and often housekeeping gene expression is used to achieve this. However, housekeeping gene expression can vary and the use of these genes as internal controls should be carefully considered in relation to cell type and the cell metabolism (Thellin et al., 1999). The large number of publications focusing on the validation of internal reference genes demonstrates the difficulty in selecting appropriate candidates. Indeed, many authors now recommend the use of at least two (Dent et al., 1997) or three (Bustin, 2000, Vandesompele et al., 2002) validated housekeeping genes to ensure the accuracy of data. In addition, the use of housekeeping genes to control for the variation in RNA extraction and cDNA synthesis is difficult when a fixed amount of blood is analysed (Johansson et al., 2000). The normal range of leucocyte counts in blood from healthy cattle is 4–12 × 109 l−1 (Blood, 1994), thus demonstrating a potential threefold variation in cell numbers. Furthermore, acute BVDV infection causes transient leucopenia, leading to a reduction in numbers of circulating leucocytes which can be pronounced. Virus virulence as well as host factors may result in the reduction of white blood cells ranging from mild to severe. In the current study all five calves were challenged with the same dose of virus and the degree of leucopenia varied from animal to animal (Fig. 1).
To facilitate virus quantification per fixed volume of blood in the face of characteristic leucopenia observed in cattle during acute BVDV infection, a novel two-step real-time RT-PCR method which utilised an external RNA reference standard was developed. A second RNA virus, Canine Enteric Coronavirus (CECov) was used as a control for the each stage of the method. To enable ease of sample handling the method was based on quantification of BVDV RNA per volume of frozen whole blood. Each bovine blood sample was spiked with a known amount of the reference CECov prior to RNA extraction. The coronavirus was co-extracted and subsequently amplified in a quantitative manner. This enabled validation of nucleic acid isolation and amplification efficiency for each sample and detection of those samples for which sample preparation or amplification failed by determining the signal generated from the CECov standard. This system of ‘spiking’ samples with a heterologous virus has been used previously for Hepatitis C virus quantification (Cleland et al., 1999, Castelain et al., 2004) and for the detection of Epstein–Bars virus and Cytomegalovirus (Niesters, 2001).
Currently there is much interest in control strategies for BVDV infection of cattle. To this end rapid and reliable diagnosis of both persistently and acutely infected cattle is imperative. Molecular diagnostic methods are being increasingly utilised as tools for the detection of numerous viral pathogens. The use of real-time RT-PCR methods to establish the presence or absence of BVDV RNA in cattle may have important implications for future diagnostic screening and control strategies.
Acknowledgement
The authors wish to thank Dr. Kerstin Erles (RVC) for kindly donating the Canine Enteric Coronavirus and corresponding primer sequences.
References
- Bae H.G., Nitsche A., Teichmann A., Biel S.S., Niedrig M. Detection of yellow fever virus: a comparison of quantitative real-time PCR and plaque assay. J. Virol. Meth. 2003;110(2):185–191. doi: 10.1016/s0166-0934(03)00129-0. [DOI] [PubMed] [Google Scholar]
- Baker J.C. The clinical manifestations of bovine viral diarrhoea infection. Vet. Clin. N. Am. Food. Anim. Pract. 1995;11(3):425–445. doi: 10.1016/s0749-0720(15)30460-6. [DOI] [PubMed] [Google Scholar]
- Bhudevi B., Weinstock D. Fluorogenic RT-PCR assay (TaqMan) for detection and classification of bovine viral diarrhea virus. Vet. Microb. 2001;83:1–10. doi: 10.1016/S0378-1135(01)00390-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhudevi B., Weinstock D. Detection of Bovine Viral Diarrhea Virus in formalin fixed paraffin embedded tissue sections by real time RT-PCR (TaqMan) J. Virol. Meth. 2003;109:25–30. doi: 10.1016/s0166-0934(03)00040-5. [DOI] [PubMed] [Google Scholar]
- Blood D.C. Balliere Tindall; London: 1994. Pocket Companion to Veterinary Medicine. [Google Scholar]
- Brownlie J. The pathways for Bovine Virus Diarrhoea virus biotypes in the pathogenesis of disease. Arch. Virol. Suppl. 1991;3:79–96. doi: 10.1007/978-3-7091-9153-8_10. [DOI] [PubMed] [Google Scholar]
- Bustin S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assay. J. Mol. Endo. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- Castelain S., Descamps V., Thibault V., François C., Bonte D., Morel V., Izopet J., Capron D., Zawadzki P., Duverlie G. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J. Clin. Virol. 2004;31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Cleland A., Nettleton P., Jarvis L.M., Simmonds P. Use of Bovine Viral Diarrhoea virus as an internal control for amplification of Hepatitis C Virus. Vox Sang. 1999;76:170–174. doi: 10.1159/000031044. [DOI] [PubMed] [Google Scholar]
- Collett M.S., Larson R., Belzer S.K., Retzel E. Proteins encoded by bovine viral diarrhea virus: the genomic organization of a pestivirus. Virology. 1988;165:200–208. doi: 10.1016/0042-6822(88)90673-3. [DOI] [PubMed] [Google Scholar]
- Dent A.L., Shaffer A.L., Yu X., Allman D., Staud L.M. Control of inflammation, cytokine expression and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- Heinz F., Collett M., Purrchell R., Gould E., Howard C., Houghton M., Moorman R., Rice C., Theil H.-J. Family Flaviviridae. In: van Regnmortel M.H.V., Fanqute C.M., Bishop D.H.L., Carstens E.V., Estes M.K., Lemon S.M., Manilott J., Mayo M.A., McGeoch D.J., Pringle C.R., Wickner R.B., editors. Virus Taxonomy, Proceedings of the Seventh Report of International Committee on Taxonomy of Viruses. Academic Press; 2000. pp. 859–878. [Google Scholar]
- Johansson M., Pisa E.K., Törmänen V., Årstrand K., Kågedal B. Quantitative analysis of tyrosinase transcripts in blood. Clin. Chem. 2000;46:921–927. [PubMed] [Google Scholar]
- Lettellier C., Kerkhofs P. Real-time PCR for simultaneous detection and genotyping of Bovine Viral Diarrhoea virus. J. Virol. Meth. 2003;114:21–27. doi: 10.1016/j.jviromet.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Niesters H.G.M. Quantitation of viral load using real-time amplification techniques. Methods. 2001;25:419–429. doi: 10.1006/meth.2001.1264. [DOI] [PubMed] [Google Scholar]
- Nobiron I., Thompson I., Brownlie J., Collins M.E. DNA vaccination against Bovine Viral Diarrhoea virus induces humoral and cellular responses in cattle with evidence for protection against viral challenge. Vaccine. 2003;21:2082–2092. doi: 10.1016/s0264-410x(02)00745-4. [DOI] [PubMed] [Google Scholar]
- Paton D.J., Sharp G., Ibata G. Foetal cross-protection experiments between type 1 and type 2 Bovine Viral Diarrhoea virus in pregnant ewes. Vet. Microbiol. 1999;64(2–3):185–196. doi: 10.1016/s0378-1135(98)00269-7. [DOI] [PubMed] [Google Scholar]
- Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucl. Acids Res. 2001;29(9):2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak M.P., Zmudzinski J.F. Experimental inoculation of calves with laboratory strains of Bovine Viral Diarrhea Virus. Comp. Immunol. Microbiol. Infect. Dis. 2000;23(3):141–151. doi: 10.1016/s0147-9571(99)00060-0. [DOI] [PubMed] [Google Scholar]
- Sandvik T. Selection and use of laboratory diagnostic assays in BVD control programmes. Prev. Vet. Med. 2005;72:3–16. doi: 10.1016/j.prevetmed.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Thellin O., Zorzi W., Lakaye B., De Borman B., Coumans B., Hennen G., Grisar T., Igout A., Heinen E. Housekeeping genes as internal standards: use and limits. J. Biotechnol. 1999;75:291–295. doi: 10.1016/s0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Whelan J.A., Russell N.B., Whelan M.A. A method for the absolute quantification of cDNA using real-time PCR. J. Immun. Meth. 2003;278:261–269. doi: 10.1016/s0022-1759(03)00223-0. [DOI] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalisation of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):1–12. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]