

Abstract
Interferons (IFNs) were discovered as antiviral agents 50 years ago, and enormous progress has been made since then. Nowadays, IFNs (specifically type I IFNs), have been ascribed as the cytokines that bridge the innate and adaptive immunity soon after the recognition of pathogen-associated molecular patterns (PAMPs) by the infected host. Notably, a unifying mechanism for type I IFN production has been established upon innate immune detection. Thus, TLR 3, 4, 7 and 9 associate endosomal recognition of PAMPs to type I IFN responses, a mechanism that has been shown in plasmacytoid dendritic cells to be dependent on the PI3K/mTOR/S6K pathway. It is worth noting that pathogen recognition triggers a fine-tuned controlled program that not only includes the production of antiviral (IFN) and pro-inflammatory cytokines to initiate the antiviral response but also signals the cessation of the response through the induction of suppressors of cytokine signaling (SOCS). SOCS in turn is under tight regulation of the TAM receptors (protein tyrosine kinase receptors TYRO3, AXL and MER), and activation of which thereby protects the host from the threats of autoimmune diseases.
Keywords: Interferons, Innate immune recognition/evasion, Antiviral
1. Introduction
Although it has been 50 years since Interferons (IFNs) were first described by Isaac and Lindenmann [1], this family of cytokines still remains at the central stage of the scientific arena. Even though IFNs were originally described as antiviral agents, Isaac and Lindenmann could not anticipate the immense impact their findings would have and the extent to which they would be relevant far beyond the field of Virology. From the discovery during the 1960s that IFNs also played a relevant role in the control exerted over cell growth and animal tumors up to the findings in more recent years that they are pivotal regulators of both innate and adaptive immune responses, the picture that emerges is that vertebrate life would be under permanent threat without IFNs. This review will focus on some aspects of the biology of the IFNs, with an emphasis on signaling, antiviral defense, and recent findings revealing remarkable strategies used by the viruses to evade host innate immune responses.
2. Recent progress in IFN signaling
There are three types of IFNs. Type I IFNs comprise the largest sub-family and include the IFN-αs (13 subtypes in humans) and IFN-β (the well-known IFNs first to be cloned, purified, and sequenced) [2], [3], [4]. All type I IFN genes diverged from a common ancestor, and it appears that IFN-β is the one from which IFN-α genes evolved to confer even more robust protection against invading pathogens [2], [5]. Indeed, IFN-β expression is required to exert positive-feedback control over the induction of IFN-α genes [6], [7]. All these IFNs share with the other members of this sub-family (e.g. ω, ɛ, κ, δ, and τ) the same ability to exert antiviral activity, are clustered on the short arm of chromosome 9 in humans, are all devoid of introns, and are both genetically and structurally very similar [2], [3], [4], [8]. Type I IFNs transduce intracellular signals through the common receptor IFNAR1/2 [9]. Type II IFNs are represented by just one member, IFN-γ. Although it also exhibits antiviral activity, IFN-γ is mainly regarded as a powerful immunomodulatory cytokine. It influences the intracellular environment by recognizing a distinct cell surface receptor named IFNGR1/2 [10].
A new sub-family of IFN has recently been identified. This family includes the type III IFNs or IFN-λs, which are comprised of three intron-containing members (λ1-3) and are known as interleukin (IL)-29, IL-28A, and IL-28B [11], [12]. A specific cellular receptor composed of the IL-10 receptor β and IL-28 receptor α chains elicits the biological effects of the IFN-λs upon receptor engagement. Type III IFNs share with the other types of IFNs the ability to control cell proliferation and interfere with virus replication [11], [13], [14], and we have recently demonstrated their abilities to protect cells against a Bunyaviridae family member known as the APEU virus [15]. A recent evolutionary study conducted by Levraud et al. suggests that Type III IFNs are the ancestral antiviral system of vertebrates [16].
All IFNs are Class II α-helical cytokines, which also includes IL-10, IL-19, and IL-20 among others [4], [17]. The signal transduction pathways initiated upon IFN binding to cognate receptors at the cell surface requires the activation, through tyrosine phosphorylation, of the intracellular receptor moiety. This role is associated with the JAKs (Janus kinases), a family of tyrosine kinases comprised of JAK1-3 and TYK2 [18]. Once phosphorylated, the receptors act as docking sites for the signal transduction and activators of transcription (STATs), which are phosphorylated upon recruitment to the receptor. Then, the STATs dissociate from the receptor, associate as homo- or heterodimers, and migrate to the nucleus. In the nucleus, they bind to cis-acting elements found at the promoter region of so-called IFN-stimulated genes (ISGs) to promote the transcription of more than 300 ISGs [3], [19], [20]. Type I IFNs typically recruit JAK1 and TYK2 to transduce their signals to STATs 1–2; in combination with IRF-9 (IFN-regulatory factor 9), these proteins form the heterotrimeric transcription factor known as ISGF3 (IFN-stimulated gene factor 3). ISGF3 in turn migrates to the nucleus, where it recognizes and binds to the IFN-stimulated response element (ISRE) to promote gene induction [18], [21]. However, a recent report revealed that ISGF3 formation and translocation to the nucleus in response to IFN-α is dependent on the acetylation of all components of the heterotrimeric complex. Upon IFN-α stimulation, the acetyltransferase CBP (CREB-binding protein)/p300 is recruited to the IFNAR2. There, it binds to two adjacent phospho-Ser residues (aa364 and aa384) and acetylates IFNAR2 on Lys399. This acetylation creates a docking site for IRF9, which in turn associates with the receptor to permit ISGF3 complex formation. Subsequently, IRF9, STAT2, and STAT1 are acetylated and the complex then migrates to the nucleus to initiate gene transcription [22]. Another recent interesting findings demonstrated that exposure of the cells to IFN-β is followed by activation of IKKɛ (inhibitor of nuclear factor κB kinase)-related kinase ɛ which in turn phosphorylates STAT 1 resulting in ISGF3 complex formation. ISGF3 then migrates to the nucleus where it binds to the promoter region of a subset of ISGs to elicit an antiviral response [23] (Fig. 1 ).
Fig. 1.
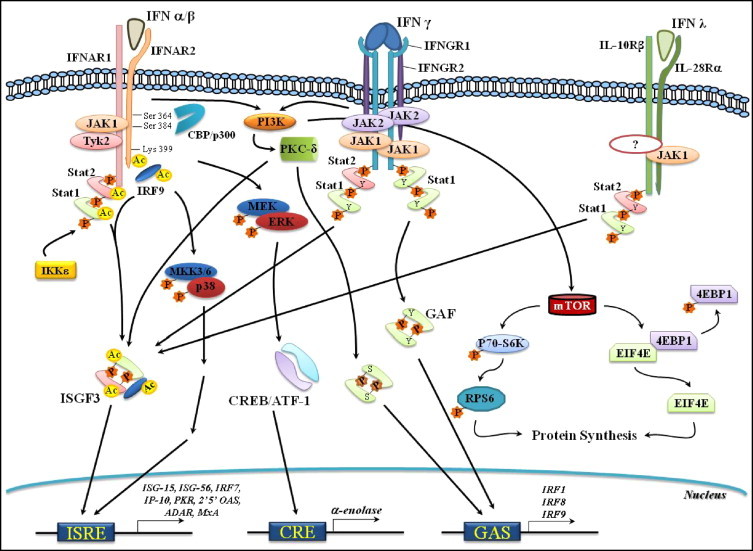
IFN-stimulated gene expression via the JAK/STAT signaling pathways. Upon recognition of its cognate receptor (IFNAR1/2), Type I IFNs (only α/β are shown) activate the Janus tyrosine kinases JAK1 and Tyk2. These kinases associate with IFNAR2 and IFNAR1, respectively. JAK1 and Tyk2 in turn tyrosine phosphorylate (Y-P) the receptor chains and create docking sites for STAT2 and STAT1. Following (Y-P), both STATs associate with IRF9 to form the transcriptional regulator ISGF3. ISGF3 then migrates to the nucleus to promote transcription via the regulatory sequence ISRE of the ISGs. IFN-α activation of ISGF3 also requires the acetyltransferase CBP/p300, which interacts with the IFNAR2 at phospho-serine (S-P) 364 and 384. This interaction is followed by lysine (Lys) acetylation at position 399. IRF9 then recognizes the acetylated Lys 399 and binds to the receptor. IRF9, STAT1, and STAT2 are all acetylated by CBP/p300 and then leave the receptor to form ISGF3, which translocates to the nucleus. IFN − β stimulation also leads to the activation of IKKɛ that in turn phosphorylates STAT1, followed by ISGF3 formation. Type II IFN (γ) engages with homodimeric receptors (IFNGR1/2s) to activate JAK1 and JAK2, which are constitutively associated with IFNGR1 and IFNGR2, respectively. The receptor chains are then (Y-P) and create docking sites for STAT1. STAT1 binds to the receptor and is (Y-P) at position 701. A homodimer of STAT1 is formed, resulting in the transcription regulator GAF. GAF in turn translocates to the nucleus, binds to the cis-acting element GAS, and initiates gene transcription. Under certain circumstances, IFN-γ also regulates gene transcription via ISGF3. Type III IFN (λ) interacts with the receptor chains IL-10Rβ and IL-28Rα to transduce signals involving JAK1, STAT1, STAT2, and ISGF3. It also regulates gene expression through ISRE and eventually GAS. IFN-stimulated gene transcription via signaling pathways other than JAK/STAT. IFN-α/β can regulate gene transcription by activating MAPKs. IFN-α stimulation of MEK/ERK leads to CREB/ATF-1 activation and the binding of the cis-acting element CRE to result in α-enolase expression. Activation of the MKK3/6-p38MAPK pathway upon IFN-α treatment leads to gene transcription via ISRE. Stimulation of PI3K by both Type I and II IFNs results in (P) of the serine kinase PKC which then acts as the downstream effector of PI3K and (P) STAT-1 at Ser (S) 727. Upon activation, STAT-1 migrates to the nucleus to initiate STAT-1-driven gene transcription through either ISRE or GAS. Activation of mTOR upon Type I or II IFN stimulation is mediated via PI3K and results in: (i) (P) of p70-S6K, followed by (P) of the ribosomal protein S6 and (ii) (P) and deactivation of the translation repressor 4E-BP1. Both events lead to the initiation of mRNA translation. Abbreviations: ATF1 (activating transcription factor 1), CBP (CREB-binding protein), CRE (cyclic-AMP response element), CREB (CRE binding protein), EIF4E (eukaryotic translation-initiation factor 4E), ERK (extracellular-signal regulated kinase), 4E-BP1 (eukaryotic translation-initiation factor 4E (EIF4E)-binding protein 1), GAF (IFN-γ-activated factor), GAS (IFN-γ-activated sequence), IKKɛ (IκB Kinase Kinase ɛ), IRF (IFN-regulatory factor), ISGF3 (IFN-stimulated gene factor 3), ISRE (IFN-stimulated response element), ISG (IFN-stimulated gene), MAPK (mitogen-activated protein kinase), MEK (MAPK/ERK kinase), MKK (MAPK Kinase Kinase), mTOR (mammalian target of rapamycin), PI3K (phosphatydilinositol-3 kinase), PKC-δ (protein kinase C-δ), p70-S6K (RPS6 kinase), and RPS6 (ribosomal protein S6).
Type II IFNs in turn rely upon the activation of JAK1-2 and STAT1. Once activated, STAT1 dimerizes to form the transcription regulator GAF (IFN-γ activated factor). This protein recognizes and binds to the regulatory sequence known as the IFN-γ activated sequence (GAS) and initiates gene transcription [10], [21]. Of note, type I IFNs can activate, on a cell-type and context-specific manner, all seven members of the STAT family of proteins. They may form homo- or heterodimeric complexes as well as heterodimeric complexes in association with other transcription factors [3], [18]. Signal transmission upon IFN-λs receptor binding requires JAK1, STAT1-2, and ISFG3 and leads to the expression of common ISGs stimulated upon treatment with type I IFNs [3], [14] (Fig. 1).
The JAK/STAT pathway was the first to be characterized as the signaling cascade that elicits the biological effects associated with the IFNs [3], [21]. However, it is clear now that a number of signaling pathways other than the JAK/STAT pathway may be activated either independently or in parallel to the JAK/STAT pathway. This variety would confer specificity, permit an even more robust response, or coordinate IFN actions [18], [24], [25]. Thus, mitogen-activated protein kinase (MAPK) family members (MEK/ERK, and p38MAPK), and phosphatidylinositol 3-kinase (PI3K) as well, have also been implicated in the generation of IFN-mediated signals. For example, David et al. described for the first time, that signals conveyed by the MEK/ERK pathway after stimulation with IFN-α and -β result in physical interaction between ERK2/STAT-1 as well as regulation of STAT-1-dependent gene expression [26]. We have recently shown that IFN-α activates MEK/ERK1/2 and the downstream transcription factor CREB/ATF-1, which leads to α-enolase gene expression that then acts as a plasminogen receptor [27] (Fig. 1). MEK/ERK stimulation is also associated with CCAAAT/enhancer-binding protein β (C/EBP)-dependent gene expression upon IFN-γ treatment [28], [29]. The C/EBP-beta-interacting protein Med1 also requires ERK phosphorylation for its activation and up-regulation of IFN-induced transcription [30]. Another report showed that Myxoma virus (MYXV) stimulation of the MEK/ERK pathway leads to IFN-β production through activation of IRF3 and STAT-1, which in turn restrain virus replication. Nonetheless, no other virus infection is reported to lead to the activation of this pathway which points to a MYXV specific mechanism [31].
Implication of p38MAPK in the regulation of type I IFN-mediated signals has firmly been established based on pharmacological and genetic approaches. Thus, incubation with SP203580, a pharmacological inhibitor of p38MAPK, blocks IFN-α-dependent gene transcription through the regulatory sequence ISRE as well as GAS elements [32], [33]. Further, gene ablation of p38 MAPK-α results in defective transcriptional regulation of IFN-α-stimulated genes [34]. It worth noting that the p38MAPK signals mediated upon type I IFN stimulation do not result in phosphorylation of STAT-1 or STAT-3 on Ser residue at position 727 [34], [35]. Antiviral activity associated with type I IFNs has also been reported to rely, at least in part, on p38MAPK [34]. Type II IFNs also appears to require p38MAPK in order to regulate the expression of genes (e.g. chemokines CCL5, CXCL9, and CXCL10, TNF-α, and inducible NO synthase) associated with the inflammatory response [36].
The PI3K signaling pathway has also been implicated with both type I and type II IFN-regulated gene expression [37], [38]. Another study has shown that p38MAPK is activated in a type I IFN-dependent manner in cells from patients with chronic myelogenous leukemia (CML), suggesting that p38 plays an important role in mediating signals required for the antileukaemic effects of type I IFN [39]. The activation of PI3K by IFN-γ results in phosphorylation of STAT-1 at Ser 727 and is followed by STAT-1-regulated gene expression [38]. Stimulation with type I or type II IFN causes the serine kinase PKC-δ to act as a downstream effector of PI3K, which consequently phosphorylates STAT-1 at Ser 727 and initiates STAT-1-driven gene transcription [40], [41]. Activation of this pathway appears to be associated with the regulation of apoptosis via STAT-1 [42]. Another downstream target of PI3K in response to stimulation with either type I or type II IFNs is the mammalian target of rapamycin (mTOR), a pivotal regulator of the initiation of mRNA translation [43], [44]. mTOR in turn regulates the downstream activation of two targets: p70-S6 kinase (p70-S6K), which subsequently phosphorylates the ribosomal protein S6 (RPS6) and initiates protein synthesis [44], and the translation repressor 4EBP1 (eukaryotic translation-initiation factor 4E (EIF4E)-binding protein 1). Upon phosphorylation, 4EBP1 dissociates from EIF4E and allows translation to start [43], [44], [45], [46] (Fig. 1).
It worth noting, that the translation repressors 4EBP1 and 4EBP2 exert a negative regulatory role over type I IFN production. Double 4EBP1/2 knockout mice showed enhanced type I IFN production as a consequence of the up-regulation of IRF-7 messenger RNA translation (Fig. 3). Under these circumstances, replication of the Encephalomyocarditis virus, Vesicular stomatitis virus, Influenza virus and Sindbis virus is markedly suppressed [47]. Thus, the biological outcomes ascribed to the IFNs should be interpreted as a result of modulation of diverse signaling pathways that actively cross-talk with one other in order to provide the host with the means to (1) mount the most adequate response to microbial infections, (2) cease or stimulate cell proliferation and (3) modulate the immune and inflammatory responses [47], [48], [49]. It is of vital importance, however, that the duration and magnitude of these responses should be under tight regulatory control; the host would otherwise be under immunopathogical threat, as illustrated by the correlation between both the increased levels of ISG and IFN expression in patients with scleroderma or systemic sclerosis (SSc) [50] as well as the augmented levels of circulating IFN-α/β found in the serum of patients with systemic lupus erythematosus (SLE) and disease severity [51]. Furthermore, plasmacytoid dendritic cells (pDcs) activated by immune complexes containing nucleic acids secrete type I IFNs in SLE. Secreted type I IFNs in turn cause the differentiation of monocytes into myeloid-derived dendritic cells (mDCs) and the activation of autoreactive T and B cells [52]. Indeed, cell stimulation with type I IFNs via the IFNAR1/2-JAK1/Tyk2-STAT-1 pathway leads to the expression of the suppressors of cytokine signaling (SOCS), a secondary-response family of genes that interrupts signal transmission and maintains immune homeostasis [48], [53]. This event is also shared by type III IFNs [54], [55] (Fig. 3). Consistent with this notion, SOCS1 knockout mice develop lupus-like disease symptoms [56].
Fig. 3.
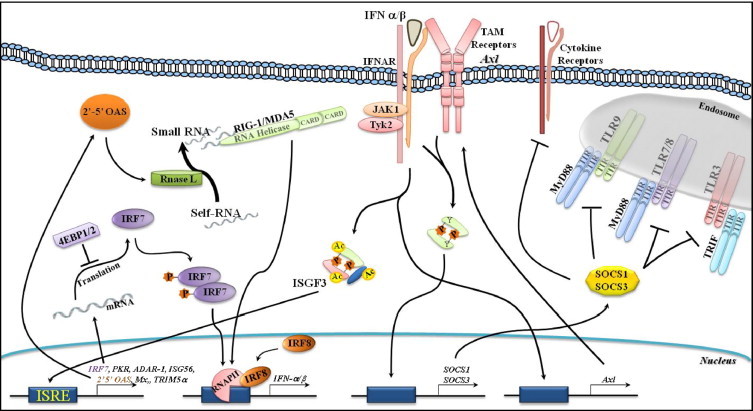
Amplifying IFN production and negative regulation exerted by the TAM receptor Ax1 and SOCS. Upon recognition of a viral nucleic acid via the endosomal TLRs, Type I IFNs are produced and secreted (early events—see Fig. 1). They then act in an autocrine/paracrine manner through the IFNAR/JAK/STAT1/IRF7 pathway to amplify both IFN production (delayed events) and antiviral effects through the expression of 2′–5′OAS, PKR, ADAR-1, TRIM 5α, ISG15, and MX. Stimulation of RNase L downstream of the 2′–5′OAS pathway is followed by the degradation of cellular ss-RNAs, recognition by RIG-1 and MDA5, and production of IFNs. Upon viral infection in DCs, IRF8 associates with RNAP II at the IFN promoters to increase IFN transcription and production. The translation repressor 4EBP1/2 exerts a negative regulatory role over IRF7 protein expression and Type I IFN production. Stimulation of the IFNAR/JAK/STAT1 pathway also leads to the up-regulation of the TAM receptor Axl, which in turn accumulates at the cell surface, physically associates with the IFNAR, and usurps the IFNAR-STAT-1 pathway to stimulate SOCS1 and SOCS3 transcription. These actions thereby exert a negative regulatory role through the inhibition of both TLR and TLR-induced cytokine receptor pathways to maintain immune homeostasis. Abbreviations: ADAR-1 (adenosine deaminase acting on RNA 1), 4EBP1/2 (eukaryotic translation-initiation factor 4E (EIF4E)-binding protein 1/2), ISG15/56 (IFN-stimulated gene 15/56), MDA5 (melanoma differentiation associated gene 5), MX (myxovirus resistance), 2′–5′OAS (2′–5′oligoadenylate synthetase), PKR (double-stranded RNA-dependent protein kinase), RNAP II (RNA polymerase II), RIG-1 (retinoic acid-induced gene 1), SOCS (suppressors of cytokine signaling), TAM receptor (protein tyrosine kinases TYRO3, AXL and MER), TRIM 5α (tripartite motif protein).
3. Viral detection by pattern recognition receptors (PRRs)
It has long been known that cytokine production is tightly regulated by the diverse stimuli that govern its expression. For instance, as a viral infection progresses, IFN production increases up to the levels required to control virus replication. Since the stimulus (virus) that triggered cytokine production (IFN) then ceases (i.e. since virus replication is controlled), IFN production returns to basal levels. Thus, microbial sensing, the mounting of host innate and adaptive responses, and the resolution of the infection are coordinated programs activated during pathogen invasion/recognition [48], [49], [57], [58], [59].
Pathogens are recognized by specialized receptors called pattern recognition receptors (PRRs). These are primarily, but not exclusively, present either at the surface or inside professional antigen-presenting cells (APCs) like macrophages and dendritic cells. These cells are localized to the portal of entry and consequently are the first cells to encounter pathogens during the infection. Recently, fibroblasts have also been found to initiate an innate immune response upon detection of viral nucleic acid (NA) in the cytosol [58], [60]. PRRs can also be secreted and circulate in the blood of vertebrates. This is the case for acute phase proteins C-reactive protein (CRP), Mannan Binding Lectin (MBL), opsonins, collectins, and pentraxins; these proteins are able to bind to molecules found at the surface of pathogens and activate both the complement and phagocytic cells [61], [62], [63]. The Toll-like receptors (TLRs) comprise the most-studied family of PRRs. They are constituted by transmembrane PRRs like TLR 1, 2, and 4–6 and endosomal TLRs like TLR3 and 7–9 [64], [65], [66]. The intracellular detection of pathogens is also mediated by the retinoic-acid-inducible gene I (RIG-I)-like helicase (RLH) family of proteins, which is comprised of RIG-I and MDA5 (melanoma differentiation associated gene 5) [57], [60] as well as the most recently described DNA-dependent activator of IFN-regulatory factors (DAI) [67].
PRRs recognize and bind to pathogen-associated molecular patterns (PAMPs), and this interaction leads to the activation of transcription factors (e.g. nuclear factor-κB, NF-κB) and IFN-regulatory factors (IRFs); these proteins regulate the expression of pro-inflammatory cytokines and type I IFNs, respectively [58], [62]. Thus, while TLR3 recognizes double-stranded (ds) RNA within the endosomal compartment, RLHs bind to dsRNA in the cytosol. TLR7 and TLR9 discriminate microbial single-stranded (ss) RNA and unmethylated ss-DNA or bacterial DNA with CpG motifs, respectively, from the host NA by sensing NA within the endosomes. This diversity of PRRs provides the host with the ability to sense unique PAMPs found in the microbial world. Through functional cooperation between PRR sub-families, the host may initiate and mount the most adequate innate and adaptive immune responses [57], [59], [60].
4. Pathogen recognition and signal transmission by the PRRs
4.1. The Toll-like receptor family of PRRs
TLRs are comprised of a family of germline-encoded transmembrane receptors with a leucine-rich repeats (LRRs) at the N-terminal and a Toll-IL-1R (TIR) domain at the C-terminal cytoplasmic moiety [64], [65]. Because the cells responsible for microbial sensing express non-overlapping sets of TLRs, it is not surprising that the recognition of a PAMPs by a given TLR will dictate the subsequent expression of cytokines and chemokines required to initiate innate responses [64], [68], [69]. For instance, plasmacytoid dendritic cells (pDCs) express TLR7 and TRL9 in the endosomal compartment; after recognizing ss-RNA or unmethylated CpG DNA, respectively, they produce enormous amounts of type I IFNs [70], [71], [72], [73] and consequently elevate systemic concentration of IFNs [73], [74], [75]. In contrast, myeloid DCs (mDCs) express a variety of TLRs capable of detecting bacterial, viral, and fungal pathogens and secrete the inflammatory cytokines IL-12, TNF-α, and IL-6 [73], [74], [75].
Within the TLRs, those of outstanding importance in viral recognition are the ones localized to the endosomal compartment and responsible for the sensing of ds-RNA (TLR3), guanosine- or uridine-rich ss-RNA (TLR 7,8), and unmethylated CpG DNA (TLR9) [64], [65], [72]. While TLR7 and TLR9 present a more restricted cellular distribution and are highly expressed in DCs (though not restricted to pDCs), TLR3 presents a wider expression that includes nonhematopoietic cells, but is preferentially expressed by conventional DCs (cDCs) [59], [64], [73].
Signaling through the TLRs has extensively been reviewed by others [20], [58], [64], [65], [66], so only a description concerning endosomal TLRs (TLR 3 and 7–9) will be given here. Upon ligand binding to TLRs, signal transmission requires the association between the receptor intracytoplasmic moiety (TIR) and a TIR-containing adaptor protein capable of binding to the homotypic sequence at the receptor in the cytosol. For example, MyD88 (myeloid-differentiation primary-response gene 88) serves as the adaptor for TLRs 7–9, whereas TRIF (TIR domain-containing adaptor protein inducing IFN-β) is the adaptor molecule used by TLR3.
Downstream targets of TLR7/9/MyD88 include IRAK1 (IL-1 receptor-associated kinase 1)/IRAK4 and TRAF6 (TNF receptor-associated factor 6), and activation of these targets results in the activation of IKK (IκB Kinase Kinase)-α and IRF7 [76], [77], [78]. TRAF6 also transmits downstream signals to TAK1 (TGF-β-activated kinase 1), TAB2 (TAK1-binding protein 2), and TAB3 and results in the activation of NF-κB and MAPKs (mitogen-activated protein kinases) [79], [80]. Accordingly, activation of these signaling pathways results in type I/III IFN production (α, β, ω, and λ), expression of pro-inflammatory cytokines (e.g. IL-6 and TNF), and expression of co-stimulatory molecules (e.g. CD80 and CD86) [53], [54], [55], [56], [57], [58], [59]. Expression of IL-6 and TNF is also mediated by IRF5, which binds directly to MyD88. On the other hand, IRF4 competes with IRF5 but not IRF7 for binding to MyD88, thereby negatively regulating cytokine production by TLRs [81], [82]. Notably, it has recently been shown that the TLR-mediated type I IFN production in pDCs is also dependent on the PI3K/mTOR/p70S6K pathway, since its pharmacological inhibition with rapamycin, during pDC activation via TLR9 stimulation, was followed by impaired IFN production. In addition, pharmacological disruption of the pathway was associated with diminished IFN production in response to Yellow fever virus infection which strongly suggests that TLR7 also recruits the mTOR pathway to confer innate immune defense [83]. TLR3/TRIF engagement leads in turn the transduction of downstream signals to TBK1 (tank-binding kinase 1) and IKKɛ kinases, which in conjunction with TRAF3 and NAP-1 (NAK-associated protein 1) connect TLR3 to the IRF3 pathway and result in IFN-β production [84], [85]. Alternatively, instead of NAP-1, TANK (TRAF family member-associated NF-κB activator) can also acts as an adaptor molecule downstream of TRIF to convey signals to TBK1 and IKKɛ that results in type I IFN production. Furthermore, TANK was also found to potentiate type I IFN production associated with TBK1/IKKɛ [86]. Full activation of IRF3 also requires phosphotidylinositol 3-kinase (PI3K) and its downstream effector kinase Akt [87]. Activation of the TLR3/IRF3 pathway is also associated with the expression of pro-inflammatory cytokines and co-stimulatory molecules [59], [88], [89], [90] in a manner quite similar to that of the cytoplasmic sensors of RNA (RLRs) and DNA (DAI) (see below) (Fig. 2 ).
Fig. 2.
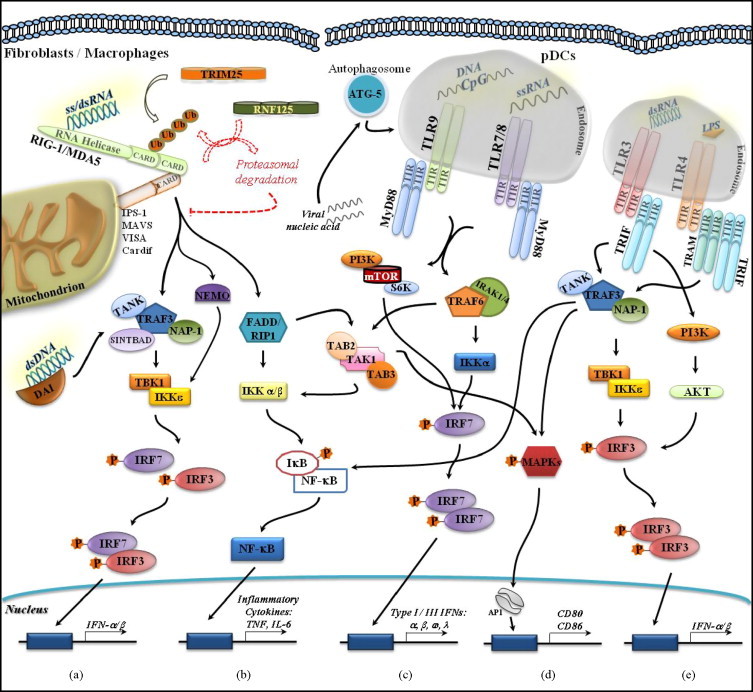
Endosomal and cytosolic detection of viral nucleic acid. Endosomal recognition of viral nucleic acid via TLR7,9/MyD88 results in the activation of the TRAF6/IKKα/IRF7 signaling pathway, which culminates in type I/III IFN production. TLR7,9/MyD88 also leads to activation of the PI3K/mTOR/S6K pathway followed by IFNα/β production (c). Downstream targets of TRAF6 also include: (i) TAK1/IKKα/β/NF-κB, a pathway that leads to the expression of inflammatory cytokines (b) and (ii) TAK1/MAPKs. Members of the latter cascade then migrate to the nucleus and activate the transcriptional regulator AP1, which in turn controls the expression of co-stimulatory molecules CD80 and 86 (d). Viral recognition via TLR3/TRIF is followed by the engagement of TRAF3/NAP1 or TANK-TBK1/IKKɛ molecules and the subsequent activation of IRF3. IRF3 translocates to the nucleus and activates transcriptional regulation of type I IFN genes. Full activation of IRF3 also requires PI3K/Akt (e). Signal transduction downstream of TRAF3 also results in the activation of MAPKs and NF-κB and leads to the expression of co-stimulatory molecules (d) and inflammatory cytokines (b). Viral recognition by endosomal TLRs is autophagy dependent. Mice deficient in the autophagy protein atg-5 failed to produce type I IFNs. Cytosolic detection of viral RNA by RIG-1/MDA5 is followed by the interaction of their CARD domains with those of the adaptor protein IPS-1 (MAVS, VISA, or Cardif). Subsequent activation of the TRAF3/NAP-1-TBK1/IKKɛ pathway results in the activation of IRF3/7 and regulation of type I IFN gene expression. Alternatively, instead of NAP-1, the scaffolding proteins TANK or SINTBAD, can also be recruited to transduce downstream signals (a). CARDs need to be ubiquitinated by the TRIM25 E3 ligase in order to transduce downstream signals and type I IFN production. In contrast, signal termination requires RIG-I ubiquitination by the E3 ligase RNF125, which leads to proteasomal degradation (a). The TRAF3/NAP-1-TBK1/IKKɛ pathway is also activated upon recognition of cytosolic DNA by the sensor DAI. Signal transmission downstream of IPS-1 also recruits FADD/RIP1 and IKKα/β to mediate the activation of NF-κB (b) and MAPKs pathways (d). Though not associated with viral recognition, TLR4 also mediates type I IFN production from the endosomal compartment (e). Abbreviations: CARD (caspase activating recruitment domain), DAI (DNA-dependent activator of IFN-regulatory factors), FADD (FAS-associated death domain), IKK (IκB Kinase Kinase), IPS (IFNβ-promoter stimulator) IRAK (IL-1 receptor-associated kinase), MAPKs (mitogen-activated protein kinases), MDA5 (melanoma differentiation associated gene 5), MyD88 (myeloid-differentiation primary-response gene 88), NAP-1 (NAK-associated protein 1), NF-κB (nuclear factor κB), (PI3K) phosphotidylinositol 3-kinase, RIG-1 (retinoic acid-induced gene 1), RIP1 (receptor-interacting protein 1), TAB (TAK1-binding protein), TAK1 (TGF-β-activated kinase 1), TBK (tank-binding kinase), TRAF (TNF receptor-associated factor), TRAM (TRIF-related adaptor molecule), TRIF (TIR domain-containing adaptor protein inducing IFN-β).
It is noteworthy that viral recognition by the endosomal TLRs is autophagy-dependent [60], [91]. It has been demonstrated that pDCs derived from knockout mice deficient in an autophagy protein (atg-5) failed to produce type I IFNs in response to VSV infection [92]. Therefore, as VSV replicates in the cytoplasm, the fusion between the autophagosome and endosome bridges the endosomal compartment and the cytosol; through autophagy, the contribution of endosomal TLRs to the detection of viral infection is no longer restricted to the endosome (Fig. 2). Interestingly, the endoplasmic reticulum membrane protein UNC-93B has been shown not only to play a vital role in endosomal TLRs signaling [93] but also to physically interact with these TLRs [94]. These interactions occurred via the TLR transmembrane domain, which has previously been shown to harbor the sorting signal required for TLR 7 and 9 endosomal localization [95], [96]. Thus, UNC-93B could act as an adaptor protein required to regulate the endosomal trafficking of TLRs. The pivotal role played by UNC-93B in viral innate immunity is further supported by the correlation between gene mutation and severe HSV encephalitis in human patients [97]. However, the precise mechanisms of how autophagy and UNC-93B-TLR interactions result in type I IFN production awaits further investigation.
Although not associated with viral recognition, TLR4, which detects LPS at the surface of Gram-negative bacteria, has also recently been found to stimulate type I IFN production from the endosomal compartment. TLR4 is unique in the sense that it transduces signals that result either in pro-inflammatory cytokine expression (TNF, IL-6, IL-12), via TIRAP/MyD88, or in type I IFN production, through TRAM/TRIF. It has been shown that while signaling from TRAM/TRIF requires endocytosis, the expression of pro-inflammatory cytokine is triggered upon recognition of LPS after engaging TLR4/TIRAP/MyD88 at the cell surface [98]. Thus, a unifying mechanism to the induction of type I IFN from an intracellular compartment has been established (Fig. 2).
Nonetheless, it is of vital importance that the induction of type I IFNs and pro-inflammatory cytokines/chemokines upon recognition by a given TLR of a pathogen PAMP be under regulatory control. It has recently been demonstrated that the TAM receptors, represented by the tyrosine kinases Tyro3, Axl and Mer [53], [99], play a negative regulatory role through the inhibition of both TLR and TLR-induced cytokine receptor pathways. The inhibitory effect is mediated via the TLR-stimulated IFNAR-STAT-1 pathway, which in turn upregulates expression of the TAM receptor Axl. Axl then accumulates on the cell surface, physically associates with the IFNAR, and usurps the IFNAR-STAT-1 pathway to stimulate SOCS1 and SOCS3 transcription upon cytokine engagement with the IFNAR. Signal transmission is consequently terminated by the interaction of the SOCS proteins with the cytoplasmic moieties of both TLR and cytokine-receptors [48], [49], [53] (Fig. 3). Furthermore, SOCS1 was also reported to exert a negative regulatory role through the interaction with the TLR2- and TLR4-adapter molecule Mal (MyD88-adaptor like)/TIRAP. The interaction was followed by Mal polyubiquitination and subsequent degradation thereby down-regulating innate immune responses [100].
4.2. The PRRs retinoic-acid-inducible gene I (RIG-I)-like helicases (RLHs)
The family of RLHs is comprised of RIG-I and MDA5, which are associated with the cytosolic (non-endosomal/non-TLR-dependent) detection of PAMPs for which viral ds-RNA is the best-characterized molecular pattern [14], [101], [102]. In addition to RIG-I and MDA5, another family member known as LGP2 has been identified. Because LGP2 lacks the caspase-recruitment and activation domains (CARDs), however, it is postulated that it might exert a negative regulatory role by competing with RIG-I and MDA5 during the detection of viral RNAs [102], [103]. Indeed, consistent with an inhibitory role, increased expression of IFN-α and -β was found in cells from LPG−/− mice challenged with the Vesicular stomatitis virus (VSV) [101]. However, infection of LPG−/− cells with the Encephalomyocarditis virus (EMCV) resulted in impaired IFN-β production; this finding points to a positive regulatory role exerted by LPG2 [104].
It has long been known that either positive- or negative-sense ss-RNA viruses, such as the EMCV and Newcastle disease virus (NDV), respectively, generate ds-RNA during their replication cycle. These viruses are capable of stimulating fibroblasts to produce and secrete type I IFNs [60], [105]. The sensing of ds-RNA by RLHs relies on the RNA helicase domain found at their C-terminal tail, because this domain provides the PRRs with the ability to bind and unwind RNA. The conformational changes that follow allow the two N-terminal domains of the RLHs (i.e. CARDs) to interact with the homotypic domain of the CARD-containing protein. These include the mitochondrial antiviral signaling (MAVS) [106] (also known as IFNβ-promoter stimulator (IPS1) [107], virus-induced signaling adaptor (VISA) [108], or CARD adaptor inducing IFN-β (CARDIF) [109]. This adaptor protein serves as is a downstream signaling molecule localized to the outer mitochondrial membrane.
Of note, the CARDs of RIG-I need to be ubiquitinated by the TRIM25 E3 ligase. This critical modification is required for the transduction of downstream signals and production of type I IFNs [110]. On the other hand, signal termination requires RIG-I ubiquitination by another E3 ligase, RNF125, that leads to proteasomal degradation of RIG-I [111]. The interaction between RLH-CARD and CARD-IPS results in the activation of: (i) TBK1 and IKKɛ via TRAF3 and one of the scaffolding proteins NAP-1, TANK or SINTBAD, a process that leads to the phosphorylation of IRF3/IRF7 and expression of IFN-α/β [107], [108], [109], [112], [113] and (ii) FADD (FAS-associated death domain) and the receptor-interacting protein 1 (RIP1), a process that recruits TAK1 and IKKα/β (IκB Kinase Kinase) and leads not only to activation of NF-κB and production of pro-inflammatory cytokines, but also to activation of IRF3 and type I IFN production [114], [115], [116]. Acting downstream of RIG-I and IPS, and upstream of TBK1 and IKKɛ, it has been demonstrated that NEMO (NF-κB essential modulator or IKKγ) also couples signal transmission to activation of IRF3/7 after a viral challenge [117] (Fig. 2).
In addition to ds-RNA, RIG-I also links type I IFN production to the recognition of the 5′-triphosphate ss-RNA of the influenza virus [118], [119]. Whether the helicase activity of RIG-I is required for the detection of 5′-triphosphate ss-RNA remains to be further clarified [120]. Genetic evidence has firmly established the fundamental role of RIG-I and MDA5 as cytosolic sensors for the detection of distinct viral RNAs [121], [122]. Thus, while RIG-I KO mice are highly vulnerable to negative-sense ss-RNA viruses like the NDV, VSV, Influenza virus and Sendai virus and positive-sense Japanese encephalitis virus, MDA5 KO mice were sensitive to Picornaviruses like the EMCV, Mengovirus, and Theiler's encephalomyelitis virus [121], [122]. The expression of RIG-I and MDA5 genes is under type I IFN regulation, which in turn stimulates type I IFN production and exerts positive-feedback control to meet the infective demand. While pDCs are devoted to robust type I IFN production via endosomal TLR 7-9, potent type I IFN production by the cytosolic pathway is associated with RLHs. RLHs play a crucial role in the detection of PRRs in fibroblasts and macrophages, but they are not functional in pDCs [123]. Experiments conducted with MAVS KO mice showed that the animals were highly susceptible to infection with the viruses detected either by RIG-I or MDA5, confirming MAVS as the common downstream RNA virus detector for both cytosolic receptors [124], [125] (Fig. 2).
4.3. The cytosolic detection of microbial ds-DNA
Accumulating evidence now supports the notion that vertebrates have also evolved a pathway capable of sensing cytosolic viral and bacterial ds-DNA, and this pathway thus links microbial detection to type I IFN production via TBK-1/IRF3 [76], [126]. The detection of ds-DNA leads to potent type I IFN production, and this is a common mechanism operating in a variety of cells analyzed (e.g. macrophages, plasmacytoid and conventional DCs, and fibroblasts). This mechanism appears to be carried out by the DNA sensor Z-DNA binding protein (ZBP1), which is also known as a DNA-dependent activator of IFN-regulatory factor (DAI) [67], [105] (Fig. 2).
5. Adjusting IFN production to meet the infective demand
To face the challenge of the infective demand, vertebrates respond with the early expression of IFN-β through diverse PRRs responsible for linking pathogen recognition to IFN-β production (“early events”). Because the amount of IFN secreted would not be sufficient to protect neighboring cells from the progeny of the invading new virus, secreted IFN-β binds to the IFN receptor (IFNAR1/2) and acts in an autocrine- and/or paracrine-manner to enhance type I IFN production [90], [127]. Thus, the binding of IFN-β to the IFNAR leads to activation of the transcription factor ISGF3, which translocates to the nucleus and regulates the transcription of IRF7 [6], [7]. The resulting homodimer of IRF7 then migrates to the nucleus, leading to further enhancement of type I IFN gene induction.
Because IRF7 is an essential regulator of type I IFN gene expression, most of these genes are only expressed in a delayed-fashion (i.e. after IRF7 production, “delayed events”) [6], [7]. This positive-feedback loop provides the host with a sufficient amount of IFN to prevent virus from spreading (Fig. 3 ). Of note, IRF8, a hematopoietic cell-specific transcription factor, is not only required for early development and final maturation of both pDCs and mDCs but can also further amplify the second wave (delayed events) of type I IFN production in pDCs. IRF8 is a nuclear protein expressed at high levels in DCs [128]. After viral infection, IRF8 associates with RNA polymerase II (RNAP II) at the IFN promoters and helps to prolong the recruitment of the basal transcription machinery to the promoters; this action thereby increases IFN transcription and production [129], [130].
An antiviral state is conferred by the transcriptional regulation of IFN-stimulated genes (ISGs), such as when adenosine deaminase acts on RNA (ADAR)-1, double-stranded RNA-dependent protein kinase (PKR), MX GTPases, tripartite motif protein (TRIM) 5α, 2′–5′oligoadenylate synthetase (2′–5′ OAS), ISG15 and ISG56 [3], [8], [86]. Stimulation of ISG56 by IFN or viral infection leads to a blockade in protein synthesis as a consequence of the binding of ISG56 to the eukaryotic initiation factor 3e (eIF3e) which prevents eIF3e from interacting with the ternary complex eIF2.GTP.Met-tRNAi [86]. In addition to this state, the cleavage of cellular RNAs by antiviral endoribonuclease RNase L also amplifies type I IFN production. The products of cleavage by RNase L (small RNAs) are in turn recognized and bound by both RIG-I and MDA5, thereby eliciting further type I IFN production [131] (Fig. 3).
Interestingly, type I IFNs also elicit effector responses that culminate not only with the activation of natural killer (NK) cells and cytotoxic T cells (CTLs) [132], [133] but also with clonal expansion and memory formation in response to viral infection by acting directly on CD8 T cells [134]. Thus, type I IFNs not only regulate the expression of chemokines required to attract cytotoxic cells to the site of infection but also stimulate their proliferation, a mechanism dependent upon the expression of the ISG IL-15 [132], [133]. The Tailor-made program carried out by the type I IFNs enables the host to counterattack intracellular microorganisms through the activation of the antiviral state. At the same time, it increases the chances that cytotoxic cells will encounter infected cells and signals them to die. Therefore, the recognition of intracellular pathogens by diverse PRRs is of utmost relevance, because it signals the cells to undergo an apoptotic program. In contrast, neighboring uninfected cells under IFN stimulation will still be preserved [3].
6. Virus counterattacks on host innate immune defenses
The mounting of an effective host antiviral response depends not only on the stimulation of type I IFN production, which impedes the virus from replicating, but also on the attraction of inflammatory cells to the site of infection, which prevents the virus from spreading. These coordinated actions elicited upon viral PAMP recognition occur in a TLR-dependent or -independent manner (RLHs/DAI). Due to genomic constraints, most viruses need to concentrate their disruptive efforts on host targets that are key players in the antiviral response. Thus, common host molecules that connect virus recognition to downstream effectors of type I IFN production and inflammatory cytokine expression are usually targeted by viral innate evasion proteins. MAVS/IPS/VISA/CARDIF illustrates the assertion. MAVS is cleaved at its C-terminal end by the NS3-4A protease of Hepatitis C virus (Hepacivirus). Since this cleavage interrupts the interaction of MAVS with the outer mitochondrial membrane, signal transmission to downstream antiviral molecules (i.e. IRF3, IFN-β) and the activation of NF-κB are disrupted [109], [135], [136]. MAVS is also affected by the Hepatitis A virus (HAV), a Picornavirus that targets the viral protein 3ABC (a precursor of 3C (pro) cysteine protease) to the mitochondria for colocalization with and degradation of MAVS [137]. The Influenza virus NS1 protein is also able to antagonize the RIG-1/MAVS pathway. Through the interaction of NS1 with RIG-1/MAVS, the viral protein blocks IRF3 activation and IFNβ expression [138]. Not surprisingly, the MDA5/IFN-β pathway was also demonstrated to be disrupted by the V proteins of the diverse Paramyxoviruses Simian virus 5, Human parainfluenza virus 2, Mumps virus, Sendai virus, and Hendra virus through direct interaction with MDA5 but not RIG-1 [101], [139]. STAT family members are also targeted by Paramyxoviruses V proteins through diverse mechanisms such as ubiquitin-ligase [140] and by preventing IFNα/β/γ transcriptional responses and STAT nuclear redistribution [141].
Since IRF3 and IRF7 are downstream effectors of PRR/PAMP interactions that lead to IFNβ/α expression, a number of viruses have focused their disruptive effort on disabling IRF3/7-activated antiviral pathways. The Human herpesvirus Kaposi's sarcoma-associated herpesvirus 8 (KSHV/HHV-8) exploits distinct strategies to block IFN production. The viral IRF homolog vIRF3 blocks IRF7 transcriptional activity through the binding of vIRF3 to the same cis regulatory element found at the promoter region where IRF7 binds to control the expression of IRF7-regulated genes; this leads to the suppression of IFN-α production and expression of antiviral ISGs [142]. The viral gene product encoded by ORF45 also targets IRF7. As a consequence, it blocks IRF7 phosphorylation and nuclear translocation as well as IFNβ/α production [143]. Yet another viral gene product is K-bZIP, a leucine zipper-containing transcription factor that binds to the positive regulatory domain (PRD III-I) region of the IFN-β promoter. This binding site is the same as that for IRF3, so the viral protein blocks IFN-β activation as well as the expression of chemokines (e.g. RANTES and CXCL11) whose promoters are also regulated by IRF3 [144].
The Rotavirus has also developed specific mechanisms to antagonize antiviral defenses. The viral NSP1 protein induces in a proteasome-dependent manner the degradation of IRF3, -5 and -7; this degradation may allow the rotavirus to replicate in specialized trafficking cells (e.g. macrophages and dendritic cells) and cross the gut barrier [145]. Nonetheless, viral infection of myeloid DCs was not sufficient to completely abrogate type I IFN production, suggesting that NSP1 activity is partially affected in a cell-type-specific manner [146]. Proteasome-dependent degradation of IRF7 also occurs after infection with the NDV [147]. The immediate-early protein BZLF-1 of the Human herpesvirus Epstein-Barr virus (EBV/HHV4) physically interacts with IRF7 and inhibits the IRF7-dependent activation of IFN-β/α, thereby antagonizing host early defenses [148].
The Severe acute respiratory syndrome coronavirus (SARS-CoV) has also developed a strategy to overcome host innate immune defenses. The viral gene products derived from ORF3b, ORF6 and N protein were able to disrupt IFN-β production through inhibition of IRF3 [149]. Similarly, blocking IRF3 activation allows the nucleoprotein of the Lymphocytic choriomeningitis virus (arenavirus) to antagonize the type I IFN response [150]. Another report showed that the sumoylation of IRF3 and IRF7 upon VSV infection leads to negative regulation of Type I IFN production [151].
Induction of type I IFNs can also be inhibited through the targeting of TLRs by some viruses. For instance, the NS3-4A protein of the HCV antagonizes the TLR3 pathway through the cleavage of the TLR-3 adaptor protein TRIF in the endosomal compartment [152]. Another interesting example is provided by the Vaccinia virus (VACV). It encodes two proteins, A46 and A52 that independently target distinct components of the TLR pathway and antagonize host innate antiviral responses. A46 interacts with the adaptor molecules MyD88, TRIF, and TRAM through its homotypic TIR domain and thereby interferes with the TLR-mediated pathways leading to IRF3 activation and type I IFN production [153]. A52 interrupts signal transmission leading to activation of NF-κB by interacting with IRAK2 and TRAF6 [154]. Notably, infection of mice with a VACV lacking the A46R or A52R gene resulted in an attenuated phenotype, demonstrating their association with VACV virulence [153], [154]. Furthermore, it has recently been demonstrated that the VACV virulence factor B14 disables pro-inflammatory responses by interacting with IKKβ, thereby inhibiting NF-κB activation [155] (Table 1 ).
Table 1.
Virus counterattacks on host innate immune defenses.
| Virus | Viral component | Cellular target | References |
|---|---|---|---|
| HCV | NS3–4A | MAVS/TRIF | [109], [135], [136], [152] |
| HAV | 3ABC | MAVS | [137] |
| Influenza virus | NS1 | RIG-1/MAVS | [138] |
| Paramyxovirus | V proteins | MDA5/STATs | [101], [139], [140], [141] |
| HHV-8 | vIRF3 | IRF7 | [142], [143], [144] |
| ORF 48 | IRF7 | ||
| K-bZIP | IRF3 | ||
| Rotavirus | NSP1 | IRF3/7 | [145], [146] |
| NDV | – | IRF7 | [147] |
| HHV-4 | BZLF-1 | IRF7 | [148] |
| SARS-CoV | OKF 3b and OKF6 N protein | IRF3 | [149] |
| Arenavirus | Nucleoprotein | IRF3 | [150] |
| VSV | NS3-4A | IRF3/7 | [151] |
| VACV | A46 | MyD88/TRIF/TRAM | [153], [154], [155] |
| A52 | IRAK2/TRAF6 | ||
| B14 | NF-κB | ||
BZLF-1: immediate-early protein of the Humanherpesvirus Epstein-Barr virus (EBV/HHV4); HAV: Hepatitis A virus; HCV: Hepatitis C virus; HHV8/KSHV: Humanherpesvirus/Kaposi's sarcoma-associated herpesvirus 8; IRAK: IL-1 receptor-associated kinase; K-bZIP: a leucine zipper-containing transcription factor; MAVS: mitochondrial antiviral signaling; MDA5: melanoma differentiation associated gene 5; MyD88: myeloid-differentiation primary-response gene 88; NDV: Newcastle disease virus; NF-κB: nuclear factor κB; NS: non-structural; NSP: NS protein; ORF: open-reading frame; RIG-1: retinoic acid-induced gene 1; TRAF: TNF receptor-associated factor; TRAM: TRIF-related adaptor molecule; TRIF: TIR domain-containing adaptor protein inducing IFN-β; vIRF: viral homolog of the interferon-regulatory factor (IRF); VSV: Vesicular stomatitis virus.
Although a large body of work suggests that viral recognition by TLRs can confer innate protection against virus infection and disease [57], [60], [65], TLRs are in some cases associated with exacerbation of the virus-induced inflammatory response and disease. TLR2 illustrates this assertion. TLR2 KO mice are significantly more resistant to lethal encephalitis and ocular lesions caused by HSV-1 [156], [157]. Notably, an additional report identified a correlation between the severity and recurrence of HSV-2 infections in humans and polymorphisms found in the TLR2 gene [158]. On the other hand, TLR2 KO mice infected with HSV-1 via intranasal inoculation fail to develop lethal encephalitis, whereas MyD88 KO mice die of severe encephalitis in response to HSV-1 infection inoculated by the same route [159]. These findings demonstrate that the outcome of a viral disease is also dependent on the route of inoculation as well.
Those who are interested in the balance between the beneficial effects elicited by TLR recognition of a viral infection and the exacerbation of inflammatory response and disease are referred to excellent reviews on the subject [57], [58], [59], [60].
7. Conclusion
In recent years, enormous progress has been made in understanding how viruses are sensed by their hosts in order to trigger the mounting of an innate immune response. This initial recognition plays a fundamental role in the sense that it will dictate the shape of the adaptive immunity. The duration and magnitude of the immune response will define whether resolution of the infection or a disease outcome will occur. In this context, the balance between the triggering of an antiviral response and its interruption is of critical relevance. Type I IFNs, since their discovery 50 years ago, continue to interfere not only with virus replication but also with the duration and magnitude of the antiviral response. Recent findings demonstrating that type I IFNs upregulate the expression of diverse PRRs and SOCS proteins testify for their role in the maintenance of homeostasis.
Acknowledgment
The authors are grateful to LUCIANA G. ANDRADE, MSc, for the excellent artwork.
References
- 1.Isaacs A., Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 2.Taniguchi T., Fujii-Kuriyama Y., Muramatsu M. Molecular cloning of human interferon cDNA. Proc Natl Acad Sci USA. 1980;77:4003–4006. doi: 10.1073/pnas.77.7.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., Schreiber R.D. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 4.Pestka S., Krause C.D., Walter M.R. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 5.Taniguchi T., Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 6.Marie I., Durbin J.E., Levy D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato M., Hata N., Asagiri M., Nakaya T., Taniguchi T., Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 8.Sadler A.J., Williams B.R. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novick D., Cohen B., Rubinstein M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell. 1994;77:391–400. doi: 10.1016/0092-8674(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 10.Farrar M.A., Schreiber R.D. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11:571–611. doi: 10.1146/annurev.iy.11.040193.003035. [DOI] [PubMed] [Google Scholar]
- 11.Kotenko S.V., Gallagher G., Baurin V.V., Lewis-Antes A., Shen M., Shah N.K. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 12.Sheppard P., Kindsvogel W., Xu W., Henderson K., Schlutsmeyer S., Whitmore T.E. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 13.Dumoutier L., Tounsi A., Michiels T., Sommereyns C., Kotenko S.V., Renauld J.C. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem. 2004;279:32269–32274. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 14.Onoguchi K., Yoneyama M., Takemura A., Akira S., Taniguchi T., Namiki H. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 15.Almeida G.M., de Oliveira D.B., Magalhaes C.L., Bonjardim C.A., Ferreira P.C., Kroon E.G. Antiviral activity of type I interferons and interleukins 29 and 28a (type III interferons) against Apeu virus. Antiviral Res. 2008;80:302–308. doi: 10.1016/j.antiviral.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Levraud J.P., Boudinot P., Colin I., Benmansour A., Peyrieras N., Herbomel P. Identification of the zebrafish IFN receptor: implications for the origin of the vertebrate IFN system. J Immunol. 2007;178:4385–4394. doi: 10.4049/jimmunol.178.7.4385. [DOI] [PubMed] [Google Scholar]
- 17.Krause C.D., Pestka S. Evolution of the Class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol Ther. 2005;106:299–346. doi: 10.1016/j.pharmthera.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 18.van Boxel-Dezaire A.H., Rani M.R., Stark G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Der S.D., Zhou A., Williams B.R., Silverman R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonjardim C.A. Interferons (IFNs) are key cytokines in both innate and adaptive antiviral immune responses—and viruses counteract IFN action. Microbes Infect. 2005;7:569–578. doi: 10.1016/j.micinf.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Darnell J.E., Jr, Kerr I.M., Stark G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 22.Tang X., Gao J.S., Guan Y.J., McLane K.E., Yuan Z.L., Ramratnam B. Acetylation-dependent signal transduction for type I interferon receptor. Cell. 2007;131:93–105. doi: 10.1016/j.cell.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 23.Tenoever B.R., Ng S.L., Chua M.A., McWhirter S.M., Garcia-Sastre A., Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 24.Kalvakolanu D.V. Alternate interferon signaling pathways. Pharmacol Ther. 2003;100:1–29. doi: 10.1016/s0163-7258(03)00070-6. [DOI] [PubMed] [Google Scholar]
- 25.Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 26.David M., Petricoin E., III, Benjamin C., Pine R., Weber M.J., Larner A.C. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science. 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 27.Sousa L.P., Brasil B.S., Silva B., de M., Nogueira S.V., Andrade A.A. Characterization of alpha-enolase as an interferon-alpha 2 alpha 1 regulated gene. Front Biosci. 2005;10:2534–2547. doi: 10.2741/1718. [DOI] [PubMed] [Google Scholar]
- 28.Roy S.K., Hu J., Meng Q., Xia Y., Shapiro P.S., Reddy S.P. MEKK1 plays a critical role in activating the transcription factor C/EBP-beta-dependent gene expression in response to IFN-gamma. Proc Natl Acad Sci USA. 2002;99:7945–7950. doi: 10.1073/pnas.122075799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H., Gade P., Xiao W., Kalvakolanu D.V. The interferon signaling network and transcription factor C/EBP-beta. Cell Mol Immunol. 2007;4:407–418. [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Gade P., Nallar S.C., Raha A., Roy S.K., Karra S. The Med1 subunit of transcriptional mediator plays a central role in regulating CCAAT/enhancer-binding protein-beta-driven transcription in response to interferon-gamma. J Biol Chem. 2008;283:13077–13086. doi: 10.1074/jbc.M800604200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F., Ma Y., Barrett J.W., Gao X., Loh J., Barton E. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5:1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 32.Uddin S., Majchrzak B., Woodson J., Arunkumar P., Alsayed Y., Pine R. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 33.Uddin S., Lekmine F., Sharma N., Majchrzak B., Mayer I., Young P.R. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275:27634–27640. doi: 10.1074/jbc.M003170200. [DOI] [PubMed] [Google Scholar]
- 34.Li Y., Sassano A., Majchrzak B., Deb D.K., Levy D.E., Gaestel M. Role of p38alpha Map kinase in Type I interferon signaling. J Biol Chem. 2004;279:970–979. doi: 10.1074/jbc.M309927200. [DOI] [PubMed] [Google Scholar]
- 35.Ramsauer K., Sadzak I., Porras A., Pilz A., Nebreda A.R., Decker T. p38 MAPK enhances STAT1-dependent transcription independently of Ser-727 phosphorylation. Proc Natl Acad Sci USA. 2002;99:12859–12864. doi: 10.1073/pnas.192264999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valledor A.F., Sanchez-Tillo E., Arpa L., Park J.M., Caelles C., Lloberas J. Selective roles of MAPKs during the macrophage response to IFN-gamma. J Immunol. 2008;180:4523–4529. doi: 10.4049/jimmunol.180.7.4523. [DOI] [PubMed] [Google Scholar]
- 37.Uddin S., Yenush L., Sun X.J., Sweet M.E., White M.F., Platanias L.C. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3′-kinase. J Biol Chem. 1995;270:15938–15941. doi: 10.1074/jbc.270.27.15938. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen H., Ramana C.V., Bayes J., Stark G.R. Roles of phosphatidylinositol 3-kinase in interferon-gamma-dependent phosphorylation of STAT1 on serine 727 and activation of gene expression. J Biol Chem. 2001;276:33361–33368. doi: 10.1074/jbc.M105070200. [DOI] [PubMed] [Google Scholar]
- 39.Mayer I.A., Verma A., Grumbach I.M., Uddin S., Lekmine F., Ravandi F. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J Biol Chem. 2001;276:28570–28577. doi: 10.1074/jbc.M011685200. [DOI] [PubMed] [Google Scholar]
- 40.Uddin S., Sassano A., Deb D.K., Verma A., Majchrzak B., Rahman A. Protein kinase C-delta (PKC-delta) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J Biol Chem. 2002;277:14408–14416. doi: 10.1074/jbc.M109671200. [DOI] [PubMed] [Google Scholar]
- 41.Deb D.K., Sassano A., Lekmine F., Majchrzak B., Verma A., Kambhampati S. Activation of protein kinase C delta by IFN-gamma. J Immunol. 2003;171:267–273. doi: 10.4049/jimmunol.171.1.267. [DOI] [PubMed] [Google Scholar]
- 42.DeVries T.A., Kalkofen R.L., Matassa A.A., Reyland M.E. Protein kinase Cdelta regulates apoptosis via activation of STAT1. J Biol Chem. 2004;279:45603–45612. doi: 10.1074/jbc.M407448200. [DOI] [PubMed] [Google Scholar]
- 43.Lekmine F., Uddin S., Sassano A., Parmar S., Brachmann S.M., Majchrzak B. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J Biol Chem. 2003;278:27772–27780. doi: 10.1074/jbc.M301364200. [DOI] [PubMed] [Google Scholar]
- 44.Lekmine F., Sassano A., Uddin S., Smith J., Majchrzak B., Brachmann S.M. Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp Cell Res. 2004;295:173–182. doi: 10.1016/j.yexcr.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 45.Kaur S., Lal L., Sassano A., Majchrzak-Kita B., Srikanth M., Baker D.P. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J Biol Chem. 2007;282:1757–1768. doi: 10.1074/jbc.M607365200. [DOI] [PubMed] [Google Scholar]
- 46.Kaur S., Sassano A., Dolniak B., Joshi S., Majchrzak-Kita B., Baker D.P. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc Natl Acad Sci USA. 2008;105:4808–4813. doi: 10.1073/pnas.0710907105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colina R., Costa-Mattioli M., Dowling R.J., Jaramillo M., Tai L.H., Breitbach C.J. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 48.Yoshimura A., Naka T., Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 49.Rothlin C.V., Ghosh S., Zuniga E.I., Oldstone M.B., Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 50.Coelho L.F., de Oliveira J.G., Kroon E.G. Interferons and scleroderma-a new clue to understanding the pathogenesis of scleroderma? Immunol Lett. 2008;118:110–115. doi: 10.1016/j.imlet.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 51.Banchereau J., Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 52.Ronnblom L., Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lemke G., Rothlin C.V. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brand S., Zitzmann K., Dambacher J., Beigel F., Olszak T., Vlotides G. SOCS-1 inhibits expression of the antiviral proteins 2′,5′-OAS and MxA induced by the novel interferon-lambdas IL-28A and IL-29. Biochem Biophys Res Commun. 2005;331:543–548. doi: 10.1016/j.bbrc.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 55.Zitzmann K., Brand S., De Toni E.N., Baehs S., Goke B., Meinecke J. SOCS1 silencing enhances antitumor activity of type I IFNs by regulating apoptosis in neuroendocrine tumor cells. Cancer Res. 2007;67:5025–5032. doi: 10.1158/0008-5472.CAN-06-2575. [DOI] [PubMed] [Google Scholar]
- 56.Hanada T., Yoshida H., Kato S., Tanaka K., Masutani K., Tsukada J. Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity. 2003;19:437–450. doi: 10.1016/s1074-7613(03)00240-1. [DOI] [PubMed] [Google Scholar]
- 57.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 58.Pichlmair A., Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 59.Gilliet M., Cao W., Liu Y.J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 60.Thompson J.M., Iwasaki A. Toll-like receptors regulation of viral infection and disease. Adv Drug Deliv Rev. 2008;60:786–794. doi: 10.1016/j.addr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holmskov U., Thiel S., Jensenius J.C. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003;21:547–578. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- 62.Hargreaves D.C., Medzhitov R. Innate sensors of microbial infection. J Clin Immunol. 2005;25:503–510. doi: 10.1007/s10875-005-8065-4. [DOI] [PubMed] [Google Scholar]
- 63.Bottazzi B., Garlanda C., Salvatori G., Jeannin P., Manfredi A., Mantovani A. Pentraxins as a key component of innate immunity. Curr Opin Immunol. 2006;18:10–15. doi: 10.1016/j.coi.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 64.Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 65.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 66.Uematsu S., Akira S. Toll-like receptors and Type I interferons. J Biol Chem. 2007;282:15319–15323. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- 67.Takaoka A., Wang Z., Choi M.K., Yanai H., Negishi H., Ban T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 68.Takeda K., Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y.J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 70.Kadowaki N., Antonenko S., Liu Y.J. Distinct CpG DNA and polyinosinic–polycytidylic acid double-stranded RNA, respectively, stimulate CD11c− type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J Immunol. 2001;166:2291–2295. doi: 10.4049/jimmunol.166.4.2291. [DOI] [PubMed] [Google Scholar]
- 71.Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 72.Krug A., Luker G.D., Barchet W., Leib D.A., Akira S., Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through Toll-like receptor 9. Blood. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 73.Fitzgerald-Bocarsly P., Dai J., Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19:3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Colonna M., Trinchieri G., Liu Y.J. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 75.Lee H.K., Iwasaki A. Autophagy and antiviral immunity. Curr Opin Immunol. 2008;20:23–29. doi: 10.1016/j.coi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 77.Honda K., Taniguchi T. Toll-like receptor signaling and IRF transcription factors. IUBMB Life. 2006;58:290–295. doi: 10.1080/15216540600702206. [DOI] [PubMed] [Google Scholar]
- 78.Hoshino K., Sugiyama T., Matsumoto M., Tanaka T., Saito M., Hemmi H. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature. 2006;440:949–953. doi: 10.1038/nature04641. [DOI] [PubMed] [Google Scholar]
- 79.Deng L., Wang C., Spencer E., Yang L., Braun A., You J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 80.Wang C., Deng L., Hong M., Akkaraju G.R., Inoue J., Chen Z.J. TAK1 is an ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 81.Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 82.Yanai H., Chen H.M., Inuzuka T., Kondo S., Mak T.W., Takaoka A. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci USA. 2007;104:3402–3407. doi: 10.1073/pnas.0611559104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cao W., Manicassamy S., Tang H., Kasturi S.P., Pirani A., Murthy N. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sasai M., Oshiumi H., Matsumoto M., Inoue N., Fujita F., Nakanishi M. Cutting Edge: NF-kappaB-activating kinase-associated protein 1 participates in TLR3/Toll-IL-1 homology domain-containing adapter molecule-1-mediated IFN regulatory factor 3 activation. J Immunol. 2005;174:27–30. doi: 10.4049/jimmunol.174.1.27. [DOI] [PubMed] [Google Scholar]
- 85.Hacker H., Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;357:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 86.Guo J., Hui D.J., Merrick W.C., Sen G.C. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J. 2000;19:6891–6899. doi: 10.1093/emboj/19.24.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sarkar S.N., Peters K.L., Elco C.P., Sakamoto S., Pal S., Sen G.C. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11:1060–1067. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- 88.Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 89.Sharma S., tenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 90.Honda K., Takaoka A., Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 91.Levine B., Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee H.K., Lund J.M., Ramanathan B., Mizushima N., Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 93.Tabeta K., Hoebe K., Janssen E.M., Du X., Georgel P., Crozat K. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 94.Brinkmann M.M., Spooner E., Hoebe K., Beutler B., Ploegh H.L., Kim Y.M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barton G.M., Kagan J.C., Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 96.Nishiya T., Kajita E., Miwa S., Defranco A.L. TLR3 and TLR7 are targeted to the same intracellular compartments by distinct regulatory elements. J Biol Chem. 2005;280:37107–37117. doi: 10.1074/jbc.M504951200. [DOI] [PubMed] [Google Scholar]
- 97.Casrouge A., Zhang S.Y., Eidenschenk C., Jouanguy E., Puel A., Yang K. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 98.Kagan J.C., Su T., Horng T., Chow A., Akira S., Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu Q., Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 100.Mansell A., Smith R., Doyle S.L., Gray P., Fenner J.E., Crack P.J. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 101.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 102.Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 103.Rothenfusser S., Goutagny N., DiPerna G., Gong M., Monks B.G., Schoenemeyer A. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 104.Venkataraman T., Valdes M., Elsby R., Kakuta S., Caceres G., Saijo S. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- 105.Takaoka A., Taniguchi T. Cytosolic DNA recognition for triggering innate immune responses. Adv Drug Deliv Rev. 2008;60:847–857. doi: 10.1016/j.addr.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 106.Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 107.Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 108.Xu L.G., Wang Y.Y., Han K.J., Li L.Y., Zhai Z., Shu H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 109.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 110.Gack M.U., Shin Y.C., Joo C.H., Urano T., Liang C., Sun L. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 111.Arimoto K., Takahashi H., Hishiki T., Konishi H., Fujita T., Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oganesyan G., Saha S.K., Guo B., He J.Q., Shahangian A., Zarnegar B. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 113.Chau T.L., Gioia R., Gatot J.S., Patrascu F., Carpentier I., Chapelle J.P. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008;33:171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 114.Saha S.K., Cheng G. TRAF3: a new regulator of type I interferons. Cell Cycle. 2006;5:804–807. doi: 10.4161/cc.5.8.2637. [DOI] [PubMed] [Google Scholar]
- 115.Balachandran S., Thomas E., Barber G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432:401–405. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- 116.Takahashi K., Kawai T., Kumar H., Sato S., Yonehara S., Akira S. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol. 2006;176:4520–4524. doi: 10.4049/jimmunol.176.8.4520. [DOI] [PubMed] [Google Scholar]
- 117.Zhao T., Yang L., Sun Q., Arguello M., Ballard D.W., Hiscott J. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- 118.Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 119.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 120.Marques J.T., Devosse T., Wang D., Zamanian-Daryoush M., Serbinowski P., Hartmann R. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006;24:559–565. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- 121.Gitlin L., Barchet W., Gilfillan S., Cella M., Beutler B., Flavell R.A. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103:8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 123.Kato H., Sato S., Yoneyama M., Yamamoto M., Uematsu S., Matsui K. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 124.Sun Q., Sun L., Liu H.H., Chen X., Seth R.B., Forman J. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 125.Kumar H., Kawai T., Kato H., Sato S., Takahashi K., Coban C. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ishii K.J., Coban C., Kato H., Takahashi K., Torii Y., Takeshita F. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 127.Levy D.E., Marie I., Prakash A. Ringing the interferon alarm: differential regulation of gene expression at the interface between innate and adaptive immunity. Curr Opin Immunol. 2003;15:52–58. doi: 10.1016/s0952-7915(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 128.Tamura T., Tailor P., Yamaoka K., Kong H.J., Tsujimura H., O'Shea J.J. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol. 2005;174:2573–2581. doi: 10.4049/jimmunol.174.5.2573. [DOI] [PubMed] [Google Scholar]
- 129.Tsujimura H., Tamura T., Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003;170:1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- 130.Tailor P., Tamura T., Kong H.J., Kubota T., Kubota M., Borghi P. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity. 2007;27:228–239. doi: 10.1016/j.immuni.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Malathi K., Dong B., Gale M., Jr, Silverman R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nguyen K.B., Salazar-Mather T.P., Dalod M.Y., Van Deusen J.B., Wei X.Q., Liew F.Y. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol. 2002;169:4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- 133.Lee C.K., Rao D.T., Gertner R., Gimeno R., Frey A.B., Levy D.E. Distinct requirements for IFNs and STAT1 in NK cell function. J Immunol. 2000;165:3571–3577. doi: 10.4049/jimmunol.165.7.3571. [DOI] [PubMed] [Google Scholar]
- 134.Kolumam G.A., Thomas S., Thompson L.J., Sprent J., Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lin R., Lacoste J., Nakhaei P., Sun Q., Yang L., Paz S. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol. 2006;80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Loo Y.M., Owen D.M., Li K., Erickson A.K., Johnson C.L., Fish P.M. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci USA. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yang Y., Liang Y., Qu L., Chen Z., Yi M., Li K. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci USA. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mibayashi M., Martinez-Sobrido L., Loo Y.M., Cardenas W.B., Gale M., Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Andrejeva J., Childs K.S., Young D.F., Carlos T.S., Stock N., Goodbourn S. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ulane C.M., Kentsis A., Cruz C.D., Parisien J.P., Schneider K.L., Horvath C.M. Composition and assembly of STAT targeting ubiquitin ligase complexes: paramyxovirus V protein carboxyl terminus is an oligomerization domain. J Virol. 2005;79:10180–10189. doi: 10.1128/JVI.79.16.10180-10189.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Palosaari H., Parisien J.P., Rodriguez J.J., Ulane C.M., Horvath C.M. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. 2003;77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Joo C.H., Shin Y.C., Gack M., Wu L., Levy D., Jung J.U. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol. 2007;81:8282–8292. doi: 10.1128/JVI.00235-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhu F.X., King S.M., Smith E.J., Levy D.E., Yuan Y. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc Natl Acad Sci USA. 2002;99:5573–5578. doi: 10.1073/pnas.082420599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lefort S., Soucy-Faulkner A., Grandvaux N., Flamand L. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J Virol. 2007;81:10950–10960. doi: 10.1128/JVI.00183-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Barro M., Patton J.T. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. 2007;81:4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Douagi I., McInerney G.M., Hidmark A.S., Miriallis V., Johansen K., Svensson L. Role of interferon regulatory factor 3 in type I interferon responses in rotavirus-infected dendritic cells and fibroblasts. J Virol. 2007;81:2758–2768. doi: 10.1128/JVI.01555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Prakash A., Levy D.E. Regulation of IRF7 through cell type-specific protein stability. Biochem Biophys Res Commun. 2006;342:50–56. doi: 10.1016/j.bbrc.2006.01.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hahn A.M., Huye L.E., Ning S., Webster-Cyriaque J., Pagano J.S. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. J Virol. 2005;79:10040–10052. doi: 10.1128/JVI.79.15.10040-10052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martinez-Sobrido L., Giannakas P., Cubitt B., Garcia-Sastre A., de la Torre J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J Virol. 2007;81:12696–12703. doi: 10.1128/JVI.00882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kubota T., Matsuoka M., Chang T.H., Tailor P., Sasaki T., Tashiro M. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem. 2008;283:25660–25670. doi: 10.1074/jbc.M804479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li K., Foy E., Ferreon J.C., Nakamura M., Ferreon A.C., Ikeda M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stack J., Haga I.R., Schroder M., Bartlett N.W., Maloney G., Reading P.C. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harte M.T., Haga I.R., Maloney G., Gray P., Reading P.C., Bartlett N.W. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen R.A., Ryzhakov G., Cooray S., Randow F., Smith G.L. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008;4:e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kurt-Jones E.A., Chan M., Zhou S., Wang J., Reed G., Bronson R. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sarangi P.P., Kim B., Kurt-Jones E., Rouse B.T. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: role of the Toll pathway in early inflammatory events in stromal keratitis. J Virol. 2007;81:11128–11138. doi: 10.1128/JVI.01008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bochud P.Y., Magaret A.S., Koelle D.M., Aderem A., Wald A. Polymorphisms in TLR2 are associated with increased viral shedding and lesional rate in patients with genital herpes simplex virus Type 2 infection. J Infect Dis. 2007;196:505–509. doi: 10.1086/519693. [DOI] [PubMed] [Google Scholar]
- 159.Mansur D.S., Kroon E.G., Nogueira M.L., Arantes R.M., Rodrigues S.C., Akira S. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol. 2005;166:1419–1426. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]