

Measles virus proteins form liquid droplets where they can encapsidate their genomic material.
Abstract
Many viruses are known to form cellular compartments, also called viral factories. Paramyxoviruses, including measles virus, colocalize their proteomic and genomic material in puncta in infected cells. We demonstrate that purified nucleoproteins (N) and phosphoproteins (P) of measles virus form liquid-like membraneless organelles upon mixing in vitro. We identify weak interactions involving intrinsically disordered domains of N and P that are implicated in this process, one of which is essential for phase separation. Fluorescence allows us to follow the modulation of the dynamics of N and P upon droplet formation, while NMR is used to investigate the thermodynamics of this process. RNA colocalizes to droplets, where it triggers assembly of N protomers into nucleocapsid-like particles that encapsidate the RNA. The rate of encapsidation within droplets is enhanced compared to the dilute phase, revealing one of the roles of liquid-liquid phase separation in measles virus replication.
INTRODUCTION
Measles virus (MeV) is a highly contagious virus that is responsible for more than 100,000 deaths every year. No treatment currently exists for MeV infection, and the impact of MeV throughout the world has recently been amplified by weakening levels of vaccine coverage (1). MeV is a nonsegmented, single-stranded negative-sense RNA virus (Mononegavirales) of the family Paramyxoviridae, whose members carry their own replication machinery comprising nucleoprotein (N), polymerase (L), and phosphoprotein (P) components. N binds to the viral genome, assembling into helical nucleocapsids, while P chaperones N before encapsidation and is thought to bring N and L in close spatial proximity during replication and transcription (2, 3). The replication process represents a promising but hitherto relatively unexplored therapeutic target.
N and P both comprise domains exhibiting high levels of intrinsic disorder, with respectively 30 and 75% of their primary sequences lacking tertiary structure. N has a 125–amino acid disordered C-terminal domain (NTAIL) (4) and a flexible 30–amino acid N-terminal arm (NARM), flanking the RNA binding domain NCORE, while P comprises disordered domains of 304 amino acids (PTAIL) (5) and 80 amino acids (PLOOP), on either side of a coiled-coil tetrameric domain (6), and is capped by a C-terminal three-helix bundle (XD) (Fig. 1A) (7). N and P exhibit numerous interactions that are essential during different stages of the viral cycle: Two distant linear motifs in PTAIL, P1–50 and P186–198, participate in chaperoning the monomeric form of N (5, 8), preventing binding to cellular RNA. In an N0P heterodimer, P1–50 binds to the same surface of N as NARM of the succeeding protomer in assembled nucleocapsids (9), so that the N0P complex efficiently inhibits assembly. XD is known to bind to NTAIL (10, 11), an interaction thought to be important for colocalizing N and L during the replication process and was recently shown to interact with L (12). The process of nucleocapsid assembly on viral RNA sequences was recently studied in real time, using nuclear magnetic resonance (NMR) and fluorescence (13), highlighting the role of NARM and P1–50, and leading to high-resolution structures revealing N-RNA interactions in assembled nucleocapsids (14).
Fig. 1. Macroscale physical characterization of MeV droplets in vitro.
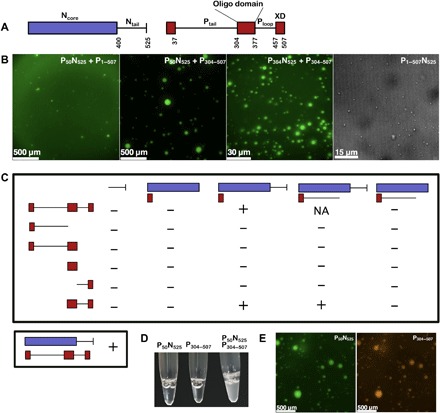
(A) Schematic diagram of the structure of N and P; intrinsically disordered regions are presented as lines and folded domains are presented as boxes. (B) Fluorescence or DIC images of mixtures where phase separation events were observed: P50N525 (80 μM, 1% labeled) + P1–507 (66 μM), P50N525 (80 μM, 1% labeled) + P304–507 (200 μM), P300N525 + P304–507 (290 μM), and N1–525 + P1–507 (coexpressed in E. coli). (C) The phase separation scaffold requires P50N525 and P304–507 domains. Mixtures of different N and P truncation mutants were tested for the ability to trigger phase separation using DIC microscopy. “−” no phase separation events were observed; “+” phase separation events were observed; “NA” not tested. All mixtures were tested at several concentrations higher than 50 μM for each protein and several ratios including 1:1. Bottom panel represents coexpression of the full-length N and P proteins. (D) Picture of tubes with P50N525 and P304–507. Sample with N and P mixed together becomes turbid. (E) Colocalization of P50N525-fluorescein (80 μM, 1% labeled) and P304–507-Alexa Fluor 594 (200 μM, 0.5% labeled) droplets.
In recent years, liquid-liquid phase separation (LLPS), driven by weak interactions often involving intrinsically disordered proteins or RNA, has been shown to offer a highly efficient means to spatially and temporally control a remarkable array of cellular processes (15). The physicochemical properties of these microenvironments are thought to provide optimal conditions to enhance specific molecular interactions within the condensate, while maintaining immiscibility with respect to other excluded molecules (16–18). A number of viruses have been shown to form cytoplasmic compartments in infected cells, known as “viral factories,” that have been associated with the viral replication process (19, 20). Colocalization of N and P in vivo is a characteristic feature of mononegaviruses such as rabies (21), vesicular stomatitis virus (22), respiratory syncytial virus (23), human parainfluenza types 3 (24) and 5 (25), human metapneumovirus (26), Nipah (27), and MeV (28, 29). Inclusion bodies from MeV have been shown to colocalize host proteins such as WDR5 (WD repeat-containing protein 5) (28) and other viral proteins [the polymerase L as well as C that is involved in controlling host immune response (30)]. Recent confirmation of the liquid-like nature of Negri bodies formed in rabies (31) and colocalization of N and P in infection-induced cytoplasmic inclusion bodies in MeV (32) suggests that LLPS represents a widespread mechanism exploited by mononegaviruses during infection.
Here, we demonstrate that mixing MeV N and P proteins in vitro triggers LLPS, allowing us to characterize the physical nature of the droplets, including the modulation of the dynamic behavior of N and P upon droplet formation, the molecular basis of the stabilizing interactions, and the thermodynamics of droplet formation. We are able to show that RNA molecules preferentially localize to the droplets and, once within the droplets, trigger assembly of nucleocapsid-like particles. The process of nucleocapsid assembly in LLPS is observed in real time using NMR spectroscopy. The rate of assembly is significantly enhanced compared to non–phase-separating conditions, providing clear evidence that LLPS has the ability to enhance viral function and supporting previous suggestions that LLPS is exploited by negative-strand RNA viruses to create viral replication factories.
RESULTS
MeV N and P proteins phase separate upon mixing in vitro
Recombinant full-length P (P1–507) and an RNA-free N0P complex (P50N525) were separately expressed and mixed together and observed to form micron-sized spherical structures (Fig. 1B). Phase separation of P1–507 and full-length N was also demonstrated by coexpressing both proteins using separate plasmids in Escherichia coli. The full N0P complex was purified in a high-salt buffer to prevent phase separation (vide infra) and diluted into low salt, resulting in phase separation as observed from differential interference contrast (DIC) microscopy (Fig. 1B).
Identification of N and P domains that are required for phase separation
To identify which domains of MeV N and P are essential for phase separation, we tested a panel of different N and P constructs for their ability to trigger phase separation (Fig. 1, A to C, and fig. S1). Several mixtures formed condensed phases: P50N525 + P1–507, P304N525 + P304–507, and P50N525 + P304–507 (Fig. 1B). P50N525 + P304–507 was thus identified as the minimal phase-separating system, forming spherical bimolecular condensates as measured by fluorescence microscopy (Fig. 1B) and showing increased turbidity (Fig. 1D). Further investigations of MeV droplets were performed using this minimal system (P50N525 and P304–507) unless otherwise stated. P50N525 and P304–507 colocalize in droplets (Fig. 1E), and their phase transition was shown to be salt dependent: Beyond 400 mM, fluorescent speckles are no longer observed (fig. S2).
The phase diagram of LLPS was established on the basis of turbidity (Fig. 2A), identifying threshold concentrations of 10 μM for both proteins, although fluorescence microscopy and negative-stain electron microscopy reveal phase separation at lower protein concentrations. Fluorescence microscopy indicates that proteins are homogeneously distributed within droplets (fig. S3).
Fig. 2. MeV droplets exhibit liquid-like behavior.
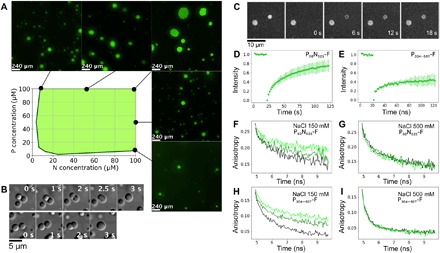
(A) Turbidity-based phase diagram as a function of P50N525 and P304–507 concentrations. Green shading represents ranges of combinations of protein concentration that form droplets. Examples of fluorescent images at several N and P concentrations that show phase separation are presented around the phase diagram. (B) Fusion events followed by relaxation into spherical droplets in a mixture comprising P304–507 (100 μM) and P50N525 (10 μM). (C) Images of FRAP shows exchange of P50N525–fluorescein amidite between phases. (D) FRAP kinetics of P50N525 (25 μM with 10 nM P50N525-fluorescein) in droplets formed with P304–507 at 1:2 ratio. (E) FRAP kinetics of P304–507 (50 μM with 50 nM P304–507-fluorescein) in droplets formed with P50N525 at 2:1 ratio. (F and G) Fluorescence anisotropy of P50N525-fluorescein (10 μM P50N525, 500 nM P50N525-fluorescein) in the dilute phase (black) and in the presence of P304–507 under phase-separating conditions [N:P = 1:2 (dark green) and N:P = 1:4 (light green)]. Fluorescence lifetimes were measured at room temperature, (F) in buffer containing 150 mM NaCl and (G) in buffer containing 500 mM NaCl that suppresses phase separation but not the interaction between proteins. (H and I) Fluorescence anisotropy of P304–507-fluorescein (10 μM, 500 nM P304–507-fluorescein) in the dilute phase (black) and in the presence of P50N525 at phase-separating conditions [P:N = 4:1 (dark green) and P:N 2:1 (light green)]. Fluorescence lifetimes were measured at room temperature, (H) in buffer containing 150 mM NaCl and (I) in buffer containing 500 mM NaCl that suppresses phase separation but not the interaction between proteins.
The liquid-like behavior of droplets was confirmed by DIC microscopy via observation of fusion events with relaxation into spherical droplets (Fig. 2B and movie S1) and further demonstrated by fluorescence recovery after photobleaching (FRAP) of fluorescein-labeled P50N525, showing recovery within seconds (Fig. 2, C and D). FRAP of P protein is measurably slower (Fig. 2E), suggesting that the branched multimeric P acts as a dynamic scaffold for the liquid droplets (33). Rotational diffusion of proteins within droplets and in the surrounding phase was measured using time-resolved fluorescence anisotropy. The rotational correlation time of P50N525 in the presence of P304–507 was significantly slowed down compared to P50N525 in free solution (Fig. 2F), while there was no difference under conditions of high-salt buffer, which suppresses phase transition but maintains interactions between the proteins (Fig. 2G). Similar observations were measured for P304–507 in the free protein and in the presence of P50N525 (Fig. 2, H and I). Both proteins therefore remain liquid in the condensed phase, although their rotational dynamics are slowed, possibly due to increased viscosity in the droplets. In summary, these results confirm that LLPS occurs under near-physiological protein and salt concentrations upon mixing of full-length N- and the C-terminal domains of P in vitro, complementing recent in vivo observations (32).
PLOOP is intrinsically disordered
The conformational behavior of the region between the tetramerization domain and XD, P377–457 (PLOOP), was analyzed using NMR spectroscopy. 13C, 15N, and 1H chemical shifts were combined with statistical coil sampling and ensemble selection approach ASTEROIDS (fig. S4), revealing that this domain is essentially devoid of significantly populated secondary structure with the exception of two regions with 20 and 10% α-helical propensity around residues 406 and 433, respectively. 15N relaxation data also show that this domain is highly dynamic (fig. S5), although relaxation rates around regions 410 and 433 increased in constructs containing the tetrameric or XD domains (P377–507 and P304–507), probably due to slower rotational tumbling. Carbon chemical shifts of P304–507 did not change significantly in comparison with PLOOP, demonstrating that PLOOP remains disordered in the context of the construct that links the tetramerization domain and XD and that is required for phase separation (see fig. S6).
The NTAIL:PXD interaction is essential for droplet formation
The C-terminal domains of N and P are known to interact via residues 487 to 501 of NTAIL, which populates a conformational equilibrium of partially folded helices in solution (34) and the XD domain of P. The affinity of the MeV NTAIL and PXD (Phosphoprotein XD domain) interaction (7, 11) under our experimental conditions is approximately 5 μM (Fig. 3, A to C). This affinity was not measurably dependent on salt concentration. The sole intermolecular hydrogen bonding interaction observed in a chimeric crystal structure of N486–505:PXD involves S491 of NTAIL interacting with K489 and D493 of PXD (7). Mutation of N (S491) has been shown to abrogate the NTAIL:PXD interaction—this is also the case under our experimental conditions (Fig. 3D)—and to significantly decrease viral transcription in vivo (11). The critical nature of this interaction for LLPS was demonstrated via the S491L mutation, which resulted in suppression of phase separation.
Fig. 3. Interaction between PXD and N460–525 is essential for MeV LLPS.
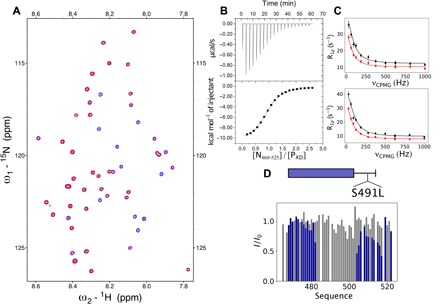
(A) 1H-15N HSQC spectrum of free PXD (blue) and upon interaction with unlabeled N460–525 (red). Concentration of PXD was 100 μM in both cases, and the concentration of N460–525 was 20 μM. Spectra were recorded at 850 MHz 1H frequency and 298 K. See Materials and Methods for additional experimental conditions. (B) Isothermal titration calorimetry of PXD and N460–525 interaction. Raw injection heats (top) and the corresponding specific binding isotherms (bottom) measured at 25°C are shown. PXD (845 μM) was sequentially injected to N460–525 (70 μM) in a microcalorimeter cell from 0 to 2.5 ratio. Fitted parameters: ∆S = −10.7 cal mol−1 deg−1, ∆H = −10,460 ± 46.47 cal mol−1, N = 0.927 sites. (C) 15N CPMG relaxation dispersion of PXD (100 μM) for residues 504 (top) and 509 (bottom) in the presence of 46% N460–525. CPMG frequencies were in the range from 31 to 1000 Hz. The experiments were recorded at a 1H frequency of 700 MHz (black) and 950 MHz (red) at 25°C. Data were fitted simultaneously for residues R465, S466, I468, R472, R477, K478, Y480, K496, I504, and E509 with the two-state model, pb = 23%, exchange rate (kex) = 285 ± 50 s−1. (D) Intensity ratio profiles of free versus PXD-bound N460–525 [wild type (WT)] (blue) and N460–525(S491L) (gray). The ratio between N and P was 1:1 at 200 μM concentration.
The PLOOP-N interaction modulates the dynamics of MeV droplets
An additional interaction between PLOOP and N was identified using NMR spectroscopy (Fig. 4, A and B), suggesting that the role of this domain, which is present in all constructs involved in phase separation, involves a direct interaction with N rather than acting as a simple spacer (35). Interactions centered on residues 409VLK411 and 433LLK435 exhibit distinct affinities [estimated dissociation constant (Kd) 264 and 606 μM, respectively]. Mutation of 409VLK411 to AAA inhibited this interaction (Fig. 4C) with N and modulated macroscale dynamic properties of droplets, as measured from FRAP, but did not completely inhibit LLPS (Fig. 4D). Both 409VLK411 and 433LLK 435 exhibit high sequence conservation over MeV phosphoproteins (fig. S7).
Fig. 4. Interaction of PLOOP with N modulates the dynamics of MeV liquid droplets.
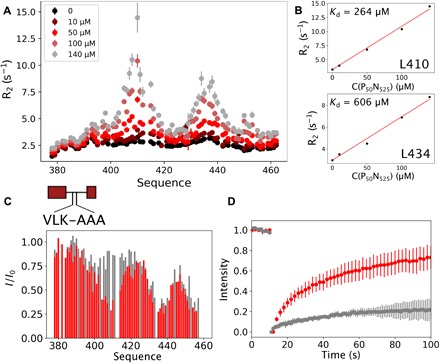
(A) 15N R2 of PLOOP (100 μM) in the presence of P50N525 at different concentrations at 600 MHz, 25°C. (B) Transverse 15N relaxation rates of the central residues of the two interaction regions in PLOOP (centered on residues 410 and 434, respectively). The rates increase linearly as a function of concentration of added P50N525. (C) Intensity ratios of free versus P50N525-bound PLOOP (red) and PLOOP 409VLK411-AAA mutant (gray). The ratio between N0P and P was 1:1; concentration was 100 μM. (D) FRAP of P50N525-fluorescein with P304–507 (WT) (red) and P304–507 409VLK411-AAA (gray) in droplets. In both cases, the concentration of P50N525 was 25 μM; concentrations of P304–507 WT and 409VLK411-AAA mutant were 50 μM, and entire droplets were photobleached.
Investigating the thermodynamics of droplet formation using NMR
To study interactions between proteins under phase-separating conditions, we compared 15N heteronuclear single-quantum coherence (HSQC) spectra of P304–507 and P50N525 separately and in different stoichiometric mixtures. Residues from 377 to 507 were observable in the P304–507 spectrum, while NTAIL and flexible residues corresponding to NARM and P1–50 were observable in P50N525 (Fig. 5). Overall peak intensity was initially observed to decrease for both N and P with increasing stoichiometric ratio (N/P), with no noticeable chemical shift differences. Similar observations were recently made in mixtures of two interacting translational regulators (36). Apparent rotational and transverse diffusion rates were similar to those measured for free proteins (fig. S8). Rotational correlation times would be expected to increase significantly within droplets due to increased viscosity (37). This result therefore suggests that observable signal derives from N and P in the dilute phase.
Fig. 5. Thermodynamics of MeV NP phase separation.
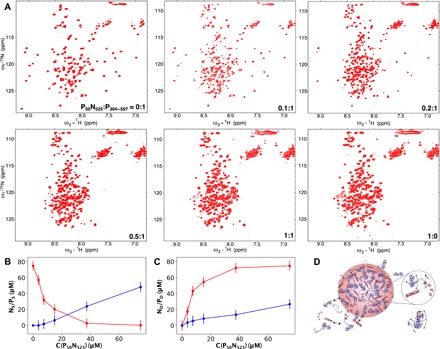
(A) 1H-15N HSQC of 15N-labeled P50N525 and P304–507 at different admixtures of the two proteins; all spectra were recorded at 850 MHz, 25°C. P304–507 concentration was 75 μM, and P50N525 concentration varies from 5 to 100% of 75 μM. (B) Concentration of P50N525 and P304–507 proteins in the dilute phase derived from the spectra shown in (A). (C) Comparison of concentrations of droplet-associated P50N525 and P304–507 in the sensitive volume, estimated on the basis of observation of protein in the dilute phase [derived from analysis of spectra shown in (A)]. Total concentration of P in the mixture was 75 μM. (D) Schematic representation of droplet formation upon mixing P50N525 and P304–507.
Decrease in resonance intensity of N and P is accompanied by broadening of resonances from PXD and N483–505, due to the expected interactions between P and N. Increasing the N:P ratio further results in a minimum of total peak intensity at a ratio of approximately 0.15, followed by continual increase (Fig. 5). To follow the apparent thermodynamics of droplet formation, intensities of 23 (N) and 9 (P) nonoverlapping peaks corresponding to residues that are not affected by the interaction under non–phase-separating conditions were followed as a function of the concentration of N (concentration of P was held constant—see Materials and Methods). The ratio of N and P in droplets was estimated according to
| (1) |
ND and PD refer to the apparent concentration of droplet-associated protein, NTOT and PTOT refer to the total concentrations of P and N, and NOBS and POBS refer to the apparent concentrations in the liquid phase that are observed by NMR. This analysis reveals a clear dependence of rD on the total concentration of N, varying by nearly a factor of 3 for total concentrations of N that vary between 7.5 and 75 μM (Fig. 5 and table S1). This dependence indicates that a simple two-state exchange mechanism cannot explain the experimental observation and suggests that at least two steps are necessary to describe the process of droplet formation.
RNA colocalizes to MeV droplets and triggers nucleocapsid formation in guttala
The functional role of MeV LLPS was examined by adding short, fluorescently labeled RNA decamers to samples containing droplets. Within seconds of adding the RNA, the fluorescence intensity of the droplets increased, starting from the perimeter (Fig. 6A and movie S2). This demonstrates that RNA localizes preferentially to the membraneless organelles where it presumably interacts with the N:P complex. After incubation for 1 hour at 37°C, the samples were examined using negative-stain electron microscopy, revealing the presence of assembled nucleocapsid-like structures (Fig. 6B).
Fig. 6. Nucleocapsid assembly occurs in liquid droplets and is enhanced by phase separation.
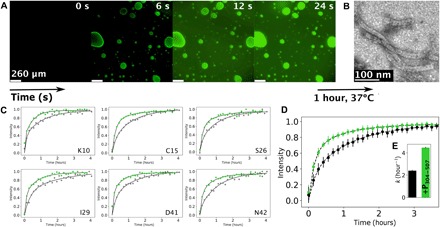
(A) Fluorescence microscopy images showing fluorescently labeled RNA diffusing into droplets preformed by mixing P50N525 (20 μM) and P304–507 (80 μM); RNA of 200 μM (with 10% fluorescently labeled) was added on one side of the coverslip and spontaneously diffused in the sample. (B) Negative-stain electron microscopy of the sample shown in (A), after 1 hour of incubation at 37°C. (C) Kinetic traces of peak intensities corresponding to individual residues of P1–50 in SOFAST 1H-15N HMQC spectra acquired at a resolution of 8.5 min (8 scans, 200 15N increments, and 1024 1H), following addition of OH-A6-OH RNA to P50N525 (gray) and under phase-separating conditions with P304–507 (green) at 293 K. Data were fitted with a biexponential function as previously described (13). (D) Averaged intensities for residues E4, K10, N11, G12, E14, C15, I16, G25, S26, L27, I29, A32, M33, A35, D41, N42, G44, and Q45 of P1–50 in P50N525 (gray) and under phase-separating conditions with P304–507 (green). Dots with SD represent experimental data, and lines represent biexponential fit. (E) Rate of assembly of nucleocapsids as measured by the appearance of resonances from P50 when assembled using P50N525 (gray) and when using P50N525 + P304–507 (green) having formed droplets. The concentration of P50N525 and RNA was constant in both cases.
To determine the rate of nucleocapsid assembly, we used real-time NMR. This approach has recently been used to observe the kinetics of assembly of nucleocapsid-like particles in the dilute phase (13). Nonfluorescently labeled RNA hexamers comprising six adenines were added to the droplet suspension comprising 15N-labeled N, resulting in the assembly of nucleocapsid-like particles. Assembly can be followed by detecting the appearance and increasing intensity of NMR signals from the P1–50 peptide that is released upon assembly of the nucleocapsids (Fig. 6, C and D). Comparison of the rate of increase of the NMR signals corresponding to P1–50 in the condensed and dilute phase (in the absence of P304–507) unambiguously shows that nucleocapsid formation is accelerated in droplets at the same global concentration of N1–525.
DISCUSSION
Nonsegmented negative-sense RNA viruses are known to form inclusion bodies comprising proteins involved in the replication machinery. The recent observation of colocalization of N and P of both rabies (31) and measles (32) into liquid-like membraneless puncta following infection suggests that this phenomenon may play a role in viral replication or transcription and that the phenomenon may be widespread throughout the Mononegavirales order and possibly related viruses. The formation of such membraneless organelles has been proposed to enhance reaction rates by concentrating interaction partners in a confined volume, for storage purposes or, in the case of viral organelles, to provide protection from the host immune system. Investigating the role of this poorly understood phenomenon is essential to understanding the viral cycle. Describing the physical mechanisms that underlie their formation and stability represents a significant challenge, particularly if one considers their potential as targets of viral inhibition. To provide molecular insight into these aspects, we aimed to recreate the phenomenon of LLPS involving MeV N and P proteins in vitro.
We first demonstrated LLPS of N and P under conditions where neither protein undergoes phase separation alone. Expression of independent domains and combinations of domains then allowed us to identify the regions of N and P that are necessary for droplet formation in vitro. This revealed that apart from the first 50 amino acids of P that chaperone N, the rest of the 300–amino acid disordered PTAIL is dispensable for LLPS, while the tetramerization domain of P, the hitherto uncharacterized disordered domain PLOOP, and the three-helix bundle XD are all essential. N cannot be purified in an isolated form, owing to the propensity of the protein to bind to cellular RNA and form nucleocapsid-like structures. We therefore purified a chaperoned form of N bound to the N-terminal peptide of P, which maintains N in a monomeric, unassembled form (N0P). LLPS only occurs when N0P comprising the full-length N protein, including both NCORE and the 125–amino acid disordered NTAIL, is present. Full-length N and P were also coexpressed, and the integral N0P complex was shown to undergo LLPS. Overall, these results verify that this minimal in vitro system reproduces the characteristic colocalization of N and P in membraneless organelles that has been observed in infected cells.
We also identified the interaction between the helical motif of NTAIL and XD to be essential for phase separation. These results align closely with data measured in vivo on cells infected with rabies (31) and more recently MeV (32). The NTAIL:PXD interaction has previously been associated with transport of the polymerase complex to the nucleocapsid before replication and transcription (10, 12, 38, 39). Here, we demonstrate that at least one role of this interaction involves the free-energy stabilization of LLPS. Mutation of a single residue (S491L) that is known to mediate the interaction between NTAIL and PXD, and to strongly affect replication in vivo (11), results in abrogation of LLPS.
Additional weak interactions between PLOOP and N are also shown to modulate the fluidity of the droplets, suggesting that polyvalency of N:P interactions plays a role in droplet formation. The importance of both disordered and folded, or partially folded domains in the polyvalent complex that stabilizes the MeV droplets invokes their possible roles as “stickers” and “spacers” (40), which assure the noncovalent binding and physical separation of interacting components, respectively. Nevertheless, the role of PLOOP appears to be more complex than this as the impact of this interaction on the nature of MeV droplets suggests that it is capable of fulfilling both roles.
Using NMR spectroscopy, we are able to investigate the thermodynamic equilibrium between dilute and condensed phases in the presence of MeV LLPS by comparing the rate of increase in signal intensity from NTAIL and the decrease in intensity of signal from PLOOP and XD, as a function of the concentration of N. Assuming that all signal derives from the dilute phase (as evidenced by rotational and translational diffusion measurements) and that signal drop results uniquely from slow exchange with an invisible form of each protein in droplets, our observations suggest that the stoichiometry of N and P within droplets depends on the ratio of the two proteins in the mixture. As the experimental data cannot be explained using a simple one-step reaction, it is interesting to speculate on the nature of the second interaction. One possible explanation involves N interacting with P to form droplets, with additional recruitment of N to existing N:P droplets. Such a model would be in agreement with the critical presence of the tetramerization domain and the polyvalency of the four branched dynamic XD interaction sites, which may form a dynamic scaffold in MeV LLPS to which additional N could be recruited. Note that both P and N remain dynamic in the context of the droplets, as evidenced by the rotational diffusion measured by fluorescence anisotropy. The proposed role of P as a dynamic scaffold is supported by the observation of differential translational mobility of P within droplets with respect to N, as reported by FRAP. More experimental measurements on this system are required to better characterize these molecular processes.
Replication and transcription of negative-strand RNA viruses are thought to occur within inclusion bodies comprising N, P, and L. The colocalization of these proteins within liquid droplets may act to increase local concentration of the components of the viral replication machinery, thereby increasing replication efficiency. We have therefore investigated whether the liquid droplets formed upon mixing of N and P in vitro are capable of supporting nucleocapsid assembly, which is an essential part of the viral replication process. We previously demonstrated that assembly can be observed in the dilute phase, by adding RNA to monomeric N0P and using NMR spectroscopy and fluorescence anisotropy to monitor the rate of assembly (13). By adding RNA to a suspension of N:P organelles, we show that RNA molecules preferentially localize to the droplets and, once within the droplets, trigger assembly of nucleocapsid-like particles. Notably, the rate of assembly is significantly increased compared to non–phase-separating conditions, providing evidence that LLPS has the ability to enhance viral function and offering a first glimpse of these viral factories in action. This also indicates that the condensed nature of the droplets does not restrict the ability to assemble these very large superstructures and demonstrates that these minimalist N:P droplets are sufficient to promote one of the essential processes of viral replication. In this context, we note that nucleocapsid structures were observed to be ejected from liquid droplets formed in cells infected with rabies virus (31).
Phosphorylation of P and N have both been reported to affect MeV function (41, 42) and, more recently, to influence the nature of liquid droplet formation in vivo (32). Although both N and P were purified from E. coli in their unmodified forms, the technology described here will allow us to investigate the impact of phosphorylation in future studies. In particular, the role of the 300–amino acid disordered PTAIL domain may become clearer as this comprises numerous phosphorylation sites but is not essential for LLPS. This experimental system will also allow us to investigate the role of host proteins that are thought to enter inclusion bodies and the viral proteins C and L.
In conclusion, we have demonstrated that purified N and P MeV proteins form liquid-like membraneless organelles upon mixing in vitro. We identify weak interactions involving intrinsically disordered domains of N and P that are implicated in this process and identify one interaction between the C-terminal domains as essential to phase separation. P protein appears to play a role as a multivalent scaffold stabilizing the droplets. The droplets exhibit both physical and functional features characteristic of paramyxoviral inclusion bodies: RNA colocalizes to the droplets and thereby triggers assembly of N protomers into nucleocapsid-like particles that encapsidate the RNA. The ability to reproduce mechanistic behavior of MeV factories in vitro will allow us to further examine the role of additional host and viral partners at the molecular level. The observation that similar puncta or inclusion bodies have been found for numerous negative-strand RNA viruses, including filoviruses and pneumoviruses, suggests that the observations made here may be of general interest in understanding the molecular basis of mononegaviral infection.
MATERIALS AND METHODS
Protein expression, purification, and mutagenesis
All P and N constructs were cloned into pET41c(+) between the Nde I and Xho I cleavage sites, where the Xho I site was ligated with a cleaved Sal I site of the insert, yielding a construct with a C-terminal 8His-tag (5). All constructs were expressed in E. coli Rosetta TM(lDE3)/pRARE (Novagen) overnight at 20°C after induction at an optical density of 0.6 with 1 mM isopropyl-β-d-thiogalactopyranoside. N525 was cotransformed with P1–507 and purified in complex. Cells were lysed by sonication and subjected to standard Ni purification in 20 mM tris (pH 8.0) and 150 mM NaCl or 500 mM NaCl for XD containing constructs. The protein was eluted with 20 mM tris (pH 8), 150/500 mM NaCl, and 500 mM imidazole and then concentrated and subjected to size exclusion chromatography (SEC, Superdex 75/200) in 50 mM Na-phosphate (pH 6.0), 150/500 mM NaCl, and 2 mM dithiothreitol (DTT) buffer (NMR buffer). The P50N525, P50N405, P300N525, and P300N405 were produced as described previously (13, 14). Mutation of S491L in N and of 409VLK411→AAA in P were inserted by site-directed mutagenesis, and P50N525 (S491L), N460–525 (S491L), P304–507 (409VLK411→AAA), and P377–507 (409VLK411→AAA) were produced and purified in the same way as wild-type proteins.
Expression of unlabeled protein was performed in LB medium. Proteins labeled for NMR (15N and 13C) were expressed under the same conditions in M9 minimal medium. Cysteine mutations for fluorescence labeling were performed by site-directed mutagenesis into the constructs where previously all native cysteines were exchanged to alanines. Mutant proteins were expressed and purified in the same way as wild type and concentrated to around 100 μM. The proteins were then labeled using maleimide chemistry (43). Briefly, the protein was incubated overnight with 10 mM DTT, then dialyzed against 50 mM Na-phosphate buffer (pH 7.0) and 150/500 mM NaCl in two steps, and mixed with 5× excess of maleimide dye (fluorescein or Alexa Fluor 594) solubilized in dimethyl sulfoxide. After 2 hours of incubation at room temperature, the labeling reaction was stopped by adding 5 mM DTT, and proteins were separated from the excess dye by SEC (Superdex 75/200) in NMR buffer. SDS–polyacrylamide gel electrophoresis (PAGE) gels are shown in fig. S1.
RNA
All RNAs were purchased from Integrated DNA Technologies (Leuven, Belgium), and all were terminated with OH at both 3′ and 5′ or with fluorescein amidite at the 3′ end.
Droplet assembly and turbidity assay
P and N constructs were first mixed and then diluted with buffer; thus, the final NaCl concentration was 150 mM (unless different NaCl concentration is stated). Samples were incubated at room temperature for 5 min, and absorbance at 395 nm was read on a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). An absorbance value higher than 0.1 was interpreted as a presence of phase separation in the system.
Microscopy imaging
Samples were imaged using either DIC or epifluorescence on an Olympus IX81 inverted microscope. Fluorescence signal was acquired using a 485(20)-nm (fluorescein) or a 560(25)-nm (Alexa Fluor 594) excitation filter combined with a 525(30)-nm or a 607(36)-nm emission filter, respectively. Excitation light (Metal-Halide X-Cite 120) and emission wavelengths are split using a 4X4M-B quadriband dichroic mirror (Semrock). Oil immersion objectives 60× [Olympus PlanAPON60, 1.42 numerical aperture (NA)] or 100× (Olympus UPLFLN, 1.3 NA) were used for imaging, and a Hamamatsu Orca Flash4.0-V2 sCMOS (scientific Complementary Metal-Oxyde Semiconductor) 2048 × 2048 camera was used for detection. The acquisition and data analysis were performed with Volocity (Quorum Technologies) software.
Fluorescence recovery after photobleaching
FRAP was measured using an Olympus IX81 spinning-disk confocal microscope equipped with a Yokogawa CSUX1 spinning disk head and using diode-pumped solid-state lasers 488 nm (50 mW Coherent Sapphire) and 561 nm (100 mW Cobolt JIVE4). Emission is filtered through a 520(28)-nm filter for fluorescein. Acquisition is performed with an iXon Ultra EMCCD (Electron Multiplying Charge-Coupled Device) (Andor) 512 × 512 pixel camera. FRAP was restricted to a region of interest, defined graphically, with a galvanometric FRAP/PA device (Andor) using the same lasers used for imaging with AOTF (Acousto-Optic Tunable Filter) 15% and with a dwell time of 50 ms for 10 repeats before bleaching and with a 2-s interval for 40 repeats after. The laser power and the dwell time during bleaching were optimized to bleach 50% of the initial fluorescence intensity. Images were analyzed using FIJI and python plugin ImageFRAP (https://imagej.net/Analyze_FRAP_movies_with_a_Jython_script); the recovery was fitted with a single exponent.
Fluorescence anisotropy
Fluorescence anisotropy decays were measured using a Mini-Tau fluorescence lifetime spectrometer (Edinburgh Instruments, UK) equipped with a 450-nm picosecond pulsed diode laser and a TCC2 electronics module for time-correlated single photon counting. Photons were detected using a high-speed photon multiplier tube (Hamamatsu H10720-01). The sample was excited with vertically polarized light, and parallel and perpendicular polarized fluorescence light was detected during 120 s sequentially. Histograms of photon arrival times were generated from every measurement, and anisotropy decays were calculated according to r(t) = (Ipar(t) − GIper(t))/(Ipar(t) + 2GIper(t)), with Ipar/Iper being the intensities of parallel/perpendicular polarized light per time unit and G being the correcting factor for detection differences between the two detection pathways. The G factor was measured for each buffer condition using the free dye.
Isothermal titration calorimetry
Isothermal titration calorimetry was measured using iMicroCal iTC200 at 25°C. The titration experiment was performed by adding 2 μl of aliquots of 500 μM PXD into the microcalorimeter cell filled with 50 μM NTAIL. PXD was titrated through 20 injections with 180-s intervals in between; the reaction mixture was continuously stirred at 750 rpm. The titration curve was fitted to the experimental data using the Origin version 7.0 software from MicroCal.
NMR spectroscopy
All experiments were acquired in NMR buffer at 25°C. The spectral assignment of P50N525 at pH 6.0 was verified on the basis of published assignment at pH 7.0 (13) using 13C, 15N-labeled samples using a set of BEST-TROSY triple resonance experiments.
The spectral assignments of 13C- and 15N-labeled P377–457 and P457–507 constructs were obtained using sets of triple resonance experiments correlating Ca, Cb, and CO resonances at a 1H frequency of 600 MHz. NMRPipe (44) was used to process the spectra, and automatic assignment was performed with the program MARS and manually verified (45). Secondary chemical shifts were calculated using the random coil values from refDB (46).
Ensemble analysis of PLOOP
The genetic algorithm ASTEROIDS selected representative ensembles [fitting 1H, 15N, and 13C(Ca, Cb, and CO) backbone chemical shifts]. A 10,000-member ensemble of PLOOP was generated using the statistical coil model Flexible Meccano (47), and 200-conformer ensembles matching experimental backbone chemical shifts were selected from this ensemble using the ASTEROIDS genetic algorithm as previously described (48, 49).
Calculation of apparent Kd values from NMR experiments for the interaction between N and PLOOP/PXD
15N R1 relaxation rates were obtained by sampling the decay of magnetization (seven delays between 0 and 1.71 s). R1ρ relaxation rates were measured using seven delay times between 0.001 and 0.24 s. The spin-lock field was 1500 Hz, and R2 was calculated from R1 and R1ρ, considering the resonance offset. The residue-specific apparent Kd values for P377–457 and N were calculated from 15N relaxation as described elsewhere (50). P377–507 concentration remained constant (100 μM), and N concentration varied from 0 to 140 μM. The rotational correlation time of the complex was assumed to be equal to τc of P50N525 alone and was estimated from its molecular weight.
15N relaxation dispersion was measured at a P457–507 concentration of 100 μM in the presence of 70 and 30 μM unlabeled NTAIL at 700 and 850 MHz and using 14 points at CPMG frequencies between 31 and 1000 Hz with a constant-time relaxation of 32 ms (51). Data for nine residues and two fields were fitted simultaneously using ChemEx (https://github.com/gbouvignies/chemex) and a two-state exchange model. The population of P457–507 bound to N460–525 (pB) and the exchange rate (kex) were obtained from the fit, and the Kd was calculated assuming a 1:1 binding stoichiometry. The error of the calculated Kd value was estimated by error propagation from the fitting error of pB and assuming a 5% error in concentration determination of both proteins.
NMR measurements of phase-separating samples
15N-labeled P50N525 and P304–507 were premixed at different ratios and diluted in buffer so that the final NaCl concentration was 150 mM and then loaded in a 3-mm NMR tube. 1H-15N HSQCs and R1ρ experiments were measured as described for non–phase-separating samples. Pulsed field gradient NMR diffusion experiments were measured using 15N-edited stimulated-echo diffusion experiments using a 300-ms diffusion delay at 16 to 20 gradient strengths ranging linearly from 0 to 0.55 T m−1 (52). 1H-15N HSQCs used for the development of the kinetic model were measured using both P50N525 and P304–507 15N-labeled samples at a constant P304–507 concentration of 75 μM and P50N525 5, 10, 20, 50, and 100%. The concentration of P50N525 and P304–507 in the dilute and condensed phase was estimated on the basis of intensities of 23 for P50N525 and 9 for P304–507 nonoverlapping and noninteracting peaks, which were summed and normalized by the intensities of the peaks in the spectra of the individual proteins. The amount of each protein in droplets was then estimated by subtraction. This allows us to estimate the concentration of droplet-associated N and P in the sensitive NMR volume (but not in droplets as we do not know the volume of droplets). We can then estimate the ratio of N and P associated to droplets as a function of concentration of N.
Nucleocapsid assembly by NMR
Kinetics of P50N525 (alone or in the presence of P304–507 or P377–507) nucleocapsid assembly were followed using NMR spectroscopy using the same procedure as previously described (13). Time resolution was approximately 8 min. The assembly process was initiated by adding six-nucleotide RNA (OH-A6-OH), and its time trace was followed by measuring intensities of resonances from P1–50 on SOFAST HMQC spectra. Assembly rates were obtained by averaging intensities of 18 peaks and fitting them with a biexponential function; mean assembly rates were calculated as follows: k1*a1 + k2*a2, where k1 and k2 are rates, a1 and a2 are their amplitudes.
Negative-stain electron microscopy
The nucleocapsid-like particles used for negative-stain electron microscopy were obtained by adding 50 μM RNA (HO-A6-OH) to droplets formed by mixing 20 μM P50N525 and 80 μM P304–507 in a final volume of 20 μl. Samples were incubated for 1 hour at 37°C, then applied to the clean side of carbon on mica (carbon/mica interface), and stained with 2% sodium silicotungstate (pH 7.0). Micrographs were taken with a T12 FEI microscope at 120 kV and a magnification of 30,000.
Supplementary Material
Acknowledgments
IBS acknowledges integration into the Interdisciplinary Research Institute of Grenoble (CEA). Funding: This work was supported by the European Research Council Advanced Grant DynamicAssemblies to M.B., GRAL (ANR-10-LABX-49-01), Finovi, and the Fondation Recherche Medicale (FRM) (Equipe DEQ20170336754). The work used the platforms of the Grenoble Instruct-ERIC Centre (ISBG; UMS 3518 CNRS-CEA-UJA-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology. S.M. acknowledges funding from an EMBO long-term fellowship (ALTF 468-2014) and European Commission (EMBOCOFUND2012, GA-2012-600394) via Marie Curie Action. The electron microscope facility was supported by the Rhône-Alpes Region, FRM, the fonds FEDER, the Centre National de la Recherche Scientifique (CNRS), the CEA, the University of Grenoble, EMBL, and the GIS-Infrastructures en Biologie Sante et Agronomie. Author contributions: S.G. carried out experiments and analyzed the data and wrote the manuscript. S.M. supervised experimental and analytical aspects. J.-P.K. assisted in the imaging experiments. D.M. prepared proteins and participated in imaging experiments. M.R.J. supervised experimental and analytical aspects. N.S. provided expertise in analytical and experimental aspects. R.W.H.R. and M.B. conceptualized and supervised the project and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz7095/DC1
REFERENCES AND NOTES
- 1.Paules C. I., Marston H. D., Fauci A. S., Measles in 2019—Going backward. N. Engl. J. Med. 380, 2185–2187 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Curran J., Kolakofsky D., Replication of paramyxoviruses. Adv. Virus Res. 54, 403–422 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Cox R., Plemper R. K., The paramyxovirus polymerase complex as a target for next-generation anti-paramyxovirus therapeutics. Front. Microbiol. 6, 459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Longhi S., Receveur-Bréchot V., Karlin D., Johansson K., Darbon H., Bhella D., Yeo R., Finet S., Canard B., The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 278, 18638–18648 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Milles S., Jensen M. R., Lazert C., Guseva S., Ivashchenko S., Communie G., Maurin D., Gerlier D., Ruigrok R. W. H., Blackledge M., An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci. Adv. 4, eaat7778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Communie G., Crépin T., Maurin D., Jensen M. R., Blackledge M., Ruigrok R. W. H., Structure of the tetramerization domain of measles virus phosphoprotein. J. Virol. 87, 7166–7169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kingston R. L., Hamel D. J., Gay L. S., Dahlquist F. W., Matthews B. W., Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. U.S.A. 101, 8301–8306 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guryanov S. G., Liljeroos L., Kasaragod P., Kajander T., Butcher S. J., Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J. Virol. 90, 2849–2857 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutsche I., Desfosses A., Effantin G., Ling W. L., Haupt M., Ruigrok R. W. H., Sachse C., Schoehn G., Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 348, 704–707 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Gely S., Lowry D. F., Bernard C., Jensen M. R., Blackledge M., Costanzo S., Bourhis J.-M., Darbon H., Daughdrill G., Longhi S., Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J. Mol. Recognit. 23, 435–447 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Bloyet L.-M., Brunel J., Dosnon M., Hamon V., Erales J., Gruet A., Lazert C., Bignon C., Roche P., Longhi S., Gerlier D., Modulation of re-initiation of measles virus transcription at intergenic regions by PXD to NTAIL binding strength. PLOS Pathog. 12, e1006058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du Pont V., Jiang Y., Plemper R. K., Bipartite interface of the measles virus phosphoprotein X domain with the large polymerase protein regulates viral polymerase dynamics. PLOS Pathog. 15, e1007995 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milles S., Jensen M. R., Communie G., Maurin D., Schoehn G., Ruigrok R. W. H., Blackledge M., Self-assembly of measles virus nucleocapsid-like particles: Kinetics and RNA sequence dependence. Angew. Chem. Int. Ed. Engl. 55, 9356–9360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desfosses A., Milles S., Jensen M. R., Guseva S., Colletier J.-P., Maurin D., Schoehn G., Gutsche I., Ruigrok R. W. H., Blackledge M., Assembly and cryo-EM structures of RNA-specific measles virus nucleocapsids provide mechanistic insight into paramyxoviral replication. Proc. Natl. Acad. Sci. U.S.A. 116, 4256–4264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin Y., Brangwynne C. P., Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nott T. J., Craggs T. D., Baldwin A. J., Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat. Chem. 8, 569–575 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Wei M.-T., Elbaum-Garfinkle S., Holehouse A. S., Chen C. C.-H., Feric M., Arnold C. B., Priestley R. D., Pappu R. V., Brangwynne C. P., Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat. Chem. 9, 1118–1125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novoa R. R., Calderita G., Arranz R., Fontana J., Granzow H., Risco C., Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 97, 147–172 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Netherton C., Moffat K., Brooks E., Wileman T., A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Adv. Virus Res. 70, 101–182 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lahaye X., Vidy A., Pomier C., Obiang L., Harper F., Gaudin Y., Blondel D., Functional characterization of Negri Bodies (NBs) in rabies virus-infected cells: Evidence that NBs are sites of viral transcription and replication. J. Virol. 83, 7948–7958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinrich B. S., Maliga Z., Stein D. A., Hyman A. A., Whelan S. P. J., Phase transitions drive the formation of vesicular stomatitis virus replication compartments. MBio 9, e02290-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rincheval V., Lelek M., Gault E., Bouillier C., Sitterlin D., Blouquit-Laye S., Galloux M., Zimmer C., Eleouet J.-F., Rameix-Welti M.-A., Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 8, 563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang S., Chen L., Zhang G., Yan Q., Yang X., Ding B., Tang Q., Sun S., Hu Z., Chen M., An amino acid of human parainfluenza virus type 3 nucleoprotein is critical for template function and cytoplasmic inclusion body formation. J. Virol. 87, 12457–12470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Precious B., Young D. F., Bermingham A., Fearns R., Ryan M., Randall R. E., Inducible expression of the P, V, and NP genes of the paramyxovirus simian virus 5 in cell lines and an examination of NP-P and NP-V interactions. J. Virol. 69, 8001–8010 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derdowski A., Peters T. R., Glover N., Qian R., Utley T. J., Burnett A., Williams J. V., Spearman P., Crowe J. E. Jr., Human metapneumovirus nucleoprotein and phosphoprotein interact and provide the minimal requirements for inclusion body formation. J. Gen. Virol. 89, 2698–2708 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omi-Furutani M., Yoneda M., Fujita K., Ikeda F., Kai C., Novel phosphoprotein-interacting region in Nipah virus nucleocapsid protein and its involvement in viral replication. J. Virol. 84, 9793–9799 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma D., George C. X., Nomburg J. L., Pfaller C. K., Cattaneo R., Samuel C. E., Upon infection, cellular WD repeat-containing protein 5 (WDR5) localizes to cytoplasmic inclusion bodies and enhances measles virus replication. J. Virol. 92, e01726-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spehner D., Drillien R., Howley P. M., The assembly of the measles virus nucleoprotein into nucleocapsid-like particles is modulated by the phosphoprotein. Virology 232, 260–268 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Nakatsu Y., Takeda M., Ohno S., Shirogane Y., Iwasaki M., Yanagi Y., Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 82, 8296–8306 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nikolic J., Le Bars R., Lama Z., Scrima N., Lagaudrière-Gesbert C., Gaudin Y., Blondel D., Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 8, 58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y., Su J. M., Samuel C. E., Ma D., Measles virus forms inclusion bodies with properties of liquid organelles. J. Virol. 93, e00948-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boke E., Ruer M., Wühr M., Coughlin M., Lemaitre R., Gygi S. P., Alberti S., Drechsel D., Hyman A. A., Mitchison T. J., Amyloid-like self-assembly of a cellular compartment. Cell 166, 637–650 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen M. R., Communie G., Ribeiro E. A. Jr., Martinez N., Desfosses A., Salmon L., Mollica L., Gabel F., Jamin M., Longhi S., Ruigrok R. W. H., Blackledge M., Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. U.S.A. 108, 9839–9844 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., Choi J.-M., Holehouse A. S., Lee H. O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D., Poser I., Pappu R. V., Alberti S., Hyman A. A., A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim T. H., Tsang B., Vernon R. M., Sonenberg N., Kay L. E., Forman-Kay J. D., Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Adamski W., Salvi N., Maurin D., Magnat J., Milles S., Jensen M. R., Abyzov A., Moreau C. J., Blackledge M., A unified description of intrinsically disordered protein dynamics under physiological conditions using NMR spectroscopy. J. Am. Chem. Soc. 141, 17817–17829 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Longhi S., Bloyet L.-M., Gianni S., Gerlier D., How order and disorder within paramyxoviral nucleoproteins and phosphoproteins orchestrate the molecular interplay of transcription and replication. Cell. Mol. Life Sci. 74, 3091–3118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunel J., Chopy D., Dosnon M., Bloyet L.-M., Devaux P., Urzua E., Cattaneo R., Longhi S., Gerlier D., Sequence of events in measles virus replication: Role of phosphoprotein-nucleocapsid interactions. J. Virol. 88, 10851–10863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posey A. E., Holehouse A. S., Pappu R. V., Phase separation of intrinsically disordered proteins. Methods Enzymol. 611, 1–30 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Sugai A., Sato H., Yoneda M., Kai C., Phosphorylation of measles virus phosphoprotein at S86 and/or S151 downregulates viral transcriptional activity. FEBS Lett. 586, 3900–3907 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Sugai A., Sato H., Yoneda M., Kai C., Phosphorylation of measles virus nucleoprotein affects viral growth by changing gene expression and genomic RNA stability. J. Virol. 87, 11684–11692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemke E. A., Site-specific labeling of proteins for single-molecule FRET measurements using genetically encoded ketone functionalities. Methods Mol. Biol. 751, 3–15 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Delaglio F., Grzesiek S., Vuister G., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX Pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Jung Y.-S., Zweckstetter M., Mars—Robust automatic backbone assignment of proteins. J. Biomol. NMR 30, 11–23 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Zhang H., Neal S., Wishart D. S., RefDB: A database of uniformly referenced protein chemical shifts. J. Biomol. NMR 25, 173–195 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Ozenne V., Bauer F., Salmon L., Huang J.-r., Jensen M. R., Segard S., Bernadó P., Charavay C., Blackledge M., Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 28, 1463–1470 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Nodet G., Salmon L., Ozenne V., Meier S., Jensen M. R., Blackledge M., Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J. Am. Chem. Soc. 131, 17908–17918 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Jensen M. R., Salmon L., Nodet G., Blackledge M., Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J. Am. Chem. Soc. 132, 1270–1272 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Milles S., Mercadante D., Aramburu I. V., Jensen M. R., Banterle N., Koehler C., Tyagi S., Clarke J., Shammas S. L., Blackledge M., Gräter F., Lemke E. A., Plasticity of an ultrafast interaction between nucleoporins and nuclear transport receptors. Cell 163, 734–745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansen D. F., Vallurupalli P., Kay L. E., An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J. Phys. Chem. B 112, 5898–5904 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Waudby C. A., Mantle M. D., Cabrita L. D., Gladden L. F., Dobson C. M., Christodoulou J., Rapid distinction of intracellular and extracellular proteins using NMR diffusion measurements. J. Am. Chem. Soc. 134, 11312–11315 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaaz7095/DC1