

The cardiac IKs (Kv7.1-KCNE1) channel assembles at ER-PM junctions before translocated to the plasma membrane.
Abstract
The potassium channel Kv7.1 associates with the KCNE1 regulatory subunit to trigger cardiac IKs currents. Although the Kv7.1/KCNE1 complex has received much attention, the subcellular compartment hosting the assembly is the subject of ongoing debate. Evidence suggests that the complex forms either earlier in the endoplasmic reticulum or directly at the plasma membrane. Kv7.1 and KCNE1 mutations, responsible for long QT syndromes, impair association and traffic, thereby altering IKs currents. We found that Kv7.1 and KCNE1 do not assemble in the first stages of their biogenesis. Data support an unconventional secretory pathway for Kv7.1-KCNE1 that bypasses Golgi. This route targets channels to endoplasmic reticulum–plasma membrane junctions, where Kv7.1-KCNE1 assemble. This mechanism helps to resolve the ongoing controversy about the subcellular compartment hosting the association. Our results also provide new insights into IKs channel localization at endoplasmic reticulum–plasma membrane junctions, highlighting an alternative anterograde trafficking mechanism for oligomeric ion channels.
INTRODUCTION
The voltage-gated potassium type 7.1 (Kv7.1) channel is the first of five members of the Kv7 subfamily and is included in the Kv superfamily (1). Although Kv7.1 is widely distributed throughout the human body, it plays a major role in cardiac tissue (2). In the heart, Kv7.1 associates with the regulatory β subunit KCNE1, giving rise to the slow delayed rectifier potassium (IKs) current, which is vital for proper repolarization of cardiac action potentials (3, 4). Cardiac channelopathies are responsible for about half of sudden cardiac death cases, and their current worldwide prevalence ranges from 1:2000 to 1:3000 in the general population. The most prevalent disorder within this group is congenital long QT syndrome. Among the 17 different types identified within this disease, Kv7.1 is associated with more than 30% of the described genetic variants, clearly underscoring its physiological importance (5).
Although Kv7.1-KCNE1 complexes are some of the most studied molecules in the ion channel field, many biological features are still elusive. For instance, the precise mechanism of the slow activation induced by KCNE1 (6), the stoichiometry of the complex (7), and the subcellular location where Kv7.1-KCNE1 complexes assemble are subjects of intense debate. Concerning this last issue, opposing evidence strongly supports two different hypotheses: (i) Kv7.1 and KCNE1 traffic separately to the plasma membrane (PM), and dynamic complexes are assembled directly at the cell surface (3, 8–10) or (ii) the association takes place early during the secretory pathway (11–17). The anterograde journey of Kv7.1 and KCNE1 subunits from the sites of their biosynthesis to the PM starts at the endoplasmic reticulum (ER). Next, the subunits should follow the classic secretory pathway, including exit from the ER and travel through the ER-to-Golgi intermediate compartment, the Golgi apparatus, and the trans-Golgi network (TGN) (18, 19). However, recent evidence suggests that some membrane proteins, including some ion channels, can reach the PM unconventionally (20–23). Deciphering Kv7.1- and KCNE1-specific secretory pathways may help to uncover the precise subcellular compartment for Kv7.1-KCNE1 complex assembly, elucidating to the pathological mechanisms of deficient Kv7.1-KCNE1 association and trafficking observed in many cardiac channelopathies (6).
Cardiomyocytes exhibit organized structures that facilitate excitation-contraction coupling. T-tubules are narrow invaginations of the sarcolemma facing adjacent sarcoplasmic reticulum that couple ion channel–mediated electrical activity with the release of calcium from intracellular stores, triggering contraction. T-tubules are the best-characterized ER-PM junctions. ER-PM junctions are ubiquitous cellular structures that play crucial roles not only in Ca2+ signaling but also in membrane trafficking (24, 25). Some cardiac Kv channels, including Kv7.1, are localized in these subcellular domains, but the targeting mechanism is still poorly understood (26) .
In the present study, we investigated Kv7.1 and KCNE1 secretory pathways. Unlike KCNE1, Kv7.1 is unconventionally routed to the cell surface, bypassing the Golgi. Upon association, Kv7.1 rerouted KCNE1 from classic anterograde transport. This route drives Kv7.1 and KCNE1 to ER-PM junctions, where the complexes assemble. This previously unidentified mechanism for Kv7.1-KCNE1 complex trafficking and assembly contributes to resolving the ongoing controversy about the subcellular compartment where the interaction takes place. We also provide new evidence for the local generation of the IKs current within ER-PM junctions at T-tubules.
RESULTS
Kv7.1/KCNE1 association recapitulates IKs currents and forms stable complexes at cardiac T-tubules
Cardiac IKs currents are crucial for heart physiology, but many questions remain regarding the localization and formation of functional Kv7.1/KCNE1 complexes. Therefore, we first investigated the colocalization and interaction of the subunits in rat cardiomyocytes. We used direct stochastic optical reconstruction microscopy (dSTORM) to locate Kv7.1 and KCNE1 in cardiomyocytes (Fig. 1). Fluorophore-coupled antibodies stained ryanodine receptor (RyR), which identified ER-PM junction structures where Kv7.1 localized (Fig. 1, A to C). Although Kv7.1 and KCNE1 showed notable whole-cell colocalization (Fig. 1, D to F), a detailed analysis demonstrated that colocalization was higher in T-tubules and PM than in other cell areas (Fig. 1, G and H). A distance of 0 nm determined by dSTORM suggests physical association (27). Coimmunoprecipitation of both proteins in protein extracts from rat hearts further confirmed this association (Fig. 1I). Reverse coimmunoprecipitation confirmed the interaction. Our data indicated that Kv7.1 and KCNE1 associate in the T-tubules of rat cardiomyocytes. This association recapitulated IKs currents when the subunits were steadily expressed in human embryonic kidney–293 (HEK-293) cells (Fig. 1, J and K). Heterologous expression of Kv7.1–cyan fluorescent protein (CFP) and KCNE1–yellow fluorescent protein (YFP) highlighted further subcellular localization features (Fig. 1, L to P). While KCNE1 was mostly present at the cell surface, Kv7.1 exhibited notable intracellular retention. As previously demonstrated in cardiomyocytes (28), the colocalization of both proteins was threefold higher at the PM than the rest of the cell (Fig. 1P), resembling the distribution within T-tubules in the heart.
Fig. 1. Kv7.1 and KCNE1 recapitulate cardiac IKs currents and associate in T-tubules in cardiomyocytes.
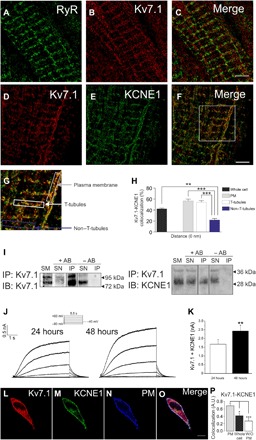
(A to H) dSTORM in rat ventricular cardiomyocytes. Fluorophore-coupled antibodies were used to stain native proteins. (A) Staining for RyR in cardiac T-tubules. (B) Kv7.1 (red) and (C) merge image. (D to F) Staining for Kv7.1 and KCNE1 in cardiac T-tubules. (D) Kv7.1, red; (E) KCNE1, green; (F) colocalization of Kv7.1 and KCNE1, yellow. Scale bars, 5 μm. (G) Magnified area from the square in (F). The insets delineate the representative regions of interest analyzed in (H). (H) Kv7.1-KCNE1 colocalization at a 0-nm distance determined by dSTORM analysis. Color code in (G) and (H): gray, PM; white, T-tubules; blue, nontubule area excluding the previous structures. The black bar represents whole-cell colocalization. Statistical analysis determined a P < 0.001 by analysis of variance (ANOVA) with a Tukey post hoc test. **P < 0.01 and ***P < 0.001 (insets, n > 10 from five independent cells). (I) Kv7.1 coimmunoprecipitates with KCNE1 in rat cardiac samples. Total crude protein extracts from rat ventricular cardiomyocytes were immunoprecipitated (IP) with anti-Kv7.1 antibodies and immunoblotted (IB) against Kv7.1 (left) and KCNE1 (right). SM, starting material; SN, supernatant; +AB, presence of antibodies; −AB, absence of antibodies during the immunoprecipitation. (J) Coexpression of Kv7.1 and KCNE1 in COS-7 cells recapitulates IKs currents. COS-7 cells were cotransfected with Kv7.1 and KCNE1 for 24 and 48 hours. The cells were held at −40 mV, and 5.5-s depolarizing pulses was applied from −80 to +60 mV. (K) Maximal intensity of currents at +60 mV. White bar, cells transfected for 24 hours; black bar, currents recorded after 48 hours of expression. The values are the means ± SEM of six to eight cells. **P < 0.01 versus 24 hours, Student’s t test. (L to P) Expression of Kv7.1 and KCNE1 in HEK-293 cells. Cells were cotransfected with Kv7.1-CFP and KCNE1-YFP for 48 hours, and representative confocal images were acquired. (L) Kv7.1, red; (M) KCNE1, green; (N) PM marker, blue; (O) merged signals. White represents triple colocalization. Scale bar, 10 μm. (P) Cellular colocalization of Kv7.1-CFP and KCNE1-YFP. The PM staining was used to create a mask to analyze the Kv7.1-KCNE1 colocalization. Whole cell, the entire cell was analyzed. Without (W/O) PM, whole-cell colocalization values were subtracted from those at the PM. The values are the means ± SEM of >25 cells. *P < 0.05 and ***P < 0.01 versus PM, Student’s t test. A.U., arbitrary units.
Kv7.1-KCNE1 complexes are assembled at the PM
Compared to entire cells, cell unroofing preparations (CUPs) enable better discrimination between cell surface and intracellular compartments (fig. S1). CUPs were obtained through hypotonic shock plus an intense burst (fig. S1, E to H; see Materials and Methods for details). Alternatively, by applying gentle rather than intense bursts, we obtained modified CUPs (mCUPs), in which part of the ER compartment remained attached to the PM through ER-PM junctions (fig. S1, I to L). Thus, by using a membrane-localized CFP-YFP tandem construct (Rho-pYC) and ER-DsRed, an ER marker, we discriminated between the cellular compartments (fig. S1M).
The colocalization between Kv7.1 and KCNE1 at the PM was higher in CUPs (89 ± 1%) than in entire cells (64 ± 1%;Fig. 2, A to G). Physical interaction was assessed by Förster resonance energy transfer (FRET) analysis and protein coimmunoprecipitation. Kv7.1-CFP/Kv7.1-YFP and Kv7.1-CFP/Kv1.5-YFP were used as positive and negative controls, respectively (29). Kv7.1-YFP and KCNE1-CFP showed a FRET efficiency comparable to that of the negative control in entire cell preparations (0.04 ± 0.02 and 0.03 ± 0.00 for Kv7.1-KCNE1 and Kv7.1-Kv1.5, respectively). In contrast, Kv7.1 and KCNE1 showed significant positive energy transfer efficiency (EF) in CUPs (0.15 ± 0.01%, P < 0.001 versus entire cell) (Fig. 2, H to R). Coimmunoprecipitation assays further supported the FRET results. Stronger coimmunoprecipitation of Kv7.1 and KCNE1 was detected in PM-enriched protein samples purified from CUPs than in samples from whole cells (Fig. 2, S and T). Reverse coimmunoprecipitation confirmed the interaction. Therefore, while the Kv7.1-KCNE1 complex is detected in entire cells, the interaction is mostly localized at the PM.
Fig. 2. Kv7.1 and KCNE1 mostly interact at the PM.
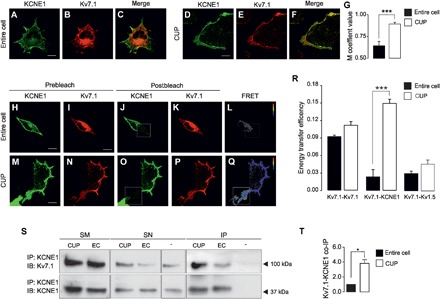
(A to F) Confocal images of KCNE1-YFP and Kv7.1-CFP in entire cells (A to C) and CUPs (D to F). Cells were cotransfected with KCNE1-YFP (A and D, in green) and Kv7.1-CFP (B and E, in red). (C and F) Merged image showing colocalization in yellow. (G) Colocalization analysis by Manders’ (M) coefficient in entire cells (black bar) or CUPs (white bar). The values show the means ± SEM. ***P < 0.001 CUP versus entire cell (n = 9 to 15, Student’s t test). (H to Q) Representative results of FRET acceptor photobleaching experiments on entire cells (H to L) and CUPs (M to Q). The prebleaching images (H to I and M and N) show the expression of KCNE1-YFP (H and M) and Kv7.1-CFP (I and N). After bleaching the acceptor molecule (KCNE1-YFP), postbleaching images were taken (J and K and O and P). The bleached area is highlighted with a white square. (L and Q) FRET ratio images from the previous panels. The calibration bar indicates the FRET ratio ranging from 0.8 (blue) to 1.4 (red). Scale bars, 10 μm. (R) Energy transfer efficiencies. The values represent the means ± SEM of the FRET measured in entire cells (black) or in CUPs (white). ***P < 0.001 CUP versus entire cell (n = 31 to 36, Student’s t test). (S) Coimmunoprecipitation of Kv7.1 with KCNE1 using anti-KCNE1 antibodies (IP: KCNE1) in CUPs and whole-cell lysates entire cells (EC; entire cells) from cultured cells. Immunoblotting was performed with antibodies against Kv7.1 (IB: Kv7.1, 100 kDa, arrow) and KCNE1 (IB: KCNE1, 37 kDa, arrow). −, immunoprecipitation in absence of anti-KCNE1 antibodies. (T) Coimmunoprecipitation (co-IP) analysis of Kv7.1 with KCNE1 in entire cells (black bar) or CUPs (white bar). The values show the means ± SEM. *P < 0.05 CUP versus entire cell (n = 3, Student’s t test).
Kv7.1-KCNE1 complexes are not assembled in early biogenesis
The ongoing debate about the subcellular location of Kv7.1-KCNE1 complex assembly has raised two possible alternative mechanisms: (i) Kv7.1 and KCNE1 traffic independently to the PM, where they form transient complexes by lateral diffusion (3, 8–10), and (ii) the assembly takes place in the ER, and the interaction is required for the efficient trafficking of KCNE1 to the PM (11–17). In this context, we analyzed the subcellular compartment hosting Kv7.1-KCNE1 association. We first studied Kv7.1 and KCNE1 colocalization with ER and PM markers (Fig. 3 and fig. S2). Both Kv7.1 and KCNE1, when expressed alone, were highly retained in the ER. However, upon coexpression of Kv7.1, KCNE1 targeting to the PM significantly increased with a concomitant reduction in ER retention. The Kv7.1 distribution remained unaltered (Fig. 3, A to G, and fig. S2, Aa to Ac, Ea to Ec, and Ia to Id). Biotin assays further supported that KCNE1 expression at the PM increased when KCNE1 was coexpressed with Kv7.1 (Fig. 3, H and I). As expected, HEK-293 and COS-7 cells showed similar expression patterns (fig. S3, A to K). These results indicated that association with Kv7.1 promoted KCNE1 trafficking to the cell surface and suggested that the Kv7.1-KCNE1 interaction might take place within the secretory pathway.
Fig. 3. Kv7.1-KCNE1 assembly within the secretory pathway.
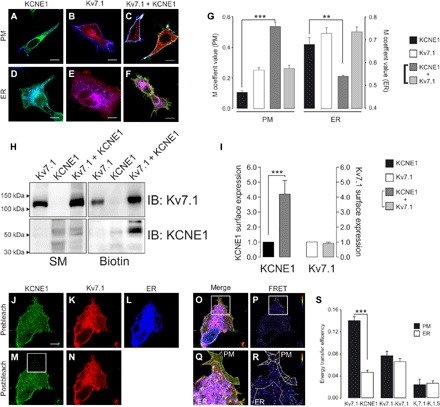
(A to F) Representative images of KCNE1-YFP (A, green), Kv7.1-CFP (B, red), and KCNE1-YFP + Kv7.1-CFP (C, green and red) coexpressed with Akt-PH-pDsRed (A to C, PM marker in blue) and ER-DsRed (D to F, blue) in HEK-293 cells. PH, pleckstrin homology. (A and D) Cyan shows colocalization between green and blue. (B and E) Magenta shows colocalization between red and blue. (C and F) Yellow shows partial colocalization between green and red. White shows triple colocalization between green, red, and blue. Scale bar, 10 μm. (G) M coefficient analysis of KCNE1 (black), Kv7.1 (white), and Kv7.1 + KCNE1 (light and dark gray) overlapping the ER and membrane (PM) markers. The bars represent the means ± SEM. **P < 0.01 and ***P < 0.001 for KCNE1 in the absence of Kv7.1 versus KCNE1 in the presence of Kv7.1 (n = 19 to 40, Student’s t test). (H) Biotin assay of KCNE1 and Kv7.1 cell surface expression. HEK-293 cells were transfected with Kv7.1-CFP, KCNE1-YFP, and Kv7.1-CFP + KCNE1-YFP. After transfection, the cells were labeled with biotin and processed as described in Materials and Methods. Western blot analysis was performed with anti-Kv7.1 (IB: Kv7.1) and anti-KCNE1 (IB: KCNE1) antibodies. Biotin, biotinylated pull down proteins. (I) Densitometric analysis of biotinylated Kv7.1 and KCNE1. KCNE1 (black), Kv7.1 (white), and Kv7.1 + KCNE1 (light and dark gray, respectively). The bars represent the means ± SEM. ***P < 0.001 for KCNE1 in the absence of Kv7.1 versus KCNE1 in the presence of Kv7.1 (n = 3, Student’s t test). (J to R) Representative results of Kv7.1-CFP and KCNE1-YFP FRET experiments in ER-containing CUPs (mCUPs). (J to L) Prebleaching images of KCNE1-YFP (J), Kv7.1-CFP (K), and ER-DsRed (L). (M and N) Postbleaching images of KCNE1-YFP (M) and Kv7.1-CFP (N). The bleached area is highlighted with a white square. (O) Merged prebleaching image (J to L). (Q) Magnified view of the bleached area in the merged image that enables the PM to be distinguished from the ER. (P and R) FRET ratio images. The calibration bar ranges from 0.8 (blue) to 1.4 (red). Scale bar, 10 μm. (S) FRET was measured independently in the PM (black bars) and the ER (white bars) within the same cell. The values indicate the means ± SEM. ***P < 0.001 for Kv7.1-KCNE1 in the PM versus the ER (n = 40, Student’s t test).
We next measured the interaction between Kv7.1 and KCNE1 either at the PM or in the ER compartment by using mCUPs (Fig. 3, J to S). The FRET efficiency between Kv7.1 and KCNE1 at the ER was low (0.05 ± 0.01), similar to that of Kv7.1 tetramers (0.07 ± 0.01), but was higher than that of the negative Kv7.1-Kv1.5 control (0.03 ± 0.01, P < 0.01). Within the same cell, positive FRET values suggested a physical interaction of Kv7.1 and KCNE1 at the PM (0.14 ± 0.02, P < 0.001 versus the ER). These findings indicated that Kv7.1 and KCNE1 assembled in a post-ER compartment rather than early in their biogenesis. Similar results were obtained in COS-7 cells (fig. S3, L to U).
Kv7.1-KCNE1 complexes traffic through an unconventional secretory pathway
To identify the intracellular compartment where Kv7.1 and KCNE1 interact, the Kv7.1 and KCNE1 post-ER secretory pathway to the cell surface (Fig. 4 and fig. S2) was examined. KCNE1 followed the classic secretory pathway, colocalizing with a cis-Golgi marker (GM130, 34 ± 6%), a TGN marker (TGN46, 20 ± 2%), and an early endosome marker (EEA1, 24 ± 2%; Fig. 4, A to C and J). However, Kv7.1 colocalization was rather low (GM130, 4 ± 1%; TGN46, 7 ± 1%; and EEA1, 4 ± 1%) (Fig. 4, D to F and J). The absence of Kv7.1 from Golgi compartments suggested that this protein traffics to the PM through a pathway bypassing this organelle. Upon KCNE1 coexpression, the Kv7.1 pattern was mainly unaltered, whereas KCNE1 distribution was changed (Fig. 4, G to J). Thus, KCNE1, when coexpressed with Kv7.1, mistargeted the cis-Golgi network, TGN, and early endosomes, mimicking the unconventional Kv7.1 behavior. Similar results were obtained by electron microscopy. While KCNE1, in the absence of Kv7.1, was detected in Golgi compartments (Fig 4K), Kv7.1, in the absence of KCNE1, mainly stained ER structures (Fig. 4L). Upon Kv7.1-KCNE1 coexpression, KCNE1 relocated in the ER (Fig. 4M). No changes in Kv7.1 ER patterns were observed either in the presence or absence of KCNE1 (Fig. 4, L and M). Collectively, these observations not only suggest that Kv7.1 traffics via an unconventional secretory pathway but also demonstrate that the Kv7.1-KCNE1 association shifts KCNE1 from its classic route to the cell surface.
Fig. 4. Kv7.1 and KCNE1 colocalization with post-ER compartments of the secretory pathway.
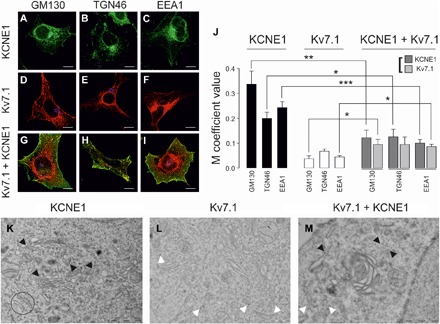
(A to C) KCNE1 colocalization with post-ER subcellular compartment markers. (A) Cis-Golgi network (GM130), (B) TGN (TGN46), and (C) early endosomes (EEA1). Green, KCNE1-YFP; blue, subcellular compartments; cyan, colocalization between green and blue. (D to F) Kv7.1 colocalization with GM130 (D), TGN46 (E), and EEA1 (F). Red, Kv7.1-CFP; blue, subcellular compartments; magenta, colocalization between red and blue. (G to I) Kv7.1 + KCNE1 colocalization with GM130 (G), TGN46 (H), and EEA1 (I). KCNE1-YFP localization is shown in green, Kv7.1-CFP is shown in red, and the corresponding subcellular compartments are shown in blue. White shows triple colocalization between green, red, and blue. Yellow shows partial green and red colocalization. Scale bars, 10 μm. (J) The values represent the means ± SEM of the M overlap coefficient between KCNE1 (black), Kv7.1 (white), or Kv7.1 + KCNE1 (light and dark gray) and the corresponding subcellular compartment. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 10 to 34, Student’s t test). (K to M) Electron micrographs of KCNE1 (K), Kv7.1 (L), and Kv7.1 + KCNE1 (M) in HEK-293 cells. (K) KCNE1 staining of Golgi cisternae as indicated by the black arrowheads. Representative ER structures are circled. (L) Kv7.1 was located in ER-like structures, indicated by the white arrowheads, and was absent from the vicinity of the Golgi. (M) KCNE1 (indicated by the black arrowheads) disappeared from the Golgi cisternae and was located in ER-like structures in the presence of Kv7.1 (indicated by the white arrowheads). Kv7.1, 12-nm gold particles; KCNE1, 18-nm gold particles. Scale bars, 1 μm.
To further decipher the Kv7.1-KCNE1 noncanonical secretory pathway, we used a dominant-negative mutant of Sar1 guanosine triphosphatase (GTPase; Sar1H79G) and brefeldin A (BFA; Fig. 5 and figs. S4 and S5). Sar1H79G blocks coat protein complex II (COPII)–mediated ER exit (30). BFA inhibits ER-to-Golgi trafficking (31). Both factors, impairing the communication between the ER and the Golgi, disrupt the Golgi apparatus (fig. S4, Ba, Ca, Ea, Fa, Ha, and Ia) (32). Blockade of ER-to-Golgi transport increased KCNE1 ER retention [control (68 ± 3%) versus Sar1H79G (94 ± 1%) and BFA (95 ± 2%), P < 0.001] (Fig. 5, A to C and J). The ER clusters of KCNE1 overlapped the Sar1H79G distribution (figs. S4B and S5B). This finding confirmed that KCNE1 is recruited as cargo by COPII vesicles at ER exit sites. In contrast, the ER localization of Kv7.1 did not change under Sar1H79G coexpression or BFA treatment (control, 73 ± 2%; Sar1H79G, 77 ± 3%; and BFA, 71 ± 3%) (Fig. 5, D to F and J). When Kv7.1 and KCNE1 were coexpressed, no treatment affected the ER localization of either subunit (Fig. 5, G to J). Coimmunoprecipitation confirmed that Kv7.1 and KCNE1 still associated after the canonical secretory pathway was disrupted, further supporting the hypothesis that association with Kv7.1 mediates KCNE1 redistribution (Fig. 5, K and L). Redirecting KCNE1 to a nonconventional pathway bypassing the Golgi could trigger changes in the glycosylation of the regulatory subunit. Therefore, we next analyzed the glycosylation pattern of Kv7.1 and KCNE1, in the presence or absence of Kv7.1, using tunicamycin as a general N-glycosylation inhibitor and a cocktail of glycosidases to identify complex N- and O-glycosylation. Unlike Kv7.1, the association of KCNE1 with Kv7.1 triggered a partial loss of larger KCNE1 bands (>50 kDa), compatible with a reduction in the glycosylation of the protein similar to that observed in the presence of either tunicamycin or the glycosidase (Gase) cocktail (Fig. 5, M and N).
Fig. 5. Kv7.1 and Kv7.1-KCNE1 complexes follow an unconventional secretory pathway.
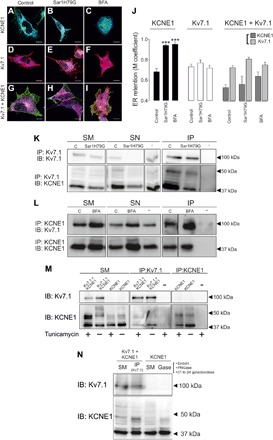
(A to I) Images of KCNE1-YFP (green, A to C), Kv7.1CFP (red, D to F), and Kv7.1-CFP + KCNE1-YFP complexes (red and green, G to I) colocalizing with ER-DsRed (blue) in the absence (A, D, and G) or the presence of either Sar1H79G (B, E, and H) or BFA (C, F, and I). Cyan, colocalization between KCNE1-YFP (green) and ER-DsRe (blue); magenta, colocalization between Kv7.1-CFP (red) and blue; and white, triple colocalization of green, red, and blue. (G to I) Partial colocalization between KCNE1 (green) and Kv7.1 (red) is also shown in yellow. Scale bars, 10 μm. (J) M coefficient values for the overlap of KCNE1 (black), Kv7.1 (white), and Kv7.1 + KCNE1 (light and dark gray) with ER marker in the absence (control) or the presence of either Sar1H79G or BFA. The values represent the means ± SEM. ***P < 0.001 for the ER overlap of KCNE1 in the presence of Sar1H79G and BFA versus that of KCNE1 under control conditions (n = 7 to 19, Student’s t test). (K) Coimmunoprecipitation of Kv7.1 with KCNE1 using anti-Kv7.1 antibodies (IP: Kv7.1) in whole-cell lysates from HEK-293 cells cotransfected with Kv7.1-CFP and KCNE1-YFP in the absence (C) or the presence of Sar1H79G. Immunoblotting was performed against Kv7.1 (IB: Kv7.1) and KCNE1 (IB: KCNE1). (L) Coimmunoprecipitation of Kv7.1 with KCNE1 using anti-KCNE1 antibodies (IP: KCNE1) in whole-cell lysates from HEK-293 cells cotransfected with Kv7.1-CFP and KCNE1-YFP and treated without (C) or with BFA. Immunoblotting was performed against Kv7.1 (IB: Kv7.1) and KCNE1 (IB: KCNE1). −, immunoprecipitation in the absence of antibodies. (M) Analysis of KCNE1 glycosylation in the presence of tunicamycin. HEK-293 cells were transfected with KCNE1-YFP and Kv7.1-CFP + KCNE1-YFP and treated (+) or not treated (−) with tunicamycin (5 μg/ml). Cell extracts were immunoprecipitated and immunoblotted with anti-Kv7.1 and anti-KCNE1 antibodies. (N) Analysis of KCNE1 glycosylation in the presence of a cocktail of glycosidases (Gase). HEK-293 cells were transfected with KCNE1-YFP and Kv7.1-CFP + KCNE1-YFP and treated with EndoH, peptide N-glycosidase F (PNGase F), and PNGase β1 to PNGase β4 galactosidase (Gase). Cell extracts from Kv7.1-CFP + KCNE1-YFP–transfected HEK-293 cells were immunoprecipitated with anti-Kv7.1 antibodies and immunoblotted with anti-Kv7.1 and anti-KCNE1 antibodies. Cell extracts from HEK-293 cells transfected with KCNE1 were immunoprecipitated with anti-KCNE1 antibodies and 50 μg of immunoprecipitate treated with Gase and immunoblotted with anti-Kv7.1 and anti-KCNE1 antibodies. Note that in both cases, heavy KCNE1 bands (>50 kDa) disappeared in the presence of either Kv7.1 or Gase.
Kv7.1 and KCNE1 assemble in ER-PM junctions
Kv7.1-KCNE1 reached the PM after interacting in a post-ER compartment and bypassing the Golgi apparatus. Similarly, the neuronal channel Kv2.1, which localizes in ER-PM junctions (24), resides at the tips of ER projections that extend close to the PM (Fig. 6, Aa to Ae). In our study, Kv7.1 and KCNE1 were highly colocalized at discrete sites in peripheral regions of the ER (Fig. 6, Ba to Be). We performed three-dimensional (3D) image reconstruction of the cell volume and surface rendering to analyze the structures (fig. S6 and movie S1). Kv2.1 was located in projections of the ER compartment, in close contact with the PM (Fig. 6, Ca to Cf). Surface rendering of Kv7.1-KCNE1 showed completely overlapping distributions in similar structures (Fig. 6, Da to Df, and movie S2). This ER-PM junction distribution was further confirmed in a 3D reconstruction video with multiple labeling of Kv7.1, KCNE1, the PM, and the ER (movie S3).
Fig. 6. ER-PM junctions are platforms for Kv7.1-KCNE1 complex assembly.
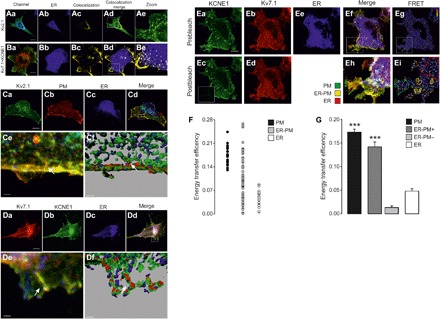
HEK-293 cells were transfected with Kv2.1-YFP and Kv7.1-CFP + KCNE1-YFP and with ER-DsRed and Akt-PH-pDsRed as ER and PM markers, respectively. (Aa to Ae) Kv2.1 colocalization with the ER compartment. (Aa) Merged image of Kv2.1-YFP (green) and ER-DsRed (blue) staining. Cyan, colocalization. (Ab) ER-DsRed. (Ac) Mask of the subcellular distribution of the Kv2.1-ER colocalization signal. (Ad) Merge image of Aa and Ac. (Ae) Magnified view [white rectangle in (Ad)]. (Ba to Be) Kv7.1 and KCNE1 subunits colocalize at discrete sites in distal ER domains. (Ba) Kv7.1-CFP (red) and KCNE1-YFP (green) coexpression. Yellow, colocalization. (Bb) ER-DsRed expression is shown in blue. (Bc) Mask of the subcellular distribution of the Kv7.1-KCNE1 colocalization signal. (Bd) Merge of the images in Bb and Bc. (Be) Magnified view [white rectangle in (Bd)]. Kv7.1-KCNE1 colocalized with the ER marker (white) in discrete areas juxtaposed with the PM. Scale bars, 10 μm. (Ca to Cf) Kv2.1 localization at ER-PM junctions in 3D surface renders. (Ca to Cd) Maximal projection of the z stack of the whole-cell volume. (Ca) Kv2.1-YFP, green; (Cb) PM, red; (Cc) ER, blue; (Cd) Merged image. Scale bars, 10 μm. The white rectangle indicates the magnified area in (Ce) and (Cf). White, triple colocalization. The arrows indicate Kv2.1 at ER-PM junctions. (Cf) 3D surface render. ER, blue; PM, red; Kv2.1, green. The white arrow points to an ER-PM contact site where Kv2.1 is localized. Scale bar, 1.5 μm. (Da to Df) Kv7.1-KCNE1 complexes localized at ER-PM junctions in 3D surface renders. (Da to Dd) Maximal projection of the z stack of the whole-cell volume. (Da) Kv7.1-CFP, red; (Db) KCNE1-YFP, green; (Dc) ER, blue; (Dd) merge. Scale bars, 10 μm. The white rectangle indicates the magnified area in (De) and (Df). (De) White, triple colocalization. The arrows highlight Kv7.1-KCNE1 at ER-PM junctions. (Df) 3D surface render. ER, blue; Kv7.1, red; KCNE1, green. The white arrow points to a Kv7.1-KCNE1 complex localized at a cortical ER projection juxtaposed with the PM. Scale bar, 2 μm. (Ea to Ei) Results of FRET experiments between Kv7.1-CFP and KCNE1-YFP in mCUPs. (Ea and Eb) Prebleaching images showing KCNE1-YFP in green (Ea) and Kv7.1-CFP in red (Eb). (Ec and Ed) Postbleaching images of KCNE1-YFP (Ec) and Kv7.1-CFP (Ed). The bleached area is highlighted with a white square. (Ee) ER-DsRed, blue. (Ef) Merged image of (Ea) to (Ee). (Eh) Magnified view of the bleached area in the merged image that enables the PM (green line) to be distinguished from the ER (red line). Kv7.1 and KCNE1 were highly colocalized in discrete ER-related structures identified as ER-PM junctions (yellow line). (Eg and Ei) Original and magnified FRET ratio images, respectively. The calibration bars range from 0.8 (blue) to 1.4 (red). Scale bar, 10 μm. (F) Plot of individual FRET measurements in the PM (black), ER-PM junctions (gray), and the ER (white) in mCUPs. (G) FRET measurements in the PM (black), positive ER-PM junctions (+, dark gray), negative ER-PM junctions (−, light gray), and the ER (white) within the same cell. The bars represent the means ± SEM (n = 20 to 38). ***P < 0.001 for Kv7.1-KCNE1 in ER-PM+ versus the ER (n = 20 to 38, Student’s t test).
Last, we wanted to determine whether Kv7.1 and KCNE1 colocalize at ER-PM junctions and assemble into complexes. Electron micrographs demonstrated that while Kv7.1, which was absent from the Golgi (fig. S7D), localized in ER structures and ER-PM junctions (fig. S7A), KCNE1, which was located in the Golgi apparatus (fig. S7E), was absent from these sites (fig. S7B). However, upon association with Kv7.1, KCNE1 mostly disappeared from the Golgi and stained ER-PM structures (fig. S7, C and F). Furthermore, FRET measurements in ER-PM junctions from mCUPs demonstrated that Kv7.1 and KCNE1 were associated in these structures (fig. S7, Ea to G). We detected highly variable FRET efficiency between Kv7.1 and KCNE1 within ER-PM junctions (Fig. 6F). While 42% of the ER-PM junctions exhibited very low FRET values (ER-PM−, 0.01 ± 0.01), 58% showed FRET efficiencies significantly higher than those of perinuclear ER regions [ER-PM+, (0.14 ± 0.01) versus ER (0.05 ± 0.01), P < 0.001) and similar to those of the PM (0.17 ± 0.01; Fig. 6G). This variability in FRET efficiencies revealed two different populations of ER-PM junctions: (i) FRET-negative structures in which association was either low or absent and (ii) FRET-positive ER-PM junctions in which interaction between Kv7.1 and KCNE1 had occurred. These data suggest that ER-PMs are specific sites where ongoing Kv7.1-KCNE1 complex assembly takes place.
DISCUSSION
In the present study, we found that Kv7.1 and KCNE1 use different secretory pathways to target the PM. While KCNE1 is driven through the Golgi from the ER using COPII-dependent anterograde transport, Kv7.1 uses an alternative COPII-independent mechanism, bypassing the Golgi apparatus. However, Kv7.1-KCNE1 association shifts KCNE1 from the conventional secretory pathway. This nonconventional trafficking route targets both subunits to ER-PM junctions in which Kv7.1-KCNE1 complexes oligomerize before reaching the cell surface. Previous evidence has indicated the existence of opposing mechanisms, suggesting that Kv7.1-KCNE1 complexes are formed either in the ER compartment or at the PM (3, 8–17) . In this context, our results contribute to resolving this debate. Kv7.1 associates with KCNE1 at ER-PM junctions, which is entirely compatible with the locations of these channels in cardiac T-tubules. Therefore, our data pave the way for resolving an ongoing controversy in cardiac physiology and elucidate an emerging unconventional mechanism for ion channel PM delivery.
Most newly synthesized proteins exit ER trafficking through the Golgi to reach the cell surface. However, some ion channels reach the PM unconventionally by using COPII-independent mechanisms and/or bypassing the Golgi. The molecular mechanisms of this noncanonical secretion pathway are still poorly understood. While Kv4.2 bypasses COPII vesicles when it is associated with Kv channel-interacting protein (KChIP) (21, 22), cystic fibrosis transmembrane conductance regulator uses both COPII-dependent and -independent routes and bypasses the Golgi under ER stress conditions (23). Here, we show that Kv7.1 reaches the PM through an unconventional secretory pathway. Several lines of evidence support this argument: (i) the absence of colocalization at post-ER subcellular compartments, (ii) ER exit via a COPII-independent mechanism, and (iii) surface expression after Golgi disruption.
The identity of the secretory compartment hosting Kv7.1-KCNE1 assembly is one of the most controversial topics regarding cardiac IKs currents (3–5). Evidence supports two opposing hypotheses: (i) Kv7.1 and KCNE1 traffic separately to the cell surface, where they form dynamic complexes with up to four KCNE1 subunits per complex (11–17) or (ii) Kv7.1-KCNE1 channels assemble early at the ER with a fixed 4:2 stoichiometry and travel together toward the PM (3, 8–10). The second hypothesis implies that KCNEs reside in the ER until they coassemble with Kv isoforms. The interaction mediates the progression of the complex through the secretory pathway (13). However, we and others have found that KCNE1 targets the PM in different situations: (i) when it is expressed alone (33), (ii) when the interaction with the α subunit is compromised (17, 34), and (iii) when PM-residing Kv7.1-KCNE1 complexes are saturated (10). Moreover, we found that KCNE1 follows the classic COPII-dependent secretory pathway and that Kv7.1-KCNE1 association occurs within the secretory pathway rather than at the PM. In this study, (i) targeting of KCNE1 to the PM was enhanced upon Kv7.1 coexpression. (ii) The presence of Kv7.1 reduced KCNE1 localization at post-ER secretory compartments. (iii) Kv7.1 rerouted KCNE1 to the nonconventional secretory pathway.
These results raise other important implications that need further debate. First, both subunits do not traffic independently to the PM to form reversible and transient complexes. The robust FRET efficiency detected between Kv7.1 and KCNE1 at the PM supports the formation of stable protein complexes rather than reversible and transient interactions. However, we cannot dismiss the possibility that additional freely diffusing KCNE1 monomers at the PM could interact with nonsaturated complexes bearing less than four β subunits (10). Second, evidence suggests that a single KCNE1 peptide may interact with a Kv7.1 dimer during the biogenesis of Kv7.1 tetramers. The subsequent dimerization of dimers would generate an oligomeric complex with a fixed stoichiometry of 4:2 (Kv7.1:KCNE1) (7). However, our data indicate that Kv7.1-KCNE1 assembles after Kv7.1 tetramerization. The results obtained with our FRET-positive control, Kv7.1-CFP/Kv7.1-YFP, suggested the existence of homotetramers of Kv7.1 in the overall ER compartment. However, Kv7.1 and KCNE1 interaction was only detected at discrete sites at the very tips of ER tubules and cisterns.
We visualized Kv7.1-KCNE1 in ER-PM membrane contact sites in patterns resembling Kv2.1 localization at ER-PM junctions. Kv2.1 plays a major role in regulating neuronal action potential frequency (35), but it is also a major component of IKs currents in rodent hearts, sharing a functional role with Kv7.1-KCNE1 in human cardiomyocytes (36). Moreover, KCNE1 can also form native cardiac complexes with Kv2.1 (37). In cardiomyocytes, Kv7.1 and Kv2.1, as well as other Kv channels, such as Kv4.2 and Kv11.1, are localized in T-tubules, which are specialized cardiac ER-PM junctions (26, 38). The short intermembrane cleft allows rapid communication between the PM and ER compartments, maximizing contraction strength and reducing response time. T-tubules are diffusion-restricted domains, and this accumulation of K+ channels probably leads to high K+ concentrations within the extracellular lumen. Kv7.1-KCNE1 complexes are resistant to the inactivation that high extracellular K+ produces on Kv7.1 homotetramers (39). In addition, ER-PM junctions are crucial in Ca2+ signaling and excitation-contraction coupling in myocytes. Kv7.1-KCNE1 complexes are regulated by Ca2+ at many different levels, such as the levels of assembly, trafficking, and function (14, 40). FRET studies on ER-PM junctions revealed that most of these structures contained perfectly assembled Kv7.1-KCNE1 complexes. However, some of the ER-PM junctions showed negative FRET efficiencies. This variability suggests that ER-PM junctions are specific platforms for the ongoing assembly of Kv7.1-KCNE1 complexes. ER-PM junctions not only participate in Ca2+ signaling but also function as PM protein trafficking hubs (24, 25). Kv7.1 and KCNE1 may traffic independently to ER-PM junctions, structures that accommodate the assembly of the complexes before delivery to the cell surface. Kv7.1-KCNE1 complex assembly and trafficking at/through ER-PM junctions may represent efficient mechanisms for localized subcellular regulation of IKs currents.
In conclusion, we have demonstrated that Kv7.1-KCNE1 complexes are not assembled in early biogenesis. Kv7.1 reroutes KCNE1 to a nonconventional secretory pathway through ER-PM junctions. The complexes are assembled at these sites and subsequently delivered to the PM. Because IKs currents are essential for cardiac repolarization, the implications of our study might be crucial for understanding many heart pathologies.
MATERIALS AND METHODS
Plasmids, cell culture, and transfection
Human Kv7.1 complementary DNA (cDNA) in a pTLN vector was provided by T. Jentsch (Leibniz-Institut für Molekulare Pharmakologie and Max-Delbrück-Centrum für Molekulare Medizin, Germany). Human KCNE1 cDNA in a pHA vector was obtained from S. de la Luna (Centro de Regulación Genómica, Spain). Rat YFP-Kv2.1HA was obtained from M. Tamkun (Colorado State University, USA). For confocal microscopy and molecular biology studies, the Kv7.1, KCNE1, and Kv1.5 cDNA sequences were cloned into pECFP-N1/pEYFP-N1, pEYFP-N1, and pEYFP-C1 vectors (Clontech), respectively. The constructs were verified by sequencing. The PDsRed-tagged pleckstrin homology (PH) domain of Akt (Akt-PH-pDsRed), used as a membrane marker, was a gift from F. Viana (Universidad Miguel Hernández, Spain). An ER marker (pDsRed-ER), a soluble CFP (pECFP-N1), and a soluble YFP (pEYFP-N1) were obtained from Clontech. Membrane-localized Rho-pYC, which consists of the C-terminal prenylation site of Rho (RQKKRRGCLLL) appended to the C terminus of the YFP-CFP fusion protein, was donated by F. Barros and P. de la Peña (Universidad de Oviedo, Spain). Hemagglutinin (HA)–Sar1H79G was provided by R. Pepperkok (European Molecular Biology Laboratory, Germany).
HEK-293 and COS-7 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin (100 U/ml). For molecular biology experiments, cells were plated in 100-mm culture dishes. For confocal microscopy studies, cells were plated on poly-d-lysine–coated coverslips. Transient transfection was performed using Lipotransfectine (AttendBio Research) when the cells had reached nearly 80% confluence. In some experiments, the cells were treated with BFA (Sigma-Aldrich) at 5 μg/ml for 4 hours to ensure the complete turnover of Kv7.1 and Kv7.1-KCNE1 complexes.
CUPs and immunocytochemistry
CUPs are membrane sheets obtained through osmotic shock (41). HEK-293 cells were seeded on poly-d-lysine–treated glass coverslips. Forty-eight hours after transfection, the cells were cooled on ice for 5 min and washed twice in phosphate-buffered saline (PBS). Next, the cells were incubated for 5 min in KHMgE buffer [70 mM KCl, 30 mM Hepes, 5 mM MgCl2, and 3 mM EGTA (pH 7.5)], diluted threefold, and then gently washed with undiluted KHMgE to induce hypotonic shock. Lysed cells were removed from the coverslips by intensively pipetting up and down. After two washes with KHMgE buffer, only the membrane sheets remained attached. We also used a milder modified protocol based on a gentler burst to prepare mCUPs. In mCUPs, a fraction of the ER compartment remains connected to the PM by the ER-PM junctions (fig. S1). The preparations were fixed with fresh 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature and mounted with homemade Mowiol mounting medium.
To detect the cis-Golgi network (GM130), the TGN (TGN46), the early endosomes (EEA1), and the ER (calnexin), we performed immunocytochemistry. After fixation, cells were further permeabilized with 0.1% Triton X-100 for 20 min. After 1 hour of incubation with blocking solution (10% goat serum, 5% nonfat powdered milk, and 0.05% Triton X-100), the cells were incubated with the following primary antibodies in 10% goat serum and 0.05% Triton X-100 overnight at 4°C: mouse anti-GM130 (1:1000; BD Transduction Laboratories), rabbit anti-TGN46 2F11 (1:100; bioNova científica), mouse anti-EEA1 (1:500; BD Transduction Laboratories), and mouse anti-calnexin (1:100; BD Transduction Laboratories). Next, the cells were incubated with either goat anti-rabbit or mouse Cy5-conjugated secondary antibodies for 2 hours at room temperature and further mounted with homemade Mowiol mounting medium.
Confocal microscopy and FRET experiments
Cell preparations were imaged under a Leica SP2 laser scanning confocal spectral microscope (Leica Microsystems GmbH) equipped with an argon multiline laser (458, 488, and 514 nm), a diode-pumped solid-state laser (561 nm), and a helium-neon laser (633 nm). Images of the YFP fluorophore were obtained using the 514-nm laser line, those of CFP were obtained using the 458-nm line, those of DsRed were obtained using the 561-nm line, and those of Cy5 were obtained using the 633-nm line. Double dichroic (458/514) and triple dichroic (488/568/633) filters were used accordingly. Images were acquired at a resolution of 1024 × 1024 pixels using a 63× oil immersion objective lens (numerical aperture, 1.32) and scanning with a pinhole aperture of 1 Airy unit at zoom 4 following the Nyquist theorem to achieve maximal lateral optical resolution. Scanning was performed sequentially for observation of each fluorescent protein.
Colocalization analysis was carried out using Manders’ (M) coefficient based on global analysis of pixel intensity distributions. The coefficient varies from 0 to 1, the former corresponding to nonoverlapping images and the latter reflecting 100% colocalization between both images [Just Another Colocalization Plugin (JACoP); https://imagej.nih.gov/ij/plugins/track/ jacop.html]. When colocalization between Kv7.1 and KCNE1 was assessed, the M coefficient reflected the proportion of Kv7.1 signal coincident with KCNE1 signal. Alternatively, we estimated the colocalization between either Kv7.1 or KCNE1 and specific subcellular markers. In that case, the M coefficient showed the overlapping distribution of a given subunit (Kv7.1 or KCNE1) with the evaluated subcellular compartment.
We measured FRET by the acceptor photobleaching method. Three scans of the region of interest (ROI) using the 514-nm line of an argon laser at 100% power intensity were performed. Before and after photobleaching, CFP and YFP images were collected. All images were sampled at 512 × 512 pixels with a 12-bit resolution. FRET EF was calculated following the formula EF = (Dpost − Dpre)/Dpost, where D is the CFP fluorescence intensity before (Dpre) or after (Dpost) photobleaching normalized by the CFP fluorescence intensity outside the ROI to monitor CFP photobleaching due to imaging. Alternatively, qualitative FRET ratio images are presented in some figures. Analysis was performed using ImageJ.
For 3D reconstructions, we obtained z-stack images of whole-cell volumes. Acquisitions were performed following the Nyquist theorem in the x, y, and z axes. Next, the confocal images were deconvoluted using Huygens Essential software (Scientific Volume Imaging B.V.) to compensate for the optical distortion during acquisition. Using Imaris software (Bitplane), we reconstructed the optical sections into a 3D representation. The volume data from the 3D reconstructions were rendered to represent the anatomical surfaces of the particular objects of interest. This information is shown in the figures as surface renders.
Superresolution imaging
Hearts were freshly obtained from wild-type male Wistar rats according to a protocol approved by the Animal Ethics Approval Committee of the University of Exeter (UK). The whole hearts were rinsed, and the ventricles were selectively dissected and frozen in optimal cutting temperature compound (O.C.T.) (Tissue-Tek). Frozen sections (10 μm thick) were cut using a Leica CM3050 cryostat and collected onto precleaned coverslips coated with 0.1% poly-l-lysine (Sigma). The sections were blocked using an Image-iT FX signal enhancer (Thermo Fisher Scientific) for 1 hour at room temperature (RT). Primary antibodies against Kv7.1 (Santa Cruz Biotechnology), KCNE1 (Alomone), and RyR2 (Thermo Scientific) were applied overnight at 4°C. Next, the samples were incubated for 2 hours at RT with secondary antibodies conjugated to Alexa Fluor 647 or Alexa Fluor 700 (Thermo Fisher Scientific). Imaging was performed in switching buffer (90% glycerol and 100 mM 2-mercaptoethylamine in PBS). For image acquisition, dSTORM images were obtained with a modified Ti Eclipse inverted fluorescence microscope (Nikon, Japan). Image processing, event localization, and grayscale rendering were performed using custom-written PYthon Microscopy Environment software (https://bitbucket.org/david_baddeley/python-microscopy). Colocalization analysis was performed as previously described (27).
Immunoprecipitation, biotinylation, and Western blotting
Hearts were freshly obtained from wild-type male Wistar rats according to a protocol approved by the Animal Ethics Approval Committee of the University of Exeter. The hearts were digested with collagenase and homogenized until cell dissociation, and the homogenates were then centrifuged for 1 min at 750 rpm. Lysis buffer [150 mM NaCl, 50 mM tris, 1 mM EDTA, 1 mM EGTA, 1 mM MgCl2, and 1% Triton X-100 (pH 7.2)] was added, and the samples were mixed, incubated for 20 min until complete disaggregation at 4°C, and centrifuged at 12,000g for 10 min at 4°C. The supernatants were kept as protein extract and prepared for Western blot analysis and immunoprecipitation.
Immunoprecipitation experiments on transfected HEK-293 cells were performed 48 hours after transfection. The cells were washed twice with ice-cold PBS and lysed in 1% Triton X-100, 10% glycerol, 5 mM Hepes (pH 7.2), and 150 mM NaCl supplemented with aprotinin (1 μg/ml), leupeptin (1 μg/ml), pepstatin (1 μg/ml), and 1 mM phenylmethylsulfonyl fluoride as protease inhibitors to isolate total protein samples. To purify proteins in CUPs, before lysis, the CUP protocol was performed in transfected HEK-293 cells cultured in 100-mm dishes. The homogenates were centrifuged at 12,000g for 15 min, and the total protein content of each supernatant was determined using a Bio-Rad Protein Assay (Bio-Rad). Protein A–Sepharose beads (GE Healthcare) were incubated with anti-KCNE1 or anti-Kv7.1 antibodies (Alomone) for 1 hour at RT. Once bead-antibody complexes were formed, they were covalently bound with dimethyl pimelimidate (DMP) (Pierce). The bead-antibody complexes were incubated with 20 mM DMP for 30 min at RT. The cross-linking reaction was quenched by adding 0.2 M glycine (pH 2.5). The cell lysates were precleared with protein A–Sepharose beads for 1 hour at 4°C. Equal amounts of protein samples were incubated overnight with the bead-antibody complexes at 4°C. After five washes, the proteins bound to the antibody-bead complexes were eluted in 100 μl of 0.2 M glycine (pH 2.5).
Cell surface protein biotinylation was carried out with the Pierce Cell Surface Protein Isolation Kit (Pierce). Briefly, cells were incubated with EZ-Link Sulfo-NHS-SS-Biotin (sulfosuccinimidyl-2-[biotinamido]ethyl-1,3-dithiopropionate) for 30 min, and surplus-free reagent was then quenched following the manufacturer’s instructions. Lysates, processed as above, were incubated overnight at 4°C in NeutrAvidin Agarose columns. After three washes, the biotinylated surface proteins were eluted in a dithiothreitol SDS–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer.
For Western blotting, protein samples were prepared in 1× Laemmli SDS loading buffer, incubated for 10 min at 65°C, and resolved by 8 to 10% SDS-PAGE. The proteins were transferred to Immobilon-P nitrocellulose membranes (Millipore), which were blocked in 5% powdered milk in PBS with 0.05% Tween-20 before immunodetection. The membranes were immunoblotted with antibodies against Kv7.1 (anti-Kv7.1; 1:500) and KCNE1 (anti-KCNE1; 1:500), both from Alomone. After washing, the blots were incubated with horseradish peroxidase (HRP)–conjugated goat anti-rabbit secondary antibodies (Bio-Rad) and developed using the Chemiluminescence Detection Kit for HRP (Biological Industries).
Transmission electron microscopy
HEK-293 cells were transfected with Kv7.1CFP, KCNE1YFP, or Kv7.1pCDNA3.1 + KCNE1YFP and cultured for 48 hours before being fixed with PFA and glutaraldehyde at RT. The proteins were labeled using polyclonal anti-Kv7.1 antibodies (1:500; Alomone) and monoclonal anti-GFP mouse antibodies (1:50; Neuromab) for KCNE1YFP. Goat anti-rabbit and goat anti-mouse secondary antibodies conjugated to 12-nm and 18-nm gold particles, respectively, were used to recognize Kv7.1CFP and KCNE1YFP. Briefly, samples were fixed for 1 hour with 4% PFA and 0.1% glutaraldehyde in 0.1 M PBS and then in 2% PFA for 30 min. High-pressure freezing cryofixation with liquid N2 and cryosubstitution, Lowicryl resin embedding, polymerization of blocks, and ultrathin sectioning at a 60-nm thickness were performed in collaboration with the Unitat de Criomicroscòpia Electrònica (CCiT, Universitat de Barcelona). The samples were mounted over Formvar-coated grids, and the sections were lastly contrasted with 2% uranyl acetate for 15 min and immunogold-labeled as abovementioned. The samples were observed using a Tecnai Spirit 120 kV microscope.
Glycosylation studies
Tunicamycin inhibits N-glycosylation. Cells treated with tunicamycin (5 μg/ml; Sigma-Aldrich) were washed twice with PBS, lysed, and processed for Western blot analysis. Furthermore, a sequential deglycosylation protocol to remove all N-linked and many common O-linked glycans from glycoproteins was also used. Transfected HEK cells were lysed as described above, and Kv7.1 and KCNE1 were immunoprecipitated. Sequential enzymatic reactions for 1 hour at 37°C followed by heat inactivation were performed following the manufacturer’s instructions (New England Biolabs). Briefly, 50 μg of immunoprecipitates was sequentially treated for EndoH, peptide N-glycosidase F (PNGase F), and PNGase β1 to PNGase β4 galactosidase digestion. Control samples were incubated with reaction buffers in the absence of glycosidases. Last, the samples were subjected to SDS-PAGE for Western blot analysis.
Electrophysiology
Currents were recorded from COS-7 cells using the perforated (amphotericin B) patch-clamp technique with an Axopatch 200B amplifier (Axon Instruments), as previously described (41). The intracellular pipette filling solution contained 80 mM K-aspartate, 50 mM KCl, 3 mM phosphocreatine, 10 mM KH2PO4, 3 mM Mg–adenosine 5′-triphosphate, 10 mM Hepes-K, and 5 mM EGTA and was adjusted to pH 7.25 with KOH. The bath solution contained 130 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM Hepes-Na, and 10 glucose and was adjusted to pH 7.40 with NaOH. Amphotericin B (20 mM; Sigma-Aldrich) was prepared in dimethyl sulfoxide and added to the internal solution to a final concentration of 0.5 mg/ml, as reported (41).
The currents were filtered at 1 kHz (with a four-pole Bessel filter) and sampled at 2 kHz. Micropipettes were pulled from borosilicate glass capillary tubes (Narishige, GD-1) on a programmable horizontal puller (Sutter Instruments Co.) and heat-polished with a microforge (Narishige). The micropipette resistance was 1 to 3 MΩ. pClamp version 9 software (Axon Instruments) was used for data acquisition and analysis. The currents were recorded at RT (21° to 23°C) at a stimulation frequency of 0.03 Hz. The Microcal Origin 2018 (Microcal Software) and Clampfit 10.8 programs were used to perform least squares fitting and for data presentation.
Statistics
The data are shown as the means ± SEM. Statistical analysis was performed by Student’s t test and one-way analysis of variance (ANOVA) with a post hoc test.
Supplementary Material
Acknowledgments
The English editorial assistance of the American Journal Experts is also acknowledged. Funding: This work was supported by the Ministerio de Economia y Competitividad (MINECO, Spain) (grants BFU2014-54928-R and BFU2017-87104-R to A.F. and SAF2016-75021-R to C.V.), Fondo Europeo de Desarrollo Regional (FEDER), the Instituto de Salud Carlos III CIBERCV program (to C.V.), and CSIC grant PIE201820E104 (to C.V.). A.O. and C.S.-N. hold fellowships from the MINECO. Author contributions: A.O., C.S.-N., C.M., and A.d.l.C. performed the experiments. C.V. and C.S. interpreted and supervised the data. N.C. and C.S.-N. analyzed and interpreted the data. A.O. and A.F. designed, interpreted, and wrote the paper. All authors discussed the work. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay4472/DC1
REFERENCES AND NOTES
- 1.Robbins J., KCNQ potassium channels: Physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 90, 1–19 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Peroz D., Rodriguez N., Choveau F., Baró I., Mérot J., Loussouarn G., Kv7.1 (KCNQ1) properties and channelopathies. J. Physiol. 586, 1785–1789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G., K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T., Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Etheridge S. P., Asaki S. Y., Niu M. C.-I., A personalized approach to long QT syndrome. Curr. Opin. Cardiol. 34, 46–56 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Bianchi L., Priori S. G., Napolitano C., Surewicz K. A., Dennis A. T., Memmi M., Schwartz P. J., Brown A. M., Mechanisms of I(Ks) suppression in LQT1 mutants. Am. J. Physiol. Heart Circ. Physiol. 279, H3003–H3011 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Haitin Y., Wiener R., Shaham D., Peretz A., Cohen E. B.-T., Shamgar L., Pongs O., Hirsch J. A., Attali B., Intracellular domains interactions and gated motions of I(KS) potassium channel subunits. EMBO J. 28, 1994–2005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulsen A. N., Klaerke D. A., The KCNE1 beta-subunit exerts a transient effect on the KCNQ1 K+ channel. Biochem. Biophys. Res. Commun. 363, 133–139 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Jiang M., Xu X., Wang Y., Toyoda F., Liu X.-S., Zhang M., Robinson R. B., Tseng G.-N., Dynamic partnership between KCNQ1 and KCNE1 and influence on cardiac IKs current amplitude by KCNE2. J. Biol. Chem. 284, 16452–16462 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakajo K., Ulbrich M. H., Kubo Y., Isacoff E. Y., Stoichiometry of the KCNQ1 - KCNE1 ion channel complex. Proc. Natl. Acad. Sci. U.S.A. 107, 18862–18867 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krumerman A., Gao X., Bian J.-S., Melman Y. F., Kagan A., McDonald T. V., An LQT mutant minK alters KvLQT1 trafficking. Am. J. Physiol. Cell Physiol. 286, C1453–C1463 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Ehrlich J. R., Zicha S., Coutu P., Hébert T. E., Nattel S., Atrial fibrillation-associated minK38G/S polymorphism modulates delayed rectifier current and membrane localization. Cardiovasc. Res. 67, 520–528 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Chandrasekhar K. D., Bas T., Kobertz W. R., KCNE1 subunits require co-assembly with K+ channels for efficient trafficking and cell surface expression. J. Biol. Chem. 281, 40015–40023 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Shamgar L., Ma L., Schmitt N., Haitin Y., Peretz A., Wiener R., Hirsch J., Pongs O., Attali B., Calmodulin is essential for cardiac IKS channel gating and assembly: Impaired function in long-QT mutations. Circ. Res. 98, 1055–1063 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Seebohm G., Strutz-Seebohm N., Ureche O. N., Henrion U., Baltaev R., Mack A. F., Korniychuk G., Steinke K., Tapken D., Pfeufer A., Kääb S., Bucci C., Attali B., Merot J., Tavare J. M., Hoppe U. C., Sanguinetti M. C., Lang F., Long QT syndrome-associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ. Res. 103, 1451–1457 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Morin T. J., Kobertz W. R., Counting membrane-embedded KCNE beta-subunits in functioning K+ channel complexes. Proc. Natl. Acad. Sci. U.S.A. 105, 1478–1482 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harmer S. C., Wilson A. J., Aldridge R., Tinker A., Mechanisms of disease pathogenesis in long QT syndrome type 5. Am. J. Physiol. Cell Physiol. 298, C263–C273 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lippincott-Schwartz J., Roberts T. H., Hirschberg K., Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol. 16, 557–589 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Appenzeller-Herzog C., Hauri H. P., The ER-Golgi intermediate compartment (ERGIC): In search of its identity and function. J. Cell Sci. 119, 2173–2183 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Nickel W., Rabouille C., Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 10, 148–155 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Hasdemir B., Fitzgerald D. J., Prior I. A., Tepikin A. V., Burgoyne R. D., Traffic of Kv4 K+ channels mediated by KChIP1 is via a novel post-ER vesicular pathway. J. Cell Biol. 171, 459–469 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flowerdew S. E., Burgoyne R. D., A VAMP7/Vti1a SNARE complex distinguishes a non-conventional traffic route to the cell surface used by KChIP1 and Kv4 potassium channels. Biochem. J. 418, 529–540 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoo J.-S., Moyer B. D., Bannykh S., Yoo H.-M., Riordan J. R., Balch W. E., Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J. Biol. Chem. 277, 11401–11409 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Fox P. D., Haberkorn C. J., Weigel A. V., Higgins J. L., Akin E. J., Kennedy M. J., Krapf D., Tamkun M. M., Plasma membrane domains enriched in cortical endoplasmic reticulum function as membrane protein trafficking hubs. Mol. Biol. Cell 24, 2703–2713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deutsch E., Weigel A. V., Akin E. J., Fox P., Hansen G., Haberkorn C. J., Loftus R., Krapf D., Tamkun M. M., Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol. Biol. Cell 23, 2917–2929 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balse E., Steele D. F., Abriel H., Coulombe A., Fedida D., Hatem S. N., Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol. Rev. 92, 1317–1358 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Hou Y., Jayasinghe I., Crossman D. J., Baddeley D., Soeller C., Nanoscale analysis of ryanodine receptor clusters in dyadic couplings of rat cardiac myocytes. J. Mol. Cell. Cardiol. 80, 45–55 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Jiang M., Wang Y., Tseng G.-N., Adult ventricular myocytes segregate KCNQ1 and KCNE1 to keep the IKs amplitude in check until when larger IKs is needed. Circ. Arrhythm. Electrophysiol. 10, e005084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveras A., Roura-Ferrer M., Sole L., de la Cruz A., Prieto A., Etxebarria A., Manils J., Morales-Cano D., Condom E., Soler C., Cogolludo A., Valenzuela C., Villarroel A., Comes N., Felipe A., Functional assembly of Kv7.1/Kv7.5 channels with emerging properties on vascular muscle physiology. Arterioscler. Thromb. Vasc. Biol. 34, 1522–1530 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Ward T. H., Polishchuk R. S., Caplan S., Hirschberg K., Lippincott-Schwartz J., Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155, 557–570 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lippincott-Schwartz J., Yuan L. C., Bonifacino J. S., Klausner R. D., Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 56, 801–813 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cutrona M. B., Beznoussenko G. V., Fusella A., Martella O., Moral P., Mironov A. A., Silencing of mammalian Sar1 isoforms reveals COPII-independent protein sorting and transport. Traffic 14, 691–708 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Manderfield L. J., George A. L. Jr., KCNE4 can co-associate with the I(Ks) (KCNQ1-KCNE1) channel complex. FEBS J. 275, 1336–1349 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Dvir M., Strulovich R., Sachyani D., Ben-Tal Cohen I., Haitin Y., Dessauer C., Pongs O., Kass R., Hirsch J. A., Attali B., Long QT mutations at the interface between KCNQ1 helix C and KCNE1 disrupt I(KS) regulation by PKA and PIP(2). J. Cell Sci. 127, 3943–3955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misonou H., Mohapatra D. P., Trimmer J. S., Kv2.1: A voltage-gated k+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26, 743–752 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Xu H., Barry D. M., Li H., Brunet S., Guo W., Nerbonne J. M., Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative Kv2 alpha subunit. Circ. Res. 85, 623–633 (1999). [DOI] [PubMed] [Google Scholar]
- 37.McCrossan Z. A., Roepke T. K., Lewis A., Panaghie G., Abbott G. W., Regulation of the Kv2.1 potassium channel by MinK and MiRP1. J. Membr. Biol. 228, 1–14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Connell K. M., Whitesell J. D., Tamkun M. M., Localization and mobility of the delayed-rectifer K+ channel Kv2.1 in adult cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H229–H237 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Larsen A. P., Steffensen A. B., Grunnet M., Olesen S.-P., Extracellular potassium inhibits Kv7.1 potassium channels by stabilizing an inactivated state. Biophys. J. 101, 818–827 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y., Zankov D. P., Jiang M., Zhang M., Henderson S. C., Tseng G.-N., [Ca2+]i elevation and oxidative stress induce KCNQ1 protein translocation from the cytosol to the cell surface and increase slow delayed rectifier (IKs) in cardiac myocytes. J. Biol. Chem. 288, 35358–35371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreno C., Oliveras A., de la Cruz A., Bartolucci C., Muñoz C., Salar E., Gimeno J. R., Severi S., Comes N., Felipe A., González T., Lambiase P., Valenzuela C., A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc. Res. 107, 613–623 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay4472/DC1