

Abstract
Purpose:
Dravet syndrome is an early-onset epileptic encephalopathy caused most often by loss-of-function SCN1A variants. Following recognition of its genetic basis and unique clinical features, Dravet syndrome has become one of the most well-studied genetic epilepsies. We sought to evaluate the genetic diversity and correlative seizure phenotype, comorbidities, and response to antiepileptic therapies of patients with clinically-diagnosed Dravet syndrome seen in a tertiary care center. The goal of this study was to examine genotype-phenotype correlations and to ascertain if specific antiepileptic therapies may be more effective on the basis of genetic test result alone.
Method:
Retrospective chart review of demographics, comorbidities, seizure types, and responses to antiepileptic therapies of all patients (n=137) with a clinical diagnosis of Dravet syndrome seen at Lurie Children’s Hospital of Chicago from 2008–2016.
Results:
Of the 96% of Dravet syndrome patients with pathogenic SCN1A variants subdivided by missense or truncating variant, there was no difference in clinical presentation. Response to antiepileptic therapies did not differ by genotype with regard to medication class.
Conclusions:
This is the largest cohort of Dravet patients from within the US to report medication response with respect to genotype. Missense variants in SCN1A were most common in the voltage sensor and pore domains. All patients were most likely to respond to the recommended medication triad compared to other antiepileptic therapies.
Keywords: Dravet syndrome, SCN1A, precision medicine, epilepsy, pharmacogenomics
Introduction:
Initially described by Dr. Charlotte Dravet over 30 years ago1, Dravet syndrome (OMIM #607208) is an infantile-onset epileptic encephalopathy associated with global developmental delays and intractable epilepsy. Hallmarks of the disease include initial normal development and seizure onset typically in the first year of life, consisting of prolonged generalized or unilateral clonic seizures often following a febrile illness or vaccination. Subsequent intractable epilepsy comprises multiple seizure types (generalized tonic-clonic, alternating hemiconvulsive, absence, myoclonic, and other focal seizures) that often requires antiepileptic polytherapy. Common comorbidities that develop after seizure onset include intellectual disability, gait abnormalities and behavioral concerns2; 3. The most common etiology identified in patients with clinical Dravet syndrome is a de novo, heterozygous, loss-of-function variant in SCN1A4; 5, the gene encoding the pore-forming (α) subunit of the voltage-gated sodium channel Nav1.1. Although the incidence of Dravet syndrome is estimated in different populations to be between 1:20,900 and 1:45,8006–9, estimates of the likelihood of Dravet syndrome attributable to pathogenic SCN1A variants have ranged from 33–100%4; 10; 11 depending largely upon subject ascertainment and gene sequencing methodology.
Among individuals in which a pathogenic SCN1A variant is identified, a range of phenotypes is possible, complicating prognostication. While asymptomatic carriers, children with febrile seizures, and risk for temporal lobe and other focal epilepsies are all associated with SCN1A12–14, the most common phenotypes are generalized epilepsy with febrile seizures plus (GEFS+) and Dravet syndrome15. It has been proposed that truncation compared to missense variants more often result in Dravet syndrome than the less severe GEFS+ phenotype16, with no substantial differences noted between specific truncation variants presumed due to a common mechanism of haploinsufficiency following nonsense-mediated decay of the SCN1A transcript. Yet, genotype-phenotype correlation is predictably more complex for interpreting missense variants, as not only the location but also the nature of the amino acid substitution impact disease phenotype. Missense variants specifically localized to the pore region have been associated with earlier seizure onset, presence of ataxia, and a more severe (i.e. higher likelihood to be refractory) epilepsy phenotype17. Yet, there are also missense pore variants to which a GEFS+ phenotype has been attributed. Another theory puts forth that missense variants in the first four transmembrane domains portend disease severity based on the change in hydrophobicity of the amino acid substitution, thus explaining missense pore variants causing conservative amino acid changes in individuals with the less severe GEFS+ phenotype18. In contrast to studies of larger patient cohorts, examples of genetic variants associated with both Dravet syndrome and GEFS+ phenotypes as well as siblings with variable phenotypes despite an identical pathogenic variant19; 20 suggest that the SCN1A genotype-phenotype correlation is nuanced and imperfect.
While prognostication has value for families facing a new diagnosis of a likely pathogenic SCN1A variant, the question of antiepileptic-genotype correlation exists may have more direct therapeutic implications. A recent retrospective study of a large cohort of Japanese patients with clinical Dravet syndrome and a presumed pathogenic SCN1A variant suggested that stiripentol, topiramate, bromide, and levetiracetam appeared effective in patients with truncating variants while clonazepam, bromide, topiramate, and phenobarbital were effective in patients with missense variants9. Yet, this study was limited in its analysis of seizure type and comorbidities as covariates. Our study seeks to extend these data by comparison with a different patient cohort from a single tertiary care center.
Methods:
Ann and Robert H. Lurie Children’s Hospital (LCH), formerly named Children’s Memorial Hospital (CMH) before its relocation in 2012, is a tertiary care epilepsy center with a broad midwestern catchment area within the United States and is a national referral center. IRB approval was obtained for a retrospective chart review of all patients seen for first and second opinions of intractable epilepsy deemed by the treating clinician to meet clinical criteria for Dravet syndrome. Medical records for all patients seen at CMH/LCH from the inception of the electronic medical record in 2007 through April 2016 were queried for “SCN1A” or “Dravet” in the Epic diagnosis or problem list fields. Patients with clinical histories and ancillary testing consistent with a diagnosis of Dravet syndrome by the treating epileptologist in accord with the nine clinical criteria described by the ILAE21, regardless of SCN1A gene testing results, were then included for further analysis. Clinical notes were reviewed to determine: age of seizure onset, as reported by parents in present time or retrospectively, family history, comorbid diagnoses, and apparent response to each pharmacotherapy trial. Seizure improvement was defined as parental report of perceived decrease in seizure frequency and/or duration, while failure was defined as increase in seizure frequency or duration; there were no paradoxical reports of increased frequency yet shortened duration. Treatment effects were defined as improved or exacerbated but limited by side effects if parental report of specific side effects introduced a dose limitation or necessitated a change in medication. Given the limitations of retrospective chart review, data were collected irrespective of antiepileptic dose or serum level, age at which the medication was trialed, history of previous medication trials, or medications combined as polytherapy.
Seizure types evident in patients were classified by the epileptologist on the basis of semiology and confirmatory EEG; seizures of unclear classification including eye fluttering, loss of posture, and dysperceptive events not captured on EEG were omitted. Degree of motor impairment was ranked based on most recent clinical exam as: unaffected, mild gait imbalance, crouch gait, or ataxic. Cognition was ranked based on age-specific evaluation of receptive and expressive speech and writing ability at time of most recent clinical exam using a clinical impression and neuropsychological testing when available to define mild, moderate, severe, or profound impairment. Psychiatric comorbidity was subcategorized for children greater than 3 years of age based upon parental report and notes from a psychiatric consultant when present as: unaffected, mild behavioral disturbances, recognized as having attention deficit hyperactivity disorder (ADHD-like), recognized as having obsessive-compulsive tendencies (OCD-like), or recognized as having features of autism spectrum disorder (ASD-like).
Genetic diagnoses were obtained from both internal and external clinical diagnostic lab reports including single-gene tests, epilepsy panels, and whole-exome sequencing, as previously ordered on a clinical basis by the treating physician. Pathogenic variants were classified by ACMG criteria on the basis of inheritance, predicted changes in the open reading frame or likelihood of introducing a stop codon, and in comparison with multiple databases of healthy individuals (ExAC, gnomad) and pathogenic variants (Gzneuro, Antwerp, Clinvar, HMGD). Definition of the functional domains of the SCN1A amino acid sequence was extracted from SWISS-PROT (accession number P35498) annotated to include the transmembrane domains, linkers, and pore region18. Comparisons are reported between patients with truncating (including frameshift, splice site, and nonsense) variants and patients with missense variants.
Results:
Characteristics of study population
Of 211 charts within the Epic framework discovered to include “Dravet syndrome” or “SCN1A” under diagnosis code or problem list, 137 patients identified by at least one of five pediatric epileptologists at Lurie Children’s Hospital tertiary care center as having clinical Dravet syndrome were included for further analyses (Fig 1). The patient population consisted of 72 males (52%) and 65 females (48%) ranging in age from 1 to 26 years old at the time of most recent clinical evaluation (median: 7 years, STD: 5.6 years).
Fig 1: Genetic landscape of clinical Dravet patients seen at Ann and Robert H. Lurie Children’s Hospital during study period.
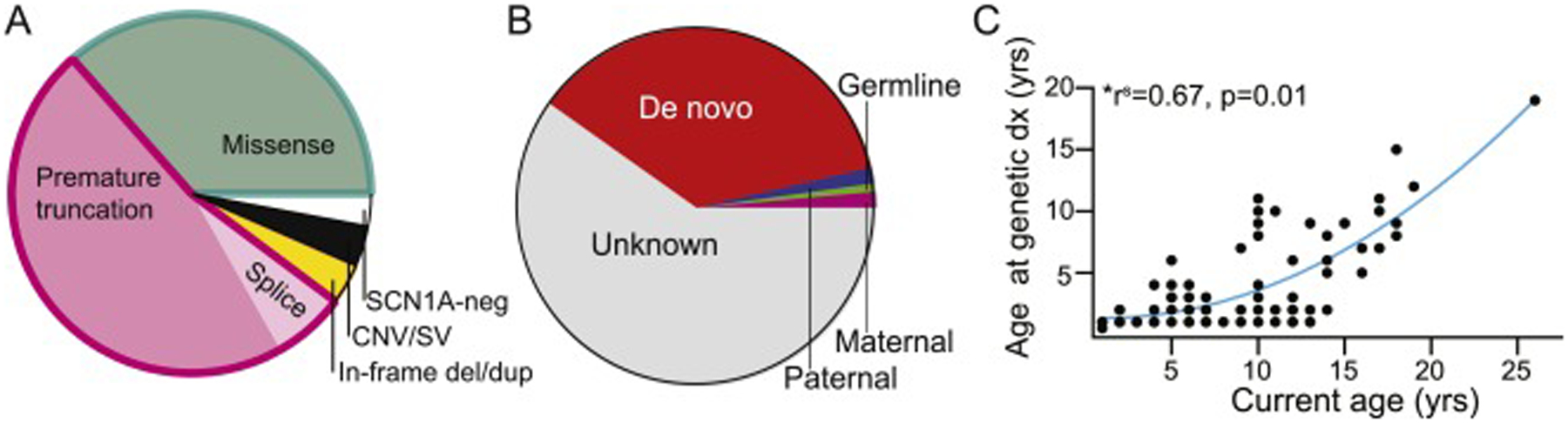
(a) Of the 137 patients evaluated with clinical Dravet syndrome, 96% were positive for pathogenic or likely pathogenic variants including 36% with missense variants and 54% with truncating variants (including 47% with nonsense/frameshift variants and 7% with splice site variants), and 4% with copy number variants including SCN1A. (b) With respect to inheritance patterns, 63% of patients had no known parental testing, 36% of variants were not found in parents and thus labeled de novo, and 1% each of variants were identified in the proband’s mother, father, or sibling. (c) A direct correlation between age of the patient at the time of study and age at which a genetic diagnosis was obtained is clear, suggesting earliest diagnosis in younger patients in our hospital system and thus improvement in speed of testing over time.
Seizure onset occurred on average at 5 ± 2.3 months of age irrespective of sex or type of pathogenic gene variant, followed by evolution in a majority of patients to include generalized tonic-clonic seizures (100%), alternating hemiconvulsions (68%), epileptic myoclonus (69%), and atypical absence (51%). Common additional seizure semiologies included focal dyscognitive, versive, or focal clonic seizures grouped as other focal seizures (53%). Of the 79% of patients with documented episodes of status epilepticus, most patients experienced prolonged generalized or hemiconvulsive seizures (64%) rather than only nonconvulsive status epilepticus (2%) or both convulsive and nonconvulsive status (12%). Though intractable epilepsy is both a presenting symptom and hallmark of Dravet syndrome, its comorbidities represent a substantial psychosocial and financial burden to caregivers22. In our population, most commonly reported impediments to daily functioning included gait disturbance, intellectual disability, and comorbid psychiatric diagnoses (Fig 2).
Fig 2: Epilepsy and comorbidity demographics of children with Dravet syndrome with SCN1A missense vs truncating variants.
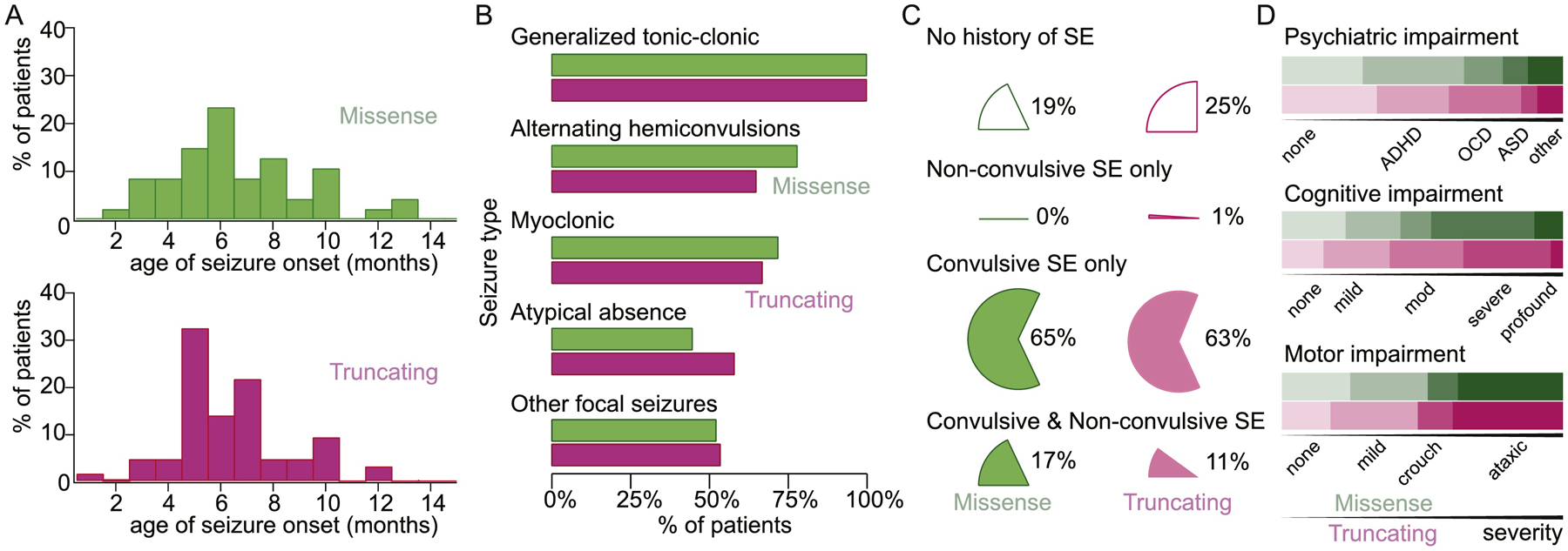
Missense variant data are displayed in green while truncation variant data are displayed in purple. (a) Seizure onset was not significantly different between patients with missense variants (n=47, mean= 5.7mos, STD =2.6) or truncating variants (n=65, mean=5.4mos, STD=2.1). (b) Percentage of children with missense variants experiencing GTCs (100%), alternating hemiconvulsions (78%), myoclonic seizures (72%), atypical absence (44%), and other focal seizures (52%) was similar to children with truncating variants (100% GTCs, 67% M, 66% AH, 58% AA, 53% OF). (c) Status epilepticus, reported as convulsive, non-convulsive, or both, was not different between SCN1A variant type. (d) Graded comorbid psychiatric impairment, cognitive impairment, or motor impairment was not different between SCN1A variant type. Top line (green) represents patients with missense variants, while bottom line (purple) represents patients with truncating variants.
Genotype diversity
Given the prevalence of SCN1A pathogenicity in patients with clinical Dravet syndrome, all but one patient (not tested due to family preference) had single-gene diagnostic Sanger or next generation DNA sequencing and deletion/duplication analysis of SCN1A. In our population including both new diagnoses and referrals to our hospital, 96% of patients with clinical Dravet syndrome had an identifiable SCN1A variant. We noted that the current age of the patient directly correlated with the age at genetic diagnosis (r2 = 0.65, p<0.001, Spearman correlation), suggesting that younger patients cared for in our hospital system are now diagnosed earlier in life (Fig 1).
Of 56 patients for whom parental genetic testing was performed, 89% were noted to be de novo variants including all missense variants. Although parental testing for SCN1A variants in the blood (or, if not available, in saliva) is now uniformly recommended following identification of a pathogenic variant in a child, the majority of patients (62%) had not completed this testing at the time of chart review; ability to obtain parental testing did not correlate with age of the child when the initial diagnosis was made or year in which the proband testing was performed. SCN1A missense variants were reported in 35% (n=47) of patients and all qualified as pathogenic or likely pathogenic by ACMG criteria (Table S1)23. Although patients with GEFS+ were not included in this study, one family carried a p.Ala1442Val variant that was maternally-inherited from a mother with febrile seizures and also present in a sibling with a history of febrile seizures, suggesting a GEFS+ phenotype in the family yet Dravet syndrome in the proband. Frameshift or nonsense variants comprised 46%, and were grouped with splice variants (7%) as “truncating” variants, as these genetic changes are all presumed to undergo nonsense-mediated decay prior to translation. Our population included 3% in-frame deletion/duplication variants which have all been reported pathogenic previously. An additional 5 patients were noted to have deletions spanning entire exons of the SCN1A gene or several neighboring genes, and were characterized as copy number variants and excluded from further analysis. The type of genetic variant present was not correlated with seizure type, occurrence of status epilepticus, or the severity of motor, cognitive, or psychiatric comorbidity (Fig 2).
Seven patients within our cohort were deceased at the time of study conclusion, including 4 due to SUDEP or status epilepticus and 3 with unknown pathologies. This patient subset included two children with the same missense variant (p.Arg101Trp), two with missense variants near the cytoplasmic c-terminus domain (p.Leu1786Pro; p.Phe1831Ser), one with a frameshift variant leading to premature truncation (p.Phe1761Thrfs*8), one with an in-frame deletion (p.Leu1269del), and finally one individual with negative SCN1A testing. To our knowledge, there are no known SCN1A variants or gene domains specifically associated with increased risk of SUDEP.
SCN1A variant ‘hotspots’
The Nav1.1 channel protein encoded by the SCN1A gene is a ~260 kD protein divided into four near-homologous homomeric domains (I-IV). Within each domain are six transmembrane domains (S1-S6) including an S4 voltage sensor, an S3-S4 intracellular loop that folds to become the inactivation gate, and an S5-S6 extracellular linker domain that translates to a hairpin-like loop integrated into the channel pore24. Although our study population was limited to 137 patients, comparison of the distribution of missense variants to either polymorphisms present in a population variant database (gnomAD) demonstrated a significantly different distribution of variants (Fig. 3; χ2 = 61.68, p<0.001). Further subdivision of each variant class into functional domains revealed that missense variants are primarily concentrated in the S4 voltage sensor and S5-S6 pore domain.
Fig 3: Distribution of missense and truncating SCN1A variants across the gene and protein domains.
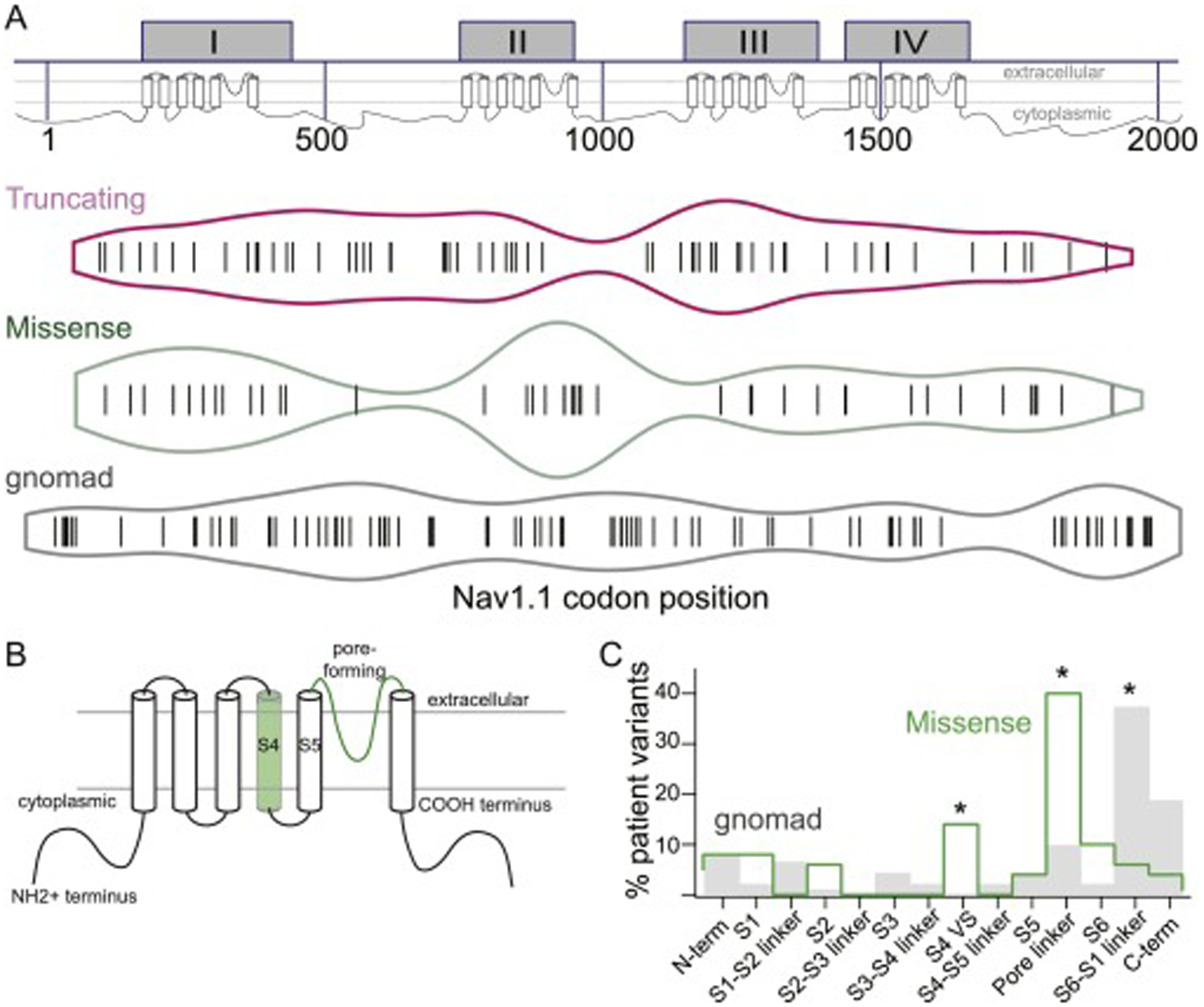
reveals (a) enrichment of missense variants in the 2nd (II) homomeric domain compared to truncating variants found in Dravet syndrome or single nucleotide polymorphisms in gnomAD (SNPs), and relative paucity of truncating variants in the N- and C-termini relative to SNPs. (b,c) Schematic and quantification of functional regions within each domain, when combined, show a higher proportion of missense variants in the S4 voltage sensor, S5-S6 linker domain that encodes the channel pore, and the S6 linker region. There is also a relative dearth of missense variants in the S6-S1 linker region compared to SNPs.
Lack of correlation of genotype with antiepileptic response
We investigated whether the genotype category (i.e. missense or truncating) was correlated with the responsiveness of patients as a cohort to antiepileptic therapies. At the time of most recent clinical assessment, Dravet syndrome patients on average with either missense or truncating variants were maintained on 3 antiepileptic medications (missense: 3.0+/− 1, n=50; truncating:3.1+/− 1.2, n=73, p>0.005) and had trialed 7 antiepileptic medications in the past (missense:7.4+/− 3.5, n=50; truncating: 7.2+/−3.6, n=73). All medications trialed by fewer than 10 patients (e.g. rufinamide, gabapentin, methsuximide, triple bromides, and hemp oil) were excluded from further analysis.
When the overall efficacy of antiepileptic medication or non-pharmacologic anti-epileptic interventions was compared between SCN1A genotypes (Fig. 4), there was no significant difference in likelihood of seizure reduction (χ2 = 11.95, p=0.747) or likelihood of seizure exacerbation (χ2 = 13.59, p=0.138). Clinical guidelines suggest use of valproic acid, clobazam, and stiripentol in patients with diagnosed Dravet syndrome25; although our data were all collected prior to publication of these guidelines, 85% of patients with truncating variants and 86% of patients with missense variants were on part or all of the recommended triad. These three medications were ranked highest in likelihood of seizure reduction and lowest in likelihood of increased seizure frequency. Clonazepam, a benzodiazepine used broadly prior to US availability of clobazam, was ranked fourth. Topiramate, whose antiepileptic properties are suggested to be a mix of sodium and calcium channel block as well as GABA-A receptor agonism and carbonic anhydrase inhibition26, was additionally noted as efficacious. Non-pharmacologic therapies including the ketogenic diet and a vagal nerve stimulator were of limited benefit. Medications such as phenobarbital and levetiracetam, often trialed near seizure onset and prior to diagnosis of Dravet syndrome, showed uniformly poor efficacy.
Fig 4: Response of Dravet patients with missense and truncating SCN1A variants to antiepileptic interventions.
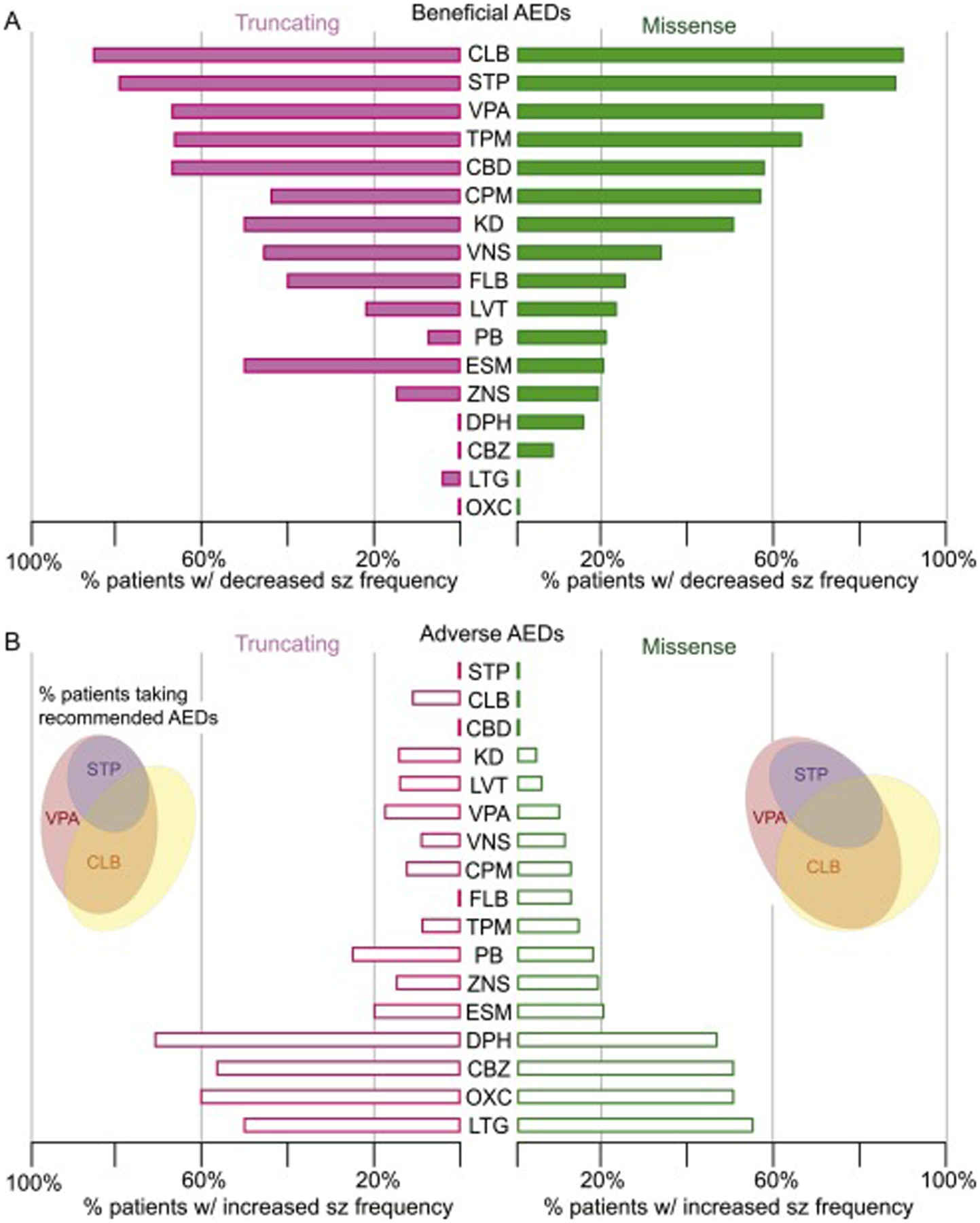
Drug responses, trialed in n patients and normalized to total responses per genotype, included: clobazam (CLB, n=32), stiripentol (STP, n=39), valproic acid (VPA, n=117), topiramate (TPM, n=101), cannabidiol (CBD, n=25), clonazepam (CPM, n=33), ketogenic diet (KD, n=55), vagal nerve stimulator (VNS, n=21), felbamate (FLB, n=26), levetiracetam (LVT, n=111), phenobarbital (PB, n=84), ethosuximide (ESM, n=22), zonisamide (ZNS, n=47), phenytoin (DPH, n=32), carbamazepine (CBZ, n=32), lamotrigine (LTG, n=51), and oxcarbazepine (OXC, n=52). Inset: Scaled Venn diagram reveals proportion of each patient population maintained at the end of the study period on one or more of the three clinically-recommended antiepileptic medications.
Discussion:
While the incidence of Dravet syndrome in the general population remains low enough to classify it as ‘rare,’ it is one of the earliest recognized genetic epilepsies with clear genotype-disease association. Prior to widespread, early genetic testing, diagnosis of patients was based on clinical history including early seizure onset, prolonged seizures provoked by heat or infection, multiple seizure types, and subsequent global developmental impairment. Our cohort of 96% SCN1A variant-positive Dravet patients included both children presenting for first evaluation to a tertiary care center as well as by referral to an epileptologist with a stated interest in Dravet syndrome. While there is likely ascertainment bias, it is also clear that early testing and physician recognition of a well-defined epilepsy syndrome has facilitated the ability to study, formulate, and evaluate guidelines for clinical care in an increasingly precise fashion.
In establishing the genetic diagnosis within our population, we relied upon previously performed state-of-the-art clinical testing for pathogenic SCN1A variants. Over the 7 years surveyed, single-gene Sanger DNA sequencing with reflex to Multiplex Ligation-dependent Probe Amplification (MLPA) to evaluate for deletions and duplications gave way to epilepsy gene panels with rapid turnaround times and whole exome sequencing performed on next-generation sequencing platforms. It is our expectation that ever-improving early genetic diagnosis to complement clinical evaluation may guide informed medication decisions and prognostication.
The majority of SCN1A variants implicated in Dravet syndrome are de novo rather than inherited, with the primary exception of families with variable expressivity represented by GEFS+ carriers whose children have Dravet syndrome. Reproductive counseling for women from these families should necessarily encompass the spectrum of clinical phenotypes but will hopefully evolve in time to be variant-specific and facilitate early effective treatment in addition to early diagnosis. With gene therapy trials for SCN1A on the horizon, this will become critically important. For patients recruited at early time points in our study and identified as having known pathogenic SCN1A variants, parental testing was not previously emphasized because neither variant resolution nor carrier status were needed to classify variant pathogenicity. Currently, we and others recommend parental testing for all variants classified as pathogenic, likely pathogenic, or variants of uncertain significance to assess risk for future conceptions. Our cohort also included siblings whereby the older child was classified as de novo following negative parental testing, then re-classified as germline upon diagnosis of his younger sister with Dravet syndrome. Recent work by Myers et al.27 indicates that up to 10% of de novo SCN1A cases are inherited from parents with somatic mosaicism, suggesting that the demonstrable risk of germline transmission necessitates a need for future development of clinical testing for somatic mosaicism.
It was previously reported that patients with a prematurely truncating SCN1A variant typically experienced seizures at an earlier age relative to patients with a missense SCN1A variant. However, we did not observe a difference in age of seizure onset between truncating and missense SCN1A Dravet patients. We further asked whether there were any notable differences in seizure types presenting in these patients. Similar to previous studies, we observed a diversity of seizure types, including GTCS, myoclonic jerks, atypical absence seizures, and focal seizures. There were no major differences between patients with missense and truncation SCN1A variants with respect to seizure types or history of SE. As we were limited by retrospective data previously entered, we did not collect data regarding age of onset of specific seizure types though age of first seizure and types of seizures present were not different between truncating and missense SCN1A variants.
This is one of the largest cohort of patients with Dravet syndrome reported from the US. Similar cohorts of Dravet patients have been published from the UK and Japan 7; 9, in addition to analyses of genotype-phenotype correlations amongst all SCN1A-positive individuals28; 29. Our study reports a similar percentage of truncation variants (47% nonsense and frameshift variants; 7% splice site variants) and missense variants (35%) as other cohorts. Our population also confirmed a significantly different distribution of missense variants compared to either truncating variants or single nucleotide polymorphisms present in gnomAD, concentrated with the second homomeric channel domain, or more specifically within the voltage sensory and pore functional domains. This result is in accordance with other patient cohorts9; 29. Truncating variants were not present in the proximal N-terminus compared to gnomAD, perhaps due to the GC content or to limitations in sequencing within the first exon; these variants were also rare in the last exon or C-terminus, attributable to escape from nonsense-mediated decay with stop codons present at this location in the transcript. Although 1800 likely pathogenic or pathogenic variants are now associated with Dravet syndrome30, 25% of missense variants we identified were not previously reported in HGMD or Clinvar, suggesting ongoing discovery and reporting of disease-causing codon alterations. We feel it is important to continue to report variants, as specific pathogenic variants with variable, non-classical phenotypes have also been reported31.
We then asked whether genotype might correlate with AED response in our cohort. All of our patients, regardless of genotype class, evidenced the highest likelihood of seizure improvement with administration of valproic acid, stiripentol, and clobazam. Although levetiracetam and phenobarbital are often used in infants due to ease of use and safety profiles, these medications are less likely to reduce seizure burden. Administration of sodium channel blockers such as carbamazepine or phenytoin, often done prior to establishing a genetic diagnosis, were nearly universally detrimental and are now known to correlate with a worse developmental outcome32. In our patient cohort, several other common antiepileptic therapies (felbamate, ethosuximide, phenobarbital, levetiracetam, and zonisamide) offered limited seizure improvement, yet compared to sodium channel blockers, they did not exacerbate seizure frequency. Our study and others also indicate a lower likelihood of achieving a beneficial antiepileptic effect using non-pharmacologic interventions such as the ketogenic diet or a vagal nerve stimulator in children with Dravet syndrome specifically. In sum, early identification of a pathogenic SCN1A variant should clearly impact choice of antiepileptic therapy and provide valuable prognostic information within a spectrum of well-characterized seizures and comorbidities.
Supplementary Material
Highlights.
Pathogenic SCN1A missense variants cluster in specific functional channel domains.
The type of pathogenic SCN1A variant does not impact seizures or comorbidities.
Seizures in Dravet syndrome respond to valproic acid, clobazam, and stiripentol.
Levetiracetam and phenobarbital have limited benefit despite frequent use.
Acknowledgements
Funding for this project included an NINDS R25 award (NS070695) and K08 (NS104237) to TSG, and an NCATS TL1 (TR001423) to JC. The authors thank Dr. Alfred L. George Jr. for critical comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Dravet C. The core Dravet syndrome phenotype. Epilepsia 2011;52 Suppl 2:3–9. [DOI] [PubMed] [Google Scholar]
- 2.Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia 2011;52 Suppl 2:44–49. [DOI] [PubMed] [Google Scholar]
- 3.Ragona F, Brazzo D, De Giorgi I, et al. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients. Brain Dev 2010;32:71–77. [DOI] [PubMed] [Google Scholar]
- 4.Fukuma G, Oguni H, Shirasaka Y, et al. Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia 2004;45:140–148. [DOI] [PubMed] [Google Scholar]
- 5.Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayat A, Hjalgrim H, Moller RS. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22,000: a population-based study from 2004 to 2009. Epilepsia 2015;56:e36–39. [DOI] [PubMed] [Google Scholar]
- 7.Brunklaus A, Ellis R, Reavey E, et al. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012;135:2329–2336. [DOI] [PubMed] [Google Scholar]
- 8.Wu YW, Sullivan J, McDaniel SS, et al. Incidence of Dravet Syndrome in a US Population. Pediatrics 2015;136:e1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii A, Watkins JC, Chen D, et al. Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia 2017;58:282–290. [DOI] [PubMed] [Google Scholar]
- 10.Claes L, Ceulemans B, Audenaert D, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat 2003;21:615–621. [DOI] [PubMed] [Google Scholar]
- 11.Mulley JC, Scheffer IE, Petrou S, et al. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–542. [DOI] [PubMed] [Google Scholar]
- 12.Ceulemans BP, Claes LR, Lagae LG. Clinical correlations of mutations in the SCN1A gene: from febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol 2004;30:236–243. [DOI] [PubMed] [Google Scholar]
- 13.Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology 2001;57:2265–2272. [DOI] [PubMed] [Google Scholar]
- 14.Kasperaviciute D, Catarino CB, Matarin M, et al. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain 2013;136:3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–490. [DOI] [PubMed] [Google Scholar]
- 16.Nabbout R, Gennaro E, Dalla Bernardina B, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology 2003;60:1961–1967. [DOI] [PubMed] [Google Scholar]
- 17.Kanai K, Hirose S, Oguni H, et al. Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology 2004;63:329–334. [DOI] [PubMed] [Google Scholar]
- 18.Kanai K, Yoshida S, Hirose S, et al. Physicochemical property changes of amino acid residues that accompany missense mutations in SCN1A affect epilepsy phenotype severity. J Med Genet 2009;46:671–679. [DOI] [PubMed] [Google Scholar]
- 19.Dravet C, Bureau M, Oguni H, et al. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol 2005;95:71–102. [PubMed] [Google Scholar]
- 20.Guerrini R, Falchi M. Dravet syndrome and SCN1A gene mutation related-epilepsies: cognitive impairment and its determinants. Dev Med Child Neurol 2011;53 Suppl 2:11–15. [DOI] [PubMed] [Google Scholar]
- 21.Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989;30:389–399. [DOI] [PubMed] [Google Scholar]
- 22.Lagae L, Irwin J, Gibson E, et al. Caregiver impact and health service use in high and low severity Dravet syndrome: A multinational cohort study. Seizure 2019;65:72–79. [DOI] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000;26:13–25. [DOI] [PubMed] [Google Scholar]
- 25.Wirrell EC, Laux L, Donner E, et al. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations From a North American Consensus Panel. Pediatr Neurol 2017;68:18–34 e13. [DOI] [PubMed] [Google Scholar]
- 26.Porter RJ, Kupferberg HJ, Hessie BJ. Mechanisms of action of anti-seizure drugs and the antiepileptic screening program of the National Institute of Neurological Disorders and Stroke. Int J Clin Pharmacol Ther 2015;53:9–12. [DOI] [PubMed] [Google Scholar]
- 27.Myers CT, Hollingsworth G, Muir AM, et al. Parental Mosaicism in “De Novo” Epileptic Encephalopathies. N Engl J Med 2018;378:1646–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Depienne C, Trouillard O, Saint-Martin C, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet 2009;46:183–191. [DOI] [PubMed] [Google Scholar]
- 29.Zuberi SM, Brunklaus A, Birch R, et al. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 2011;76:594–600. [DOI] [PubMed] [Google Scholar]
- 30.Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 2003;21:577–581. [DOI] [PubMed] [Google Scholar]
- 31.Sadleir LG, Mountier EI, Gill D, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology 2017;89:1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Lange IM, Gunning B, Sonsma ACM, et al. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A-related seizure phenotypes. Epilepsia 2018;59:1154–1165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.