

Abstract
Introduction:
The association between Gaucher disease and Parkinson’s disease was first recognized in the clinic, where it was noted that patients with Gaucher disease, caused by the inherited deficiency of the lysosomal enzyme glucocerebrosidase, and their relatives with mutations in the glucocerebrosidase gene (GBA1) had a higher than expected incidence of Parkinson’s disease. Mutations in GBA1 are now accepted as the most commonly known genetic risk factor for Parkinson’s disease and dementia with Lewy bodies, and there appears to be an inverse relationship between glucocerebrosidase and α-synuclein, the key factor in Parkinson pathogenesis. The hypothesis that therapeutic enhancement of brain glucocerebrosidase levels might reduce the aggregation, accumulation or spread of α-synuclein has spurred great interest in glucocerebrosidase as a novel therapeutic target.
Area covered:
This article explores the potential molecular mechanisms underlying the association between GBA1 mutations and Parkinson’s disease and outlines therapeutic strategies to increase brain glucocerebrosidase, including gene therapy, targeted delivery of recombinant glucocerebrosidase to the brain, small-molecule chaperones to rescue mutant glucocerebrosidase, and small-molecule modulators to activate wild-type glucocerebrosidase.
Expert opinion:
Although an improved understanding of the mechanistic basis for GBA1-associated parkinsonism is still imperative, enzyme enhancement in the brain may have wide therapeutic implications. While gene therapy may ultimately be the more dramatic and effective approach, the development of less expensive and invasive small-molecule non-inhibitory chaperones or activators could significantly impact the disease course.
Keywords: Parkinson’s disease, Gaucher disease, α-synuclein, glucocerebrosidase, lysosome, chaperones, gene therapy
1. Introduction
Parkinson’s disease (PD), first described by James Parkinson 200 years ago, is the second most common neurodegenerative disorder of aging, affecting 1% of the population age 65 and older, and 4-5% of the population age 85 and older worldwide[1]. PD is characterized by motor symptoms, including rigidity, resting tremor, bradykinesia, and postural instability, and by non-motor features such as cognitive impairment, impaired olfaction, sleep disturbances, anxiety, and depression. The motor symptoms are due to the progressive loss of dopamine neurons in the substantia nigra, with approximately 50% of dopamine neurons lost in the midbrain by the onset of motor symptoms[2]. Although many details of PD pathophysiology remain unclear, its hallmark is the aggregation of α-synuclein in Lewy bodies and Lewy neurites present in neurons of the substantia nigra, cerebral cortex, and hippocampus, as well as the selective loss of dopaminergic neurons in the midbrain[3]. α-Synuclein, an intrinsically disordered 140-amino-acid protein encoded by the gene SNCA, is abundantly expressed in the nervous system and appears to control neurotransmitter release through its effects on the soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) protein complex. α-Synuclein forms α-helical structures upon binding to negatively charged lipids on the cell membrane and has a propensity to aggregate into β-sheet structures similar to those of β-amyloid. The direct role of α-synuclein in PD pathogenesis is clear from cases of familial PD resulting from mutations, duplications, and triplications of SNCA, all of which lead to pathologic aggregation of α-synuclein and neurodegeneration[4]. In addition to SNCA, highly penetrant mutations resulting in monogenic forms of PD are also found in genes such as Parkin, DJ-1, PINK, LRRK2, and VPS35[5]. PINK1 and Parkin work together to govern mitochondrial quality control. PINK1 detects mitochondrial dysfunction and recruits Parkin to damaged mitochondria. Parkin ubiquitinates mitochondria with compromised function to instigate their removal by mitophagy. Loss-of-function mutations and deletions in Parkin and PINK are associated with inherited early-onset PD [6]. Vacuolar protein sorting-associated protein 35 (VPS35) is a core functional component of retromer complex that acts to prevent mis-sorting of selected transmembrane cargo proteins into the lysosomal degradation pathway. Mutations in VPS35 cause a rare form of autosomal-dominant PD [7]. LRRK2 encodes for leucine-rich repeat kinase 2 (LRRK2). The most prevalent pathogenic mutation, G2019S, increases LRRK2 kinase activity and impairs the autophagy process, leading to a significant accumulation of α-synuclein, as assessed by in vitro and in vivo experiments [8,9]. Although monogenic forms of PD tend to have a high penetrance, they account for only about 10% of all cases of PD, with the remainder considered as idiopathic PD. Unique variants with incomplete penetrance in LRRK2 and GBA1 are recognized as strong risk factors for PD, while over 20 other common variants with small effect sizes also modulate the risk for PD[10]. Although most risk loci associated with PD were identified by largely unbiased genome-wide studies, the association between mutations in GBA1 and the development of PD was first appreciated in the clinic, with the identification of rare patients with Gaucher disease who also developed PD.
2. Gaucher disease
Gaucher disease was first described by Philippe Gaucher in 1882. It results from the inherited deficiency of glucocerebrosidase due to mutations in GBA1 and is an autosomal recessively inherited rare disorder. Glucocerebrosidase breaks down the glycolipid glucocerebroside into ceramide and glucose, and glucosylsphingosine into sphingosine and glucose inside lysosomes (Fig. 1)[11]. Its deficiency leads to the accumulation of undegraded glucocerebroside and glucosylsphingosine inside lysosomes. Gaucher disease primarily affects the mononuclear phagocyte system since glucocerebroside is enriched in the cell membrane of erythrocytes, which are cleared by macrophages via phagocytosis. In Gaucher disease, macrophages with accumulated glucocerebrosidase substrates appear engorged and are often referred to as “Gaucher cells”. Gaucher cells usually populate the spleen, liver and bone marrow, resulting in inflammation and organomegaly, whereas a subgroup of patients with Gaucher disease may also have brain involvement of varying severity[12–14]. Although Gaucher disease is rare, it is one of the most common lysosomal storage diseases, with an estimated frequency of 1:40 000-60 000 live births in the general population[15] and an exceptionally high prevalence of 1:850 in the Ashkenazi Jewish population[16]. Thus far, more than 495 known GBA1 mutations have been associated with Gaucher disease, including point mutations, frameshift mutations, splice-site alterations, and recombinant alleles that encompass segments of the pseudogene sequence[17]. Although some phenotypic predictions can be made based on the genotype, genotype–phenotype correlation is incomplete in Gaucher disease. Different clinical manifestations are often encountered in patients with the same genotype, implicating the contribution of disease modifiers. Given the wide spectrum of phenotypes encountered, Gaucher disease is typically divided into three types based on the absence (type 1) or rate of progression (types 2 and 3) of neuronopathic involvement[14]. However, the associated phenotypes can also be considered a continuum, ranging from asymptomatic individuals to infants who have Gaucher disease-associated hydrops fetalis in utero.
Figure 1.
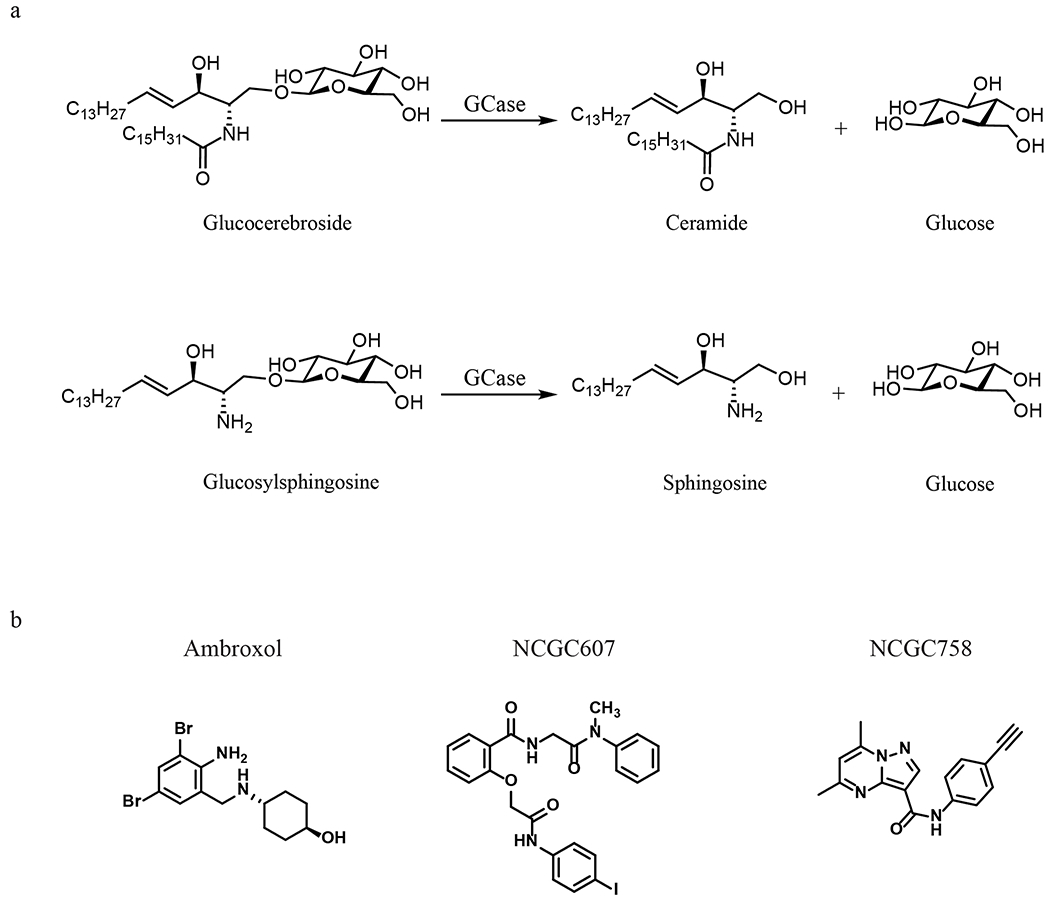
Chemical reaction catalyzed by glucocerebrosidase (GCase) and chemical structures of small-molecule chaperones of glucocerebrosidase including ambroxol, NCGC607 and NCGC758.
After several decades of preclinical work, the first effective therapy for Gaucher disease, enzyme replacement therapy (ERT) became available in 1991[15]. While initially a placental derived product, a recombinant form of the enzyme, imiglucerase was soon developed, and currently there are several approved forms of the recombinant enzyme. Each successfully reverses the anemia, thrombocytopenia and hepatosplenomegaly associated with Gaucher disease, remarkably improving the current clinical course for patients with Gaucher disease. However, ERT is a treatment, but not a cure, and it requires regular intravenous infusions of the costly enzyme. Furthermore, it does not cross the blood-brain -barrier (BBB), and thus is not effective in treating manifestations specifically encountered in patients with neuronopathic forms of the disease. A second therapeutic approach is substrate reduction therapy (SRT) targeting glucosylceramide synthesis, which also effectively reduces non-neurologic manifestations of GD. However, since both ERT and SRT are extremely expensive and do not reverse neuronopathic GD, there remains a strong rationale for developing new therapeutic strategies for patients with Gaucher disease.
3. Association between Gaucher disease and Parkinson’s disease
The association between mutations in GBA1 and the development of PD was first appreciated in the 1990’s with the diagnosis of PD among sporadic patients with Gaucher disease [18,19]. Subsequently, it was recognized that PD was also more frequent in heterozygote family members of GD probands[20]. Following these initial observations, the frequency of mutations in GBA1 was assessed in different PD centers worldwide and was found to be consistently higher in patients with PD than in control individuals. Ultimately, a large multicenter collaborative study, which incorporated 16 research centers from four continents with 5691 patients with PD and 4898 control individuals without PD, concluded that in patients with PD, the odds ratio (OR) for carrying a GBA1 mutation was 5.43 (95% CI 3.89-7.57), confirming that mutations in GBA1 are a common risk factor for PD[21]. Subsequent genome-wide association studies that focused on specific single nucleotide polymorphisms in GBA1 also confirmed the GBA1 locus as a risk factor for PD[22,23]. Now it is widely accepted that the frequency of GBA1 mutations in subjects with PD from varied ethnicities is greater than any other genetic risk factor for PD, once common risk variants of low effect are excluded[10].
4. An inverse relationship between glucocerebrosidase and α-synuclein
The exact molecular mechanisms involved in the interaction between glucocerebrosidase and α-synuclein remain unresolved, but experimental data indicate that there is a reciprocal relationship between glucocerebrosidase and α-synuclein[13,24]. The key element in this reciprocal relationship model is the lysosome, which is the main organelle responsible for the degradation of proteins, lipids and organelles[25,26]. Lysosomal degradation of α-synuclein occurs through both macroautophagy and chaperone-mediated autophagy. When destined for chaperone-mediated autophagy, α-synuclein forms a complex with heat shock cognate 70, and the complex interacts with lysosomal associated membrane protein 2A to translocate into the lysosome[27]. When the delicate balance of α-synuclein homeostasis is disturbed in the process of aging, it is speculated that increased levels of α-synuclein can inhibit the translocation of glucocerebrosidase from the endoplasmic reticulum (ER) to the lysosome[28]. In turn, less lysosomal glucocerebrosidase leads to a gradual increase of glucocerebroside inside the lysosome. Accumulated glucocerebroside can then induce oligomerization and accumulation of α-synuclein in the lysosomes, potentially by stabilizing soluble oligomeric intermediates of α-synuclein[29,30]. Eventually, the lysosomes become dysfunctional, and autophagy-mediated α-synuclein turnover is impaired, leading to α-synuclein aggregates in the cytoplasm. Cytoplasmic α-synuclein aggregates then further inhibit glucocerebrosidase trafficking from the ER to the lysosome. This positive feedback loop of dysfunctional glucocerebrosidase trafficking, impaired lysosomal function, and progressive α-synuclein accumulation will eventually cause neurodegeneration[31,32]. In fact, even in the absence of GBA1 mutations, patients with idiopathic PD also appear to have lower levels of glucocerebrosidase activity in multiple brain regions [33,34] as well as in the blood [35]. The decreased levels of glucocerebrosidase in patients with PD without GBA1 mutations suggest that the reduction of glucocerebrosidase activity may contribute to disease progression of idiopathic PD as well.
In addition to this loss of enzymatic function, glucocerebrosidase mutants may also induce α-synuclein accumulation and aggregation and death of dopaminergic neurons directly, independent of the presence of accumulated substrates. It is postulated that misfolded glucocerebrosidase mutants can increase the burden on the ubiquitin-proteasome system (UPS) and induce ER stress. Prolonged activation of the unfolded protein response (UPR) and/or endoplasmic reticulum-associated degradation (ERAD) in the presence of ER stress may lead to increased apoptosis of dopaminergic neurons[36]. Mutated glucocerebrosidase may also induce α-synuclein accumulation and aggregation by blocking the access of lysosomal proteases to α-synuclein[37,38].
5. Glucocerebrosidase as a therapeutic target for Parkinson’s disease
Given the reciprocal association between glucocerebrosidase and α-synuclein, glucocerebrosidase has become a promising therapeutic target to explore as a novel treatment strategy for PD. A therapy to restore neural glucocerebrosidase levels in the brain has the potential to prevent PD onset in patients susceptible to Gaucher disease-associated PD or even idiopathic PD, and could also drastically improve the quality of life for patients with neuronopathic Gaucher disease. Preliminary experiments in mouse models provided evidence that enhancing brain glucocerebrosidase activity can impact PD. For example, adeno-associated virus (AAV)-mediated expression of glucocerebrosidase not only corrected substrate accumulation, cognitive impairment, and α-synuclein aggregation in the CNS of mice with features of neuronopathic GD[39], but also reduced the α-synuclein level in transgenic mice over-expressing A53T α-synuclein without GBA1 mutations[40].
There are multiple potential strategies to enhance glucocerebrosidase activity in lysosomes in the patient brain. One mechanism is gene therapy that aims to deliver wild-type GBA1 into neurons directly. AAV is a popular viral vector for gene delivery to the nervous system because it has a strong CNS tropism, and AAV-based gene therapy vectors form episomal concatemers in the host cell nucleus without integration into the host genome[41]. In non-dividing cells such as post-mitotic neurons, these concatemers remain intact for the life of the host cells. GBA1 has been successfully delivered into the brains of animal models using AAV, and the resulting augmented glucocerebrosidase expression exhibited impressive neuroprotective effects[39,40,42].
Alternatively, methodologies have been investigated to improve the delivery of recombinant glucocerebrosidase across the BBB. Tat is an 11 amino acid peptide derived from the transactivator protein of HIV. Tat-linked cargo proteins are shown to be sequestered in endosomes and lysosomes after uptake by micropinocytosis, independent of cell surface receptors[43,44]. Rabies virus (RABV) is strictly neurotropic and binds specifically to neuronal cells through its glycoprotein. Peptides derived from RABV glycoprotein (RDPs) have successfully delivered several proteins such as BDNF, GDNF and β-galactosidase across the BBB [45–47]. Glucocerebrosidase tagged with Tat or RDP shows enhanced delivery to a neuronal cell line compared to control glucocerebrosidase, Alglucerase (Genzyme), and extended treatment resulted in reduction of the lipid substrate[48]. Further in vivo studies of these methods are needed to assess their potential to deliver glucocerebrosidase across the BBB.
Since they have the potential to penetrate the BBB effectively, pharmacological chaperones of glucocerebrosidase currently under development to treat Gaucher disease have become promising candidates for the treatment of PD with GBA1 mutations, or even idiopathic PD. Pharmacological chaperones directly bind to mutant glucocerebrosidase, stabilizing the misfolded enzymes in the ER and diverting them from proteasomal degradation to the lysosome (Fig. 2). For many years, competitive inhibitors of glucocerebrosidase, such as iminosugar derivatives, have been explored as potential glucocerebrosidase chaperones, and current efforts are focusing on improving their cell and the ER permeability for better delivery and on enhancing their selectivity towards glucocerebrosidase [49–51]. Because these inhibitors bind to the active site of the misfolded glucocerebrosidase, glucocerebroside will need to out-compete the inhibitors in order to gain access to the enzyme in the lysosomes. This competition between the substrates and the inhibitors requires that the drug dosage be optimized so that the inhibitors only function as chaperones to facilitate the delivery of glucocerebrosidase from the ER to lysosomes, but not as inhibitors in the lysosomes[52,53]. Ambroxol (Figure 1)is a novel inhibitory chaperone of glucocerebrosidase identified in a high-throughput screening (HTS) of a library of FDA-approved drugs[54]. Widely used as a cough medicine, ambroxol exhibits maximal inhibitory activity at the neutral pH found in the ER and has undetectable inhibition at the acidic pH of lysosomes. The pH dependence of its inhibitory activity makes ambroxol a desirable pharmacological chaperone that does not block substrate access to glucocerebrosidase in the lysosome. Ambroxol treatment improved lysosomal delivery of mutant glucocerebrosidase in patient cells[54,55], mice[55,56] and patients[57]. Importantly, oral administration of ambroxol increased brain glucocerebrosidase activity in mice[56] and non-human primates[58]. An early clinical trial of ambroxol for GBA1-associated PD (ClinicalTrials.gov identifier NCT02941822)[59] demonstrated that ambroxol was detectable in cerebrospinal fluid (CSF) and increased CSF glucocerebrosidase protein levels as well as the CSF α-synuclein concentration, indicating successful in vivo engagement of glucocerebrosidase and α-synuclein pathways by ambroxol. Another clinical trial of ambroxol to assess its effects on cognitive and motor symptoms of PD is currently underway in Canada (ClinicalTrials.gov NCT02914366).
Figure 2.
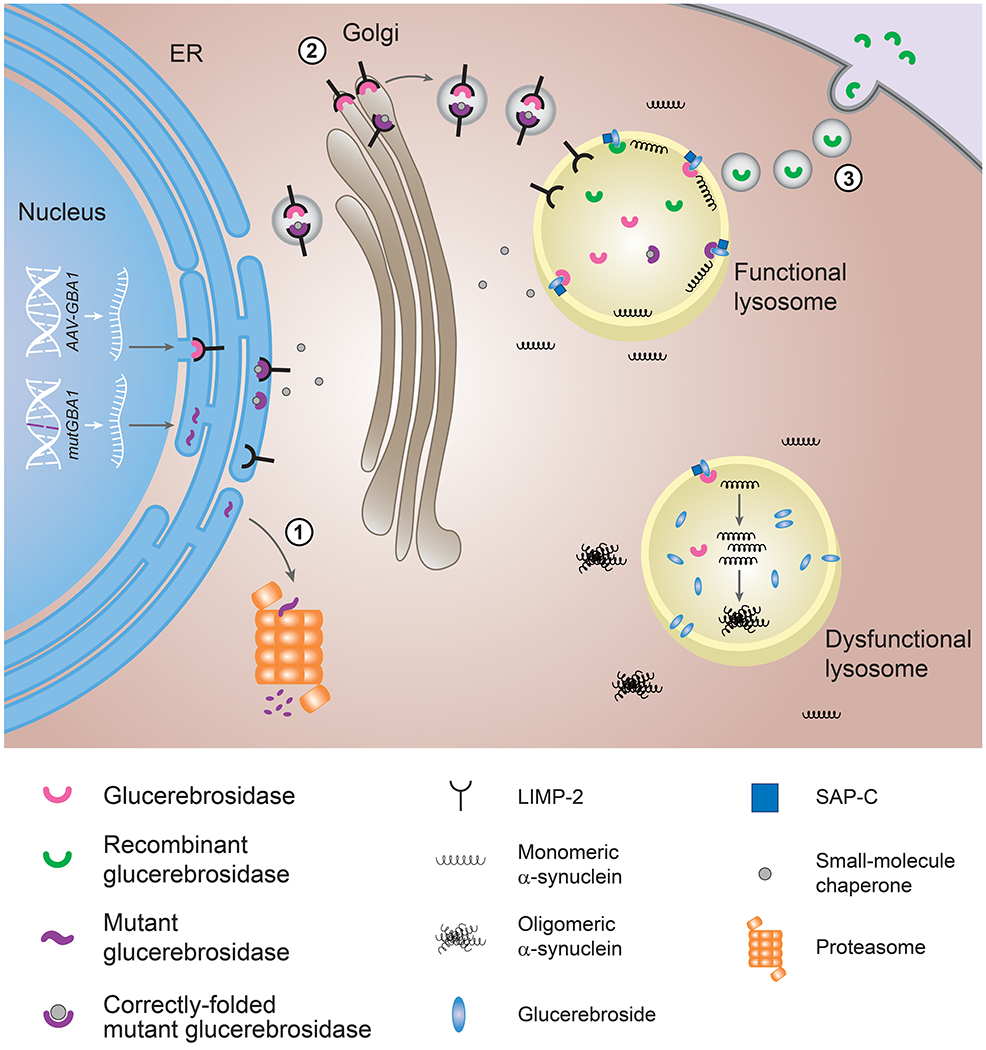
Therapeutic strategies to enhance glucocerebrosidase activity in lysosomes.
Mutant glucocerebrosidase is misfolded in the ER and degraded through proteasomes. Small-molecule chaperones (gray) bind to mutant glucocerebrosidase (purple) and facilitate its delivery to lysosomes. Glucocerebrosidase activity can also be restored by gene therapy to deliver the GBA1 gene into the host genome (AAV-GBA1) or by enzyme replacement therapy to supply recombinant glucocerebrosidase (green). LIMP-2 (black) aids to transport wildtype (pink) or mutant (purple) glucocerebrosidase to the lysosome and SAP-C (blue) is a cofactor on the lysosomal membrane necessary to activate glucocerebrosidase. α-Synuclein (black squiggles), mostly present as a monomer, tends to aggregate in dysfunctional lysosomes.
In collaboration with colleagues at the National Center for Advancing Translational Sciences (NCATS), our group has employed HTS to identify additional candidate chaperones. Initially a screen was performed using wild-type recombinant glucocerebrosidase, and several classes of chaperones were found, although most bound to the active site and were competitive inhibitors[60]. To find non-inhibitory chaperones, a novel strategy was developed. Tissue samples from a patient with Gaucher disease were used as the source of mutant enzyme in a second HTS of a small-molecule library of 250,000 compounds[61]. This led to the identification of compounds that enhanced enzymatic activity by non-competitively binding to glucocerebrosidase, including: NCGC607 and NCGC758 (Figure1). In order to better establish the efficacy of these lead compounds and to facilitate drug development, we then focused on developing relevant cell models including macrophage, midbrain dopaminergic neurons and astrocytes differentiated from induced pluripotent stem cells (iPSCs) generated using fibroblasts from patients with Gaucher disease[62–64]. Administration of NCGC607, a salicylic acid derivative, to human midbrain dopamine neurons differentiated from iPSCs from patients with both Gaucher disease and PD increased the delivery of glucocerebrosidase to lysosomes, reduced substrate accumulation, and decreased α-synuclein levels, indicating its potential as a treatment for PD[63]. Treatment with NCGC758, a pyrazolopyrimidine, similarly resulted in reduction of glucocerebrosidase substrates and the clearance of pathological α-synuclein in iPSC-derived human midbrain dopamine neurons including those carrying distinct mutations in SNCA, GBA1 or PARK9[65]. Importantly, the reduction of α-synuclein by NCGC758 was sufficient to reverse cellular pathologies downstream of pathological α-synuclein, including perturbations in hydrolase maturation and lysosomal dysfunction[65].
Recently, S-181, a small-molecule glucocerebrosidase modulator, was shown to increase wild-type glucocerebrosidase activity in iPSC-derived dopaminergic neurons from sporadic PD patients, as well as patients carrying a c.84insG frame-shift mutation in GBA1, and those with mutations in LRRK2, DJ-1, or PARKIN who had decreased glucocerebrosidase activity[66]. Treatment of these iPSC-derived dopaminergic neurons with S-181 partially restored lysosomal function and lowered levels of glucocerebroside, α-synuclein and oxidized dopamine. Moreover, S-181 treatment of mice heterozygous for the D409V GBA1 mutation (Gba1D409V/+) resulted in activation of wild-type glucocerebrosidase and consequent reduction of glucocerebrosidase substrates and α-synuclein levels in mouse brain tissue.
Overall, these preclinical studies of pharmacological chaperones of glucocerebrosidase indicate that using chemical chaperones to promote translocation of mutant glucocerebrosidase from the ER into lysosomes or to directly activate glucocerebrosidase in lysosomes may provide new therapeutic strategies with great potential for PD.
6. Conclusion
In summary, we have discussed the most recent advances in leveraging therapeutic strategies that have been developed for Gaucher disease to treat PD. The inverse relationship between glucocerebrosidase and α-synuclein suggest that modulation of brain glucocerebrosidase should be a promising approach for the treatment of PD with GBA1 mutations, as well as other forms of PD with decreased brain glucocerebrosidase activity.
7. Expert opinion
PD is a debilitating neurodegenerative disease with no disease modifying treatments available, and thus there is an urgent need to develop new therapies for this disease. The lysosomal enzyme glucocerebrosidase is extensively studied and has the potential to be an appropriate therapeutic target for developing effective treatments for PD. An inverse relationship between glucocerebrosidase and α-synuclein has emerged, based on the increased risk of PD in patients with Gaucher disease as well as GBA1 mutation carriers, and the reduced glucocerebrosidase activity observed in brain tissue and neurons from patients with idiopathic or other genetic forms of PD not linked to GBA1 mutations. Based on this relationship, it is speculated that enhancement of glucocerebrosidase activity or levels in lysosomes may reverse α-synuclein pathology and slow down PD progression.
While considerable progress has been made in therapeutic development, a thorough understanding of the mechanistic basis of the reciprocal relationship between glucocerebrosidase and α-synuclein is still essential in order to conceive rational treatment plans. Both loss- and gain-of-function theories have been proposed for this relationship. The loss-of-function hypothesis proposes that GBA1 mutations lead to a loss of- enzymatic activity and consequent decreased lysosomal activity and function, which might negatively affect α-synuclein processing. Gain-of-function theory posits that mutated glucocerebrosidase promotes α-synuclein aggregation and accumulation by disrupting proteostasis in the ER and/or compromising the accessibility of α-synuclein to lysosomal proteases in lysosomes. The contribution of these two mechanisms to the inverse relationship between glucocerebrosidase and α-synuclein need to be determined in order to evaluate different therapeutic options properly. For instance, gene therapy often focuses on delivering wild-type GBA1 to compensate the loss in activity of endogenous glucocerebrosidase. However, it is still unclear how the continued presence of mutated glucocerebrosidases will influence the therapeutic outcome. Given the versatility of small-molecule chaperones and activators, targeted therapies can be developed based on the underlying genetics of PD, and they are likely to have a major impact on this neurodegenerative disease. Several of the currently identified small-molecule chaperones appear to have therapeutic potential. However, it is not clear how these molecules interact with glucocerebrosidase and where they bind to the enzyme. Co-crystallization of the enzyme with the chaperones would enhance our understanding of these compounds and could help guide drug optimization. Further characterization of these small-molecule chaperones on their effects on specific GBA1 mutations would help to tailor treatment plans based on the genotypes of individual patients [67,68].
Although considered a strong risk factor for PD, mutations in GBA1 have rather limited penetrance and the vast majority of patient with Gaucher disease and GBA1 mutation carriers do not develop PD. The discordance for PD between siblings with the same GBA1 genotypes emphasizes the contribution of other modifiers[69]. Novel concepts and strategies are needed to better understand other pathways or proteins that may impact the disease penetrance in GBA1- associated PD. These unknown genetic or epigenetic modifiers may illuminate parts of the story that are yet uncovered.
One further challenge involves how lysosomal glucocerebrosidase activity is evaluated. Most laboratories depend on artificial substrates like 4-methylumbelliferyl-β-D-glucopyranoside (4MU-β-glc). However, since the relationship between GBA1 and PD likely involves the lysosomes, better methods to assess glucocerebrosidase activity specifically in this acidic compartment are needed. Improved assays will greatly enhance our ability to conduct additional small-molecule screens and to undertake SAR for drug design.
In recent years the number of publications regarding the association between mutations in GBA1 and PD have risen exponentially, and now far exceed the number of studies focused on Gaucher disease. The new models, cohorts and approaches being described are opening new avenues for investigation. We can only hope that a deeper probe into the aspects of protein mis-folding, lysosomal biology and lipidomics will provide the necessary insights to better guide logical drug development.
Thus, although an improved understanding of the mechanistic basis for GBA1-associated parkinsonism is still imperative, successfully enhancing glucocerebrosidase levels in the brain may have wide therapeutic implications. In the long run, gene therapy may ultimately provide the most dramatic and permanent result. However, currently, while the challenges related to gene therapy are still being addressed in different disorders, the development of less expensive and invasive small-molecule non-inhibitory chaperones or activators could significantly impact the disease course.
Article highlights.
Glucocerebrosidase, encoded by GBA1, breaks down glucocerebroside and glucosylsphingosine in lysosomes, and its deficiency leads to Gaucher disease, where undegraded glucocerebroside and glucosylsphingosine accumulate inside lysosomes.
After the initial observation in the clinics that Parkinson’s disease is more frequent in patients with Gaucher disease, as well as in their heterozygote family members, mutations in the GBA1 are now recognized as a common risk factor for Parkinson’s disease.
An inverse relationship exists between glucocerebrosidase and α-synuclein, whose aggregation in Lewy bodies and Lewy neurites is the hallmark of Parkinson’s disease pathophysiology.
While enzyme replacement therapy (ERT) greatly improves visceral symptoms of Gaucher disease, it does not alleviate neurological manifestations because recombinant glucocerebrosidase does not cross the blood-brain barrier (BBB). Gene therapy and small-molecule chaperones and activators are promising approaches to treat patients with neurological involvement.
It is imperative to better understand the mechanistic basis for neuronopathic Gaucher disease and GBA1-associated parkinsonism in order to devise rational treatment plans.
Acknowledgements
The authors acknowledge the assistance of Julia Fekecs for the preparation of Figure 2.
Funding
This research was supported by the Intramural Research Program of the National Human Genome Research Institute and the National Institutes of Health.
Declaration of interests
The authors all declare funding support from the Intramural Research Program of the National Human Genome Research Institute and the National Institutes of Health. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Footnotes
Reviewer disclosures
A peer reviewer on this manuscript has closely collaborated with one of the authors and has worked with them on results described in this manuscript. The paper was independently reviewed by four further referees and the peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- [1].Poewe W, Seppi K, Tanner CM, et al. Parkinson’s disease. Nat. Rev. Dis. Prim 2017;3:1–21. [DOI] [PubMed] [Google Scholar]
- [2].Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson’s disease and the neurobiology of axons. Ann. Neurol 2010;67:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes RGM. a -Synuclein in Lewy bodies. Nature. 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- [4].Stefanis L Alpha synuclein in parkinson’s disease. Cold Spring Harb Perspect Med. 2014;2:691–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bellomo G, Paciotti S, Gatticchi L, et al. The vicious cycle between α-synuclein aggregation and autophagic-lysosomal dysfunction. Mov. Disord 2019;35:34–44. [DOI] [PubMed] [Google Scholar]
- [6].Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson’s disease. Am. J. Hum. Genet 2011;89:162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. [DOI] [PubMed] [Google Scholar]
- [9].Anand VS, Braithwaite SP. LRRK2 in Parkinson’s disease: Biochemical functions. FEBS J. 2009;276:6428–6435. [DOI] [PubMed] [Google Scholar]
- [10].Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Large meta-analysis of current GWAS evaluations in subjects with Parkinson’s disease
- [11].Brady R, Kanfer J, Shapiro D. the metabolism of Glucocerebrosides. J. Biol. Chem 1965;240:39–43. [PubMed] [Google Scholar]
- [12].Siebert M, Sidransky E, Westbroek W. Glucocerebrosidase is shaking up the synucleinopathies. Brain. 2014;137:1304–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aflaki E, Westbroek W, Sidransky E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron. 2017;93:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Comprehensive review detailing different theories regarding the basis of the link between GBA1 and Parkinsonism
- [14].Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11:986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mistry PK, Lopez G, Schiffmann R, et al. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab 2017;120:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Beutler E, Nguyen NJ, Henneberger MW, et al. Gaucher disease: Gene frequencies in the Ashkenazi Jewish population. Am. J. Hum. Genet 1993;52:85–88. [PMC free article] [PubMed] [Google Scholar]
- [17].Hruska KS, LaMarca ME, Scott CR, et al. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat 2008;29:567–583. [DOI] [PubMed] [Google Scholar]
- [18].Tayebi N, Callahan M, Madike V, et al. Gaucher disease and parkinsonism: A phenotypic and genotypic characterization. Mol. Genet. Metab 2001;73:313–321. [DOI] [PubMed] [Google Scholar]
- [19].Tayebi N, Walker J, Stubblefield B, et al. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab 2003;79:104–109. [DOI] [PubMed] [Google Scholar]; *Early observation in the clinics of the association between Gaucher disease and Parkinson’s disease.
- [20].Goker-Alpan O, Schiffmann R, LaMarca ME, et al. Parkinsonism among Gaucher disease carriers. J. Med. Genet 2004;41:937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sidransky E, Nalls M, Aasly J, et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. 2009;1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** First multicenter analysis of GBA1 mutations in subjects with Parkinson’s disease.
- [22].Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet 2009;41:1303–1307. [DOI] [PubMed] [Google Scholar]
- [23].Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet 2009;41:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mazzulli JR, Xu Y-H, Sun Y, et al. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell. 2011;146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Important paper describing bi-directional loop theory in GBA1-associated Parkinson’s disease
- [25].Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol 2016;32:223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol 2019;21:133–142. [DOI] [PubMed] [Google Scholar]
- [27].Sala G, Marinig D, Arosio A, et al. Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front. Mol. Neurosci 2016;9:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mazzulli JR, Zunke F, Isacson O, et al. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. P.N.A.S 2016;113:1931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yap TL, Gruschus JM, Velayati A, et al. α-Synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and gaucher diseases. J. Biol. Chem 2011;286:28080–28088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Taguchi YV, Liu J, Ruan J, et al. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated parkinson’s disease. J. Neurosci 2017;37:9617–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stojkovska I, Krainc D, Mazzulli JR. Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 2018;373:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Balestrino R, Schapira AHV. Glucocerebrosidase and Parkinson’s disease: Molecular, Clinical, and Therapeutic Implications. Neuroscientist. 2018;24:540–559. [DOI] [PubMed] [Google Scholar]
- [33].Gegg ME, Burke D, Heales SJR, et al. Glucocerebrosidase deficiency in substantia nigra of Parkinson’s disease brains. Ann. Neurol 2012;72:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Murphy KE, Gysbers AM, Abbott SK, et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain. 2014;137:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138:2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Maor G, Rapaport D, Horowitz M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet 2019;00:1–14. [DOI] [PubMed] [Google Scholar]
- [37].McGlinchey RP, Dominah GA, Lee JC. Taking a Bite out of Amyloid: Mechanistic Insights into α-Synuclein Degradation by Cathepsin L. Biochemistry. 2017;56:3881–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yap TL, Jiang Z, Heinrich F, et al. Structural features of membrane-bound glucocerebrosidase and α-synuclein probed by neutron reflectometry and fluorescence spectroscopy. J. Biol. Chem 2015;290:744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sardi SP, Clarke J, Kinnecom C, et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. U. S. A 2011;108:12101–12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sardi SP, Clarke J, Viel C, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. U. S. A 2013;110:3537–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Work in animal model supporting the development of gene therapy stratedies for Parkinson’s disease
- [41].Hudry E, Vandenberghe LH. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron. 2019;101:839–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rocha EM, Smith GA, Park E, et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis 2015;82:495–503. [DOI] [PubMed] [Google Scholar]
- [43].Chauhan A, Tikoo A, Kapur AK, et al. The taming of the cell penetrating domain of the HIV Tat: Myths and realities. J. Control. Release. 2007;117:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gillmeister MP, Betenbaugh MJ, Fishman PS. Cellular trafficking and photochemical internalization of cell penetrating peptide linked cargo proteins: A dual fluorescent labeling study. Bioconjug. Chem 2011;22:556–566. [DOI] [PubMed] [Google Scholar]
- [45].Fu A, Wang Y, Zhan L, et al. Targeted delivery of proteins into the central nervous system mediated by rabies virus glycoprotein-derived peptide. Pharm. Res 2012;29:1562–1569. [DOI] [PubMed] [Google Scholar]
- [46].Fu A, Zhang M, Gao F, et al. A Novel Peptide Delivers Plasmids across Blood-Brain Barrier into Neuronal Cells as a Single-Component Transfer Vector. PLoS One. 2013;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fu A, Zhao Z, Gao F, et al. Cellular uptake mechanism and therapeutic utility of a novel peptide in targeted-delivery of proteins into neuronal cells. Pharm. Res 2013;30:2108–2117. [DOI] [PubMed] [Google Scholar]
- [48].Gramlich PA, Westbroek W, Feldman RA, et al. A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: Design, production, and assessment. J. Biotechnol 2016;221:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Martínez-Bailén M, Carmona AT, Patterson-Orazem AC, et al. Exploring substituent diversity on pyrrolidine-aryltriazole iminosugars: Structural basis of β-glucocerebrosidase inhibition. Bioorg. Chem 2019;86:652–664. [DOI] [PubMed] [Google Scholar]
- [50].Mena-Barragán T, García-Moreno MI, Sevšek A, et al. Probing the inhibitor versus chaperone properties of sp2-iminosugars towards human β-glucocerebrosidase: A picomolar chaperone for gaucher disease. Molecules. 2018;23:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Baudoin-Dehoux C, Castellan T, Rodriguez F, et al. Selective targeting of the interconversion between glucosylceramide and ceramide by scaffold tailoring of iminosugar inhibitors. Molecules. 2019;24:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Khanna R, Benjamin ER, Pellegrino L, et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of β-glucosidase. FEBS J. 2010;277:1618–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sun Y, Liou B, Xu YH, et al. Ex vivo and in vivo effects of isofagomine on acid β-glucosidase variants and substrate levels in Gaucher disease. J. Biol. Chem 2012;287:4275–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Maegawa GHB, Tropak MB, Buttner JD, et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem 2009;284:23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Luan Z, Li L, Higaki K, et al. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. 2013;35:317–322. [DOI] [PubMed] [Google Scholar]
- [56].Migdalska-Richards A, Daly L, Bezard E, et al. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol 2016;80:766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells, Mol. Dis 2013;50:134–137. [DOI] [PubMed] [Google Scholar]
- [58].Migdalska-Richards A, Ko WKD, Li Q, et al. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse. 2017;71:17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mullin S, Smith L, Lee K, et al. Ambroxol for the Treatment of Patients with Parkinson’s disease with and without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zheng W, Padia J, Urban DJ, et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc. Natl. Acad. Sci. U. S. A 2007;104:13192–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Goldin E, Zheng W, Motabar O, et al. High throughput screening for small molecule therapy for gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLoS One. 2012;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Aflaki E, Stubblefield BK, Maniwang E, et al. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci. Transl. Med 2014;6:240ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Aflaki E, Borger DK, Moaven N, et al. A New Glucocerebrosidase Chaperone Reduces -Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci 2016;36:7441–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Describes the effect of the glucocerebrosidase chaperone NCGC607 in neurons from subjects with Gaucher disease with and without Parkinson’s disease.
- [64].Aflaki E, Stubblefield BK, McGlinchey RP, et al. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis 2020;134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mazzulli JR, Zunke F, Tsunemi T, et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci 2016;36:7693–7706. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Detatils the effect of chaperone NCGC758 in dopamnnergic neurons generated from patients with Parkinson’s disease
- [66].Burbulla LF, Jeon S, Zheng J, et al. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci. Transl. Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Thirumal Kumar D, Iyer S, Christy JP, et al. A comparative computational approach toward pharmacological chaperones (NN-DNJ and ambroxol) on N370S and L444P mutations causing Gaucher’s disease 1st ed. Adv. Protein Chem. Struct. Biol Elsevier Inc.; 2019. [DOI] [PubMed] [Google Scholar]
- [68].Ivanova MM, Changsila E, Turgut A, et al. Individualized screening for chaperone activity in gaucher disease using multiple patient derived primary cell lines. Am. J. Transl. Res 2018;10:3750–3761. [PMC free article] [PubMed] [Google Scholar]
- [69].Lopez G, Steward A, Ryan E, et al. Clinical Evaluation of Sibling Pairs With Gaucher Disease Discordant for Parkinsonism. Mov. Disord 2019;1–7. [DOI] [PubMed] [Google Scholar]