

Abstract
Aim: Inflammation plays a significant role in the pathogenesis of human abdominal aortic aneurysm (AAA). AEBP1 can promote activation of the NF-κB pathway, subsequently affecting the expression of NF-κB target genes, including inflammatory cytokines and matrix metalloproteinases (MMPs). Our objective was to examine the role of AEBP1 in the development of AAA and characterize the underlying mechanism.
Methods: ITRAQ, RT-PCR, western blot, immunohistochemistry, and ELISA were used to compare different experimental groups with the controls and to determine the differentially expressed genes. We generated an AAA model using porcine pancreatic elastase in Sprague-Dawley rats and silenced their AEBP1 in vivo by adenoviruses injected intra-adventitially. We also silenced or overexpressed AEBP1 in human vascular smooth muscle cells in vitro in the presence and in the absence of NF-κB inhibitor BAY 11-7082.
Results: Proteome iTRAQ revealed a high expression of AEBP1 in AAA patients, which was verified by qRT-PCR, western blot, immunohistochemistry, and ELISA. The mean expression level of AEBP1 in AAA patients was higher than that in controls. Along with AEBP1 upregulation, we also verified mis-activation of NF-κB in human AAA samples. The in vivo studies indicated that AEBP1 knockdown suppressed AAA progression. Finally, the in vitro studies illustrated that AEBP1 promotes activation of the NF-κB pathway, subsequently upregulating pro-inflammatory factors and MMPs.
Conclusions: Our results indicate a role of AEBP1 in the pathogenesis of AAA and provide a novel insight into how AEBP1 causes the development of AAA by activating the NF-κB pathway.
Keywords: AEBP1, Abdominal aortic aneurysm, Inflammation, VSMC, NF-κB pathway
Introduction
Abdominal aortic aneurysm (AAA) is a vascular disease characterized by permanent, localized full-thickness dilation of the abdominal aorta, with high incidences of morbidity and mortality1). Parallel to the increase in the global mean age, the incidence of AAA has increased seven-fold in the past 30 years2, 3). The typical pathological signatures of AAA include inflammatory infiltration in the adventitia and tunica media, aortic elastin proteolytic degradation, and pathological remodeling4). Vascular inflammation has been regarded as the primary determinant of AAA5). It proceeds with gradual infiltration of many inflammatory cell types, including macrophages, lymphocytes, mast cells, and neutrophils, into the intima from the adventitia. These cells subsequently secrete various inflammatory cytokines6–9) and matrix metalloproteinases (MMPs)10, 11) and induce smooth muscle cell apoptosis12). These events are considered key factors directly associated with AAA formation and progression.
Adipocyte enhancer binding protein 1 (AEBP1) is a transcriptional repressor that modulates the activity of the adipogenesis aP2 gene, which was first identified during adipocyte differentiation13). Recently, AEBP1 has been identified in macrophages as a novel pro-inflammatory mediator involved in macrophage cholesterol homeostasis and inflammatory responsiveness14, 15). More recently, Majdalawieh et al. reported that AEBP1 manifests its pro-inflammatory effects by promoting NF-κB activity through impeding the inhibitory function of IκBα in macrophages, which likely occurs through AEBP1-IκBα physical interaction16).
NF-κB is a nuclear transcriptional factor that is widely expressed in eukaryotic cells and has a wide range of biological functions. It can identify and bind κB sequences in gene promoters, thereby promoting the expression of the corresponding target genes17). In the canonical NF-κB pathway, the various stimuli that activate NF-κB cause phosphorylation of IκBα, followed by IκBα's ubiquitination and subsequent degradation. This results in the exposure of the nuclear localization signals on NF-κB subunits and their subsequent translocation to the nucleus. In the nucleus, NF-κB binds to the κB sequence of various genes, thereby activating their transcription. The NF-κB pathway can promote chronic inflammation of vascular walls and regulate transcription of MMPs18). Animal model experiments have revealed that inhibiting the activity of NF-κB can delay occurrence and development of AAA19, 20).
Aim
Given the abundance of AEBP1 in AAA patients and its association with two AAA inducers, inflammation and NF-κB, the present study aimed to examine the cause–effect relationship between AEBP1 and AAA.
Materials and Methods
Clinical Samples
Patients at The First Affiliated Hospital of China Medical University with an abdominal aorta diameter > 5.5 cm (n = 38) were recruited for this study. Patients with Ehlers–Danlos syndrome, Marfan syndrome, and other known vascular disorders were excluded. The healthy control (HC) group (n = 38) comprised age- and sex-matched healthy individuals. They had a relatively healthy peripheral vascular system, as determined by ultrasonography, and indicated no evidence or medical history of aneurysm or other vascular disorders. Because the AAA patients also had prominent atherosclerosis21), we identified age- and sex-matched patients with infrarenal aortic atherosclerotic occlusive disease (AOD) in the same hospital (n = 12) as a second control group. These patients had no medical history of aneurysmal disease, other vascular disorders, and connective tissue disorders. AOD was diagnosed by enhanced CT scanning or 3D-CTA, and this group was used as a control for the involvement of atherosclerosis, especially in early lesions (Table 1).
Table 1. Comparison of the Clinical Features Between AAA , AOD and HC group.
|
P value |
|||||
|---|---|---|---|---|---|
| AAA (n = 38) | AOD (n = 12) | HC (n = 38) | AAA vs AOD | AAA vs HC | |
| Gender (Male) | 32 (84.1%) | 10 (83.3%) | 30 (78.9%) | 0.94 | 0.55 |
| Age (years) | 61.95 ± 9.69 | 61.00 ± 11.35 | 58.11 ± 11.66 | 0.72 | 0.17 |
| Smoking history | 16 (42%) | 5 (42%) | 14 (37%) | 0.97 | 0.63 |
| Hypertension history | 18 (47%) | 6 (50%) | 16 (42%) | 0.87 | 0.65 |
| Diabetes history | 7 (18%) | 3 (25%) | 5 (13%) | 0.62 | 0.53 |
| WBC count (*109/L) | 6.65 ± 1.82 | 7.01 ± 0.87 | 6.99 ± 1.36 | 0.58 | 0.57 |
| Lymph count (*109/L) | 1.62 ± 0.62 | 1.53 ± 0.60 | 1.65 ± 0.42 | 0.57 | 0.40 |
| Triglyceride (mmol/L) | 1.47 ± 0.56 | 1.35 ± 0.87 | 1.29 ± 0.44 | 0.24 | 0.17 |
| Total cholesterol (mmol/L) | 4.10 ± 1.23 | 4.05 ± 0.77 | 4.13 ± 0.65 | 0.84 | 0.87 |
| HDL-C (mmol/L) | 1.09 ± 0.29 | 1.01 ± 0.25 | 1.02 ± 0.21 | 0.48 | 0.31 |
| LDL-C (mmol/L) | 2.92 ± 1.08 | 2.52 ± 0.68 | 2.63 ± 0.54 | 0.22 | 0.09 |
| Diameter (mm) | 57.12 ± 13.16 | 18.67 ± 0.88 | 18.52 ± 0.95 | < 0.001 | < 0.001 |
Continuous data are presented as mean ± SD. Data from three groups showed no significant difference in associated risk factors. The diameter of abdominal aorta in AAA group is significantly different to those in AOD group and HC group.
AAA tissues (n = 20) and AOD tissues (n = 8) were respectively obtained from the patients in the AAA group (n = 38) and the AOD group (n = 12), who then underwent open surgery. HC tissues (n = 12) were derived from organ donors (HC group, n = 38). Tissues were either frozen at −80°C for qRT-PCR and western blot analyses or dehydrated and embedded in paraffin for immuno-histochemistry/fluorescence analysis. The clinical procedures followed in the study were approved by the Institutional Review Board of China Medical University and the local ethical committee. All participants provided written informed consent.
Proteome iTRAQ
Vascular wall tissues from three individuals from each of the AAA, AOD, and HC groups were collected for proteome iTRAQ analysis. The collected samples were processed for iTRAQ analysis in KangChen Bio-tech. The output was visualized using R and CummeRbund. The heatmaps were generated in R using pHeatmap and R ColorBrewer. The results were analyzed according to uniprot20160315_human database and displayed using Thermofisher Proteome Discoverer 1.3/1.4.
Rat Models
The animals were handled according to the guidelines described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All the experimental protocols were approved by the Animal Research Ethics Committee of China Medical University. Fifty-five male SD rats (Liaoning Changsheng Biotechnology, Dalian, China) weighing 250–280 g were used in this study. The rats were randomly divided into four groups and subjected to the following treatments: the sham group (operation only, n = 10), the control group (elastase perfusion only, n = 25), the con + AV group (elastase perfusion + empty adenovirus, n = 10) and the con + shAEBP1 group (elastase perfusion + adenovirus with AEBP1 shRNA, n = 10). The AAA rat experimental model was established using elastase infusion following established protocols22, 23). Briefly, the rats were first given general anesthesia (a gas mixture of 2.0%–2.5% isoflurane and oxygen), and a long midline abdominal incision was then made to expose the abdominal aorta. The aorta was then isolated from the level of the left renal vein toward the bifurcation. All lumbar branches of the exposed infrarenal aorta were ligated. An aortotomy was performed, and a PE10 catheter was inserted into the aorta. The upper and the lower ends of the catheter were ligated temporarily to create a closed lumen, and porcine pancreatic elastase (20 U/mL) (Solarbio, Beijing, China) was perfused for 15 min. Before closing the aortotomy, heparin (50 U) was injected in to prevent thrombosis. For the con + AV and con + shAEBP1 groups, 40 µL adenovirus (1 × 1011/mL) was injected into the intra-adventitial space from at least four positions. The specimens were collected after 28 days.
Cell Culture
293A, 293T, and human VSMC cells (ATCC CRL1999) were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (PAN-biotech, Leipzig, Germany). Adenovirus and lentivirus were packaged in 293A and 293T cells, respectively. Adenovirus and Lentivirus were concentrated through high-speed centrifugation. The shRNA sequences targeting human AEBP1 lentivirus and rat AEBP1 adenovirus were 5- GCGATGACATGGACTATTACctc-gagGTAATAGTCCATGTCATCGc-3 and 5-cgaaaGGAGGAAAGGAAGGAAGTTGActcgagTCAACTTCCTTCCTTTCCTCCAAAA-3, respectively.
For lentivirus transfection, VSMC cells were first seeded into a 24-well plate with DMEM. After 24 h, the culture medium was replaced with a fresh medium containing 2 µg/mL polybrene (Solarbio, Beijing, China) and concentrated lentivirus. Forty-eight hours later, the positive transfected cells were subjected to selection using 8 µg/mL puromycin (Solarbio, Beijing, China) for 1 week. The selected positive cells were then used for the subsequent functional studies. The NF-κB pathway inhibitor BAY11-7082 was purchased from MCE (MedChemExpress USA, New Jersey, USA) and used at a final concentration of 10 µM in cell culture. It was dissolved in DMSO [< 0.1% (v/v) to avoid any toxicity to the cells].
qRT-PCR
Total RNA was extracted from the cells using the TRIzol reagent (Invitrogen, Carlsbad, USA), as per the manufacturer's instructions. RNA quality and quantity were assessed using NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, USA). The total RNA was reverse-transcribed using PrimeScript reverse transcriptase (Takara) and oligo-dT primers according to the manufacturer's instructions. Gene expression was measured by qPCR using a MyiQTM real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). The sequences of the primers used are as follows: for human AEBP1: Forward: 5- GAGGAGTTGGAGGAGGAGTGGAC -3, Reverse: 5-AGGAGGCTCGGATCTGGTTGTC -3; for human NF-κB (p65): Forward: 5- AGGCTCCTGTGCGTGTCTCC -3, Reverse: 5- TCGTCTGTATCTGGCAGGTACTGG -3; for human IκBα: Forward: 5-TCCACTCCATCCTGAAGGCTACC -3, Reverse: 5- AGGTCCACTGCGAGGTGAAGG -3; and for human GAPDH: Forward: 5″- CATCACTGCCACCCAGAAGACTG -3″, Reverse: 5- ATGCCAGTGAGCTTCCCGTTCAG -3. GAPDH was used as the internal control for normalization of the relative expression values. The fold change in gene expression was calculated using the 2−ΔΔCT formula.
Western Blot
Cell and tissue samples were lyzed with RIPA lysis buffer (Solarbio, Beijing, China) including 1:100 PMSF (Solarbio, Beijing, China) and 1:100 protease inhibitor cocktail (Roche, Indianapolis, USA), and the lyzed samples were quantified with the BCA assay (Thermo Scientific, Waltham, MA, USA). After adding the 4X SDS loading buffer followed by boiling, 30 µg total protein from each sample was separated on Bis-Tris polyacrylamide gels and transferred onto PVDF membranes (Millipore Corporation, Bedford, MA, USA). The samples were then immunoblotted overnight at 4°C. The following antibodies were used: AEBP1 (SC-271374) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); IκBα (4812S) (Cell Signaling Technology, Inc., Danvers, MA, USA); NF-κB p65 (ab16502), Phospho-NF-κB p65 (ab86299), MMP2 (ab92536), MMP9 (ab38898), TNF-α (ab6671), IL-1β (ab9722), and IL-6 (ab9324) (Abcam, Cambridge, USA); MCP-1 (Waileibio, Shenyang, China); GAPDH and Lamin B1 (Proteintech, Wuhan, China). GAPDH and Lamin B1 were used as internal controls.
Immunohistochemical Staining
Paraffin-embedded sections were blocked with 5% normal goat serum for 30 min and then incubated overnight with rabbit anti-AEBP1 (ab222147, Abcam, Cambridge, USA) (1:1000) in PBS at 4°C. The sections were then incubated with goat anti-rabbit secondary antibody (1:500) for 20 min. All the sections were incubated for 20 min with the avidin-biotin peroxidase complex (Vectastain ABC kit, Burlingame, CA, USA) at room temperature. Labeling was visualized using a 0.075 mg/mL diaminobenzidine solution activated with 0.1% H2O2. All the sections were thoroughly rinsed with PBS between the staining steps. The integrated optical density (IOD) of the positively stained area was analyzed at 400× magnification using an image analysis software (Image-Pro Plus 6.0).
Immunofluorescence Staining
Paraffin-embedded sections were blocked with 5% normal goat serum for 30 min and then incubated overnight with the corresponding antibody (1:1000) in PBS at 4°C. The sections were rinsed with PBS-Tween 20 and then incubated with a CY3- or FITC-labeled secondary antibody (1:500) for 50 min. Thereafter, the sections were rinsed three times with PBS-Tween 20 and counterstained with DAPI, followed by rinsing as before. The sections were dehydrated, mounted with an anti-fade mounting medium, and covered with a coverslip. Nuclear P-p65 was evaluated at 400× magnification using an image analysis software (Image-Pro Plus 6.0).
Histological Studies
All rats were sacrificed after 28 days. The aortas were fixed in polyformaldehyde and processed for paraffin embedding. Aortic tissue sections (5 µm) were stained with normal hematoxylin and eosin, Elastic Van Gieson (EVG) staining (for elastin) and Masson staining (for collagen) followed standard procedures. Images of the sections were captured with a microscope system (IX71; Olympus, Tokyo, Japan)
Enzyme-Linked Immunosorbent Assay
For the detection of serum AEBP1- (CLOUDCLONE CORP, Wuhan, China) and VSMC-secreted AEBP1, TNF-α (R&D Systems, Minneapolis, MN), IL-10 (R&D Systems, Minneapolis, MN), IL-6 (Sino Biological, Beijing, China), and IL-1β (Sino Biological, Beijing, China) in the cell culture after 24 h, we used commercial enzyme-linked immunosorbent assay (ELISA) kits following the manufacturer's guidelines. Briefly, 100 µL of the sample was used per well, and the plate was incubated with the antibodies for 1 h at room temperature. After the incubation, the wells were washed three times, and 100 µL of each chromogenic substrates A and B was added. The plate was then incubated in the dark for 20 min at 37°C. Finally, 50 µL of a stop solution was added to each well to stop the reaction, and the absorbance was measured at 450 nm. Expression values were calculated using a standard curve.
Co-Immunoprecipitation
The Pierce Classic IP Kit (Thermo Scientific, Waltham, MA, USA) was used to perform co-immunoprecipitation (Co-IP) assays following the manufacturer's protocols. The Pierce AminoLink Plus Coupling Resin was washed twice with 200 mL of the coupling buffer and then mixed with an anti-AEBP1 or anti-IκBα antibody in a spin column and incubated for 2 h with shaking at 4 °C. Untreated VSMCs were lyzed with IP lysis buffer and ultrasonic decomposition on ice for 30 min; the lysate was then precleared with a control agarose resin for 1 h according to the manufacturer's protocol. Next, the pre-cleared lysate was added into the pre-treated spin columns mentioned above and incubated overnight at 4 °C. On the next day, after several wash and centrifugation steps, IP samples were eluted from the spin columns with 40 µL of the elution buffer. The samples were boiled and analyzed with western blot as described above.
Statistical Analysis
A statistical analysis was carried out using the SPSS 20.0 (SPSS Inc, Chicago, IL, USA) software, and graphs were prepared using the Graph Prism 7.0 (GraphPad, San Diego, CA, USA) software. The Kolmogorov–Smirnov test was used to verify whether the data were normally distributed. Continuous variables were analyzed using one-way ANOVA for univariate analysis and two-way ANOVA for multivariate analysis. A nonparametric Mann–Whitney U test was used in the cases where the data were not normally distributed. All data are presented as mean ± SEM. In all the analyses, p < 0.05 was considered statistically significant.
Results
AEBP1 is upregulated in AAA patients
To evaluate the expression pattern of AEBP1 in the abdominal aorta during an aneurysm, we analyzed the vascular wall tissues of three AAA patients with proteome iTRAQ, in comparison with those obtained from three AOD and three HC individuals. AEBP1 was found to be significantly upregulated in AAA patients compared to AEBP1 in AOD and HC individuals (Fig. 1).
Fig. 1.
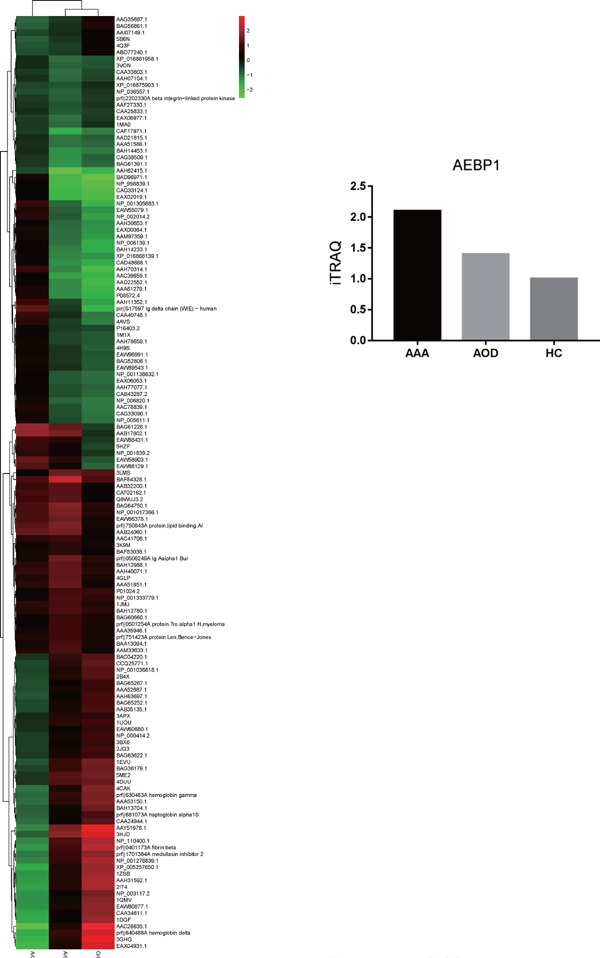
Summary of changes in the global protein expression profiles, as revealed by proteome iTRAQ
The vascular wall was sampled from AAA patients, AOD patients, and HC individuals (n = 3) and processed for proteome iTRAQ. The heatmap indicates the global protein expression profile for AAA, AOD, and HC individuals. The protein level in the HC group was used as the reference.
To elaborate the relationship between AEBP1 and AAA, we evaluated AEBP1 expression in the vascular wall tissues of 20 AAA, 8 AOD, and 12 HC individuals using qRT-PCR and the western blot. Both analyses indicated that AEBP1 was significantly upregulated in AAA patients compared with AEBP1 in AOD and HC individuals (Fig. 2A–C). The ELISA analysis of serum samples from AAA (n = 38), AOD (n = 12), and HC (n = 38) groups indicated that the level of circulating AEBP1 was significantly upregulated in AAA patients (Fig. 2D). The immunohistochemistry (IHC) analysis also indicated a high AEBP1 expression in the vascular wall of AAA patients (Fig. 2E, F). These results revealed that AEBP1 expression (cellular mRNA/protein and circulating protein levels) was significantly upregulated in AAA patients compared with that in AOD and HC individuals. Overall, these results suggest that AEBP1 may play a role in the development of AAA.
Fig. 2.
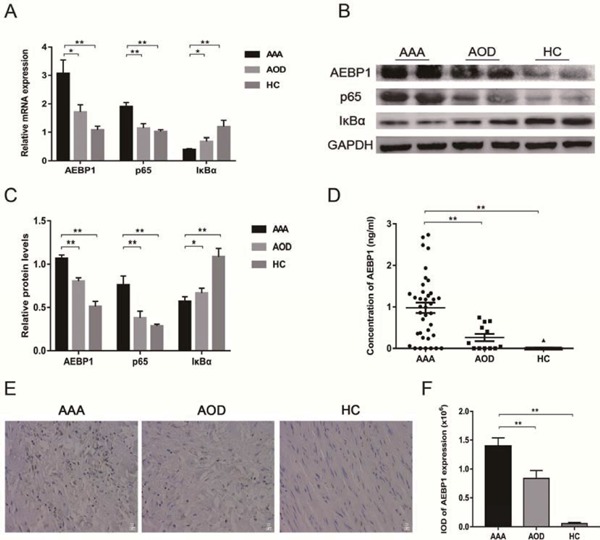
AEBP1 expression is upregulated and the NF-κB pathway is activated in the clinical samples of AAA patients
The vascular wall tissues were sampled from 20 AAA patients, 8 AOD patients, and 12 HC individuals for qRT-PCR, the western blot, and IHC staining, in addition to the blood samples collected from them for ELISA. (A) mRNA expression levels of AEBP1, NF-κB subunit p65, and NF-κB inhibitor IκBα were measured by qRT-PCR. The relative expression against GAPDH is presented (mean ± SD; Mann–Whitney, *p < 0.05; **p < 0.01). (B) The corresponding protein levels of AEBP1, NF-κB p65, and IκBα were evaluated using the western blot (with GAPDH as the loading control). (C) Quantification of the protein bands from three independent groups. The relative expression against GAPDH is presented (mean ± SD; Mann–Whitney, *p < 0.05; **p < 0.01). (D) Circulating levels of AEBP1 were measured using an ELISA kit (AAA group n = 38; AOD group n = 12; HC group n = 38) (mean ± SD; Mann-Whitney, **p < 0.01). (E) IHC staining and micrographs (400×) indicating AEBP1 expression (n = 5). (F) Quantification of IOD in AEBP1 using IHC staining (mean ± SD; one-way ANOVA, **p < 0.01).
The NF-κB pathway is mis-activation in AAA patients
We hypothesized that inhibition of NF-κB activity suppressed the occurrence and development of AAA. Accordingly, we first examined the NF-κB pathway in AAA patients. The NF-κB subunit p65 and its inhibitor IκBα were used as markers for the NF-κB pathway. qRT-PCR (Fig. 2A) and the western blot (Fig. 2B, C) revealed that p65 and IκBα were respectively up- and down-regulated in AAA patients, indicating that the NF-κB pathway is induced during aneurysm. Activation of the NF-κB pathway proceeds with translocation of NF-κB subunits into the nucleus. Therefore, we next assessed translocation of P-p65 in AAA, AOD, and HC groups. The western blot analysis indicated that P-p65 levels were higher in the vascular nuclear extracts of AAA patients relative to those of AOD and HC individuals (Fig. 3A, B). These results were confirmed with immunofluorescence (IF) analysis of the vascular wall tissue samples (Fig. 3C, D). Thereby, the samples were found to be in the order of AAA > AOD > HC in terms of their nuclear P-p65 content. Overall, these results indicated that the NF-κB pathway was aberrantly activated in AAA patients.
Fig. 3.
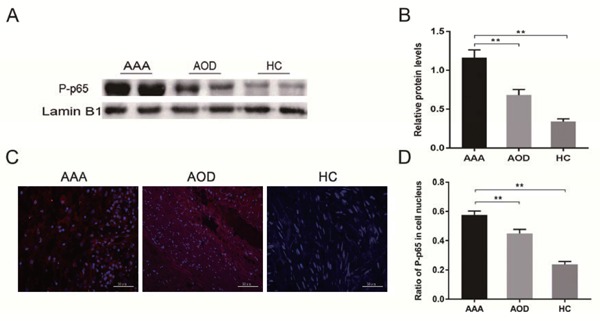
The NF-κB pathway is activated in the clinical samples of AAA patients
(A) Nuclear proteins were extracted from the vascular wall tissues of AAA, AOD, and HC individuals, and P-p65 expression was evaluated using the western blot. (B) Quantification of the protein bands from three independent groups. The relative expression against Lamin B1 is indicated (mean ± SD; Mann–Whitney, **p < 0.01). (C) Immunofluorescent staining and micrographs (400×) indicated P-p65 expression in the tissues (n = 5). (D) Quantification of the proportion of P-p65 positive nuclei (mean ± SD; one-way ANOVA, **p < 0.01).
Konck down of AEBP1 inhibits the development of AAA in an elastase-induced rat model
To elaborate the relationship between AEBP1 and the development of AAA, we generated an AAA SD rat model using porcine pancreatic elastase. We successfully established aortic aneurysm in this model (Table 2), as the aorta was clearly dilated in the control (only elastase-treated) and empty adenovirusinfected (con + AV) groups compared with the aorta in the sham group 28 days after the surgery. However, after transfection of the aorta with AEBP1 shRNA (con + shAEBP1), the dilatation was almost completely suppressed (Fig. 4A). Hematoxylin-eosin staining indicated that arterial smooth muscle cells were disorganized and considerably infiltrated with inflammatory cells in the control and con + AV groups compared with those in the sham group. Notably, shAEBP1 transfection suppressed the inflammatory cell infiltration. EVG staining revealed that elastic fibers in the media layers of the control and con + AV aortas were badly damaged and displayed irregular, discontinuous morphology with fractures, fragmentation, and loss of normal curvature. Importantly, elastic fibers remained relatively intact in the con + shAEBP1 group and displayed distinct layers with a regular, continuous wave-like pattern and fine curvature. In addition, Masson staining indicated deposition of collagen fibrils in the control and con + AV groups, whereas shAEBP1 partly suppressed the symptoms (Fig. 4B). These results indicated that AEBP1 silencing partly suppressed elastase-induced AAA development in the rat.
Table 2. Dilatation ratio for AEBP1 knockdown in AAA rat models.
| Group | Day 0 (mm) | Day 28 (mm) | Dilatation ratio (%) |
|---|---|---|---|
| sham | 1.18 ± 0.14 | 1.21 ± 0.13 | 3.43 ± 4.75a |
| control | 1.20 ± 0.13 | 2.44 ± 0.19 | 104.19 ± 14.96ab |
| con + AV | 1.19 ± 0.10 | 2.41 ± 0.16 | 103.64 ± 9.53ab |
| con + shAEBP1 | 1.18 ± 0.10 | 1.90 ± 0.13 | 62.60 ± 43.38b |
The diameters of infrarenal abdominal aorta in sham, control, con + AV and con + shAEBP1 were measured by vernier caliper on day 0 and day 28 after surgery. Group sham, rats were opened as a control; Group control, rats received elastase perfusion (20 U/ml) (Solarbio, Beijing, China) for 15 min; Group con + AV, rats received elastase perfusion and 40ul empty adenovirus (5x1011/ml) was injected into the intra-adventitial via at least four positions; Group con + shAEBP1, rats received elastase perfusion and adenovirus with AEBP1 shRNA.
Dilation ratio (%) was calculated according to the following formula: Dilation ratio (%) = (diameter of D28-diameter of D0)/ diameter of D0 x 100%.
p < 0.01 vs group con + shAEBP1.
p < 0.01 vs group sham.
Fig. 4.
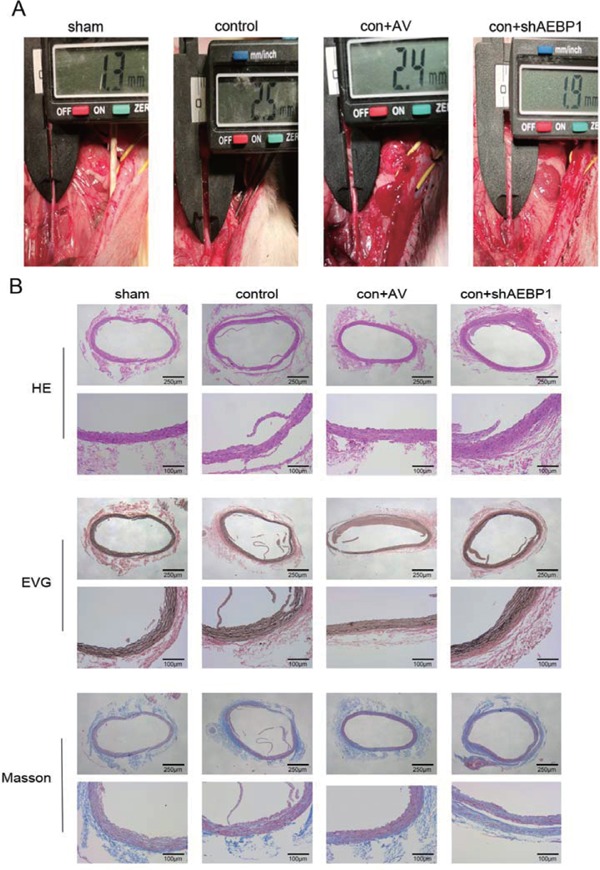
AEBP1 silencing impairs development of AAA
An AAA rat model generated by elastase perfusion was used to investigate the role of AEBP1 in the development of AAA. (A) Diameter of the infrarenal abdominal aorta in sham, control, con + AV, and con + sh AEBP1 group. (B) HE, EVG, and Masson staining of the vascular wall of the infrarenal abdominal aorta in different groups.
We analyzed the vascular wall tissue at different time points of AAA development (4, 7, 14, 21, and 28 days after the surgery). The western blot analysis indicated that AEBP1 was upregulated in a time-dependent manner (Fig. 5A), further verifying the correlation between AEBP1 and AAA. As the development of AAA is always accompanied with local inflammation and fibrillar disorders, we measured the levels of metalloproteases MMP-2/9 and inflammatory cytokines TNFα, MCP-1, IL-6, and IL-1β in the vascular wall of the rat model. We found that the levels of MMPs and inflammatory cytokines were clearly elevated in the control and con + AV groups, but AEBP1 silencing suppressed this effect (Fig. 5B, C). Overall, these results indicated that AEBP1 was important for the development of AAA, and silencing AEBP1 could suppress the development of AAA.
Fig. 5.
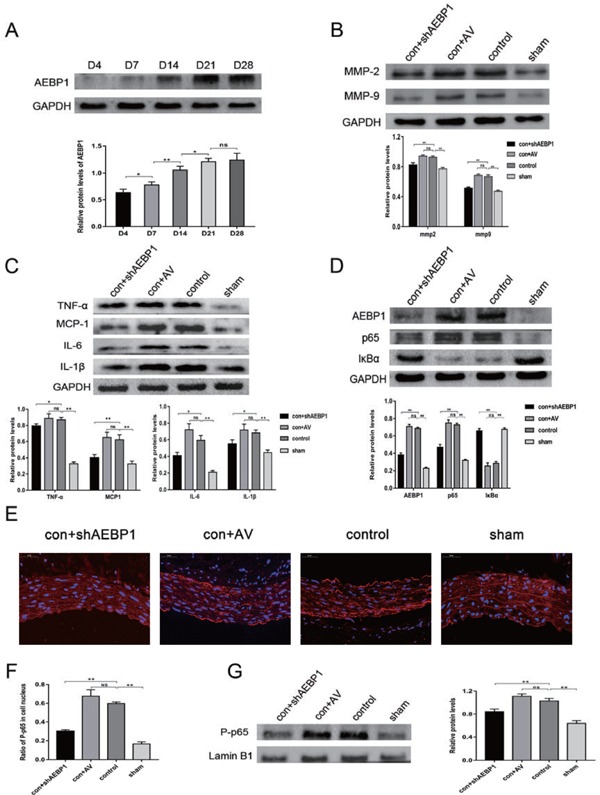
Expression profiles of AEBP1, the NF-κB pathway factors, inflammatory cytokines, and MMPs in the established AAA model (n = 10)
(A) AEBP1 expression was measured using the western blot at days 4, 7, 14, 21, and 28 (n = 3) after the surgery. (B) MMP2 and MMP9 expression were evaluated using the western blot (with GAPDH as the loading control). (C) Evaluation of the levels of inflammatory cytokines TNFα, MCP-1, IL-6, and IL-1β by the western blot. (D) Protein levels of AEBP1, NF-κB p65, and IκBα were evaluated by western blot in the established AAA model. (E) Immunofluorescence staining and micrographs (400X) indicated P-p65 expression in the infrarenal abdominal aorta in different groups (n = 4). (F) Quantification of the proportion of P-p65 positive nuclei. (G) Nuclear proteins were extracted from the infrarenal abdominal aorta (n = 4), and P-p65 expression was evaluated using the western blot. All the relative expression levels are presented as mean ± SD; one-way ANOVA, ns > 0.05,*p < 0.05; **p < 0.01. GAPDH was used as the loading control in C and D, whereas Lamin B1 was used in G.
AEBP1 mediates the aberrant activation of the NF-κB pathway, thereby promoting AAA development in the rat model
Activation of the NF-κB pathway in AAA patients has previously been reported24). Accordingly, we checked for the activation of the NF-κB pathway in the AAA rat model. In both the control and con + AV groups, NF-κB p65 and IκBα were found to be up- and down-regulated, respectively. These effects were suppressed upon AEBP1 silencing (Fig. 5D). Similar results were obtained with immunofluorescence (Fig. 5E, F) and the western blot (Fig. 5G) assessment of nuclear translocation of NF-κB P-p65 in the vascular walls. These results suggested that AEBP1 promoted the development of AAA by activating the NF-κB pathway.
The role of AEBP1 in the development of AAA relies on the activation of the NF-κB pathway in VSMC
Both clinical samples and rat models indicated that AEBP1 was closely correlated with the development of AAA, which was accompanied by activation of the NF-κB pathway. AEBP1 regulated the expression of MMPs and inflammatory cytokines, whose expression can be regulated by the NF-κB pathway. Previous studies have also reported that AEBP1 could promote activation of the NF-κB pathway through degradation of IκBα16). These observations led us to hypothesize that AEBP1 promoted the development of AAA through activation of the NF-κB pathway.
We first examined the localization of AEBP1 relative to that of the neointima marker CD34, medialayer smooth muscle cell marker SMA, adventitia fibroblast marker FSP1, and macrophage marker CD68 in AAA samples. The results indicated that AEBP1 was co-expressed with SMA in the vascular wall (Fig. 6).
Fig. 6.
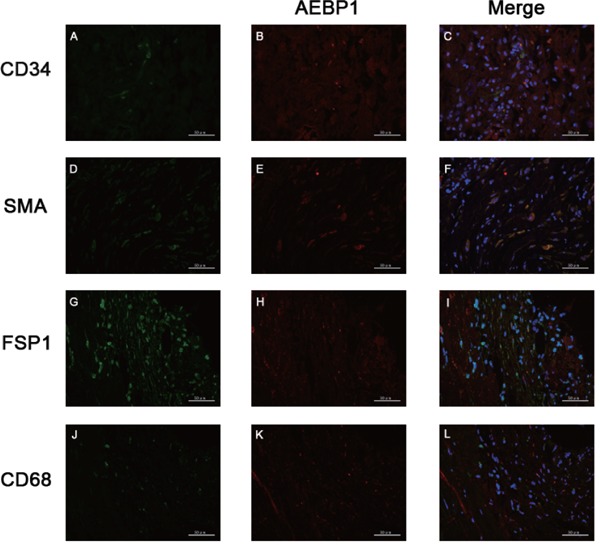
Co-localization of AEBP1 with different vascular wall markers
Immunofluorescence staining and micrographs (400×) indicated AEBP1 expression with a neointima marker CD34 (A–C), a media-layer smooth muscle cell marker SMA (D–F), an adventitia fibroblast marker FSP1 (G–I), and a macrophage marker CD68 (K–L). AEBP1 and each corresponding marker are depicted in red and green, respectively. DAPI was used for marking the nucleus.
Thereafter, we used VSMCs to investigate the relationship between AEBP1 and the NF-κB pathway. AEBP1-sh and AEBP1-Over VSMCs were established by lentivirus-mediated AEBP1 knockdown and overexpression, respectively. AEBP1 expression was measured by qRT-PCR and western blot analyses in AEBP1-sh, AEBP1-Over, and control VSMCs. AEBP1 expression was found to be significantly upand down-regulated in AEBP1-over and AEBP1-sh VSMCs, respectively (Fig. 7A–C). Subsequently, NF-κ B inhibitor BAY11-7082 was used to assess whether the role of AEBP1 in the development of AAA relied on activation of the NF-κB pathway. The secretion of inflammatory cytokines TNFα, IL-10, IL-6, and IL-1β was measured by ELISA, and the results indicated that AEBP1 overexpression and knockdown respectively upregulated and down-regulated the secretion of TNFα, IL-10, IL-6, and IL-1β. By contrast, NF-κB inhibitor BAY11-7082 suppressed the effect of AEBP1 on inflammatory cytokine secretion (Fig. 7D–G). Subsequent quantification of the inflammatory cytokines TNFα, MCP-1, IL-6, and IL-1β and the metalloproteases MMP-2 and MMP-9 by western blot revealed results consistent with the ELISA results reported previously (Fig. 8A, B).
Fig. 7.
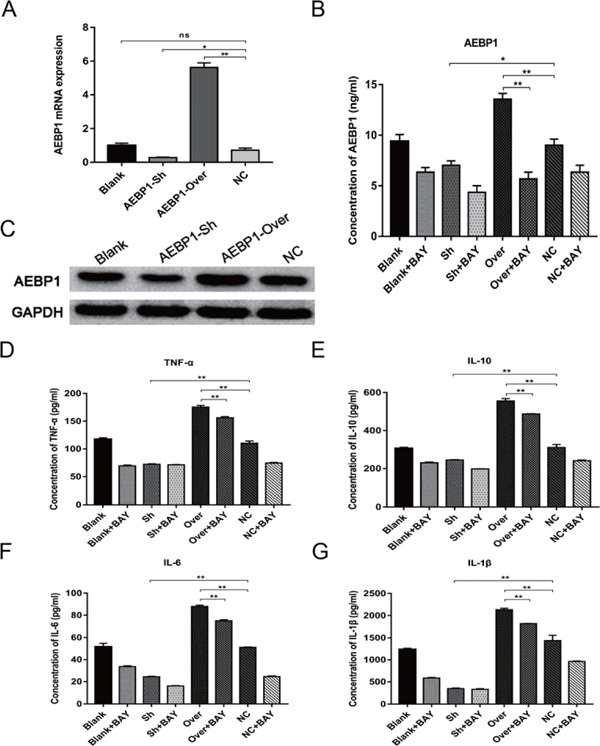
AEBP1 is important for the secretion of inflammatory cytokines
Established VSMC cell lines with AEBP1 overexpression or knockdown were used to analyze AEBP1 function in relation to the NF-κB pathway. AEBP1 expression was measured using qRT-PCR (A) (mean ± SD; one-way ANOVA, ns > 0.05, *p < 0.05; **p < 0.01) and western blot (C). ELISA was used to measure the levels of AEBP1 (B), TNFα (D), IL-10 (E), IL-6 (F), and IL-1β (G) in the supernatant of VSMC cells cultured in the presence or absence of the NF-κB pathway inhibitor BAY11-7082 (10 µM) (mean ± SD; two-way ANOVA, *p < 0.05; **p < 0.01, ns: not significant when it is > 0.05).
Fig. 8.
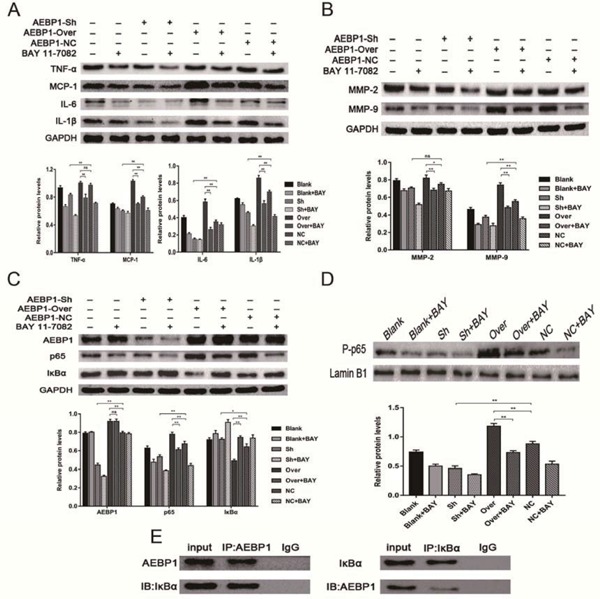
The role of AEBP1 in the development of AAA depends on the activation of the NF-κB pathway
Total (A, B, C, and E) and nuclear proteins (D) were extracted from the VSMC cell lines treated with BAY11-7082 for the following western blot quantifications. (A) Inflammatory cytokines TNFα, MCP-1, IL-6, and IL-1β. (B) MMP2 and MMP9. (C) AEBP1, NF-κB p65, and IκBα. (D) P-p65. (E) Co-immunoprecipitation was performed to assess whether AEBP1 interacted with IκBα. Quantification of the bands (A-D) was performed with three independent experiments. Relative expression against GAPDH (A, B, and C) or LaminB1 (D) is shown (mean ± SD; two-way ANOVA, ns > 0.05 * p < 0.05; ** p < 0.01).
To further test whether the role of AEBP1 in secretion of the inflammatory cytokines and expression of the MMPs relies on the NF-κB pathway, NF-κB pathway activity was measured in AEBP1-sh and AEBP1-over VSMCs. To this end, the levels of NF-κB p65, IκBα, and nuclear P-p65 were evaluated. We found that AEBP1 knockdown in VSMC cells down- and up-regulated NF-κB p65 and IκBα, respectively, while decreasing translocation of P-p65 into the nucleus. Contrarily, AEBP1 overexpression elicited the opposite effects. As expected, all these differential expression patterns caused by AEBP1 knockdown or overexpression were negated with the addition of NF-κB pathway inhibitor BAY11-7082 (Fig. 8C, D). Given that AEBP1 had previously been demonstrated to promote activation of the NF-κB pathway through degradation of IκBα16), we examined whether AEBP1 could directly bind to IκBα. We found that AEBP1 co-immunoprecipitated with IκBα from VSMC extracts, indicating that AEBP1 could successfully bind to IκBα, thereby activating the NF-κB pathway (Fig. 8E). Overall, these results suggested that AEBP1 could bind to IκBα and subsequently induce activation of the NF-κB pathway, thereby promoting the expression of inflammatory cytokines and MMPs.
Discussion
Abdominal aortic aneurysm is a common degenerative cardiovascular disorder, characterized in part by an inflammatory response in the aortic wall25, 26). In recent years, there have been rapid developments in endovascular aortic aneurysm repair (EVAR), and favorable clinical effects have been achieved, but the associated long-term complications severely restrict the outcome of EVAR. Moreover, surgery is not a suitable option for a large proportion of AAA patients, such as asymptomatic cases with an aortic diameter < 5.5 cm or those who cannot tolerate surgery because of various factors. Therefore, identifying non-surgical interventions that can control and repair AAA, such as gene therapy, has important clinical benefits. In this study, we provide sufficient evidence for the first time that upregulation of AEBP1 contributes to the development of AAA via the NF-κB pathway.
As a transcriptional repressor, AEBP1 has been reported to be associated with the development of several diseases, especially cardiovascular disorders. Moreover, AEBP1 is involved in cholesterol homeostasis and inflammatory responses in macrophages and formation of foam cells14, 15, 27). Using proteome iTRAQ, we first indicated that AEBP1 was upregulated in AAA clinical samples. Subsequent qRT-PCR and western blot analyses confirmed these results. Additionally, ELISA and IHC revealed that AEBP1 levels were significantly elevated in the circulating blood and vascular wall tissues, respectively. Although the increased expression of AEBP1 in varied tissues has been reported in other diseases28–30), the increased expression of AEBP1 in serum has not been reported. In some tumor-related studies, high expression of AEBP1 suggested poor prognosis, such as gastric cancer31) and bladder cancer32). We make a conjecture that AEBP1 may serve as a serum biomarker for diagnosing AAA, but this needs further verification by a future study.
Chronic infiltration of inflammatory cells is the most important pathological feature in AAA development5). In this process, inflammatory cells secrete a variety of inflammatory cytokines, such as TNF-α, MCP-1, IL-6, and IL-1β. These cytokines can directly damage the aortic wall33) and cause degradation of the elastic fibers by promoting the VSMCs to secrete proteolytic enzymes and MMPs34–36). Moreover, chronic infiltration of inflammatory cells can induce VSMCs to undergo apoptosis by activating the Fas/FasL pathway12, 37). Although a large number of potential targets for AAA treatment have been identified, very few of them have been investigated in AAA animal models. We successfully established an AAA rat model using the elastase method and found that AEBP1 was upregulated during the development of AAA in a timedependent manner, further confirming that AEBP1 is closely related to the development and progression of AAA. AEBP1 knockdown suppressed AAA development and inhibited the expression of inflammatory cytokines and MMPs, which are considered key enzymes directly related to AAA formation and progression11, 38).
Majdalawieh et al. reported that AEBP1 exerts its pro-inflammatory effects by promoting NF-κB activity through impeding the inhibitory function of Iκ Bα. In the canonical pathway, the activation of NF-κ B p65 requires phosphorylation and subsequent degradation of its inhibitor IκBα. NF-κB is known to mediate inflammatory reactions and regulate the transcription of MMP-1, MMP-2, MMP-3, and MMP-924, 39–41). Pre-clinical studies have indicated that inhibiting the activity of NF-κB can delay the onset and development of AAA19, 20). Our results were consistent with the previous reports, and we found that the NF-κB pathway was activated in both the clinical AAA samples and the elastase-induced AAA animal model. Previous studies have also reported that AEBP1 can promote activation of the NF-κB pathway through degradation of IκBα16). Overall, these results suggest that AEBP1 may promote the development of AAA by regulating the NF-κB pathway.
VSMC cells are the main cellular component of the vascular wall. Their apoptosis and phenotypic transformation play significant roles in the development of AAA42, 43). Previous studies have demonstrated that p53 expression in AAA arteries is four times higher than that in normal arteries. In AAA arteries, the surviving VSMC cells are still active, but their ability to proliferate and replicate is considerably reduced44, 45). During the development of AAA, abnormal metabolites and inflammatory factors stimulate VSMCs to transform from the contractile type to synthetic type, which affects vascular remodeling and leads to formation of AAA46, 47). In our studies, the double-immunofluorescence technique revealed that AEBP1 was mainly expressed in the mid-layer VSMCs. In human VSMCs, AEBP1 overexpression and knockdown respectively up- and down-regulated inflammatory cytokines and MMPs, but all these effects were negated upon addition of the NF-κB pathway inhibitor BAY11-7082. Co-immunoprecipitation confirmed that AEBP1 could bind to IκBα, further suggesting that AEBP1 could induce the degradation of IκBα, thereby activating the NF-κB pathway and inducing AAA development.
Although we confirmed that AEBP1 could promote the canonical NF-κB pathway in AAA, the effects of AEBP1 on the non-canonical NF-κB pathway were not addressed in this study. Future studies should investigate the relationship between AEBP1 and the non-canonical NF-κB pathway in AAA. Furthermore, there are other mechanisms of a correlation between AEBP1 and AAA, and their role in the development and progression of AAA cannot be ruled out. Although further studies are still required to establish a pharmacological approach, the current studies provide reliable evidence that AEBP1 is a strong determinant of AAA development, suggesting AEBP1 to be a potential target for the treatment of AAA.
Conclusions
Overall, the results of our study demonstrate that AEBP1 has a very strong correlation with AAA in clinical samples. This is in line with the previous observations in mouse models, where AEBP1 was demonstrated to promote AAA. The effect of AEBP1 was transduced with activation of the NF-κB pathway. Perturbation of AEBP1 levels affected the expression of inflammatory cytokines and MMPs, and these effects were negated by the NF-κB pathway inhibitor BAY11-7082. Altogether, these results suggest that AEBP1 promotes AAA formation by modulating the NF-κB pathway.
Acknowledgments
The research was conducted in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals (China) and approved by the China Medical University Animal Ethics Committee.
This research was funded by Basic Research Program of the Liaoning Provincial Institution of 550 Higher Learning 2017 (grant number LZDK 201701)
Conflicts of Interests
The authors declare no conflict of interests.
Author Contributions
Jian Zhang and Shijie Xin designed the experiments and provided the financial support; Jiancong Ren performed the experiments; Tongming Ren, Hong Fang, and Xiaohan Xu contributed to the rat care; Yu Lun and Han Jiang collected tissue samples; Yanshuo Han, Jiancong Ren and Jian Zhang analyzed the results.
References
- 1). Kent KC: Clinical practice. Abdominal aortic aneurysms. N Engl J Med, 2014; 371: 2101-2108 [DOI] [PubMed] [Google Scholar]
- 2). Ahmed R, Ghoorah K, Kunadian V: Abdominal Aortic Aneurysms and Risk Factors for Adverse Events. Cardiol Rev, 2016; 24: 88-93 [DOI] [PubMed] [Google Scholar]
- 3). Kuivaniemi H, Elmore JR: Opportunities in abdominal aortic aneurysm research: epidemiology, genetics, and pathophysiology. Ann Vasc Surg, 2012; 26: 862-870 [DOI] [PubMed] [Google Scholar]
- 4). Bode-Janisch S, Schmidt A, Gunther D, Stuhrmann M, Fieguth A: Aortic dissecting aneurysms--histopathological findings. Forensic Sci Int, 2012; 214: 13-17 [DOI] [PubMed] [Google Scholar]
- 5). Li H, Bai S, Ao Q, Wang X, Tian X, Li X, Tong H, Hou W, Fan J: Modulation of Immune-Inflammatory Responses in Abdominal Aortic Aneurysm: Emerging Molecular Targets. J Immunol Res, 2018; 2018: 7213760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR, Jr.: Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation, 2005; 112: 232-240 [DOI] [PubMed] [Google Scholar]
- 7). Wang J, Lindholt JS, Sukhova GK, Shi MA, Xia M, Chen H, Xiang M, He A, Wang Y, Xiong N, Libby P, Wang JA, Shi GP: IgE actions on CD4+ T cells, mast cells, and macrophages participate in the pathogenesis of experimental abdominal aortic aneurysms. EMBO Mol Med, 2014; 6: 952-969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Rateri DL, Howatt DA, Moorleghen JJ, Charnigo R, Cassis LA, Daugherty A: Prolonged infusion of angiotensin II in apoE(-/-) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am J Pathol, 2011; 179: 1542-1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Takahara Y, Tokunou T, Ichiki T: Suppression of Abdominal Aortic Aneurysm Formation in Mice by Teneligliptin, a Dipeptidyl Peptidase-4 Inhibitor. J Atheroscler Thromb, 2018; 25: 698-708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Keeling WB, Armstrong PA, Stone PA, Bandyk DF, Shames ML: An overview of matrix metalloproteinases in the pathogenesis and treatment of abdominal aortic aneurysms. Vasc Endovasc Surg, 2005; 39: 457-464 [DOI] [PubMed] [Google Scholar]
- 11). Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT: Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest, 2002; 110: 625-632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P: Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation, 1999; 99: 96-104 [DOI] [PubMed] [Google Scholar]
- 13). He GP, Muise A, Li AW, Ro HS: A eukaryotic transcriptional repressor with carboxypeptidase activity. Nature, 1995; 378: 92-96 [DOI] [PubMed] [Google Scholar]
- 14). Majdalawieh A, Zhang L, Fuki IV, Rader DJ, Ro HS: Adipocyte enhancer-binding protein 1 is a potential novel atherogenic factor involved in macrophage cholesterol homeostasis and inflammation. Proc Natl Acad Sci U S A, 2006; 103: 2346-2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Majdalawieh A, Ro HS: LPS-induced suppression of macrophage cholesterol efflux is mediated by adipocyte enhancer-binding protein 1. Int J Biochem Cell Biol, 2009; 41: 1518-1525 [DOI] [PubMed] [Google Scholar]
- 16). Majdalawieh A, Zhang L, Ro HS: Adipocyte enhancer-binding protein-1 promotes macrophage inflammatory responsiveness by up-regulating NF-kappaB via IkappaBalpha negative regulation. Mol Biol Cell, 2007; 18: 930-942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Sen R, Baltimore D: Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell, 1986; 46: 705-716 [PubMed] [Google Scholar]
- 18). Chase AJ, Bond M, Crook MF, Newby AC: Role of nuclear factor-kappa B activation in metalloproteinase-1, -3, and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler Thromb Vasc Biol, 2002; 22: 765-771 [DOI] [PubMed] [Google Scholar]
- 19). Nakashima H, Aoki M, Miyake T, Kawasaki T, Iwai M, Jo N, Oishi M, Kataoka K, Ohgi S, Ogihara T, Kaneda Y, Morishita R: Inhibition of experimental abdominal aortic aneurysm in the rat by use of decoy oligodeoxynucleotides suppressing activity of nuclear factor kappaB and ets transcription factors. Circulation, 2004; 109: 132-138 [DOI] [PubMed] [Google Scholar]
- 20). Miyake T, Aoki M, Nakashima H, Kawasaki T, Oishi M, Kataoka K, Tanemoto K, Ogihara T, Kaneda Y, Morishita R: Prevention of abdominal aortic aneurysms by simultaneous inhibition of NFkappaB and ets using chimeric decoy oligonucleotides in a rabbit model. Gene Ther, 2006; 13: 695-704 [DOI] [PubMed] [Google Scholar]
- 21). Miyazaki T, Miyazaki A: Dysregulation of Calpain Proteolytic Systems Underlies Degenerative Vascular Disorders. J Atheroscler Thromb, 2018; 25: 1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michel JB: Elastase-induced experimental aneurysms in rats. Circulation, 1990; 82: 973-981 [DOI] [PubMed] [Google Scholar]
- 23). Azuma J, Asagami T, Dalman R, Tsao PS: Creation of murine experimental abdominal aortic aneurysms with elastase. J Vis Exp, 2009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Mi T, Nie B, Zhang C, Zhou H: The elevated expression of osteopontin and NF-kappaB in human aortic aneurysms and its implication. J Huazhong Univ Sci Technolog Med Sci, 2011; 31: 602. [DOI] [PubMed] [Google Scholar]
- 25). Wang Q, Ren J, Morgan S, Liu Z, Dou C, Liu B: Monocyte chemoattractant protein-1 (MCP-1) regulates macrophage cytotoxicity in abdominal aortic aneurysm. PLoS One, 2014; 9: e92053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). Ramella M, Bertozzi G, Fusaro L, Talmon M, Manfredi M, Catoria MC, Casella F, Porta CM, Boldorini R, Fresu LG, Marengo E, Boccafoschi F: Effect of Cyclic Stretch on Vascular Endothelial Cells and Abdominal Aortic Aneurysm (AAA): Role in the Inflammatory Response. Int J Mol Sci, 2019; 20: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Majdalawieh A, Ro HS: PPARgamma1 and LXRalpha face a new regulator of macrophage cholesterol homeostasis and inflammatory responsiveness, AEBP1. Nucl Recept Signal, 2010; 8: e004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Hu W, Jin L, Jiang CC, Long GV, Scolyer RA, Wu Q, Zhang XD, Mei Y, Wu M: AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death & Disease, 2013; 4: e914-e914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29). Holloway RW, Bogachev O, Bharadwaj AG, McCluskey GD, Majdalawieh AF, Zhang L, Ro H-S: Stromal Adipocyte Enhancer-binding Protein (AEBP1) Promotes Mammary Epithelial Cell Hyperplasia via Proinflammatory and Hedgehog Signaling. J Biol Chem, 2012; 287: 39171-39181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30). Shijo M, Honda H, Suzuki SO, Hamasaki H, Hokama M, Abolhassani N, Nakabeppu Y, Ninomiya T, Kitazono T, Iwaki T: Association of adipocyte enhancer-binding protein 1 with Alzheimer's disease pathology in human hippocampi. Brain Pathol, 2018; 28: 58-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Liu J-y, Jiang L, Liu J-j, He T, Cui Y-h, Qian F, Yu P-w: AEBP1 promotes epithelial-mesenchymal transition of gastric cancer cells by activating the NF-κB pathway and predicts poor outcome of the patients. Scientific Reports, 2018; 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32). Li S, Liu X, Liu T, Meng X, Yin X, Fang C, Huang D, Cao Y, Weng H, Zeng X, Wang X: Identification of Biomarkers Correlated with the TNM Staging and Overall Survival of Patients with Bladder Cancer. Front Physiol, 2017; 8: 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Blomkalns AL, Gavrila D, Thomas M, Neltner BS, Blanco VM, Benjamin SB, McCormick ML, Stoll LL, Denning GM, Collins SP, Qin Z, Daugherty A, Cassis LA, Thompson RW, Weiss RM, Lindower PD, Pinney SM, Chatterjee T, Weintraub NL: CD14 directs adventitial macrophage precursor recruitment: role in early abdominal aortic aneurysm formation. J Am Heart Assoc, 2013; 2: e000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Rabkin SW: The Role Matrix Metalloproteinases in the Production of Aortic Aneurysm. Prog Mol Biol Transl Sci, 2017; 147: 239-265 [DOI] [PubMed] [Google Scholar]
- 35). Qin Y, Cao X, Yang Y, Shi GP: Cysteine protease cathepsins and matrix metalloproteinases in the development of abdominal aortic aneurysms. Future Cardiol, 2013; 9: 89-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Yamashiro Y, Yanagisawa H: Crossing Bridges between Extra- and Intra-Cellular Events in Thoracic Aortic Aneurysms. J Atheroscler Thromb, 2018; 25: 99-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Zhang J, Bockler D, Ryschich E, Klemm K, Schumacher H, Schmidt J, Allenberg JR: Impaired Fas-induced apoptosis of T lymphocytes in patients with abdominal aortic aneurysms. J Vasc Surg, 2007; 45: 1039-1046 [DOI] [PubMed] [Google Scholar]
- 38). Manicone AM, McGuire JK: Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol, 2008; 19: 34-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Bond M, Baker AH, Newby AC: Nuclear factor kappaB activity is essential for matrix metalloproteinase-1 and -3 upregulation in rabbit dermal fibroblasts. Biochem Biophys Res Commun, 1999; 264: 561-567 [DOI] [PubMed] [Google Scholar]
- 40). Takeshita H, Yoshizaki T, Miller WE, Sato H, Furukawa M, Pagano JS, Raab-Traub N: Matrix metalloproteinase 9 expression is induced by Epstein-Barr virus latent membrane protein 1 C-terminal activation regions 1 and 2. J Virol, 1999; 73: 5548-5555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Li YF, Wang H, Fan Y, Shi HJ, Wang QM, Chen BR, Khurwolah MR, Long QQ, Wang SB, Wang ZM, Wang LS: Epigallocatechin-3-Gallate Inhibits Matrix Metalloproteinase-9 and Monocyte Chemotactic Protein-1 Expression Through the 67-kappaDa Laminin Receptor and the TLR4/MAPK/NF-kappaB Signalling Pathway in Lipopolysaccharide-Induced Macrophages. Cell Physiol Biochem, 2017; 43: 926-936 [DOI] [PubMed] [Google Scholar]
- 42). Mack CP: Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol, 2011; 31: 1495-1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Shioi A, Ikari Y: Plaque Calcification During Atherosclerosis Progression and Regression. J Atheroscler Thromb, 2018; 25: 294-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Cao X, Cai Z, Liu J, Zhao Y, Wang X, Li X, Xia H: miRNA504 inhibits p53dependent vascular smooth muscle cell apoptosis and may prevent aneurysm formation. Mol Med Rep, 2017; 16: 2570-2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). Holmes DR, Lopez-Candales A, Liao S, Thompson RW: Smooth muscle cell apoptosis and p53 expression in human abdominal aortic aneurysms. Ann N Y Acad Sci, 1996; 800: 286-287 [DOI] [PubMed] [Google Scholar]
- 46). Peng H, Zhang K, Liu Z, Xu Q, You B, Li C, Cao J, Zhou H, Li X, Chen J, Cheng G, Shi R, Zhang G: VPO1 Modulates Vascular Smooth Muscle Cell Phenotypic Switch by Activating Extracellular Signal-regulated Kinase 1/2 (ERK 1/2) in Abdominal Aortic Aneurysms. J Am Heart Assoc, 2018; 7: e010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47). Riches K, Clark E, Helliwell RJ, Angelini TG, Hemmings KE, Bailey MA, Bridge KI, Scott DJA, Porter KE: Progressive Development of Aberrant Smooth Muscle Cell Phenotype in Abdominal Aortic Aneurysm Disease. J Vasc Res, 2018; 55: 35-46 [DOI] [PubMed] [Google Scholar]