

Abstract
Objectives
The aims were to demonstrate pharmacokinetic (PK) similarity between DRL_RI, a proposed rituximab biosimilar, and two reference innovator products (Rituxan® [RTX-US] and MabThera® [RTX-EU]) and compare their pharmacodynamics (PD), efficacy, safety, and immunogenicity in rheumatoid arthritis (RA) patients with inadequate response to methotrexate (MTX)-based therapy and no prior biologic administration.
Methods
In this randomized, double-blind, parallel-group study, 276 patients with moderate-to-severe active RA were randomized to receive DRL_RI, RTX-US, or RTX-EU on days 1 and 15. The primary PK end points included area under the concentration–time curve from time 0 to 336 h after first infusion (AUC0–14 days, first infusion), AUC from day 1 through week 16 (AUC0–∞, entire course), and AUC from time 0 to time of last quantifiable concentration after the second dose (AUC0–t, second infusion). Secondary end points included other PK parameters, such as maximum concentration (Cmax), time to Cmax after each infusion, terminal half-life, systemic clearance, and volume of distribution after the second infusion; PD parameters and efficacy until week 24; safety and immunogenicity at week 24 and 52; and B cell recovery until week 52. AUC from time 0 to time of last quantifiable concentration after the first dose and over the entire course from day 1 through week 16 (AUC0–t, entire course) was analyzed as an exploratory end point.
Results
The 91% confidence intervals (CIs) of the geometric mean ratios (GMRs) for the primary end point of AUC0–∞, entire course were within the bioequivalence limits of 80–125% for all comparisons: DRL_RI versus RTX-US 100.37% (92.30–109.14), DRL_RI versus RTX-EU 93.58% (85.98–101.85), and RTX-US versus RTX-EU 93.24% (85.62–101.54). PD outcomes (peripheral blood B-cell depletion and mean change in Disease Activity Score [28 joints]–C-reactive protein), efficacy, safety, and immunogenicity were also comparable between DRL_RI and the reference products.
Conclusion
DRL_RI, a proposed biosimilar, demonstrated three-way PK similarity with RTX-EU and RTX-US, the reference innovator products, with comparable efficacy, PD, safety, and immunogenicity.
Clinical Trials Registration Number
ClinicalTrials.gov identifier: NCT02296775.
Electronic supplementary material
The online version of this article (10.1007/s40259-020-00406-1) contains supplementary material, which is available to authorized users.
Key Points
| DRL_RI, a proposed rituximab biosimilar, shows pharmacokinetic similarity with the US- and EU-approved reference innovator products. |
| The pharmacodynamics, efficacy, safety, and immunogenicity profiles were also comparable between the proposed biosimilar and reference innovator products. |
| No clinically meaningful differences in adverse events, B-cell recovery, and immunogenicity were identified between the proposed biosimilar and the reference innovator products during the longer-term follow-up period (up to 52 weeks). |
Introduction
Comparative studies designed to demonstrate the similarity of a biosimilar and the reference medicinal product in key pharmacokinetic (PK) parameters are an essential part of biosimilar development programs [1].
For PK similarity assessment of biosimilars, a study in healthy subjects is considered more sensitive than a study in patients due to the lack of potential confounding factors such as underlying and/or concomitant disease and concomitant medications [2]. However, because rituximab cannot be safely administered to healthy subjects, such studies for rituximab are conducted in patients. Amongst the patient populations treated with rituximab, rheumatoid arthritis (RA) patients are more appropriate than patients with lymphoid malignancies because of the simpler administration schedule and fewer confounding factors. The simpler dosing schedule with administration of two doses, 2 weeks apart, and no re-dosing for a minimum of 16 weeks [3] also has an advantage (over the repeated dosing in malignancies), as it allows a full PK profile characterization, including its late elimination phase, which is considered preferable [1].
Hence, this study was conducted in RA patients with the aim to establish PK similarity of DRL_RI with the US and EU reference products, while also comparing their pharmacodynamics (PD), safety, immunogenicity, and efficacy.
RA is a chronic systemic inflammatory disease affecting approximately 0.24% of the population globally, and is the 42nd highest among 291 contributors to global disability [4]. The introduction of biologic disease-modifying anti-rheumatic drugs (DMARDs) has been a major advance in the treatment of RA [5]. Rituximab, used in RA as a biologic DMARD, is a chimeric murine/human anti-CD20 monoclonal antibody and acts by depleting the CD20-expressing peripheral B cells and causing variable B-cell depletion in synovium and other sites, including lymphoid tissue and bone marrow [6].
The proposed rituximab biosimilar, DRL_RI (Dr. Reddy’s Laboratories Ltd.), has completed an extensive comparison of structural, physicochemical, analytical, and functional characteristics with the reference products approved in the United States, Rituxan® (RTX-US; Genentech, Inc.) and the European Union, MabThera® (RTX-EU; Roche Pharmaceuticals) (data on file). This study was designed to demonstrate PK similarity between DRL_RI and both reference products (RTX-US and RTX-EU) in patients with moderate-to-severe active RA and an inadequate response to methotrexate (MTX)-based therapy. The study also compared the PD, efficacy, safety, and immunogenicity over 24 weeks between DRL_RI and the reference products. The extended follow-up period (from week 24 until week 52) examined the longer-term safety, immunogenicity, and B-cell recovery.
Methodology
The study protocol (ClinicalTrials.gov identifier: NCT02296775) was approved by the Regulatory Authorities and Ethics Committees of all participating centers. The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines [7, 8]. All patients provided written informed consent before study participation.
Study Population
Patients aged 18–65 years with active RA (revised 1987 American College of Rheumatology [ACR] classification criteria [9]) for at least 6 months, not previously treated with rituximab or any other biologic DMARD treatment and with an inadequate response to MTX were eligible for inclusion. Active RA was defined as six or more tender joints and six or more swollen joints from the 66/68-joint count system, with a high-sensitivity C-reactive protein (CRP) level greater than the upper limit of normal, or an erythrocyte sedimentation rate ≥ 28 mm, and positive rheumatoid factor and/or anticyclic citrullinated peptide antibody results, and a Disease Activity Score in 28 joints (DAS28) of > 3.2 at screening [10]. Patients had to be on an accepted MTX-based therapy for at least 6 months; on a stable MTX dose for at least 3 months; and on a stable dose of folic acid supplement (≥ 5 mg/week) for at least 4 weeks. Accepted MTX based therapies included MTX alone 15–25 mg/week, MTX 10–25 mg/week + hydroxychloroquine 200–400 mg/day, MTX 10–25 mg/week + sulfasalazine 2–3 g/day. Prednisone/prednisolone ≤ 10 mg/day, or an equivalent and stable dose for at least 4 weeks was allowed. Key exclusion criteria were systemic manifestations of RA, any confounding disease (secondary Sjogren’s syndrome, inflammatory diseases, HIV, etc.), pregnancy, lactation, or unwillingness to use contraception. Detailed exclusion criteria are provided in the electronic supplementary material (ESM), Table S1. Patients were withdrawn if they required rituximab retreatment before week 24, had a clinically significant adverse event (AE) that outweighed the benefit of the study drug in the opinion of the investigators, had severe mucocutaneous reactions, or had disease progression. Treatment was discontinued for patients who withdrew their consent, were lost to follow-up, or had a significant protocol violation.
Study Design and Treatments
This was a three-arm, randomized, double-blind, parallel-group, comparative study in patients with active RA conducted at 29 centers across India and Ukraine between November 2014 and November 2016. On completion of the screening phase (up to 6 weeks), patients were randomly assigned (1:1:1) using computer-generated blocks (stratified by gender and region [India/Ukraine]) to receive one of the three products: DRL_RI, RTX-US, or RTX-EU (13 patients were randomized under blinded conditions between the two investigational products available at the center at that time).
The study included a 24-week treatment period, wherein patients received two study drug intravenous infusions of 1000 mg, administered on days 1 and 15.
All patients continued their stable DMARD treatment regimen throughout the study period. Standard premedication was administered before rituximab dosing [3, 11]. Patients having an inadequate clinical response at or after 16 weeks as determined by the investigator were allowed to receive rescue therapy with leflunomide, and were followed up for safety, immunogenicity, and B-cell recovery.
All patients were followed up until week 52 for long-term safety, immunogenicity, and B-cell recovery. After week 24, sampling for B-cell count was discontinued before week 52 in patients whose B-cell counts recovered to values above the lower limit of normal (LLN). Patients requiring high doses of corticosteroids or retreatment with rituximab were discontinued from the study, but were followed up for B-cell recovery up to week 52.
Study End Points and Assessments
Pharmacokinetic and Pharmacodynamic End Points
The PK sampling schedule is provided in the ESM, online supplementary Table S2. Rituximab was quantitated in plasma using a validated enzyme-linked immunosorbent assay (ELISA) method using plates coated with monoclonal rat anti-rituximab antibodies to capture rituximab, followed by detection with biotinylated goat antihuman IgG (Fc-specific) antibody. In the second step, streptavidin–horseradish–peroxidase was bound to the biotinylated antihuman antibody. The addition of chromogenic substrate resulted in development of color that was measured at 450 nm. The lower limit of quantitation was 0.313 µg/mL.
PK parameters were calculated from the plasma concentration–time profiles by standard non-compartmental methods with estimation of the area under the plasma concentration–time curve (AUC) from time 0 to the time of last quantifiable concentration (AUC0–t) by the linear trapezoidal rule using Phoenix WinNonlin version 6.4.0.768 (Certara LLP, Princeton, NJ, USA).
The primary PK end points were AUC from time 0 to day 14 (AUC0–14 days)1 after the first infusion (AUC0–14 days, first infusion); AUC from time 0 extrapolated to infinity (AUC0–∞) over the entire course (AUC0–∞, entire course); and AUC0–t after the second dose (AUC0–t, second infusion).
Secondary PK parameters were maximum concentration (Cmax) after the first and second infusion1 (Cmax, first infusion and Cmax, second infusion), time to Cmax (tmax) after each infusion, AUC0–t after the first dose (considered exploratory and numerically equal to AUC0–14 days, first infusion; thus not detailed separately) and over the entire course (AUC0–t, entire course) (considered exploratory), volume of distribution (Vz), systemic clearance (CL), and terminal half-life (t½) after the second infusion.
PD was assessed by (1) the dual PD/efficacy parameter mean change in DAS28-CRP from baseline to weeks 4, 8, 12, and 16 [10] (examination of joints was conducted by an independent blinded evaluator); and (2) proportion of patients with peripheral blood B-cell depletion (B-cell counts were determined using a validated flow cytometry assay for counting of CD3 − CD19 + cells) at 48 h after dose 1 and at weeks 16 and 24. The time to B-cell depletion and repletion were evaluated. The sampling schedule for B-cell counts is provided in the ESM, online supplementary Table S3.
Efficacy, Safety, and Immunogenicity End Points
Efficacy end points included ACR20/50/70 response [9, 12], mean change from baseline in DAS28-CRP [10], and the Health Assessment Questionnaire–Disability Index (HAQ-DI) score at week 24 [13].
Safety was assessed as AEs, including infusion reactions, neutrophil counts, serious adverse events (SAEs), and treatment-related AEs, and proportion of patients with B-cell recovery at weeks 24 and 52.
Presence of antidrug antibodies (ADAs) was tested at baseline and weeks 4, 16, 24, and 52. Determination of ADAs was achieved using a validated affinity capture and elution ELISA method including three components: (1) a screen assay, which identified initial putative positive samples; (2) a confirmation assay, which assessed the specificity of the putative positive samples; and (3) a titration assay, which estimated the level of antibody in the confirmed positive samples. Samples were pretreated with acid to dissociate immune complexes, and the free ADAs were captured in solid-phase bound drug. Bound ADAs were eluted and transferred to a fresh plate, where ADAs were detected using F(ab)2 fragments of biotin–rituximab. The neutralizing capacity of the ADAs was tested in an in vitro cell-based assay using WIL2-S cells (B-lymphoblast expressing CD20) as responding cells and changes in cell viability, using complement-dependent cytotoxicity as the end point.
Statistical Analysis
Statistical analyses were conducted using SAS (SAS Institute, Cary, NC, USA) version 9.2. The PK population included all patients who received both study drug doses (on days 1 and 15) and had sufficient plasma samples for PK assessments. Patients with major protocol deviations (e.g., incomplete dosing and missed samples for critical time points) or confirmed ADAs were not included in the PK population.
For the efficacy analysis, the intention-to-treat (ITT) population included all randomly assigned patients who received at least one dose of study drug and had baseline and post-baseline efficacy data for at least one end point and at least one time point during treatment. The per-protocol (PP) population included patients who completed week 24 of the study without any major protocol deviations that could have affected study outcomes.
The PD population included all patients who received at least one dose of study drug, had at least one baseline and one post-baseline assessment of PD end points, and had no protocol deviations thought to affect the specific PD end point evaluation. For safety and immunogenicity analyses, the safety population (all dosed) was considered.
The sample size was calculated as follows: assuming a true geometric mean ratio (GMR) between 0.95 and 1.05 and an anticipated 43% coefficient of variation for the comparisons of AUC0-∞, entire course and Cmax after the second dose, a sample size of 246 patients (82 per treatment group) was determined to provide at least 83% statistical power to demonstrate for the GMR between treatments for each parameter a 91% confidence interval (CI) completely between 80% and 125% for each pairwise comparison. A 10% dropout rate was considered, resulting in a sample size of 276 patients. At the time of study design, only estimates for the variability of rituximab non-compartmental PK parameters based on single-arm evaluations were available in the literature. Such estimates did not provide a sufficient basis to calculate the final sample size for the intended comparative PK evaluation [2]. For this reason, a blinded sample size re-estimation (BSSR) was planned to re-confirm the final sample size, and was conducted by evaluating the pooled variability of Cmax and AUC0–∞ in terms of geometric coefficient of variation (GCV) from the data of approximately 40% of the scheduled patients (N = 115) [14]. Reduction of the sample size was not allowed (Wittes and Brittain adjustment rule) [14]. Had the pooled GCV been higher than initially estimated, the needed sample size for 85% power for the pairwise comparisons would have had to have been calculated. A 91% rather than 90% CI was used to protect against eventual type I error inflation due to the use of a BSSR. This value was sufficiently protective in simulations. The BSSR outcome confirmed that the initially calculated sample size was sufficient to fulfill the study objectives.
Continuous data were summarized by treatment group and time using descriptive statistics. Categorical data were summarized by treatment group and time using frequency tables. Percentages were based on the total category count excluding the missing category, if not otherwise mentioned.
The primary variables and the secondary PK variables Cmax after each infusion and AUC0–t, entire course were analyzed after logarithmic transformation using an analysis of variance (ANOVA) model including as fixed effects treatment, region (to account for possible regional variations in medical practice or environmental influences), and gender (which has been reported to affect rituximab PK parameters in RA patients [15, 16] and to influence rituximab exposure in diffuse large B-cell lymphoma patients [17]) to calculate 91% CIs for the GMR of the parameter between each pair of treatments. Similarity in PK was considered demonstrated if the 91% CI for the GMR between treatments of the primary parameters was within 80% and 125%.
The analysis of DAS28-CRP (both for PD and efficacy) is based on a generalized estimating equation (GEE) model, where the response variable is the mean change from baseline at each time point (every 4 weeks between weeks 4 and 24). Baseline score, treatment, and time were included as factors in the model. The robust (sandwich) variance estimate was used. The goodness of fit to link distributions like normal, log-normal, and gamma distributions was evaluated graphically and by residual analysis. The best fit link function was considered for the analysis. The adjusted mean change with differences and 95% CIs by treatment group are presented.
Peripheral blood B-cell counts were compared between treatment arms by evaluating the 95% CI for the difference in the proportion of patients with B-cell depletion at 48 h after dose 1, week 16, and week 24. The proportion of patients with B-cell repletion at each evaluation time was reported and compared between treatment groups by evaluating the 95% CI for the difference (Newcombe–Wilson score method). The proportion of patients with B-cell depletion/repletion in each treatment group, with corresponding two-sided 95% CIs, was calculated using the exact Clopper–Pearson method. The Kaplan–Meier method was used to summarize the time to depletion/repletion data.
The proportion of patients meeting the ACR20/50/70 improvement definitions at week 24 was analyzed using logistic regression including treatment, gender, and region as fixed effects. The adjusted difference, with 95% CI, was derived using the normal approximation, and the standard error (SE) was computed using the delta method. Efficacy parameters were reported descriptively, and equivalence margins were not defined a priori, because efficacy was evaluated as a secondary objective.
Results
Demographics and Patient Disposition
A total of 470 patients were screened, of which 276 were randomized to treatment with DRL_RI (n = 91), RTX-US (n = 92), or RTX-EU (n = 93). At week 24, 254 patients (DRL_RI, n = 87 [95.6%]; RTX-US, n = 84 [91.3%]; and RTX-EU, n = 83 [89.2%]) completed the treatment phase and continued to the follow-up phase until week 52. Fourteen patients discontinued in the follow-up phase. There were no differences in the rate of treatment compliance and trial discontinuation between treatment groups. Patient disposition, treatment compliance, and patient analysis populations are presented in Fig. 1 and online supplementary Tables S4 and S5. Reasons for study withdrawal until week 24 included AEs (DRL_RI, n = 1; RTX-US, n = 3; and RTX-EU, n = 6), withdrawal of consent (DRL_RI, n = 3; RTX-US, n = 3; and RTX-EU, n = 2), progressive or relapsed disease (RTX-US, n = 1), and failure to meet randomization criteria (RTX-EU, n = 1). From week 24 to week 52, reasons included AEs (DRL_RI, n = 1, and RTX-US, n = 1), withdrawal of consent (DRL_RI, n = 1, and RTX-EU, n = 2), progressive or relapsed disease (RTX-US, n = 2), and study visits stopped due to B-cell recovery (RTX-EU, n = 1).
Fig. 1.

Patient disposition (all randomized patients). RTX-EU MabThera®, RTX-US Rituxan®. aThe number of patients analyzed for each outcome is presented in the electronic supplementary material, Table S5. bOne patient had primary reason for discontinuation as “B-cell recovery”; for this reason, the patient did not have all safety visits
Rescue medications administered from week 16 to week 52 included leflunomide (DRL_RI six patients [6.6%], RTX-US four patients [4.3%], and RTX-EU two patients [2.2%]), followed by systemic corticosteroids (DRL_RI three patients [3.3%] and RTX-EU one patient [1.1%]) and sulfasalazine (DRL_RI two patients [2.2%]). High-dose glucocorticosteroids were given to six patients during the study (two patients during the treatment period [DRL_RI one patient and RTX-EU one patient] and four patients during the follow-up period [DRL_RI three patients and RTX-EU one patient]).
Baseline demographics and disease characteristics were comparable among treatment groups (Table 1). The mean (SD) age of patients was 44.5 (10.56) years, with female predominance (88.0%). Most patients were Asian (84.8%), and the mean (SD) body mass index (BMI) was 24.86 (5.23) kg/m2. In addition to MTX, the other most common concomitant medication was hydroxychloroquine (151 [54.7%]), and 13.8% of patients were on glucocorticoids (online supplementary Table S6).
Table 1.
Baseline demographics and disease characteristics (all patients enrolled)
| Category | DRL_ RI N = 91 |
RTX-US N = 92 |
RTX-EU N = 93 |
|---|---|---|---|
| Age, mean ± SD, years | 44.1 ± 10.96 | 44.0 ± 10.36 | 45.5 ± 10.41 |
| Age group, n (%) | |||
| 18–30 years | 12 (13.2) | 10 (10.9) | 11 (11.8) |
| 31–60 years | 72 (79.1) | 76 (82.6) | 76 (81.7) |
| 61–65 years | 7 (7.7) | 6 (6.5) | 6 (6.5) |
| Gender, n (%) | |||
| Female | 81 (89.0) | 81 (88.0) | 81 (87.1) |
| Male | 10 (11.0) | 11 (12.0) | 12 (12.9) |
| Race, n (%) | |||
| Asian | 78 (85.7) | 78 (84.8) | 78 (83.9) |
| White | 13 (14.3) | 14 (15.2) | 15 (16.1) |
| Ethnicity, n (%) | |||
| Not Hispanic or Latino | 84 (92.3) | 89 (96.7) | 89 (95.7) |
| Other | 7 (7.7) | 3 (3.3) | 4 (4.3) |
| Weight, mean ± SD, kg | 61.77 ± 13.87 | 61.76 ± 17.23 | 62.95 ± 15.82 |
| BMI, mean ± SD, kg/m2 | 24.61 ± 4.63 | 24.80 ± 5.73 | 25.17 ± 5.29 |
| Total swollen joints 66-joint count, mean ± SD | 16.2 ± 7.87 | 13.5 ± 5.97 | 13.5 ± 5.99 |
| Total tender joints 68-joint count, mean ± SD | 24.2 ± 10.50 | 20.5 ± 11.34 | 18.6 ± 9.18 |
| DAS28-CRP score, mean ± SD | 6.03 ± 0.67 | 5.72 ± 0.76 | 5.74 ± 0.70 |
| HAQ-DI score, mean ± SD | 1.56 ± 0.63 | 1.44 ± 0.51 | 1.46 ± 0.58 |
| LTBI status, n (%) | |||
| Positive | 19 (20.9) | 20 (21.7) | 16 (17.2) |
| Negative | 66 (72.5) | 63 (68.5) | 61 (65.6) |
| Indeterminate | 6 (6.6) | 9 (9.8) | 16 (17.2) |
Percentages are based on the number of patients within each treatment group under the all patients randomized set (N)
BMI body mass index, CRP C-reactive protein, DAS28 Disease Activity Score in 28 joints, HAQ-DI Health Assessment Questionnaire–Disease Index, LTBI latent tuberculosis infection, RTX-EU MabThera®, RTX-US Rituxan®, SD standard deviation
Pharmacokinetics
All tested products exhibited comparable plasma concentration–time profiles after both study drug infusions (Fig. 2). The 91% CI for the GMR of all primary PK end points, i.e., AUC0–14 days, first infusion, AUC0–∞, entire course, and AUC0–t, second infusion, were within the predefined similarity acceptance range of 80–125% for all pairwise treatment comparisons. The 91% CI for the GMR of the secondary PK end points Cmax after the first and second infusions and AUC0–t, entire course were also completely within the 80–125% range for all comparisons, indicating three-way PK similarity among DRL_RI, RTX-US, and RTX-EU (Table 2). Other secondary PK parameters were comparable between treatment groups (online supplementary Table S15). The comparisons of the PK end points in PK sensitivity analysis population 1 (including both ADA-positive patients and patients randomized only between the two available products) and PK sensitivity analysis population 2 (excluding both ADA-positive patients and patients randomized only between the two available products) also met the pre-specified acceptance criteria for all comparisons (online supplementary Tables S7 and S8).
Fig. 2.

Mean (± SD) plasma rituximab concentration versus time profiles after infusion 1 (a) and 2 (b) (main PK population). PK population excluding ADA-positive are considered. ADA antidrug antibody, PK pharmacokinetic, MabThera RTX-EU, Rituxan RTX-US, SD standard deviation
Table 2.
Summary of key primary and secondary PK parameters (main PK population excluding ADA-positive patients, N = 230)
| Parameter (units) | Treatment | GLS mean | GLS mean ratio, % (91% CI) | ||
|---|---|---|---|---|---|
| DRL_RI/RTX-US | DRL_RI/RTX-EU | RTX-US/RTX-EU | |||
| PK population—primary end points | |||||
| AUC0–14 days, first infusion (µg h/mL) | DRL_RI | 42,380 | 100.80 (94.62–107.38) | 95.45 (89.60–101.68) | 94.69 (88.85–100.92) |
| RTX-US | 42,040 | ||||
| RTX-EU | 44,400 | ||||
| AUC0–∞, entire course (µg h/mL) | DRL_RI | 162,000 | 100.37 (92.30–109.14) | 93.58 (85.98–101.85) | 93.24 (85.62–101.54) |
| RTX-US | 161,500 | ||||
| RTX-EU | 173,200 | ||||
| AUC0–t, second infusion (µg h/mL) | DRL_RI | 118,100 | 101.55 (92.60–111.36) | 94.83 (86.52–103.93) | 93.38 (85.08–102.50) |
| RTX-US | 116,300 | ||||
| RTX-EU | 124,600 | ||||
| PK population—secondary end points | |||||
| AUC0–t, entire course (µg h/mL) | DRL_RI | 160,600 | 100.66 (92.71–109.30) | 94.24 (86.71–102.42) | 93.61 (86.11–101.77) |
| RTX-US | 159,600 | ||||
| RTX-EU | 170,400 | ||||
| Cmax, first infusion (µg/mL) | DRL_RI | 348.229 | 105.31 (98.70–112.37) | 100.25 (93.95–106.97) | 95.19 (89.17–101.61) |
| RTX-US | 330.659 | ||||
| RTX-EU | 347.370 | ||||
| Cmax, second infusion (µg/mL) | DRL_RI | 420.740 | 104.68 (98.17–111.63) | 102.26 (95.94–109.00) | 97.69 (91.53–104.26) |
| RTX-US | 401.926 | ||||
| RTX-EU | 411.447 | ||||
Main PK population, N = 230 (DRL_RI, n = 79; RTX-US, n = 73; RTX-EU, n = 78) The values are back-transformed from the log scale Results based on an ANOVA model with treatment (DRL_RI, RTX-US, and RTX-EU) region and gender being considered as fixed effects
ANOVA analysis of variance, AUC0–14 days area under the plasma concentration–time curve from time 0 to day 14, AUC0–t area under the plasma concentration–time curve from time 0 to last quantifiable concentration, AUC0–∞ area under the plasma concentration–time curve from time 0 extrapolated to infinite time, CI confidence interval, Cmax peak plasma concentration, GLS geometric least-squares, PK pharmacokinetic, RTX-US Rituxan®, RTX-EU MabThera®
Pharmacodynamics
The reduction in mean DAS28-CRP from baseline to weeks 4, 8, 12, and 16 was comparable between DRL_RI and RTX-US, between DRL_RI and RTX-EU, and between RTX-US and RTX-EU (online supplementary Figure S3). Mean DAS28-CRP change from baseline to week 24 is detailed in the efficacy section.
B-cell depletion was rapid. Most patients (DRL_RI 100%, RTX-US 95.2%, and RTX-EU 98.7%) showed levels below 20% of LLN within 10 h after infusion 1 (online supplementary Figure S1). B-cell depletion (to below 20% of the LLN or below the limit of detection [LOD]) continued through week 24 (Fig. 3 and online supplementary Table S9). The proportion of patients with B-cell depletion (to below 20% of the LLN) at week 24 was 67.1% for DRL_RI, 69.1% for RTX-US, and 78.5% for RTX-EU. The 95% CI for the differences at week 24 between DRL_RI and RTX-US or RTX-EU was −2.0% (95% CI −16.22 to 12.19) and −11.4% (95% CI −24.73 to 2.50), respectively. The proportion of patients with B-cell depletion (below LOD) at week 24 was 0 for DRL_RI, 3.7% for RTX-US, and 1.3% for RTX-EU. The 95% CI for the differences at week 24 between DRL_RI and RTX-US or RTX-EU was −3.7% (95% CI −10.33 to 1.44) and −1.3% (95% CI −6.83 to 3.38); all 95% CIs included the value zero, and hence, differences were not significant.
Fig. 3.

Proportion of patients with B-cell depletion below 20% of the LLN a after infusion 1 up to 52 h and b after infusion 2 up to week 24 (PD population). The LLN for B-cell counts in this study was 107.0 cells/μL. 0 (h) at infusion 2 is pre-infusion sample day 15; 3700 (h) at infusion 2 is week 24 with respect to infusion 1. Upper and lower bounds indicated the confidence interval. LLN lower limit of normal, PD pharmacodynamic, MabThera RTX-EU, Rituxan RTX-US
Efficacy
The proportion of patients meeting ACR20, ACR50, and ACR70 response criteria were comparable across treatment groups (Fig. 4). In the PP population (N = 242), 68.4–72.0% of patients achieved ACR20 response, 39.5–43.9% achieved ACR50 response, and 14.8–17.1% achieved ACR70 response by week 24. The differences in adjusted ACR response rates (95% CI) between treatments in the PP population were not significant (Table 3).
Fig. 4.
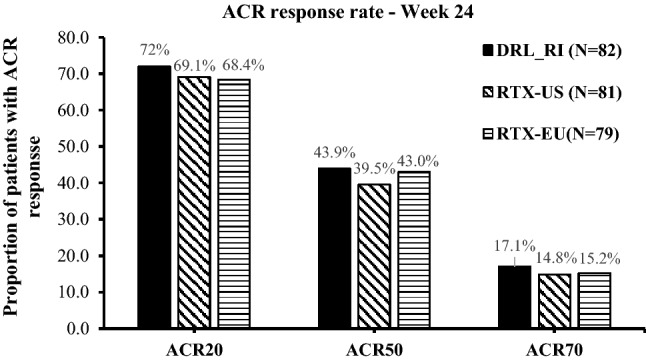
Proportions of patients achieving clinical response (PP population). ACR20, ACR50, and ACR70 are the proportion of the patients with at least 20%, 50%, or 70% improvement in counts of tender and swollen joints and in 3 of the following: patient’s assessment of pain, patient’s global assessment of disease activity, patient’s assessment of physical function, the physician’s global assessment of disease activity, and acute phase reactant. ACR American College of Rheumatology, PP per-protocol, RTX-EU MabThera®, RTX-US Rituxan®
Table 3.
Difference in adjusted ACR response rates (PP population, N = 230)
| Treatment | N | Adjusted response rate (%) | Comparison | Difference in percent adjusted response rate (95% CI) | |
|---|---|---|---|---|---|
| ACR20 | DRL_RI | 82 | 72.0 | DRL_RI–RTX-US | 2.8 (−11.18 to 16.81) |
| RTX-US | 81 | 69.1 | DRL_RI–RTX-EU | 3.6 (−10.54 to 17.73) | |
| RTX-EU | 79 | 68.4 | RTX-US–RTX-EU | 0.8 (−13.58 to 15.15) | |
| ACR50 | DRL_RI | 82 | 43.9 | DRL_RI–RTX-US | 4.4 (−10.73 to 19.52) |
| RTX-US | 81 | 39.5 | DRL_RI–RTX-EU | 0.9 (−14.45 to 16.18) | |
| RTX-EU | 79 | 43.0 | RTX-US–RTX-EU | −3.5 (−18.78 to 11.72) | |
| ACR70 | DRL_RI | 82 | 17.1 | DRL_RI–RTX-US | 2.3 (−8.97 to 13.49) |
| RTX-US | 81 | 14.8 | DRL_RI–RTX-EU | 1.9 (−9.47 to 13.24) | |
| RTX-EU | 79 | 15.2 | RTX-US–RTX-EU | −0.4 (−11.44 to 10.69) |
Adjusted response rates for the treatment arms using the logistic regression analysis including treatment, gender, and region as fixed effects and patients as a random effect in the model
ACR20, ACR50, and ACR70: proportion of the patients with at least 20%, 50%, or 70% improvement in counts of tender and swollen joints and in 3 of the following: patient’s assessment of pain, patient’s global assessment of disease activity, patient’s assessment of physical function, the physician’s global assessment of disease activity, and acute phase reactant
Percentages = (number of responders/number of patients in the corresponding visit) × 100
ACR American College of Rheumatology, CI confidence interval, PP per-protocol, RTX-EU MabThera®, RTX-US Rituxan®
The GEE-adjusted mean change from baseline in DAS28-CRP in the PP population (N = 241) was comparable in all treatment groups (Fig. 5 and online supplementary Table S10). The mean (SE) DAS28-CRP change from baseline to week 24 was − 1.63 (0.13) for DRL_RI, − 1.57 (0.12) for RTX-US, and − 1.78 (0.12) for RTX-EU. The 95% CI for the differences at week 24 between DRL_RI and either RTX-US (− 0.06 [0.14] [95% CI − 0.33 to 0.21]) or RTX-EU (0.16 [0.15] [95% CI − 0.13 to 0.44]) included the value zero; thus, differences were not significant.
Fig. 5.
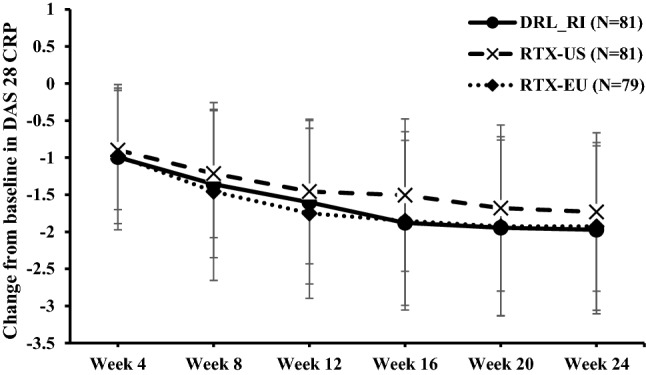
Change in mean DAS28-CRP score up to 24 weeks from baseline (PP population). The geometric mean calculation for change from baseline columns is based on the ratio of visit value to baseline value. DAS28-CRP Disease Activity Score (28 joints)–C-reactive protein, PP per-protocol, RTX-EU MabThera®, RTX-US Rituxan®
The changes from baseline in mean (SD) HAQ-DI score at week 24 were comparable in all treatment groups (online supplementary Figure S4), with values of − 0.68 (0.59), −0.62 (0.61), and −0.68 (0.64) in the DRL_RI, RTX-US, and RTX-EU groups, respectively.
Safety
All 276 patients received at least one dose of study medication and were included in the safety analysis. Safety follow-up was further continued until week 52 for most patients (N = 240). No clinically meaningful differences among treatment groups were observed in the incidence of all and treatment-related AEs, SAEs, and AEs leading to discontinuation of the drug (Table 4). Until week 24, 214 treatment-emergent AEs (TEAEs) were reported for 100 patients (36.2%): 33 patients (36.3%) in the DRL_RI arm reported 71 TEAEs; 30 patients (32.6%) in the RTX-US arm reported 67 TEAEs; and 37 patients (39.8%) in the RTX-EU arm reported 76 TEAEs. One hundred thirty-three TEAEs were reported in 84 patients (30.4%) in the follow-up phase: 27 patients (29.7%) in the DRL_RI arm reported 42 TEAEs; 28 patients (30.4%) in the RTX-US arm reported 40 TEAEs, and 29 patients (31.2%) in the RTX-EU arm reported 51 TEAEs. Most AEs (DRL_RI 63 of 71 events, RTX-US 55 of 67 events, and RTX-EU 60 of 67 events) reported until 24 weeks were mild in severity.
Table 4.
Summary of treatment-emergent AEs (safety analysis population)
| Part 1 | Part 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| DRL_RI N = 91 |
RTX-US N = 92 |
RTX-EU N = 93 |
Total N = 276 |
DRL_RI N = 91 |
RTX-US N = 92 |
RTX-EU N = 93 |
Total N = 276 |
|
| Patients with at least 1 AE, n (%) | 33 (36.3) | 30 (32.6) | 37 (39.8) | 100 (36.2) | 27 (29.7) | 28 (30.4) | 29 (31.2) | 84 (30.4) |
| Patients with at least 1 treatment-related AE, n (%) | 10 (11.0) | 10 (10.9) | 12 (12.9) | 32 (11.6) | 8 (8.8) | 9 (9.8) | 12 (12.9) | 29 (10.5) |
| Patients with at least 1 SAE, n (%) | 2 (2.2) | 4 (4.3) | 6 (6.5) | 12 (4.3) | 1 (1.1) | 1 (1.1) | 4 (4.3) | 6 (2.2) |
| Patients with at least 1 treatment-related SAE, n (%) | 1 (1.1) | 3 (3.3) | 4 (4.3) | 8 (2.9) | 0 | 0 | 3 (3.2) | 3 (1.1) |
| Patients died from AE, n (%) | 1 (1.1) | 0 | 0 | 1 (0.4) | 1 (1.1) | 0 | 0 | 1 (0.4) |
| Patients discontinued study due to AE, n (%) | 1 (1.1) | 3 (3.3) | 6 (6.5) | 10 (3.6) | 1 (1.1) | 1 (1.1) | 0 | 2 (0.7) |
Part 1: All treatment-emergent AEs occurring up to week 24 of the study
Part 2: Treatment-emergent AEs ongoing at the week 24 and AEs occurring after week 24 up to week 52
Patients experiencing multiple events were counted only once within the treatment group
Percentages were based on number of patients within each treatment group under safety analysis population (N)
AE adverse event, RTX-EU MabThera®, RTX-US Rituxan®, SAE serious adverse event
The AEs reported in > 2% of patients in any treatment group until week 52 included upper respiratory tract infection, urinary tract infection, cough, pyrexia, mouth ulceration, B-lymphocyte count decrease, body tinea, anemia, infusion-related reactions (IRRs), upper abdominal pain, headache, increased aspartate aminotransferase, increased alanine aminotransferase, RA, and hypertension. The AEs with an incidence > 2% until week 24 and from week 24 to week 52 are presented in online supplementary Tables S11 and S12, respectively. The most commonly reported treatment-related AEs were IRR (one [1.1%] in the DRL_RI group, two [2.2%] in the RTX-US group, and one [1.1%] in the RTX-EU group), urinary tract infection (two [2.2%] in the DRL_RI group and one [1.1%] in the RTX-EU group), blood and lymphatic system disorders (one patient [1.1%] each in the DRL_RI, RTX-US, and RTX-EU groups), and musculoskeletal and connective tissue disorders (two [2.2%] in the RTX-US group and one [1.1%] in the RTX-EU group).
A total of 21 treatment-emergent SAEs were reported in 18 patients until week 52 (three patients [3.3%] in the DRL_RI group, five patients [5.4%] in the RTX-US group, and ten patients [10.8] in the RTX-EU group) (online supplementary Table S13). All SAEs resolved completely, except for two events with a fatal outcome in patients in the DRL_RI arm, which occurred on days 155 and 354 and were considered unrelated to the study drug, and one event in the RTX-EU arm, which occurred 2 days after the administration of the first dose of study drug; the patient recovered with sequelae, and the event was considered related to the study drug.
Ten patients (DRL_RI one [1.1%], RTX-US three [3.3%], and RTX-EU six [6.5%]) withdrew from the study because of AEs until week 24. The AEs leading to treatment discontinuation (all reported once) were H1N1 influenza, herpes simplex infection, upper respiratory tract infection, choking sensation, interstitial lung disease, rash, urticaria, cardiorespiratory arrest, IRR, and spinal compression fracture. Until week 24, fewer patients withdrew because of AEs in the DRL_RI group (one patient [1.1%]) as compared with the RTX-EU (six patients [6.5%]) and RTX-US groups (three patients [3.3%]). Between weeks 24 and 52, one patient each discontinued treatment in the DRL_RI and RTX-US groups; no patient discontinued treatment in the RTX-EU group. No reactivation or new cases of tuberculosis were reported during the study. No SAEs related to deep venous thrombosis or pulmonary embolism were reported. Only a mild thrombophlebitis event was reported in a patient treated with RTX-EU. There were no atypical infections reported during the study.
The proportions of patients achieving B-cell repletion ≥ LLN by week 52 were comparable across all treatment groups (DRL_RI 33.3%, RTX-US 23.2%, and RTX-EU 23.7%). The 95% CI for the differences between DRL_RI and RTX-US or RTX-EU was 10.1% (95% CI −6.31 to 25.68) and 9.6% (95% CI −6.59 to 25.12). A similar trend was seen for B-cell repletion to ≥ 80% of baseline through week 52 across all treatment groups, except at week 52, where the proportion of patients in the DRL_RI group achieving repletion was higher compared with RTX-US and RTX-EU (21.7% vs. 14.3% and 6.8%). The 95% CI for the differences between DRL_RI and RTX-US or RTX-EU was 7.4% (95% CI −6.91 to 21.17) and 14.9% (95% CI 2.19–27.53); most 95% CIs included the value zero, and hence, most differences were not significant (online supplementary Table S14 and Figures S2a and S2b).
Immunogenicity
The proportion of ADA-positive patients by assessment and treatment group is summarized in Table 5. The proportion (95% CI) of ADA-positive patients at any time until week 52 was 18.7% (11.28–28.22) in the DRL_RI group, 30.4% (21.27–40.90) in the RTX-US group, and 28.0% (19.14–38.22) in the RTX-EU group. At week 24, one patient in each treatment group had neutralizing ADA. No neutralizing ADAs were detected by week 52 in the DRL_RI group; whereas two patients (2.5%) in the RTX-US group and one patient (1.2%) in the RTX-EU group tested positive for neutralizing ADAs. The median (minimum − maximum) titers of ADA by time point are given in online supplementary Table S16.
Table 5.
Proportion of ADA-positive patients (PP population)
| Baseline n (%) | Week 4 n (%) | Week 16 n (%) | Week 24 n (%) | Week 52 n (%) | |
|---|---|---|---|---|---|
| DRL_RI (N = 82) | 3 (3.3) | 1 (1.1) | 2 (2.3) | 4 (4.6) | 12 (14.6) |
| RTX-US (N = 81) | 2 (2.2) | 0 | 3 (3.4) | 12 (14.0) | 22 (27.5) |
| RTX-EU (N = 79) | 0 | 1 (1.2) | 2 (2.4) | 7 (8.0) | 22 (26.8) |
ADA anti-drug antibody, PP per-protocol, RTX-EU MabThera®, RTX-US Rituxan®
Discussion
This study established the PK similarity of the proposed biosimilar DRL_RI and the reference products from the USA and the EU by meeting its primary end points, while comparing the PD, efficacy, safety, and immunogenicity of the same products in RA patients (the most sensitive population for detection of PK differences between rituximab products).
All primary (AUC0–14 days, first infusion, AUC0–t, second infusion, and AUC0–∞, entire course) and secondary (Cmax, first infusion, Cmax, second infusion, AUC0–t, entire course) PK end point comparisons among DRL_RI, RTX-US, and RTX-EU showed the 91% CIs of their GMRs were contained within the predefined equivalence margins of 80–125%. Sensitivity analyses performed by including patients with confirmed ADA and by excluding patients randomized only between the two available products at the site and time confirmed the above results, supporting the robustness of the conclusions.
The PK results are comparable with prior publications. The geometric mean AUC0–∞, entire course for the tested products was 161,500–173,200 µg·h/mL in the different groups and 162,377–196,000 µg·h/mL in several published PK similarity studies of proposed rituximab biosimilars in RA patients. Geometric mean Cmax values were also comparable with published values. Geometric mean Cmax, first infusion and Cmax, second infusion values were 331–348 µg/mL and 402–421 µg/mL, respectively, in the different groups, and 320–362 µg/mL and 367–478 µg/mL, respectively, in several published PK similarity studies of proposed rituximab biosimilars in RA patients [18–21].
A comparable reduction was seen in DAS28-CRP from baseline to weeks 4, 8, 12, and 16 between DRL_RI and reference rituximab (RTX-US and RTX-EU). Additionally, all treatment groups showed B-cell depletion below 20% of LLN within 10 h after first infusion, with nearly complete depletion occurring within 48 h. B-cell suppression was sustained until week 24 and was comparable across the three treatment groups. B-cell depletion kinetics observed with DRL_RI and the reference products in this study were consistent with those published for rituximab in RA patients in the REFLEX and DANCER trials [22, 23] and in candidate rituximab biosimilars studies [18–20].
The findings of the present study suggest that DRL_RI efficacy in terms of ACR20, ACR50, and ACR70 rates is comparable to that of RTX-US and RTX-EU, with 95% CIs including no difference (a value of 0) and excluding differences by more than 20%.
The ACR20, ACR50, and ACR70 rates at week 24 with DRL_RI were 72%, 43.9%, and 17.1%, respectively. These values are on the higher side of the reported range for rituximab in combination with MTX in patients with RA pivotal trials, which were 51–73% (ACR20), 25–43% (ACR50), and 10–23% (ACR70) at week 24 [22–25]. Prior treatment with Tumor Necrosis Factor alpha (TNF-α) inhibitors varied across these studies; some of them [22–24] included TNF-α inhibitor–exposed/failed patients, while our study had only TNF-α inhibitor–naïve patients, which could have contributed to the observation of higher ACR response rates in this study.
All 95% CIs for the difference between DRL-RI and either reference product in the mean change in DAS28 from baseline to week 24 included the value 0, and their limits are not broader than 0.44 Units. Interestingly, this difference is narrower than 0.6 Units, thought to be the minimum detectable difference in DAS28 [26], a posteriori supporting that the efficacy of DRL_RI measured in DAS28-CRP terms is comparable with that of the reference products.
The observed mean change in DAS28 for DRL_RI in this study is in line with previously published results [21]. The mean change in DAS28-CRP (−1.63) observed with DRL_RI at week 24 was comparable to improvements reported in DAS28 from baseline to week 24 in the REFLEX (−1.90) and DANCER (−2.05) trials [22, 23]. A comparable mean reduction (−2.13) in DAS28-CRP at week 24 from baseline was published for another potential biosimilar [18]. As mentioned earlier, given the different patient populations included in the various studies, these values cannot be directly compared across studies.
Changes in the mean HAQ-DI quality-of-life scores between baseline and week 24 were comparable between DRL_RI (−0.68) and the reference products RTX-US (−0.62) and RTX-EU (−0.68), and with historical data in the REFLEX (−0.4) and DANCER (−0.5) trials [22, 23].
No relevant differences in the incidence, profile, or severity of AEs between treatment groups were observed. In this study, the most frequently reported AEs until week 24 were under the System Organ Class of infections and infestations (14.1%), an expected finding for rituximab. In the follow-up phase until week 52, the incidence of infections and infestations was 6.9%. However, the overall incidence of infection-related AEs was lower in all treatment groups in this study than the incidences reported in the pivotal trials [22, 23]. The overall incidence of SAEs was similar across all treatment groups and consistent with that observed in other studies with rituximab in RA [22, 23, 25]. The incidence of IRRs was comparable among the three treatment groups, with 1.1% of patients in DRL_RI and RTX-EU groups and 2.2% of patients in the RTX-US group experiencing IRRs. The incidence of IRRs observed in this study is lower than that observed in historical studies of rituximab, where the incidence of IRRs following the first infusion was 23–32%, and declined after the second infusion (6–9%) [22, 23, 25]. A numerically lower incidence of AEs leading to treatment discontinuation was observed in the DRL_RI group compared with the other treatment groups (DRL_RI 1.1%, RTX-US 3.3%, and RTX-EU 6.5%).
B-cell recovery was not completed by week 52 in any treatment group, and most patients had low B-cell counts. There was no significant difference between DRL_RI and the reference products in the proportion of patients who had B-cell repletion (either ≥ LLN or ≥ 80% of baseline) at most time points, except at week 52, when the proportion of patients with B-cell recovery ≥ 80% of baseline was higher in the DRL_RI group than that in the RTX-EU group. This isolated and inconsistent observation is likely a random finding, and no clinical implications of the same were evident in the study.
The incidence of ADAs at week 52 in the DRL_RI group compared with both reference groups was numerically lower, with overlapping 95% CIs. Most observed ADAs were low titer and non-neutralizing. The ADA incidence, titer values, and neutralizing capacity were not different between treatment groups, suggesting a comparable immunogenicity between DRL_RI and reference rituximab (RTX-US and RTX-EU).
The present study with its randomized, double-blind design and high patient retention rates established the proposed biosimilar DRL_RI had similar PK and comparable PD, safety, efficacy, and immunogenicity to reference rituximab.
Limitations
This study was conducted in a biologics-naïve population that differs from the labeled population for rituximab treatment in many regions; however, rituximab is still considered suitable therapy for this population of patients. For a comparative study, with the objectives to establish biosimilarity in the clinical setting, given that the background was matched for both arms, this aspect does not influence the study outcome. With the primary end point of the study being PK equivalence, the study was not designed to provide a statistically powered evaluation of efficacy similarity, and an a posteriori analysis based on literature has been performed and discussed.
Conclusion
DRL_RI, a proposed rituximab biosimilar, was compared with the reference products RTX-US and RTX-EU in biologics-naïve patients with moderate-to-severe RA who had inadequate response to MTX-based therapy. The study met its primary objective by demonstrating the three-way PK similarity of DRL_RI, RTX-EU, and RTX-US. The study also revealed comparable PD, efficacy, safety, and immunogenicity between DRL_RI and both reference products.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank medical writers, Mr. Lalit Doshi and Ms. Prajakta Nachane, from Sciformix Technologies Pvt. Ltd., for providing writing assistance in the development of this manuscript; Mr. Pramod Kumar Reddy, from Dr. Reddy’s Laboratories Ltd., for assistance in the generation of pharmacokinetic and pharmacodynamic plots and review of the CONSORT flow chart; and Mr. Naveen Kumar SR (who worked for Dr. Reddy’s Laboratories Ltd. during the manuscript writing time) for providing assistance in the development of this manuscript. We also thank the entire project team at Dr. Reddy’s Laboratories Ltd. for their contribution to the study.
Compliance with Ethical Standards
Funding
The study and the manuscript writing have been funded by Dr. Reddy’s Laboratories.
Conflict of interest
Luis Lopez-Lazaro, Sonica Sachdeva Batra, and Suresh Kankanwadi are employees of Dr. Reddy’s Laboratories Ltd. Vikram Muralidhar Haridas, Rahul Katta, Ajit Nalawade; Sandeep Kharkar, Vyacheslav Zhdan, and Olena Garmish were the investigators involved in the study and have no conflicts of interest to disclose.
Footnotes
Maximum plasma concentration (Cmax) after the second infusion was initially a primary end point, later changed to secondary, while AUC0–14 days after the first infusion was a secondary end point changed to a primary one before unblinding, based upon Regulatory Authority discussions.
A part of this work has been previously published as two poster presentations (abstract #513 for pharmacokinetics and pharmacodynamics and abstract #518 for safety, immunogenicity, and efficacy) in the 2018 Edition of the Canadian Rheumatology Association Annual Scientific Meeting and AHPA meeting (February 21–24, 2018), Vancouver, Canada.
Contributor Information
Vikram Muralidhar Haridas, Email: haridasvikram@yahoo.co.in.
Rahul Katta, Email: rahulkatta@hotmail.com.
Ajit Nalawade, Email: drajitnalawade1975@gmail.com.
Sandeep Kharkar, Email: drsandeep2k@yahoo.com.
Vyacheslav Zhdan, Email: vyacheslav.zhdan@i.ua.
Olena Garmish, Email: garmish.elena@gmail.com.
Luis Lopez-Lazaro, Email: llopezlazaro@drreddys.com.
Sonica Sachdeva Batra, Email: sonicabatra@drreddys.com.
Suresh Kankanwadi, Email: sureshkankanwadi@drreddys.com.
References
- 1.European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues dated 18 December 2014. Document Reference EMEA/CHMP/BMWP/42832/2005 Rev1. http://www.ema.europa.eu/docs/en_GB/document library/Scientific guideline/2015/01/WC500180219.pdf. Accessed 13 Aug 2019.
- 2.US Food and Drugs Administration. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. https://www.fda.gov/media/88622/download Accessed 21 Nov 2019.
- 3.Rituxan (rituximab) 100 mg/10 mL and 500 mg/50 mL, injection for intravenous use—United States prescribing information, 2019. https://www.gene.com/ download/pdf/rituxan prescribing.pdf. Accessed 13 Aug 2019.
- 4.Cross M, Smith E, Hoy D, et al. The global burden of rheumatoid arthritis: estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis. 2014;73:1316–1322. doi: 10.1136/annrheumdis-2013-204627. [DOI] [PubMed] [Google Scholar]
- 5.Curtis JR, Singh JA. Use of biologics in rheumatoid arthritis: current and emerging paradigms of care. Clin Ther. 2011;33:679–707. doi: 10.1016/j.clinthera.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen MD, Keystone E. Rituximab for rheumatoid arthritis. Rheumatol Ther. 2015;2(2):99–111. doi: 10.1007/s40744-015-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Medical Association World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 8.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonized Tripartite Guideline. Guideline for Good Clinical Practice E6(R1) 1996. http://www.ich.org/fileadmin/PublicWeb_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed on 13 Augt 2019. [PubMed]
- 9.American College of Rheumatology. ACR endorsed criteria. From 1987 ACR criteria. http://www.rheumatology.org/practice/clinical/classification/ra/ra.asp. Accessed 27 Dec 2018.
- 10.DAS Score Website. https://www.das-score.nl/das28/en/ Accessed 5 Jan 2020.
- 11.MabThera 100 mg concentrate for solution for infusion—Summary of Product Characteristics (SPC). https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf. Accessed on 13 Aug 2019.
- 12.Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–735. doi: 10.1002/art.1780380602. [DOI] [PubMed] [Google Scholar]
- 13.Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2):137–145. doi: 10.1002/art.1780230202. [DOI] [PubMed] [Google Scholar]
- 14.Wittes J, Brittain E. The role of internal pilot studies in increasing the efficiency of clinical trials. Stat Med. 1990;9(1–2):65–71. doi: 10.1002/sim.4780090113. [DOI] [PubMed] [Google Scholar]
- 15.Ng CM, Bruno R, Combs D, Davies B. Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol. 2005;45:792–801. doi: 10.1177/0091270005277075. [DOI] [PubMed] [Google Scholar]
- 16.Lioger B, Edupuganti SR, Mulleman D, et al. Antigenic burden and serum IgG concentrations influence rituximab pharmacokinetics in rheumatoid arthritis patients. Br J Clin Pharmacol. 2017;83:1773–1781. doi: 10.1111/bcp.13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller C, Murawski N, Wiesen MH, et al. The role of sex and weight on rituximab clearance and serum elimination half-life in elderly patients with DLBCL. Blood. 2012;5(119):3276–3284. doi: 10.1182/blood-2011-09-380949. [DOI] [PubMed] [Google Scholar]
- 18.Park W, Božić-Majstorović L, Milakovic D, et al. Comparison of biosimilar CT-P10 and innovator rituximab in patients with rheumatoid arthritis: a randomized controlled Phase 3 trial. MAbs. 2018;10:934–943. doi: 10.1080/19420862.2018.1487912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo DH, Suh CH, Shim SC, et al. A multicenter randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:566–570. doi: 10.1136/annrheumdis-2016-209540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen S, Emery P, Greenwald M, et al. A phase I pharmacokinetics trial comparing PF-05280586 (a potential biosimilar) and rituximab in patients with active rheumatoid arthritis. Br J Clin Pharmacol. 2016;82:129–138. doi: 10.1111/bcp.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smolen JS, Cohen SB, Tony HP, et al. A randomised, double-blind trial to demonstrate bioequivalence of GP2013 and reference rituximab combined with methotrexate in patients with active rheumatoid arthritis. Ann Rheum Dis. 2017;76:1598–1602. doi: 10.1136/annrheumdis-2017-211281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 23.Emery P, Fleischmann R, Filipowicz-Sosnowska A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–1400. doi: 10.1002/art.21778. [DOI] [PubMed] [Google Scholar]
- 24.Edwards JCW, Szczepanski L, Szechinski J, et al. Efficacy of B-cell targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 25.Emery P, Deodhar A, Rigby WF, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE) Ann Rheum Dis. 2010;69:1629–1635. doi: 10.1136/ard.2009.119933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Gestel AM, Prevoo ML, van’t Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39:34–40. doi: 10.1002/art.1780390105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.