

Abstract
The electrical control and readout of molecular spin states are key for high-density storage. Expectations are that electrically-driven spin and vibrational excitations in a molecule should give rise to new conductance features in the presence of magnetic anisotropy, offering alternative routes to study and, ultimately, manipulate molecular magnetism. Here, we use inelastic electron tunneling spectroscopy to promote and detect the excited spin states of a prototypical molecule with magnetic anisotropy. We demonstrate the existence of a vibron-assisted spin excitation that can exceed in energy and in amplitude a simple excitation among spin states. This excitation, which can be quenched by structural changes in the magnetic molecule, is explained using first-principles calculations that include dynamical electronic correlations.
Subject terms: Magnetic properties and materials, Molecular electronics, Spintronics
Electrically manipulating molecular magnetism is a challenge to overcome for applications in high-density storage. Here, the authors use inelastic electron tunneling spectroscopy to show that a vibron-assisted spin excitation in a nickel-nickelocene complex exceeds a pure spin excitation in energy and amplitude.
Introduction
Electrically-driven excitations among spin states1, or spin excitations, are increasingly observed in organometallic molecules coupled to metallic electrodes2–10, and show great potential in view of manipulating the molecular spin4,11. In these molecules, the reduced symmetry of the metal center aligns the magnetic moment of the molecule along certain favorable directions and spin excitations can be produced in the absence of a magnetic field owing to this magnetic anisotropy12. Along with spin excitations, electrons may also trigger molecular vibrational modes, or vibrons, that modify13–18 or even suppress19,20 molecular conductance. Even if the presence of both excitations could be evidenced in a molecular system21,22, experimental observations regarding their interplay have remained surprisingly elusive.
Individual molecular spins are also prone to the Kondo screening by the host electrons of the metal electrode. In this case, the electron–vibron interaction can produce resonances in the molecular conductance at the bias of the vibron’s excitation energy23–27. These resonances are ascribed to tunneling electrons that have their spin flipped when elastically scattering off the molecular spin, but with sufficient energy to activate a vibrational mode in the molecule28. In principle, a similar scattering mechanism can be expected for tunneling electrons losing energy to both the molecular spin states and a vibrational mode.
With this in mind, here we use scanning tunneling microscopy (STM) to study a molecular complex with magnetic anisotropy, which includes a nickelocene molecule [Ni(C5H5)2, see Fig. 1d; noted Nc hereafter] and a Ni atom. When the complex (noted NiNc hereafter) is embedded in a Nc layer on Cu(100), sizable spin and vibrational excitations can be electrically-driven in NiNc at distinct threshold energies. With the help of density functional theory (DFT) calculations, we demonstrate that a joint spin-vibration excitation is also present in NiNc resulting from the concomitant excitation of a spin and a vibration. This excitation should be quite common to magnetic molecules.
Fig. 1. NiNc adsorption in a nickelocene layer.
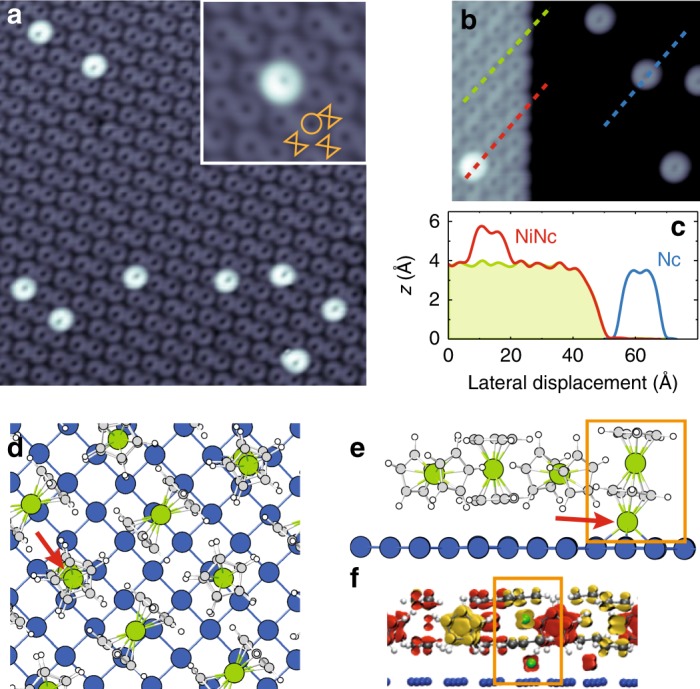
a Self-assembled layer (paired configuration) and NiNc complex on Cu(100) (image size: 16 × 16 nm2, sample bias: 20 mV, tunneling current: 20 pA). Inset: Close-up view of a NiNc complex in the paired layer. The molecular arrangement is sketched in orange as a circle for the vertical Nc and as a hourglass for the horizontal Nc (4 × 4 nm2, 20 mV, 100 pA). b Edge of a paired layer and isolated Nc molecules on Cu(100) (10 × 10 nm2, 20 mV, 100 pA) and c their corresponding height profiles along the dashed lines in b. d Top view, e side view of the DFT optimized structure of the NiNc complex embedded in a Nc paired layer—some molecules have been removed for clarity (H: white, C: gray, Ni: green). The red arrow indicates the position of the Ni adatom. f Side view of the spin density map of the paired structure containing two Ni atoms. Orange rectangles mark the NiNc complex. Yellow: Spin up, red: Spin down.
Results
Layer-integrated NiNc
After deposition of Nc onto the Cu(100) surface (see Methods section), well-ordered molecular assemblies on the surface were found (Fig. 1a), along with isolated Nc molecules (Fig. 1b). The ring-shaped pattern in the images is produced by a cyclopentadienyl (Cp hereafter) ring and indicates that Nc is adsorbed with its principal axis perpendicular to the surface29. In the molecular layer, however, these “vertically” adsorbed molecules coexist with “horizontally” adsorbed molecules (principal axis parallel to the surface), as sketched in the inset of Fig. 1a. This T-shaped configuration is governed by van der Waals interactions30 and results in two possible molecular configurations, known as paired (Fig. 1a) and compact (not shown)29. Our experimental observations regarding the formation and properties of the NiNc complex showed no significant difference between the two configurations, therefore in the following we will only focus on the paired configuration.
To build NiNc complexes, we proceeded as in earlier work on ferrocene31 and exposed the Nc layer to single Ni atoms (Fig. 1a). The molecular complex is imaged as a ring with an apparent height of 5.8 ± 0.2 Å relative to the underlying copper surface (Fig. 1b, c), while the neighboring Nc molecules have an apparent height of 4.1 ± 0.2 Å. Similar to previous experiments in which cobalt was deposited onto a ferrocene layer31, the ring-like shape demonstrates that the atom is positioned beneath a Nc molecule. This assignment is confirmed by our DFT calculations (Fig. 1d–f); details of the calculation are given in the Methods section] showing a 3 eV energy difference in favor of the deposited Ni atom beneath Nc rather than on top. The Ni atom, which for clarity we refer to as Ni adatom hereafter, is located 2.4 Å above the copper surface. The NiNc complex displays a lower symmetry with a 0.5 Å misalignment between the two Ni atoms (Fig. 1d) and a tilt of the principal axis of Nc (Fig. 1e). This tilt is confirmed by close-up STM images [see inset of Fig. 1a and line profile in Fig. 1c], differentiating the present NiNc complex from those investigated numerically in previous studies, where the Ni adatom is centered on the ring32,33. We show below that NiNc adopts instead this structure outside the molecular layer.
Figure 2a presents a typical dI/dV spectrum acquired above the Cp ring of a NiNc complex in the paired layer, while the d2I/dV2 spectrum is shown in Fig. 2b. The dI/dV spectrum is dominated by stepped features, symmetric with respect to zero bias, which point to inelastic excitations. The energy onset of these steps, as determined over a collection of NiNc complexes, are ∣ϵ1∣ = 4.3 ± 0.4 meV (the excitation is noted 1 hereafter), ∣ϵ2∣ = 33.9 ± 0.5 meV (noted 2) and ∣ϵ3∣ = 38.1 ± 0.6 meV (noted 3). The data exhibited a negligible tip dependence.
Fig. 2. Local spectroscopy of NiNc.

a dI/dV spectrum and b d2I/dV2 spectrum acquired above the Cp ring of a NiNc complex (feedback loop opened at −80 mV and 200 pA). The solid red line is a fit based on a dynamical scattering model34. The data exhibited a negligible tip dependence as reflected by the measurement uncertainties over a collection of NiNc complexes. The energy onsets are labeled 1, 2, and 3 in b. The dashed curve in a is the dI/dV spectrum acquired above the Cp ring of a Nc in the layer (feedback loop opened at −70 mV and 100 pA). Inset: Arrow indicates the position where the spectrum was acquired. c d2I/dV2 spectra acquired above the center of a NiNc complex at various tip-molecule distances. The feedback loop was opened at 50 pA (solid black line), 500 pA (solid blue line) and 1 nA (solid red line) with a bias set to −20 mV—higher opening currents correspond to smaller tip-molecule distances. The spectra were normalized by the opening current. The vertical lines indicate the peak position of excitation 2. d–f Spatial variation of the d2I∕dV2 signal acquired at a constant-height and at a bias of 4.5 mV, 32.5 mV, and 37 mV, respectively (feedback loop opened at −60 mV and 500 pA above a Nc molecule of the layer; 3 mV rms modulation amplitude). These biases correspond to the excitation thresholds highlighted in b. g dI/dV spectra acquired at four locations above NiNc [indicated by colored dots in the line profile of NiNc presented in the inset]. The feedback loop was opened at −30 mV (300 pA) above all locations in order to have the same amplitude for step 1 in the dI∕dV spectra.
Spectroscopic assignment
Given the similarity to the spin excitation spectrum measured above Nc [dashed line in Fig. 2a], which is known from previous studies8,34, we assign 1 to a spin excitation. To confirm this assignment, we carried out DFT calculations (see Methods section). Using the relaxed structure of NiNc determined above, we find that the dxz and dyz frontier orbitals of Nc in the complex are spread out in a range of ±1 eV around the Fermi level (Supplementary Fig. 1). The NiNc complex has a total magnetic moment of 1.4 μB, corresponding to an antiferromagnetic coupling between the Ni adatom (−0.2 μB) and Nc (+1.6 μB) with a charge transfer of 0.1 electrons from the substrate. The DFT calculations point therefore to an effective spin of S = 1. The strong resemblance with the spin excitation of single Nc molecules8,35 further allows us to identify the spin spectrum with the one originating from a S = 1 system. Consequently, we model 1 via a spin Hamiltonian that includes axial magnetic anisotropy
| 1 |
where is an Anderson Hamiltonian involving a single Nc orbital of the NiNc complex (see Methods section). The z-axis is chosen along the molecular axis running through the center of the Cp rings. Within this viewpoint, 1 is assigned to a spin excitation occurring between the ground state and the doubly degenerate excited states of NiNc [see Fig. 2a], the onset ∣ϵ1∣ corresponding then to D. The fit to the line shape based on Eq. (1) is highly satisfactory [solid red line in Fig. 2a]34, and yields D = 4.6 ± 0.2 meV. The inclusion of many-body interactions in the fit through , i.e., the inclusion of Kondo-like phenomena, is crucial for reproducing the cusp observed above the energy threshold of the spin excitation9,36,37. The cusp is associated to a Kondo fitting parameter that is typical for Nc on copper substrates (Supplementary Fig. 2 and Supplementary Note 1).
Excitation 2 corresponds instead to a vibrational excitation of energy ∣ϵ2∣ = ℏω = 33.9 ± 0.5 meV. When approaching the tip towards NiNc, we observe a red shift of the peak (dip) associated to 2 as high as 1.5 meV (Fig. 2c and Supplementary Table 1), which is characteristic of a vibrational excitation38,39—the onset of 1 is instead nearly constant. The spatial dependence of the d2I∕dV2 signal above the NiNc molecule also hints to a vibrational excitation. While excitation 1 is located on the Cp ring (Fig. 2d), excitation 2 is instead maximal in the center of the ring (Fig. 2e). To elucidate this difference, we computed the vibrational modes for the relaxed NiNc structure determined above. We found three vibrational modes at 29.1, 31.8 meV (Fig. 3a), and 35.5 meV (Fig. 3b) close to the experimental energy ∣ϵ2∣, all of them being robust to the finite atomic displacements used in the calculation. The first mode corresponds to a molecular frustrated rotation, which we discard as is at variance with the experimentally observed spatial location of the vibration. The second and third mode correspond to a translational motion of the Ni atom inside Nc. The tilted adsorption geometry of Nc on the Ni adatom breaks the degeneracy of these two modes observed in the gas phase. While the 31.8 meV mode only gives a negligible change in the simulated conductance across the molecule, we find instead a dominant contribution for the 35.5 meV mode with a spatial dependence matching experimental observations (Fig. 3c). The simulated STM image of NiNc is presented in Fig. 3d40. Excitation 2 is then assigned to a Ni-Cp mode (Fig. 3b), where the internal Ni atom moves parallel to the tilted Cp ring.
Fig. 3. Active vibrational modes and simulated spectrum of NiNc.
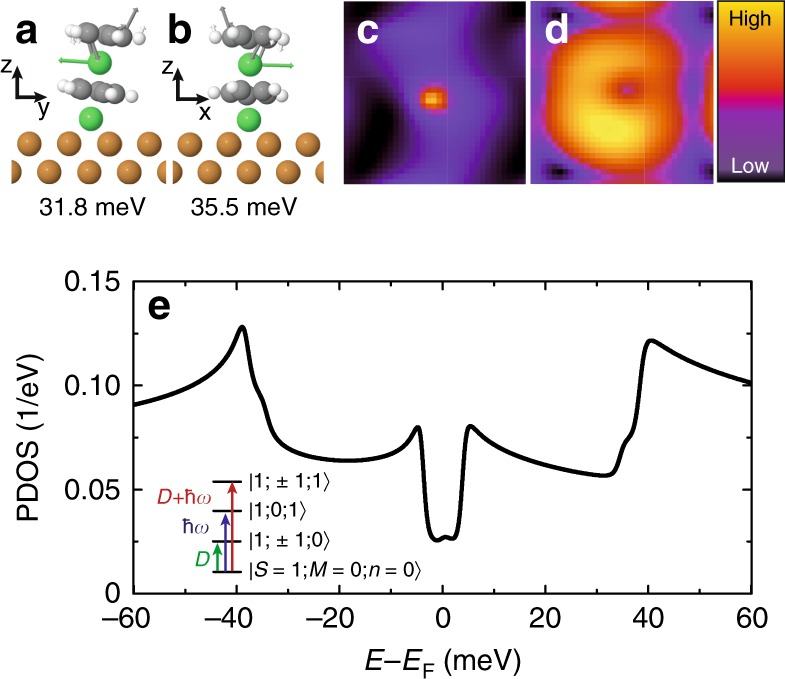
a, b Calculated vibrational modes close to ∣ϵ2∣. c Computed ratio between the inelastic and elastic tunneling conductance using the 35.5 meV mode. The maximum change represents 5% of the elastic conductance and is located in the center of the molecule. d Calculated local density of states of a NiNc complex at the Fermi level. e Projected density of states (PDOS) on the occupied molecular orbital as a function of electron energy calculated from Eq. (2). In the tunneling regime, the differential conductance is proportional to the PDOS42. Inset: State diagram of a NiNc complex based on Eq. (2); the eigenstates are noted as and the arrows depict the three transitions leading to the excitation steps in the dI/dV spectrum.
Excitation 3 shares spectroscopic fingerprints with excitations 1 and 2: (i) all the spectra recorded so far showed that the energy onset of 3 is the sum of the energy onsets of 1 and of 2, ∣ϵ3∣ = ∣ϵ1∣ + ∣ϵ2∣ (Supplementary Table 2); (ii) the line shape of 1 and 3 exhibit a cusp above their corresponding excitation energies, which, as stressed above, is a characteristic feature of a spin excitation; (iii) a red shift is observed for 3 when approaching the tip towards NiNc as seen for excitation 2 (Fig. 2c); (iv) 2 and 3 have same spatial distribution over the NiNc complex (d2I/dV2 maps of Fig. 2e, f), their step amplitudes in the dI/dV spectra varying proportionally to one another across the NiNc molecule with a ratio of 3 ± 1 (Fig. 2g). These results are remarkable and raise the question of how a second spin excitation can be present in the NiNc complex and how it relates to a vibrational excitation.
Model Hamiltonian and electronic transport
In order to model the experimentally observed dI/dV spectrum, we extend the spin Hamiltonian of Eq. (1) to include vibrational effects
| 2 |
As previously, we take a single d-level for the molecule and introduce the states d annihilated and created by and with spin σ, respectively. We also assume that a molecular vibration of frequency ω is annihilated and created by and , respectively, and, moreover, that the electron and vibration couple with a strength W when the state d is populated. We use a weak electron-vibration coupling of W = 20 meV as estimated from the DFT calculations presented above41. Equation (2), which does not include a spin-vibron coupling, shows that a vibron is excited when an electron tunnels into the molecule. This in turn affects the electronic correlation included in , as well as the probability of spin excitation, which strongly depends on the occupation of the molecular orbital37. Figure 3e presents the computed spectrum based on Eq. (2) (see Methods section)42, where D = 4.6 meV and ℏω = 35.5 meV. The computed spectrum showed little dependence on the parameters used for the molecular orbital. The electron–phonon interaction in the presence of spin excitations is sufficient to reproduce the experimental spectrum, but the inclusion of dynamical electronic correlations in the calculation is essential to correctly grasp the amplitude of the vibrational step.
The inset of Fig. 3e sketches the corresponding eigenenergies and allowed excitations. The first and second excited states correspond to a spin excitation and to a vibrational excitation , respectively. The third excited state, , corresponds to a spin excitation energetically displaced upward in energy by a vibron. This excitation mechanism is similar to the coupled-spin vibrational Kondo effect and its replicas of the Kondo resonance in the tunneling spectra at energies close to ℏω. Consistent with this assignment, the relation between the three excitation thresholds simply reflects ∣ϵ3∣ ≃ D + ℏω. The distortion of the NiNc molecule is negligible when the molecular vibration is active as expected for a weak electron-vibration coupling. A renormalized value of D would be observed otherwise leading to ∣ϵ3∣ ≠ ∣ϵ1∣ + ∣ϵ2∣43,44. The relative amplitudes of the steps in the dI∕dV spectrum of NiNc can also be explained using this framework. Noting the step amplitudes by σ1, σ2, and σ3 (Supplementary Fig. 3 and Supplementary Note 2), we find that all spectra recorded obey σ3/σ0 = (σ2/σ0)(σ1/σ0) (Supplementary Table 3), where the vacuum barrier thickness is accounted for by the elastic contribution σ0. This relation indicates that the spin and the vibrational excitations occur independently one from another with transition rates proportional to σ1/σ0 and σ2/σ0, respectively, while the transition rate of the combined excitation σ3/σ0 is their product. This can lead, eventually, to a vibron-assisted spin excitation that exceeds in intensity the spin excitation (Fig. 2g). The same relation among step amplitudes was observed for single and double spin excitations produced by one electron tunneling across two magnetic molecules8.
Isolated NiNc
For completeness, we highlight the importance of the Nc layer for observing the inelastic excitations in the NiNc complex. For this purpose, we engineered the NiNc complex outside the layer via a tip-assisted manipulation31. To do so, we first transferred an isolated Nc to the tip8,35 and then transferred it back atop an isolated Ni adatom on the surface (Fig. 4a, b). The newly formed molecule (Fig. 4b) has an apparent height of 5.6 ± 0.2 Å exceeding that of an isolated Nc molecule, 3.5 ± 0.2 Å (Fig. 4c), and presents a perfectly ring-shaped pattern, indicating that this complex is not tilted but lies straight. The dI/dV spectrum changes completely compared to NiNc in the layer, showing now a broad resonance centered near the Fermi level (Fig. 4d); no inelastic excitation could be evidenced. DFT calculations give a clear picture of the new adsorption configuration adopted by NiNc. The Ni adatom, which is adsorbed in a hollow position of Cu(100), is now centered on the Cp ring (Fig. 4e) and located at a distance of 2.47 Å from the copper surface. This higher adsorption symmetry for NiNc follows from the absence of steric constraints with neighboring Nc. The lowest unoccupied molecular orbital of an isolated NiNc can be qualitatively represented by the mixing of the pz orbitals of C and of the dxz and dyz orbitals of Ni placed around the Fermi level resulting in a calculated magnetic moment of 1.17 μB. The resonance of Fig. 4c is well described by a Frota-Fano fit45 and is then assigned to a spin-1/2 Kondo effect, the fit yielding a Kondo temperature of TK = 68 ± 7 K. The Kondo effect is carried by the dxz and dyz frontier orbitals of Nc as 94% of the spin density of NiNc is located on Nc. Compared to the NiNc complex of the layer, the Ni adatom likely provides a hybridization pathway to the copper surface due to its central adsorption in the Cp ring, causing the effective spin of NiNc to drop, but simultaniously promoting the Kondo physics35. The change of symmetry for the isolated NiNc compared to layer-integrated NiNc may also explain the absence of vibrational signature in Fig. 4d.
Fig. 4. Isolated NiNc on Cu(100).
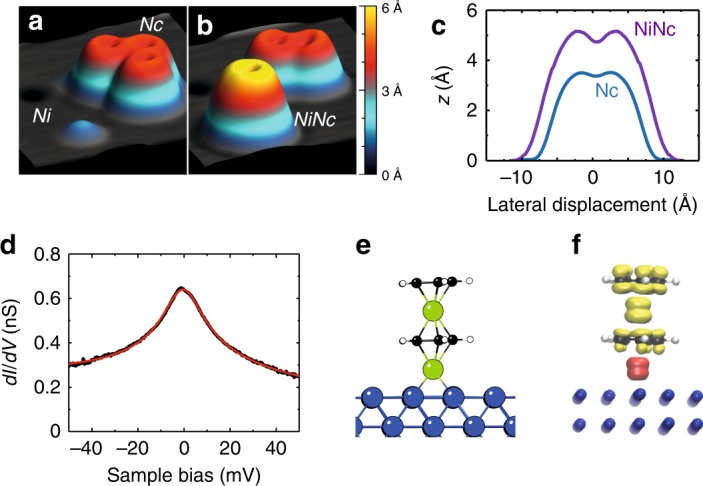
a, b Pseudo 3D images showing the tip-assisted assembly of an isolated NiNc complex (5 × 5 nm2, −20 mV, 20 pA): a before and b after the transfer of Nc atop an isolated Ni adatom, and c height profiles of isolated NiNc and of isolated Nc. d dI/dV spectrum measured above the Cp ring of an isolated NiNc on Cu(100) (feedback loop opened at −50 mV and 20 nA). The solid red line is a Frota-Fano fit yielding a resonance centered at −0.2 ± 0.2 meV and TK = 68 ± 7 K. e DFT optimized structure of isolated NiNc on Cu(100), and f Side view of the spin density map.
Discussion
To summarize, we have shown that Nc adsorbed on a Ni atom yields a vibron-assisted spin excitation at an energy that is one order of magnitude higher than usual spin-excitation energies. We have demonstrated the general character of this excitation through a model that includes magnetic anisotropy, intramolecular correlations and electron-phonon coupling. Our findings suggest that the vibron-assisted spin excitation can be present in molecules with magnetic anisotropy and that it is easier to detect than the vibrational excitation associated to it. With this in mind, we assign the excitation detected above the center of layer-integrated Nc molecules (Supplementary Fig. 4) to a vibron-assisted spin excitation.
While preparing the revised manuscript, a related study to the present one was published by another group46.
Methods
Experimental details
The measurements were performed in an ultra-high vacuum STM operating at 2.4 K. The Cu(100) surface was cleaned in vacuo by sputter/anneal cycles, while a sputter-cleaned etched tungsten tip was employed for tunneling. The tip was further prepared by controlled tip-surface contacts to ensure a monoatomically sharp copper apex. All the spectra were recorded with a lock-in amplifier (200 μV rms and 716 Hz) using a tip that was verified to have a negligible electronic structure in the bias range investigated. The d2I/dV2 spectra were numerically derived. Nickelocene was deposited onto the cold (<100 K) Cu(100) surface. A molecular flux of 2.5 × 10−2 monolayer/min was used in order to obtain well-ordered molecular assemblies along with isolated Nc molecules. To build NiNc complexes, we then exposed the surface to a small amount of Ni atoms (0.05 monolayers). The single-nickel atoms were deposited from a Ni wire source (99.99% purity) onto the cold surface (≈10 K) through an opening in the cryostat shields.
Computational details
We have continued the work of previous publications8,35 treating the adsorption, chemical and physical properties of adsorbed Nc molecules on Cu(100). In the present case, a Ni atom was added to the substrate and the adsorption of Nc on this nucleation center was studied. Electron transmission calculations were also performed at the density functional theory (DFT) level. In this way, two implementations of DFT were used: (i) VASP47–52 for the adsorption, (ii) TRANSIESTA for the transport calculations53. The molecular geometry was optimized using DFT at the spin-polarized generalized gradient approximation (GGA-PBE) level, as implemented in VASP47–52. In order to introduce long-range dispersion corrections, we employed the so-called DFT-D2 approach proposed by Grimme54. We used a plane wave basis set and the projected augmented wave (PAW) method with an energy cutoff of 400 eV. The two surfaces representing substrate and tip were modeled using a slab geometry with a 4 × 4 surface unit cell and six layers for the surface holding the tip-apex and the molecule and five layers for the approaching surface electrode. The k-point sampling was converged at 3 × 3, although the sampling was 11 × 11 for the transmission calculations. For these last calculations, The valence-electron wave functions were double-ζ plus polarization (DZP) basis sets for Nc and diffuse orbitals were used to improve the surface electronic description and a single-ζ plus polarization (SZP) basis set for the copper electrodes. The use of a DZP basis set to describe the adsorbate states is mandatory in order to yield correct transmission functions55,56. The vibrational modes of NiNc were calculated by diagonalizing the dynamical matrix obtained from VASP while only taking the degrees of freedom of the Nc molecule into account.
Simulations
The electronic structure is modeled by one single molecular orbital. In order to model the experimentally observed dI/dV spectrum, we then write an Anderson Hamiltonian for a S = 1 molecular system with a single molecular orbital in the presence of a molecular vibration of frequency ω and axial magnetic anisotropy, D
| 3 |
where
| 4 |
k represents the quantum numbers for the metal states, assumed to be one-electron (ϵk are the spinless bands and the corresponding annihilation operator for that one-electron state). For the molecule we take a single level ϵd and the states d destroyed and created by and with spin σ respectively. The molecular vibration is local and generated or destroyed by or . And the electron and vibration couple with a strength W when the state d is populated. Also the electron and spin are coupled via the axial anisotropy term that considers the total spin of the molecule projected on the z axis. We assume that there is already one spin on the molecule, S2z, and that the fluctuations of charge in the molecular level leads to changes in .
By solving the above Hamiltonian, we can realistically approach the experimental situation where inelastic spin-flip transitions affect the electron transmission through the molecule. In order to do this, we use the multi-orbital non-crossing approximation (MONCA) in the implementation by Korytár and Lorente37,57 that has been shown to reproduce inelastic spectra correctly37. Vibrations can be included using the self-consistency loops of MONCA as explained in the works by Roura-Bas and collaborators58,59. The vibron is excited when an electron jumps into the molecule during the transport process. This is strongly affected by the electronic correlation included in the Anderson Hamiltonian. But it also affects the probability of spin excitation, that strongly depends on the occupation of the molecular orbital37.
Supplementary information
Acknowledgements
We thank M. Ternes for providing his fitting program. This work was supported by the Agence Nationale de la Recherche (grants No. ANR-13-BS10-0016, ANR-11-LABX-0058 NIE and ANR-10-LABX-0026 CSC) and by the Agencia Española de Investigación (grants Nos. MAT2016-78293-C6-1-R and MDM-2016-0618). D.-J.C. and N.L. thank the MICINN (project RTI2018-097895-B-C44). M.-L.B. thanks the national computational center CINES and TGCC (GENCI project: A0030807364).
Author contributions
L.L. conceived the experiments. N.B., B.V., L.G., J.Z., M.O., D.-J.C., and L.L. performed the STM measurements. B.V., C.R.-V. analyzed and fitted the data. P.A., M.-L.B., and N.L. performed first principles density functional theory calculations. P.A. calculated the vibrational modes. N.L. developed the transport theory and simulated the differential conductance. B.V., J.I.P., N.L., and L.L. drafted the manuscript; all authors discussed the results and contributed to the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Code availability
The code used for the calculations of this study is available from M.-L.B. and N.L. upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewers are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
B. Verlhac, Email: verlhac@ipcms.unistra.fr
L. Limot, Email: limot@ipcms.unistra.fr
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-15266-0.
References
- 1.Heinrich AJ, Gupta JA, Lutz CP, Eigler DM. Single-atom spin-flip spectroscopy. Science. 2004;306:466–469. doi: 10.1126/science.1101077. [DOI] [PubMed] [Google Scholar]
- 2.Chen X, et al. Probing superexchange interaction in molecular magnets by spin-flip spectroscopy and microscopy. Phys. Rev. Lett. 2008;101:197208. doi: 10.1103/PhysRevLett.101.197208. [DOI] [PubMed] [Google Scholar]
- 3.Tsukahara N, et al. Adsorption-induced switching of magnetic anisotropy in a single iron(ii) phthalocyanine molecule on an oxidized Cu(110) surface. Phys. Rev. Lett. 2009;102:167203. doi: 10.1103/PhysRevLett.102.167203. [DOI] [PubMed] [Google Scholar]
- 4.Heinrich BW, Braun L, Pascual JI, Franke KJ. Protection of excited spin states by a superconducting energy gap. Nat. Phys. 2013;9:765–768. doi: 10.1038/nphys2794. [DOI] [Google Scholar]
- 5.Choi T, et al. Magnetism in single metalloorganic complexes formed by atom manipulation. Nano Lett. 2014;14:1196–1201. doi: 10.1021/nl404054v. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson P, et al. Quantum engineering of spin and anisotropy in magnetic molecular junctions. Nat. Commun. 2015;6:8536. doi: 10.1038/ncomms9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgess JAJ, et al. Magnetic fingerprint of individual Fe4 molecular magnets under compression by a scanning tunnelling microscope. Nat. Commun. 2015;6:8216. doi: 10.1038/ncomms9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ormaza M, et al. Efficient spin-flip excitation of a nickelocene molecule. Nano Lett. 2017;17:1877–1882. doi: 10.1021/acs.nanolett.6b05204. [DOI] [PubMed] [Google Scholar]
- 9.Rubio-Verdú C, et al. Orbital-selective spin excitation of a magnetic porphyrin. Commun. Phys. 2018;1:15. doi: 10.1038/s42005-018-0015-6. [DOI] [Google Scholar]
- 10.Li J, et al. Electrically addressing the spin of a magnetic porphyrin through covalently connected graphene electrodes. Nano Lett. 2019;19:3288–3294. doi: 10.1021/acs.nanolett.9b00883. [DOI] [PubMed] [Google Scholar]
- 11.Loth S, et al. Controlling the state of quantum spins with electric currents. Nat. Phys. 2010;6:340–344. doi: 10.1038/nphys1616. [DOI] [Google Scholar]
- 12.Hirjibehedin CF, et al. Large magnetic anisotropy of a single atomic spin embedded in a surface molecular network. Science. 2007;317:1199–1203. doi: 10.1126/science.1146110. [DOI] [PubMed] [Google Scholar]
- 13.Stipe BC, Rezaei MA, Ho W. Single-molecule vibrational spectroscopy and microscopy. Science. 1998;280:1732–1735. doi: 10.1126/science.280.5370.1732. [DOI] [PubMed] [Google Scholar]
- 14.Park H, et al. Nanomechanical oscillations in a single-C60 transistor. Nature. 2000;407:57. doi: 10.1038/35024031. [DOI] [PubMed] [Google Scholar]
- 15.Flensberg K. Tunneling broadening of vibrational sidebands in molecular transistors. Phys. Rev. B. 2003;68:205323. doi: 10.1103/PhysRevB.68.205323. [DOI] [PubMed] [Google Scholar]
- 16.LeRoy BJ, Lemay SG, Kong J, Dekker C. Electrical generation and absorption of phonons in carbon nanotubes. Nature. 2004;432:371. doi: 10.1038/nature03046. [DOI] [PubMed] [Google Scholar]
- 17.Mitra A, Aleiner I, Millis AJ. Phonon effects in molecular transistors: Quantal and classical treatment. Phys. Rev. B. 2004;69:245302. doi: 10.1103/PhysRevB.69.245302. [DOI] [Google Scholar]
- 18.Ganzhorn M, Klyatskaya S, Ruben M, Wernsdorfer W. Strong spin-phonon coupling between a single-molecule magnet and a carbon nanotube nanoelectromechanical system. Nat. Nanotechnol. 2013;8:165. doi: 10.1038/nnano.2012.258. [DOI] [PubMed] [Google Scholar]
- 19.Koch J, von Oppen F, Andreev AV. Theory of the Franck-Condon blockade regime. Phys. Rev. B. 2006;74:205438. doi: 10.1103/PhysRevB.74.205438. [DOI] [Google Scholar]
- 20.Burzurí E, et al. Franck-Condon blockade in a single-molecule transistor. Nano Lett. 2014;14:3191–3196. doi: 10.1021/nl500524w. [DOI] [PubMed] [Google Scholar]
- 21.Fu Y-S, et al. Identifying charge states of molecules with spin-flip spectroscopy. Phys. Rev. Lett. 2009;103:257202. doi: 10.1103/PhysRevLett.103.257202. [DOI] [PubMed] [Google Scholar]
- 22.Dubout Q, et al. Controlling the spin of Co atoms on Pt(111) by hydrogen adsorption. Phys. Rev. Lett. 2015;114:106807. doi: 10.1103/PhysRevLett.114.106807. [DOI] [PubMed] [Google Scholar]
- 23.Yu LH, et al. Inelastic electron tunneling via molecular vibrations in single-molecule transistors. Phys. Rev. Lett. 2004;93:266802. doi: 10.1103/PhysRevLett.93.266802. [DOI] [PubMed] [Google Scholar]
- 24.Parks JJ, et al. Tuning the Kondo effect with a mechanically controllable break junction. Phys. Rev. Lett. 2007;99:026601. doi: 10.1103/PhysRevLett.99.026601. [DOI] [PubMed] [Google Scholar]
- 25.Fernández-Torrente I, Franke KJ, Pascual JI. Vibrational Kondo effect in pure organic charge-transfer assemblies. Phys. Rev. Lett. 2008;101:217203. doi: 10.1103/PhysRevLett.101.217203. [DOI] [PubMed] [Google Scholar]
- 26.Choi T, et al. A single molecule Kondo switch: Multistability of tetracyanoethylene on Cu(111) Nano Lett. 2010;10:4175–4180. doi: 10.1021/nl1024563. [DOI] [PubMed] [Google Scholar]
- 27.Rakhmilevitch D, Korytár R, Bagrets A, Evers F, Tal O. Electron-vibration interaction in the presence of a switchable Kondo resonance realized in a molecular junction. Phys. Rev. Lett. 2014;113:236603. doi: 10.1103/PhysRevLett.113.236603. [DOI] [PubMed] [Google Scholar]
- 28.Paaske J, Flensberg K. Vibrational sidebands and the Kondo effect in molecular transistors. Phys. Rev. Lett. 2005;94:176801. doi: 10.1103/PhysRevLett.94.176801. [DOI] [PubMed] [Google Scholar]
- 29.Bachellier N, et al. Unveiling nickelocene bonding to a noble metal surface. Phys. Rev. B. 2016;93:195403. doi: 10.1103/PhysRevB.93.195403. [DOI] [Google Scholar]
- 30.Ormaza M, et al. Assembly of ferrocene molecules on metal surfaces revisited. J. Phys. Chem. Lett. 2015;6:395–400. doi: 10.1021/jz5026118. [DOI] [PubMed] [Google Scholar]
- 31.Ormaza M, et al. On-surface engineering of a magnetic organometallic nanowire. Nano Lett. 2016;16:588–593. doi: 10.1021/acs.nanolett.5b04280. [DOI] [PubMed] [Google Scholar]
- 32.Yi Z, et al. Tuning the magneto-transport properties of nickel-cyclopentadienyl multidecker clusters by molecule-electrode coupling manipulation. ACS Nano. 2010;4:2274–2282. doi: 10.1021/nn901784t. [DOI] [PubMed] [Google Scholar]
- 33.Morari C, Allmaier H, Beiuşeanu F, Jurcuţ T, Chioncel L. Electronic structure and magnetic properties of metallocene multiple-decker sandwich nanowires. Phys. Rev. B. 2012;85:085413. doi: 10.1103/PhysRevB.85.085413. [DOI] [Google Scholar]
- 34.Ternes M. Spin excitations and correlations in scanning tunneling spectroscopy. N. J. Phys. 2015;17:063016. doi: 10.1088/1367-2630/17/6/063016. [DOI] [Google Scholar]
- 35.Ormaza M, et al. Controlled spin switching in a metallocene molecular junction. Nat. Commun. 2017;8:1974. doi: 10.1038/s41467-017-02151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurley A, Baadji N, Sanvito S. Perturbative approach to the Kondo effect in magnetic atoms on nonmagnetic substrates. Phys. Rev. B. 2011;84:115435. doi: 10.1103/PhysRevB.84.115435. [DOI] [Google Scholar]
- 37.Korytár R, Lorente N, Gauyacq J-P. Many-body effects in magnetic inelastic electron tunneling spectroscopy. Phys. Rev. B. 2012;85:125434. doi: 10.1103/PhysRevB.85.125434. [DOI] [Google Scholar]
- 38.Lauhon LJ, Ho W. Single-molecule vibrational spectroscopy and microscopy: Co on Cu(001) and Cu(110) Phys. Rev. B. 1999;60:R8525–R8528. doi: 10.1103/PhysRevB.60.R8525. [DOI] [Google Scholar]
- 39.Vitali L, et al. Surveying molecular vibrations during the formation of metal-molecule nanocontacts. Nano Lett. 2010;10:657–660. doi: 10.1021/nl903760k. [DOI] [PubMed] [Google Scholar]
- 40.Lorente N. Mode excitation induced by the scanning tunnelling microscope. Appl. Phys. A. 2004;78:799–806. doi: 10.1007/s00339-003-2434-8. [DOI] [Google Scholar]
- 41.Monturet S, Lorente N. Inelastic effects in electron transport studied with wave packet propagation. Phys. Rev. B. 2008;78:035445. doi: 10.1103/PhysRevB.78.035445. [DOI] [Google Scholar]
- 42.Choi D-J, Abufager P, Limot L, Lorente N. From tunneling to contact in a magnetic atom: The non-equilibrium Kondo effect. J. Chem. Phys. 2016;146:092309. doi: 10.1063/1.4972874. [DOI] [Google Scholar]
- 43.May F, Wegewijs MR, Hofstetter W. Interaction of spin and vibrations in transport through single-molecule magnets. Beilstein J. Nanotechnol. 2011;2:693–698. doi: 10.3762/bjnano.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruiz-Tijerina DA, Cornaglia PS, Balseiro CA, Ulloa SE. Dynamical magnetic anisotropy and quantum phase transitions in a vibrating spin-1 molecular junction. Phys. Rev. B. 2012;86:035437. doi: 10.1103/PhysRevB.86.035437. [DOI] [Google Scholar]
- 45.Pruser H, et al. Long-range Kondo signature of a single magnetic impurity. Nat. Phys. 2011;7:203–206. doi: 10.1038/nphys1876. [DOI] [Google Scholar]
- 46.Czap G, et al. Detection of spin-vibration states in single magnetic molecules. Phys. Rev. Lett. 2019;123:106803. doi: 10.1103/PhysRevLett.123.106803. [DOI] [PubMed] [Google Scholar]
- 47.Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 1993;47:558. doi: 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- 48.Kresse G, Hafner J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B. 1993;48:13115. doi: 10.1103/PhysRevB.48.13115. [DOI] [PubMed] [Google Scholar]
- 49.Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996;6:15. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 50.Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54:11169. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 51.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59:1758. doi: 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- 52.Hafner J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008;29:2044–2078. doi: 10.1002/jcc.21057. [DOI] [PubMed] [Google Scholar]
- 53.Papior N, Lorente N, Frederiksen T, García A, Brandbyge M. Improvements on non-equilibrium and transport Green function techniques: The next-generation TRANSIESTA. Comput. Phys. Commun. 2017;212:8–24. doi: 10.1016/j.cpc.2016.09.022. [DOI] [Google Scholar]
- 54.Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006;27:1787. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 55.Abufager P, Robles R, Lorente N. FeCoCp3 molecular magnets as spin filters. J. Phys. Chem. C. 2015;119:12119. doi: 10.1021/acs.jpcc.5b01839. [DOI] [Google Scholar]
- 56.García-Fernández C, Lorente N. Tunneling electronic currents simulated with localized basis sets. Surf. Sci. 2018;678:201–205. doi: 10.1016/j.susc.2018.05.021. [DOI] [Google Scholar]
- 57.Korytár R, Lorente N. Multi-orbital non-crossing approximation from maximally localized Wannier functions: The Kondo signature of copper phthalocyanine on Ag(100) J. Phys. Condens. Matter. 2011;23:355009. doi: 10.1088/0953-8984/23/35/355009. [DOI] [PubMed] [Google Scholar]
- 58.Roura-Bas P, Tosi L, Aligia AA. Nonequilibrium transport through magnetic vibrating molecules. Phys. Rev. B. 2013;87:195136. doi: 10.1103/PhysRevB.87.195136. [DOI] [Google Scholar]
- 59.Roura-Bas P, Tosi L, Aligia AA. Replicas of the Kondo peak due to electron-vibration interaction in molecular transport properties. Phys. Rev. B. 2016;93:115139. doi: 10.1103/PhysRevB.93.115139. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
The code used for the calculations of this study is available from M.-L.B. and N.L. upon reasonable request.