

Abstract
The fruit is the most important economical organ in the grape; accordingly, to investigate the grapevine genomic methylation landscape and examine its functional significance during fruit development, we generated whole genome DNA methylation maps for various developmental stages in the fruit of grapevine. In this study, thirteen DNA methylation-related genes and their expression profiles were identified and analyzed. The methylation levels for mC, mCG, mCHG, and mCHH contexts in 65 days after flowering (65DAF) fruit (véraison stage) were higher than those in 40DAF (green stage) and 90DAF (mature stage) fruits. Relative to methylation in the mC context, methylation levels in the mCHH context were higher than those of mCG and mCHG. The DNA methylation level in the ncRNA regions was significantly higher than that in exon, gene, intron, and mRNA regions. The differentially methylated regions (DMRs) and differentially methylated promoters (DMPs) in 65DAF_vs_40DAF were both higher than those in 90DAF_vs_65DAF and 90DAF_vs_40DAF. Most DMRs (or DMPs) were involved in metabolic processes and cell processes, binding, and catalytic activity. These results indicated that DNA methylation represses gene expression during grape fruit development, and it broadens our understanding of the landscape and function of DNA methylation in grapevine genomes.
Electronic supplementary material
The online version of this article (10.1007/s12298-020-00759-5) contains supplementary material, which is available to authorized users.
Keywords: Grapevine, DNA methylation, Fruit development, Epigenetics, Gene expression
Introduction
Epigenetic modification refers to the stable changes in gene expression (between cell divisions, and sometimes between generations); however, it does not involve changes in the underlying DNA sequence (Bird 2007). Epigenetic mechanisms regulate the interpretation of genetic information and are intricately linked to cellular differentiation and tissue specification (Raddatz et al. 2013). Major epigenetic modifications include DNA methylation, histone modification, chemical modification and chromatin remodeling changes in gene expression caused by microRNAs (miRNAs). During cell development and differentiation, these mechanisms work in an integrated manner to execute specific gene expression programs that enable the cells to respond adequately to the changes in environment (Barter et al. 2012; Xie et al. 2013). DNA methylation is the addition of a methyl group to the DNA molecule at cytosine to form 5-methylcytosine; this typically happens at CpG, CpHpG, and CpHpH sites (where H can be A, C or T) in eukaryotes (Barter et al. 2012). Methylation is a conserved epigenetic silencing mechanism and is involved in numerous biological processes, such as defense against transposon proliferation and control of genomic imprinting and gene expression (Bird 2002; Goll and Bestor 2005). DNA methylation is influenced by DNA methylases and demethylases. Three methyltransferases (MET), two chromomethylase 2 (CMT2), one chromomethylase 3 (CMT3), one chromomethylase 1 (CMT1), and one domain rearranged methyltransferase 2 (DRM2) were isolated from grapevine genome, which play significant roles in abiotic (heat, drought, or cold) stress reponse (Ren et al. 2017). Moreover, studies have isolated and characterized methylases and demethylases of several plant species, such as Arabidopsis (Ashapkin et al. 2016), rice (Ahmad et al. 2014), tomato (Cao et al. 2014), eggplant (Moglia et al. 2019), carrot (Bernacchia et al. 1998), peach (Bernacchia et al. 1998), strawberry (Gu et al. 2016), pear (Liu et al. 2018), and wheat (Dai et al. 2005).
In the past, DNA sequencing was dominated by Sanger sequencing. Currently, various high-throughput methods are used for the large-scale detection of methylation (Jones 2012; Laird 2010; Suzuki and Bird 2008). In plants, whole-genome bisulfite sequencing (WGBS) was first applied to Arabidopsis thaliana, which revealed genome-wide patterns of both CG-gene body methylation and RNA-directed DNA methylation (Cokus et al. 2008; Lister et al. 2008; Zhang et al. 2006). A DNA methylation study of 86 A. thaliana silenced mutants identified novel components required for CG, CHG, or CHH methylation; however, additional components still remain unidentified (Stroud et al. 2013). WGBS has been used to analyze DNA methylation patterns in many species, such as A. thaliana (Yang et al. 2015), Glycine max (Song et al. 2013a; Wang et al. 2015b), Malus domestica (Kumar et al. 2016), Oryza sativa (Li et al. 2012), Populus trichocarpa (Liang et al. 2014), Ricinus communis (Xu et al. 2016), Solanum lycopersicum (Zhang et al. 2016; Zhong et al. 2013) and Zea mays (Eichten et al. 2013). Previous studies have indicated the role of DNA methylation in transposon silencing (Rabinowicz et al. 2003), plant vernalization (Burn et al. 1993; Khan et al. 2013), hybrid vigor (Kawanabe et al. 2016; Shen et al. 2012), gene silencing (Cao and Jacobsen 2002), flower development (Song et al. 2013b; Yang et al. 2015), and endosperm development (Xu et al. 2016). In addition, studies have demonstrated DNA methylation during fruit development and their role in ripening. In 1991, differential DNA methylation was first reported in two tomato species by Messeguer et al. (1991). The levels of DNA methylation govern fruit development in tomatoes. An epigenetic change in the SBP box-Cnr (SQUAMOSA promoter binding protein-like-Colorless non-ripening) promoter inhibited tomato fruit ripening (Manning et al. 2006). Moreover, Teyssier et al.(2008) found that the global DNA methylation level changed during fruit growth and one CMT (chromomethylase) and two DRM genes (domain-rearranged methyltransferases) genes were preferentially expressed in the pericarp during fruit growth. Zhong et al. (2013) confirmed that epigenome status was not static during fruit development, and the demethylation of RIN (a ripening inhibitor) binding sites ensured the fidelity of fruit developmental stages, such as ripening in tomato. Two years later, Liu et al. (2015a) demonstrated an association between tomato fruit development and genomic DNA demethylation that was governed by DNA demethylase 2 (DML2). In addition, a transient increase in DNA methylation was observed during chilling of tomato fruits (Zhang et al. 2016). Transcriptome and methylome analyses revealed a decrease in the expression levels of some key elements (e.g., genes encoding volatile synthesis enzymes and ripening-associated transcription factors) accompanied by a transient increase in DNA methylation of the promoter regions of these genes during chilling (Zhang et al. 2016).
Grapevine (Vitis vinifera L.) is an economically important fruit crop. It is used as a model plant for fruit crop genomics studies, and has an estimated genome size of 500 megabases (Mb) that was sequenced in 2007 (Jaillon et al. 2007; Velasco et al. 2007). Grape is a non-climacteric fresh fruit; it only shows weak changes in endogenous ethylene around véraison. During ripening, berries undergo irreversible changes in texture, sugar content, organic acid content, color, flavor, and aroma (Lister et al. 2008). Transcriptomic studies have indicated that more than 6000 genes are expressed in a stage-specific manner, and these genes are related to several pathways, such as secondary metabolite synthesis and transport, cell wall metabolism, organic acid and sugar metabolism (Pilati et al. 2007; Sweetman et al. 2012; Zenoni et al. 2010).
However, the status of DNA methylation during grapevine fruit growth is unclear. Furthermore, it is not known how DNA methylation affects gene expression patterns during fruit development. Therefore, we aim to investigate the whole genome DNA methylation pattern during grapevine fruit developmental stages. The results will provide an important foundation for future research on the mechanism of fruit development.
Results
DNA methylation-related genes in grapevine
The protein sequences of ten DNA methyltransferases and four demethylases of A. thaliana were retrieved from TAIR. We identified and characterized the sequence structure of ten genes encoding DNA methyltransferases and three genes encoding demethylases from the grapevine genome. Basic information on these DNA methyltransferases and demethylases is presented in Table 1. Compared with the DNA methylation-related genes of Arabidopsis, castor bean (Xu et al. 2016), and tomato (Cao et al. 2014), the chromosomal distribution of genes encoding DNA methyltransferases and demethylases was different in grape. MET2 and DNMT2 were found in Arabidopsis and grape but not in caster bean or tomato. DML2 was identified in Arabidopsis and tomato but not in caster bean or grape. The difference in distribution of DNA methyltransferases and demethylases may explain the difference among species in DNA methylation profiles. Pfam analysis revealed that all the DNA methyltransferases contained a C-5 cytosine-specific DNA methylase domain. In addition, VvMET2a and VvMET2b contained two cytosine-specific DNA methyltransferase replication foci domains and two bromo-adjacent homology (BAH) domains, while VvCMT1, VvCMT2a, and VvCMT3 contained one BAH domain and one chromo domain each. All the three identified DNA demethylase genes (VvROS1, VvDME, and VvDML3) contained one permuted single zf-CXXC unit domain and one RRM in Demeter domain each (Supplementary file Fig. S1). In the table grapevine ‘Fujiminori’, the expression level of some DNA methylation-related genes (VvROS1, VvDRM2, VvDRM1, VvDNMT2, and VvDME) decreased during fruit development. VvDRM2 and VvDRM1 were significantly down-regulated during fruit development (Fig. 1 and Supplementary file Table S1). In the ‘Norton berry’ exocarp, VvDRM2 and VvDME expression levels were higher, and their expression decreased throughout fruit development (Supplementary files Fig. S2a and Table S1). Meanwhile, we detected no significant difference in DNA methylation-related genes between mesocarp and exocarp, except for VvDME (Supplementary files Fig. S2b and Table S1).
Table 1.
Identification of genes encoding DNA methyltransferases and demethylases in Vitis vinifera
| Gene ID | Protein length (aa) | Location (chromosome: start site–end site) | Homologs | Annotation |
|---|---|---|---|---|
| Cytosine-5 DNA methyltransferases | ||||
| VIT_07s0130g00390 | 1549 | 7:20695409–20704468 | At4g14140 | VvMET2a |
| VIT_07s0130g00380 | 1530 | 7:20683940–20689960 | At4g08990 | VvMET2b |
| VIT_12s0035g01770 | 460 | 12:21905231–21909960 | At4g13610 | VvMET3 |
| VIT_14s0066g01040 | 532 | 14:27478378–27484652 | At5g15380 | VvDRM1 |
| VIT_05s0020g00450 | 712 | 5:2361509–2376608 | At5g14620 | VvDRM2 |
| VIT_08s0007g06800 | 827 | 8:20455686–20461582 | At1g80740 | VvCMT1 |
| VIT_02s0033g00610 | 767 | 2:14943456–14983872 | At4g19020 | VvCMT2a |
| VIT_16s0039g02460 | 477 | 16:2651867–2668098 | At4g19020 | VvCMT2b |
| VIT_06s0004g01080 | 782 | 6:1171054–1183851 | At1g69770 | VvCMT3 |
| VIT_04s0008g05060 | 393 | 4:4545259–4550148 | At5g25480 | VvDNMT2 |
| DNA demethylases | ||||
| VIT_08s0007g03920 | 1775 | 8:17914203–17923902 | At2g36490 | VvROS1 |
| VIT_13s0074g00450 | 1873 | 13:8239220–8249559 | At5g04560 | VvDME |
| VIT_06s0061g01270 | 932 | 6:19096083–19101208 | At4g34060 | VvDML3 |
Fig. 1.
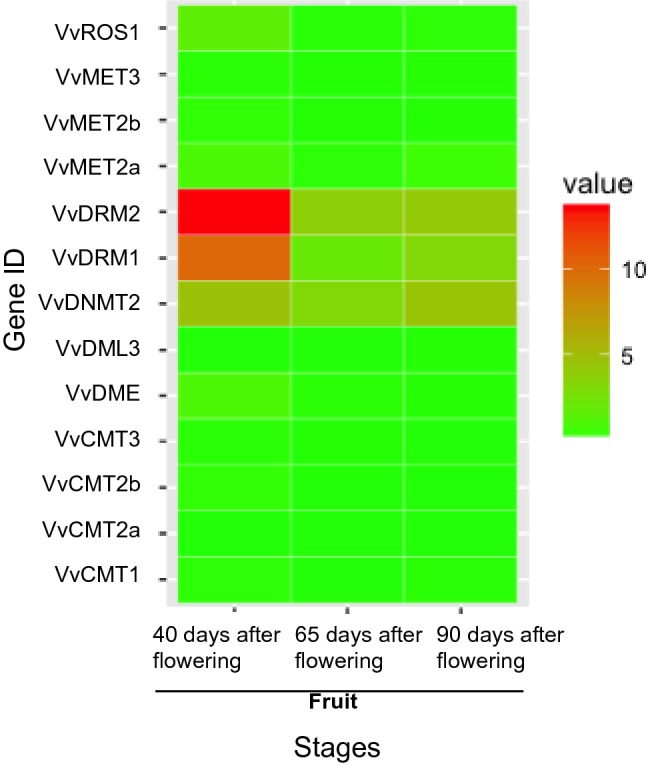
The expression profiles of DNA methylation-related genes in fruits at different developmental stages. The expression value was represented by different colors (color figure online)
To clarify how DNA methylation is established and maintained, we assayed the expression profiles of DNA methylation-related genes in different species (Vitis vinifera cv. ‘Cabernet Sauvignon’ and Vitis quinquangularis accession ‘Danfeng-2’), cultivars (‘Sangiovese’, ‘Barbera’, ‘Negro amaro’, ‘Refosco’, and ‘Primitivo’), or different tissues/stages. The expression levels of VvROS1, VvDRM2, VvDRM1, VvDNMT2, VvDME were more than that of the other genes in different species or tissues/stages (Supplementary files Fig. S2c–e, and Table S1). VvMET3, VvMET2b, VvDML3, and VvCMT2a were expressed at very low levels in the 54 grapevine tissue samples (Supplementary files Fig. S2e and Table S1). Among the genes encoding DNA methyltransferases, VvMET2a, VvDRM2, VvDRM1, and VvDNMT2 were expressed in nearly all tissues (e.g., leaf, fruit mesocarp, fruit exocarp, and seed), while VvCMT3, VvCMT2b, and VvCMT1 were only expressed in the bud, fruit mesocarp, inflorescence, root, leaf, pollen, seed, stem, and tendril. With respect to fruit development, the expression levels of the above DNA methyltransferase genes exhibited no change (such as VvDRM2 and VvCMT2b), increased slightly (VvDNMT2), or decreased (VvMET2a). Among the genes encoding the demethylases, VvDME and VvROS1 were expressed in nearly all tissues (e.g., leaf, fruit mesocarp, fruit exocarp, and seed). VvDME and VvROS1 were repressed from fruit set to ripening in the fruit pericarp. The expression level of VvROS1 decreased from the post-harvest withering I to III stages in the fruit pericarp, while the expression level of VvDME increased in the fruit pericarp. VvROS1 and VvDME demonstrated similar expression levels at the post-fruit set stage in the mesocarp, while the expression level of VvROS1 decreased and the expression of VvDME increased from véraison to post-harvest withering III stages in the mesocarp. Similar expression patterns were observed for VvROS1 and VvDME in the exocarp. At seed maturity, the expression levels of VvROS1 and VvDME were high, and these levels were more than the levels in pericarp, mesocarp, and exocarp at the same stage.
Methylation landscapes among fruits at different developmental stages
We further examined the whole genome DNA methylation levels in ‘Fujiminori’ fruits at three different developmental stages by high-throughput sequencing of bisulfite-converted DNA. In total, 181–189 million sequencing reads were produced from the three samples. After removal of adapter and low-quality sequences from the raw data, 170–179 million clean reads were retained for each of the three grape fruit samples, which yielded 21.10–22.50 giga base pairs (Gbp) of data covering 90.40–90.75% of the reference grape genome (International Grape Genome Program, 12×, 2007) (Table 2 and Supplementary files Tables S2–4). Approximately 94.18–94.97% of clean reads were obtained, and the average read depth ranged from 34.59× to 37.95× (Table 2 and Supplementary file Fig. S3). The basic summary statistics indicated the suitability of our sequencing data for further analysis.
Table 2.
The basic information of BS-sequencing in fruits at different developmental stages
| Type | 40DAF | 65DAF | 90DAF |
|---|---|---|---|
| Raw reads number | 184,278,812 | 189,500,082 | 181,000,026 |
| Clean reads number | 174,264,285 | 179,975,266 | 170,463,158 |
| Clean reads size (Gbp) | 21.10 | 22.50 | 21.31 |
| Clean reads rate (%) | 94.57 | 94.97 | 94.18 |
| Genome coverage (%) | 90.40 | 90.75 | 90.70 |
| Average read depth (×) | 35.34 | 37.95 | 34.59 |
The ratio of reads covering every methylated-cytosine to the entire reads covering the site defines the methylation level of an explicit cytosine. Global DNA methylation profiles demonstrated that all three sequence contexts, CG, CHG, and CHH, were methylated in grapevine fruits at different developmental stages (Table 3). Intriguingly, the methylation level at 65 days after flowering (65 DAF) was more than that at 40 DAF, and similar to that at 90 DAF. The methylation levels of CG and CHG were decreased from 40 DAF to 65 DAF, then maintained the similar level until 90 DAF. However, CHH methylation increased at 65 DAF and was consistent at 90 DAF (Table 3).
Table 3.
The DNA methylation level in fruits at different developmental stages
| Sample | Methylation level (%) | |||
|---|---|---|---|---|
| C | CG | CHG | CHH | |
| 40DAF | 8.3 | 41.1 | 21.65 | 2.96 |
| 65DAF | 9.29 | 36.34 | 18.99 | 4.74 |
| 90DAF | 9.24 | 36.46 | 19.36 | 4.6 |
The relative methylation rates for mCG, mCHG, and mCHH contexts at 40 DAF, 65 DAF, and 90 DAF were distinct. The rates of mCG, mCHG, and mCHH methylation at 65 DAF were almost identical to those at 90 DAF. The rates of mCG and mCHG methylation at 40 DAF were higher compared with those at 90 DAF, whereas an opposite pattern was observed for mCHH methylation in fruits (Fig. 2a). The seqLogo for mCHG/CHG showed that the relative frequency of A/T/C did not differ at the G site, while approximately three-fold increase in the relative frequency of A/T (or A/C) was found at the last H site of mCHH/CHH (Fig. 2b). The distribution of cytosine methylation indicated that the majority of mCHH sites had a lower methylation level compared with the majority of mCG sites (Fig. 2c).
Fig. 2.


The distribution of DNA methylation levels in grape fruits at three different developmental stages. a Relative proportions of mCs in mCG, mCHG, and mCHH contexts in grape fruits at three different developmental stages. b Seqlogo of the DNA sequences near to DNA methylation positions. One pile for each position in CHG and CHH contexts. The total height of the pile indicates the sequence conservation rate at each position, while the height of letters within the pile indicates the frequency (relatively) of each letter at each position. c Distribution of methylation level of mCs in mCG, mCHG, and mCHH contexts in fruits at different developmental stages. The x-axis indicates the percentage of methylated cytosine at a given cytosine site, while the y-axis indicates the portion of overall mCs. d Methyl-cytosine density distribution of chromosome 1 in fruits at different developmental stages. The methyl-cytosine density in 10-kb windows throughout chromosome were represented by the blue dots. The different mCs contexts densities were represented by different colored smoothed lines (color figure online)
The methylation levels in three sequence contexts exhibited a specific chromosomal distribution. The methylation levels of cytosine drastically varied across the genome (Fig. 2d, Supplementary file Fig. S4), and the methylation pattern represented a mosaic feature with relatively thick methylated domains interrupted by non-methylated regions. Moreover, chromosome 15 exhibited the highest methylation level for all sequence contexts among the chromosomes (Supplementary file Table S5).
DNA methylation distribution in different genomic regions
The distribution of DNA methylation in the genome is not usually uniform and is affected by the local characteristics of chromosomal regions (Song et al. 2013a). To investigate the distribution of DNA methylation (mCG, mCHG, and mCHH) in different regions of the grape genome, we analyzed the distribution of DNA methylation in fruits at 40 DAF, 65 DAF, and 90 DAF. We detected a low CpG density in the mCG context, a medium CpG density in the mCHG context, and a high CpG density in the mCHH context (Fig. 3, Supplementary files Figs. S5 and S6). A high proportion of CpGs had either low (10%) or high (100%) DNA methylation level in the mCG context, whereas more than 70% and 80% of CpGs had low (10%) DNA methylation level in mCHG and mCHH contexts, respectively (Fig. 3, Supplementary files Figs. S5 and S6). The methylation pattern of fruits at 40 DAF, 65 DAF, and 90 DAF indicated that regions with a low CpG density had a high DNA methylation level, while those with a high CpG density had a low DNA methylation level in the mCG context. For the mCHG and mCHH contexts, most methylation sites had a low DNA methylation level, while sites were abundant in regions of intermediate and high CpG density (Fig. 3, Supplementary files Figs. S5 and S6).
Fig. 3.


Distinct CpG density patterns and mCG (a), mCHG (b), and mCHH (c) methylation level of 40DAF fruit in different genomic regions. Different features were represented by different panels, and n in each panel refers to the total number of analyzed CpGs. The x-axis (CpG density) indicates total number of CpG dinucleotides in each window (200 bp), and the y-axis (methylation level) indicates the average methylation level of cytosines in CpGs. The mesial methylation level of CpGs at the specified local density was represented by the thin black lines, the plenty CpGs that belongs to the bin of specified CpG densities and methylation levels were represented by the red gradient, and the distribution of CpG densities were represented by the blue bar charts above each heatmap, which projected onto the x-axis of the heatmaps. The distribution of methylation level was represented by the green bar charts to the right of the heatmaps, which also projected onto the y-axis of the heatmaps (color figure online)
The highest (9.89%) and lowest (4.66%) methylation levels were found in ncRNA and exon regions, whereas the methylation level in mRNA regions was only 6.11% in fruits at 40 DAF. The methylation level in exon regions was lower than that in introns, whereas the methylation level in gene regions was intermediate between exon and intron levels. Similar patterns were observed in fruits at 65 DAF and 90 DAF (Table 4). For the mC context, the methylation level increased in each region with fruit development. For the mCG context, the methylation level decreased and was lowest at 65 DAF in intron, mRNA, and ncRNA regions. The methylation level increased in gene and and decreased in exon regions. For the mCHG context, the methylation level was decreased and showed the lowest value at 65DAF in gene, intron, mRNA, and ncRNA regions. The methylation level in exon regions decreased and was lowest at 90DAF. For the mCHH context, the methylation level increased and was highest at 65DAF in exon, gene, intron, and mRNA regions. The methylation level in ncRNA regions increased and was highest at 90DAF (Table 4).
Table 4.
The DNA methylation level of different gene structure in fruits at different developmental stages
| Type | 40DAF | 65DAF | 90DAF | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | CG | CHG | CHH | C | CG | CHG | CHH | C | CG | CHG | CHH | |
| Exon | 4.66 | 33.92 | 5.31 | 0.99 | 4.7 | 29.44 | 4.68 | 1.49 | 4.38 | 27.24 | 4.34 | 1.31 |
| Gene | 5.23 | 31.5 | 10.28 | 1.59 | 5.98 | 32.56 | 9.51 | 2.62 | 6.05 | 34.07 | 9.83 | 2.57 |
| Intron | 6.47 | 59.1 | 13.41 | 1.89 | 7.1 | 55.05 | 11.89 | 3.04 | 7.05 | 56.44 | 12 | 2.91 |
| mRNA | 6.11 | 52.06 | 11.52 | 1.74 | 6.77 | 47.96 | 10.6 | 2.82 | 6.77 | 50.13 | 10.78 | 2.72 |
| ncRNA | 9.89 | 66.68 | 23.28 | 3.05 | 11.13 | 65.19 | 19.96 | 4.91 | 11.77 | 67.75 | 21.37 | 4.99 |
Characterization of DMRs among fruits at different developmental stages
DMRs are regions with at least five CG, CHG, or CHH in one bin. The total number of DMRs between 65 DAF and 40 DAF (65DAF_vs_40DAF) was larger than the numbers of DMRs in 90DAF_vs_40DAF or 90DAF_vs_65DAF comparisons (Supplementary file Table S6), indicating that the hypomethylation and hypermethylation of DNA mostly occurred between 65DAF and 40DAF, respectively. Approximately 0.88%, 3.43%, and 4.06% of chromosomes had DMRs for 90DAF_vs_65DAF, 90DAF_vs_40DAF, and 65DAF_vs_40DAF comparisons, respectively (Supplementary file Table S6). The number of DMRs was more for the CHH context than for the CHG and CG contexts in all comparisons, and the number of CG, CHG, and CHH DMRs for 90DAF_vs_65DAF was significantly less than those for 90DAF_vs_40DAF and 65DAF_vs_40DAF (Fig. 4a and Supplementary file Table S7). After annotation, the DMRs located in gene regions were identified. Nearly 30% of CG, 40% of CHG, and 10% of CHH DMRs (Gene_DMRs) were located in gene regions (Fig. 4b and Supplementary file Table S7). The percentage of CHH Gene_DMRs was significantly less than the percentage of CG and CHG Gene_DMRs. Most CG and CHG Gene_DMRs exhibited hypomethylation, while nearly all CHH Gene_DMRs exhibited hypermethylation in 90DAF_vs_40DAF (or 65DAF_vs_40DAF). Interestingly, more than half of CHG and CHH Gene_DMRs exhibited hypomethylation, while more than half of CG Gene_DMRs exhibited hypermethylation in 90DAF_vs_65DAF (Fig. 4c and Supplementary file Table S7).
Fig. 4.


DMRs distribution between any of fruits at different developmental stages. a The number of DMRs distribute between any of the fruits at different developmental stages. b The percentage of Gene_DMRs distribute between any of the fruits at different developmental stages. c The percentage of hypomethylation and hypermethylation of Gene_DMRs between any of the fruits at different developmental stages. d Analysis of hypermethylated and hypomethylated DMR associated genes of 65DAF_vs_40DAF using WEGO analysis. Green bar indicates the hypomethylated DMR associated gene, and red bar indicates the hypermethylated DMR associated gene. e Top 20 statistics of pathway enrichment for 65DAF_vs_40DAF. The circle size represented the gene number and the circle color represented the q-value of pathway enrichment analysis. f The comparison of DNA methylation level of DMR associated genes in 65DAF_vs_40DAF (color figure online)
We further used BGI WEGO (Web Gene Ontology Annotation Plotting) to functionally categorize the hypermethylated and hypomethylated DMR-associated genes. A total of 4881 hypermethylated and 359 hypomethylated DMR-associated genes identified between 65 DAF and 40 DAF fruits were assigned to 51 GO functional groups, including cellular components, molecular functions, and biological processes (Fig. 4d and Supplementary file Fig. S7). For the three major GO categories, the dominant subcategories were cell process, metabolic process, cell, cell part, binding, and catalytic activity (Fig. 4d). The same subcategories were also obtained for 90DAF_vs_65DAF and 90DAF_vs_40DAF (Supplementary file Fig. S7). To demonstrate the potential role of DMR-associated genes, biochemical pathways were also examined for the gene collection. The “sesquiterpenoid and triterpenoid biosynthesis” pathway was significantly enriched in 65DAF_vs_40DAF and 90DAF_vs_40DAF (Fig. 4e and Supplementary file Fig. S8). The top five pathways in 65DAF_vs_40DAF belonged to “spliceosome,” “RNA transport,” “pyrimidine metabolism,” “mRNA surveillance pathway,” and “Flavone and flavonol biosynthesis” categories (Fig. 4e). In 90DAF_vs_65DAF and 90DAF_vs_40DAF, the top five pathways fell into “Plant-pathogen interaction,” “RNA transport,” “Spliceosome,” “Purine metabolism,” and “Pyrimidine metabolism” categories (Supplementary file Fig. S8).
We also compared the DNA methylation patterns of DMR-associated genes between fruits at different developmental stages. Between 65 DAF and 40 DAF fruits, CG and CHG methylation levels in the promoter and exon regions at 40 DAF were more than those at 65 DAF, and similar methylation levels were observed in the intron regions at 40 DAF and 65 DAF. However, the CHH methylation levels in promoter, exon, and intron regions at 65 DAF were more than those at 40 DAF (Fig. 4f). Between 90DAF and 65DAF, only the CHH methylation levels in the promoter region and intron regions at 65 DAF were slightly more than those at 90 DAF (Supplementary file Fig. S9a). Interestingly, the CG and CHG methylation levels at 40 DAF were slightly more than those at 90 DAF. However, the CHH methylation level was significantly more at 90 DAF than at 40 DAF (Supplementary file Fig. S9b).
Identification of DMPs among fruits at different developmental stages
To analyze the methylation levels in promoter regions, only the promoters with P < 0.05 and a difference in methylation level > 0.1 between fruits at different development stages were treated as DMPs. In total, 96, 1202, 479, and 192 DMPs were identified in C, CG, CHG, and CHH sites, respectively, for 65DAF_vs_40DAF. The number of DMPs identified in 65DAF_vs_40DAF and 90DAF_vs_40DAF was significantly more than that in 90DAF_vs_65DAF (Table 5). In 65DAF_vs_40DAF, the methylation levels of DMPs decreased in C, CHG, and CHH contexts. The methylation level of CG DMPs decreased from 2 K upstream of transcriptional start site (TSS) site to TSS site, then increased and reached highest between 1 K downstream of TSS site and 2 K downstream of TSS site. Finally, the methylation level of CG DMPs decreased (Fig. 5a). A similar methylation pattern was found in 90DAF_vs_65DAF and 90DAF_vs_40DAF (Supplementary file Fig. S10). In 65DAF_vs_40DAF, the methylation levels of C and CHH DMPs at 65 DAF were more than those at 40 DAF, while an opposite pattern was observed for CG and CHG DMPs (Fig. 5a). In 90DAF_vs_65DAF, the methylation levels of C and CHH DMPs at 65 DAF were slightly more than those at 90 DAF, while the methylation levels of CG and CHG were similar at 90 DAF and 65 DAF (Supplementary file Fig. S8a). In 90DAF_vs_40DAF, the methylation levels of C and CHH DMPs at 90 DAF were more than those at 40 DAF, while the opposite was observed for CG and CHG DMPs (Supplementary file Fig. S8b).
Table 5.
The DMP distribution between any of fruits at different developmental stages
| Type | 90DAF_vs_65DAF | 90DAF_vs_40DAF | 65DAF_vs_40DAF |
|---|---|---|---|
| C | 41 | 71 | 96 |
| CG | 349 | 1373 | 1202 |
| CHG | 235 | 483 | 479 |
| CHH | 28 | 132 | 192 |
Fig. 5.
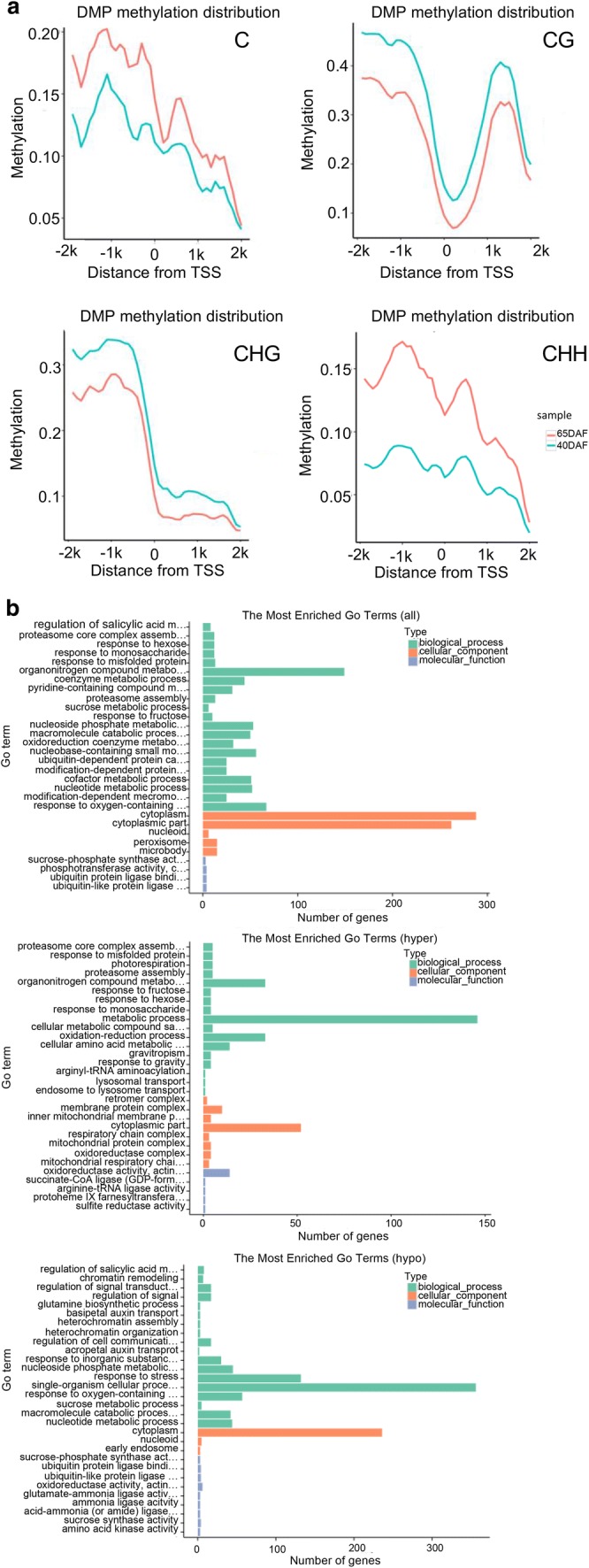
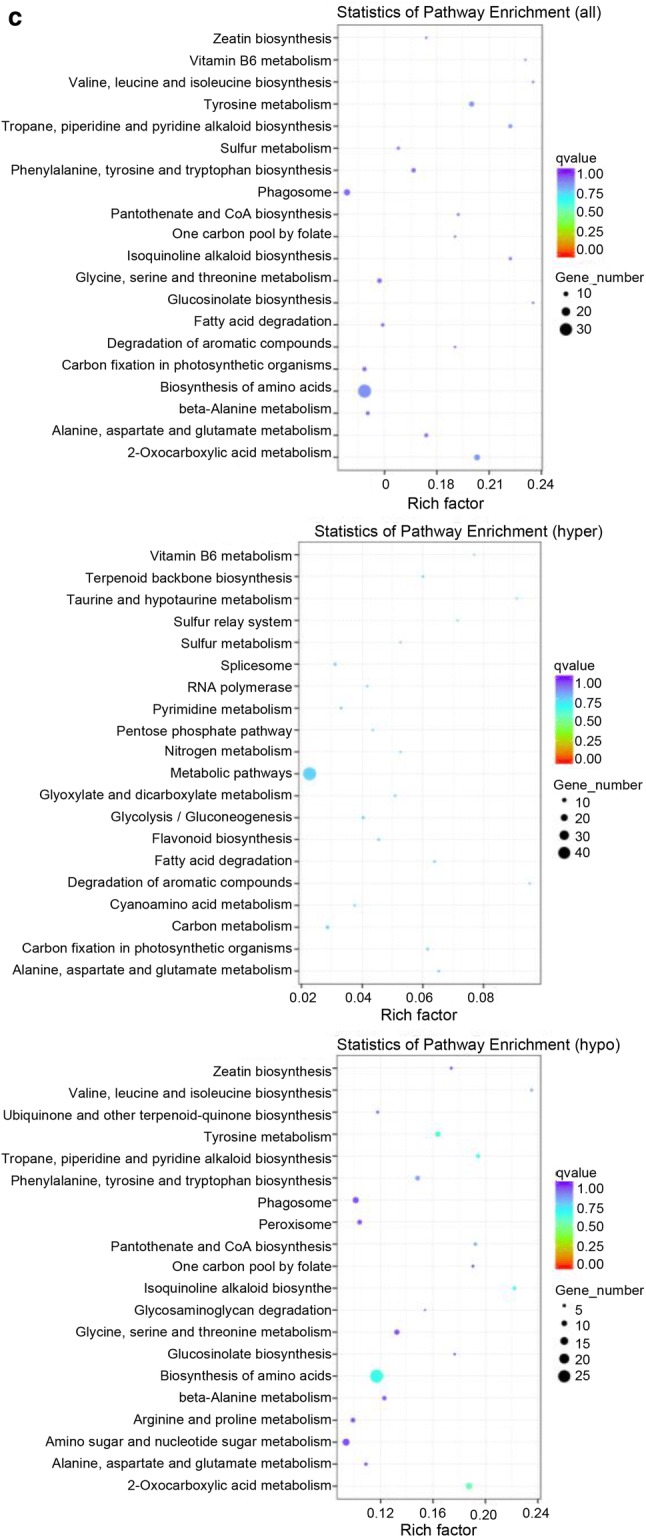
DMP comparison and annotation between any of fruits at different developmental stages. a Methylation level comparison of DMPs between 65DAF and 40DAF fruits. b GO enrichment analysis between 65DAF and 40DAF fruits (from upper to downer was all DMP, hyper-DMP, and hypo-DMP). c KEGG pathway enrichment analysis between 65DAF and 40DAF fruits (from upper to downer was all DMP, hyper-DMP, and hypo-DMP). The circle size represented the gene number and the circle color represented the q-value of pathway enrichment analysis (color figure online)
A GO enrichment analysis indicated that all-DMPs, hyper-DMPs, and hypo-DMPs in 65DAF_vs_40DAF were enriched in “organonitrogen compound metabolism,” “metabolic process,” and “single-organism cellular process” categories (Fig. 5b). KEGG pathway enrichment analysis showed that “biosynthesis of amino acids,” “metabolic pathway,” and “biosynthesis of amino acids” pathways were enriched in the sets of all-DMPs, hyper-DMPs, and hypo-DMPs of 65DAF_vs_40DAF (Fig. 5c). The GO and KEGG enrichment analyses of 90DAF_vs_65DAF and 90DAF_vs_40DAF indicated that most DMPs were related to metabolic, coenzyme biosynthesis, amino acid biosynthesis, and cellular component assembly processes or pathways (Supplementary files Figs. S9 and S10).
Effect of DNA methylation on gene expression
To investigate the effects of DNA methylation on gene expression in grape fruits at different developmental stages, transcriptome sequencing data (retrieved from the same fruit stage samples) was used to confirm the expression levels of unmethylated and methylated genes (Shangguan et al. 2017). We classified the genes into four groups according to their expression levels as follows: none (FPKM < 1), low (1 < FPKM < lower quartiles), medium (lower quartiles < FPKM < upper quartiles), and high (FPKM > upper quartiles). The comparative results of gene expression and methylation level indicated that the genes with no and low expression had higher DNA methylation levels, while medium and highly expressed genes had relatively lower DNA methylation levels in gene body and downstream_2k regions in all contexts for 40 DAF fruits. However, in upstream_2k regions, only genes with no expression had higher DNA methylation levels, while genes with high, medium, and low expression had similar DNA methylation levels in CG and CHG contexts for 40 DAF fruits. Interestingly, the DNA methylation in CG gene body regions was more than those in CG upstream_2k and downstream_2k regions (Fig. 6a). In 90DAF_vs_65DAF and 90DAF_vs_40DAF, a similar phenomenon was observed (Supplementary files Fig. S13a and S13b). Different regions had different DNA methylation levels. We also compared the methylation patterns between gene and promoter regions. For most DEGs, the difference in methylation was less in gene regions than in promoter regions (Fig. 6b, Supplementary file Fig. S13c and S13d). DNA methylation and gene expression levels were also compared between TSS and transcriptional end site (TES) regions among 90 DAF, 65 DAF, and 40 DAF fruits. DNA methylation levels were correlated with gene expression levels in TSS and TES region in these fruits (Fig. 6c, d, Supplementary files Fig. S13e–S13h). In 40 DAF fruits as an example, the relationship between DNA methylation and gene expression levels in TSS regions was similar to that in TES regions. The percentage of unmethylated genes was more than that of methylated genes in the low gene expression group, whereas the percentage of methylated genes was more than that of unmethylated genes in high gene expression group (Fig. 6c, d).
Fig. 6.


The effect of DNA methylation on gene expression. a The relationship of DNA methylation level and gene expression in 40DAF fruit. Genes were divided into four groups: none (FPKM < 1); low (1 < FPKM < lower quartiles); medium (lower quartiles < FPKM < upper quartiles); high (FPKM > upper quartiles). b The relationship of gene expression level and DNA methylation level in promoter and gene regions (65DAF_vs_40DAF). c The relationship of DNA methylation level and gene expression level of 40DAF fruit in TSS region. d The relationship of DNA methylation level and gene expression level of 40DAF fruit in TES region. e The Venn figure of DMR associated genes and differential expressed genes in 65DAF_vs_40DAF. f GO enrichment analysis of common genes between DMR associated genes and DEGs in 65DAF_vs_40DAF. g KEGG pathway analysis of shared genes between DMR associated genes and DEGs in 65DAF_vs_40DAF. The circle size represented the gene number and the circle color represented the q-value of pathway enrichment analysis (color figure online)
To analyze the effect of DNA methylation on gene expression, we obtained the common genes in the sets of DMR-associated genes and differentially expressed genes. In total, we obtained 1272, 492, and 966 genes for 65DAF_vs_40DAF, 90DAF_vs_65DAF, 90DAF_vs_40DAF, respectively (Fig. 6e, Supplementary files Fig. S14a and S14d). GO and KEGG pathway enrichment results indicated that most common genes were involved in the “cellular processes,” “cellular localization,” and “metabolic processes” categories and “carbon metabolism,” “biosynthesis of amino acids,” “biosynthesis of secondary metabolites,” and “metabolic” pathways (Fig. 6f, g, Supplementary files Fig. S14b, S14c, S14e, S14f). RNA-seq also revealed some highly expressed DEGs between fruits at different developmental stages. The comparative results also indicated that all of the genes had low DNA methylation levels in promoter and transcript regions (Supplementary file Fig. S15).
‘Fujiminori’ is one of the most widely cultivated grapevine cultivar in China. Its fruit color significantly changes during ripening process. We further isolated 38 DEGs including structural genes and transcriptional factors of anthocyanin biosynthesis pathway from 3381 to 5620 ripening-related DEGs. The methylation level and gene expression value were obtained (Supplementary file Table S8). The methylation levels in promoter and transcript regions, and the expression levels of anthocyanin related genes were changed during ripening process (Supplementary files Fig. S16a and Table S8). However, the relationship between methylation level and expression level was not positive or negative. The expression level of MYBPA1 (VIT_15s0046g00170) decreased continuously; however, the methylation level did not decreased continuously. The highest methylation level was observed in 65 DAF fruits in promoter and transcript regions. Moreover, we spliced the upstream 2 k, transcript, and downstream 2 k regions of these genes into 60 bins, and calculated the methylation levels of mCG/CG, mCHG/CHG, and mCHH/CHH groups. Only mCHH/CHH group showed difference among the three fruit developmental stages, and 40 DAF fruits demonstrated the lowest methylation level (Supplementary file Fig. S16b).
Discussion
Difference in methylation landscapes between grape and other plants
Studies have explored the relationship between gene expression and DNA methylation in plants and animals. Methylation in promoters represses gene expression, whereas methylation in gene-body is positively associated with gene expression (Cokus et al. 2008; Li et al. 2012; Popp et al. 2010; Raddatz et al. 2013; Song et al. 2013a). Nevertheless, the effects of DNA methylation in grapevine fruit development are still unknown. In this study, we obtained the global and single-base resolution methylome of grapevine and investigated the changes in DNA methylation profiles among three fruit developmental stages by high-throughput sequencing.
Ren et al. (2017) has identified eight DNA methyltransferases from the grapevine genome, including three METs, four CMTs, and one DRM. Two more genes (one DRM and one DNMT) were identified in this study. Interestingly, the high methylation level of CG was observed in grapevine fruits at different development stages; however, no studies have identified MET1 or DNMT1 genes in grapevine fruits so far. Currently, four grapevine genomes databases are available, including Genoscope (8X and 12X, https://www.genoscope.cns.fr/externe/GenomeBrowser/Vitis/), Grape CRIBI database (http://genomes.cribi.unipd.it/grape/), NCBI grape genome database (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Vitis_vinifera/101/), and Ensembl Plants grape genome database (http://plants.ensembl.org/Vitis_vinifera/Info/Index). The incomplete genome sequence of grapevine (Shangguan et al. 2013; Tang and Bassham 2018) and the different versions of grape genome database used may have led to the differences between Ren’s work (2017) and our study and the mis-annotation of MET1 in grapevine genome. Moreover, the expression levels of DRM1, DRM2, and DNMT2 were higher during fruit development in grapevine (Fig. 1). Previous reports in Arabidopsis have indicated the role of AtDRM2 in de novo methylation on CpG, CpNpG, and CpNpN, and to maintain CpG, CpNpG, and CpNpN methylation (Cao et al. 2003; Cao and Jacobsen 2002). DRM2 may play similar functions in grapevine. However, we need to further investigate on the detailed function of grapevine methyltransferases.
We observed that the methylation levels were different between grapevine and tomato fruits. Zhong et al. (2013) detected 73.97–79.16%, 51.99–53.88%, and 13.52–14.20% methylation of CpG, CHG, and CHH, respectively, in tomato. Meanwhile, we detected only 36.34–41.1%, 19.6–21.35%, and 2.96–4.74% methylation of CpG, CHG, and CHH, respectively, in grapevine (Table 3). The genome size of tomato is approximately 900 Mb, which is twice that of grapevine (427 Mb). Moreover, the genome-wide methylation levels differ among plants. We found that the genome-wide methylation level in V. vinifera fruits (~ 8.3%) was slightly more than that in Arabidopsis flower buds (5.26%) (Lister et al. 2008) and less than those in Populus leaves (10.04%) and rice samples from panicle initiation to booting (15.40%) (Li et al. 2012; Liang et al. 2014). Obviously, methylation level is related to genome size; that of A. thaliana is 135 Mb, V. vinifera is 427 Mb, Populus trichocarpa is 510 Mb, and Oryza sativa is ~ 500 Mb. A positive correlation between methylation rate and genome size has been found in metazoans (Lechner et al. 2013). In plants, Alonso et al. (2015) demonstrated that the whole genome methylation level and genome size are evolutionarily correlated, and a 10-fold increase in genome size is associated with an increase of about 10% in the whole genome methylation level. The transposition of repeated elements is the main cause of plant genome expansion (Bennetzen et al. 1998; SanMiguel et al. 1998). Large genomes have a high portion of transposable elements (e.g., the maize genome has > 50% transposable elements), and small genomes have relatively few transposable elements (e.g., the Arabidopsis genome has < 10% transposable elements) (Bennetzen et al. 1998; Fedoroff 2012). Cytosines are always more densely methylated in transposons than in genes. Therefore, the global DNA methylation level of plant genome should be directly related to the repeat sequences in the genome (Bender 2004; Rabinowicz et al. 2003; Vaughn et al. 2007). The grape genome has abundant transposable elements (Jaillon et al. 2007; Velasco et al. 2007) compared with poplar, Arabidopsis, and rice (Goff et al. 2002; Tuskan et al. 2006). The positive correlation between the DNA methylation level and genome transposable element density seems to be applicable in grapevine and other plants. The mC sites have a high density in or near the centers of each chromosome consistent with the location of the centromere in each chromosome (Fig. 2d and Supplementary file Fig. S4).
Different trends in methylation contexts were observed among grapevine fruits at different developmental stages. The methylation level of mC sites increased by 1% from 40 DAF fruits (8.3%) to 65 DAF fruits (9.29%); the methylation level at 65 DAF was similar to that at 90 DAF (9.24%). This trend was different among all sequence contexts (mCG, decreased by 4.76%; mCHG, decreased by 2.66%; mCHH, increased by 1.78%; Table 3). Moreover, nearly 60% of mC sites and 70–90% of DMRs belonged to the mCHH context at all stages of fruit development (Figs. 2a, 4a). Extensive CHH methylation occurred in grape fruits, which appears to be similar to the patterns observed in the seeds of castor bean and the leaves of cassava (Wang et al. 2015a; Xu et al. 2016). The relatively high proportion of newly identified mCHH sites and 10% mCHH_DRM-associated genes suggest that mCHH may be important for the regulation of gene expression during fruit development in the grapevine.
DNA methylation and gene expression
Studies have analyzed the relationship between DNA methylation and gene expression has been studied in many plants. In general, DNA methylation represses gene expression. In grapevine also, DNA methylation repressed gene expression (Fig. 6a, Supplementary files Fig. S13a and S13b). These results are consistent with the previous findings in Populus (Liang et al. 2014), A. thaliana (Zilberman et al. 2007), rice (Li et al. 2012), and humans (Lister et al. 2009),. DNA methylation affects the binding of RNA polymerase to DNA and in turn affects gene transcription (Zilberman et al. 2007). We identified few top DEGs by RNA-sequencing, and a comparison between gene expression level and DNA methylation level indicated a significantly lower DNA methylation level in promoter and transcript regions (Supplementary file Fig. S15).
For gene bodies, the CG context had a much high methylation level compared with CHG and CHH contexts (Fig. 4f and Supplementary file Fig. S7). Meanwhile, the methylation level for the three contexts in exon regions was less than that in intron regions, especially for the CHG and CHH contexts (Fig. 4f and Supplementary file Fig. S7). It was hypothesized that DNA methylation in intron regions mainly affects gene expression. Most DMRs were detected in the CHH context in this study. MET1 controls the CG methylation level, whereas DRM1, CMT3, and DRM2 control the CHH methylation level (Aufsatz et al. 2004; Bartee et al. 2001; Zilberman et al. 2004). This is not consistent with the RNA-seq results, which indicated that the expression levels of CMT3, DMR1 and DMR2 were more than those of METs, and no MET1 homologue was found in grapevine.
The DNA methylation level in the upstream and downstream flanking sequences increased rapidly with increased in distance from the TSS and TTS in all contexts (Fig. 6a, Supplementary files Fig. S13a and S13b). DNA methylation levels in TSS and TTS were positively corrected with gene expression (Fig. 6c, d, and Supplementary files Fig. S13e–S13h). Interestingly, previous reports have shown that methylation in TTS and TSS can significantly repressed gene expression in rice (Li et al. 2012), Arabidopsis (Zhang et al. 2006), and Populus (Liang et al. 2014). The mechanism by which the gene expression level is affected by DNA methylation in TSS and TTS is unclear.
DNA methylation and fruit development
Fruit development is controlled by many factors, such as light, temperature, and water (Adams et al. 2001; Dokoozlian and Kliewer 1996; Li et al. 1989). DNA methylation also plays an key role in this process, and the gene expression has tissue-specific features and show variation among stages during plant growth (Gehring and Henikoff 2007). In tomato, methylation level was more in mature tissues, such as leaves, seeds, and fruits, than that in immature tissues (Messeguer et al. 1991; Teyssier et al. 2008). In this study, the DNA methylation level increased from 8.3% (40 DAF fruits, green stage) to 9.24% (90 DAF fruits, mature stage), and the difference in methylation levels between 90 DAF (mature stage) and 65 DAF (véraison stage) was only 0.05%, which indicates the role of DNA methylation in fruit development (Table 3). The number of DMRs for various pairwise comparisons were 33,631 (90DAF_vs_65DAF), 157,675 (90DAF_vs_40DAF), and 186,638 (65DAF_vs_40DAF) (Fig. 4a and Supplementary file Table S6). Moreover, the comparison of DNA methylation levels for DMR-associated genes between fruits at different developmental stages indicated a significant difference in the CHH context between 65 DAF and 40 DAF fruits (or 90 DAF and 40 DAF fruits, Fig. 4f and Supplementary file Fig. S9). These results indicate that DNA methylation was the most frequent between the green stage (40 DAF) and the véraison stage (65 DAF), and the CHH context may contribute substantially to the regulation of gene expression by DNA methylation. A similar methylome pattern was also found in the shoots of chestnut (Castanea sativa) (Hasbun et al. 2005). In tomato, hypermethylation of Cnr arrested fruit development (Manning et al. 2006). The overall methylation levels differ among tomato fruits (23.45–31.41%), and the genes involved in ripening, shelf-life, and fruit-quality were also affected by DNA methylation (Zhong et al. 2013). Gene functional annotation results showed that DMR-related genes were mainly involved in metabolic processes and cellular processes, binding, and catalytic activity (Fig. 4d, e, and Supplementary files Figs. S7 and S8).
Venn diagrams identified some genes as both DEGs and DMR-associated genes (Fig. 6f, g, and Supplementary file Fig. S14). The functional analysis of these genes indicated that most of the genes were involved in carbon metabolism and the biosynthesis of amino acids between 65 DAF and 40 DAF fruits and in the biosynthesis of secondary metabolites between 90 DAF and 65 DAF fruits (Fig. 6f, g, and Supplementary file Fig. S14). RNA-seq results also indicated that the DEGs were involved in primary metabolism, secondary metabolism, and defense (Ali et al. 2011; Pilati et al. 2007). The methylation level compared with the expression level indicated no significant relationship in the anthocyanin biosynthesis-related genes of this study, which proves that DNA methylation is involved in fruit development, however, it is not the only regulator of gene expression. Moreover, the fruit mCHH/CHH level in 40 DAF fruits was less than it in 65 DAF fruits and 90DAF fruits; however, the regulatory mechanism is still unknown (Supplementary files Fig. S16 and Table S8). Combining the WGBS and RNA-seq results, we conclude that fruit development is influenced by different factors such as mRNA expression and DNA methylation. The complex relationship between mRNA expression and DNA methylation during fruit development, especially at the véraison stage, needs further investigation.
Conclusion
We globally characterized the DNA methylation-related genes encoding DNA methyltransferases and demethylases and comprehensively investigate the association of DNA methylation and gene expression during fruit development in grapevine. The DNA methylation pattern changed during ripening in grapevine. The methylation levels for mC, mCG, mCHG, and mCHH contexts in 65 DAF fruits (véraison stage) were more than those in fruit at the other two stages (green and mature stages). The difference in the methylation profile between fruits at different development stages was primarily detected in the CHH context. The differentially methylated regions (DMRs) and the differentially methylated promoters (DMPs) in 65DAF_vs_40DAF were both more than those in 90DAF_vs_65DAF and 90DAF_vs_40DAF. Most DMRs (or DMPs) were involved in metabolic processes and cell processes, binding, and catalytic activity. These results reveal variations in whole genome DNA methylation during grapevine fruit development, and will also be helpful for the research on fruit development in other species.
Methods
Identification and analysis of DNA methylation-related genes
We performed a comprehensive search on the Vitis vinifera genome database (EnsemblPlants, http://plants.ensembl.org/Vitis_vinifera) to detect all DNA methyltransferase genes (CMT, chromomethyltransferase; MET, methyltransferase; DRM, domains rearranged methyltransferase) and demethylase genes (ROS1, repressor of silencing 1; DME, demeter; DML2, demeter-like 2; DML3, demeter-like 3). The protein sequences of ten Arabidopsis DNA methyltransferase genes and four demethylase genes were downloaded from the TAIR database (The Arabidopsis Information Resource, https://www.arabidopsis.org). BLASTP (http://plants.ensembl.org/Vitis_vinifera/Tools/Blast?db=core) compared the Arabidopsis protein sequences used as queries against the grape protein database using 1e−10, 30% and 60% as the thresholds for e-value, sequence identity, and sequence coverage, respectively. The candidate protein sequences (putative DNA methylation-related grape proteins) were confirmed by analyzing the characterized domains using Pfam and InterProScan online softwares (http://pfam.xfam.org/ and http://www.ebi.ac.uk/interpro/search/sequence-search) (Finn et al. 2006; Quevillon et al. 2005). The protein structure of DNA methylation-related genes was illustrated using the IBS online tool (http://ibs.biocuckoo.org/online.php) (Liu et al. 2015b).
To investigate the expression profiles of DNA methylation-related genes, grape microarray and RNA-sequencing datasets (GEO no. GSE36128, GSE24561, GSE41206, GSE76256, GSE62744, and GSE77218) were retrieved and analyzed from NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/). The differentially expressed DNA methylation-related genes during ‘Fujiminori’ fruit development were obtained from Shangguan’s paper (2017), and the expression profiles were confirmed by RT-qPCR. We used Excel 2019 (one way ANOVA test) to determine statistically significant difference in DNA methylation-related genes between fruit mesocarp and exocarp or between Vitis vinifera cv. ‘Cabernet Sauvignon’ and Vitis quinquangularis accession ‘Danfeng-2’. When the gene was compared, the expression value in each tissue/cultivar was treated as one replicate. R packages (edgeR and ggplot2) were used to calculate the expression levels and create the heat maps of grape DNA methylation-related genes (Kim et al. 2013; Langmead and Salzberg 2012; Li et al. 2009; Robinson et al. 2010; Team 2009; Trapnell et al. 2012; Wickham 2009).
Grape berry sampling
Berries from V. vinifera (cv. ‘Fujiminori’) were sampled at the first growth (40 days after flowering, represented as 40 DAF), véraison (65 days after flowering, represented as 65 DAF), and maturity (90 days after flowering, represented as 90 DAF) stages. Furthermore, to obtain a representative biological repeats at these time-point, DNA for sequencing was purified from tissue samples obtained from 40 berries sampled from 10 bunches.
Illumina sequencing
Total DNA was extracted from the fruits using Illustra Nucleon Phytopure Genomic DNA Extraction Kit (GE Life Sciences, Pittsburgh, PA, USA) following the manufacturer’s instructions. Bioruptor (Diagenode, Liege, Belgium) was used to fragment the DNA to an appropriate size (~ 250 bp). Then, blunt ending, dA and adaptor addition were performed. According to Hayatsu’s study (2006), the bisulfite conversion of the DNA was performed. DNA sequencing was performed using the Illumina HiSeq 2000 system according to the manufacturer’s instructions. Illumina base-calling pipeline was used to process the raw data.
Mapping and processing of bisulfite sequence-Seq reads
Vitis vinifera genome sequence was obtained from the EnsemblPlants database (http://plants.ensembl.org/Vitis_vinifera), and was used as the reference sequence to be aligned with the methylation sequencing data. Reads were mapped using BSMAP version 2.73 (Xi and Li 2009). Methylation ratio was determined using methratio.py script, with a cytosine sequencing depth in CpG context equal to or greater than tenfold coverage was retained for further analysis.
Gene ontology (GO) and KEGG pathway analyses
AgriGO database (http://bioinfo.cau.edu.cn/agriGO/download/item2term_56), BGI WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl), and Blast2GO were used for the GO analysis of interested grapevine genes (Du et al. 2010). KEGG database (http://www.genome.jp/kegg/ko.html) was used for pathway mapping of interested grapevine genes.
Identification of differentially methylated regions (DMRs) and differentially methylated promoters (DMPs)
Only cytosines with more than three reads in both samples were considered for further analysis. The gene region with more than twofold difference in methylation level between different developmental stages was confirmed as differentially methylated cytosine. Only those regions with the sliding-window (200 bp window with 100 bp interval) that contained more than six differentially methylated cytosines and with a p value less than 0.005 with Fisher’s exact test were confirmed as differentially methylated regions (DMRs). The 2000 bp upstream sequence of the transcription start site (TSS) was used to identify differentially methylated promoters (DMPs). Only those regions with the window that contained more than five methylated cytosines, a p value less than 0.05 with Fisher’s exact test, and a methylation level difference larger than 0.1 were considered as DMPs. Python scripts were used for data processing, and R and ggplot2 were used for data visualization (Wickham 2009). We have deposited the datasets in NCBI database (accession number GSE77066).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 2 (DOCX 22010 kb)
Acknowledgements
This work was supported by grants from the Natural Science Foundation of China (NSFC) (No. 31772283), and Key R&D projects in Jiangsu Province (BE2018389). We thank Novogene for helping the joint analysis of RNA sequencing and Methylation sequencing data.
Abbreviations
- CMT
Chromomethylase
- DAF
Days after flowering
- DME
Demeter
- DML2
Demethylase 2
- DML3
Demeter-like 3
- DMPs
DNA methylation promoters
- DMRs
DNA methylation regions
- DRM
Domain-rearranged methyltransferases
- Gbp
Giga base pairs
- GO
Gene ontology
- Mb
Megabase
- RIN
Ripening inhibitor
- RNA-seq
RNA-sequencing
- ROS1
Repressor of silencing 1
- SBP box-Cnr
SQUAMOSA promoter binding protein-like-Colorless non-ripening
- TAIR
The Arabidopsis Information Resource
- TSS
Transcription start site
- TTS
Transcription termination sites
- WGBS
Whole-genome bisulfite sequencing
Authors’ contributions
LFSG and JGF conceived and designed the experiments. LFSG, XF, and KKZ analyzed the data. LFSG and HFJ, helped in manuscript write-up. LFSG, MXC and JGF revised the manuscript. All authors read and approved the final manuscript.
Data Archiving Statement
The sequencing datasets were deposited in NCBI database with accession number GSE77066.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adams S, Cockshull K, Cave C. Effect of temperature on the growth and development of tomato fruits. Ann Bot Lond. 2001;88:869–877. [Google Scholar]
- Ahmad F, Huang X, Lan HX, Huma T, Bao YM, Huang J, Zhang HS. Comprehensive gene expression analysis of the DNA (cytosine-5) methyltransferase family in rice (Oryza sativa L.) Genet Mol Res. 2014;13:5159–5172. doi: 10.4238/2014.July.7.9. [DOI] [PubMed] [Google Scholar]
- Ali M, Howard S, Chen S, Wang Y, Yu O, Kovacs L, Qiu W. Berry skin development in Norton grape: distinct patterns of transcriptional regulation and flavonoid biosynthesis. BMC Plant Biol. 2011;11:7. doi: 10.1186/1471-2229-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso C, Pérez R, Bazaga P, Herrera CM. Global DNA cytosine methylation as an evolving trait: phylogenetic signal and correlated evolution with genome size in angiosperms. Front Genet. 2015;6:4. doi: 10.3389/fgene.2015.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashapkin VV, Kutueva LI, Vanyushin BF. Plant DNA methyltransferase genes: multiplicity, expression, methylation patterns. Biochemistry (Moscow) 2016;81:141–151. doi: 10.1134/S0006297916020085. [DOI] [PubMed] [Google Scholar]
- Aufsatz W, Mette MF, Matzke AJM, Matzke M. The role of MET1 in RNA-directed de novo and maintenance methylation of CG dinucleotides. Plant Mol Biol. 2004;54:793–804. doi: 10.1007/s11103-004-0179-1. [DOI] [PubMed] [Google Scholar]
- Bartee L, Malagnac F, Bender J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Gene Dev. 2001;15:1753–1758. doi: 10.1101/gad.905701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter MJ, Bui C, Young DA. Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthr Cartill. 2012;20:339–349. doi: 10.1016/j.joca.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Bender J. DNA methylation and epigenetics. Annu Rev Plant Biol. 2004;55:41–68. doi: 10.1146/annurev.arplant.55.031903.141641. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, SanMiguel P, Chen M, Tikhonov A, Francki M, Avramova Z. Grass genomes. Proc Natl Acad Sci USA. 1998;95:1975–1978. doi: 10.1073/pnas.95.5.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernacchia G, Primo A, Giorgetti L, Pitto L, Cella R. Carrot DNA-methyltransferase is encoded by two classes of genes with differing patterns of expression. Plant J. 1998;13:317–329. doi: 10.1046/j.1365-313x.1998.00034.x. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Gene Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- Burn J, Bagnall D, Metzger J, Dennis E, Peacock W. DNA methylation, vernalization, and the initiation of flowering. Proc Natl Acad Sci USA. 1993;90:287–291. doi: 10.1073/pnas.90.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002;12:1138–1144. doi: 10.1016/s0960-9822(02)00925-9. [DOI] [PubMed] [Google Scholar]
- Cao X, Aufsatz W, Zilberman D, Mette MF, Huang MS, Matzke M, Jacobsen SE. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol. 2003;13:2212–2217. doi: 10.1016/j.cub.2003.11.052. [DOI] [PubMed] [Google Scholar]
- Cao D, et al. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum lycopersicum. Gene. 2014;550:230–237. doi: 10.1016/j.gene.2014.08.034. [DOI] [PubMed] [Google Scholar]
- Cokus SJ, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Ni Z, Dai J, Zhao T, Sun Q. Isolation and expression analysis of genes encoding DNA methyltransferase in wheat. Biochim Biophys Acta BBA Gene Struct Expr. 2005;1729:118–125. doi: 10.1016/j.bbaexp.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Dokoozlian N, Kliewer W. Influence of light on grape berry growth and composition varies during fruit development. J Am Soc Hortic Sci. 1996;121:869–874. [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:w64. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichten SR, et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell. 2013;25:2783–2797. doi: 10.1105/tpc.113.114793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff NV. Transposable elements, epigenetics, and genome evolution. Science. 2012;338:758–767. doi: 10.1126/science.338.6108.758. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Henikoff S. DNA methylation dynamics in plant genomes. Biochim Biophys Acta. 2007;1769:276–286. doi: 10.1016/j.bbaexp.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Goff SA, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica) Science. 2002;296:92–100. doi: 10.1126/science.1068275. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Gu T, Ren S, Wang Y, Han Y, Li Y. Characterization of DNA methyltransferase and demethylase genes in Fragaria vesca. Mol Genet Genom. 2016;291:1333–1345. doi: 10.1007/s00438-016-1187-y. [DOI] [PubMed] [Google Scholar]
- Hasbun R, et al. In vitro proliferation and genome DNA methylation in adult chestnuts. Acta Hortic. 2005;693:333–340. [Google Scholar]
- Hayatsu H, Tsuji K, Negishi K. Does urea promote the bisulfite-mediated deamination of cytosine in DNA? Investigation aiming at speeding-up the procedure for DNA methylation analysis. Nucleic Acids Symp Ser (Oxf) 2006;50:69. doi: 10.1093/nass/nrl034. [DOI] [PubMed] [Google Scholar]
- Jaillon O, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–465. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Kawanabe T, et al. Role of DNA methylation in hybrid vigor in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2016;113:E6704–E6711. doi: 10.1073/pnas.1613372113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AR, Enjalbert J, Marsollier A-C, Rousselet A, Goldringer I, Vitte C. Vernalization treatment induces site-specific DNA hypermethylation at the VERNALIZATION-A1 (VRN-A1) locus in hexaploid winter wheat. BMC Plant Biol. 2013;13:1. doi: 10.1186/1471-2229-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:1. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G, Rattan UK, Singh AK. Chilling-mediated DNA methylation changes during dormancy and its release reveal the importance of epigenetic regulation during winter dormancy in apple (Malus x domestica Borkh.) PLoS ONE. 2016;11:e0149934. doi: 10.1371/journal.pone.0149934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, Marz M, Ihling C, Sinz A, Stadler PF, Krauss V. The correlation of genome size and DNA methylation rate in metazoans. Theory Biosci. 2013;132:47–60. doi: 10.1007/s12064-012-0167-y. [DOI] [PubMed] [Google Scholar]
- Li S, Huguet J, Schoch P, Orlando P. Response of peach tree growth and cropping to soil water deficit at various phenological stages of fruit development. J Hortic Sci. 1989;64:541–552. [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012;13:300. doi: 10.1186/1471-2164-13-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, et al. Single-base-resolution methylomes of populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014;15:S9. doi: 10.1186/1471-2156-15-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc Natl Acad Sci USA. 2015;112:10804–10809. doi: 10.1073/pnas.1503362112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–3361. doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li H, Lin J, Wang Y, Xu X, Cheng ZM, Chang Y. Genome-wide characterization of DNA demethylase genes and their association with salt response in Pyrus. Genes (Basel) 2018 doi: 10.3390/genes9080398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning K, et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38:948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- Messeguer R, Ganal MW, Steffens JC, Tanksley SD. Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear-DNA. Plant Mol Biol. 1991;16:753–770. doi: 10.1007/Bf00015069. [DOI] [PubMed] [Google Scholar]
- Moglia A, Gianoglio S, Acquadro A, Valentino D, Milani AM, Lanteri S, Comino C. Identification of DNA methyltransferases and demethylases. PLoS ONE. 2019;14:e0223581. doi: 10.1371/journal.pone.0223581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilati S, et al. Genome-wide transcriptional analysis of grapevine berry ripening reveals a set of genes similarly modulated during three seasons and the occurrence of an oxidative burst at veraison. BMC Genom. 2007;8:428. doi: 10.1186/1471-2164-8-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp C, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowicz PD, Palmer LE, May BP, Hemann MT, Lowe SW, McCombie WR, Martienssen RA. Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res. 2003;13:2658–2664. doi: 10.1101/gr.1784803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz G, et al. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenet Chromatin. 2013;6:36. doi: 10.1186/1756-8935-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F, Yang L, Su L, Gong L, Wang P, Wang Y. Genome-wide identification and analysis of DNA methyltransferases in grape. Agric Sci Technol. 2017;18:1781–1794. [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. The paleontology of intergene retrotransposons of maize. Nat Genet. 1998;20:43–45. doi: 10.1038/1695. [DOI] [PubMed] [Google Scholar]
- Shangguan L, et al. Evaluation of genome sequencing quality in selected plant species using expressed sequence tags. PLoS ONE. 2013;8:e69890. doi: 10.1371/journal.pone.0069890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan L, et al. RNA-Sequencing reveals biological networks during table grapevine (‘Fujiminori’) fruit development. PLoS ONE. 2017;12:e0170571. doi: 10.1371/journal.pone.0170571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell. 2012;24:875–892. doi: 10.1105/tpc.111.094870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q-X, et al. Genome-wide analysis of DNA methylation in soybean. Mol Plant. 2013;6:1961–1974. doi: 10.1093/mp/sst123. [DOI] [PubMed] [Google Scholar]
- Song Y, Ma K, Ci D, Chen Q, Tian J, Zhang D. Sexual dimorphic floral development in dioecious plants revealed by transcriptome, phytohormone, and DNA methylation analysis in Populus tomentosa. Plant Mol Biol. 2013;83:559–576. doi: 10.1007/s11103-013-0108-2. [DOI] [PubMed] [Google Scholar]
- Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 2013;152:352–364. doi: 10.1016/j.cell.2012.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Sweetman C, Wong DC, Ford CM, Drew DP. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genom. 2012;13:691. doi: 10.1186/1471-2164-13-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Bassham DC. Autophagy in crop plants: what’s new beyond Arabidopsis? Open Biol. 2018 doi: 10.1098/rsob.180162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team D. R: A language and environment for statistical computing. Computing. 2009;14:12–21. [Google Scholar]
- Teyssier E, Bernacchia G, Maury S, Kit AH, Stammitti-Bert L, Rolin D, Gallusci P. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta. 2008;228:391–399. doi: 10.1007/s00425-008-0743-z. [DOI] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- Vaughn MW, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007;5:e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco R, et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE. 2007;2:e1326. doi: 10.1371/journal.pone.0001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc Natl Acad Sci USA. 2015;112:13729–13734. doi: 10.1073/pnas.1519067112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, et al. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in maize (Zea may) BMC Genom. 2015;16:1. doi: 10.1186/s12864-014-1204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer; 2009. [Google Scholar]
- Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang T, Dong X, Li D-Z, Liu A. Genomic DNA methylation analyses reveal the distinct profiles in castor bean seeds with persistent endosperms. Plant Physiol. 2016;171:1242–1258. doi: 10.1104/pp.16.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, et al. Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J. 2015;81:268–281. doi: 10.1111/tpj.12726. [DOI] [PubMed] [Google Scholar]
- Zenoni S, et al. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010;152:1787–1795. doi: 10.1104/pp.109.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Zhang B, et al. Chilling-induced tomato flavor loss is associated with altered volatile synthesis and transient changes in DNA methylation. Proc Natl Acad Sci USA. 2016;113:12580–12585. doi: 10.1073/pnas.1613910113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol. 2013;31:154–159. doi: 10.1038/nbt.2462. [DOI] [PubMed] [Google Scholar]
- Zilberman D, Cao XF, Johansen LK, Xie ZX, Carrington JC, Jacobsen SE. Role of arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr Biol. 2004;14:1214–1220. doi: 10.1016/j.cub.2004.06.055. [DOI] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran R, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 2 (DOCX 22010 kb)
Data Availability Statement
The sequencing datasets were deposited in NCBI database with accession number GSE77066.