

Abstract
Objective
Bombesin-like receptor 3 (BRS3) is an orphan receptor and Brs3 knockout mice develop obesity with increased food intake and reduced resting metabolic rate and body temperature. The neuronal populations contributing to these effects were examined.
Methods
We studied energy metabolism in mice with Cre-mediated recombination causing 1) loss of BRS3 selectively in SIM1- or MC4R-expressing neurons or 2) selective re-expression of BRS3 from a null background in these neurons.
Results
The deletion of BRS3 in MC4R neurons increased body weight/adiposity, metabolic efficiency, and food intake, and reduced insulin sensitivity. BRS3 re-expression in these neurons caused partial or no reversal of these traits. However, these observations were confounded by an obesity phenotype caused by the Mc4r-Cre allele, independent of its recombinase activity. The deletion of BRS3 in SIM1 neurons increased body weight/adiposity and food intake, but not to the levels of the global null. The re-expression of BRS3 in SIM1 neurons reduced body weight/adiposity and food intake, but not to wild type levels. The deletion of BRS3 in either MC4R- or SIM1-expressing neurons affected body temperature, with re-expression in either population reversing the null phenotype. MK-5046, a BRS3 agonist, increases light phase body temperature in wild type, but not Brs3 null, mice and BRS3 re-expression in either population restored response to MK-5046.
Conclusions
BRS3 in both MC4R- and SIM1-expressing neurons contributes to regulation of body weight/adiposity, insulin sensitivity, food intake, and body temperature.
Keywords: Bombesin-like receptor 3, BRS3, Obesity, MC4R neurons, SIM1 neurons, Body temperature, Food intake, Energy homeostasis
Abbreviations: DMH, dorsomedial hypothalamus; HFD, high fat diet; PVH, paraventricular nucleus of the hypothalamus; Tb, core body temperature; MeA, medial amygdala; MC4R, melanocortin receptor 4; SIM1, single-minded homolog 1
Graphical abstract

Highlights
-
•
BRS3 in MC4R neurons regulates adiposity and food intake.
-
•
BRS3 in both SIM1+ and SIM1− neurons regulates adiposity and food intake.
-
•
BRS3 in both MC4R and SIM1 neurons regulates body temperature.
-
•
BRS3 in either MC4R or SIM1 neurons confers body temperature response to agonist.
1. Introduction
Bombesin-like receptor 3 (BRS3, bombesin receptor subtype 3, BB3) is a G protein-coupled receptor regulating energy homeostasis. It belongs to a subfamily including the neuromedin B (NMB) and gastrin-releasing peptide (GRP) receptors [1]. An endogenous ligand for BRS3 has not been detected in placental mammals [[2], [3], [4]], although NMB has modest efficacy in a subset of assays [5] and both NMB and GRP are high affinity ligands in nonplacental vertebrates [3,6]. BRS3 may also function through receptor crosstalk [7]. The BRS3 null mouse has a decreased resting metabolic rate and light phase core body temperature (Tb), increased food intake, and develops obesity [[8], [9], [10], [11], [12]]. While obesity occurs on a chow diet [8], it is amplified by a high fat diet (HFD) [13]. BRS3 in glutamatergic, but not GABAergic, neurons is responsible for the metabolic changes of the BRS3 global knockout mice [14]. Stimulation of BRS3 neurons in the dorsomedial hypothalamus (DMHBrs3) increases energy expenditure and Tb, and activates brown adipose tissue, but does not alter food intake or physical activity, whereas stimulation of BRS3 neurons in the paraventricular hypothalamus (PVHBrs3) reduces food intake, with no effect on energy expenditure, Tb, or physical activity [15]. Rat studies also support a role for PVHBrs3 neurons in food intake and DMHBrs3 neurons in energy expenditure [16].
Mutations in melanocortin receptor 4 (MC4R) cause obesity in humans and mice. Mouse Mc4r ablation causes hyperphagia, reduced energy expenditure, reduced sympathetic tone, increased fat mass, and impaired glucose homeostasis, leading to overt early-onset obesity [[17], [18], [19]]. MC4R is localized in the thalamus, hypothalamus, and hippocampus among other brain and peripheral sites [20,21].
Single-minded homolog 1 (SIM1) is a member of the bHLH-PAS (basic helix-loop-helix Per-Arnt-Sim) family of transcription factors and is required for the proper formation of the basal forebrain [22,23]. SIM1 homozygous knockout mice fail to properly form at least the paraventricular (PVH), supraoptic (SON), and anterior periventricular (aPV) hypothalamic nuclei and die perinatally [24]. SIM1 heterozygous (Sim1+/−) mice survive and develop early-onset obesity with increased linear growth, hyperinsulinemia, and hyperleptinemia. Sim1+/− mice are hyperphagic but their energy expenditure is not significantly decreased [25]. In humans, germline haploinsufficiency of SIM1 causes hyperphagic obesity, sometimes with Prader-Willi-like features [26,27].
To further define the neurons relevant for BRS3 function in energy intake and expenditure [28], we assessed the effects of the loss and re-expression of BRS3 in MC4R- and SIM1-expressing neurons. We report a role for BRS3 in these neurons in regulating body weight/adiposity, insulin sensitivity, and Tb.
2. Materials and methods
2.1. Animals
Male mice were singly housed on a 12:12-h dark–light cycle (lights on at 0600), at ∼22 °C, with Teklad bedding (7090, Envigo Inc.) and ad libitum access to food and water. At 8 weeks of age, they were singly housed and placed on an HFD (D12492, 60% kcal fat, 5.24 metabolizable kcal/g; Research Diets, New Brunswick, NJ). Procedures were approved by the NIDDK Animal Care and Use Committee (protocol K016-DEOB-17).
Mc4rtm3.1(cre)Lowl (JAX 030759 [29], hereafter Mc4r-Cre) mice were supplied by Dr. Michael Krashes, NIDDK. Tg(Sim1-cre)1Lowl/J (hereafter Sim1-Cre) mice were purchased from the Jackson laboratory (JAX 006395 [18]). Brs3 inactivation studies used littermate male progeny of female Brs3flox/flox (JAX 031353 [14]) × male Brs3+/y;Cre/+ matings, on a congenic C57BL/6J background. Brs3 re-expression studies used littermate male progeny of female Brs3loxTB/loxTB (JAX 032580 [14]) × male Brs3+/y;Cre/+ matings, also on a congenic C57BL/6J background. Littermate mice differing only by Cre status were compared.
2.2. Phenotyping
Body weight and food intake were measured weekly and body composition was assessed every two weeks by EchoMRI (EchoMRI LLC, Houston, TX) [14]. Mice were studied on an HFD (rather than chow) because it amplifies the obesity caused by the loss of BRS3 [13].
2.3. Glucose and insulin tolerance tests, hormone and metabolite profiles
Intraperitoneal glucose (1 g/kg, with AUC calculated from the baseline) tolerance tests were performed at 0930, following an overnight (16 h) fast. Glucose was measured with a Glucometer Contour (Bayer, Mishawaka, IN). Insulin (0.75 unit/kg, i.p.) tolerance tests were performed at 0930, in nonfasted mice, with AUC calculated from 0 mg/dl. Blood was collected at 0930 by tail bleed at 17 weeks of age for measurements of fed glucose and insulin, triglycerides, free fatty acids, cholesterol, leptin, and adiponectin. Free fatty acids (Fujifilm Waco Diagnostics, Mountain View, CA, reagents # 999-34691, 995-34791, 991-34891, 993-35191), triglycerides (Pointe Scientific Inc., Canton, MI, # T7532-120), and cholesterol (Thermo Scientific, Middletown, VA, # TR13421) were measured using the indicated colorimetric assays. Leptin (R&D Systems, Minneapolis, MN, # MOB00), insulin (Crystal Chem, Downers Grove, IL, # 90010 using mouse insulin standard # 90020), and adiponectin (Alpco, Salem, NH, #47-ADPMS-E01) were measured by ELISA.
2.4. Telemetric monitoring of body temperature (Tb)
Tb and physical activity were measured continuously by telemetry using intraperitoneally implanted G2 E-mitters, ER4000 energizer/receivers, and VitalView software (Starr Life Sciences, Oakmont, PA) with data collected each minute. Tb span is defined as the difference between the Tb 95th and 5th percentiles from 24-h measurement intervals (i.e., 24, 48, or 72 h).
2.5. Effects of MK-5046 on food intake and Tb
For food intake response, mice were fasted overnight (1700–0930), dosed, and HFD was resupplied at 1000 with food intake measured over the next 6 h. Mice were treated with MK-5046 (10 mg/kg, i.p., MedChemExpress, Monmouth Junction, NJ) [30] or vehicle (10% Tween 80 in 0.25% methylcellulose in saline) in random order two days apart. The food intake response to MK-5046 (ΔFI) is the 6-h food intake after MK-5046 minus the food intake after vehicle, within mouse.
For Tb response, mice in their home cages with bedding at usual vivarium temperature (∼22 °C) were started on telemetric monitoring of Tb and activity, then fasted overnight and dosed as detailed for food intake response. The baseline- and vehicle-corrected Tb response to MK-5046 (ΔTb) was calculated within the mice as: effect of MK-5046 (mean Tb of 60–180 min minus mean Tb of −150 to −30 min) minus the effect of vehicle (mean Tb of 60–180 min minus mean Tb of −150 to −30 min), with all times relative to drug/vehicle dosing.
2.6. Dissection of paraventricular hypothalamic nuclei (PVH)
Briefly, brains were rapidly removed and sectioned (300 μm) with a Leica VT1000 vibratome (Wetzlar, Germany) using ice-cold 194 mM sucrose, 20 mM NaCl, 4.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1.2 mM NaH2PO4, 10 mM glucose, and 26 mM NaHCO3 saturated with 95% O2/5% CO2. A triangle around the PVH was dissected from a slice on a microscope slide using an Axioskop 2 microscope (Zeiss), transferred to a tube on dry ice, and stored at −80 °C until processed for RNA.
2.7. Quantitative RT-PCR
RNA from whole hypothalamus (for Mc4r-Cre cohorts) or micro-dissected PVH (for Sim1-Cre cohorts) was extracted and processed as reported previously [31]. Brs3 mRNA was quantitated by RT-PCR using primers x573 (5′-CTGCTGACTTGTGTGCCTGT) and x574 (5′-AGTGGCTTCACGACTGCTTT). Brs3 mRNA from the unrecombined Brs3flox allele was present at 61 ± 2% of the level of the wild type allele. Mc4r mRNA was quantitated by RT-PCR using primers x581 (5′-ATCTGTAGCTCCTTGCTCGC) and x582 (5′-TGCAAGCTGCCCAGATACAA), using QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA). Expression was normalized using 18S RNA levels.
2.8. Statistical analysis
Data are presented as mean ± SEM. Since mice with different Brs3 background genotypes were not studied contemporaneously, statistical testing was limited to the effect of ±Cre within Brs3 background genotype. Statistical significance was determined by two-tailed t-test at P < 0.05 using Prism v8.2.1 (GraphPad, San Diego, CA) or Excel.
3. Results
3.1. BRS3 in MC4R-expressing neurons regulates body weight/adiposity
BRS3 and MC4R are expressed in overlapping locations in the hypothalamus. Mc4r-Cre-mediated deletion of Brs3 caused a 10% reduction in hypothalamic BRS3 mRNA (Figure 1A). In Brs3loxTB;Mc4r-Cre mice the BRS3 mRNA recovered to 31% of wild type levels (Figure 1A). Thus Mc4r-Cre is expressed in a small subset of BRS3-expressing hypothalamic neurons.
Figure 1.

Brs3 and Mc4r expression in Mc4r-Cre cohorts of mice. Hypothalamic Brs3 (A) and Mc4r (B) mRNA were quantified by RT-PCR. Within each background genotype pair (indicated by dashed lines), the relative mRNA expression is reported with the higher valued at 100 (except that Brs3 mRNA in loxTB;Mc4r-Cre is normalized to the mean of WT, WT;Mc4r-Cre, and flox). Data are mean ± SEM, N = 5–6/group. ND indicates not detected, with a limit of detection of ∼1% of loxTB;Mc4r-Cre.
Mc4r mRNA levels in heterozygous Mc4r-Cre (Mc4rCre/+) mice averaged 76 ± 3% of wild type (Mc4r+/+) mice (Figure 1B). Thus, Mc4r mRNA levels produced by the Mc4r-Cre allele are about 52% (i.e., [76-50]/50) of the wild type allele. On an HFD, heterozygous Mc4r-Cre mice with wild type BRS3 had increased body weight, adiposity, food intake, and metabolic efficiency demonstrating that the Mc4rCre allele produces a mild obesity phenotype (Figure S1), which was less prominent at 28 weeks of age (Figure S2A–E).
Previous experiments demonstrated that Brs3flox/y mice have a wild type phenotype and Brs3loxTB/y mice have the expected null phenotype [14]. Selective deletion of BRS3 in MC4R-expressing neurons produced an increase in body weight (+20%), adiposity, and food intake on an HFD (Figure 2, flox vs flox;Mc4r-Cre, red color). Re-expression of BRS3 only in Mc4r-Cre-expressing cells had no detectable effect on body weight (−1%) or adiposity (Figure 2, loxTB vs loxTB;Mc4r-Cre, blue color). At 29 weeks, compared to Brs3flox mice, the Brs3flox;Mc4r-Cre mice had increased weight (+14%) and adiposity, and liver, BAT, and iWAT masses, and decreased eWAT mass (Figure S3A–E). Deletion of BRS3 by Mc4r-Cre increased metabolic efficiency, while re-expression reduced it (Figure 2E). Thus BRS3 in MC4R-expressing neurons contributes to these adiposity phenotypes.
Figure 2.

Role of BRS3 in MC4R-expressing neurons on body weight and energy intake. Mice were singly housed and fed a high fat diet (HFD) starting at 8 weeks of age. (A) Body weight, (B) fat mass, (C) lean mass, (D) food intake, and (E) metabolic efficiency over 8–16 weeks of age. (F) P values by t-test for the littermate comparisons in the indicated panels and times. Data are mean ± SEM, N = 8–13/group.
3.2. BRS3 in MC4R-expressing neurons regulates insulin sensitivity and glucose metabolism
We investigated the role of BRS3 in MC4R-expressing neurons in glucose tolerance and insulin sensitivity. Mc4r-Cre by itself did not affect insulin sensitivity or glucose tolerance (Figure S4). BRS3 deletion in MC4R-expressing neurons impaired insulin tolerance and increased fed insulin levels and BRS3 re-expression reduced fed glucose (Figure 3A–D). While no difference in glucose tolerance or fasted glucose levels were seen (Figure 3F–H), BRS3 deletion increased fasted and glucose stimulated insulin levels (Figure 3I). These results suggest that BRS3 in MC4R-expressing neurons regulates insulin sensitivity and glucose metabolism, possibly both due to increased body weight and independent of it (Figure 3E, J).
Figure 3.

Effect of BRS3 manipulation in MC4R-expressing neurons on glucose homeostasis. (A) Insulin tolerance test (ITT) at 19 weeks of age (B) glucose area under the curve (AUC), (C) fed glucose, (D) fed insulin, and (E) body weight (BW) at the time of the test. (F) Glucose tolerance test (GTT) at 18 weeks of age (G) glucose area under the curve (AUC), (H) pre-test fasting glucose, (I) insulin at baseline (0 min) and 15 min after glucose dosing, and (J) body weight (BW) at the time of the test. Data are mean ± SEM, N = 8–13/group. P values were determined by t-test for the effect of Mc4r-Cre within each Brs3 genotype.
Neither Mc4r-Cre by itself (Figure S2F–J) nor BRS3 manipulation in MC4R-expressing neurons (Figure S3F–J) had a consistent, obvious effects on serum triglyceride, free fatty acid, cholesterol, adiponectin, or leptin levels.
3.3. BRS3 in SIM1-expressing neurons regulates body weight/adiposity and food intake
We next explored the contribution of BRS3 in SIM1-expressing neurons, which are predominantly in the paraventricular nucleus of the hypothalamus (PVH). There was a 90% reduction of Brs3 mRNA levels in PVH of the Brs3flox;Sim1-Cre mice. In the PVH of Brs3loxTB;Sim1-Cre mice, the Brs3 mRNA levels recovered to that of wild type mice (Figure 4).
Figure 4.
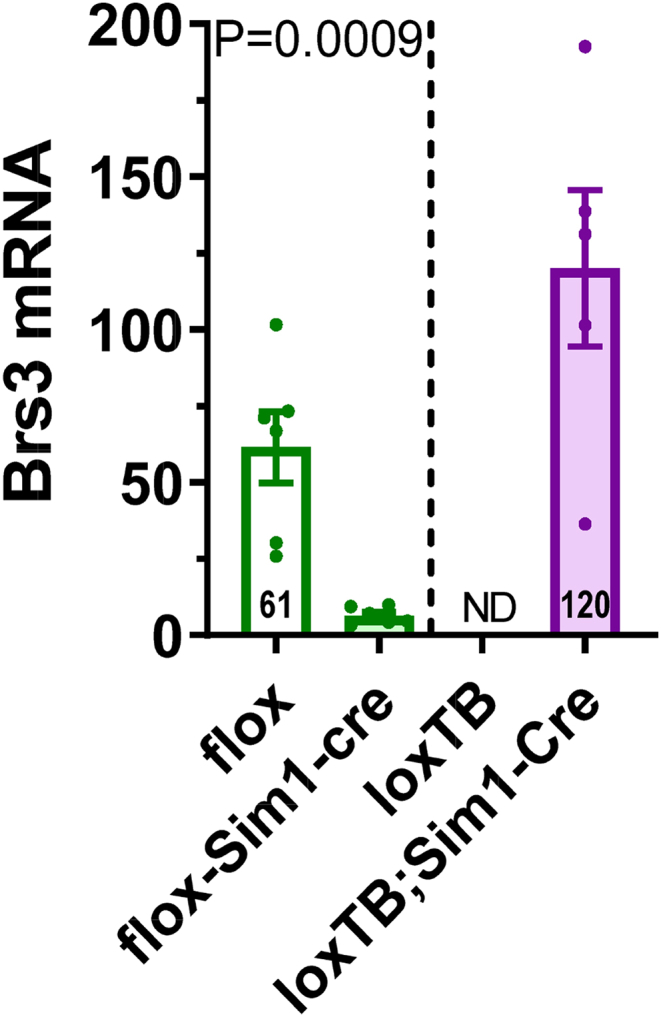
Brs3 expression in Sim1-Cre cohorts of mice.Brs3 mRNA was quantified by RT-PCR in micro-dissected paraventricular nuclei of hypothalamus (PVH). Expression is normalized to WT = 100. Data are mean ± SEM, N = 3–6/group. ND, not detected.
The Sim1-Cre allele itself does not affect body weight [18]. Mice with BRS3 deleted in SIM1-expressing neurons had increased body weight (+9%), fat mass, food intake, and metabolic efficiency (Figure 5, flox vs flox;Sim1-Cre, green color). Each of these changes was less than that seen in global null (Brs3loxTB/y) mice (body weight +24%) and each was partially and significantly reversed (body weight −12%) with selective re-expression of BRS3 in SIM1 neurons (Figure 5, loxTB vs loxTB;Sim1-Cre, purple color). At 45 weeks of age, the effect of BRS3 deletion in SIM1 neurons was no longer statistically significant (body weight +4%), although liver and eWAT weights were changed. Re-expression of BRS3 in SIM1 neurons reduced body (−9%), liver, and iWAT weight, and increased eWAT weight at 27 weeks of age (Figure S5A–E). BRS3 manipulation in SIM1-expressing neurons had no consistent, obvious effects on serum triglyceride, free fatty acid, cholesterol, adiponectin, or leptin levels (Figure S5F–J).
Figure 5.

Role of BRS3 in SIM1-expressing neurons on body weight and energy intake. Mice were singly housed and fed a high fat diet (HFD) starting at 8 weeks of age. (A) Body weight, (B) fat mass, (C) lean mass, (D) food intake, and (E) metabolic efficiency over 8–16 weeks of age. (F) P values by t-test for the littermate comparisons in the indicated panels and times. Data are mean ± SEM, N = 8–13/group.
3.4. BRS3 in SIM1-expressing neurons regulates insulin sensitivity
BRS3 deletion in SIM1-expressing neurons impaired insulin tolerance and increased insulin levels. BRS3 re-expression in only SIM1 neurons lowered insulin levels but did not have a statistically significant effect on insulin tolerance. Manipulation of BRS3 in SIM1 neurons had little effect on glucose levels or on glucose tolerance (Figure 6). These results suggest that BRS3 in SIM1-expressing neurons regulates insulin sensitivity, possibly with body weight effects contributing.
Figure 6.

Effect of BRS3 manipulation in SIM1-expressing neurons on glucose homeostasis. (A) Insulin tolerance test (ITT) at 19 weeks of age (B) glucose area under the curve (AUC), (C) fed glucose, (D) fed insulin, and (E) body weight (BW) at the time of the test. (F) Glucose tolerance test (GTT) at 18 weeks of age (G) glucose area under the curve, (H) pre-test fasting glucose, (I) insulin at baseline (0 min) and 15 min after glucose dosing, and (J) body weight (BW) at the time of the test. Data are mean ± SEM, N = 8–13/group. P values were determined by t-test for the effect of Sim1-Cre within each Brs3 genotype.
3.5. BRS3 in both MC4R and SIM1 neurons regulates baseline core body temperature
Brs3−/y mice have a slightly lower Tb during resting periods in the light phase, but their Tb is comparable to wild type mice during active intervals in the dark phase [11], resulting in an increased Tb range. This increased Tb range is robustly quantified as Tb span, the difference between the 95th and 5th Tb percentiles during 24-h periods [14]. Selective deletion of BRS3 in either MC4R- or SIM1-expressing neurons had little or no effect on mean light and dark phase Tb, but increased the Tb span, while BRS3 re-expression reduced it (Figure 7, Table S1). This result suggests that BRS3 in both MC4R- and SIM1-expressing neurons contributes to regulating Tb.
Figure 7.
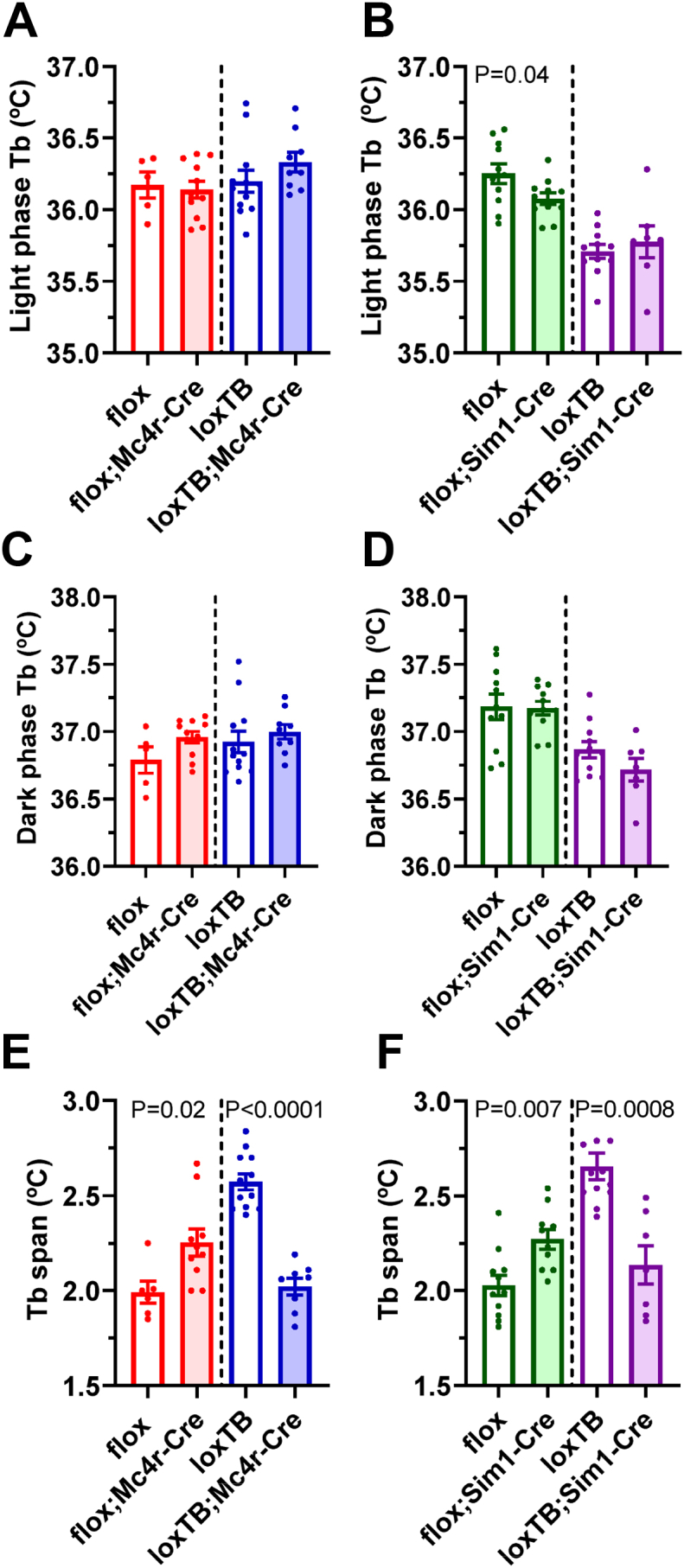
Effect of BRS3 in MC4R- and SIM1-expressing neurons on body temperature (Tb). Tb span, the difference between the 95th and 5th percentiles of daily Tb, was measured in 72 h of continuous telemetric Tb data from singly-housed, free-ranging mice in their home cage at 23–26 weeks of age. Ambient temperature was 22 °C and compared groups were studied simultaneously. (A, C & E) Effect of BRS3 in MC4R-expressing neurons. (B, D & F) Effect of BRS3 in SIM1-expressing neurons. Data are mean ± SEM, N = 8–13/group. P values were determined by t-test for the effect of Mc4r-Cre or Sim1-Cre within each Brs3 genotype.
3.6. Tb and food intake responses to BRS3 agonist MK-5046
Treatment with a BRS3 agonist increases Tb during the light phase by ∼0.5 °C [32]. This effect using the BRS3 agonist MK-5046 was intact in Brs3flox and Brs3flox;Mc4r-Cre mice, lost in Brs3loxTB, and recovered in Brs3loxTB;Mc4r-Cre mice (Figure 8A). Similarly, MK-5046 increased Tb in Brs3flox and probably in Brs3flox;Sim1-Cre mice, was lost in Brs3loxTB, and recovered in Brs3loxTB;Sim1-Cre mice (Figure 8B). Thus BRS3 in neurons expressing either MC4R or SIM1 is sufficient, but neither appears to be necessary for the Tb-raising effect of MK-5046.
Figure 8.

Response to BRS3 agonist after manipulation of BRS3 in MC4R- and SIM1-expressing neurons. (A–B) Tb response to BRS3 agonist MK-5046 (10 mg/kg, i.p.). (C–D) Food intake (FI) response to BRS3 agonist MK-5046 (10 mg/kg, i.p.). See Materials and Methods for experimental details. Data are mean ± SEM, N = 8–13/group. P values were determined by t-test for the effect of Mc4R-Cre or Sim1-Cre within each Brs3 genotype.
Treatment with BRS3 agonists inhibits food intake [13]. The inhibition of food intake by MK-5046 was intact in Brs3flox and lost in Brs3flox;Mc4r-Cre mice. It was lost in Brs3loxTB, but not clearly recovered in Brs3loxTB;Mc4r-Cre mice (Figure 8C). This suggests that BRS3 in MC4R-expressing neurons is necessary, and possibly sufficient for the food intake-suppressing effect of MK-5046. In both Brs3flox and Brs3flox;Sim1-Cre mice, the inhibition of food intake was intact. This effect was lost in Brs3loxTB, and probably recovered, in Brs3loxTB;Sim1-Cre mice (Figure 8D). These results suggest that BRS3 in SIM1 neurons is not necessary but is possibly sufficient for MK-5046-induced suppression of food intake.
4. Discussion
4.1. Manipulation of BRS3 in MC4R- or SIM1-expressing neurons
The loss of BRS3 from MC4R-expressing neurons reproduced much of the global BRS3 null phenotype for body weight/adiposity, insulin sensitivity, food intake, metabolic efficiency, and body temperature (Figure 9). However, BRS3 re-expression in MC4R neurons only modestly reversed some of these phenotypes, possibly due to confounding by the recombinase-independent obesity phenotype of the Mc4rCre allele. Similarly, the effect of BRS3 deletion in MC4R neurons may be exaggerated by the effects of the Mc4rCre allele (see 4.4).
Figure 9.

Effects of manipulating BRS3 in MC4R- and SIM1-expressing neurons. Summary of results. Food intake refers to the effect upon initiating a high fat diet.
The loss of BRS3 from SIM1-expressing neurons also produced features of the BRS3 global null, including body weight/adiposity, insulin sensitivity, food intake, metabolic efficiency, and body temperature. Unlike with Mc4r-Cre, the phenotypes showed reversal with BRS3 re-expression in Sim1-Cre-expressing neurons. Furthermore, the effects of loss or re-expression of BRS3 on body weight/adiposity and food intake are partial, suggesting that BRS3 in both SIM1+ and SIM1- neurons contributes.
4.2. BRS3 neuronal populations and food intake
The BRS3 [15] and SIM1 expression patterns in adult mice overlap principally in the medial amygdala (MeA) and PVH. During embryogenesis, SIM1 is expressed in two discrete regions of the diencephalon and mesencephalon and peripherally in the developing somites, mesonephric duct, and foregut [22,23]. In adult mice, both SIM1 and Sim1-Cre are strongly expressed in the paraventricular hypothalamic nucleus (PVH), nucleus of the lateral olfactory tract (NLOT), supraoptic nucleus (SON), posterior hypothalamic nuclei (PH), and MeA [18].
There are historical data that various lesions in the amygdala affect food intake [33]. Focusing on the MeA, optogenetic activation of inhibitory ARCAGRP neurons projecting to the MeA increased food intake, associated with reduced attention to territoriality [34]. The MeA may contribute to action prioritization, such as when eating competes with other behaviors [35].
A larger body of evidence demonstrates the major contribution of the PVH in regulating food intake [28]. Lesions that targeted the PVH increase food intake and body weight [36] and optogenetic activation of inhibitory ARCAGRP neurons projecting to the PVH stimulates food intake [37]. PVH neurons that inhibit food intake include those expressing SIM1 [37], MC4R [18,29,38], Nitric Oxide Synthase 1 (NOS1) [39], GLP1R [40], BRS3 [15], and prodynorphin (PDYN) [38], but not oxytocin, corticotrophin-releasing hormone, or vasopressin [39,40]. Activation of pituitary adenylate cyclase-activating polypeptide (PACAP) or thyrotropin releasing hormone (TRH) neurons in the PVH stimulates ARCAGRP neurons, thereby increasing food intake [41]. There is incomplete information about the co-expression of these markers. Most PVH neurons express SIM1, and recent work identified that ARCAGRP neurons project to non-overlapping subsets of PVH neurons (PVHMC4R and PVHPDYN) that inhibit food intake through separate pathways [38]. While the number of distinct PVH neuron types is not known, understanding this diversity will be advanced by single cell RNA profiling. Reduced food intake caused by MK-5046 in the Brs3flox;Sim1-Cre mice could also be due to BRS3 neurons in other regions, such as the parabrachial nucleus.
MC4R is expressed widely across the brain, including the cortex, thalamus, hypothalamus, brainstem, and spinal cord, specifically including the PVH and medial preoptic area [21,42,43]. Most nuclei expressing BRS3 also express MC4R but overlap at the neuronal level is not well studied. The PVHMC4R neurons that regulate feeding are glutamatergic and express SIM1 [44]. Since the BRS3 neurons regulating food intake are glutamatergic [14] and chemogenetic activation of PVHBRS3 neurons decreases food intake [15], it seems plausible that Mc4r-Cre-driven deletion or re-expression of BRS3 produces its food intake and obesity effects at least in part via neurons in the PVH that express both BRS3 and MC4R.
4.3. Energy expenditure, body temperature, and BRS3 neuronal populations
For singly-housed mice under typical conditions, 1/3 or more of total energy expenditure is dedicated to cold-induced thermogenesis, devoted to maintaining the Tb [45]. Under some circumstances (for example, light phase, resting state, cool ambient temperature, and single housing), body temperature can be a surrogate for metabolic rate in mice [46]. Thus neurons controlling Tb do so in part by regulating energy expenditure. The preoptic area (POA) is an important site for regulating body temperature and energy expenditure [[47], [48], [49], [50], [51], [52]], and BRS3 is expressed in many and MC4R is expressed in some POA subregions. Ablation of SIM1-expressing neurons reduced energy expenditure [53], but inactivation of the SIM1 gene itself did not change energy expenditure [54]. During development, Sim1-Cre is expressed in other hypothalamic nuclei, including the DMH/DHA, areas where BRS3 is also expressed.
BRS3 agonists regulate energy expenditure and Tb by sympathetic activation of brown adipose tissue [11,13,16,55]. Activation of DMHBRS3 (but not PVHBRS3) neurons increases energy expenditure and Tb [15]. However, the effects of BRS3 on energy expenditure are likely to be multifaceted, since preliminary observations in our laboratory suggest that BRS3-expressing neurons in other sites also contribute to Tb regulation.
The Tb span is increased in Brs3−/y mice due to a lower Tb during resting light phase periods combined with a normal Tb during dark phase active intervals [11]. The loss of BRS3 from either Mc4r-Cre- or Sim1-Cre-expressing neurons caused an increased Tb span, which was normalized by re-expression in either population. It is not known which brain nuclei contribute to this phenotype. Speculatively, BRS3 loss in MC4R and SIM1 neurons in the DMH/DHA could contribute to this phenotype.
Brs3−/y mice fail to increase their Tb upon treatment with a BRS3 agonist. This Tb increase was recovered when BRS3 was re-expressed in either Mc4r-Cre- or Sim1-Cre-expressing neurons. It is not clear how many populations of neurons account for these observations.
4.4. The Mc4rCre allele causes an obesity phenotype
The Mc4rCre allele produces lower Mc4r mRNA levels than a wild type Mc4r allele. Additionally, MC4R produced from Mc4rCre has a 17-amino acid C-terminal extension, which may reduce receptor function [15]. These effects presumably explain the mild obesity seen in Mc4rCre/+ mice fed an HFD. Thus, phenotypes in mice carrying a Mc4rCre allele may be due to reduced MC4R expression/function in addition to the desired gene expression changes caused by Cre-mediated recombination.
4.5. Study considerations
Potential limitations or confounders of this study include the following: 1) Since both Mc4r-Cre and Sim1-Cre express the recombinase constitutively, recombinase activity in progenitor cells could cause BRS3 deletion/re-expression in differentiated neurons that no longer express the driver Cre (for example, [56]; for a SIM1 example see [57] vs [58]). 2) Obesity causes insulin resistance, obscuring primary, adiposity-independent effects on insulin sensitivity. 3) While acute effects on Tb and energy expenditure can correlate [46], indirect calorimetry experiments were not performed. 4) When parsing attribution of phenotypes to multiple neuronal subpopulations, effect sizes become smaller, reducing statistical power.
4.6. Conclusions
BRS3 in both MC4R and SIM1 neurons regulates energy homeostasis. Prior experiments demonstrated that the relevant BRS3 neurons are glutamatergic (Vglut2+) and not GABAergic (Vgat+). While the hypomorph status of the Mc4rCre allele can confound phenotypes, we demonstrate that BRS3 in both MC4R- and SIM1-expressing neurons contributes to the regulation of body weight/adiposity, insulin sensitivity, food intake, and body temperature.
Acknowledgments
We thank Dr. Michael Krashes for helpful discussions and Yinyan Ma and Yuning Huang for assistance with experiments. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases [ZIA DK075057, ZIA DK075062; ZIA DK075063].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2020.02.012.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the supplementary data to this article:
Table S1. Body temperature (Tb) and physical activity.
Figure S1. Effect of heterozygous Mc4rCre on body weight and energy intake. Mice were singly housed and fed a high fat diet (HFD) starting at 8 weeks of age. (A) Body weight, (B) fat mass, (C) lean mass, (D) food intake, and (E) metabolic efficiency over 8–16 weeks of age. (F) P values were determined by t-test. Data are mean ± SEM, N = 9–13/group.
Figure S2. Anatomic parameters and serum analytes of heterozygous Mc4rCre mice. (A) Body, (B) liver, (C) brown adipose tissue (BAT), (D) inguinal white adipose tissue (iWAT), and (E) epididymal WAT (eWAT) were quantitated in WT and WT;Mc4r-Cre mice at 28 weeks of age. Serum samples were obtained from ad lib fed mice at 17 weeks of age for measurement of (F) triglycerides (TG), (G) non-esterified fatty acids (FFA), (H) cholesterol, (I) adiponectin, and (J) leptin. Data are mean ± SEM, N = 9–13/group. P values were determined by t-test.
Figure S3. Anatomic parameters and serum analytes of mice with manipulation of BRS3 in MC4R-expressing neurons. (A) Body, (B) liver, (C) brown adipose tissue (BAT), (D) inguinal white adipose tissue (iWAT), and (E) epididymal WAT (eWAT) were quantitated in Brs3flox and Brs3flox;Mc4r-Cre mice at 29 weeks of age, and Brs3loxTB and Brs3loxTB;Mc4r-Cre mice at 32 weeks of age. Serum samples were obtained from ad lib fed mice at 17 weeks of age for measurement of (F) triglycerides (TG), (G) non-esterified fatty acids (FFA), (H) cholesterol, (I) adiponectin, and (J) leptin. Data are mean ± SEM, N = 8–13/group. P values were determined by t-test for the effect of Mc4r-Cre within each Brs3 genotype.
Figure S4. Effect of heterozygous Mc4rCre on glucose homeostasis. (A) Insulin tolerance test (ITT) at 19 weeks of age (B) glucose area under the curve (AUC), (C) fed glucose, (D) fed insulin, and (E) body weight (BW) at the time of the test. (F) Glucose tolerance test (GTT) at 18 weeks of age (G) glucose area under the curve (AUC), (H) pre-test fasting glucose, (I) insulin at baseline (0 min) and 15 min after glucose dosing, and (J) body weight (BW) at the time of the test. Data are mean ± SEM, N = 9–13/group, with P values determined by t-test.
Figure S5. Anatomic parameters and serum analytes of mice with manipulation of BRS3 in SIM1-expressing neurons. (A) Body, (B) liver, (C) brown adipose tissue (BAT), (D) inguinal white adipose tissue (iWAT), and (E) epididymal WAT (eWAT) were quantitated in Brs3flox and Brs3flox;Sim1-Cre mice at 45 weeks of age, and Brs3loxTB and Brs3loxTB;Sim1-Cre mice at 27 weeks of age. Serum samples were obtained from ad lib fed mice at 17 weeks of age for measurement of (F) triglycerides (TG), (G) non-esterified fatty acids (FFA), (H) cholesterol, (I) adiponectin, and (J) leptin. Data are mean ± SEM, N = 8–13/group. P values were determined by t-test for the effect of Sim1-Cre within each Brs3 genotype.
References
- 1.Jensen R.T., Battey J.F., Spindel E.R., Benya R.V. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacological Reviews. 2008;60(1):1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Civelli O., Reinscheid R.K., Zhang Y., Wang Z., Fredriksson R., Schioth H.B. G protein-coupled receptor deorphanizations. Annual Review of Pharmacology and Toxicology. 2013;53:127–146. doi: 10.1146/annurev-pharmtox-010611-134548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang H., Shu C., Chen H., Zhang X., Zang Z., Deng C. Constitutively active BRS3 is a genuinely orphan GPCR in placental mammals. PLoS Biology. 2019;17(3) doi: 10.1371/journal.pbio.3000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lateef D.M., Xiao C., Reitman M.L. Search for an endogenous bombesin-like receptor 3 (BRS-3) ligand using parabiotic mice. PLoS One. 2015;10(11) doi: 10.1371/journal.pone.0142637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster S.R., Hauser A.S., Vedel L., Strachan R.T., Huang X.P., Gavin A.C. Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell. 2019;179(4):895–908. doi: 10.1016/j.cell.2019.10.010. e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mo C., Huang L., Cui L., Lv C., Lin D., Song L. Characterization of NMB, GRP and their receptors (BRS3, NMBR and GRPR) in chickens. Journal of Molecular Endocrinology. 2017;59(1):61–79. doi: 10.1530/JME-17-0020. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y., Liu Y., Wu L., Fan C., Wang Z., Zhang X. Receptor-specific crosstalk between prostanoid E receptor 3 and bombesin receptor subtype 3. FASEB J. 2018;32(6):3184–3192. doi: 10.1096/fj.201700337RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohki-Hamazaki H., Watase K., Yamamoto K., Ogura H., Yamano M., Yamada K. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature. 1997;390(6656):165–169. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- 9.Ladenheim E.E., Hamilton N.L., Behles R.R., Bi S., Hampton L.L., Battey J.F. Factors contributing to obesity in bombesin receptor subtype-3-deficient mice. Endocrinology. 2008;149(3):971–978. doi: 10.1210/en.2007-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brommage R., Desai U., Revelli J.P., Donoviel D.B., Fontenot G.K., Dacosta C.M. High-throughput screening of mouse knockout lines identifies true lean and obese phenotypes. Obesity. 2008;16(10):2362–2367. doi: 10.1038/oby.2008.361. [DOI] [PubMed] [Google Scholar]
- 11.Lateef D.M., Abreu-Vieira G., Xiao C., Reitman M.L. Regulation of body temperature and brown adipose tissue thermogenesis by bombesin receptor subtype-3. American Journal of Physiology. Endocrinology and Metabolism. 2014;306(6):E681–E687. doi: 10.1152/ajpendo.00615.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao C., Reitman M.L. Bombesin-like receptor 3: physiology of a functional orphan. Trends in Endocrinology and Metabolism. 2016;27(9):603–605. doi: 10.1016/j.tem.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan X.M., Chen H., Dobbelaar P.H., Dong Y., Fong T.M., Gagen K. Regulation of energy homeostasis by bombesin receptor subtype-3: selective receptor agonists for the treatment of obesity. Cell Metabolism. 2010;11(2):101–112. doi: 10.1016/j.cmet.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Xiao C., Pinol R.A., Carlin J.L., Li C., Deng C., Gavrilova O. Bombesin-like receptor 3 (Brs3) expression in glutamatergic, but not GABAergic, neurons is required for regulation of energy metabolism. Molecular Metabolism. 2017;6(11):1540–1550. doi: 10.1016/j.molmet.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinol R.A., Zahler S.H., Li C., Saha A., Tan B.K., Skop V. Brs3 neurons in the mouse dorsomedial hypothalamus regulate body temperature, energy expenditure, and heart rate, but not food intake. Nature Neuroscience. 2018;21(11):1530–1540. doi: 10.1038/s41593-018-0249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maruyama M., Hotta N., Nio Y., Hamagami K., Nagi T., Funata M. Bombesin receptor subtype-3-expressing neurons regulate energy homeostasis through a novel neuronal pathway in the hypothalamus. Brain and Behaviour. 2018;8(1) doi: 10.1002/brb3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huszar D., Lynch C.A., Fairchild-Huntress V., Dunmore J.H., Fang Q., Berkemeier L.R. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 18.Balthasar N., Dalgaard L.T., Lee C.E., Yu J., Funahashi H., Williams T. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 19.Sohn J.W., Harris L.E., Berglund E.D., Liu T., Vong L., Lowell B.B. Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons. Cell. 2013;152(3):612–619. doi: 10.1016/j.cell.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tao Y.X. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocrine Reviews. 2010;31(4):506–543. doi: 10.1210/er.2009-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mountjoy K.G., Mortrud M.T., Low M.J., Simerly R.B., Cone R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Molecular Endocrinology. 1994;8(10):1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 22.Ema M., Morita M., Ikawa S., Tanaka M., Matsuda Y., Gotoh O. Two new members of the murine Sim gene family are transcriptional repressors and show different expression patterns during mouse embryogenesis. Molecular and Cellular Biology. 1996;16(10):5865–5875. doi: 10.1128/mcb.16.10.5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan C.M., Kuwana E., Bulfone A., Fletcher C.F., Copeland N.G., Jenkins N.A. Expression patterns of two murine homologs of Drosophila single-minded suggest possible roles in embryonic patterning and in the pathogenesis of Down syndrome. Molecular and Cellular Neuroscience. 1996;7(1):1–16. doi: 10.1006/mcne.1996.0001. [DOI] [PubMed] [Google Scholar]
- 24.Michaud J.L., Rosenquist T., May N.R., Fan C.M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes & Development. 1998;12(20):3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michaud J.L., Boucher F., Melnyk A., Gauthier F., Goshu E., Levy E. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Human Molecular Genetics. 2001;10(14):1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 26.Ramachandrappa S., Raimondo A., Cali A.M., Keogh J.M., Henning E., Saeed S. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. Journal of Clinical Investigation. 2013;123(7):3042–3050. doi: 10.1172/JCI68016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnefond A., Raimondo A., Stutzmann F., Ghoussaini M., Ramachandrappa S., Bersten D.C. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. Journal of Clinical Investigation. 2013;123(7):3037–3041. doi: 10.1172/JCI68035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andermann M.L., Lowell B.B. Toward a wiring diagram understanding of appetite control. Neuron. 2017;95(4):757–778. doi: 10.1016/j.neuron.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garfield A.S., Li C., Madara J.C., Shah B.P., Webber E., Steger J.S. A neural basis for melanocortin-4 receptor-regulated appetite. Nature Neuroscience. 2015;18(6):863–871. doi: 10.1038/nn.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebhat I.K., Franklin C., Lo M.-C., Chen D., Jewell J.P., Miller R. Discovery of MK-5046, a potent, selective bombesin receptor subtype-3 agonist for the treatment of obesity. ACS Medicinal Chemistry Letters. 2011;2:43–47. doi: 10.1021/ml100196d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao C., Goldgof M., Gavrilova O., Reitman M.L. Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity (Silver Spring) 2015;23(7):1450–1459. doi: 10.1002/oby.21124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan X.M., Metzger J.M., Yang L., Raustad K.A., Wang S.P., Spann S.K. Antiobesity effect of MK-5046, a novel bombesin receptor subtype-3 agonist. Journal of Pharmacology and Experimental Therapeutics. 2011;336(2):356–364. doi: 10.1124/jpet.110.174763. [DOI] [PubMed] [Google Scholar]
- 33.Rogers Q.R., Leung P.M. The influence of amino acids on the neuroregulation of food intake. Federation Proceedings. 1973;32(6):1709–1719. [PubMed] [Google Scholar]
- 34.Padilla S.L., Qiu J., Soden M.E., Sanz E., Nestor C.C., Barker F.D. Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nature Neuroscience. 2016;19(5):734–741. doi: 10.1038/nn.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burnett C.J., Li C., Webber E., Tsaousidou E., Xue S.Y., Bruning J.C. Hunger-driven motivational state competition. Neuron. 2016;92(1):187–201. doi: 10.1016/j.neuron.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leibowitz S.F., Hammer N.J., Chang K. Hypothalamic paraventricular nucleus lesions produce overeating and obesity in the rat. Physiology & Behavior. 1981;27(6):1031–1040. doi: 10.1016/0031-9384(81)90366-8. [DOI] [PubMed] [Google Scholar]
- 37.Atasoy D., Betley J.N., Su H.H., Sternson S.M. Deconstruction of a neural circuit for hunger. Nature. 2012;488(7410):172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M.M., Madara J.C., Steger J.S., Krashes M.J., Balthasar N., Campbell J.N. The paraventricular hypothalamus regulates satiety and prevents obesity via two genetically distinct circuits. Neuron. 2019;102(3):653–667. doi: 10.1016/j.neuron.2019.02.028. e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutton A.K., Pei H., Burnett K.H., Myers M.G., Jr., Rhodes C.J., Olson D.P. Control of food intake and energy expenditure by Nos1 neurons of the paraventricular hypothalamus. Journal of Neuroscience. 2014;34(46):15306–15318. doi: 10.1523/JNEUROSCI.0226-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C., Navarrete J., Liang-Guallpa J., Lu C., Funderburk S.C., Chang R.B. Defined paraventricular hypothalamic populations exhibit differential responses to food contingent on caloric state. Cell Metabolism. 2019;29(3):681–694. doi: 10.1016/j.cmet.2018.10.016. e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krashes M.J., Shah B.P., Madara J.C., Olson D.P., Strochlic D.E., Garfield A.S. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014;507(7491):238–242. doi: 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kishi T., Aschkenasi C.J., Lee C.E., Mountjoy K.G., Saper C.B., Elmquist J.K. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. Journal of Comparative Neurology. 2003;457(3):213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 43.Liu H., Kishi T., Roseberry A.G., Cai X., Lee C.E., Montez J.M. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. Journal of Neuroscience. 2003;23(18):7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah B.P., Vong L., Olson D.P., Koda S., Krashes M.J., Ye C. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(36):13193–13198. doi: 10.1073/pnas.1407843111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abreu-Vieira G., Xiao C., Gavrilova O., Reitman M.L. Integration of body temperature into the analysis of energy expenditure in the mouse. Molecular Metabolism. 2015;4(6):461–470. doi: 10.1016/j.molmet.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metzger J.M., Gagen K., Raustad K.A., Yang L., White A., Wang S.P. Body temperature as a mouse pharmacodynamic response to bombesin receptor subtype-3 agonists and other potential obesity treatments. American Journal of Physiology. Endocrinology and Metabolism. 2010;299(5):E816–E824. doi: 10.1152/ajpendo.00404.2010. [DOI] [PubMed] [Google Scholar]
- 47.McKinley M.J., Yao S.T., Uschakov A., McAllen R.M., Rundgren M., Martelli D. The median preoptic nucleus: front and centre for the regulation of body fluid, sodium, temperature, sleep and cardiovascular homeostasis. Acta Physiologica (Oxford, England) 2015;214(1):8–32. doi: 10.1111/apha.12487. [DOI] [PubMed] [Google Scholar]
- 48.Yu S., Qualls-Creekmore E., Rezai-Zadeh K., Jiang Y., Berthoud H.R., Morrison C.D. Glutamatergic preoptic area neurons that express leptin receptors drive temperature-dependent body weight homeostasis. Journal of Neuroscience. 2016;36(18):5034–5046. doi: 10.1523/JNEUROSCI.0213-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song K., Wang H., Kamm G.B., Pohle J., Reis F.C., Heppenstall P. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science. 2016;353(6306):1393–1398. doi: 10.1126/science.aaf7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan C.L., Knight Z.A. Regulation of body temperature by the nervous system. Neuron. 2018;98(1):31–48. doi: 10.1016/j.neuron.2018.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Z.D., Yang W.Z., Gao C., Fu X., Zhang W., Zhou Q. A hypothalamic circuit that controls body temperature. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(8):2042–2047. doi: 10.1073/pnas.1616255114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morrison S.F., Nakamura K. Central mechanisms for thermoregulation. Annual Review of Physiology. 2019 doi: 10.1146/annurev-physiol-020518-114546. [DOI] [PubMed] [Google Scholar]
- 53.Xi D., Gandhi N., Lai M., Kublaoui B.M. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0036453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tolson K.P., Gemelli T., Meyer D., Yazdani U., Kozlitina J., Zinn A.R. Inducible neuronal inactivation of Sim1 in adult mice causes hyperphagic obesity. Endocrinology. 2014;155(7):2436–2444. doi: 10.1210/en.2013-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nio Y., Hotta N., Maruyama M., Hamagami K., Nagi T., Funata M. A selective bombesin receptor subtype 3 agonist promotes weight loss in male diet-induced-obese rats with circadian rhythm change. Endocrinology. 2017;158(5):1298–1313. doi: 10.1210/en.2016-1825. [DOI] [PubMed] [Google Scholar]
- 56.Padilla S.L., Reef D., Zeltser L.M. Defining POMC neurons using transgenic reagents: impact of transient Pomc expression in diverse immature neuronal populations. Endocrinology. 2012;153(3):1219–1231. doi: 10.1210/en.2011-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghosal S., Packard A.E.B., Mahbod P., McKlveen J.M., Seeley R.J., Myers B. Disruption of glucagon-like peptide 1 signaling in Sim1 neurons reduces physiological and behavioral reactivity to acute and chronic stress. Journal of Neuroscience. 2017;37(1):184–193. doi: 10.1523/JNEUROSCI.1104-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J., Conde K., Zhang P., Lilascharoen V., Xu Z., Lim B.K. Enhanced AMPA receptor trafficking mediates the anorexigenic effect of endogenous glucagon-like peptide-1 in the paraventricular hypothalamus. Neuron. 2017;96(4):897–909. doi: 10.1016/j.neuron.2017.09.042. e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.