

Abstract
Background
Reversible ε‐amino acetylation of lysine residues regulates transcription as well as metabolic flux; however, roles for specific lysine acetyltransferases in skeletal muscle physiology and function are unknown. In this study, we investigated the role of the related acetyltransferases p300 and cAMP response element‐binding protein‐binding protein (CBP) in skeletal muscle transcriptional homeostasis and physiology in adult mice.
Methods
Mice with skeletal muscle‐specific and inducible knockout of p300 and CBP (PCKO) were generated by crossing mice with a tamoxifen‐inducible Cre recombinase expressed under the human α‐skeletal actin promoter with mice having LoxP sites flanking exon 9 of the Ep300 and Crebbp genes. Knockout of PCKO was induced at 13–15 weeks of age via oral gavage of tamoxifen for 5 days to both PCKO and littermate control [wildtype (WT)] mice. Body composition, food intake, and muscle function were assessed on day 0 (D0) through 5 (D5). Microarray and tandem mass tag mass spectrometry analyses were performed to assess global RNA and protein levels in skeletal muscle of PCKO and WT mice.
Results
At D5 after initiating tamoxifen treatment, there was a reduction in body weight (−15%), food intake (−78%), stride length (−46%), and grip strength (−45%) in PCKO compared with WT mice. Additionally, ex vivo contractile function [tetanic tension (kPa)] was severely impaired in PCKO vs. WT mice at D3 (~70–80% lower) and D5 (~80–95% lower) and resulted in lethality within 1 week—a phenotype that is reversed by the presence of a single allele of either p300 or CBP. The impaired muscle function in PCKO mice was paralleled by substantial transcriptional alterations (3310 genes; false discovery rate < 0.1), especially in gene networks central to muscle contraction and structural integrity. This transcriptional uncoupling was accompanied by changes in protein expression patterns indicative of impaired muscle function, albeit to a smaller magnitude (446 proteins; fold‐change > 1.25; false discovery rate < 0.1).
Conclusions
These data reveal that p300 and CBP are required for the control and maintenance of contractile function and transcriptional homeostasis in skeletal muscle and, ultimately, organism survival. By extension, modulating p300/CBP function may hold promise for the treatment of disorders characterized by impaired contractile function in humans.
Keywords: Acetylation, Acetyltransferases, Transcriptomics, Proteomics, Muscle contraction
Introduction
The reversible acetylation of lysine ε‐amino groups is a prevalent, highly conserved post‐translational modification that regulates diverse cell physiological processes, including transcription, cell cycle progression, and energy metabolism.1, 2, 3, 4, 5 Recent studies of complete acetyl‐lysine proteomes reveal that numerous proteins involved in substrate metabolism, mitochondrial electron transport, and muscle contraction are acetylated in skeletal muscle,6 suggesting central although enigmatic roles for lysine acetylation in skeletal muscle function.4 In general, protein lysine acetylation is controlled by balance between the addition of acetyl groups from acetyl coenzyme A to a lysine residue by lysine acetyltransferases and their removal by lysine deacetylases.2, 3, 4, 5 While a large body of work has elucidated roles for deacetylases in skeletal muscle regulation,7, 8, 9, 10, 11 contributions of acetyltransferases to skeletal muscle physiology and function remain to be determined.
The E1A binding protein p300 (p300; also known as KAT3B) and cAMP response element‐binding protein‐binding protein (CBP; also known as KAT3A) are related lysine acetyltransferases,12, 13 sharing an extensive network of over 400 interaction partners.12 Several p300 and CBP binding proteins are key transcriptional regulators that ubiquitously recruit p300/CBP to enhancer and promoter regions throughout the genome,9, 14 where p300 and CBP mediate transcription factor recruitment and acetylation of histone proteins.12, 13, 15 p300/CBP have critical roles in transcriptional regulation—particularly during development—as evident by embryonic lethality of whole‐animal knockout (KO) of p300 or CBP or combined heterozygous KO of p300 and CBP.16, 17 In addition to numerous p300/CBP acetylation substrates associated with transcriptional regulation in the nucleus,12, 13, 15 several proteins in the cytosol have also been identified as targets of p300/CBP, suggesting further complexity and importance of p300/CBP in diverse cellular functions.
Studies of skeletal muscle cells describe a central role for p300 in transcriptional regulation of myotube differentiation and muscle integrity in vitro.18, 19, 20 However, we recently found that in vivo, muscle‐specific deletions of p300 in mice do not affect skeletal muscle development, morphology, contractile function, or exercise capacity.21 This lack of impact of loss of p300 may stem from compensation by CBP, a phenomenon that is often described in other cell types but has not yet been demonstrated for p300 and CBP in muscle.22, 23, 24 Here, to better delineate the contributions of p300 and CBP to muscle physiology in vivo, we generated mouse models with inducible skeletal muscle‐specific double KO of p300 and CBP (PCKO)—as well as PCKO mice heterozygous for either p300 (PZ) or CBP (CZ) that allow for single allele studies. We find that the combined loss of p300 and CBP in adult skeletal muscle rapidly alters gene expression patterns fundamental to skeletal muscle function, in parallel with loss of contractile function and, ultimately, lethality within 1 week. Remarkably, muscle weakness and lethality phenotypes were prevented by the presence of one allele of either p300 or CBP. Together, our work demonstrates critical, cooperative roles for p300 and CBP in mediating transcriptional homeostasis in skeletal muscle physiology, in a manner necessary for organism survival.
Materials and methods
Animals
Inducible, skeletal muscle‐specific Ep300 and Crebbp double‐knockout (PCKO) mice were generated by crossing mice with a tamoxifen (TMX)‐inducible Cre recombinase expressed under the human α‐skeletal actin promoter25 with mice harbouring LoxP sites flanking exon 9 of the Ep300 gene22 and exon 9 of the Crebbp gene.26 TMX (2 mg) was administered via oral gavage to all mice for five consecutive days, starting at 13–15 weeks of age. At either 1, 3, or 5 days after TMX start, tissues were excised from fasted (4 h) and anaesthetized animals, snap frozen in liquid nitrogen, and stored at −80°C for subsequent analysis. Mice were kept in a conventional facility with a 12 h light/12 h dark cycle and had free access to food and water unless otherwise noted. All animal experiments were approved by, and conducted in accordance with, the Animal Care Program at the University of California, San Diego.
Food intake and body composition
Food intake was measured in ad libitum fed, single‐housed mice. Food weight and mouse body weight was recorded at 12 p.m., every day during a 1 week period. Body composition was measured using an EchoMRI‐100TM analyser (EchoMRI Medical Systems, Houston, TX, USA) 1 day before TMX and at 3 and 5 days post‐TMX start.
Gait analysis
Gait testing was performed using a TreadScan (CleverSys, Reston, VA, USA) digital analysis system. Mice walked on a transparent treadmill belt kept at a speed of 5 cm/s and were imaged from below using a high‐speed digital camera. A total of five videos, 800 frames per video, were collected per day for each mouse and videos were analysed using TreadScan software so as to get the following gait characteristics: stance time [milliseconds (ms) that the is paw on the belt], swing time (ms that the paw is in the air), and total stride length [millimeters (mm)]. The tester was blinded to the genotype of the mice.
Ex vivo contractile testing
Muscle contractile function and mechanical properties were assessed in the fifth‐toe extensor digitorum long (EDL) muscle as previously described.27 Briefly, muscles were isolated and incubated in Ringer solution at 25°C, and muscle sarcomere length was normalized using laser diffraction.28 All soleus experiments were performed at optimal muscle length. Supramaximal stimulation conditions were established for each experiment by single 0.5 ms twitch pulses of increasing current, using +50% more than the value where the force plateaued for experimental testing. Muscles were electrically stimulated (model S88; Astro‐Med, West Warwick, RI, USA) via parallel platinum electrodes (300 ms train duration and 0.5 ms pulse duration) to assess twitch characteristics (twitch tension, half‐relaxation time, and time‐to‐peak tension). After a 10 min rest, muscles were stimulated at increasing frequencies (1–120 Hz) with 120 s intervals between contractions to determine the force–frequency relationship. From this, fusion frequency was calculated as the lowest frequency where oscillations in force are no longer evident. For all analysis, force was normalized to the muscle physiological cross‐sectional area.29 For each day (D1, D3, and D5), the appropriate wildtype (WT) control mouse was analysed alongside the PCKO mice. For force–frequency curves, there was no significant differences between WT mice at the different timepoints, and the WT data were collapsed.
Statistics
For data other than microarray and proteomics analysis (see respective sections for a detailed description of statistical analysis), an unpaired Student's t‐test, one‐way or two‐way analysis of variance (using repeated measurement where appropriate), followed by either Bonferroni's or Tukey's post hoc test, was used. Significance was set at P < 0.05. Statistical analyses were performed using Prism 6 (GraphPad Software Incorporated, La Jolla, CA, USA). All data are expressed as mean ± SEM.
Additional materials and methods may be found online in the Supporting Information, Data S1.
Results
Knockout of both p300 and cAMP response element‐binding protein‐binding protein in adult mouse skeletal muscle
To generate PCKO mice, we crossed mice carrying a TMX‐inducible Cre recombinase (Cre) expressed under the human α‐skeletal actin promoter25 with mice harbouring LoxP sites flanking exon 9 of the Ep300 gene22 and exon 9 of the Crebbp gene.26 To activate Cre recombinase in skeletal muscle and initiate PCKO in adult mice, 13‐ to 15‐week‐old floxed and Cre negative [i.e. ‘wildtype’/control (WT)] and PCKO mice were administered TMX orally for five consecutive days (D0–D4), where D1 is considered the first day after starting TMX (Figure 1A). Protein abundance of p300 and CBP was robustly reduced in skeletal muscle of PCKO vs. WT mice at D1, D3, and D5 (p300: −60%, −69%, and −68%; CBP: −70%, −78%, and −80%, respectively) (Figure 1B). There was no difference in body weight between WT and PCKO mice between D0 and D3; however, at D4 and D5, PCKO mice had lost ~6% and ~14% of their starting body weight, respectively (Figure 1C). PCKO mice had a ~75% reduction in overall fat mass and ~10% reduction in lean mass at D5 compared with D0 (Figure 1D). Body water content was not altered over time for either WT or PCKO mice but was slightly higher in the PCKO group during all timepoints (Figure 1E). Epididymal fat pad weight was ~70% lower and liver weight trended (P = 0.057) to be lower in PCKO vs. WT mice at D5 (Table 1). Heart and skeletal muscle weights were comparable between WT and PCKO mice at D1, D3, and D5 (Table 1), and there was no difference in tibialis anterior protein concentration in PCKO vs. WT mice at D5 (Table 1). At the D4 and D5 timepoints, PCKO mice consumed less food over the previous 24 h compared with WT mice (Figure 1F), which was likely a major contributing factor for the reduced body and fat mass in PCKO mice at D5 (Figure 1C and 1D).
Figure 1.
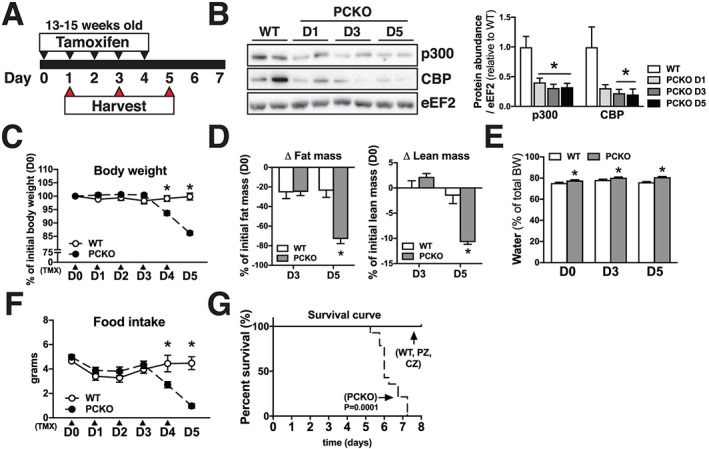
Knockout of p300 and CBP in PCKO mice is lethal. (A) Schematic representation. Tamoxifen (TMX) was administered to the mice via gavage at day 0 (D0) through D4. Tissues were harvested at D1, D3, and D5. (B) Representative blots of p300 and CBP protein abundance in gastrocnemius. Bar graphs represent quantification of p300 and CBP protein abundance in WT and PCKO mice (n = 6 WT, n = 6 D1 PCKO, n = 6 D3 PCKO, and n = 6 D5 PCKO), normalized to eEF2. (C) Body weight expressed as % of initial weight of WT and PCKO mice at D0 (n = 7 WT and n = 9 PCKO). (D) Loss of fat and lean mass (%) at D3 and D5 in PCKO and WT mice compared with D0 (n = 7 WT and n = 9 PCKO). (E) Water content as % of body weight (n = 7 WT and n = 9 PCKO). (F) Food intake of WT and PCKO over the previous 24 h at D0–D5 (n = 7 WT and n = 9 PCKO). (G) WT and PCKO mice survival over time, from the start of TMX administration until D8 (n = 6 WT, n = 14 PCKO, n = 8 PZ, and n = 8 CZ). Statistics: Data reported as means ± SEM. (B) One‐way analysis of variance (ANOVA), Bonferroni post hoc test, * P < 0.05, PCKO vs. WT. (C–F) Two‐way ANOVA, repeated measures, Bonferroni post hoc test, * P < 0.05, PCKO vs. WT within each day; (G) Log‐rank (Mantel–Cox) test.
Table 1.
Body and tissue weights of WT and PCKO mice at days 1, 3, and 5
| Day 1 | Day 3 | Day 5 | ||||
|---|---|---|---|---|---|---|
| WT | PCKO | WT | PCKO | WT | PCKO | |
| BW (g) | 28.7 ± 1.7 (a) | 29.2 ± 1.7 (a) | 28.4 ± 2.0 (a) | 28.3 ± 2.7 (a) | 28.5 ± 1.7 (a) | 24.2 ± 2.6 (b) |
| GA (mg) | 136.9 ± 10.6 (a) | 138.1 ± 10.6 (a) | 137.8 ± 12.4 (a) | 138.3 ± 13.0 (a) | 127.4 ± 9.9 (a) | 127.5 ± 10.6 (a) |
| TA (mg) | 51.5 ± 3.5 (a) | 51.9 ± 5.1 (a) | 53.0 ± 4.4 (a) | 49.7 ± 8.3 (a) | 48.5 ± 4.4 (a) | 48.2 ± 4.8 (a) |
| TA protein conc. (mg/g muscle) | N/A | N/A | N/A | N/A | 144.4 ± 30.9 (a) | 143.7 ± 20.9 (a) |
| QUAD (mg) | 199.6 ± 10.1 (a) | 206.8 ± 17.8 (a) | 218.8 ± 17.9 (a) | 224.6 ± 19.7 (a) | 195.9 ± 20.6 (a) | 193.9 ± 23.1 (a) |
| Heart (mg) | 135.1 ± 21.5 (a) | 133.0 ± 10.7 (a) | 131.2 ± 16.7 (a) | 121.2 ± 10.2 (a) | 126.9 ± 10.7 (a) | 122.8 ± 16.4 (a) |
| Liver (mg) | 1497.9 ± 65.7 (a) | 1498.6 ± 98.2 (a) | 1344.3 ± 88.8 (a,b) | 1377.8 ± 164.9 (a) | 1345.8 ± 89.2 (a,b) | 1155.3 ± 136.7 (b) |
| eWAT (mg) | 394.6 ± 153.1 (a) | 418.5 ± 109.1 (a) | 350.9 ± 24.6 (a) | 379.9 ± 110.7 (a) | 270.8 ± 37.7 (a) | 86.3 ± 42.5 (b) |
BW, body weight; eWAT, epididymal white adipose tissue (day 1/3/5; n = 7/5/7 WT and 7/5/9 PCKO, respectively); GA, gastrocnemius; QUAD, quadriceps; TA, tibialis anterior. Total protein concentration normalized to muscle wet weight (day 5; n = 6 WT and n = 6 PCKO). Statistics: Data reported as means ± SEM. Within rows, means followed by the same letter, are not significantly different (Student's t‐test or two‐way analysis of variance, Tukey's post hoc test, P < 0.05).
Skeletal muscle p300 and cAMP response element‐binding protein‐binding protein are fundamental to survival
Remarkably, 6–7 days after initiating TMX treatment in PCKO and WT mice, all PCKO mice either died spontaneously or were considered to be in such a moribund state that they were euthanized (Figure 1G). Equally remarkable, the lethality phenotype in PCKO mice was reversed in PZ and CZ mice (i.e. mice with one functional allele of p300 or CBP, respectively), which survived for at least 4 weeks (longest time these mice were observed) after initiating TMX treatment (Figure 1G). In light of the lethality phenotype in PCKO mice, all further experiments were conducted in mice at D5 or earlier.
Compromised mobility and neuromuscular function of p300 and cAMP response element‐binding protein‐binding protein knockout mice
A striking phenotype of PCKO mice is progressive muscle weakness, such that they are essentially unable to move their hind limbs and have a limited capacity to use their fore limbs to move around the cage. To quantify this phenomenon, we performed gait analysis in WT and PCKO mice at D3, D4, and D5. PCKO mice demonstrated an altered movement pattern, with longer stance time at D5, shorter swing time at D4 and D5, and shorter stride length at D5 compared with WT mice (Figure 2A). Furthermore, neuromuscular function, as assessed by rotarod, was severely impaired in PCKO vs. WT mice at D5 (Figure 2B), grip strength was significantly reduced in PCKO mice vs. WT mice at D4 and D5 (Figure 2C), and the ability of PCKO mice to stay on the grid during an inverted screen test was abrogated at D4 (Figure 2D). Altogether, these findings demonstrate that PCKO mice develop a rapid, general loss of mobility and neuromuscular function in the days after initiating knockout of p300 and CBP.
Figure 2.
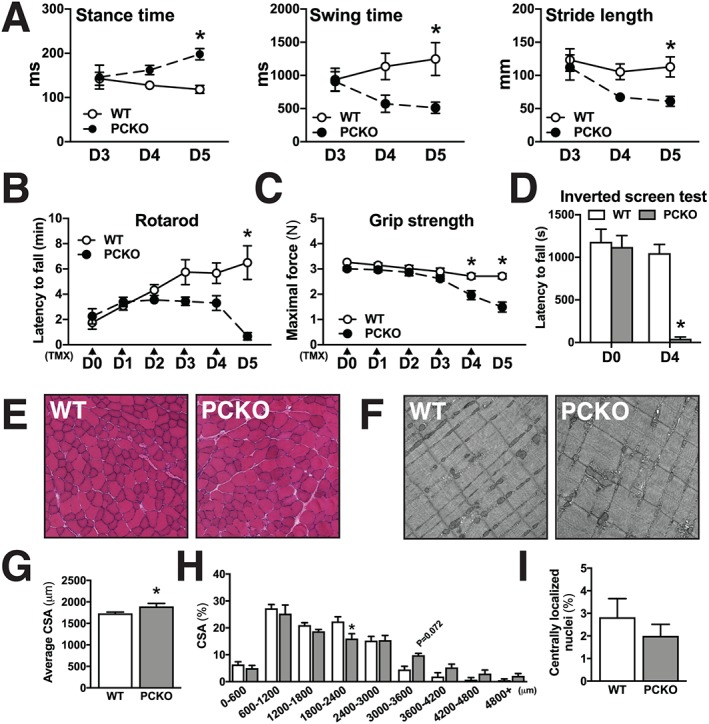
Muscle weakness, but minor changes in muscle histology in PCKO mice. (A) Stance time, swing time, and stride length of WT and PCKO mice as analysed by treadmill gait analysis at day (D)3, D4, and D5 (n = 9 WT and n = 10 PCKO). (B–D) Functional tests in WT and PCKO mice. (B) Rotarod test at D0–D5 (n = 12 WT and n = 9 PCKO), (C) grip strength test at D0–D5 (n = 7 WT and n = 9 PCKO), and (D) inverted screen test at D0 and D4 (n = 7 WT and n = 9 PCKO). (E) Representative pictures of haematoxylin & eosin‐stained tibialis anterior (TA) muscle from WT and PCKO mice. (F) Representative electron micrographs (×5000 magnification) of extensor digitorum longus (EDL) muscle from WT and PCKO mice (n = 2 WT and n = 2 PCKO). (G) Average cross‐sectional area (CSA, μm2) and (H) distribution of fibre CSAs (μm2) presented as per cent (%) fibres within each size bin (n = 8 WT and n = 7 PCKO). (I) Centrally localized nuclei in TA, presented as % fibres with centrally localized nuclei from WT and PCKO mice (n = 4 WT and n = 4 PCKO). Statistics: Data reported as means ± SEM. (A) Two‐way analysis of variance (ANOVA), Bonferroni post hoc test, * P < 0.05, PCKO vs. WT. (B–D) Two‐way ANOVA, repeated measures, Bonferroni post hoc test, * P < 0.05, PCKO vs. WT. (G and I) Student's t‐test, * P < 0.05, PCKO vs. WT. (H) Two‐way ANOVA, Bonferroni post hoc test, * P < 0.05, PCKO vs. WT. TMX, tamoxifen.
No histological abnormalities in skeletal muscle of p300 and cAMP response element‐binding protein‐binding protein knockout mice
To gain insight into how functional impairments develop in PCKO animals, we performed histological assessments of skeletal muscle from WT and PCKO mice. Tibialis anterior and diaphragm histology in D5 PCKO mice did not differ from WT mice (Figure 2E and Supporting Information, Figure S1A). Electron micrographs of EDL show normal muscle ultrastructure in D5 PCKO vs. WT mice (Figure 2F and Supporting Information, Figure S1B). Average fibre cross‐sectional area was higher in PCKO vs. WT mice (~9%) (Figure 2G and Supporting Information, Figure S1C). Specifically, there was a 28% reduction in the number of medium‐sized muscle fibres (1800–2400 μm) in PCKO vs. WT mice and a trend towards more large muscle fibres (3000+ μm) (Figure 2H and Supporting Information, Figure S1C). There was no difference in the number of centrally nucleated fibres in PCKO vs. WT mice (Figure 2I), which is a proxy for muscle damage and regeneration. To assess oxidative capacity in PCKO skeletal muscle, we performed a succinate dehydrogenase staining in tibialis anterior and diaphragm muscles; there were no genotype differences in oxidative fibre type distribution in either muscle type (Supporting Information, Figure S1D–E).
Skeletal muscle contractile function is severely impaired in p300 and cAMP response element‐binding protein‐binding protein knockout mice
Given the loss of neuromuscular coordination and strength in PCKO mice, we next assessed muscle contractile function. Ex vivo contractility measurements in the EDL at D1 showed comparable maximum twitch (Figure 3A) and tetanic (Figure 3B and 3C) tension between genotypes. Remarkably, at D3 and D5, maximum twitch (Figure 3A) and tetanic (Figure 3B and 3C) tension in PCKO mice were reduced by ~80–95%. At D3 and D5, PCKO muscles generated a fused tetanic contraction at ~50–100% higher stimulation frequency compared with WT mice (Figure 3D). Importantly, these differences in muscle function were not due to an inability to respond to electrical stimulation; when expressing tetanic tension at a given stimulation frequency relative to maximum tetanic tension, PCKO mice show increased force with higher frequency of stimulation (Figure 3E), demonstrating that muscle fibres are activated in PCKO mice. Because the lethality phenotype of PCKO mice was reversed in PZ and CZ mice, we assessed if contractile function was normalized as well. Strikingly, ~93% of twitch (Figure 3F) and ~90% of max tetanic (Figure 3G) tension was retained in PZ vs. PCKO mice at D5. Similarly, ~73% of twitch (Figure 3F) and ~86% of tetanic (Figure 3G) tension was maintained in CZ vs. PCKO mice at D5. The reduced contractile function of PCKO mice was also evident in soleus, with maximum twitch (Figure 3H) and tetanic (Figure 3I and 3J) tension reduced by ~80–95%, which indicates that the effect of loss of p300 and CBP on muscle function is not fibre‐type dependent. Overall, these results reveal the absolute requirement of p300 and CBP in skeletal muscle for contractile function and that one allele of CBP or p300 is sufficient for normal skeletal muscle force production.
Figure 3.
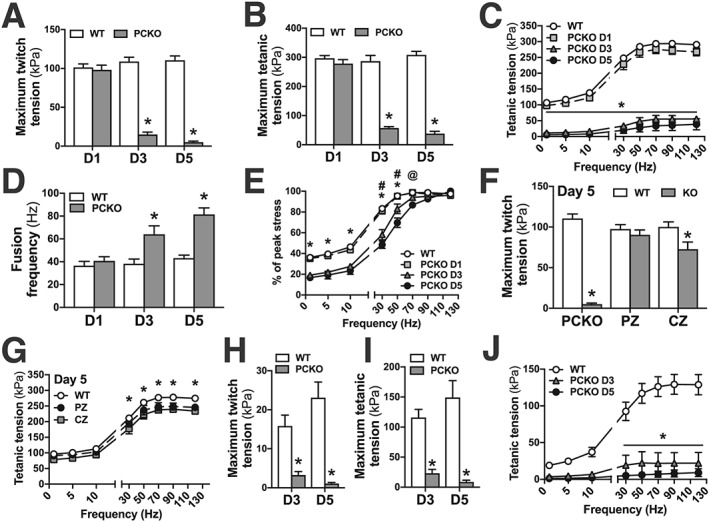
Abrogated force production at D3 and D5 in PCKO mice. Ex vivo twitch and force–frequency (F‐F) measurements in extensor digitorum longus (EDL) fifth‐toe muscle and soleus from mixed male and female PCKO, PZ, CZ, and WT mice. EDL: (A) maximum twitch tension (D1/D3/D5; n = 8/6/5 WT and 7/9/6 PCKO, respectively) and (B) maximum tetanic tension (D1/D3/D5; n = 6/5/5 WT and 6/7/6 PCKO, respectively). (C) F‐F curves (n = 16 WT, n = 6 D1 PCKO, n = 7 D3 PCKO, and n = 6 D5 PCKO). (D) Fusion frequency (D1/D3/D5; n = 6/5/5 WT and 6/7/6 PCKO, respectively). (E) F‐F curves normalized to max force (n = 16 WT, n = 6 D1 PCKO, n = 7 D3 PCKO, and n = 6 D5 PCKO). (F) Maximum twitch tension (WT/PCKO, n = 5/6; WT/PZ, n = 10/6; and WT/CZ, n = 7/8). (G) F‐F curves (n = 13 WT, n = 5 PZ, and n = 7 CZ). Soleus: (H) maximum twitch tension (D3/D5; n = 6/4 WT and n = 6/5 PCKO, respectively) and (I) maximum tetanic tension (D3/D5; n = 6/4 WT and 6/5 PCKO, respectively). (J) F‐F curves (n = 10 WT, n = 6 D3 PCKO, and n = 5 D5 PCKO). Statistics: Data reported as means ± SEM. (A, B, D, H, and I) Two‐way analysis of variance (ANOVA), Bonferroni post hoc test, * P < 0.05, PCKO vs. WT. (C) Two‐way ANOVA, repeated measures, Tukey's post hoc test, * P < 0.05, PCKO D3 and D5 vs. WT and PCKO D1, respectively. (E and J) Two‐way ANOVA, Tukey's post hoc test, repeated measures, * P < 0.05, PCKO D3 and D5 vs. WT and PCKO D1, respectively; # P < 0.05, PCKO D3 vs. PCKO D5; @ P < 0.05, D5 PCKO vs. WT and PCKO D1, respectively. (F) Two‐way ANOVA, Bonferroni post hoc test, * P < 0.05, PCKO/PZ/CZ vs. respective WT littermate group. (G) Two‐way ANOVA, Tukey's post hoc test, repeated measures, * P < 0.05, CZ vs. WT.
Transcriptional changes in skeletal muscle of k p300 and cAMP response element‐binding protein‐binding protein knockout mice
For insight into why PCKO mice develop severe impairments in muscle contractile function, we performed a microarray analysis in the EDL (i.e. the muscle used for measuring ex vivo contractility) of D5 WT and PCKO mice. Within 5 days of initiating TMX treatment, 3310 genes (1763 up‐regulated and 1547 down‐regulated) were differentially expressed (false discovery rate < 0.1) in PCKO vs. WT muscle (Figure 4A and Supporting Information, Table S1). This rapid and extensive disruption of transcriptional patterns suggest that p300 and CBP are important for transcriptional homeostasis in skeletal muscle. In line with our findings of impaired contractile function in PCKO mice, gene ontology (GO) enrichment revealed that the differentially expressed genes were predominately enriched for processes central to skeletal muscle contraction, muscle development, and carbohydrate metabolism [Figure 4B and Supporting Information, Table S2 (GO:BP)]. In line with a robust ‘muscle contraction’ signature within the differentially expressed genes, there was strong enrichment in myocellular compartments central to skeletal muscle contractile function, including sarcoplasmic reticulum, sarcomere, striated muscle thin filaments, and ion channel complexes [Figure 4C and Supporting Information, Table S2 (GO:CC)], with most genes in these categories (~48–100%) being down‐regulated in PCKO vs. WT muscles (Figure 4D). Together, these data indicate that p300/CBP control the transcription of genes associated with central aspects of skeletal muscle contraction, such as action potential potentiation, calcium release/reuptake, the troponin/tropomyosin complex, and actin/myosin kinetics. Importantly, for genes down‐regulated in PCKO vs. WT muscle, there was an enrichment for motifs predicted to be bound by transcription factors central to skeletal muscle development and homeostasis [e.g. myocyte enhancer factor 2A (MEF2A) and myogenic differentiation 1 (MYOD1)] (Figure 4E). To validate the microarray findings, we performed qPCR on a subset of genes specific to contractile function (i.e. sarcoplasmic reticulum/Ca2+, troponin/tropomyosin complex, actin/myosin kinetics, and cytoskeleton/extracellular matrix) that were down‐regulated/up‐regulated in the microarray data, in two additional skeletal muscle depots, quadriceps (Figure 4F) and gastrocnemius (Figure 4G). As part of this, we assessed temporal changes in gene expression at D1, D3, and D5 in PCKO and WT mice. In line with reduced contractile function in D3 PCKO mice, genes associated with muscle function were down‐regulated >50% in PCKO muscle by D3 and, in some cases, reduced already by D1 (Figure 4G), which implies that these transcriptional changes parallel the contractile impairments in PCKO mice.
Figure 4.
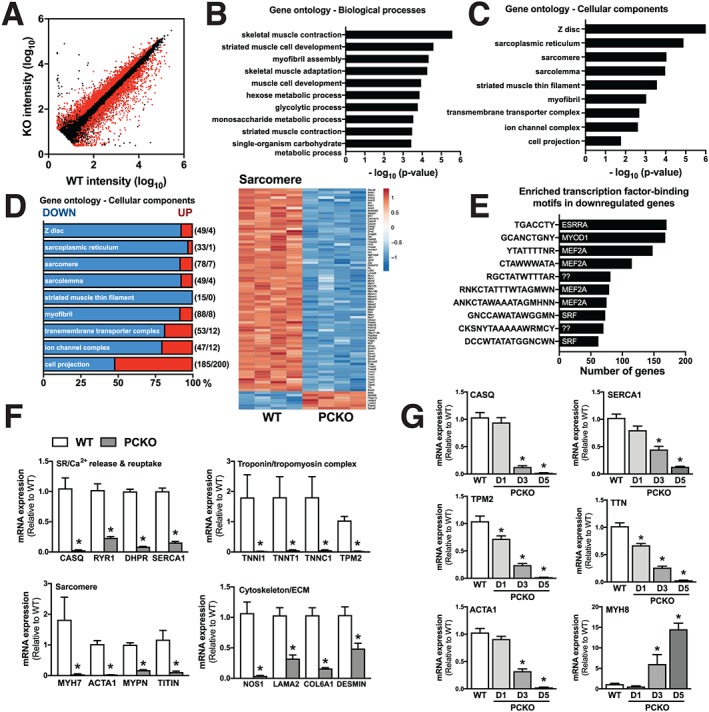
Down‐regulation of major muscle gene programs in PCKO mice. (A) Scatter plot of gene expression profiles in WT and PCKO muscles. The x and y axes indicate the gene expression levels (log10 transformation). Red dots represent differentially expressed genes in PCKO vs. WT muscle [n = 4 WT, n = 4 PCKO; false discovery rate (FDR) < 0.1]. (B–C) Top 10 overrepresented gene ontology (GO) terms (local FDR < 0.1) for differentially expressed genes in PCKO vs. WT muscle, (B) biological processes and (C) cellular components. (D) Expression profile [down (blue) and up (red)] for genes associated with cellular components GO categories in panel C. Numbers in parentheses indicate the number of altered genes (DOWN/UP, FDR < 0.1). Heat map generated using the ClustVis online tool using log10 transformed probe set intensities for genes differentially expressed (FDR < 0.1) in PCKO vs. WT mice and associated with the GO:cellular components categories ‘GO:0030017, sarcomere’. The colour ranges from deep red (high abundance) to deep blue (low abundance); white is no change. (E) Top 10 transcription factor binding motifs enriched in genes down‐regulated (FDR < 0.1) in PCKO vs. WT mice. Bar graph lists enriched motifs (left) and the transcription factors (TFs) predicted to be associated with these motifs (right). Question marks indicate that the motif is not associated with a TF. (F) mRNA levels of indicated genes in quadriceps normalized to Tbp, in D5 PCKO mice vs. WT mice (n = 6 WT and n = 6 PCKO). (G) mRNA levels of indicated genes in gastrocnemius muscle normalized to Tbp, in D1, D3, and D5 PCKO mice compared with WT mice (n = 12 WT, n = 6 D1 PCKO, n = 6 D3 PCKO, and n = 6 D5 PCKO). Statistics: Data reported as means ± SEM. (F) Student's t‐test, * P < 0.05, PCKO vs. WT. (G) One‐way analysis of variance, Bonferroni post hoc test, * P < 0.05, PCKO vs. WT.
Protein alterations in skeletal muscle of p300 and cAMP response element‐binding protein‐binding protein knockout mice
To gain insights into how rapid transcriptional changes may relate to impairments in skeletal muscle function, we next carried out proteome analyses to quantify alterations in protein content in skeletal muscle of WT, D3, and D5 PCKO mice. Replicate samples of mouse skeletal muscle tissues were lysed and proteolytically digested. Following reduction, alkylation, and solid phase extraction, purified tryptic peptides were labelled with 10plex tandem mass tag reagents and then combined for 2D LC–MS/MS analysis. Peptides representative of 2745 mouse skeletal muscle proteins were quantified across all three experimental groups. Of these, 446 proteins were determined to be significantly altered (fold‐change >1.25; false discovery rate < 0.1) between either WT vs. D3 PCKO or WT vs. D5 PCKO (Figure 5A and Supporting Information, Table S3). To systematically compare protein expression patterns and associated biomolecular processes potentially altered in D3 and D5 PCKO vs. WT mice, we performed unsupervised hierarchical clustering of all significantly altered proteins, followed by GO enrichment analysis. We determined three primary protein expression patterns in skeletal muscle of PCKO vs. WT mice, such that proteins were down‐regulated at D5 only (cluster 1), down‐regulated at D3 and D5 (cluster 2), or up‐regulated at D5 only (cluster 3) (Figure 5B). GO enrichment analysis using WebGestalt30 revealed that proteins in each of these clusters were associated with distinct cellular processes, supporting a model whereby different cellular functions are temporally affected in D3 vs. D5 PCKO mice. Proteins in cluster 1 were mainly associated with peptide metabolism and translation, while proteins in cluster 2 were associated with cytoskeleton organization and skeletal muscle development. Proteins in cluster 3 were associated with intracellular transport and protein localization (Figure 5B and Supporting Information, Table S4).
Figure 5.
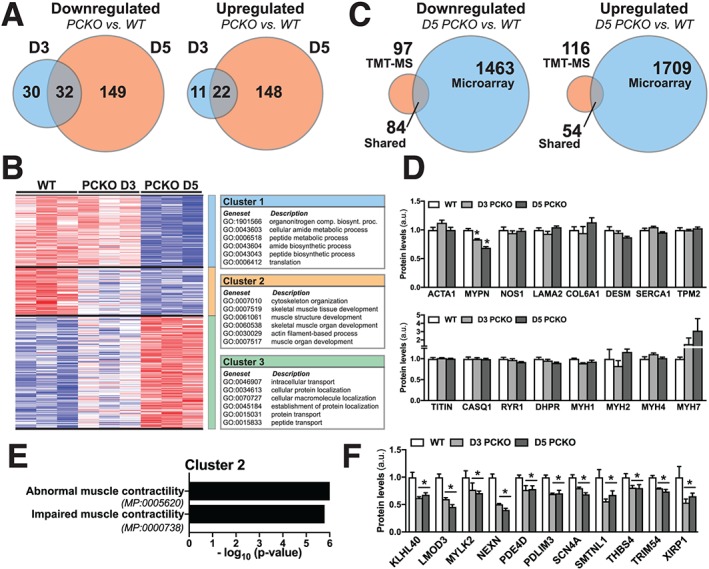
Proteomics analysis of skeletal muscle from PCKO and WT mice. (A) VENN diagrams comparing number of unique or overlapping proteins differentially expressed [fold‐change (FC) >1.25; false discovery rate (FDR) <0.1] in WT vs. D3 PCKO or WT vs. D5 PCKO mice (n = 3 D5 WT, n = 3 D3 PCKO, and n = 3 D5 PCKO). (B) Heatmap of differentially expressed proteins in WT vs. D5 PCKO mice. The colour ranges from deep red (high abundance) to deep blue (low abundance); white is no change. Data were log‐2 transformed and subjected to hierarchical cluster analysis, resulting in three distinct clusters; proteins down‐regulated only in D5 (cluster 1) or both D3 and D5 (cluster 2) or up‐regulated only in D5 (cluster 3). Boxes present the top functional annotation terms (gene ontology, biological processes, and FDR < 0.05) associated with each distinct cluster (full list in Supporting Information, Table S4). (C) VENN diagrams depicting the unique and overlapping differentially expressed proteins and genes either up‐regulated or down‐regulated in both the proteomics (FC > 1.25, FDR < 0.1) and microarray analysis (FDR < 0.1) of WT and PCKO muscles. (D) Bar graphs show levels of proteins from TMT‐MS data, from D3 and D5 PCKO mice vs. WT mice (n = 3 WT, n = 3 D3 PCKO, and n = 3 D5 PCKO). (E) Enrichment analysis of cluster 2 (proteins down‐regulated in both D3 and D5 PCKO vs. WT mice) using the Mammalian Phenotype Ontology Database (FDR < 0.05). (F) Bar graph shows proteins from TMT‐MS data found within these phenotype ontology terms. Statistics: Data reported as means ± SEM. (D and F) One‐way analysis of variance, Bonferroni post hoc test, *PCKO D3 or D5 differentially expressed (FDR < 0.1) compared with WT.
Comparing transcriptomic to proteomic data, only ~3–5% of the genes that were up‐regulated or down‐regulated at D5 in PCKO mice were matched by a significant change in protein abundance (Figure 5C). For contractile genes down‐regulated in D5 PCKO muscle (Figure 4F), 13 out of 16 proteins were detected using tandem mass tag‐MS (Figure 5D); myopalladin protein levels were reduced in D3 and D5 PCKO; and myosin heavy chain 7 (MYH7) trended towards being up‐regulated in D5 PCKO mice; however, the remaining 11 proteins were unchanged (Figure 5D). There was also no difference in the other main MYHs (MYH2, MYH4, or MYH1) between WT and PCKO mice (Figure 5D). Consequently, changes in these proteins could not explain the phenotype of PCKO mice. Considering global changes in the PCKO muscle proteome, we sought to determine whether any of these could explain the contractile dysfunction in PCKO mice at D3 and D5. Thus, we mapped the proteins in cluster 2 to the Mammalian Phenotype Ontology Database31 using WebGestalt.30 Interestingly, in line with the abrogated contractile phenotype at D3 and D5 in PCKO mice (Figure 3A–3C), proteins down‐regulated at both the D3 and D5 timepoints were associated with terms such as abnormal muscle contractility (MP:0005620) and impaired muscle contractility (MP:0000738) (Figure 5E). Cross‐referencing the proteins belonging to these categories (Figure 5F) with the microarray data (Supporting Information, Table S1) revealed that all of these proteins, except for xin actin binding repeat containing 1, were down‐regulated at the transcriptional level in PCKO D5 skeletal muscle (Supporting Information, Table S1). Taken together, these data suggest that reduced levels of a discreet set of mRNA and proteins involved in skeletal muscle contractile function underlie the impaired contractile phenotype in PCKO mice at D3 and D5.
Discussion
Acetylation of the ε‐amino group of lysine residues is a common post‐translational modification that regulates various aspects of cellular homeostasis, including mitochondrial energy metabolism and gene transcription.1, 2, 3, 4, 5 Over the past decade, a significant body of work has investigated the regulation of skeletal muscle acetylation by lysine deacetylases, including sirtuin (SIRT) 1 and SIRT3.7, 8, 32, 33, 34, 35 However, surprisingly, little is known about the contribution of lysine acetyltransferases to skeletal muscle biology and function. To address this gap in knowledge, we studied the functionally related proteins p300 and CBP, which are acetyltransferases with a large interactome (i.e. over 400 bindings partners).12 Using a novel, inducible and skeletal muscle‐specific mouse model, we demonstrate a requirement for p300 together with CBP in skeletal muscle contractile function, transcriptional homeostasis, and organism survival.
The essential role of p300/CBP in development is well established.36 Humans with autosomal mutations in p300 or CBP are characterized by severe developmental abnormalities36; likewise, whole‐body KO of p300 or CBP16 or heterozygous whole‐body KO of both p300 and CBP17 in mice is embryonically lethal. Importantly, while mice with germline KO of p30021 using muscle creatine kinase‐driven Cre‐expression (MCK‐Cre) are viable, concurrent MCK‐Cre germline PCKO is embryonically lethal (unpublished observation, K. Svensson, S.A. LaBarge & S. Schenk). Thus, the embryonic lethality in whole‐body p300/CBP mouse models could, at least in part, be due to developmental deficits in skeletal and/or cardiac muscle, considering that MCK‐Cre impacts both cardiac and skeletal muscle. The lethality phenotype is not surprising, because at least 190 of the interaction partners of p300/CBP are encoded by genes considered critical to survival in mice,22 including some specific to skeletal muscle development, such as (MEF2C, MYOD, and myogenin).18, 19, 20 To circumvent these developmental complications and lethality, we used an inducible Cre approach specific to skeletal muscle (i.e. cardiac muscle is not affected),25 which allowed us to temporally KO p300/CBP in adult mice. Despite this, we unexpectedly found that ablation of p300 and CBP in skeletal muscle of adult mice is lethal. This lethality phenotype is also present in a pancreatic p300/CBP double‐KO model, although these mice were germline KOs and died during embryonic development.37
Skeletal muscle strength and contractile function is intimately linked to health span and organismal survival.38 In line with this, the rapid lethality of PCKO mice is paralleled by profound impairments in grip strength, neuromuscular function, and ability to move, which at the muscle level presents as an ~85–95% reduction in intrinsic force production. This force decrement is independent of muscle fibre type, as it occurs to the same extent in both fast‐twitch (EDL) and slow‐twitch (soleus) muscles, and is not accompanied by a fibre type shift (i.e. altered oxidative capacity). Ultimately, we believe that the lethal phenotype in PCKO mice is driven by the rapid decline in muscle function, which severely limits the ability of the mice to move, feed, drink water, and, perhaps most importantly, their ability to breathe, due to weakness of diaphragm and intercostal muscles, a phenotype reminiscent of muscle dystrophy models.39 The connection between p300/CBP and a muscular dystrophy‐like phenotype is further supported by studies in zebrafish. For example, skeletal muscle p300 and CBP expression is reduced in a zebrafish muscular dystrophy model,40 while chemical inhibition of p300 and CBP activity in zebrafish results in a muscular dystrophy‐like phenotype.18 In fact, overexpression of CBP alone in a zebrafish dystrophy model is sufficient to ameliorate muscle damage.40 Additionally, our studies using PZ and CZ mice show that when one allele of either p300 or CBP is present in skeletal muscle, both the functional and lethality phenotype of PCKO mice are prevented. These findings underscore the potential clinical importance of p300 or CBP in skeletal muscle, not only to muscle function but also survival.
p300 and CBP interact with over 400 bindings partners12 and are central transcriptional regulators.9, 12, 13, 14, 15 Strikingly, just 5 days after initiating TMX treatment, there were rapid and substantial transcriptional alterations in PCKO skeletal muscle, with 3310 differentially expressed genes. Similar to previous in vitro studies,18 loss of p300 and CBP impacted gene programs crucial to contractile function and muscle integrity, with qPCR analysis of select genes revealing that these changes manifest as early as D1 post‐TMX in PCKO mice. In fact, gene programs critical to all aspects of skeletal muscle contraction, including signal propagation via ion channel complexes, calcium release and reuptake at the sarcoplasmic reticulum, calcium binding at the thin filaments, and sarcomere structure and function, were down‐regulated in PCKO mice, which indicates that the presence of p300 and CBP are required for transcriptional homeostasis in adult skeletal muscle. Based on genes down‐regulated in PCKO mice, the activity of two central transcription factors in muscle, MEF2A and MYOD, was predicted to be reduced. Both p300 and CBP have previously been described in in vitro studies to interact with and coactivate MYOD and the MEF2 family member MEF2C.18, 19, 20, 41, 42, 43, 44, 45 Thus, we speculate that the broad down‐regulation of muscle gene programs in PCKO mice could stem from the role of p300 and CBP in coactivating these, and other, myogenic transcription factors.
Considering the relatively slow contractile proteins turnover in skeletal muscle,46 a key question was whether the transcriptional changes seen in PCKO muscle at D5 after TMX underlie the contractile defects and lethality in PCKO mice. Through quantitative mass spectrometry analysis, we identified that 95 and 351 proteins were differentially expressed at D3 and D5, respectively. Despite the small (~3–5%) overlap between the transcriptional and protein changes in PCKO mice, phenotype ontology analysis revealed that proteins down‐regulated at both D3 and D5 in PCKO muscle were enriched in terms associated with impaired muscle contractility and abnormal muscle physiology. In fact, KO mouse models for some of the proteins down‐regulated in PCKO muscle have been associated with muscle weakness and impaired contractility (e.g. KLHL4047 and LMOD348). While reduced levels of these proteins in PCKO mice could potentially explain the muscle weakness phenotype, the levels of these proteins are retained at ~70% in PCKO mice and thus differ substantially from the complete ablation in KO models.47, 48 Accordingly, it is perhaps unlikely that these changes are the sole mediators of the rapid impairment in muscle function of PCKO mice. It is also possible that the major disruption of gene transcription in PCKO muscle could lead to reduced proteolysis in order to maintain sarcomere protein homeostasis. Because mature muscle cells rely on proteolysis to clear damaged proteins, this would suggest that sarcomere contractile function could be compromised even though sarcomere protein levels in PCKO muscles are unaltered.
While mainly considered to be nuclear proteins,12, 13, 15, 49 p300/CBP have a strong cytosolic presence and acetylate numerous cytosolic proteins.50, 51, 52, 53 Considering that >80% of contractile proteins in striated muscle are acetylated,6 and acetylation positively correlates with muscle contractile function in cardiac54, 55 and skeletal muscle,56, 57, 58 it is possible that direct acetylation of contractile proteins by p300/CBP contributes to the phenotype of PCKO mice. Along this line, a recent study found that addition of recombinant p300 to contracting rat myofibres ex vivo improved relaxation parameters59 supporting a direct role for p300 and acetylation in regulation of contractile function. Additionally, aside from acetylation, p300/CBP can regulate intracellular crotonylation,60, 61 propionylation, and butyrylation.62 Thus, it will be interesting in future studies to address whether these types of acylations occur in adult mouse skeletal muscle and whether they, or lysine acetylation by p300/CBP, can directly impact muscle contractile function.
In conclusion, there are direct clinical implication that can be drawn from our findings. First, the essential role for p300 and CBP, and by extension protein lysine acetylation, in skeletal muscle contractile function described in our study cautions the current state of thinking in the field, which implicates hyperacetylation of muscle proteins as negatively affecting skeletal muscle physiology and metabolic health.63, 64, 65 Second, considering that activators of protein deacetylases, such as SIRT1 and SIRT3, and inhibitors of p300/CBP are actively being pursued as treatments or prophylactic interventions for host of diseases, including diabetes, sarcopenia, and cancer, it is imperative that they are thoroughly vetted so as to ensure that they do not have unwanted consequences on the force‐generating capacity in skeletal muscle. For example, this is particularly relevant for cancer‐related treatments focused on inhibiting p300/CBP66, 67, 68 as this could further exacerbate impairments in skeletal muscle function found in many cancer patients.69 Finally, in line with recent studies correlating reduced p300/CBP content40 or activity18 with development of dystrophic phenotype in zebrafish, our study highlights p300 and CBP as central regulators of skeletal muscle integrity and function in mice. Considering the beneficial effect seen with inhibition of histone deacetylases on muscle function in both mice and humans with muscular dystrophy,70, 71 one goal of future research should be to help elucidate the potential beneficial effects of increasing acetyltransferase activity, especially p300/CBP, in the context of muscle dystrophies, as well as other diseases that are characterized by impaired muscle function, such as sarcopenia.
Author contributions
K.S., S.A.L., and S.S. conceived the study. K.S. and S.S. wrote the manuscript. K.S., S.A.L., and S.S. designed the experiments. K.S., S.A.L., V.F.M., and S.T. performed all food intake, survival, and performance tests in the mice. K.S. and A.S. performed all ex vivo contractility experiments. S.K.M. generated transmission electron micrographs. S.A.L. and A.P. generated microarray data. G.A.M., R.S., and K.S. analysed microarray data. J.M.C. and L.L.D. generated tandem mass‐tag mass spectrometry (TMT‐MS) data. J.M.C., L.L.D., and K.S. analysed TMT‐MS data. S.R.W., C.E.M., and J.E.A. were advisors on the project. All authors reviewed the manuscript and provided input.
Conflict of interest
The authors have declared that no conflict of interest exists.
Supporting information
Data S1: Supporting Information
Figure S1. Muscle histology in PCKO mice
Table S1. Microarray data from D5 WT and PCKO mice.
Table S2. Functional annotation of differentially expressed genes from microarray.
Table S3. TMT‐MS data from WT, D3 PCKO and D5 PCKO mice.
Table S4. Functional annotation of Clusters from TMT‐MS data.
Table S5. Primers used in qPCR analysis.
Acknowledgements
This work was supported by National Institutes of Health (NIH) grant numbers R21 AR072882 and R01 AG043120 to S.S., T32 AR060712 and F30 DK115035 to V.F.M., and R01 HL146549 to J.E.A., a grant from the UC San Diego Frontiers of Innovation Scholars Program to S.S., postdoctoral fellowships from the Swiss National Science Foundation and the American Federation for Aging Research to K.S., American Heart Association grant number 17SDG33350075 to J.E.A., and Graduate Student Research Support from the UC San Diego Institute of Engineering in Medicine and the Office of Graduate Studies to V.F.M. This research was conducted while K.S. was a Glenn Foundation for Medical Research Postdoctoral Fellow. The authors are grateful to John M. Bucci for help with hierarcical clustering and heatmap generation, to Shannon Bremner for help with ex vivo contractility experiments, and to Mary Esparza for help with histological staining. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.72
Svensson K., LaBarge S. A., Sathe A., Martins V. F., Tahvilian S., Cunliffe J. M., Sasik R., Mahata S. K., Meyer G. A., Philp A., David L. L., Ward S. R., McCurdy C. E., Aslan J. E., and Schenk S. (2020) p300 and cAMP response element‐binding protein‐binding protein are essential for skeletal muscle homeostasis, contractile function, and survival, Journal of Cachexia, Sarcopenia and Muscle, 11, 464–477. 10.1002/jcsm.12522.
References
- 1. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 2015;16:258–264. [DOI] [PubMed] [Google Scholar]
- 2. Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta ‐ Proteins Proteomics 1864;2016:1372–1401. [DOI] [PubMed] [Google Scholar]
- 3. Ali I, Conrad RJ, Verdin E, Ott M. Lysine acetylation goes global: from epigenetics to metabolism and therapeutics. Chem Rev 2018;118:1216–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Philp A, Rowland T, Perez‐Schindler J, Schenk S. Understanding the acetylome: translating targeted proteomics into meaningful physiology. Am J Physiol Cell Physiol 2014;307:C763–C773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Menzies KJ, Zhang H, Katsyuba E, Auwerx J. Protein acetylation in metabolism‐metabolites and cofactors. Nat. Rev. Endocrinol. 2016;12:43–60. [DOI] [PubMed] [Google Scholar]
- 6. Lundby A, Lage K, Weinert BT, Bekker‐Jensen DB, Secher A, Skovgaard T, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2012;2:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013;62:3404–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, et al. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high‐fat‐fed mice. Diabetes 2015;64:3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome‐wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009;138:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moresi V, Carrer M, Grueter CE, Rifki F, Shelton JM, Richardson JA, et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. PNAS 2012;109:1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Renzini A, Marroncelli N, Noviello C, Moresi V, Adamo S. HDAC4 regulates skeletal muscle regeneration via soluble factors. Front Physiol 2018;9:1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev 2015;115:2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996;87:953–959. [DOI] [PubMed] [Google Scholar]
- 14. Ramos YFM, Hestand MS, Verlaan M, Krabbendam E, Ariyurek Y, van Galen M, et al. Genome‐wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res 2010;38:5396–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 2001;114:2363–2373. [DOI] [PubMed] [Google Scholar]
- 16. Tanaka Y, Naruse I, Hongo T, Xu MJ, Nakahata T, Maekawa T, et al. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB‐binding protein. Mech Dev 2000;95:133–145. [DOI] [PubMed] [Google Scholar]
- 17. Yao T‐P, Oh S‐P, Fuchs M, Zhou N‐D, Ch'ng L‐E, Newsome D, et al. Gene dosage‐dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998;93:361–372. [DOI] [PubMed] [Google Scholar]
- 18. Fauquier L, Azzag K, Parra MAM, Quillien A, Boulet M, Diouf S, et al. CBP and P300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity. Sci Rep 2018;8:12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puri PL, Sartorelli V, Yang X‐JJ, Hamamori Y, Ogryzko VV, Howard BH, et al. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell 1997;1:35–45. [DOI] [PubMed] [Google Scholar]
- 20. Polesskaya A, Naguibneva I, Fritsch L, Duquet A, Ait‐Si‐Ali S, Robin P, et al. CBP/p300 and muscle differentiation: no HAT, no muscle. EMBO J 2001;20:6816–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. LaBarge SA, Migdal CW, Buckner EH, Okuno H, Gertsman I, Stocks B, et al. P300 is not required for metabolic adaptation to endurance exercise training. FASEB J 2016;30:1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T‐cell development. Mol Cell Biol 2006;26:789–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu W, Fukuyama T, Ney PA, Wang D, Rehg J, Boyd K, et al. Global transcriptional coactivators CREB‐binding protein and p300 are highly essential collectively but not individually in peripheral B cells. Blood 2006;107:4407–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hennig AK, Peng GH, Chen S. Transcription coactivators p300 and CBP are necessary for photoreceptor‐specific chromatin organization and gene expression. PLoS One 2013;8:e69721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCarthy JJ, Srikuea R, Kirby TJ, Peterson CA, Esser KA. Inducible Cre transgenic mouse strain for skeletal muscle‐specific gene targeting. Skelet Muscle 2012;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang‐Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, et al. Loss of CBP causes T cell lymphomagenesis in synergy with p27Kip1 insufficiency. Cancer Cell 2004;5:177–189. [DOI] [PubMed] [Google Scholar]
- 27. Svensson K, Dent JR, Tahvilian S, Martins VF, Sathe A, Ochala J, et al. Defining the contribution of skeletal muscle pyruvate dehydrogenase alpha 1 (Pdha1) to exercise performance and insulin action. Am J Physiol Endocrinol Metab 2018;315:E1034–E1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Young KW, Dayanidhi S, Lieber RL. Polarization gating enables sarcomere length measurements by laser diffraction in fibrotic muscle. J Biomed Opt 2014;19:117009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chleboun GS, Patel TJ, Lieber RL. Skeletal muscle architecture and fiber‐type distribution with the multiple bellies of the mouse extensor digitorum longus muscle. Acta Anat (Basel) 1997;159:147–155. [DOI] [PubMed] [Google Scholar]
- 30. Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017;45:W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith C, Goldsmith C, Eppig J. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol 2004;6:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dittenhafer‐Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, et al. SIRT3 mediates multi‐tissue coupling for metabolic fuel switching. Cell Metab 2015;21:637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Svensson K, LaBarge SA, Martins VF, Schenk S. Temporal overexpression of SIRT1 in skeletal muscle of adult mice does not improve insulin sensitivity or markers of mitochondrial biogenesis. Acta Physiol 2017;221:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator‐activated receptor‐gamma coactivator‐1alpha (PGC‐1alpha) deacetylation following endurance exercise. J Biol Chem 2011;286:30561–30570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schenk S, McCurdy CE, Philp A, Chen MZ, Holliday MJ, Bandyopadhyay GK, et al. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J Clin Invest 2011;121:4281–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev 2000;14:1553–1577. [PubMed] [Google Scholar]
- 37. Wong CK, Wade‐Vallance AK, Luciani DS, Brindle PK, Lynn FC, Gibson WT. The p300 and CBP transcriptional coactivators are required for β‐cell and α‐cell proliferation. Diabetes 2018;67:412–422. [DOI] [PubMed] [Google Scholar]
- 38. Navas‐Enamorado I, Bernier M, Brea‐Calvo G, de Cabo R. Influence of anaerobic and aerobic exercise on age‐related pathways in skeletal muscle. Ageing Res Rev 2017;37:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burns DP, Roy A, Lucking EF, McDonald FB, Gray S, Wilson RJ, et al. Sensorimotor control of breathing in the mdx mouse model of Duchenne muscular dystrophy. J Physiol 2017;595:6653–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bajanca F, Vandel L. Epigenetic regulators modulate muscle damage in duchenne muscular dystrophy model. PLoS Curr 2017;Dec 21;9: ecurrents.md.f1e2379fa632f8135577333dd92ca83b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Polesskaya A, Naguibneva I, Duquet A, Bengal E, Robin P, Harel‐Bellan A. Interaction between acetylated MyoD and the bromodomain of CBP and/or p300. Mol Cell Biol 2001;21:5312–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khilji S, Hamed M, Chen J, Li Q. Loci‐specific histone acetylation profiles associated with transcriptional coactivator p300 during early myoblast differentiation. Epigenetics 2018;13:642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roth JF, Shikama N, Henzen C, Desbaillets I, Lutz W, Marino S, et al. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J 2003;22:5186–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol 1997;17:1010–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma K, Chan JKL, Zhu G, Wu Z. Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol 2005;25:3575–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Obinata T, Maruyama K, Sugita H, Kohama K, Ebashi S. Dynamic aspects of structural proteins in invertebrate skeletal muscle. Muscle Nerve 1981;4:456488. [DOI] [PubMed] [Google Scholar]
- 47. Garg A, O'Rourke J, Long C, Doering J, Ravenscroft G, Bezprozvannaya S, et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J Clin Invest 2014;124:3529–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tian L, Ding S, You Y, Li T, Liu Y, Wu X, et al. Leiomodin‐3‐deficient mice display nemaline myopathy with fast‐myofiber atrophy. Dis Model Mech 2015;8:635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holmqvist P‐H, Mannervik M. Genomic occupancy of the transcriptional co‐activators p300 and CBP. Transcription 2013;4:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sebti S, Prébois C, Pérez‐Gracia E, Bauvy C, Desmots F, Pirot N, et al. BAT3 modulates p300‐dependent acetylation of p53 and autophagy‐related protein 7 (ATG7) during autophagy. Proc Natl Acad Sci U S A 2014;111:4115–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi D, Pop MS, Kulikov R, Love IM, Kung AL, Kung A, et al. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci U S A 2009;106:16275–16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aslan JE, Rigg RA, Nowak MS, Loren CP, Baker‐Groberg SM, Pang J, et al. Lysine acetyltransfer supports platelet function. J Thromb Haemost 2015;13:1908–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fermento ME, Gandini NA, Salomón DG, Ferronato MJ, Vitale CA, Arévalo J, et al. Inhibition of p300 suppresses growth of breast cancer. Role of p300 subcellular localization. Exp Mol Pathol 2014;97:411–424. [DOI] [PubMed] [Google Scholar]
- 54. Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem 2008;283:10135–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Samant SA, Pillai VB, Sundaresan NR, Shroff SG, Gupta MP. Histone deacetylase 3 (HDAC3)‐dependent reversible lysine acetylation of cardiac myosin heavy chain isoforms modulates their enzymatic and motor activity. J Biol Chem 2015;290:15559–15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Viswanathan MC, Blice‐Baum AC, Schmidt W, Foster DB, Cammarato A. Pseudo‐acetylation of K326 and K328 of actin disrupts Drosophila melanogaster indirect flight muscle structure and performance. Front Physiol 2015;6:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM, Judge AR. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J Cell Sci 2014;127:1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ryder DJ, Judge SM, Beharry AW, Farnsworth CL, Silva JC, Judge AR. Identification of the acetylation and ubiquitin‐modified proteome during the progression of skeletal muscle atrophy. PLoS One 2015;10:e0136247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jeong MY, Lin YH, Wennersten SA, Demos‐Davies KM, Cavasin MA, Mahaffey JH, et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med 2018;10:eaao0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sabari BR, Tang Z, Huang H, Yong‐Gonzalez V, Molina H, Kong HE, et al. Intracellular crotonyl‐CoA stimulates transcription through p300‐catalyzed histone crotonylation. Mol Cell 2015;58:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu X, Wei W, Liu Y, Yang X, Wu J, Zhang Y, et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase‐deficient and crotonyltransferase‐competent CBP/p300. Cell Discov 2017;3:17016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, et al. Lysine propionylation and butyrylation are novel post‐translational modifications in histones. Mol Cell Proteomics 2007;6:812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age‐related diseases. Trends Pharmacol. Sci. 2014;35:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol Mech Dis 2010;5:253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guarente L. Sirtuins as potential targets for metabolic syndrome. Nature 2006;444:868–874. [DOI] [PubMed] [Google Scholar]
- 66. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage‐specific tumours. Nature 2017;550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jin L, Garcia J, Chan E, De La Cruz C, Segal E, Merchant M, et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration‐resistant prostate cancer. Cancer Res 2017;77:5564–5575. [DOI] [PubMed] [Google Scholar]
- 68. Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res 2015;75:5106–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Prim 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 70. Bettica P, Petrini S, D'Oria V, D'Amico A, Catteruccia M, Pane M, et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord 2016;26:643–649. [DOI] [PubMed] [Google Scholar]
- 71. Consalvi S, Mozzetta C. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med 2013;19:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information
Figure S1. Muscle histology in PCKO mice
Table S1. Microarray data from D5 WT and PCKO mice.
Table S2. Functional annotation of differentially expressed genes from microarray.
Table S3. TMT‐MS data from WT, D3 PCKO and D5 PCKO mice.
Table S4. Functional annotation of Clusters from TMT‐MS data.
Table S5. Primers used in qPCR analysis.