

Abstract
Cyanines derived from heptamethines were mainly discussed regarding their functionalization to broaden the solubility in different surroundings exhibiting either hydrophilic or hydrophobic properties and to tailor made the ΔGet photopysical properties with respect to absorption and fluorescence. Electrochemical properties were additionally considered for some selected examples. The cyanines chosen comprised as end groups either indolenine, benzo[e]- or benzo[cd]indolium pattern, which facilitated to shift the absorption between 750–1000 nm. This enabled their use in applications with light sources emitting in the near-infrared (NIR) region selected from high power LEDs or lasers with line-shaped focus. The absorbers considered were discussed regarding their function as sensitizer for applications related to Chemistry 4.0 standards. These were mainly photopolymer coatings, which can be found for applications in the graphic industry or to protect selected substrates. The huge release of heat on demand upon turning ON or OFF the NIR light source enables them for photothermal treatment in processes requesting heat to initiate either chemical (activated reactions) or physical (melting, evaporation) events.
Keywords: chemistry 4.0, cyanine, near infrared, photopolymer, polymer synthesis, sensitizer
Introduction
Cyanines have received in the class of polymethines big attention within the last hundred years [1–4]. Their substitution pattern easily facilitates shift of the absorption from the ultra violet part into the near infrared region while they exhibit huge extinction coefficients [3]. The connection of two nitrogens with an odd number of methine groups results in a positive charge of the final molecule if the cyanine comprises no additional substituent exhibiting a negative charge at the molecular skeleton. Furthermore, it requires to combine this cation with appropriate anions to achieve a good compatibility with the surrounding matrix. Compatibility in these examples preferably refers to the solubility in the surrounding matrix, while undesirable events such as aggregation are of minor importance. In the worst case, it can also result in crystallization of the cyanine in the matrix under certain storage conditions such as high humidity and/or elevated temperatures available in some geographic areas where these materials have been used in practice. Such anions can be either bis(trifluoromethyl)sulfonylimides [5], aluminates [6] or sulfonates comprising long alkyl chains [5]. Alternatively, a barbiturate group positioned at the meso-position of the cyanine can also sometimes lead to unexpectedly high solubility in aprotic polar surroundings [5]. Nevertheless, a variation between the cation of the cyanine with distinct anions may move such materials to applications where either an aqueous surrounding being available in biological applications [7–9] or a dipolar aprotic matrix takes the function of the matrix with absorber embedded. Typically, digital imaging in Computer to Plate technology (CtP) [10–14], curing of liquid coatings [5,15], powder coatings [16–17], laser drying [18–20], laser welding [21–25], or laser marking of plastics [26–31] represent some practical examples. Nevertheless, cyanines have held a long history regarding their practical use, which started to use such materials as sensitizers in silver halide photography [32–34] until the point when electronic media took their place in imaging sciences to save pictures. Many fundamental knowledge was grown up in this period as shown for example by several reviews [35–40]. Particularly, the use of model systems and the knowledge obtained by exploration systems in silver halide photography [32–34,41–45] also helped to understand the function and formation H and J-aggregates [46–49] being aligned parallel or in series, respectively. The capability of cyanines to form J-aggregates exhibiting a huge bathochromic shift, which was reported for the first time by Scheibe [50] and Jelley in 1937 [51], enabled spectral sensitization of AgX photography over a long spectral range [41–45]. Though AgX photography has not been alive at large scale, research pursued in this field helped to develop theories regarding the design of absorbers/sensitizers and their function in other industrial applications.
Cyanines possess an odd number 2n + 3 of π-centers and 2n + 4 π-electrons [1]. The variable n represents the number of vinylene groups in the methine chain. The absorption of cyanines can be tailor made by extension of the methine chain by two methine groups of the vinylene moiety resulting in a bathochromic shift of the absorption of about 100 nm [3]. Substitution of the terminal groups comprising by either substituted indolium, benzo[e]indolium or benzo[c,d]indolium patterns helps to finetune the absorption wavelength with respect to the light source used. Furthermore, different substituents at the meso-position comprising either electron-donating or electron-withdrawing substituents complement the design of tailor made absorption [52–54]. The fact that many investigations were pursued in the visible range assigned these materials in general as dyes, which should not be correct. The term dye relates to the visible part covering the range between 400–700 nm considering the longest absorption maximum [55]. The use of the term absorber fits better as a general assignment that also relates to the absorption range below and above this range. Furthermore, one can also name the function of a cyanine in a given application which can be either a sensitizer, initiator, staining, activator or a filter material just to name a few possible examples. Unfortunately, literature often does not clearly distinguish between their functionality.
Besides cyanines, there were made huge efforts to synthesize alternatives such as rylenes [56] or conjugated polymers [57] with the focus to receive materials covering a broad absorption range up to the near infrared (NIR) part. Nevertheless, they have not reached the necessary practical use as cyanines explainable by the modest solubility in many industrial matrix materials used for different applications. These facts advantageously demonstrate the benefits of cyanines where tailor-made synthesis of the unsaturated cation and the selection of an appropriate anion facilitated sufficient solubility with many matrix materials [5–6]. In comparison, cyanines depict from this point of view outstanding properties since the aforementioned benefits enabled their use in photopolymerization to pattern light sensitive coatings [11–14], to cure liquid coatings [15], and powder coatings [16–17] or to initiate either a chemical and/or thermal curing process [58].
Although some photophysical studies disclose the absorption and emission properties [13,59–62], comparison of available data may lead sometimes to contradictory conclusions. This article first summarizes possibilities for synthesis of different patterns of cyanines exhibiting indolenine and benzindolenine moiety at the ends of the methine chain while the cyanine pattern also changed at the center of the molecule. A summary of determined and almost published photophysical and electrochemical data should complement available data available [3,13–14,59–62] to provide a brief idea regarding their use in applications based on sensitized photoinduced electron transfer. This can be NIR-sensitized photopolymerization resulting in formation of initiating radicals and conjugate acid [5–6,13–15,63–64]. Recently, the use of NIR-LEDs exhibiting high excitation intensity brought more light in this field and helped to understand the function of existing intrinsic barrier in such systems comprising cationic cyanines in photoinduced electron transfer systems [65]. Furthermore, the huge amount of heat released by nonradiative deactivation of the excited state brings up to use them as “molecular oven/furnace” in heat-based physical and/or chemical processes. There, they can generate heat in an ON/OFF procedure on demand [58].
Many examples of cyanines focus on the use of absorption in the NIR [64]. Among of the disclosed applications, photopolymerization depicts one representative method where the absorber uptakes the function of a sensitizer to generate initiating radicals and conjugate acid [13–17,58,65]. The fact that these systems work upon turning ON/OFF cycles facilitates their use in Industry 4.0 related applications where printing represents one feasible application [66]. Data collection [67] in combination with self-leaning software related to artificial intelligence (A.I.) drives their use in applications with more efficiency in large workflows. Such tools can recognize failures in industrial processes just by collection of data in a big database where data analysis of global production may solve issues related to the materials.
Review
Synthesis of cyanines absorbing in the NIR
Cyanines comprising the patterns I–V (Scheme 1) have received much importance as NIR absorbers applied in different technologies [1,8–9,11–20,32–34]. They exhibit a flexible chain (I–III) or bridged chain (IV, V) where the cyclopentene pattern keeps the system planar while the introduction of the non-planar cyclohexene moiety results in improvement of compatibility of the surrounding matrix [5]. Indolium, benzo[e]indolium, and benzo[c,d]indolium (Scheme 2) represent the most widely used substitution patterns of NIR absorbers. Their structures formed can be easily build together following the procedures disclosed [2] while patent procedures also worked well [68–69].
Scheme 1.

Structural patterns of several symmetric cyanines relating to trimethines (I), pentamethines (II), and heptamethines (III–V). While I-III exhibit an open methine chain, functionalization by bridged moieties results in either a planar (IV) or non-planar (V) structure. The terminal group A/A’ belongs to indolium, benzo[e]indolium, and benzo[c,d]indolium (Scheme 2). I–V comprise a further substituent in the meso-position.
Scheme 2.

1-Substituted 2,3,3-trimethylindolium-, 2,3,3-benzo[e]indolium-, and 2,3,3-benzo[c,d]indolium salts lead to the patterns a/a’, b/b’ and c/c’ of the terminal substituent A, respectively.
These structures depict only one valence pattern while the positive charge typically distributes over the entire conjugated system. There are much more valence patterns possible to draw. For a sake of simplicity, we operate with one feasible structure. The availability of these precursors bases on easy pursuable chemical reactions resulting in huge libraries of cyanines exhibiting different substitution patterns.
Thus, I–V belong to symmetric cyanines whose synthesis has been known for a long time [2]. This also explains why there exist plenty of procedures facilitating the synthesis with high yields and acceptable purity of the crude reaction products. These efforts helped to enable such materials to technologies requesting a higher demand on materials; that is for example the graphic industry where these absorbers have been successfully worked in Computer to Plate (CtP) applications for more than 25 years [11–14]. Nowadays, manufacturing of symmetric cyanines approaches up to several tons worldwide/year in the aforementioned applications. On the other hand, cyanines exhibiting asymmetric structural patterns have also reached practical fields but this relates more or less to medical and biological uses [7–9] with rather low but exclusive quantities. These requests enforced to introduce special functional groups facilitating their use as marker in the aforementioned fields. Nevertheless, applications relating to biology or medicine demand less economic pressure because the use of these compounds typically resides at the milligram or gram-scale.
The methine chain can be either open as in I–III facilitating a certain flexibility in the main chain or bridged resulting in either a five-membered (IV) or six-membered (V) moiety. IV exhibits almost a planar structure while the more flexible 6-membered unit in the center of V results in a non-planar geometry [63]. Such a geometry affects compatibility with the surrounding matrix. Thus, aggregation phenomena dominate in the case of IV. On the other hand, absorbers related to V often exhibit a much better solubility in organic surroundings such as multi-functional acrylates [5]. This can be seen as a huge challenge – obtaining a sufficient solubility of such ionic materials in a surrounding being aprotic polar and highly viscous. Many absorbers failed to meet these requirements in practice although their fundamental research pursued in highly diluted solutions showed promising directions [5]
The substitution of either I, II, III, IV or V with distinct terminal groups A/A’ results in cyanines exhibiting either a cationic, zwitterionic or anionic pattern. Their solubility covers a wide range including aprotic polar organic solvents or also water. The latter requests to introduce functional groups promoting their water solubility; that is the sulfobutyl group in the case of R1 or -SO3− in the case of either R2 or R3. Water soluble cyanines could feasibly favor physical drying of aqueous dispersions upon irradiation with a NIR-LED [70]. In addition, chemical drying of coatings requires the use of strong anions promoting the solubility in organic surroundings. Representative anions in the case of cationic absorbers relate to tosylate [5], [n-C12H25-Ph-SO3−] [5], FAP ([(C2F5)3PF3]−)[71], NTf2 ([(CF3SO2)2N]−) [5] or aluminates ([Al(t-C4F9O)4]−) [6] – just to count a few possible examples. Thus, replacement of the counter ion can sometimes unexpectedly improve the solubility as shown for some cationic absorbers comprising either the NTf2-anion [5] or the aforementioned aluminate anion [6] where the solubility of the respective absorber can approach 10–30 g/L in multi-functional acrylic esters [5]. In addition, small structural changes of the alkyl substituent R1 even caused big differences regarding the solubility in the surrounding matrix. This field, namely driving of absorber solubility with the surrounding matrix by the structural pattern of both the cyanine and the respective counter ion, has not been well understood yet. It depicts rather semi-empiric approaches nowadays. More theoretical works would definitely bring more light in this field while the vide supra mentioned applications would definitively benefit from such studies.
Synthesis of I–V proceeds according to the reactions shown in Scheme 3. It uses the indolium derivatives shown in Scheme 2 as starting materials. They can react either with ortho esters, tris-halogenated precursors or diphenylformamidines resulting in trimethine cyanines [2]. The latter reference depicts much more reagents resulting in the desired trimethine patterns. Moreover, synthesis of pentamethine cyanines II follows a route requesting cb (Scheme 4) as precursor. There serve some aldehydes as source to synthesize this precursor by reaction with aniline. Similar reaction philosophy also follows synthesis of cb-0 (Scheme 4) to synthesize open heptamethines III. Particularly, an additional substituent at the meso-position introduces more freedoms to functionalize such materials to obtain an absorption being tailor made with respect to the light source applied. This can be a semiconductor laser [14–16,18–20] or high-power NIR-LED [65] with emission between 800–1100 nm. Such findings enforce activities to make absorbers exhibiting internal barriers in photoinduced electron transfer reactions [72] resulting in a certain white light stability under ambient light conditions. Such properties can be seen as a big benefit from a practical point of view in comparison with UV-sensitive materials.
Scheme 3.

Substitution of the chlorine substituent at the meso-position by a stronger nucleophilic moiety B [68].
Scheme 4.

Structure of alternative chain builders for synthesis of heptamethines.
Synthesis of the bridged heptamethines (IV and V) follows the general route shown in Scheme 3 [2,68–69]. Scheme 4 discloses the use of the chain builders cb and cb0-cb-6 [68–69]. The materials cb-1 and cb-2 were available by reaction of the respective cyclic ketone with POCl3 and DMF in CH2Cl2 in the case of the amino substituted derivatives in high yields. This reference also describes the synthesis of the chain builders cb-3 and cb-4. It occurred by a similar procedure. Synthesis of cb5 and cb6 was already reported [68].
The heptamethine pattern also facilitated replacement of the substituent at the meso-position. Scheme 3 shows the activation of the carbon at this position by the chlorine substituent. This explains why functionalization of cyanine pattern by either electron withdrawing or electron donating substituents easily proceeds. It also enabled the introduction of reactive groups such as -N=C=S facilitating them to function as marker in biological/imaging applications [9]. This reaction occurred with good yields and extended the use of such NIR absorbers in more fields [1–2,7–9]. In general, this functionalization resulted in changes of absorption maxima and electrochemical potentials appearing sometimes with rather low quantities, see Table 1 for explanation of the absorbers and Table 2 for the data vide infra.
Table 1.
Summary of structure elements of absorbers related to the structural patterns I–V. Table 2 summarizes their photophysical data. FEW Chemicals GmbH provided samples for the investigations.
| # | groupa | A/A’ b | Bc | R1 | R2 | R3 | counter ionc |
| 1 | I | c/c’ | H | C4H9 | H | – | BF4− |
| 2 | II | c/c’ | H | C4H9 | H | – | BF4− |
| 3 | II | c/c’ | H | C4H9 | OC4H9 | – | BF4− |
| 4 | II | c/c’ | H | t-C4H9 | t-BuPSc | t-BuPSc | NTf2− |
| 5 | III | c/c’ | H | C4H9 | H | H | PF6− |
| 6 | IV | c/c’ | (Ph)2N- | C4H9 | H | H | BF4− |
| 7 | IV | c/c’ | Ph- | C4H9 | H | H | BF4− |
| 8 | IV | c/c’ | Ph- | C4H9 | H | H | DoPhSO3− |
| 9 | V | c/c’ | Ph- | C4H9 | H | H | BF4− |
| 10 | V | c/c’ | Ph- | C4H9 | H | H | DoPhSO3− |
| 11 | V | c/c’ | Cl | C4H9 | H | H | BF4− |
| 12 | III | a/a’ | H | C4H9 | H | – | PF6− |
| 13 | III | a/a’ | H | (CH2)4SO3− | SO3− | – | 3 K+ |
| 14 | V | a/a’ | Ph-O- | C4H9 | H | – | PF6− |
| 15 | V | a/a’ | Ph-O- | (CH2)4SO3− | H | – | Na+ |
| 16 | III | a/a’ | H | CH3 | H | – | ClO4− |
| 17 | IV | a/a’ | N(CH3)2 | C4H9 | H | – | PF6− |
| 18 | V | a/a’ | Ph- | C4H9 | H | – | PF6− |
| 19 | V | a/a’ | Ph- | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 20 | V | a/a’ | Ph- | (CH2)4SO3− | H | – | Na+ |
| 21 | V | a/a’ | H | CH3 | H | – | ClO4− |
| 22 | V | a/a’ | Ph-O- | (CH2)4SO3− | SO3− | – | 3Na+ |
| 23 | IV | a/a’ | Ph-O- | C4H9 | H | – | PF6− |
| 24 | V | b/b’ | Barb1 | C2H5 | H | – | – |
| 25 | IV | a/a’ | Ph-O- | (CH2)4SO3− | H | – | Na+ |
| 26 | IV | a/a’ | Barb2 | CH3 | H | – | – |
| 27 | IV | a/a’ | Barb2 | C4H9 | H | – | – |
| 28 | V | a/a’ | Ph- | CH3 | H | – | Cl− |
| 29 | IV | a/a’ | Barb2 | (CH2)4SO3− | H | – | 2 Na+ |
| 30 | V | a/a’ | Ph-O- | CH3 | H | – | Cl− |
| 31 | IV | a/a’ | Ph- | C4H9 | SO3− | – | Na+ |
| 32 | IV | a/a’ | Ph- | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 33 | V | a/a’ | Barb1 | iC5H11 | Cl | – | – |
| 34 | IV | b/b’ | Barb2 | CH3 | H | – | – |
| 35 | IV | a/a’ | (Ph)2N- | C4H9 | H | – | PF6− |
| 36 | IV | a/a’ | (Ph)2N- | C4H9 | H | – | BF4- |
| 37 | IV | a/a’ | (Ph)2N- | C4H9 | H | – | [Al(O-t-C4F9)4]− |
| 38 | V | a/a’ | Ph(Ph-CH2)N- | C4H9 | H | – | PF6− |
| 39 | IV | a/a’ | (Ph)2N- | (CH2)4SO3− | H | – | Na+ |
| 40 | IV | a/a’ | (Ph)2N- | (CH2)4SO3− | SO3− | – | 3Na+ |
| 41 | IV | a/a’ | (Ph)2N- | (CH2)4SO3− | Cl | – | HN(C2H5)3+ |
| 42 | IV | a/a’ | Barb2 | C4H9 | SO3− | – | 2 Na+ |
| 43 | V | a/a’ | Barb2 | C4H9 | H | – | – |
| 44 | IV | a/a’ | Cl | C4H9 | H | – | BF4− |
| 45 | V | a/a’ | Cl | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 46 | IV | a/a’ | Cl | CH3 | H | – | MePhSO3− |
| 47 | V | a/a’ | Barb2 | C2H5 | H | – | – |
| 48 | V | b/b’ | Barb2 | C2H5 | H | – | – |
| 49 | V | a/a’ | Barb2 | (CH2)4SO3− | H | – | 2 Na+ |
| 50 | IV | a/a’ | Barb2 | (CH2)4SO3− | SO3− | – | 4 Na+ |
| 51 | V | b/b’ | Barb4 | C2H5 | H | – | – |
| 52 | IV | a/a’ | Cl | C4H9 | SO3− | – | Na+ |
| 53 | IV | a/a’ | Cl | (CH2)4SO3− | H | – | Na+ |
| 54 | V | a/a’ | Cl | (CH2)4SO3− | H | – | Na+ |
| 55 | V | a/a’ | Cl | C4H9 | H | – | PF6− |
| 56 | IV | a/a’ | Cl | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 57 | V | a/a’ | Cl | C4H9 | Cl | – | PF6− |
| 58 | V | a/a’ | Cl | C4H9 | SO3− | – | Na+ |
| 59 | III | b/b’ | H | (CH2)4SO3− | H | – | Na+ |
| 60 | V | a/a’ | Tria | C4H9 | H | – | BF4− |
| 61 | IV | a/a’ | Ph-S- | (CH2)4SO3− | H | – | Na+ |
| 62 | V | a/a’ | Barb3 | CH3 | H | – | – |
| 63 | V | a/a’ | Cl | CH3 | H | – | Cl− |
| 64 | V | a/a’ | Ph-S- | CH3 | H | – | Cl− |
| 65 | V | a/a’ | Ph-S- | C4H9 | H | – | ClO4− |
| 66 | V | a/a’ | Ph-S- | (CH2)4SO3− | H | – | Na+ |
| 67 | V | a/a’ | Ph-S- | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 68 | IV | a/a’ | Ph-S- | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 69 | V | a/a’ | C6H13 | (CH2)4SO3− | SO3− | – | 3Na+ |
| 70 | V | a/a’ | Barb2 | (CH2)4SO3− | SO3− | – | 4 Na+ |
| 71 | V | a/a’ | Barb5 | (CH2)4SO3− | SO3− | – | 4 Na+ |
| 72 | V | a/a’ | (Ph)HN- | C4H9 | H | – | PF6− |
| 73 | V | a/a’ | (Ph)HN- | (CH2)4SO3− | H | – | Na+ |
| 74 | V | a/a’ | IM+ | C4H9 | H | – | 2 PF6− |
| 75 | V | a/a’ | CH3 | (CH2)4SO3− | SO3− | – | 3Na+ |
| 76 | V | a/a’ | C6H13 | CH3 | H | – | ClO4− |
| 77 | V | a/a’ | CH3 | CH3 | H | – | Cl− |
| 78 | V | a/a’ | PMT | CH3 | H | – | Cl− |
| 79 | V | a/a’ | PMT | iC5H11 | H | – | DoPhSO3− |
| 80 | V | a/a’ | PMT | C2H5 | Cl | – | NTf2− |
| 81 | V | a/a’ | PMT | C2H5 | Cl | – | DoPhSO3− |
| 82 | V | a/a’ | PMT | C2H5 | Cl | – | ViPhSO3− |
| 83 | V | b/b’ | PMT | C2H5 | H | – | NTf2− |
| 84 | V | a/a’ | IM | C4H9 | H | – | PF6− |
| 85 | V | a/a’ | (Ph)HN- | C4H9 | Cl | – | PF6− |
| 86 | V | a/a’ | (Ph)HN- | (CH2)4SO3− | SO3− | – | 3 Na+ |
| 87 | V | a/a’ | (Ph)HN- | C4H9 | SO3− | – | Na+ |
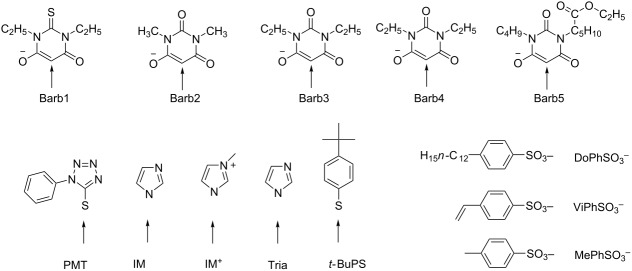
Table 2.
Summary of photophysical (absorption maximum λmaxabs, fluorescence maximum λmaxflu, extinction coefficient ε, fluorescence quantum yield Φf) and electrochemical data (oxidation potential Eox, and reduction potential Ered). Data of absorbersa relate to the structural patterns I–V. Supporting Information File 1 gives information of the electrochemical measurements (data were taken in CH3CN), and the determination of photophysical data. The latter complements previously published data [5–6] helping to receive a more completed pattern about these NIR absorbers.
| # | λmaxabs (nm) | λmaxflu (nm) | Φf (%) | ε (105 × M−1 cm−1) | solvent | Eox (V) | Ered (V) | Reference |
| 1 | 758 | 777 | 0.5 | 1.26 | MeOH | b | ||
| 2 | 858 | 877 | 0.3 | 2.09 | MeOH | 0.73 | −0.27 | b |
| 3 | 916 | MeOH | b | |||||
| 4 | 946 | 986 | 0.1 | 2.07 | MeOH | 0.69 | −1.01 | [5] |
| 5 | 973 | 2.08 | MeOH | b | ||||
| 6 | 995 | c | 0.001 | 1.76 | MeOH | 0.61 | −0.25 | [5] |
| 7 | 1024 | MeOH | b | |||||
| 8 | 1024 | MeOH | b | |||||
| 9 | 997 | MeOH | b | |||||
| 10 | 997 | 0.001 | 2.23 | MeOH | 0.66 | −0.34 | [5] | |
| 11 | 1014 | MeOH | b | |||||
| 12 | 746 | 774 | 26 | 2.80 | MeOH | b | ||
| 13 | 752 748 |
781 774 |
25 15 |
2.40 | MeOH H2O |
b | ||
| 14 | 766 | 784 | 20 | 3.04 | MeOH | b | ||
| 15 | 768 | 786 | 19 | 2.43 | MeOH | b | ||
| 16 | 740 | 766 | 19 | 2.22 | MeOH | b | ||
| 17 | 657 | 783 | 19 | 1.29 | MeOH | b | ||
| 18 | 760 | 778 | 19 | 3.28 | MeOH | b | ||
| 19 | 767 759 |
785 778 |
19 9 |
3.02 2.26 |
MeOH H2O |
b | ||
| 20 | 761 | 779 | 18 | 2.41 | MeOH | b | ||
| 21 | 744 | 765 | 18 | 2.08 | MeOH | b | ||
| 22 | 772 769 |
790 789 |
17 7 |
1.61 1.38 |
MeOH H2O |
b | ||
| 23 | 790 | 812 | 16 | 3.13 | MeOH | b | ||
| 24 | 792 | 815 | 16 | 2.88 | MeOH | 0.49 | −1.11 | [5] |
| 25 | 790 | 812 | 15 | 2.63 | MeOH | b | ||
| 26 | 777 | 800 | 15 | 3.58 | MeOH | 0.40 | −0.80 | [5] |
| 27 | 784 | 806 | 15 | 3.00 | MeOH | b | ||
| 28 | 755 | 772 | 15 | 2.79 | MeOH | b | ||
| 29 | 784 | 806 | 14 | 2.57 | MeOH | b | ||
| 30 | 763 | 781 | 14 | 2.34 | MeOH | b | ||
| 31 | 796 | 817 | 14 | 2.98 | MeOH | b | ||
| 32 | 796 790 |
818 813 |
13 6 |
2.84 2.30 |
MeOH H2O |
b | ||
| 33 | 763 | 787 | 13 | 3.15 | MeOH | 0.47 | −0.87 | [5] |
| 34 | 811 | 834 | 13 | 3.17 | MeOH | 0.42 | −0.87 | [5] |
| 35 | 794 | 818 | 13 | 2.60 | MeOH | b | ||
| 36 | 792 | d | 2.41 | MeOH | 0.57 | −0.60 | [5] | |
| 37 | d | b | d | d | d | [6] | ||
| 38 | 795 | 821 | 13 | 2.00 | MeOH | b | ||
| 39 | 796 | 819 | 11 | 1.57 | MeOH | d | d | b |
| 40 | 796 786 |
824 818 |
11 2 |
2.14 1.72 |
MeOH H2O |
d | d | b |
| 41 | 796 | 11 | 1.63 | MeOH | 0.60 | −0.57 | [5] | |
| 42 | 788 | 810 | 11 | 2.44 | MeOH | d | d | b |
| 43 | 759 | 781 | 11 | 2.92 | MeOH | d | d | b |
| 44 | 804 | 823 | 11 | 2.39 | MeOH | b | ||
| 45 | 789 782 |
807 800 |
11 5 |
2.49 2.15 |
MeOH H2O |
b | ||
| 46 | 773 | 797 | 10 | 1.97 | MeOH | 0.68 | −0.50 | [5] |
| 47 | 756 | 778 | 10 | 2.62 | MeOH | 0.41 | −0.86 | [5] |
| 48 | 791 | 811 | 10 | 2.76 | MeOH | 0.48 | −0.97 | [5] |
| 49 | 759 760 |
781 780 |
10 6 |
2.17 1.70 |
MeOH H2O |
d | d | b |
| 50 | 790 790 |
811 811 |
10 6 |
1.96 1.50 |
MeOH H2O |
d | d | b |
| 51 | 791 | 810 | 10 | 2.80 | MeOH | d | d | b |
| 52 | 812 | 831 | 10 | 2.66 | MeOH | d | d | b |
| 53 | 806 | 825 | 10 | 2.66 | MeOH | d | d | b |
| 54 | 782 | 801 | 10 | 2.63 | MeOH | d | d | b |
| 55 | 781 | 800 | 10 | 3.07 | MeOH | 0.68 | −0.50 | [5] |
| 56 | 813 804 |
832 824 |
9 4 |
2.41 2.27 |
MeOH H2O |
d | d | b |
| 57 | 790 | 807 | 9 | 2.89 | MeOH | d | d | b |
| 58 | 788 | 807 | 9 | 2.57 | MeOH | d | d | b |
| 59 | 784 | 819 | 9 | 2.33 | MeOH | b | ||
| 60 | 791 | 809 | 9 | 2.81 | MeOH | b | ||
| 61 | 820 | 842 | 8 | 2.78 | MeOH | d | d | b |
| 62 | 753 | 774 | 8 | 2.72 | MeOH | d | d | b |
| 63 | 775 | 792 | 7 | 2.37 | MeOH | d | d | b |
| 64 | 787 | 810 | 7 | 2.41 | MeOH | 0.75 | −0.49 | [5] |
| 65 | 793 | 815 | 7 | 2.77 | MeOH | b | ||
| 66 | 795 788 |
819 814 |
7 3 |
2.33 | MeOH H2O |
b | ||
| 67 | 801 794 |
824 818 |
6 3 |
2.46 2.04 |
MeOH H2O |
b | ||
| 68 | 827 819 |
850 842 |
6 3 |
2.24 2.29 |
MeOH H2O |
b | ||
| 69 | 781 | 799 | 6 | 2.51 | MeOH | b | ||
| 70 | 765 766 |
786 785 |
6 6 |
2.33 2.18 |
MeOH H2O |
b | ||
| 71 | 765 | 785 | 6 | 2.55 | MeOH | b | ||
| 72 | 745 | 791 | 6 | 1.66 | MeOH | b | ||
| 73 | 745 | 791 | 5 | 0.12 | MeOH | b | ||
| 74 | 798 | 816 | 5 | 2.13 | MeOH | b | ||
| 75 | 775 767 |
797 789 |
4 3 |
2.42 1.98 |
MeOH H2O |
b | ||
| 76 | 768 | 787 | 4 | 2.70 | MeOH | b | ||
| 77 | 763 | 785 | 3 | 2.60 | MeOH | b | ||
| 78 | 799 | 817 | 3 | 2.26 | MeOH | b | ||
| 79 | 813 | d | d | 1.62 | MeOH | d | d | b |
| 80 | 810 | d | d | d | MeOH | d | d | b |
| 81 | 807 | d | d | 2.40 | MeOH | 0.52 | −0.52 | [5] |
| 82 | 809 | d | d | 2.13 | MeOH | 0.64 | −0.52 | [5] |
| 83 | 844 | d | 1.35 | 0.56 | −0.57 | [5] | ||
| 84 | 785 | 803 | 2 | 2.93 | MeOH | 0.67 | −0.51 | [5] |
| 85 | 744 | 800 | 2 | 1.43 | MeOH | b | ||
| 86 | 739 731 |
795 788 |
1 0.1 |
1.01 0.90 |
MeOH H2O |
b | ||
| 87 | 736 | 795 | 0.8 | 1.10 | MeOH | b | ||
aFEW Chemicals GmbH provided those materials as samples, which were not previously investigated. bData were taken in this work to complement previously published data [5–6]. See Supporting Information File 1 for experimental details. cNot possible to determine. dNot determined due to similarity of above explored cyanine pattern, similar quantity was expected.
These synthetic efforts resulted in many distinct absorbers whose absorption and electrochemical properties differ. It facilitates their use with modern light sources such LEDs and lasers with emission in the NIR between 800–1100 nm. A purposeful use requires a more detailed understanding of the photochemistry occurring in such compounds.
Photophysics and photochemistry
General aspects
Figure 1 depicts photochemistry and photophysics of NIR absorbers from the simplest point of view. Excitation based on one-photon excitation (OP) results in population of higher vibrational levels of the first excited singlet state (S1), which fast relax by vibrational relaxation into the lowest vibrational level of the S1. The excited state possesses several competitive reaction pathways for deactivation. This is a photochemical reaction such as photoinduced electron transfer serving as source to generate reactive intermediates such as initiating radicals or conjugate acid [5]. Photophysical events of S1 occurring after one-photon excitation (OP) include internal conversion (IC), fast vibrational relaxation (VR) from higher vibrational modes of the S1 (v’ = 2, v’ = 1) into its lowest vibrational mode (v’ = 0), vibrational relaxation from lowest vibrational mode of the S1 into higher vibrational modes (for example v = 2) of the ground state (S0), intermolecular energy transfer from vibrationally hot molecules to their cold surrounding (vibrational cooling = VC) [73–74], and fluorescence (F) [75–76]. Most likely, nonradiative deactivation of such a hot molecule could be the transfer of its energy by collision with matrix molecules to the surrounding (VC) occurring from higher vibrational modes of the ground state formed by IC. Photochemical reactions typically occur from the lowest vibrational level (v’ = 0) of the S1. This figure does not contain an intersystem crossing route resulting in triplet states because this does not play any role considering the absorbers explored in this study. It was omitted because from our best knowledge there were no triplet reactions reported for heptamethines. In general, vibrational cooling may be seen as the main contributing to heat release (ΔT) into the surrounding matrix caused by collision with matrix molecules. Although deactivation by vibrational coupling competitively occurs as well, this reaction should play only a minor function in the deactivation scheme. There exists too much excessive energy, which would destroy the NIR absorber if the deactivation would mainly proceed by vibrational deactivation within the molecule. Therefore, transfer of the excessive energy by collision with matrix molecules appears as the most likely alternative which does not exclude the aforementioned vibrational deactivation within the molecule. The heat generated is large enough to initiate physical processes such as melting of powder coatings [16] or activation of thermal reaction such as blocked isocyanates [58]. It can exceed, depending on excitation intensity, several 100 °C. Particularly, embedded pigments with higher heat capacity can drive such systems to temperatures greater than 350 °C [77].
Figure 1.

Simplified process chart of photophysical processes occurring in NIR absorbers.
Nonradiative deactivation represents the main route of NIR absorbers to transfer the S1 to the S0. Its efficiency is higher than 85% [14] exhibiting an absorption between 750–850 nm, see also Table 2 vide infra. The non-radiative deactivation of absorbers with an absorption above 900 nm occurs almost quantitatively. This can be seen as one big feature of NIR absorbers; that is the function of a “molecular oven/furnace” whose operation bases on a photonic process, which can be easily turned ON or OFF by operation of the light source. Medical applications with focus on cancer therapy named this phenomenon photothermal treatment [78–80] In addition, heat generation on demand also explains the function of laser drying developed for the graphic industry [11–14] or laser welding [21–25] where the release of heat by a photonic event responsibly patterns a positive image on lithographic materials [11–14]. Moreover, traditional processes based on furnace techniques have been widely used in industry but their uneconomic efficiency has called a demand for more energy efficient drying technologies [18–20,54]. This can be physical and/or chemical drying of coatings. Particularly, photoinduced electron transfer enables chemical drying. For this purpose, systems based on photoinduced electron transfer with internal barrier facilitate their practical use in industrial systems. The thermal energy released helps to overcome the internal barrier of the system.
Equations 1–4 summarize these events discussed above whose rate constants quantify the individual processes. They occur from the S1 while no triplet states have been reported yet for the cyanines shown in Table 1. A modification of these patterns with more heavy atoms would definitively drive these into the triplet state and brings benefits known from UV systems to NIR systems either. This can be the generation of singlet oxygen and would enable the use of NIR absorbers in photodynamic therapy. Another benefit can be seen to increase the efficiency of photoinduced electron transfer needed in imaging applications. This would definitively decrease the efficiency of electron back transfer. Nowadays, typical electron acceptors (AC) applied in such systems related to either iodonium salts or triazines also possess a high capability of electron back transfer resulting in a decrease of the overall efficiency. PA relates to a photoactive compound, which functions in the examples as sensitizer Sens vide infra.
| [1] |
| [2] |
| [3] |
| [4] |
In practice, addition of an electron-donating compound (DN) often reduces tendency of electron back transfer in such singlet systems as shown in Equation 5 because the photo-oxidized sensitizer formed in Equation 4 reacts with DN resulting in back formation of PA while a new reaction intermediate DN+ is formed, Equation 6. This fast decomposes resulting in generation of reactive intermediates and makes the system irreversible.
| [5] |
| [6] |
The oxidized species (PA+) also competitively decomposes resulting in lower molecular weight products caused by cleavage of the methine chain and conjugate acid, see Equation 7 [5,63]. The latter can initiate ring opening polymerization of aziridines [5] and oxiranes [63]. We also prefer to use the term conjugate acid instead of proton. The use of the latter widely distributes in literature but is wrong because protons do not exist alone and data related to pH-values relate only to aqueous solutions. Ring opening by rhodamine B lactone quantitatively probed formation conjugate acid by formation of rhodamine B [5]. We interpret the term conjugate acid as a species in which the matrix binds H+ released by PA+.
| [7] |
Table 2 summarizes photophysical and electrochemical data of several dozens of absorbers. Some of the data may serve as reference for future studies since the availability of such data could be seen as more or less rare. The benzo[c,d]indolium group (compounds 1–11) offers the most interesting features regarding spectral variation covering a range between 750–1000 nm considering these examples. Structures 1, 2, and 5, which fit into the structural group of I, II and III, respectively. They demonstrate the expected absorption shift of about 100 nm by extension of the methine chain by one (CH=CH) unit. The fluorescence quantum yield of 1 and 2 was low while the extinction coefficient resided around 2 × 105 M−1 cm−1. The anions used were mostly derived from BF4−, PF6−, NFT2−, and n-C12H25-Ph-SO3−. This helps to improve the solubility in different surroundings and brings them to practice. Particularly, absorbers comprising the C12H25-Ph-SO3− anion for laser drying in offset printing depict one example [54]. For comparison, indolium (structure a/a’) and benzo[e]indolium end groups required more methine groups to obtain a comparable absorption in the NIR (compare structures 12–87). The planar c/c’-pattern helped to drive the desired absorption easier in the NIR. Nevertheless, applications using such absorbers in a sensitized oxidation mechanism benefit from their use. Such photoreactions typically result in cleavage of the methine chain. Nevertheless, the obtained photoproducts should not exhibit such a strong brownish color as observed in photochemical studies to explore the heptamethines 48 [63]. Nowadays, this arises as a big challenge to use such absorbers in applications where the color formed can be seen as an issue.
Absorbers comprising indolium and benzo[e]indolium derivatives resulted in materials exhibiting an observable fluorescence. The fluorescence quantum yield (Φf) obtained approached for some absorbers around 25%. This related to materials comprising the general pattern III. The Φf-data sometimes dropped by changing the substituent at the indolium nitrogen resulting in a decrease of the molar extinction coefficient (ε) as well. Aggregation/dimerization can cause such phenomena, which typically leads to a decrease of absorption at the considered absorption maximum and a pronounced non-radiative deactivation resulting in a decrease of Φf. Moreover, structures based on IV and V additionally provided absorbers with fluorescence quantum yields of up to 20% yield. Cyanines comprising at the meso-position either a phenyl or phenoxy group resulted in a Φf of about 15–20% (derivatives comprising PhO-: 14, 15, 22, and 30; derivatives comprising Ph-: 18 – 20, 23, 25, 28, 31, and 32). Absorbers with a barbiturate group at the same position resulted in fluorescence quantum yields between 10–16% (compare 24, 26, 27, 29, 33, 34, 42, 43, 47–51). Diphenylamino substitution at the meso-position showed a slight decrease of Φf (compare 35). Replacement with thiol moieties such as phenylmercapto and phenylmercapto-tetrazole substituents resulted in Φf of 7% (compare 64–68) and 3% (compare 78), respectively. Thus, one can roughly draw the following order for the decrease fluorescence quantum yields located at the meso-position:
PhO-; Ph- ≥ barbiturate ≥ Ph2N- > PhS- > phenylmercapto tetrazole.
Thus, it would be difficult to draw conclusions based on these substituents with respect to their electronic properties. While Ph-O, Ph2N-, and Ph-S- exhibit electron donating properties, barbiturate and phenyl substituents can be seen more or less as electron withdrawing moieties. Quantum chemical calculations showed only small contributions of the substituent placed at the meso-position to the electron density in the HOMO/LUMO pattern [81]. Presumably, coupling of lower occupied molecular orbitals with the HOMO on the one hand side and higher unoccupied MOs with the LUMO on the other side may give an answer regarding the electronic interaction of this substituent with the methine chain.
In general, these absorbers combine interesting features; that is emission on one hand side and the release of heat on the other side. The latter uptakes the major part; that is more than 80–95% depending on substitution. Thus, such absorbers can also work in systems where typically thermal processes initiated by furnace techniques play the major role. These absorbers generate heat on demand just by switching on and off the light source. Thus, they can be seen more or less as aforementioned molecular ovens. Recently development of light sources in the NIR based on LEDs brought new hope in this field [65]. Furthermore, the fact that still some fluorescence occurs makes them interesting as fluorescent probes for imaging applications. NIR radiation possesses a scattering coefficient being a third with respect to those measured at 400 nm [82]. Particularly, biological materials have received increased interest since they benefit from deeper penetration of radiation into the sample.
Absorbers with an absorption maximum >900 nm (compare 2–11) exhibit fluorescence quantum yields of <<1% showing that more than 99% of the absorbed light energy was converted to heat. From this point of view, they appear more or less as efficient molecular ovens, which was practically approved by melting of powder coatings applying lasers with line-shaped focus [16]. Thus, these absorbers possess large potential to replace thermal processes based on furnaces by introduction of such molecular ovens in combination with modern light sources [64–65,67]. The development of modern LED systems with emission in the NIR will enforce such developments [64].
Compatibility with the surroundings is the biggest challenge in practical applications of these systems. Thus, counter ions such as n-C12H25-Ph-SO3− (8, 10, 79, 81), NTf2− (4) or [Al(OC4F9)4]− (37) depict alternatives resulting in a solubility of 10–30 g/L in reactive monomers such as multifunctional acrylates [5], printing inks or powder coatings. Alternatively, the introduction of alkyl groups with branched moieties (79) interestingly improved solubility with the surrounding matrix as well [5]. As aforementioned noticed, this area appears more or less as semi-empirically disclosed and would require even more theoretical work based on molecular modeling to understand relationships/interactions between absorbers and a surrounding matrix in the near future.
The introduction of functional groups promoting the solubility in water results in absorbers exhibiting an appropriate water solubility, see Table 1 for structures. This can be either -N+RR’R’’ or a SO3− group. The fluorescence quantum yields obtained in water exhibit smaller values compared to those derivatives with no SO3− groups taken in MeOH. Nevertheless, the emission released should appear large enough for imaging application of biological samples.
Photoinduced electron transfer and internal barriers
Sensitized generation of reactive intermediates such as radicals and conjugate acid [5] also followed in the case of NIR sensitive materials a photoinduced electron transfer (PET). However, it did not work as smooth as disclosed for UV systems [83] because there was often no reaction between a cationic sensitizer and an electron acceptor; i.e. an iodonium salt [5]. However, the use of stronger emitting LEDs resulted sometimes in successful PET [67]. Particularly, light sources with low emission intensity arose these issues. This supports the idea that the system possesses an internal activation barrier resulting in a system having a certain energy threshold. Equation 8 shows how temperature affects the rate constant for electron transfer ket The free activation enthalpy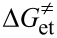 [67,72] controls the internal activation barrier, Equation 8 [65]. The free enthalpy of electron transfer (ΔGet) and the reorganization energy λ depict additional parameters affecting ket. Knowledge about redox potentials and excitation energy provides information about ΔGet needed for Equation 8 [72]. The dielectric constant ε and refractive index n of the surrounding matrix contribute to the outer sphere coordinates (λo) while geometric changes of the starting materials responsible tune inner-sphere coordinates λi (λ = λo + λi) [72].
[67,72] controls the internal activation barrier, Equation 8 [65]. The free enthalpy of electron transfer (ΔGet) and the reorganization energy λ depict additional parameters affecting ket. Knowledge about redox potentials and excitation energy provides information about ΔGet needed for Equation 8 [72]. The dielectric constant ε and refractive index n of the surrounding matrix contribute to the outer sphere coordinates (λo) while geometric changes of the starting materials responsible tune inner-sphere coordinates λi (λ = λo + λi) [72].
| [8] |
Many NIR sensitive systems possess an internal activation barrier although ΔGet < 0 using either the positively charged iodonium compound 88 (Ered = −0.64 V [5,84]) or the triazine 89 (Ered = −0.77 V [5]) carrying no charge as acceptor (Scheme 5). Thus, the positive charge of the iodonium cation is likely not the reason because 89 exhibits no charge. Both possess similar reduction potentials but the efficiency to initiate radical polymerization was higher in the case of 88 [5]. Experimentally, there is often no reaction under ambient light conditions. However, strong sources such as lasers with line-shaped focus or high-power NIR-LEDs helped to initiate photochemical reactions [64–65,67] . Recent studies confirmed these findings [85–88].
Scheme 5.

Chemical structure of the electron acceptors that were from iodonium cations 88 and triazines 89.
Crossing of the potential energy curves of the starting materials comprising Sens and 88 with those of the products (Sens+• and 88−•) results in the scenario shown in Figure 2 [65,67]. It requires under some circumstances to introduce additional heat also if ΔGet < 0 to cross the intrinsic barrier. NIR-sensitive systems comprising 12–87 and 13 or 14 result in ΔGet being +/−0.5 eV using 88 as acceptor [5]. The reorganization energy λ can approach values of 1–1.5 eV [65,70]. This helps to overcome the internal activation barrier. Non-radiative deactivation of the NIR absorber provides enough thermal energy to overcome the activation barrier. It can easily achieve temperatures >100 °C and may be certainly seen as a new concept also to drive thermally controlled reactions [56]. Again, the intrinsic activation barrier exhibits in all three examples a similar value while a) relates to endothermal conditions, b) corresponds to thermoneutral conditions and c) displays the scenario for exothermal conditions. The inverse case with extremely large ΔGet is not considered in this example. It also relates to large intrinsic .
Figure 2.

Photoinduced electron transfer under different scenarios in which each example exhibits an intrinsic activation barrier resulting in threshold systems a) endothermal conditions, b) thermoneutral conditions, c)exothermal conditions.
Only some of the absorbers showed chemical reactivity in the case of a low intensity (<100 mW/cm2) NIR-LED resulting in radicals and conjugate acid [5]. Most of them comprised a barbiturate group. 26, 34, 47, and 48 depict some representative examples. On the other hand, positively charged sensitizers showed no reactivity with such a low intensity LED [5]. Switching the LED source to a device providing significant higher exposure intensity resulted in a remarkable reactivity of even positively charged sensitizers [65]. This helped the system to travel over the internal activation barrier and facilitated access to photopolymerization reactions. Moreover, it also improved handling of such NIR sensitive materials under ambient room light conditions where the formulations appeared as stable.
Future developments should also focus on systems resulting in less yellowing upon exposure in the presence of an acceptor. As aforementioned discussed, the brown color formed may be seen as an issue. Particularly 48 showed remarkable polymerization efficiency in combination with 88 [5,63]. Photoinduced electron transfer typically followed an exothermal reaction route resulting in the formation of the cation radical of the sensitizer and the iodyl radical (Ar2-I•) formed by reduction of the diaryl iodonium salt, Scheme 6. This occurred from the singlet state while no indication has been available regarding the involvement of triplet states [89]. Sometimes singlet oxygen was believed to play a major role to explain the bleaching of cyanines [90–91]. This report does not consist with our findings where no formation of singlet oxygen was confirmed [89]. Other pathways request attention which could be formation of the O2-• radical to propose an alternative [90]. Such intermediates exhibit high reactivity resulting in similar photoproducts as disclosed for photoinduced electron transfer vide infra [63]. The cation radical formed cleaved at the methine chain resulting in the reaction products 33a–f. They appear as yellow/brownish materials and cause the partially unpleasant color appearance of thick clear coats.
Scheme 6.

Photoexcited absorber 33 results in reaction with an iodonium cation in the respective cation radical. Cleavage in the methine chain leads to the reaction products 33a–f.
Furthermore, the reaction products comprise nucleophilic nitrogen that inhibits cationic polymerization of epoxides [5]. Though conjugate acids are formed according to the routes shown in Scheme 6, cationic photopolymerization only succeeded with aziridines [5]. The different reaction mechanism occurring with such monomers explains these different observations [92]. Aziridines tolerate nucleophiles because chain growth requests the ammonium ion [93–96]. On the other hand, chain propagation includes the carbocation in the case of oxiranes and oxetanes [92].
More of interest can be seen absorbers, which do not follow such reaction routes. Scheme 7 depicts a general structure following not mainly the path shown in Scheme 7. They derive from the general structural pattern IV comprising a five-membered moiety in the center of the conjugated system. Compounds 35–37 represent some absorbers fitting in this scheme [65]. Particularly, the five-membered moiety opens new deactivation channels, which is the formation of an additional double bond [95–96]. This leads to a change of the conjugated system from a cyanine motif to a fulvene type. The latter shows, although if it contains one additional double bond and thus two additional π-electrons more, a hypsochromic shifted absorption of about 100 nm. Practically, such systems appear blue after exposure, which brings them to color on demand applications [97–98]. Furthermore, cleavage of the methine chain resulting in nucleophilic products seems to occur with minor importance. This explains why no brownish photoproducts were formed and cationic photopolymerization even of epoxides succeeded with good conversions vide infra. Additional absorbers fitting in this scheme comprise at the meso-position alternative substituents such as Phenyl or Chlorine (see Table 1 for comparison). Surprisingly, this reaction has not received remarkable attention in the academic community although industrial applications use this reaction to pattern imaged areas [97–98]. The structure was confirmed by direct synthesis following a procedure using MnO2[63].
Scheme 7.

Reaction scheme of absorbers comprising in the molecules center a five ring bridged moiety. This leads to two competitive pathways (from [65]. Schmitz, C.; Pang, Y.; Gülz, A.; Gläser, M.; Horst, J.; Jäger, M.; Strehmel, B., New High-Power LEDs Open Photochemistry for Near-Infrared-Sensitized Radical and Cationic Photopolymerization. Angew. Chem., Int. Ed. 2019, 58 (13), 4400-4404. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission).
Polymer synthesis
NIR sensitized radical photopolymerization
Photosensitized formation of reactive intermediates such as initiating radicals follows the Equations 1–7 vide supra. The system comprises as sensitizer an absorber derived from structure IV or V, and an electron acceptor such as 88 or 89. The main challenge can be seen to drive the system somehow that Equation 5 relating to electron back transfer occurs with minor importance. As vide infra mentioned, involvement of triplet states in such sensitized systems, which could decrease the rate for this reaction, does not play a major role. Thus, no change of spin multiplicity may cause a high rate of Equation 5 as long as the systems remains in the singlet state. There must be found an opportunity to make the system irreversible so that the electron back transfer in Equation 5 receives minor importance. Thus, cleavage of the main chain [63] (Scheme 6) or formation of distinct conjugated patterns [65] (Scheme 7) depict alternative reaction routes. In this case, the formation of initiating radicals follows an oxidative mechanism with respect to the sensitizer resulting in irreversibility of the system.
Sometimes, it was practiced adding a third component into the system exhibiting electron donating properties to keep the efficiency of Equation 5 on a low level. Thus, the donor transfers an electron into the SOMO of the Sens+•, Equation 6, resulting in back formation of the Sens. The oxidized donor unimolecular cleaves in a consecutive reaction resulting in formation of additional initiating radicals. Typical donors applied in such systems are borates 90 [99–114], phenyl imino acetic acid 91 [115] or mercapto triazoles 92 [116]. The latter often exists in its tautomeric thione form. Scheme 8 shows the respective structures.
Scheme 8.

Structure of donor compounds used in a three component system.
References [11–14,63–65] provide more details regarding the mechanistic details. Such systems have been successfully applied in CtP systems for more than 20 years [11–12]. Some pioneering works also disclose their use for chemical solidification of varnishes [5]. Remarkable has been still the function in powder coating, where they properly helped to melt and cure the applied powder monomers nearly just in one step [16]. Typical monomers used in CtP, liquid coatings and powder coatings derive from urethane (meth)acrylates, epoxy (meth)acrylates, polyester(meth)acrylates or polyether (meth)acrylates. Literature provides some reviews and original articles discussing mechanistic details [5–6,11–14,54,63–64,67,85].
NIR sensitized cationic photopolymerization
Equations 1–7 responsibly affect the efficiency of cationic photopolymerization. This was first demonstrated in 2016 [5] taking a combination of the sensitizer 48 and the iodonium cation 88 (used as PF6−-salt) to photopolymerize aziridines. The photoproducts formed according to Scheme 6 do not significantly inhibit chain growth of such monomers while no polymerization occurred with epoxides. Again, the strong nucleophilic properties of the photoproducts 33a–f did not permit successful run of cationic polymerization neither of oxiranes nor oxetanes. This became possible with the sensitizers 35–37 differing only with respect to their anion; that is [BF4]−, [PF6]− and [Al(O-t-C4F9)]− [6] after the first successful report of NIR-sensitized cationic photopolymerization of epoxides [65]. Though [Al(O-t-C4F9)4]− possesses the lowest nucleophilicity in this series, its high molecular weight requires a higher loading to obtain similar OD compared with sensitizers carrying anions with lower molecular weight. There exists still the issue regarding HF formation in the case of [PF6]–-salts while no issues have been reported for the aluminate [6]. The oxidized intermediate PA+• (Equation 7) forms the conjugate acid by decomposition of this instable intermediate resulting in nucleophilic photoproducts inhibiting ring opening polymerization mechanism where carbocations are involved.
Another strategy followed Scheme 7 in which formation of a fulvene like pattern prevented formation of nucleophilic photoproducts. This five ring pattern in the molecules center surprisingly prevents cleavage of the methine chain resulting in the oxidation mechanism in a fulvene pattern. The nitrogens in the cyanine chain still exhibit low nucleophilicity since electron density distributes over the entire methine chain. Thus, protonation of the nitrogen in the cyanine moiety occurs with less efficiency compared to amines [117]. This strategy helped to enable cationic photopolymerization with cyanine as sensitizers combined with 88 as PF6−-salt. Exposure with a high power NIR LED emitting at 805 nm initiated cationic photopolymerization of the epoxide Epikote 357, Figure 3. Decomposition of the oxidized sensitizer/oxidized photoactive compound PA+• (Equation 7) provided the conjugate acid needed for initiation. Thus, the higher the concentration of PA, the higher the conversion. Figure 3 nicely documents this scenario.
Figure 3.

Cationic photopolymerization of an epoxide (Epikote 828) initiated by excitation of the absorber 36 and bis(4-tert-butyl phenyl)iodonium hexafluorophosphate (2 wt %) applying different NIR sensitizer concentrations with an NIR-LED source emitting at 805 nm with an intensity of 1.2 W cm–2. (From [65]. Schmitz, C.; Pang, Y.; Gülz, A.; Gläser, M.; Horst, J.; Jäger, M.; Strehmel, B., New High-Power LEDs Open Photochemistry for Near-Infrared-Sensitized Radical and Cationic Photopolymerization. Angew. Chem., Int. Ed. 2019, 58 (13), 4400–4404. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission).
In 2019, the first report about NIR-sensitized cationic photopolymerization appeared in combination with a high intensity NIR LED [65]. Such a combination facilitated the NIR photoinitiating system to move over the internal reaction barrier as explained in Figure 2. Until this time, NIR LEDs exhibiting low intensity served as exposure source where only 36 showed photochemical activity in combination with an iodonium cation. The fact that such systems comprising a cationic absorber as sensitizer can also react was not recognized until this time since strong emitting devices were not available. This technology based on NIR absorbers possess big potential in the near future since the development of more devices with emission centering between 800–1000 nm has been in progress.
Tailor-made synthesis by photo-ATRP with NIR sensitizers
NIR absorbers/sensitizers have also reached the field of polymer synthesis; that is controlled/living radical polymerization (CLRP). This facilitates well-defined polymeric materials with pre-design architectures, predetermined molar masses and narrow molecular weight distributions. These techniques allow linear increase of molar mass by time and monomer conversion and low dispersity (principally between 1.2 to 1.5). The most common methods of controlled radical polymerization are namely the nitroxide-mediated radical polymerization (NMP) processes, reversible addition-fragmentation chain transfer (RAFT) and atom transfer radical polymerization (ATRP) [118]. While there has been no report available with NIR-sensitized NMP, there exist a few reports for RAFT polymerization with NIR light [119–121]. Recently, ATRP with Cu(II)-catalyst scaling in ppm amount in combination with a NIR sensitizer facilitated tailor-made synthesis of block copolymers [81]. This helped to reduce the heavy metal concentration in the polymerization system at a low level and complements therefore several methods developed to operate at low catalyst concentration by continuous regeneration of Cu(I) species using Cu(II) as starting material. These methods include initiators for continuous activator regeneration (ICAR) ATRP, activators regenerated by electron transfer (ARGET) ATRP, supplemental activators and reducing agent (SARA) ATRP, and electrochemically mediated ATRP (eATRP) [122–128]. In this content, various reducing agents, and electrochemical redox processes, and copper comprising nanoparticles have been accomplished to conduct ATRP by generating the required Cu(I) by the reduction of Cu(II) complexes. A well written review provides available references disclosing controlled polymer synthesis via photoinduced electron transfer reactions [129].
In addition to those approaches, photo-induced ATRP has been widely used to synthesize well-defined polymers by controlled radical polymerization either with or without heavy metal ions. In this process, basically, in the presence of photoactive materials (PAs) such as photoinitiators, photosensitizers or photoredox catalysts, the photoexcitation of a photoredox system results in formation of reactive radicals. Those reactive radicals add monomer and the polymerization proceeds. Several UV and visible light sensitive compounds in conjunction with or without Cu(II) complexes were used to initiate and control the photo-ATRP process (Scheme 9) [130–137]. In the absence of these photoactive materials, the direct irradiation of Cu(II) was presented for in situ generation of Cu(I) to initiate the polymerization with the alkyl bromide. In this system, the Cu(II) complex was exposed to UV light to form Cu(I) which can react with alkyl halide resulting in generation of reactive radicals and Cu(II), Scheme 9. Those reactive radicals add monomer and deactivated by reaction with Cu(II) resulting in formation of dormant species and Cu(I). Altogether, these reactions must be seen as equilibrium coexisting together.
Scheme 9.

Different modes of photoinitiated ATRP using UV, visible and NIR light.
Researchers also spent much time to develop photo-ATRP procedures working without any metal catalyst; that is the photoinduced metal-free ATRP strategy [132–137]. Latter reference provided a comprehensive overview demonstrating the function of phenothiazine derivatives, perylene, diaryl dihydrophenazines, anthracene or pyrene. In addition, type II photoinitiators including benzophenone, and thioxanthones were shown to realize photoinitiated controlled/living radical polymerization of various monomers in metal free conditions. In the visible region, several dyes (fluorescein, eosin Y, erythrosin B) worked well to mediate ATRP in the presence of amines [72]. The reversibility of the electron transfer steps provides the living nature of the process as well as control over the control of functionality as confirmed by the spectroscopic analyses, and chain extension and block copolymerization experiments [138].
The same strategies were also performed in NIR region using NIR sensitizers comprising cationic, zwitterionic and anionic patterns shown in Scheme 10. The polymers obtained exhibited quite high molecular weights (more than 2 × 105 g·mol−1) and failed to exhibit living character [81]. However, this system exhibited a successful route with Cu(II) catalysts at the ppm scale [81]. Quite recently, NIR light-induced ATRP has been performed at low catalyst concentrations using NIR sensitizers (Sens) as photo-reducing agents with NIR LEDs [81]. Among the NIR sensitizers investigated with cationic, zwitterionic, or anionic structures, only the zwitterionic structure 43 comprising a barbiturate moiety exhibited the successful activity under NIR light resulting in formation of polymers with controlled molecular weight characteristics and functionalities. Scheme 10 depicts the respective structural patterns.
Scheme 10.

The structure of Sens used in photo-ATRP.
UV–vis–NIR spectrum of the reaction components showed strong absorption of Sens in the NIR as shown by the huge extinction coefficients in Table 2 vide supra. Additionally, polymerization was not successful in the absence of Sens showing the necessity to polymerize NIR-absorber upon exposure with NIR light. This system facilitated control over molecular weight with low dispersity exhibition. Furthermore, chain extension and block copolymerization experiments confirmed the chain end fidelity and therefore the living character of polymerization (Table 3, Figure 4). On/Off experiments demonstrated the light dependency of polymerization [81].
Table 3.
NIR sensitized photo-ATRP of MMA under different experimental conditions (L: tris(2-pyridylmethyl)amine) with different sensitizers (Sens) [81].
| Sens | [MMA]/[EBPA]/[CuBr2]/[L]/[Sens] | Cu(II) (ppm) |
time (h) |
conversion (%) |
Mn (g·mol−1) |
Mw/Mn |
| 48 | 300/1/0.15/1.65/1.5 | 500 | 2 | 27.2 | 14000 | 1.47 |
| 48 | 300/1/0.03/0.135/0.3 | 100 | 24 | 44.3 | 13000 | 1.17 |
| 48 | 300/0/0.03/0.135/0.3 | 100 | 24 | – | – | – |
| 48 | 300/1/0.03/0.135/0 | 100 | 24 | – | – | – |
| 48 | 300/1/0/0/0.3 | 100 | 24 | 26.7 | 400000 | 2.47 |
| 78 | 300/1/0.03/0.135/0.3 | 100 | 24 | – | – | – |
| 24 | 300/1/0.03/0.135/0.3 | 100 | 24 | – | – | – |
| 93 | 300/1/0.03/0.135/0.3 | 100 | 24 | – | – | – |
| 94 | 300/1/0.03/0.135/0.3 | 100 | 24 | – | – | – |
Figure 4.

Comparison of the GPC traces of precursor PMMA with a) chain extended PMMA and b) PMMA-b-PS. Conditions for MMA macroinitiator).
The proposed mechanism (Figure 5) involves the excited state of the sensitizer which directly reduces Cu(II) to Cu(I). The combination of Cu(I) and R–X continues as usual in ATRP generating reactive radicals and halide anion [81]. These radicals add monomer resulting in formation of polymer radicals. The electron transfer from the formed halide anion to the cation radical Sens+• forms Sens back in the cycle. The system exhibited photocatalytic behavior with no big changes of absorption for 90 min under aerobic and anaerobic conditions. However, no polymerization occurred under air showing the inhibition of polymerization by oxygen [81].
Figure 5.

Spectral changes of the solution of 48 in the presence of [Cu(L)]Br2 (L: tris(2-pyridylmethyl)amine) and ethyl α-bromophenylacetate (EBPA) in DMF upon irradiation at 790 nm under nitrogen).
This described system opens the possibility to synthesize well-defined polymeric materials in a green way due to the photochemical stability, low copper-catalyst concentration and the use of NIR light. Taking advantage of good penetration of NIR light, this approach provides also the opportunity to embed UV absorbing materials in coatings and new pathways for bio-applications. Remarkable has been still the fact that this kind of photo-ATRP only worked with 48 carrying a barbiturate group. Such moieties interact with Cu(II) resulting in the formation of complexes as reported by Zwikker already in the 1930’s [139]. This special interaction occurring in particular between Sens 48 and Cu(II) helps to bring the reactants together and may explain the high selectivity in the NIR-sensitized photo-ATRP of this system. Again, the other sensitizers studied namely 78, 24, 93 and 94 showed no activity.
NIR light sensitized copper catalyzed azide–alkyne click reactions
In 2001, the 1,3-dipolar cycloaddition reaction of organic azides and alkynes has gained considerable attention in the fact they are simple to perform and work well under many conditions with high yields [140–142]. The different applications of Cu(I) catalyzed click chemistry have previously reviewed [142–145] while the general system was introduced more than 15 years ago [140–141]. Additional work was performed to develop methods for photoinduced generation of Cu(I) using Cu(II) without any sensitizer or in combination with different sensitizers for this type of click chemistry [143–146]. This mainly worked with either UV or visible light and helped to overcome the requirement of Cu(I) catalyst as substrate. Scheme 11 summarizes the reaction mechanism that includes NIR exposure as well [147] .
Scheme 11.

Photoinduced CuAAC reactions in which photochemical reactions result in formation of the Cu(I) catalyst. UV exposure photolysis of Cu(II) results in direct formation of the catalyst for the click reaction while photolysis with visible or NIR light requires the use of a sensitizer resulting in reduction of Cu(II) to Cu(I).
NIR sensitizers can also participate in photo-induced mediated click chemistry based on Cu(II), see Scheme 11. The successful electron transfer reaction from the excited NIR sensitizer to the Cu(II) complex for photoinduced ATRP [81] brought the idea to use NIR sensitizers as photoreducing agents for the generation of Cu(I) complex. We applied this system for NIR-light-induced copper–azine–alkyne (CuAAC) reactions for the synthesis of low molar-mass organic compounds and polymeric materials [147]. Different cyanines comprising either neutral or cationic structures were used to activate CuAAC reaction under NIR light (Scheme 11). Again, the sensitizer comprising a cyanine with a barbiturate group (2) showed the best activity for the formation of Cu(I) by photoreduction of Cu(II) and worked for the synthesis of low molecular weight compounds and polymers with high yields [147].
Initially, the successful reaction was observed with benzyl azide and phenylacetylene as model compounds. The mixture including NIR sensitizers together with CuBr2/PMDETA, benzyl azide and phenylacetylene were irradiated at 790 nm under anaerobic conditions for 2 hours (Scheme 12). Expectedly, the reaction using 1 as sensitizer failed to show the activity in this system. On the other hand, using 2 as photosensitizer in conjunction with Cu(II) complex showed successful reactivity with almost complete conversion (99%) confirmed by 1H NMR.
Scheme 12.

Model reaction between benzyl azide and phenyacetylene using the absorber 48 as NIR sensitizer at 790 nm.
Extension of this model reaction to polymers opens the possibility for tailor-made polymer synthesis by click chemistry resulting in formation of block copolymers. This type of copolymer synthesis was performed by reacting polystyrene-azide (PS-N3) with alkyne-terminated poly(ε-caprolactone) (Alkyne-PCL) in the presence of the barbiturate sensitizer 48 and CuBr2/PMDETA. The structure of block copolymer was confirmed by 1H NMR analysis and GPC chromatograms of precursors and the resulting block copolymer displayed a unimodal distribution indicating the absence of any other reactions during the click process. The shift of the molecular weight region also showed the successful photoinduced blocking process (Figure 6). This reaction facilitated to connect a hydrophilic and a hydrophobic block by the disclosed click chemistry.
Figure 6.

Block copolymerization of the precursors PS-N3 and Alkyne-PCL results in the block copolymer PS-b-PCL as shown by the respective GPC curves.
Figure 7 proposes the mechanism based on experimental and spectroscopic investigations. The photoinduced electron transfer from the excited state of the absorber reduces the Cu(II) complex resulting in Cu(I). This catalyzes the CuAAC reaction showing successful formation of the triazole moiety [147]. Photolysis of a mixture comprising benzyl azide, phenylacetylene, and Cu(II)Br2/PMDETA and 43 was also studied in a photobleaching experiment. Though the absorption decreases, its lowering is relatively small in this time frame since the reaction with 43 and an iodonium cation resulted in a disappearance of the absorption within less than a minute using the same light source. This smaller decrease of the absorption maximum indicated the capability of the formed cation radical either to decompose in side reactions.
Figure 7.

UV–vis–NIR absorption changes of the solution of 48 in the presence of PMDETA, phenylacetylene and benzyl azide in DMSO upon exposure at 790 nm (under nitrogen).
In this work, the use of NIR sensitizer in the photo-CuAAC reaction was shown in the near infrared region from model organic compounds to various macromolecular reactions including polymer end-group functionalization and block copolymer formation with the use of appropriate azide and alkyne click components. This system can be easily implemented in biological and synthetic materials particularly when surface patterning and incorporation of specific groups are required as NIR light is used to induce the click reaction.
This photoreaction possesses also potential to build up polymer networks with almost unique length of the distance between junctions. Such materials should exhibit less brittleness compared to those obtained in a process where chain propagation and termination control polymer formation; that is free radical polymerization. First attempts were made to synthesize such networks applying UV radiation, but this still required some heat treatment after exposure to finalize network formation. The systems disclosed in this contribution also offer enough heat provided in the non-radiative deactivation process, which is higher than ≈80%, Table 2. In addition, it also facilitates embedding of fillers covering an absorption in the UV.
Chemistry 4.0
General remarks [67]
Chemistry 4.0 bases on the idea of Industry 4.0 as introduced by the German association VCI (“Verband der Chemischen Industrie e.V.”). It discloses the industrial development starting with Chemistry 1.0 [148] in the 18th century where fossil energy (coal) and chemical processes (firing) generated mechanical power for industrial manufacturing. This is known as founder’s time or “Gründerzeit” relying on coal chemistry. Together with petrochemistry, Chemistry 2.0 was introduced for scale up of production and introduction of new classes of materials such as polymers. Modern development of the industrial society started production of fine chemicals resulting in opening of new ways of industrial chemistry in the early 1980s, which is known as Chemistry 3.0. The development of the internet and new powerful hard- and software where also mobile devices can compete in some applications with desktop computers have moved the focus to develop digital-based production technologies; that is Chemistry 4.0. This digital revolution of the chemical industry stands for sustainable recycling or sometimes called circular economy where waste serves as renewable source or feedstock of new production cycles. Such rapid developments will help to double the production volume as forecasted by 2040. Digitalization will contribute to get these goals. It also uses artificial intelligence (A.I.); that is software helping to learn from huge data sets. Machine learning (ML) represents a sub-group in these tools available. It also helps bring the classical design of experiment (DoE) into a next step which is named intelligent design of experiment (iDoE). NIR absorbers may uptake a function in these workflows since they have been used in coating formulations to design light sensitive materials used in CtP [10–14,66]. Scheme 13 depicts a typical setup of an iDoE [67].
Scheme 13.

Workflow to design and process new materials in a setup based on an intelligent DoE to develop technologies. Combination of chemistry (theoretical and synthetic chemistry), engineering and informatics creating artificial intelligence results in intelligent tools for development of new materials. NIR technology may benefit from these developments.
Absorber parameters based on concentration, absorption, and geometry importantly influence the workflow while the radiation source such as an LED or a laser provide the necessary excitation wavelengths and radiation intensity. High Throughput Formulation screening (HTFS), which still relates to automation and therefore Industry 3.0 standards, helps to process them in coating formulations where engineering parameters such as mixing, stirring, and characterization add as more variables to define parameter settings. The measured data train a defined digital model, which correlates input factors and characterization answers. Data analysis of this large data set occurs in a relatively short time frame where each experiment contributes to the model resulting in improvement. A classical DoE ends at this step resulting in information about the success of this experiment. However, an intelligent DoE applies advanced algorithms and improves by several iteration steps the model while its uncertainty becomes smaller. Such routines can predict results in smaller confidence intervals while the model improves in each iteration step to give the definition of new formulations. In other words, the program learns from failures where artificial intelligence and big data sciences uptake a major function. Such routines tolerate variations where robust formulations were applied in production cycles [66].
NIR sensitizers can be also found in Industry 4.0 applications, which relates to Chemistry 4.0 with focus on digitalization of chemical processes. It was translated from the more general term Industry 4.0. The objective is the acceleration and automization of activities like research and development of new materials, quality control, manufacturing processes, service for the customer and so on by devices setting up communication network called internet of things. Acquisition and sharing of data in its digital form for the data analysis is the key task introducing Chemistry 4.0 concepts. Including the characteristics of the materials such as photophysical and photochemical data of the cyanine based NIR absorbers can be seen as one illustrative example, which shows the benefits from the analysis of these data explaining the influence of the large numbers of factors of the absorber material on the processes being described in the preceding chapters. This considers small changes on the molecular pattern resulting in evidence alteration of material characteristics like deactivation processes, solubility, electrochemical properties, etc. and parameters in the area of application [67].
Digitalization, machine learning and Industry 4.0
Data analysis is the key aspect in the digitalization in chemistry since it derives models from the real existing chemical system. If the data cloud cover the entire area of interest and all influencing parameters, the model, also called digital twin, is capable to predict characteristics of a material or a process based on the experience from real experiments. The far field of machine learning algorithm has already been expanded to application areas like theoretical chemistry and organic synthesis to train models on data sets derived from experimental data and common rules in computational chemistry [149–151]. It has been shown that these models can compete with calculations from chemical laws and increment methods [151–153].
Further development of algorithms led to innovations, which are useful for the design-of-experiment (DoE) [154] rather than typically known factorial DoE. First reports claim a faster converging of the prognosis quality due to an intelligent design of experiments (iDoE) within the chosen boundaries even if a high number of experimental factors are included [155]. Scheme 14 shows a schematic diagram of the iDoE planning experiments as each adaption with a smaller number of experiments proposed by the A.I. Usually the sampling of the experiments in a DoE is done entirely before execution of the experiments based on the chosen model and chemical system to be analyzed. In contrast, the sampling of experiments during the execution of experiments and training of the model in an adaptive way offers a development of the iDoE based on the history of experiments being already executed. The parameters of the input will be set in spaces of the iDoE with a high uncertainty increasing the prognosis quality. In this direction, a predictive model can be achieved with a number of experiments as low as possible. The execution and training have to be aborted if the prognosis quality does not further increase. At best, this gives a valid model with a prognosis quality close to unity. Based on accurate data, a digital twin can be created for predicting characteristics varied by the input parameters also giving a confidence interval of the prediction.
Scheme 14.

Illustration of the iDoE setting up experiments suggested and analyzed by the A.I. After defining the space of the iDoE by the scientist the algorithm will suggest the starting points (a) and check the validity of the model. In case of a low prognosis quality the A.I. set new data points in experimental spaces with a lack of information (b) improving the model. The task will be finished if the model is capable to predict the outputs based on the inputs within a desired boundary (c).
In engineering processes, like photonic or thermal activation in a photoreactor or applied as coating films, the final properties are influenced by material properties and process parameter. Both types of influences can be introduced into one digital twin and analyzed by the iDoE. In case of applications using heptamethine cyanine absorbers inducing physical processes [16–17,54,58,64] or chemical reactions [5,81,147] the properties of the absorber are important factors besides other coming from distinct ingredients [5–6,10–14,54,63–65]. On the other hand, the analytical solution fails by generating a robust model because such complex relations cannot be described by one mathematical expression available from laws. However, a valid mathematical expression, with Y as the characteristic of the product, according to Equation 9 can be acquired by the empirical method applying machine learning algorithms. For this reason, a graphical description in Scheme 15 takes up again the complexity of the photopolymerization by photoelectron transfer reactions using near infrared LEDs as described vide supra.
Scheme 15.

Classification of the factors for the formation of polymer networks by NIR-photocuring depending on the absorber material, the monomers, parameters of the light source, the coinitiator and surrounding conditions. Each position consists of factors directly influencing the process and properties of the material chosen for the formulation at the 2nd layer. A digital twin created by data analysis using machine learning algorithms can describe the relation of the factors in sum.
| [9] |
xprocess,i = process parameters; xmaterial property,j = parameters from material properties of the ingredients/substrates; xconcentration,k = concentration parameters from the ingredients/substrates
The factors can be divided into certain sub-layers to influence the desired polymer properties. The formulation (e.g., concentrations) and the processing parameters (e.g., intensity, temperature) influence these results directly. However, properties of the material being chosen are factors in the second layer and have an impact on the predictability of the digital twin, too. Every detail can be explained by the experimental findings for each factor but is limited at forecasting the sum of these interacting factors.
On the other side the digital twin from this chemical operation derived by the machine learning algorithm will adaptively increase the prognosis quality if data of each parameter have been integrated into the iDoE. Also, the interactions of factors like the overlap of emission range of the light source and absorption range of the cyanine are taken into account. Again, NIR absorbers play a crucial role in these complex flow charts. Their material properties such as absorption wavelength, absorption coefficient, emission characteristics and electrochemical data enable them in large workflows used for ML.
Conclusion
This overview clearly demonstrates the potential of NIR absorbers for distinct applications although NIR has been still in its infancy in the field of photopolymerization. A certain existing lack regarding the function of light furthermore confirms such opinions since NIR light has been always considered as source to provide heat and not to initiate chemical and physical processes of molecules. This has surprised since several applications were almost reported until 2013 [57] after the first commercialization of CtP-technology applying NIR-lasers [11,54]. It will hopefully not need such a long period to expand the applicability of NIR absorbers/sensitizers for different purposes.
The compiled photophysical data in this overview will be a helpful toolbox for future design of NIR absorbers; that is absorption and emission maxima, extinction coefficient, and emission quantum yield. Electrochemical data complement the pattern. In addition, the compatibility of the surrounding matrix possesses a major function to transfer NIR absorbers for different applications. This has been often not considered with necessary care. From this point of view, organic chemistry possesses a key to tailor made the properties regarding absorption and solubility as shown by the examples in this overview. Since cyanines exhibit a positive charge in the methine chain, there exist huge possibilities to provide NIR absorbers whose solubility can nicely fit for distinct applications just be change of the anion. This can exhibit either more hydrophobic or hydrophilic properties. Modern tools based on machine learning and A.I. will help to accelerate the development of this field in a shorter time frame. Thus, prediction of molecule properties by quantum chemical methods, organic synthesis, the combination of photophysical data and the functions of NIR absorbers in several applications favors digitalization of chemistry based on the aforementioned described possibilities of machine learning.
Further progress will appear regarding additional uses in industrial applications. Though such materials have been almost available at large scale, more synthetic efforts based on ML in combination with theoretical tools to design molecules will drive this field towards more practical use.
Supporting Information
Information of the electrochemical measurements and the determination of photophysical data.
Acknowledgments
We acknowledge Chemspeed Technologies AG for graphical material showing their automization modules in Scheme 13. We additionally acknowledge Wiley to use graphical materials adapted in the graphical abstract from [67], graphical materials adapted in Scheme 13 from [67] (Strehmel, B.; Schmitz, C.; Cremanns, K.; Göttert, J., Photochemistry with Cyanines in the Near Infrared: A Step to Chemistry 4.0 Technologies. Chemistry – A European Journal 2019, 25 (56), 12855-12864. Copyright: the authors; article published by Wiley-VCH), graphical materials adapted from [81] (Kütahya, C.; Schmitz, C.; Strehmel, V.; Yagci, Y.; Strehmel, B., Near-Infrared Sensitized Photoinduced Atom-Transfer Radical Polymerization (ATRP) with a Copper(II) Catalyst Concentration in the ppm Range. Angew. Chem., Int. Ed. 2018, 57 (26), 7898-7902. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.) used in Figures 4 and 5 and graphical materials adapted from [147] (Kütahya, C.; Yagci, Y.; Strehmel, B., Near-Infrared Photoinduced Copper-Catalyzed Azide-Alkyne Click Chemistry with a Cyanine Comprising a Barbiturate Group. ChemPhotoChem 2019, 3 (11), 1180-1186. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.) for Figures 6 and 7.
Funding Statement
BS acknowledges the project D-NL-HIT carried out in the framework of INTERREG-Program Deutschland-Nederland, which is co-financed by the European Union, the MWIDE NRW, the Ministerie van Economische Zaken en Klimaat and the provinces of Limburg, Gelderland, Noord-Brabant und Overijssel. BS and CS additionally thank the county of North Rhine-Westphalia for funding of the project REFUBELAS (grant 005-1703-0006). Furthermore, AD acknowledges the support of the Federal Ministry of Economics and Technology of Germany funding the project NIR-Fluo (ZIM KF2108618 AK1).
References
- 1.Mustroph H. Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2014. Dyes, General Survey; pp. 1–35. [DOI] [Google Scholar]
- 2.Hamer F E. The Cyanine Dyes and Related Compounds. New York: Interscience Publishers, John Wiley & Sons; 1964. [Google Scholar]
- 3.Fabian J, Nakazumi H, Matsuoka M. Chem Rev. 1992;92:1197–1226. doi: 10.1021/cr00014a003. [DOI] [Google Scholar]
- 4.Sturmer D M. Syntheses and Properties of Cyanine and Related Dyes. In: Taylor E C, editor. Chemistry of Heterocyclic Compounds. Vol. 30. John Wiley & Sons; 1977. pp. 441–587. [DOI] [Google Scholar]
- 5.Schmitz C, Halbhuber A, Keil D, Strehmel B. Prog Org Coat. 2016;100:32–46. doi: 10.1016/j.porgcoat.2016.02.022. [DOI] [Google Scholar]
- 6.Kocaarslan A, Kütahya C, Keil D, Yagci Y, Strehmel B. ChemPhotoChem. 2019;3:1127–1132. doi: 10.1002/cptc.201900170. [DOI] [Google Scholar]
- 7.James N S, Chen Y, Joshi P, Ohulchanskyy T Y, Ethirajan M, Henary M, Strekowski L, Pandey R K. Theranostics. 2013;3:692–702. doi: 10.7150/thno.5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi H S, Nasr K, Alyabyev S, Feith D, Lee J H, Kim S H, Ashitate Y, Hyun H, Patonay G, Strekowski L, et al. Angew Chem, Int Ed. 2011;50:6258–6263. doi: 10.1002/anie.201102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipowska M, Patonay G, Strekowski L. Synth Commun. 1993;23:3087–3094. doi: 10.1080/00397919308011153. [DOI] [Google Scholar]
- 10.Vollmann H W. Angew Chem. 1980;92:95–106. doi: 10.1002/ange.19800920206. [DOI] [Google Scholar]
- 11.Baumann H, Hoffmann-Walbeck T, Wenning W, Lehmann H-J, Simpson C D, Mustroph H, Stebani U, Telser T, Weichmann A, Studenroth R. Ullmann's Encyclopedia of Industrial Chemistry. Weinheim, Germany: Wiley-VCH Verlag GmbH; 2015. Imaging Technology, 3. Imaging in Graphic Arts; pp. 1–51. [DOI] [Google Scholar]
- 12.Baumann H. Chem Unserer Zeit. 2015;49(1):14–29. doi: 10.1002/ciuz.201400642. [DOI] [Google Scholar]
- 13.Strehmel B, Ernst S, Reiner K, Keil D, Lindauer H, Baumann H. Z Phys Chem. 2014;228:129–153. doi: 10.1515/zpch-2014-0451. [DOI] [Google Scholar]
- 14.Brömme T, Schmitz C, Oprych D, Wenda A, Strehmel V, Grabolle M, Resch-Genger U, Ernst S, Reiner K, Keil D, et al. Chem Eng Technol. 2016;39:13–25. doi: 10.1002/ceat.201500453. [DOI] [Google Scholar]
- 15.Strehmel B, Schmitz C, Bromme T, Halbhuber A, Oprych D, Gutmann J S. J Photopolym Sci Technol. 2016;29(1):111–121. doi: 10.2494/photopolymer.29.111. [DOI] [Google Scholar]
- 16.Schmitz C, Strehmel B. ChemPhotoChem. 2017;1(1):26–34. doi: 10.1002/cptc.201600009. [DOI] [Google Scholar]
- 17.Schmitz C, Gökce B, Jakobi J, Barcikowski S, Strehmel B. ChemistrySelect. 2016;1:5574–5578. doi: 10.1002/slct.201601288. [DOI] [Google Scholar]
- 18.Pitz H, inventor. Vorrichtung und Verfahren zur Zuführung von Strahlungsenergie auf einen Bedruckstoff in einer Flachdruckmaschine. EP 1302735 A2. 2003
- 19.Schlörholz M, Keil D, Pitz H, inventors. Verfahren zum Trocknen von Druckfarbe auf einem Bedruckstoff. DE 102008028533 A1. 2008
- 20.Ernst S, Peiter G D, Pitz H, Reiner K, Mistol J, Schlörholz M, inventors. IR dyes and laser markable articles comprising such IR dyes. DE 2008013312 A1. 2007
- 21.Nakhaei M R, Mostafa Arab N B, Kordestani F. Adv Mater Res. 2012;445:454–459. doi: 10.4028/www.scientific.net/amr.445.454. [DOI] [Google Scholar]
- 22.Brunnecker F, Sieben M. Laser Tech J. 2010;7:24–27. doi: 10.1002/latj.201090072. [DOI] [Google Scholar]
- 23.Wissemborski R, Klein R. Laser Tech J. 2010;7:19–22. doi: 10.1002/latj.201090070. [DOI] [Google Scholar]
- 24.Jones I A, Taylor N S, Sallvanti R, Griffith J. Int J Res Eng Technol. 2014;2:191–206. [Google Scholar]
- 25.Ernst S, Keil D, Reiner K, Senns B, inventors. NIR-Absorber Additive für das Laserstrahlschweißen von Kunststoffen. DE102016213372A1 . 2018
- 26.Pérez-Barrado E, Darton R J, Guhl D. MRS Commun. 2018;8(03):1070–1078. doi: 10.1557/mrc.2018.144. [DOI] [Google Scholar]
- 27.Patel C, Patel A J, Patel R C. Int J Sci Res Dev. 2017;5:147–150. doi: 10.18203/2349-3259.ijct20162796. [DOI] [Google Scholar]
- 28.Loccufier J, inventor. Developing Agent Precursor for Laser Markable Composiitons. WO2018228857A1. 2018
- 29.Waumans B, Geuens I, Callant P, Van Aert H, inventors. Security documents and colour laser marking methods for securing them. US8975211B2. 2015
- 30.Thaker J, inventor. Laser fusible coating compositions used for laser marking systems and methods. WO2011162814A2. 2011
- 31.Koenemann M, Boehm A, Pschirer N G, Qu J, Mattern G, inventors. Substituierte Rylenderivate. WO2007006717A1. 2007
- 32.Brooker L G S. Sensitizing and Desensitizing Dyes. In: Mees C E K, James T H, editors. The Theory of the Photographic Process. 3rd ed. New York: Macmillan; 1966. pp. 198–232. [Google Scholar]
- 33.Sturmer D M, Heseltine D W. Sensitizing and Desensitizing Dyes. In: James T H, editor. The Theory of the Photographic Process. 4th ed. New York: Macmillan; 1977. pp. 194–234. [Google Scholar]
- 34.Keller K, Kampfer H, Matejec R, Lapp O, Krafft W, Frenken H, Lührig H, Scheerer R, Heilmann M, Meckl H, et al. Ullmann's Encyclopedia of Industrial Chemistry. Weinheim, Germany: Wiley-VCH Verlag GmbH; 2000. Photography. [DOI] [Google Scholar]
- 35.Ficken G E. Cyanine Dyes. In: Venkataraman K, editor. The Chemistry of Synthetic Dyes. Vol. 4. New York: Academic Press; 1971. pp. 211–340. [DOI] [Google Scholar]
- 36.Berlin L, Riester O. Methoden der organischen Chemie. 5/1d. Stuttgart: Thieme; 1972. Methoden zur Herstellung von Cyaninen (Polymethinen) pp. 229–299. [Google Scholar]
- 37.Sturmer D M. Syntheses and Properties of Cyanine and Related Dyes. In: Weissberger A, Taylor E C, editors. The Chemistry of Heterocyclic Compounds. Vol. 30. New York: Wiley; 1977. pp. 441–587. [DOI] [Google Scholar]
- 38.Sturmer D M. Kirk-Othmer Encyclopedia of Chemical Technology. 3rd ed. Vol. 7. New York: Wiley; 1979. Cyanine Dyes; pp. 335–358. [Google Scholar]
- 39.Raue R, Riester O. Ullmanns Enzyklopädie der Technischen Chemie. 5th ed. Vol. 16. Weinheim: Verlag Chemie; 1979. Methinfarbstoffe; pp. 635–669. [Google Scholar]
- 40.Mishra A, Behera R K, Behera P K, Mishra B K, Behera G B. Chem Rev. 2000;100:1973–2012. doi: 10.1021/cr990402t. [DOI] [PubMed] [Google Scholar]
- 41.Steiger R, Hediger H, Junod P. Photogr Sci Eng. 1980:185–195. [Google Scholar]
- 42.Beretta P, Jaboli A. Photogr Sci Eng. 1974;18:197–206. [Google Scholar]
- 43.Herz A H. Photogr Sci Eng. 1974;18:323–335. [Google Scholar]
- 44.Herz A H. Photogr Sci Eng. 1974;18:207–215. [Google Scholar]
- 45.Norland K, Ames A, Taylor T. Photogr Sci Eng. 1970;14:295–307. [Google Scholar]
- 46.McRae E G. Aust J Chem. 1961;14(3):329–343. doi: 10.1071/ch9610329. [DOI] [Google Scholar]
- 47.Kasha M, Rawls H R, Ashraf El-Bayoumi M. Pure Appl Chem. 1965;11:371–392. doi: 10.1351/pac196511030371. [DOI] [Google Scholar]
- 48.McRae E G, Kasha M. J Chem Phys. 1958;28:721–722. doi: 10.1063/1.1744225. [DOI] [Google Scholar]
- 49.Hestand N J, Spano F C. Chem Rev. 2018;118:7069–7163. doi: 10.1021/acs.chemrev.7b00581. [DOI] [PubMed] [Google Scholar]
- 50.Scheibe G. Angew Chem. 1937;50:212–219. doi: 10.1002/ange.19370501103. [DOI] [Google Scholar]
- 51.Jelley E E. Nature. 1937;139:631. doi: 10.1038/139631b0. [DOI] [Google Scholar]
- 52.Dewar M J S. J Chem Soc. 1950:2329–2334. doi: 10.1039/jr9500002329. [DOI] [Google Scholar]
- 53.Knott E B. J Chem Soc. 1951:1024–1028. doi: 10.1039/jr9510001024. [DOI] [Google Scholar]
- 54.Strehmel B, Brömme T, Schmitz C, Reiner K, Ernst S, Keil D. Dyes and Chromophores in Polymer Science. Hoboken, NJ, U.S.A.: John Wiley & Sons, Inc.; 2015. NIR-Dyes for Photopolymers and Laser Drying in the Graphic Industry; pp. 213–249. [DOI] [Google Scholar]
- 55.Color relates to the spectral sensitivity of the human eye covering 400–700 nm. The cones comprise three sensitive pigments for blue, green and red. The combined stimulus signal of the three visual pigments results in perception of color. An equally strong stimulus signal in the green and red areas produces the color sensation yellow if these colors possess the same intensity. The fact that one interprets 770 nm still as red relates to the fact the eye translates this heat related radiation into a slight red.
- 56.Yuan Z, Lee S-L, Chen L, Li C, Mali K S, De Feyter S, Müllen K. Chem – Eur J. 2013;19:11842–11846. doi: 10.1002/chem.201302086. [DOI] [PubMed] [Google Scholar]
- 57.Wang Z Y. Near-Infrared Organic Materials and Emerging Applications. Boca Raton: CRC Press; 2013. [DOI] [Google Scholar]
- 58.Schmitz C, Strehmel B. J Coat Technol Res. 2019;16(6):1527–1541. doi: 10.1007/s11998-019-00197-3. [DOI] [Google Scholar]
- 59.Lee H, Berezin M Y, Henary M, Strekowski L, Achilefu S. J Photochem Photobiol, A. 2008;200:438–444. doi: 10.1016/j.jphotochem.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benson R C, Kues H A. J Chem Eng Data. 1977;22:379–383. doi: 10.1021/je60075a020. [DOI] [Google Scholar]
- 61.Pouradier J. J Chim Phys. 1964;61:1107–1114. doi: 10.1051/jcp/1964611107. [DOI] [Google Scholar]
- 62.Kim J S, Kodagahally R, Strekowski L, Patonay G. Talanta. 2005;67:947–954. doi: 10.1016/j.talanta.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 63.Brömme T, Schmitz C, Moszner N, Burtscher P, Strehmel N, Strehmel B. ChemistrySelect. 2016;1(3):524–532. doi: 10.1002/slct.201600048. [DOI] [Google Scholar]
- 64.Schmitz C, Oprych D, Kutahya C, Strehmel B. NIR Light for Initiation of Photopolymerization. In: Lalevée J, Fouassier J-P, editors. Photopolymerisation Initiating Systems. Royal Society of Chemistry; 2018. pp. 431–478. [DOI] [Google Scholar]
- 65.Schmitz C, Pang Y, Gülz A, Gläser M, Horst J, Jäger M, Strehmel B. Angew Chem, Int Ed. 2019;58:4400–4404. doi: 10.1002/anie.201813696. [DOI] [PubMed] [Google Scholar]
- 66.Plümer L, Korolik P, Koifman D, Baumann H, Strehmel B, inventors. US20110189600A1. Method for automated control of processing parameters. 2011
- 67.Strehmel B, Schmitz C, Cremanns K, Göttert J. Chem – Eur J. 2019;25(56):12855–12864. doi: 10.1002/chem.201901746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen M T, Locas M A, inventors. Thermally reactive near-infrared absorbing acetal copolymers, methods of preparation and methods of use. US20060275698A1. 2006
- 69.Roschger P, Michaelis S, Hassenrueck K, Berneth H, Callant P, inventors. Verfahren zur Herstellung von Cyaninfarbstoffen. DE4331162A1. 1995
- 70.Baumann H, Ernst U, Goez M, Griesbeck A, Oelgemöller M, Oppenländer T, Schlörholz M, Strehmel B. Nachr Chem. 2014;62:507–512. doi: 10.1002/nadc.201490153. [DOI] [Google Scholar]
- 71.Ignatyev N D, Kucheryna A D, Welz-Biermann U D, Willner H P D, inventors. FAP-Farbstoffe. DE10357360 A1. 2005
- 72.Kavarnos G J, Turro N J. Chem Rev. 1986;86:401–449. doi: 10.1021/cr00072a005. [DOI] [Google Scholar]
- 73.Aulin Y V, Liu M, Piotrowiak P. J Phys Chem Lett. 2019;10:2434–2438. doi: 10.1021/acs.jpclett.9b00406. [DOI] [PubMed] [Google Scholar]
- 74.Dlott D D. J Lumin. 1990;45:397–400. doi: 10.1016/0022-2313(90)90204-o. [DOI] [Google Scholar]
- 75.Mustroph H. ChemPhysChem. 2016;17:2616–2629. doi: 10.1002/cphc.201600243. [DOI] [PubMed] [Google Scholar]
- 76.Mustroph H, Ernst S. Chem Unserer Zeit. 2011;45:256–269. doi: 10.1002/ciuz.201100547. [DOI] [Google Scholar]
- 77.Explorative curing studies of powder coatings as pursued in reference [16] showed temperature increase up to 200 °C while no pigment was embedded. Addition of iron oxide pigments significantly resulted in an increase of the temperature during processing up to 350 °C using the same setup.
- 78.Lee S, George Thomas R, Ju Moon M, Ju Park H, Park I-K, Lee B-I, Yeon Jeong Y. Sci Rep. 2017;7:2108. doi: 10.1038/s41598-017-01108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan Y, Chen J, Yang Z, Zhang X, Liu Z, Hua J. J Mater Chem B. 2018;6:7420–7426. doi: 10.1039/c8tb01750f. [DOI] [PubMed] [Google Scholar]
- 80.Xu Q, Shen Y, Zhang Y, Shao X. Bioorg Med Chem Lett. 2019;29:2398–2404. doi: 10.1016/j.bmcl.2019.05.057. [DOI] [PubMed] [Google Scholar]
- 81.Kütahya C, Schmitz C, Strehmel V, Yagci Y, Strehmel B. Angew Chem, Int Ed. 2018;57:7898–7902. doi: 10.1002/anie.201802964. [DOI] [PubMed] [Google Scholar]
- 82.Uo M, Kudo E, Okada A, Soga K, Jogo Y. J Photopolym Sci Technol. 2009;22:551–554. doi: 10.2494/photopolymer.22.551. [DOI] [Google Scholar]
- 83.Schwalm R. UV Coatings. Basics, Recents Developments and New Applications. Oxford: Elsevier; 2007. [DOI] [Google Scholar]
- 84.Romańczyk P P, Kurek S S. Electrochim Acta. 2017;255:482–485. doi: 10.1016/j.electacta.2017.09.166. [DOI] [Google Scholar]
- 85.Bonardi A H, Dumur F, Grant T M, Noirbent G, Gigmes D, Lessard B H, Fouassier J-P, Lalevée J. Macromolecules. 2018;51(4):1314–1324. doi: 10.1021/acs.macromol.8b00051. [DOI] [Google Scholar]
- 86.Bonardi A-H, Bonardi F, Morlet-Savary F, Dietlin C, Noirbent G, Grant T M, Fouassier J-P, Dumur F, Lessard B H, Gigmes D, et al. Macromolecules. 2018;51(21):8808–8820. doi: 10.1021/acs.macromol.8b01741. [DOI] [Google Scholar]
- 87.Bonardi A, Bonardi F, Noirbent G, Dumur F, Dietlin C, Gigmes D, Fouassier J-P, Lalevée J. Polym Chem. 2019;10(47):6505–6514. doi: 10.1039/c9py01447k. [DOI] [Google Scholar]
- 88.Bonardi A H, Bonardi F, Dumur F, Gigmes D, Fouassier J P, Lalevée J. Macromol Rapid Commun. 2019;40(23):1900495. doi: 10.1002/marc.201900495. [DOI] [PubMed] [Google Scholar]
- 89.In 2013, samples of 34 and 48 were transferred to the lab of Prof. P. R. Ogilby in Åarhus (Denmark) to quantify singlet oxygen formed. He confirmed that there was no singlet oxygen formed. This rules out formation of triplet states that could build singlet oxygen.
- 90.Samanta A, Vendrell M, Das R, Chang Y-T. Chem Commun. 2010;46:7406–7408. doi: 10.1039/c0cc02366c. [DOI] [PubMed] [Google Scholar]
- 91.Gorka A P, Nani R R, Zhu J, Mackem S, Schnermann M J. J Am Chem Soc. 2014;136:14153–14159. doi: 10.1021/ja5065203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Odian G. Principles of Polymerization. Hoboken: John Wiley & Sons; 2004. [DOI] [Google Scholar]
- 93.Weyts K F, Goethals E J. Polym Bull. 1988;19:13–19. doi: 10.1007/bf00255018. [DOI] [Google Scholar]
- 94.Goethals E J, Van de Velde M, Eckhaut G, Bouquet G. ACS Symp Ser. 1985;286:219–229. doi: 10.1021/bk-1985-0286.ch017. [DOI] [Google Scholar]
- 95.Goethals E J, Vlegels M A. Polym Bull. 1981;4:521–525. doi: 10.1007/bf00284064. [DOI] [Google Scholar]
- 96.Goethals E J, Schacht E H, Bruggeman P, Bossaer P. ACS Symp Ser. 1977;59:1–12. doi: 10.1021/bk-1977-0059.ch001. [DOI] [Google Scholar]
- 97.Iwai Y, Kunita K, inventors. Compound having polymethine-chain structure, image forming material, planographic printing plate precursor, and image forming method using the same, method of making planographic printing plate, and planographic printing method. US20070212643A1. 2007
- 98.Iwai Y, Kunita K, inventors. Compound having polymethine-chain structure, image forming material, planographic printing plate precursor, and image forming method using the same, method of making planographic printing plate, and planographic printing method. EP1849836 A2. 2006
- 99.Simpson P, Baumann H, Strehmel B, inventors. Sensitizer/initiator combination for negative-working thermal-sensitive compositions usable for lithographic plates. WO2009109579 A1. 2009
- 100.Kropp M A, Baillargeon M, Park K M, Bhamidapaty K, Schuster G B. J Am Chem Soc. 1991;113:2155–2163. doi: 10.1021/ja00006a038. [DOI] [Google Scholar]
- 101.Schuster G B. Pure Appl Chem. 1990;62:1565–1572. doi: 10.1351/pac199062081565. [DOI] [Google Scholar]
- 102.Chatterjee S, Davis P D, Gottschalk P, Kurz M E, Sauerwein B, Yang X, Schuster G B. J Am Chem Soc. 1990;112:6329–6338. doi: 10.1021/ja00173a022. [DOI] [Google Scholar]
- 103.Kabatc J, Zasada M, Paczkowski J. J Polym Sci, Part A: Polym Chem. 2007;45:3626–3636. doi: 10.1002/pola.22112. [DOI] [Google Scholar]
- 104.Bruder F-K, Fäcke T, Rölle T. Polymers (Basel, Switz) 2017;9(12):472. doi: 10.3390/polym9100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bruder F-K, Hagen R, Rölle T, Weiser M-S, Fäcke T. Angew Chem. 2011;123:4646–4668. doi: 10.1002/ange.201002085. [DOI] [PubMed] [Google Scholar]
- 106.Bruder F-K, Deuber F, Facke T, Hagen R, Honel D, Jurberg D, Kogure M, Rolle T, Weiser M-S. J Photopolym Sci Technol. 2009;22(2):257–260. doi: 10.2494/photopolymer.22.257. [DOI] [Google Scholar]
- 107.Sarker A M, Kaneko Y, Neckers D C. J Photochem Photobiol, A. 1999;121:83–90. doi: 10.1016/s1010-6030(98)00444-4. [DOI] [Google Scholar]
- 108.Kaneko Y, Sarker A M, Neckers D C. Chem Mater. 1999;11:170–176. doi: 10.1021/cm980618w. [DOI] [Google Scholar]
- 109.Sarker A M, Lungu A, Mejiritski A, Kaneko Y, Neckers D C. J Chem Soc, Perkin Trans 2. 1998:2315–2322. doi: 10.1039/a800469b. [DOI] [Google Scholar]
- 110.Sarker A M, Kaneko Y, Nikolaitchik A V, Neckers D C. J Phys Chem A. 1998;102:5375–5382. doi: 10.1021/jp972855c. [DOI] [Google Scholar]
- 111.Popielarz R, Sarker A M, Neckers D C. Macromolecules. 1998;31:951–954. doi: 10.1021/ma971287w. [DOI] [Google Scholar]
- 112.Feng K, Zang H, Martin D, Marino T L, Neckers D C. J Polym Sci, Part A: Polym Chem. 1998;36(10):1667–1677. doi: 10.1002/(sici)1099-0518(19980730)36:10<1667::aid-pola20>3.0.co;2-d. [DOI] [Google Scholar]
- 113.Sarker A M, Lungu A, Neckers D C. Macromolecules. 1996;29:8047–8052. doi: 10.1021/ma960818y. [DOI] [Google Scholar]
- 114.Hassoon S, Sarker A, Polykarpov A Y, Rodgers M A J, Neckers D C. J Phys Chem. 1996;100:12386–12393. doi: 10.1021/jp960931q. [DOI] [Google Scholar]
- 115.Hauck G, Savariar-Hauck C, Timpe H-J, inventors. Kodak Polychrome Graphics. DE 19906823 A1. 2000
- 116.Berdzinski S, Strehmel B, Strehmel V. Photochem Photobiol Sci. 2015;14:714–725. doi: 10.1039/c4pp00386a. [DOI] [PubMed] [Google Scholar]
- 117.Strekowski L, Lee H, Mason J C, Say M, Patonay G. J Heterocycl Chem. 2009;44:475–477. doi: 10.1002/jhet.5570440233. [DOI] [Google Scholar]
- 118.Matyjaszewski K, Davis T P, editors. Handbook of Radical Polymerization. Hoboken, NJ, U.S.A.: John Wiley & Sons, Inc.; 2002. [DOI] [Google Scholar]
- 119.Jiang J, Ye G, Wang Z, Lu Y, Chen J, Matyjaszewski K. Angew Chem, Int Ed. 2018;57:12037–12042. doi: 10.1002/anie.201807385. [DOI] [PubMed] [Google Scholar]
- 120.McKenzie T G, Fu Q, Uchiyama M, Satoh K, Xu J, Boyer C, Kamigaito M, Qiao G G. Adv Sci. 2016;3(9):1500394. doi: 10.1002/advs.201500394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shanmugam S, Xu J, Boyer C. Angew Chem. 2016;128:1048–1052. doi: 10.1002/ange.201510037. [DOI] [PubMed] [Google Scholar]
- 122.Matyjaszewski K. Macromolecules. 2012;45(10):4015–4039. doi: 10.1021/ma3001719. [DOI] [Google Scholar]
- 123.Ribelli T G, Fantin M, Daran J-C, Augustine K F, Poli R, Matyjaszewski K. J Am Chem Soc. 2018;140:1525–1534. doi: 10.1021/jacs.7b12180. [DOI] [PubMed] [Google Scholar]
- 124.Enciso A E, Fu L, Russell A J, Matyjaszewski K. Angew Chem, Int Ed. 2018;57(4):933–936. doi: 10.1002/anie.201711105. [DOI] [PubMed] [Google Scholar]
- 125.Yang Y, Liu X, Ye G, Zhu S, Wang Z, Huo X, Matyjaszewski K, Lu Y, Chen J. ACS Appl Mater Interfaces. 2017;9:13637–13646. doi: 10.1021/acsami.7b01863. [DOI] [PubMed] [Google Scholar]
- 126.Matyjaszewski K. Chem Int. 2017;39(4):7–11. doi: 10.1515/ci-2017-0404. [DOI] [Google Scholar]
- 127.Ribelli T G, Konkolewicz D, Pan X, Matyjaszewski K. Macromolecules. 2014;47:6316–6321. doi: 10.1021/ma501384q. [DOI] [Google Scholar]
- 128.Ribelli T G, Konkolewicz D, Bernhard S, Matyjaszewski K. J Am Chem Soc. 2014;136:13303–13312. doi: 10.1021/ja506379s. [DOI] [PubMed] [Google Scholar]
- 129.Dadashi-Silab S, Doran S, Yagci Y. Chem Rev. 2016;116:10212–10275. doi: 10.1021/acs.chemrev.5b00586. [DOI] [PubMed] [Google Scholar]
- 130.Tasdelen M A, Uygun M, Yagci Y. Macromol Rapid Commun. 2011;32:58–62. doi: 10.1002/marc.201000351. [DOI] [PubMed] [Google Scholar]
- 131.Tasdelen M A, Uygun M, Yagci Y. Macromol Chem Phys. 2010;211:2271–2275. doi: 10.1002/macp.201000445. [DOI] [Google Scholar]
- 132.Treat N J, Sprafke H, Kramer J W, Clark P G, Barton B E, Read de Alaniz J, Fors B P, Hawker C J. J Am Chem Soc. 2014;136:16096–16101. doi: 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]
- 133.Pan X, Fang C, Fantin M, Malhotra N, So W Y, Peteanu L A, Isse A A, Gennaro A, Liu P, Matyjaszewski K. J Am Chem Soc. 2016;138:2411–2425. doi: 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]
- 134.Kutahya C, Allushi A, Isci R, Kreutzer J, Ozturk T, Yilmaz G, Yagci Y. Macromolecules. 2017;50:6903–6910. doi: 10.1021/acs.macromol.7b01335. [DOI] [Google Scholar]
- 135.Aydogan C, Kutahya C, Allushi A, Yilmaz G, Yagci Y. Polym Chem. 2017;8:2899–2903. doi: 10.1039/c7py00069c. [DOI] [Google Scholar]
- 136.Kutahya C, Aykac F S, Yilmaz G, Yagci Y. Polym Chem. 2016;7:6094–6098. doi: 10.1039/c6py01417h. [DOI] [Google Scholar]
- 137.Jockusch S, Yagci Y. Polym Chem. 2016;7:6039–6043. doi: 10.1039/c6py01410k. [DOI] [Google Scholar]
- 138.Stenzel M H, Barner-Kowollik C. Mater Horiz. 2016;3:471–477. doi: 10.1039/c6mh00265j. [DOI] [Google Scholar]
- 139.Zwikker J L. Pharm Weekbl. 1933;70:551–559. [Google Scholar]
- 140.Kolb H C, Finn M G, Sharpless K B. Angew Chem, Int Ed. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 141.Rostovtsev V V, Green L G, Fokin V V, Sharpless K B. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 142.Wu P, Feldman A K, Nugent A K, Hawker C J, Scheel A, Voit B, Pyun J, Fréchet J M J, Sharpless K B, Fokin V V. Angew Chem, Int Ed. 2004;43(30):3928–3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- 143.Tasdelen M A, Yagci Y. Angew Chem, Int Ed. 2013;52:5930–5938. doi: 10.1002/anie.201208741. [DOI] [PubMed] [Google Scholar]
- 144.Gacal B, Akat H, Balta D K, Arsu N, Yagci Y. Macromolecules. 2008;41:2401–2405. doi: 10.1021/ma702502h. [DOI] [Google Scholar]
- 145.Tasdelen M A, Yilmaz G, Iskin B, Yagci Y. Macromolecules. 2012;45:56–61. doi: 10.1021/ma202438w. [DOI] [Google Scholar]
- 146.Temel G, Aydogan B, Arsu N, Yagci Y. Macromolecules. 2009;42:6098–6106. doi: 10.1021/ma901162y. [DOI] [Google Scholar]
- 147.Kütahya C, Yagci Y, Strehmel B. ChemPhotoChem. 2019;3:1180–1186. doi: 10.1002/cptc.201900012. [DOI] [Google Scholar]
- 148.More about Chemistry 4.0 can be found in “Growth through innovation in a transforming world” published by Verband der Chemischen Industrie VCI, 2017. https://www.vci.de/vci-online/services/publikationen/broschueren-faltblaetter/vci-deloitte-study-chemistry-4-dot-0-short-version.jsp (11 January 2020).
- 149.Lelièvre T, Stoltz G. Acta Numerica. 2016;25:681–880. doi: 10.1017/s0962492916000039. [DOI] [Google Scholar]
- 150.Li L, Baker T E, White S R, Burke K. Phys Rev B. 2016;94(24):245129. doi: 10.1103/physrevb.94.245129. [DOI] [Google Scholar]
- 151.Grajciar L, Heard C J, Bondarenko A A, Polynski M V, Meeprasert J, Pidko E A, Nachtigall P. Chem Soc Rev. 2018;47:8307–8348. doi: 10.1039/c8cs00398j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rupp M. Int J Quantum Chem. 2015;115(16):1003–1139. doi: 10.1002/qua.24955. Special issue on machine learnin and quantum mechanics. [DOI] [Google Scholar]
- 153.Grisafi A, Fabrizio A, Meyer B, Wilkins D M, Corminboeuf C, Ceriotti M. ACS Cent Sci. 2019;5:57–64. doi: 10.1021/acscentsci.8b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cremanns K, Roos D. arXiv. 2017:No. 1710.06202. https://arxiv.org/abs/1710.06202v2 (16 November 2019). [Google Scholar]
- 155.Cremanns K, Schmitz C, Wagner L. Farbe und Lack. 2019;11:50–55. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Information of the electrochemical measurements and the determination of photophysical data.