

Highlights
-
•
A method to prepare viruses for identification by mass spectrometry was developed.
-
•
The sample preparation method was rapid, simple and robust.
-
•
That allowed for generic and sensitive identification of cultured respiratory viruses.
-
•
Also simultaneous identification of mixed viral cultures was possible.
Keywords: Mass spectrometry, MALDI-TOF MS, LC–MS/MS, Influenza virus, Identification, Virological diagnosis
Abstract
The rapid identification of existing and emerging respiratory viruses is crucial in combating outbreaks and epidemics. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is a rapid and reliable identification method in bacterial diagnostics, but has not been used in virological diagnostics. Mass spectrometry systems have been investigated for the identification of respiratory viruses. However, sample preparation methods were laborious and time-consuming. In this study, a reliable and rapid sample preparation method was developed allowing identification of cultured respiratory viruses. Tenfold serial dilutions of ten cultures influenza A strains, mixed samples of influenza A virus with human metapneumovirus or respiratory syncytial virus, and reconstituted clinical samples were treated with the developed sample preparation method. Subsequently, peptides were subjected to MALDI-TOF MS and liquid chromatography tandem mass spectrometry (LC–MS/MS). The influenza A strains were identified to the subtype level within 3 h with MALDI-TOF MS and 6 h with LC–MS/MS, excluding the culturing time. The sensitivity of LC–MS/MS was higher compared to MALDI-TOF MS. In addition, LC–MS/MS was able to discriminate between two viruses in mixed samples and was able to identify virus from reconstituted clinical samples. The development of an improved and rapid sample preparation method allowed generic and rapid identification of cultured respiratory viruses by mass spectrometry.
1. Introduction
Respiratory viruses are a major cause of infections, natural outbreaks, and epidemics. Approximately 200 million cases of viral community-acquired pneumonia occur every year: 100 million in children and 100 million in adults (Ruuskanen et al., 2011). A wide range of viruses, including current circulating subtypes of influenza A virus (influenza A(H1N1)pdm09 and influenza A H3N2), influenza B virus, respiratory syncytial virus (RSV), parainfluenza viruses, adenovirus, and rhinovirus, have been implicated in respiratory tract infections in past decades. In the last ten years, new viruses have emerged as severe acute respiratory syndrome (SARS) virus, Middle East respiratory syndrome (MERS), avian influenza A (H5N1) virus, human metapneumovirus (hMPV), coronaviruses NL63 and HKU1, and human bocavirus. Rapid identification of existing and emerging respiratory viruses is of the utmost importance in combating outbreaks and epidemics, and starting early therapy and prophylaxis. Currently, cell culture, serology, and real-time reverse transcription-polymerase chain reaction (rRT-PCR) are used in virological diagnostics. However, there are a few drawbacks with these techniques: (i) serology and rRT-PCR-based assays are target directed and thus potentially miss non-selected or emerging pathogenic viruses (Binnicker et al., 2013, Yang et al., 2014); (ii) culturing is time consuming; (iii) serology-based assays are not applicable in the acute phase and/or have low sensitivity. Furthermore, multiple tests are needed to detect and subtype mixed infections.
Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) is a generic technique that can rapidly identify cultured microorganisms (Seng et al., 2009, van Veen et al., 2010). The analysis of bacteria by mass spectrometry (MS) has made great progress over the last two decades, and it is employed in many hospitals as a rapid and reliable alternative to traditional identification methods. Thus far, mass spectrometry-based methods have not been used for virological diagnostics. However, it has been shown that purified influenza virus particles could be identified with either MALDI-TOF MS or MALDI Fourier Transform Ion Cyclotron Resonance MS (MALDI-FT-ICR MS). Identification was based on the proteolytic digestion of concentrated and purified viral samples and on mass spectrometry analysis of the specific peptide mass profile. The sample preparation methods used in these studies included virus concentration and/or purification with either ultracentrifugation, differential centrifugation, precipitation, filtration, isolation of viral particles or protein(s) with an affinity capture immunoassay or gel electrophoresis (Downard et al., 2009, Schwahn et al., 2009a, Schwahn et al., 2009b, Schwahn et al., 2010a, Schwahn et al., 2010b, Chou et al., 2011, Jang et al., 2011, Nguyen and Downard, 2013, Fernandes and Downard, 2014). These sample preparation procedures were laborious and often time consuming (up to 24 h) and are therefore not applicable for high-throughput virological diagnostics.
The aim of this study was to develop a rapid, generic and robust sample preparation method for MALDI-TOF MS and LC–MS/MS that will enable reliable and fast identification of respiratory viruses. For this purpose, cultured influenza A virus strains and mixed samples of influenza A virus with hMPV or RSV were treated with the developed preparation method. Subsequently, obtained peptides were subjected to MALDI-TOF MS and LC–MS/MS for analyses. The identification of the respiratory viruses was based on peptide sequence differences in abundant viral proteins. To confirm correct identification of peptides by mass spectrometry, the amino acid sequences were compared to the corresponding DNA sequences, obtained by sequencing of the viruses. Finally, to determine the sensitivity of the in-house developed method the titers of cultured viruses were determined with rRT-PCR.
2. Materials and methods
2.1. Ethics statement
All of the clinical influenza strains, anonymized from routine diagnostics, originated from a collection at the University Medical Center Utrecht (UMCU). Collection of the samples and analysis of the isolated virus strains were approved by the local Medical Ethics Committee of the UMCU. The institutional review board (IRB) confirmed (protocol 12/320) that the viral strains were not regarded as patient-owned material; consequently, the use of these strains was not restricted by Dutch law (“Law Medical Scientific Research with People”, WMO; art. 1b).
2.2. Viruses
Nasopharyngeal swabs and tracheal aspirates were collected between 2009 and 2011 from patients who were hospitalized with respiratory distress symptoms in the wards of the UMCU (Table S1). The collected samples were cultured immediately in either LLC-MK2 or HEp2 cell lines and were routinely checked for cytopathological effect (CPE) formation. The samples exhibiting specific CPE in 80% of the cells and testing positive for respiratory viruses by real-time PCR were harvested and stored at −80 °C.
Next, eight influenza A H1N1 strains (designated here as BM1456, BM1457, and BM1480 through BM1485) and two influenza A H3N2 strains (designated here as BM1454 and BM1455) were cultured in LLC-MK2 cells in the presence of EMEM (Eagle's minimal essential medium) with trypsin at 37 °C (Table S1). In addition, hMPV (designated here as BM1460) and RSV type A (designated here as BM1450) were cultured in LLC-MK2 cells in the presence of EMEM with trypsin at 37 °C and in HEp2 cells in the presence of DMEM (Dulbecco's modified Eagle medium) with 5% FBS (fetal bovine serum) at 33 °C, respectively (Table S1). Subsequently, the culture supernatants were collected, and the cell debris was removed by centrifugation (239 × g, 10 min). Aliquots were frozen at −80 °C and were used without further purification steps.
2.3. Viral RNA extraction, cDNA synthesis, and rRT-PCR
To determine the sensitivity of the in-house developed sample preparation method the titers of cultured viruses were determined with rRT-PCR by using a standard curve. This standard curve was developed by counting virus particles using electron microscopy and by subsequently performing PCR.
Viral RNA extraction and PCR were performed according to previously described protocols (Tan et al., 2012). In short, viral genomic RNA was isolated using a MagnaPure LC total nucleic acid kit, according to the manufacturer's guidelines (Roche Diagnostics, Mannheim, Germany). Murine encephalomyocarditis virus was used as an internal control. Reverse transcription of the isolated viral RNA was performed using a MultiScribe reverse transcriptase kit and random hexamers (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's guidelines. PCR primers and probes were designed on the basis of highly conserved genomic regions of the M1 gene for influenza A and the N gene for both hMPV and RSV A, and these primers and probes were used for the typing of the viral strains. The primers and probes used are listed in Table S2.
cDNA samples were analyzed in a 25 μl reaction mixture, containing 10 μl of cDNA, TaqMan universal PCR master mix (Applied Biosystems, ABI), primers and fluorogenic probes labeled with the 5′ reporter dye 6-carboxy-fluorescein (FAM), and the 3′ quencher dye 6-carboxy-tetramethyl-rhodamine (TAMRA). Amplification and detection were performed with an ABI 7500 system for 2 min at 50 °C, 10 min at 95 °C, and 45 cycles of 15 sec at 95 °C and 1 min at 60 °C. The samples were assessed for the presence of possible inhibitors of the amplification reaction using the indicated internal control, the signals of which had to range within a clear-cut interval.
2.4. Viral genomic cDNA synthesis and sequencing
To confirm the presence and amino acid sequence accuracy of the identified peptides, six of the clinical isolates were sequenced. The RSV strain (BM1450) with accession number JQ901450 was sequenced previously (Tan et al., 2012). For sequencing purposes, hMPV (BM1460) and influenza (BM1454, BM1456, BM1457, BM1480, and BM1483) PCR fragments were obtained by fractional amplification of MagNAPure LC genomic RNA isolates, using the Superscript III one-step RT-PCR System with Platinum Taq High Fidelity kit (Invitrogen) and a 9800 Fast thermal cycler (ABI), according to the manufacturers’ protocols. Unlike hMPV and RSV, influenza virus contains a segmented genome consisting of 8 segments. All of the segments were completely amplified and purified from an agarose gel prior to fractional amplification, as previously described (Zhou et al., 2009). The PCR products were applied to a 1% agarose gel and were purified from the gel with a Gene JET Gel Extraction Kit (Thermo Fisher Scientific, Landsmeer, the Netherlands), according to the manufacturer's protocol. The isolated fragments were used for whole-genome sequencing.
The hMPV (BM1460) and influenza strains (BM1454, BM1456, BM1457, BM1480, and BM1483) listed in Table S1 were sequenced according to the whole-genome sequence protocol described recently, using the conventional Sanger technique (Tan et al., 2012). Fragments ranging between 650 and 1400 nucleotides were sequenced with an ABI 3730 48-capillary DNA analyzer, using Big-Dye Terminator 3.1 (ABI). The resulting sequence information was assembled into an hMPV whole-genome sequence through alignment with the corresponding reference sequence (GenBank: FJ168779; http://www.ncbi.nlm.nih.gov/genbank/index.html) using Seqman software (DNASTAR lasergene 8); a similar approach was followed for the influenza strains. Table S3 provides an overview of the primers used for influenza, hMPV, and RSV sequencing. The DNA and amino acid sequences of the sequenced strains are provided in Supplementary Data.
2.5. Sample preparation of cultured viruses
The starting titers of the viral cultures are shown in Table S1. The starting titers for the influenza strains and RSV appeared roughly similar (∼3 × 1011 copies/ml), but the starting titer for hMPV was 100-fold lower. Tenfold serial dilutions for all of the strains (influenza A virus, RSV, and hMPV) were prepared in CyMol medium (Copan Italia, Brescia, Italy). CyMol is a collection, transport, and preservation medium for cells (cytology analysis), viruses, and nucleic acids (PCR-based assays) (Luinstra et al., 2011a). Subsequently, 200 μl of each tenfold dilution step was treated according to the in-house developed sample pretreatment protocol, as follows. A 200 μl aliquot was mixed with 25 μl of a 0.5% stock solution of RapiGest (Waters Corporation, Milford, CT, USA) and was incubated for 7 min at 95 °C. Next, DL-dithiothreitol (DTT, Sigma–Aldrich) and acetonitrile (LC–MS Chromasolv, Sigma–Aldrich) were added to 5 mM and to 25% final concentration, respectively. Reduction and precipitation of the proteins were performed under controlled microwave radiation in a Rapid Enzymatic Digestion System (REDS; Hudson Surface Technology [HST] Inc., Old Tappan, NJ, USA) at 60 °C and 400 W for 10 min. The subsequent carbamidomethylation of cysteine residues was executed at 37 °C and 400 W for 5 min in REDS, after the addition of iodoacetamide (IAA; Sigma–Aldrich) to a 12.5 mM final concentration. Next, the proteinaceous particles were precipitated at 20,800 × g for 7 min, and the supernatants were discarded. The pellets were suspended in 40 μl of 50 mM ammonium bicarbonate (Sigma–Aldrich) containing 4 μg/ml modified and TPCK-treated trypsin from bovine pancreas (Sigma–Aldrich). Digestion was performed for 10 min in REDS at 37 °C and 400 W and was terminated by the addition of trifluoroacetic acid (TFA) to 1% final concentration. After incubation at 37 °C for 30 min, the RapiGest precipitates were removed by centrifugation, and the supernatants were filtered through a Microcon YM-10 filter (10 MWCO, Merck Millipore Ltd.; Co. Cork, Ireland). All of the samples were prepared at least twice.
For the analysis of mixtures of two different viral culture samples, each tenfold dilution of influenza A H3N2 (BM1454) or H1N1 (BM1456) was mixed with the corresponding dilution of the RSV (BM1450) or hMPV (BM1460) culture samples, respectively, followed by the exact sample preparation procedure described above. All the samples were prepared at least twice. To test whether human proteins present in clinical material interfere with the developed sample preparation method or identification by LC–MS/MS, reconstituted throat swabs were included. A swab that was rubbed around the tonsils of a volunteer was washed by vortexing the swab in CyMol medium. Subsequently, this so-called clinical sample was spiked with dilutions of cultured influenza A virus (BM1454, BM1456), RSV (BM1450), or hMPV (BM1460). The same sample preparation and LC–MS/MS analysis procedure as described above were performed on these reconstituted clinical samples. All tested samples were prepared at least twice.
2.6. MALDI-TOF MS analysis and database search
A 10 μl aliquot of each sample was desalted on a C18 ZipTip (Merck Millipore Ltd.), and the peptides were eluted with 1 μl of 50% acetonitrile containing 10 mg/ml α-cyano-4-hydroxycinnamic acid (HCCA) and 2.5% trifluoroacetic acid (TFA). Elution was performed directly onto the MALDI target (MTP Polished steel, Bruker Daltonics, GmbH, Bremen, Germany), and the droplets were air-dried. Spectra were recorded in reflector mode, using a MALDI-TOF MS instrument (Autoflex III, Bruker). Each spectrum was acquired by averaging 2000 laser shots (at 40% laser power) across a mass range of m/z 500–4000 followed by noise reduction. The instrument was mass calibrated with an external peptide standard (Peptide Calibration Standard, Bruker). The spectra were analyzed with FlexAnalysis and Biotools (Bruker). Internal calibration was performed using identified peptides derived from viral nucleoprotein or trypsin. From the spectra, peak lists were generated and searched against the NCBI virus database (database collection of viruses: 2013.06.29) using MASCOT, version 2.2.04 (Matrix Science, London, UK) with a mass tolerance of 70 ppm (Perkins et al., 1999, Koenig et al., 2008). The carbamidomethylation of cysteine residues and oxidation of methionine were included in the calculations, and up to two missed cleavages were permitted. The MASCOT scoring system was used to determine protein expectation values that corresponded to the number of matches that were expected to occur by chance alone. The highest expectation value was considered the best match. All of the assays were performed at least twice.
In separate experiments, the peaks that were identified initially as nucleoprotein (NP) peptides of influenza A were subsequently confirmed by MALDI-TOF MS/MS. The MS/MS spectra were recorded in the reflector mode using LIFT technology (Bruker).
2.7. LC–MS/MS analysis and database search
The digests were analyzed by LC–MS/MS using a nano-Advance LC system (Bruker) coupled to a Q-TOF mass spectrometer (maXis impact, Bruker). A 5 μl aliquot of each digested sample was injected onto a Magic C18AQ UHPLC NanoTrap column (C18, 100 μm ID × 100 mm, 5 μm, 200 Å, Bruker) and was washed with loading solvent H2O (0.2% formic acid) for 5 min at a flow rate of 4 μl/min. Following valve switching, the peptides were separated on a Magic C18AQ analytical column (100 μm ID × 150 mm, C18, 3 μm, 200 Å, Bruker) at a constant flow of 800 nl/min. The peptide elution gradient was from 98% A (H2O with 0.1% formic acid) to 45% B (CH3CN) in 30 min, followed by an increase to 95% B for 5 min. Optima LC–MS H2O (0.1% formic acid; Fisher Scientific) was used for the LC–MS/MS studies.
The nanoLC system was coupled to the mass spectrometer using a CaptiveSpray (Bruker) ionization source. The spray voltage was set at 1.4–1.6 kV, and the temperature of the heated capillary was set at 150 °C. The eluting peptides were analyzed using the data-dependent MS/MS mode. The ten most abundant ions (charge state 2+, 3+, and 4+) in an MS spectrum (400–1400 m/z) were selected for data-dependent MS/MS analysis by collision-induced dissociation, using nitrogen as the collision gas. The MS/MS scans were acquired over a mass range of 100–2000 m/z.
Peak lists were generated using DataAnalysis software, version 4.1 (Bruker), and the lists were exported as MASCOT Generic (MGF) files. These files were searched against the NCBI database (taxonomy: viruses) with the MASCOT search algorithm (MASCOT 2.2.04, Matrix Science). A mass tolerance of 50 ppm and an MS/MS tolerance of 25 mmu were used. Up to two missed cleavages for trypsin were allowed; carbamidomethylcysteine was selected for fixed modification and oxidation of methionine for variable modification. Only significant protein hits with at least two unique peptides with a score greater than 20 were selected.
3. Results
3.1. Sample preparation and identification of influenza A viruses by MALDI-TOF MS
Ten influenza A virus strains, isolated from hospitalized patients and characterized to the subtype level (eight H1N1 and two H3N2), were cultured in LLC-MK2 cells, resulting in viral titers ranging from 2.1 to 4.3 × 1011 genome copies/ml (Table S1). After the removal of cell debris, tenfold dilution series of the viral cultures were prepared in CyMol medium. Each dilution was treated according to the in-house developed sample preparation procedure. Subsequently, peptides were subjected to MALDI-TOF MS and were analyzed by MASCOT software. In two independent experiments, all of the tested influenza A strains (eight H1N1 and two H3N2) were correctly identified based on the recognition of peptides derived from nucleoprotein (Table 1 and Fig. 1 ). The ten influenza A strains were not only identified to their type, but also correctly identified to their subtype (Table 1). The entire procedure, i.e. the dilution series preparation followed by the treatment according to the in-house protocol, data analysis and the obtaining of results, was performed within 3 h for ten samples.
Table 1.
Identified influenza A viruses based on nucleoprotein peptide detection by MALDI-TOF MS.
| Straina | Subtype | Id. scoreb | Seq. cov.c (%) | Identified as nucleoprotein ofd (subtype/host) |
|---|---|---|---|---|
| BM1454 | H3N2 | 90 | 30 | Influenza A virus (H3N2/human) |
| BM1455 | H3N2 | 108 | 35 | Influenza A virus (H3N2/human) |
| BM1456 | H1N1 | 91 | 37 | Influenza A virus (H1N1/human) |
| BM1457 | H1N1 | 89 | 31 | Influenza A virus (H1N1/human) |
| BM1480 | H1N1 | 95 | 41 | Influenza A virus (H1N1/swine) |
| BM1481 | H1N1 | 102 | 36 | Influenza A virus (H1N1/human) |
| BM1482 | H1N1 | 89 | 34 | Influenza A virus (H1N1/human) |
| BM1483 | H1N1 | 93 | 36 | Influenza A virus (H1N1/swine) |
| BM1484 | H1N1 | 95 | 32 | Influenza A virus (H1N1/swine) |
| BM1485 | H1N1 | 103 | 37 | Influenza A virus (H1N1/human) |
Viral strains were tested at approximately 1 × 109 genome copies in the total volume of each sample deposited onto the MALDI plate and analyzed.
Protein identification score given by MASCOT. A score ≥76 was considered significant identification.
Sequence coverage of nucleoprotein, as specified by MASCOT in percentage, based on the number of amino acids covered by mass values (peptides) matched with the identified protein.
The nucleoprotein that received the highest MASCOT score is shown. The influenza subtype should be considered as an indication because the identification was based on detection of nucleoprotein peptides.
Fig. 1.
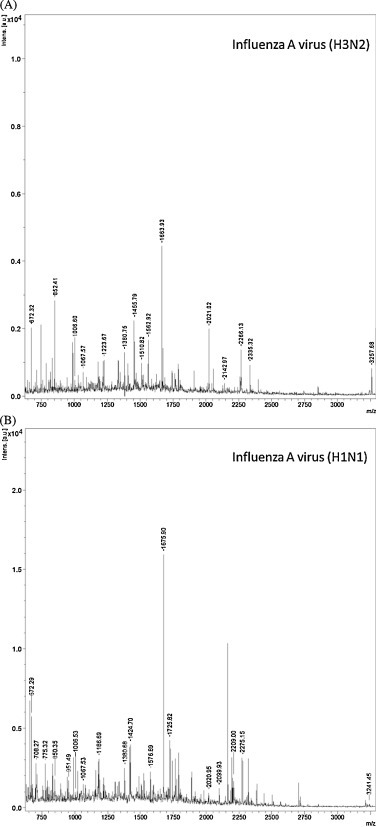
Examples of MALDI-TOF mass spectra from whole influenza A virus tryptic digests of (A) H3N2 and (B) H1N1. The shown m/z ratios are examples of the detected peptides derived from influenza nucleoprotein.
The identification limit at which the viral strains were correctly typed and subtyped appeared to be 1 × 109 viral genome copies in the total sample volume deposited onto a MALDI plate for analysis (corresponding to a CT value of approximately 24; Table 1). At this viral titer, more than 20 mass peaks could be assigned with high significance to peptides derived from nucleoprotein (Table 2 ). The identity of approximately half of these peptide peaks was confirmed by MALDI-TOF MS/MS (data not shown). The intensities of the remaining peptide peaks assigned to nucleoprotein were too low for MS/MS analysis. The genomes of five influenza A strains, namely BM1454, BM1456, BM1457, BM1480, and BM1483, were sequenced, and the amino acid sequences of all of the identified peptides were found to be encoded in the corresponding genome sequences (Supplementary Data, genomic and amino acid sequences of sequenced strains).
Table 2.
Peptides derived from influenza A nucleoprotein and detected by MALDI-TOF MS.
| H1N1 strains | Observed [M+H]+ | H3N2 strains | Observed [M+H]+ |
|---|---|---|---|
| SALILR | 672.4 | SALILR | 672.4 |
| NLPFER | 775.4 | DS | 2184.1 |
| QDATEIR | 832.4 | QNATEIR | 831.4 |
| AMMDQVR | 850.4 | AMVDQVR | 818.4 |
| KTGGPIYR | 891.5 | KTGGPIYR | 891.5 |
| SRYWAIR | 951.5 | SGYWAIR | 852.4 |
| GKFQTAAQR | 1006.5 | GKFQTAAQR | 1006.5 |
| MVLSAFDER | 1067.5 | MVLSAFDER | 1067.5 |
| GVGTIAMELIR | 1159.7 | ND | – |
| LIQNSITIER | 1186.7 | DS | 2207.2 |
| MVLSAFDERR | 1223.6 | MVLSAFDERR | 1223.6 |
| MCSLMQGSTLPR | 1380.6 | MCSLMQGSTLPR | 1380.6 |
| ELILYDKEEIR | 1420.7 | ELVLYDKEEIR | 1406.7 |
| ATVMAAFSGNNEGR | 1424.6 | STIMAAFTGNTEGR | 1455.7 |
| ELILYDKEEIRR | 1576.9 | ELVLYDKEEIRR | 1562.8 |
| NPGNAEIEDLIFLAR | 1671.9 | NPGNAEIEDLIFLAR | 1671.9 |
| ASAGQISVQPTFSVQR | 1675.8 | ASAGQTSVQPTFSVQR | 1663.9 |
| MMESAKPEDLSFQGR | 1725.8 | MMEGAKPEEVSFR | 1510.7 |
| SQLVWMACHSAAFEDLR | 2020.9 | SQLVWMACHSAAFEDLR | 2020.9 |
| ESRNPGNAEIEDLIFLAR | 2044.0 | ESRNPGNAEIEDLIFLAR | 2044.0 |
| SYEQMETGGERQDATEIR | 2099.9 | SYEQMETDGDRQNATEIR | 2142.9 |
| SCLPACVYGLAVASGHDFER | 2209.0 | ND | |
| DS | 775.4; 1424.6 | NLPFEKSTIMAAFTGNTEGR | 2184.1 |
| DS | 1186.7; 1067.5 | LIQNSLTIEKMVLSAFDER | 2207.2 |
| LLQNSQVVSLMRPNENPAHK | 2275.2 | LLQNSQIYSLIRPNENPAHK | 2335.2 |
| GVQIASNENVETMDSNTLELR | 2320.2 | GVQIASNENMDNMGSSTLELR | 2266.0 |
Peptides with a S/N of ≥6 are listed. These peptides were identified in tested viral strains at titers of 1 × 109 genome copies in the total volume of each sample, deposited onto a MALDI plate and analyzed.
ND, peptide not detected.
DS, detected sequence in the tested strains but in another peptide due to missed cleavage by trypsin.
Of the other proteins that constitute the viral proteome, only M1-derived peptides were identified occasionally (data not shown). The MS spectra also contained peaks from abundant non-viral proteins derived from cell lysates, such as actin and trypsin auto-digestion fragments (shown as un-annotated peaks in Fig. 1).
3.2. Identification of influenza A viruses by LC–MS/MS
The same sample preparation procedure was followed as described above, after which the peptides were subjected to LC–MS/MS. Virus identification, including the sample preparation and analysis, was achieved in less than 6 h for ten samples. In two independent experiments, all ten influenza strains were correctly identified as either H1N1 or H3N2 influenza A virus, with an identification limit of 7 × 106 genome copies in the total sample volume subjected to LC–MS/MS analysis (corresponding to a CT value of approximately 30–32; Table 3 ). At this viral titer, the influenza viruses were typed and subtyped based on the identification of peptides derived from the nucleoprotein protein, indicated in boldface in Table 4 . The nucleoprotein-derived peptides that were identified repeatedly in at least 90% of the samples containing ≥7 × 106 viral genome copies are also shown in Table 4. In addition to the peptides derived from NP, the LC–MS/MS analysis identified peptides derived from M1, non-structural protein (NS1), HA, and NA (Table S4). The amino acid sequence coverage of these proteins (for each tenfold dilution of viral culture, an average value of two independent experiments was calculated) is shown in Table 3. Similar to the MALDI-TOF MS results, the identified amino acid sequences of the five sequenced strains (BM1454, BM1456, BM1457, BM1480, and BM1483) were in accordance with their respective genome sequences. The LC–MS/MS spectra also demonstrated proteins derived from cell lysates or culture additives, such as actin, lectin, histone, and enolase (data not shown). Moreover, most of the peptides detected with MALDI-TOF MS were also identified with LC–MS/MS (Table 2, Table 4).
Table 3.
Influenza A viruses identified by LC–MS/MS.
| Strains | Subtype | Titera | Sequence coverageb (%) |
Identified asc | ||||
|---|---|---|---|---|---|---|---|---|
| NP | M1 | NS1 | HA | NA | ||||
| BM1454-55 | H3N2 | 109 | 70 | 75 | 56 | 21 | 15 | Influenza A H3N2 |
| 108 | 49 | 47 | 34 | 17 | 11 | |||
| 107 | 25 | 18 | 0 | 2 | 0 | |||
| 106 | 10 | 0 | 0 | 0 | 0 | |||
| BM1456-57 | H1N1 | 109 | 72 | 71 | 61 | 18 | 5 | Influenza A H1N1 |
| BM1480-85 | 108 | 56 | 49 | 39 | 10 | 4 | ||
| 107 | 44 | 30 | 15 | 0 | 0 | |||
| 106 | 14 | 0 | 0 | 0 | 0 | |||
Tenfold dilution series of each tested influenza strain was prepared and analyzed by LC–MS/MS. Indicated are the total genome copies of the analyzed influenza virus subjected to LC–MS/MS.
Amino acid sequence coverage (%) of proteins determined by MASCOT, as based on identified peptides. Percentages are the averages of combined results for the H3N2 or H1N1 strain. Proteins were considered significantly identified when the MASCOT score was ≥50 and when a minimum of three peptides each with a score of ≥20 was identified. NP, nucleoprotein; M1, matrix protein; NS1, non-structural protein 1; HA, hemagglutinin; NA, neuraminidase;
Identification of type and subtype based on the highest MASCOT scores of nucleoprotein; thus, the given subtypes should be considered as indicative.
Table 4.
Repeatedly LC–MS/MS-identified influenza A virus peptides derived from nucleoprotein at titers ≥7 × 106 genome copies.
| H1N1 strains | Observed m/z | H3N2 strains | Observed m/z |
|---|---|---|---|
| FQTAAQR | 820.4 | FQTAAQR | 820.4 |
| LSDYDGR | 824.4 | ND | – |
| QDATEIR | 831.4 | QNATEIR | 830.4 |
| AMMDQVR | 849.4 | AMVDQVR | 817.4 |
| KTGGPIYR | 890.5 | KTGGPIYR | 890.5 |
| TRVAYER | 893.5 | TRSAYER | 881.4 |
| IDGKWMR | 904.5 | RVDGKWMR | 1046.5 |
| TGGPIYRR | 918.5 | TGGPIYRR | 918.5 |
| SRYWAIR | 950.3 | SGYWAIR | 851.4 |
| MCNILKGK | 962.5 | MCNILKGK | 962.5 |
| GKFQTAAQR | 1005.5 | GKFQTAAQR | 1005.5 |
| GVFELSDEK | 1022.5 | GVFELSDEK | 1022.5 |
| KTGGPIYRR | 1046.6 | DS | 891.5 |
| MVLSAFDER | 1066.5 | MVLSAFDER | 1066.5 |
| YLEEHPSAGK | 1129.5 | YLEEHPSAGK | 1129.5 |
| GVGTIAMELIR | 1158.7 | GIGTMVMELIR | 1218.6 |
| LIQNSITIER | 1185.7 | LIQNSLTIEK | 1157.7 |
| DS | 849.4; 2044.0 | AMVDQVRESR | 1189.6 |
| TSDMRTEVIR | 1206.6 | TSDMRAEIIR | 1190.6 |
| DS | 1022.5 | GRGVFELSDEK | 1235.6 |
| MVLSAFDERR | 1222.6 | MVLSAFDERR | 1222.6 |
| SYEQMETGGER | 1285.5 | SYEQMETDGDR | 1329.5 |
| HSNLNDATYQR | 1317.6 | HSNLNDATYQR | 1317.6 |
| EGYSLVGIDPFK | 1323.7 | EGYSLVGIDPFK | 1323.7 |
| FYIQMCTELK | 1331.6 | FYIQMCTELK | 1331.6 |
| NKYLEEHPSAGK | 1371.7 | NKYLEEHPSAGK | 1371.7 |
| MCSLMQGSTLPR | 1379.6 | MCSLMQGSTLPR | 1379.6 |
| LLQNSQVVSLMR | 1386.8 | DS | 2334.2 |
| ELILYDKEEIR | 1419.7 | ELVLYDKEEIR | 1405.7 |
| ATVMAAFSGNNEGR | 1423.6 | STIMAAFTGNTEGR | 1454.7 |
| YLEEHPSAGKDPK | 1469.7 | YLEEHPSAGKDPK | 1469.7 |
| MCSLMQGSTLPRR | 1535.7 | MCSLMQGSTLPRR | 1535.7 |
| ELILYDKEEIRR | 1575.9 | ELVLYDKEEIRR | 1561.8 |
| YLEEHPSAGKDPKK | 1597.8 | YLEEHPSAGKDPKK | 1597.8 |
| NPGNAEIEDLIFLAR | 1670.9 | NPGNAEIEDLIFLAR | 1670.9 |
| ASAGQISVQPTFSVQR | 1674.8 | ASAGQTSVQPTFSVQR | 1662.9 |
| NKYLEEHPSAGKDPK | 1711.9 | NKYLEEHPSAGKDPK | 1711.9 |
| MMESAKPEDLSFQGR | 1724.8 | MMEGAKPEEVSFR | 1509.7 |
| MCNILKGKFQTAAQR | 1764.9 | MCNILKGKFQTAAQR | 1764.9 |
| SQLVWMACHSAAFEDLR | 2019.9 | SQLVWMACHSAAFEDLR | 2019.9 |
| ESRNPGNAEIEDLIFLAR | 2043.0 | ESRNPGNAEIEDLIFLAR | 2043.0 |
| SYEQMETGGERQDATEIR | 2098.9 | SYEQMETDGDRQNATEIR | 2141.9 |
| SCLPACVYGLAVASGHDFER | 2208.0 | SCLPACAYGPAVSSGYDFEK | 2177.9 |
| RSYEQMETGGERQDATEIR | 2255.0 | ND | – |
| LLQNSQVVSLMRPNENPAHK | 2274.2 | LLQNSQIYSLIRPNENPAHK | 2334.2 |
| GVQIASNENVETMDSNTLELR | 2319.2 | GVQIASNENMDNMGSSTLELR | 2265.0 |
| QANNGEDATAGLTHIMIWHSNLNDATYQR | 3240.5 | QANNGEDATAGLTHIMIWHSNLNDATYQR | 3256.5 |
| DS | 1323.7; 2208.0 | SCLPACAYGPAVSSGYDFEKEGYSLVGIDPFK | 3483.6 |
Identification data were extracted from MASCOT.
ND, peptide sequence not identified.
DS, identified sequence in another peptide.
The influenza viruses (≥7 × 106 genome copies) were typed and subtyped based mainly on the identification of peptides derived from nucleoprotein (shown in bold).
3.3. Identification of respiratory viruses in mixed samples
Tenfold serial dilutions of strain BM1454 (H3N2) were mixed with the corresponding dilution factor of RSV (mixture A), resulting in an approximate 1:1 ratio of both strains. Similarly, in mixture B, tenfold serial dilutions of strain BM1456 (H1N1) were mixed with the corresponding dilution factor of hMPV, although the starting titer of hMPV was 100 times lower than those of influenza and RSV. The mixtures were treated according to the in-house developed sample preparation procedure, subjected to LC–MS/MS, and subsequently analyzed as described for the samples containing a single virus.
Using LC–MS/MS, both viruses in mixtures A and B were identified simultaneously based on the peptides derived from nucleoprotein proteins. The influenza strains were correctly typed and subtyped from sample dilutions containing approximately ≥ 5.3 × 107 genome copies in the total sample volume subjected to LC–MS/MS analysis. For influenza, peptides derived from M1, NS1, HA, and NA proteins were also identified (Table 5 ). For the hMPV and RSV strains, in addition to nucleoprotein-derived peptides, those of matrix protein (M), matrix protein 2–1 (M2-1), phosphoprotein (P), and fusion glycoprotein (F) were also identified (Tables 5, S5, and S6). The identification limit at which the RSV strain was identified correctly, was 3.6 × 107 genome copy equivalents in the total sample volume subjected to LC–MS/MS analysis (corresponding to a CT value of approximately 28; Tables 5 and S1). For hMPV, the identification limit was 1.1 × 107 genome copy equivalents in the total sample volume subjected to LC–MS/MS analysis (corresponding to a CT value of approximately 28; Tables 5 and S1). All of the identified peptide sequences of the tested influenza A, RSV, and hMPV strains were confirmed with their respective genome sequence data (Supplementary Data, genomic and amino acid sequences of sequenced strains).
Table 5.
Identified proteins of two different respiratory viruses from mixed samples. Mixture A contained H3N2 (BM1454) and RSV (BM1450). Mixture B contained H1N1 (BM1456) and hMPV (BM1460).
| Mix | Titera | Id. virusb | Average sequence coverage of identified proteinsc (%) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NP | M1 | NS1 | HA | NA | M2-1 | F | P | |||
| A | 109 | Influenza A/H3N2 | 63 | 74 | 16 | 15 | 4 | |||
| RSV | 21 | 43 | 53 | 9 | 55 | |||||
| 108 | Influenza A/H3N2 | 48 | 49 | 20 | 13 | 7 | ||||
| RSV | 23 | 40 | 46 | 3 | 53 | |||||
| 107 | Influenza A/H3N2 | 18 | 34 | 0 | 5 | 0 | ||||
| RSV | 14 | 0 | 23 | 0 | 0 | |||||
| B | 109 | Influenza A/H1N1 | 73 | 67 | 60 | 0 | 0 | |||
| hMPVd | 14 | 29 | 10 | 15 | 16 | |||||
| 108 | Influenza A/H1N1 | 49 | 33 | 39 | 0 | 0 | ||||
| hMPVd | 8 | 18 | 10 | 9 | 18 | |||||
| 107 | Influenza A/H1N1 | 24 | 10 | 11 | 0 | 0 | ||||
| hMPVd | 0 | 0 | 0 | 0 | 0 | |||||
Total genome copy number of the tested influenza virus and RSV in a total sample volume subjected to LC–MS/MS analysis.
Identified virus.
An average sequence coverage (%) of the identified protein was calculated from two independent experiments. A protein was considered identified when the MASCOT score was ≥50 and when a minimum of three peptides with a score ≥20 was identified in the protein.
The starting concentration of hMPV was 100 times lower than that for influenza and RSV.
NP, nucleoprotein; M1, matrix protein; NS1, non-structural protein 1; HA, hemagglutinin; NA, neuraminidase; M2-1, matrix protein; F, fusion glycoprotein; P, phosphoprotein.
3.4. Identification of reconstituted clinical samples by LC–MS/MS
Tenfold serial dilutions of influenza A H1N1 (BM1456), H3N2 (BM1454), RSV (BM1450), and hMPV (BM1460) were spiked in CyMol medium, containing material from a throat swab. These reconstituted clinical samples were treated according to the in-house developed sample preparation method, subjected to LC–MS/MS and analyzed as described above. All the spiked respiratory viruses were identified. In both influenza A and RSV samples, peptides derived from nucleoprotein, matrix protein M1, and non-structural protein NS1 were identified. In addition, in RSV samples also peptides derived from matrix protein M2-1 and phosphoprotein were identified. In one of the two tested hMPV samples, two peptides derived from phosphoprotein were identified (Table 6 ). The identification limit at which influenza A H3N2 was detected, was 1 × 107 genome copies in the total sample volume subjected to LC–MS/MS; for influenza A H1N1 and RSV this was approximately 1 × 108 genome copies; for hMPV 1 × 107 genome copies.
Table 6.
Identification of viral proteins from reconstituted clinical material spiked with viruses.
| Virus | Titera | Average sequence coverage of identified proteinsb (%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NP | M1 | NS1 | HA | NA | PA | M2-1 | F | P | ||
| H3N2 | 109c | 69 | 71 | 32 | 13 | 12 | 0 | ND | ND | ND |
| 108 | 25 | 27 | 0 | 0 | 0 | 0 | ND | ND | ND | |
| 107 | 4 | 4 | 0 | 0 | 0 | 0 | ND | ND | ND | |
| H1N1 | 109c | 68 | 77 | 71 | 0 | 7 | 3 | ND | ND | ND |
| 108 | 24 | 14 | 9 | 0 | 0 | 0 | ND | ND | ND | |
| RSV | 109c | 54 | 54 | 27.3 | ND | ND | ND | 35 | 9 | 31 |
| 108 | 3 | 35 | ND | ND | ND | ND | 15 | 0 | 17 | |
| hMPV | 107c | 25 | 48 | ND | ND | ND | ND | 16 | 28 | 19 |
| 106 | 0 | 0 | ND | ND | ND | ND | 0 | 0 | 6d | |
| Neg. control | Throat swab | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Total genome copy number of the tested influenza virus and RSV in a total sample volume subjected to LC–MS/MS analysis.
An average sequence coverage (%) of the identified protein was calculated from two independent experiments. A protein was considered identified when the MASCOT score was ≥50 and when a minimum of two peptides with a score ≥20 was identified in the protein. NP, nucleoprotein; M1, matrix protein; NS1, non-structural protein 1; HA, hemagglutinin; NA, neuraminidase; M2-1, matrix protein; F, fusion glycoprotein; P, phosphoprotein.
Positive control; isolation of the virus was without throat swab material.
Only in one of the two tested samples peptides were detected.
4. Discussion
The accurate and rapid identification of influenza and other respiratory viruses is important in combating outbreaks and epidemics, detecting newly emerging viruses, and initiating prophylaxis and early therapy. Antigen-detection and rRT-PCR tests are used most frequently for the detection of viruses in the acute phase. PCR-based methods can yield results within 3–8 h with high sensitivity and specificity (Centers for Disease Control and Prevention: Guidance for Clinicians on the Use of rRT-PCR and Other Molecular Assays for Diagnosis of Influenza Virus Infection, 2013), whereas the rapid antigen-detection tests can even yield results within 15 min. However, both methods feature drawbacks. (i) They are target-directed and can potentially miss non-selected or emerging pathogenic viruses that are not covered by the PCR primer sets and antibodies used in the antigen-detection assays. For instance, it was shown that a new type of human papillomavirus (HPV) was not detected by a broad HPV-primer PCR system (Johansson et al., 2013) and newly emerging mutations in the conserved region of M1 prevented rRT-PCR detection of influenza A H1N1 and H3N2 from clinical specimens (Binnicker et al., 2013, Yang et al., 2014). (ii) Multiple tests (different PCRs or PCR followed by sequencing) are needed for (sub)typing and for detection of mixed infections. (iii) The sensitivity of rapid antigen-detection tests is low. The commonly used Rapid Influenza Diagnostic Test (RIDT), although fast, lacks accuracy and sensitivity (40–70%), resulting in a high percentage of false-negative results (Beck et al., 2012). Theoretically, mass spectrometry-based systems could overcome these drawbacks and they were already investigated for the identification of respiratory viruses. However, until now the used sample preparation methods were too laborious and time-consuming for mass spectrometry to be an identification method of clinical potential. Therefore, in this study a rapid, generic and robust sample preparation method for mass spectrometry-based virus identification was developed. Subsequently, the sensitivity of the combined procedure of this sample preparation and mass spectrometry analysis was determined.
Tenfold serial dilutions of ten cultured influenza A strains of two different subtypes, mixed samples of influenza A virus with either hMPV or RSV, and reconstituted clinical samples were treated with the in-house developed sample preparation method without prior purification by laborious procedures, apart from standard removal of cell debris. Subsequently, peptides were subjected to MALDI-TOF MS and LC–MS/MS and identified by use of a web-based MASCOT search algorithm. To confirm correct identification, the amino acid sequences were compared to the corresponding DNA sequences, obtained by sequencing.
All of the influenza A strains were successfully identified to the subtype level by both mass spectrometry systems. Identification of viruses was achieved within 3 h with MALDI-TOF MS and within 6 h with LC–MS/MS, excluding the time necessary for culturing the viruses. Sensitivity of MALDI-TOF MS was lower (detection limit of 1 × 109; Table 1) compared to LC–MS/MS (detection limit of 7 × 106; Table 3); the latter corresponding to a CT value of 30–32.
Earlier studies have already shown that it is possible to identify highly pure and/or concentrated influenza A viruses with MALDI-TOF MS, MALDI-FT-ICR MS or LC–MS/MS (Schwahn et al., 2009a, Schwahn et al., 2009b, Schwahn et al., 2010a, Schwahn et al., 2010b, Chou et al., 2011, Jang et al., 2011, Nguyen and Downard, 2013, Fernandes and Downard, 2014, Li et al., 2014). However, the sample preparation methods used were time-consuming (up to 24 h) and laborious, and are therefore not suitable for rapid applications. In these studies, the same nucleoprotein-derived peptides were found among the identified viral peptides as detected here (Schwahn et al., 2009a, Schwahn et al., 2009b, Li et al., 2014), which indicates that with the in-house developed sample preparation method, comparable results could be achieved in a much shorter time. Furthermore, in the developed sample preparation method, prior purification or extended concentration of cultured viruses, for example by applying ultracentrifugation, is not required for the identification of viral proteins in the presence of other viral or non-viral proteins. The sample preparation time was significantly reduced to approximately 1.5 h by the introduction of REDS for rapid reduction, alkylation, precipitation, and trypsin digestion. The tenfold serial dilutions of cultured viruses were prepared in CyMol, a collection, transport and preservation medium known for its compatibility with multiple diagnostic platforms including cytology and molecular diagnostics (Luinstra et al., 2011). Suspending viruses in CyMol significantly (approximately 100-fold) improved the identification limit compared to the other tested solutions, such as ammonium bicarbonate, NTE buffer (100 mM NaCl, 10 mM Tris–HCl [pH 7.5], 1 mM EDTA), PBS (phosphate buffered saline), and a universal transport medium (UTM-RT; data not shown).
Besides monocultures of influenza A strains, mixed samples of influenza A virus with either hMPV or RSV were subjected to MALDI-TOF MS and LC–MS/MS. Viruses were identified simultaneously and specifically by LC–MS/MS (Table 5), with a detection limit of 5.3 × 107 for influenza A (H1N1 and H3N2), 3.6 × 107 for RSV, and 1.1 × 107 genome copies for hMPV. Identification was not possible by MALDI-TOF MS due to its low sensitivity. In addition to monocultures and mixed samples, throat swabs spiked with influenza A, hMPV or RSV were subjected to LC–MS/MS. All the spiked viruses were identified, with detection limits of 107, 108, and 107 genome copies for influenza A H3N2, influenza A H1N1 and RSV, and hMPV, respectively. These results demonstrate that LC–MS/MS is not only able to identify monocultures of viruses, but can also discriminate between two viruses in the same sample and can identify a virus from a sample containing human proteins originating from a throat swab. Sensitivity of LC–MS/MS appeared to be lower when mixed samples or reconstituted samples were tested.
Viral loads in clinical samples vary depending on the type of respiratory material, subtype of the virus, severity and phase of infection, and host factors, such as immunity. In general, viral loads fall within the range of 104–106 genome copies/ml (Ngaosuwankul et al., 2010, Lee et al., 2011). In addition, in a recent pan-European survey (Nuttall et al., 2011), the average CT values for influenza virus, RSV, and hMPV detected in patients were 29, 29, and 30, respectively (Coenjaerts, F.E.J., personal communication). Therefore, the ultimate identification limit of a mass spectrometry method should reach 104 genome copies/ml for direct clinical application. The sensitivity of the developed sample preparation method in combination with LC–MS/MS is 100-fold lower and therefore not good enough for direct application in virological diagnostics. For detection of viruses from clinical samples, viral culture is needed before samples can be prepared and analyzed. Because culturing is required, viable virus has to be recovered and successfully grown. For application in virological diagnostics, sensitivity should be increased by developing techniques that enable fast and efficient virus concentration and purification from respiratory samples. Furthermore, mass spectrometry systems should be improved to increase sensitivity. In the meanwhile, keeping the drawbacks into mind this in-house developed sample preparation method in combination with LC–MS/MS could be experimentally used for patients with suspicion of a respiratory infection where routine diagnostics are negative.
The identification of the influenza A strains, RSV and hMPV was based on detection of peptides derived from different proteins. All these identified peptides were found to be encoded in the corresponding genome sequences. MALDI-TOF MS and LC–MS/MS both detected nucleoprotein-derived peptides. In addition, LC–MS/MS-identified peptides derived from other viral proteins, namely M1, NS1, HA, and NA proteins of influenza viruses and M, M2-1, P, and F proteins of RSV and hMPV. The identification of peptides from different proteins not only accomplishes identification but also potentially delivers information about virulence, resistance to antiviral medications, and/or transmission efficiency. For instance, an amino acid substitution in nucleoprotein (M136L) was shown to enhance human-to-human transmission (Zhou et al., 1999). Interestingly, one of the identified nucleoprotein-derived peptides, i.e., QANNGEDATAGLTHIMIWHSNLNDATYQR, contained this substitution. This peptide was found in all of the tested influenza strains (Table 4), suggesting that these strains might exhibit increased human-to-human transmission efficiency.
In conclusion, mass spectrometry systems have been investigated for the identification of highly pure and/or concentrated influenza A viruses. However, used sample preparation methods were laborious and time-consuming. The developed sample preparation method in combination with LC–MS/MS allowed rapid and reliable identification of cultured respiratory viruses and viruses spiked in throat swabs. To introduce this LC–MS/MS based identification method in virological diagnostics, sensitivity must be improved up to 100-fold and clinical samples have to be tested. The combination of an improved mass spectrometry technique and/or clinical sample preparation method allowing direct identification of viruses from clinical samples will be an important new diagnostic tool in clinical virology.
Conflict of interest
The authors declare that they have no competing interests.
Acknowledgements
This work was supported financially by the Dutch Ministry of Defense, grant number V1036.
CyMol medium was kindly provided by Copan Italia S.p.A., of Brescia, Italy.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jviromet.2014.11.014.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- Beck E., Fan J., Hendrickson J.K., Kumar S., Kramp W., Villanueva J., Jeringan D., Shively R., Kimov A., Chen L.M., Donis R., Williams R., Prikle J., Barr J. Evaluation of 11 commercially available rapid influenza diagnostic tests—United States, 2011–2012. Morb. Mortal. Wkly Rep. 2012:873–876. [PubMed] [Google Scholar]
- Binnicker M.J., Baddour L.M., Grys T.E., Espy M.J., Hata D.J., Wotton J.T., Patel R. Identification of an influenza A H1N1/2009 virus with mutations in the matrix gene causing a negative result by a commercial molecular assay. J. Clin. Microbiol. 2013;51:2006–2007. doi: 10.1128/JCM.00446-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.C., Hsu W., Wang C.H., Chen Y.J., Fang J.M. Rapid and specific influenza virus detection by functionalized magnetic nanoparticles and mass spectrometry. J. Nanobiotechnol. 2011;9:52–3155. doi: 10.1186/1477-3155-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downard K.M., Morrissey B., Schwahn A.B. Mass spectrometry analysis of the influenza virus. Mass Spectrom. Rev. 2009;28:35–49. doi: 10.1002/mas.20194. [DOI] [PubMed] [Google Scholar]
- Fernandes N.D., Downard K.M. Incorporation of a proteotyping approach using mass spectrometry for surveillance of influenza virus in cell-cultured strains. J. Clin. Microbiol. 2014;52:725–735. doi: 10.1128/JCM.02315-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H.B., Sung H.W., Nho S.W., Park S.B., Cha I.S., Aoki T., Jung T.S. Enhanced reliability of avian influenza virus (AIV) and Newcastle disease virus (NDV) identification using matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) Anal. Chem. 2011;83:1717–1725. doi: 10.1021/ac102846q. [DOI] [PubMed] [Google Scholar]
- Johansson H., Bzhalava D., Ekstrom J., Hultin E., Dillner J., Forslund O. Metagenomic sequencing of “HPV-negative” condylomas detects novel putative HPV types. Virology. 2013;440:1–7. doi: 10.1016/j.virol.2013.01.023. [DOI] [PubMed] [Google Scholar]
- Koenig T., Menze B.H., Kirchner M., Monigatti F., Parker K.C., Patterson T., Steen J.J., Hamprecht F.A., Steen H. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J. Proteome Res. 2008;7:3708–3717. doi: 10.1021/pr700859x. [DOI] [PubMed] [Google Scholar]
- Lee C.K., Lee H.K., Loh T.P., Lai F.Y., Tambyah P.A., Chiu L., Koay E.S., Tang J.W. Comparison of pandemic (H1N1) 2009 and seasonal influenza viral loads. Singapore. Emerg. Infect. Dis. 2011;17:287–291. doi: 10.3201/eid1702.100282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Sun W., Wu D., Gao X., Sun N., Liu N. Mass spectrometry analysis coupled with de novo sequencing reveals amino acid substitutions in nucleocapsid protein from influenza A virus. Int. J. Mol. Sci. 2014;15:2465–2474. doi: 10.3390/ijms15022465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luinstra K., Petrich A., Castriciano S., Ackerman M., Chong S., Carruthers S., Ammons B., Mahony J.B., Smieja M. Evaluation and clinical validation of an alcohol-based transport medium for preservation and inactivation of respiratory viruses. J. Clin. Microbiol. 2011;49:2138–2142. doi: 10.1128/JCM.00327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngaosuwankul N., Noisumdaeng P., Komolsiri P., Pooruk P., Chokephaibulkit K., Chotpitayasunondh T., Sangsajja C., Chuchottaworn C., Farrar J., Puthavathana P. Influenza A viral loads in respiratory samples collected from patients infected with pandemic H1N1, seasonal H1N1 and H3N2 viruses. Virol. J. 2010;7:75–422. doi: 10.1186/1743-422X-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A.P., Downard K.M. Subtyping of influenza neuraminidase using mass spectrometry. Analyst. 2013;138:1787–1793. doi: 10.1039/c3an00086a. [DOI] [PubMed] [Google Scholar]
- Nuttall J., Hood K., Verheij T.J., Little P., Brugman C., Veen R.E., Goossens H., Butler C.C. Building an international network for a primary care research program: reflections on challenges and solutions in the set-up and delivery of a prospective observational study of acute cough in 13 European countries. BMC Fam. Pract. 2011;12:78–2296. doi: 10.1186/1471-2296-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ruuskanen O., Lahti E., Jennings L.C., Murdoch D.R. Viral pneumonia. Lancet. 2011;377:1264–1275. doi: 10.1016/S0140-6736(10)61459-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwahn A.B., Wong J.W., Downard K.M. Rapid differentiation of seasonal and pandemic H1N1 influenza through proteotyping of viral neuraminidase with mass spectrometry. Anal. Chem. 2010;82:4584–4590. doi: 10.1021/ac100594j. [DOI] [PubMed] [Google Scholar]
- Schwahn A.B., Wong J.W., Downard K.M. Typing of human and animal strains of influenza virus with conserved signature peptides of matrix M1 protein by high resolution mass spectrometry. J. Virol. Methods. 2010;165:178–185. doi: 10.1016/j.jviromet.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Schwahn A.B., Wong J.W., Downard K.M. Signature peptides of influenza nucleoprotein for the typing and subtyping of the virus by high resolution mass spectrometry. Analyst. 2009;134:2253–2261. doi: 10.1039/b912234f. [DOI] [PubMed] [Google Scholar]
- Schwahn A.B., Wong J.W., Downard K.M. Subtyping of the influenza virus by high resolution mass spectrometry. Anal. Chem. 2009;81:3500–3506. doi: 10.1021/ac900026f. [DOI] [PubMed] [Google Scholar]
- Seng P., Drancourt M., Gouriet F., La Scola B., Fournier P.E., Rolain J.M., Raoult D. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis. 2009;49:543–551. doi: 10.1086/600885. [DOI] [PubMed] [Google Scholar]
- Tan L., Lemey P., Houspie L., Viveen M.C., Jansen N.J., van Loon A.M., Wiertz E., van Bleek G.M., Martin D.P., Coenjaerts F.E. Genetic variability among complete human respiratory syncytial virus subgroup A genomes: bridging molecular evolutionary dynamics and epidemiology. PLoS ONE. 2012;7:e51439. doi: 10.1371/journal.pone.0051439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen S.Q., Claas E.C., Kuijper E.J. High-throughput identification of bacteria and yeast by matrix-assisted laser desorption ionization-time of flight mass spectrometry in conventional medical microbiology laboratories. J. Clin. Microbiol. 2010;48:900–907. doi: 10.1128/JCM.02071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.R., Kuo C.Y., Huang H.Y., Wu F.T., Huang Y.L., Cheng C.Y., Su Y.T., Chang F.Y., Wu H.S., Liu M.T. Newly emerging mutations in the matrix genes of the human influenza A(H1N1)pdm09 and A(H3N2) viruses reduce the detection sensitivity of real-time reverse transcription-PCR. J. Clin. Microbiol. 2014;52:76–82. doi: 10.1128/JCM.02467-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Donnelly M.E., Scholes D.T., St George K., Hatta M., Kawaoka Y., Wentworth D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza a viruses. J. Virol. 2009;83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N.N., Shortridge K.F., Claas E.C., Krauss S.L., Webster R.G. Rapid evolution of H5N1 influenza viruses in chickens in Hong Kong. J. Virol. 1999;73:3366–3374. doi: 10.1128/jvi.73.4.3366-3374.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.