

Highlights
-
•
dsRNA species are byproducts of RNA virus replication and/or transcription.
-
•
Prompt detection of dsRNA by RIG-I like receptors (RLRs) is a hallmark of the innate immune response.
-
•
RLRs activation triggers production of the type I interferon (IFN)-based antiviral response.
-
•
Highly pathogenic RNA viruses encode proteins that block the RLRs pathway.
-
•
Hide, mask and hit are 3 strategies of RNA viruses to avoid immune system activation.
Keywords: Innate immune system evasion, RIG-I, MDA5, LGP2, Viral dsRNA detection, Interferon alpha/beta
Abstract
Double-stranded RNA (dsRNA) is synthesized during the course of infection by RNA viruses as a byproduct of replication and transcription and acts as a potent trigger of the host innate antiviral response. In the cytoplasm of the infected cell, recognition of the presence of viral dsRNA as a signature of “non-self” nucleic acid is carried out by RIG-I-like receptors (RLRs), a set of dedicated helicases whose activation leads to the production of type I interferon α/β (IFN-α/β). To overcome the innate antiviral response, RNA viruses encode suppressors of IFN-α/β induction, which block RLRs recognition of dsRNA by means of different mechanisms that can be categorized into: (i) dsRNA binding and/or shielding (“hide”), (ii) dsRNA termini processing (“mask”) and (iii) direct interaction with components of the RLRs pathway (“hit”). In light of recent functional, biochemical and structural findings, we review the inhibition mechanisms of RLRs recognition of dsRNA displayed by a number of highly pathogenic RNA viruses with different disease phenotypes such as haemorrhagic fever (Ebola, Marburg, Lassa fever, Lujo, Machupo, Junin, Guanarito, Crimean-Congo, Rift Valley fever, dengue), severe respiratory disease (influenza, SARS, Hendra, Hantaan, Sin Nombre, Andes) and encephalitis (Nipah, West Nile).
1. Introduction
1.1. Viral commandment: stay unseen, do not let your dsRNA be detected
The intrinsic innate immune system is the primary line of defense against virus invasion, which detects the presence of a virus in an infected cell through a set of proteins named pathogen recognition receptors (PRRs). PRRs recognize viral components, both proteins and nucleic acids, as signatures of “non-self” that are the sign of an infection in place and are named pathogen-associated molecular patterns (PAMPs) (Kumar et al., 2011). Upon PAMPs recognition, PRRs initiate a signaling cascade which culminates with the induction of type I alpha/beta interferons (IFN-α/β). These cytokines act in both an autocrine and paracrine manner to promote viral clearance or induce apoptosis in infected cells, to establish an antiviral state in non-infected cells and to modulate the priming of optimal antigen-specific T cell and antibody adaptive response (Randall and Goodbourn, 2008).
Viral PAMPs mainly consist of nucleic acids that may originate from the uncoating process of newly-infecting virions, the transcription of viral genes and the replication of genomic intermediates. Among these, double-stranded RNA (dsRNA) moieties, especially those with the atypically-featured 5′-triphosphate (5′-ppp) termini, have no homologues in the cytoplasm and therefore represent the most efficient triggers for PRRs activation (Gerlier and Lyles, 2011). Given the central dsRNA role in stimulating IFN-α/β induction, a subset of PRRs is specifically dedicated to its detection and, depending on their cell compartments localization, can be ascribed to two families. dsRNA recognition in endosomes, lysosomes and even at the extracellular surface is accomplished by the Toll-like receptor 3 (TLR-3), a member of the TLR family (Barbalat et al., 2011), while dsRNA recognition in the cytosolic environment is carried out by RNA helicases belonging to the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) family (Wilkins and Gale, 2010), named after the first characterized member, RIG-I (Yoneyama et al., 2004).
Because RNA viruses tend to accumulate nucleic acid intermediates and byproducts in the host cytoplasm during their replication cycle, preventing recognition by host PRRs of such PAMPs is of crucial importance. Therefore, most, if not all, RNA viruses encode proteins that display IFN-antagonism properties aimed to circumvent the host innate immune system. Among those proteins, most have been revealed as involved in targeting the RLR pathway at several levels (Versteeg and Garcia-Sastre, 2010). The impact of this counteraction becomes particularly evident for those RNA viruses that cause severe diseases in humans, since in several cases the fatal outcome has been related to their ability to subvert the type I IFN-mediated innate immune response (Bray, 2005). It is therefore not surprising that, among the different viral proteins identified as inhibitors of IFN-α/β production and signaling, most are also molecular determinants of virulence and pathogenesis (Bowie and Unterholzner, 2008, Versteeg and Garcia-Sastre, 2010).
This review focuses on the strategies adopted by several highly pathogenic RNA viruses to suppress type I IFN induction at the level of dsRNA detection by RLRs. Even relying on the extremely variegated arsenal found in the biodiversity of viral proteins, these strategies can be categorized into three main lines of action:
-
(i)
hiding the dsRNA to make it inaccessible to RLRs,
-
(ii)
masking the dsRNA PAMP signatures to avoid their recognition by RLRs,
-
(iii)
hitting the components of the RLRs pathway to destroy their functionalities (Fig. 1 ).
Fig. 1.
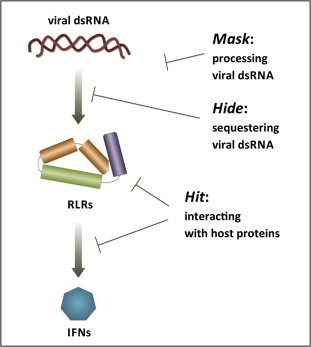
Schematic representation of RLR-mediated type I IFN induction and viral strategies aimed at its suppression. Recognition of viral dsRNA by RLRs leads to the production of type I IFN-α/β, triggering an innate immune antiviral response. Viruses prevent it by processing dsRNA (Mask), sequestering dsRNA (Hide) or physically interacting with the host proteins involved in the RLRs pathway (Hit).
As representatives of the three different strategies, some prominent human RNA viruses have been here chosen on the basis of their lethality, their status of emerging or re-emerging pathogens and the substantial lack of therapeutics to counter the deadly diseases they cause. In light of recent functional, biochemical and structural findings, the mechanisms of action by which these virally-encoded IFN-antagonists cause a failure in RLR activation and signaling are discussed.
2. The origin of dsRNA as a viral PAMP
The existence of dsRNA as a byproduct of replication and transcription steps in the replication cycle of all viruses was proposed about half a century ago (Field et al., 1972a, Montagnier and Sanders, 1963) and it was hypothesized that it could serve as a potent stimulus for IFN-α/β induction (Field et al., 1970). Accordingly, it was demonstrated that the addition of dsRNA, of either viral (Nemes et al., 1969, Tytell et al., 1967) or synthetic origin (Field et al., 1972b), to the culture medium of several cell lines, as well as its intracellular delivery, resulted in a robust type I IFN response (Marcus 1983). Thereafter, several studies aimed to dissect the cellular IFN-based antiviral response by using polyinosinic:polycytidylic acid (polyI:C) to mimic the dsRNA intermediates that are present during viral infections. After more than two decades, such efforts led to the identification of TLR3 as the receptor responsive to dsRNA internalized by endocytosis (Alexopoulou et al., 2001) and, later, to the discovery of RLRs as the class of molecules that detects dsRNA in the cytoplasm (Andrejeva et al., 2004, Yoneyama et al., 2004, Yoneyama et al., 2005).
Indeed, how and when base-paired RNA sequences were generated and/or were freely exposed during infection by different viruses has represented the main question of several studies. On the one hand, the use of polyI:C as viral mimetic, while demonstrating that the double-helical nature of RNA is itself a potent IFN inducer (Hausmann et al., 2008), somehow depicted an artificial recognition process by RLRs. On the other hand, while viral dsRNAs of genomic length were able to activate RLRs pathway to induce IFN-α/β (DeWitte-Orr et al., 2009, Kato et al., 2008), immunofluorescence analysis with dsRNA-specific monoclonal antibody showed detectable amounts of dsRNA only in cells infected by some positive-stranded (+)-RNA viruses, whereas no dsRNA was detected in cells infected by negative-stranded (−)-RNA viruses (Weber et al., 2006). Currently, apart from dsRNA viruses for which the dsRNA PAMP is the viral genome itself, it is generally assumed that other viruses produce this PAMP as a replicative byproduct. In DNA viruses, for example, dsRNA may be generated through the self-annealing of convergent bidirectional transcription products (Weber et al., 2006). (+)-RNA viruses, whose single-stranded genomes directly serve for the translation of viral proteins, generate (−)-ssRNA antigenome templates and newly-synthesized (+)-ssRNA genomes during their replication, and both these products are susceptible of base-pairing into dsRNA with their complementary template strand (Ahlquist, 2006). (−)-RNA viruses transcribe monocistronic mRNAs from every gene and during the replication step synthesize viral genomes and antigenomes from a template that is tightly encapsidated by nucleoproteins. This encapsidation occurs concomitantly to RNA synthesis, since it is strictly required by the viral RNA-dependent RNA polymerase (RdRp) complex for its attachment and progression along the template (Morin et al., 2013). As a result, RNA encapsidation also prevents double-strand formation, since no genomic or antigenomic intermediates are normally found naked in the cytoplasm and they therefore cannot anneal to each other to produce dsRNA moieties (Gerlier and Lyles, 2011).
Notably, prevention of dsRNA formation may also involve the recruitment of host factors, as the downregulation of the cellular helicase UAP56, which was found to unwind self-paired transcripts from influenza A virus (IAV), resulted in cytosolic accumulation of viral dsRNA (Wisskirchen et al., 2011). However, viral replication and transcription are both processes susceptible to errors, which may lead to occasional production of defective transcripts and abortive 5′-genome and antigenome ends that, annealing one to each other or self-pairing into panhandle-like secondary structures, become ideal IFN-inducing viral PAMP candidates (Gerlier and Lyles 2011). In line with this, defective interfering (DI) RNAs generated through genome copyback during Sendai virus (SeV) infection, as well as subgenomic DI particles from IAV were found to potently activate the IFN response and act as RLR ligands (Strahle et al., 2006, Baum et al., 2010).
Proteins having dsRNA-binding motifs recognize this nucleic acid in a sequence-independent manner and on the basis of its A-type helical grooves and negative charge (Chang and Ramos, 2005, Tian et al., 2004). Moreover, the presence of an RNA double helix is essential for RLRs to induce a type I IFN-based response (Schlee and Hartmann, 2010), as ssRNA molecules which did not undergo self-pairing and double-stranded secondary structures were unable to bind and activate RIG-I (Schlee et al., 2009, Schmidt et al., 2009). However, the fact that some endogenous short dsRNAs of annealed antisense transcripts and microRNA precursors with secondary-stem loops – that are occasionally present at low levels in mammalian cells – do not elicit any IFN response, demonstrated that RNA double-strandness alone may be not sufficient for proper RLRs stimulation (Lehner et al., 2002, Marques et al., 2006, Werner, 2005).
In fact, other features are also important, as the ability of viral dsRNA species to trigger a type I IFN-induced antiviral response is dictated by their atypical length, abundance, cytoplasmic localization (DeWitte-Orr et al., 2009, Kato et al., 2008) and, (only for RIG-I), by the presence of a 5′-ppp at their termini (Hornung et al., 2006, Pichlmair et al., 2006, Schlee et al., 2009, Schmidt et al., 2009). 5′-ppp moieties originate from the first-added nucleotide during the primer-independent initiation of viral RNA synthesis, within both transcription and replication processes (Choi, 2012). At the end of transcription, covalent attachment of a cap structure assures the complete removal of 5′-ppp (Decroly et al., 2011), whereas the triphosphates at 5′-genome termini are either processed or enwrapped by viral nucleoproteins into homo-polymeric nucleocapsids (Choi, 2012). However, 5′-ppp groups may be inadvertently exposed to the cytosolic environment in byproducts such as leader RNAs, as well as defective replicative intermediates that remain uncapped (Gerlier and Lyles, 2011). In addition to viral genome replication and transcription, the uncoating process to which viral nucleocapsids are subjected immediately after infection represents another critical moment for the recognition of 5′-ppp and panhandle structures. In line with this, a recent study showed that RIG-I was able to physically interact with, and to be activated by, nucleocapsids from several negative-stranded (−)-RNA viruses, independently from viral RNA synthesis (Weber et al., 2013).
Thus, even though it remains to be shown if such interaction involves intact nucleocapsids, or rather implies the exposure of dsRNA tracts accessible within them, these findings establish the cytosolic release of incoming virions as the possible earliest event for RIG-I detection of viral dsRNA (Weber et al., 2013). In summary, byproducts of viral replication and transcription (Rehwinkel et al., 2010) or DI RNAs (Baum et al., 2010, Strahle et al., 2006), as wells as unmodified 5′-ppp ends and dsRNA panhandle structures that come from newly-infecting virions (Weber et al., 2013) are all theoretically prone to provide double-stranded structures, originated through self-pairing or annealing with complementary sequences. Therefore, it is conceivable that all these together may represent the overall source of dsRNA ligands for RLRs activation.
3. RLRs: structure and pathway
The RLRs are a set of cytosolic sensors for dsRNA belonging to the superfamily 2 (SF2) clade of RNA helicases (Fairman-Williams et al., 2010). In addition to RIG-I (Yoneyama et al., 2004), this group includes also the melanoma differentiation associated factor-5 (MDA5) and the laboratory of genetics and physiology-2 (LGP2) proteins (Andrejeva et al., 2004, Yoneyama et al., 2005).
RLRs share a similar structural organization (Fig. 2 ), consisting of an RNA-binding C-terminal domain (CTD) (Cui et al., 2008, Takahasi et al., 2008, Takahasi et al., 2009) and a central DExD/H-box helicase domain comprising two RecA-like helicase domains, namely Hel1 and Hel2, separated by the insert domain Hel2i (Luo et al., 2011). In RIG-I and MDA5, but not in LGP2, the helicase domain is preceded by two tandemly-repeated N-terminal caspase activation and recruitment domains (CARDs) (Yoneyama et al., 2005). Unlike other members of the SF2 helicases, which bind one strand of the nucleic acid to unwind its double helix (Fairman-Williams et al., 2010), RLRs have double-strand specificity and do not seem to display unwinding activity. Indeed, a weak unwinding was initially described for a RIG-I deletion mutant (Marques et al., 2006) as well as for the full length protein in the presence of dsRNA substrates with overhangs at the 3′ strand longer than 5 nts (Takahasi et al., 2008). However, lack of RIG-I helicase activity was reported by subsequent studies (Myong et al., 2009, Jiang et al., 2012). Instead, RLRs bind to both dsRNA strands and translocate onto it in an ATP hydrolysis-dependent manner (Bamming and Horvath, 2009, Myong et al., 2009).
Fig. 2.
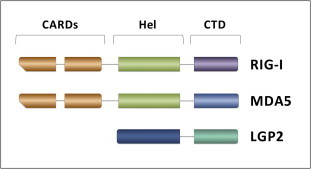
Structural organization of RLRs. RIG-I, MDA5 and LGP2 share an overall domain organization that consists of two tandemly-repeated N-terminal CARDs (orange boxes, absent in LGP2) followed by an Hel domain (green box in RIG-I and MDA5, blue box in LGP2) and a C-terminal RNA binding CTD (violet, azure and cyan box in RIG-I, MDA5 and LGP2 respectively).
According to the current model (schematically depicted in Fig. 3 ), physiologically inactive RIG-I is found in an auto-inhibited state, in which the two CARDs are kept engaged in intramolecular interactions with the Hel2i domain by a repressor pincer motif located between the CTD and the helicase domains (Kowalinski et al., 2011). Upon dsRNA binding to the CTD (Civril et al., 2011), the pincer motif is relieved from CARDs that are then displaced from the Hel2i allowing its binding to dsRNA (Luo et al., 2011). Subsequent to dsRNA enwrapping to Hel domains, RIG-I dimerization and activation of its ATPase activity take place (Cui et al., 2008). ATP hydrolysis propels the translocation of the RIG-I dimer along dsRNA (Myong et al., 2009) and an overall conformational change releases N-terminal CARDs for signaling (Jiang et al., 2011). Freely exposed RIG-I CARDs then interact with unanchored Lys-63-linked poly-ubiquitin (poly-Ub) chains (Zeng et al., 2010). Next, RIG-I homo-oligomerization and ATP-ase-driven translocation along dsRNA take place (Patel J.R. et al., 2013), leading to additional conformational changes that provide the signaling platform for interaction with the CARDs of the IFN-β promoter stimulator 1 (IPS-1) (Kawai et al., 2005). This is a mitochondrion-associated protein, now called mitochondrial antiviral signaling protein (MAVS) (Seth et al., 2005), formerly also known as IFN-β inducing CARD adaptor (Cardif) (Meylan et al., 2005) or virus-induced signaling adaptor (VISA) (Xu et al., 2005).
Fig. 3.
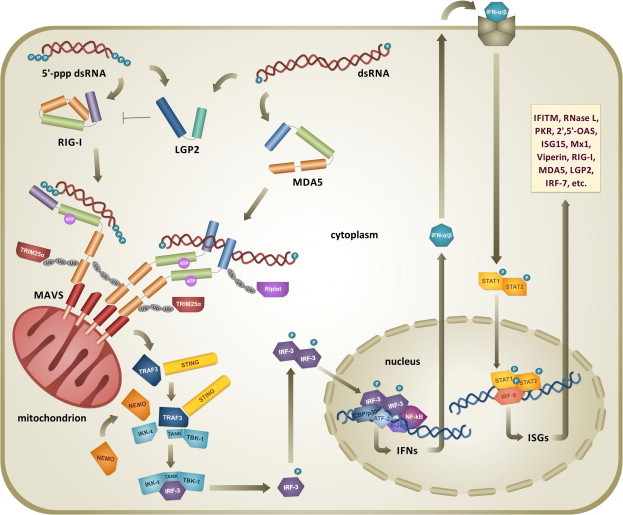
Activation of the type I IFN antiviral response triggered by the RLRs pathway upon dsRNA detection. Viral short 5′-ppp dsRNA and long dsRNA are preferentially recognized by the CTD of RIG-I (violet) and of MDA5 (azure) respectively, with LGP2 modulating the activity of RIG-I helicase. Upon dsRNA binding to their Hel domain (green), ATP-mediated homo-oligomerization, translocation onto dsRNA, ubiquitination, and TRIM25- or Riplet-mediated ubiquitination, RLRs interact through their CARDs (orange) with the CARD of mitochondrion-associated MAVS (red). Signaling prosecution involves recruitment of TRAF3, NEMO and STING adaptors and the assembly of TBK1-IKK-ε complex, which phosphorylates IRFs. Activated IRF dimers translocate to the nucleus and, together with other transcription factors, induce the expression of IFN-α/β. Type I IFNs are secreted and bind to their cognate receptor, activating STAT transcription factors for the induction of several ISG products with antiviral activity and the overexpression of RLRs pathway components.
Essential for the prosecution of immune signaling, the redistribution of RIG-I from the cytosol to mitochondrial membranes is sustained by the formation of a translocon that involves the mitochondrion-targeting chaperone 14-3-3 ε and the tripartite motif 25 alpha (TRIM25α) E3 ligase (Liu et al., 2012). Furthermore, in addition to RIG-I activation via unanchored polyubiquitin chains, Lys-63-linked polyubiquitination of RIG-I CARDs by TRIM25α was found to be important for the interaction with MAVS (Gack et al., 2007). In turn, such activity of TRIM25α requires another E3 ligase, namely RING finger protein leading to RIG-I activation (Riplet) (Oshiumi et al., 2009, Oshiumi et al., 2010), whose polyubiquitination of RIG-I CTD facilitates the release of CARDs and their association with TRIM25α (Oshiumi et al., 2013). At this point, the described mechanism, even though proposed as a general model for dsRNA recognition by RLRs, is mostly based on the characterization of RIG-I. However, the activation and signaling of MDA5 shows some differences. For instance, CARDs are not sequestered in the MDA5 resting form, and its activation – for which both ATPase activity and Lys-63-linked ubiquitination are also important – results in the cooperative assembly of MDA-5 protomers onto dsRNA helical filaments (Berke and Modis, 2012, Jiang et al., 2012, Peisley et al., 2011, Wu et al., 2013). Next, prosecution of the pathway involves MAVS prion-like polymerization (Hou et al., 2011) and the recruitment of two adaptors, tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3) and the nuclear factor-kB (NF-kB) essential modifier (NEMO), which connect and regulate signaling between the upstream sensory domain of MAVS and the downstream complex formed by TRAF family member-associated NF-kB activator (TANK)-binding kinase 1 (TBK-1) and inducible IkB kinase epsilon (IKK-ε) (Oganesyan et al., 2005, Zhao et al., 2007, Belgnaoui et al., 2012, Wang et al., 2012). The two kinases TBK-1 and IKK-ε carry out the phosphorylation of latent IFN regulatory factors 3 and 7 (IRF-3/7), leading to their dimerization and nuclear translocation (Fitzgerald et al., 2003).
It is worth noting that the recruitment of RIG-I to MAVS (Liu et al., 2012) and the interaction of TBK1-IKK-ε complex with MAVS are two critical events for IRF phosphorylation. In particular, in the latter was found involved a newly-identified adaptor that acts as an RLR signaling enhancer, independently discovered by three research groups and variously termed stimulator of interferon genes (STING), mediator of IRF-3 activation (MITA) and endoplasmic reticulum IFN stimulator (ERIS), respectively (Ishikawa and Barber, 2008, Sun et al., 2009, Zhong et al., 2008). Once in the nucleus, IRF-3 and 7 homo- or hetero-dimers associate into an enhanceosome with other transcription factors, such as cAMP response element-binding (CREB) binding protein/p300, NF-kB, activating transcription factor 2 (ATF-2) and c-Jun to drive the transcription of IFN-α/β genes (Panne et al., 2007). In turn, secreted type I IFNs act towards the same cell, or the neighboring ones, by binding to their ubiquitous cognate receptors and activating a signaling cascade that ultimately leads to the expression of hundreds of IFN-stimulated genes (ISGs) with antiviral properties (Sadler and Williams, 2008). Some ISGs encode for effectors that are able to directly interfere with the viral replication cycle, such as the interferon-induced transmembrane (IFITM), ribonuclease L (RNase L), 2′, 5′-oligoadenylate synthetase (OAS), dsRNA-dependent protein kinase R (PKR), Viperin, myxovirus resistance 1 (Mx1) and interferon stimulated gene 15 (ISG15) proteins (Liu et al., 2011). In addition, since among the ISGs products are also the PRRs, their adaptor molecules and transcription factors such as the IRFs, the expression of these proteins determines an amplification loop that increases both IFN-α/β and ISGs production, thereby establishing an overall antiviral state to limit virus spread and eventually clear the infection (Sadler and Williams, 2008).
4. RLRs: differential recognition of dsRNA agonists
RIG-I and MDA5 helicases have been shown to preferentially sense different types of dsRNA PAMPs, based on their length, bluntness and end overhangs. RIG-I optimal ligands are short 5′-ppp-containing dsRNA molecules less than 20 bp in length (Lu et al., 2010, Jiang et al., 2011, Wang et al., 2010) and, while it can also bind 5′-hydroxyl (OH) blunt-ended dsRNA (Lu et al., 2011, Luo et al., 2011, Kowalinski et al., 2011), the presence of the 5′-ppp group is essential for its proper activation (Hornung et al., 2006, Pichlmair et al., 2006, Rehwinkel et al., 2010). The 5′-ppp recognition is imparted by a set of key basic residues that form a positively-charged pocket within the RIG-I CTD, thus allowing efficient accommodation of the three phosphates (Cui et al., 2008, Lu et al., 2010, Takahasi et al., 2009). However, even though RIG-I recognizes 5′-ppp dsRNA by end-capping it (Lu et al., 2011, Luo et al., 2011), the 5′-ppp feature alone is not sufficient for efficient IFN induction, and at least small regions of dsRNA terminal base-pairing are required for proper interaction with the helicase (Lu C. et al., 2011). In particular, double-strandness must encompass the 5′ nucleotide carrying the triphosphate and 5′ overhang at the 5′-ppp end abolishes RIG-I activity (Schlee et al., 2009). Instead, 3′ overhangs are tolerated, although decreasing RIG-I binding (Schlee et al., 2009) or supporting its ATPase activity without inducing IFN response (Schmidt et al., 2009).
Moreover, despite the fact that 5′-monophosphate and 5′-OH groups at dsRNA ends are correlated with diminished RIG-I binding affinity and IFN induction (Schlee et al., 2009, Schmidt et al., 2009), such detrimental effect of 5′-ppp lack tends to vanish as the length of dsRNA substrate increases. In fact, provided the substrate is long enough, multiple internal initiation binding sites can compensate for the absence of the 5′-ppp at dsRNA ends (Binder et al., 2011). In agreement with the cooperative oligomerization of RIG-I along dsRNA, whose efficiency is in turn correlated with the strength of IFN induction (Patel J.R. et al., 2013), such flexible behavior allows RIG-I to integrate the two distinct signals of high-affinity motif and dsRNA length, thereby displaying the highest possible sensitivity in response to diverse PAMPs to be detected (Binder et al., 2011).
Regarding MDA5, its optimal ligands were initially identified as blunt-ended dsRNA moieties more than 1 kbp in length (Kato et al., 2008), while a subsequent study showed that this helicase is activated by an RNA web formed by branched dsRNA stretches (Pichlmair et al., 2009). Moreover, such high molecular weight moieties are bound by MDA5 regardless of the nature of the dsRNA 5′-ends, since the MDA5 CTD has a flat basic surface that lacks the RIG-I-like 5′-ppp-lodging pocket and does not cap dsRNA terminus (Li et al., 2009a, Takahasi et al., 2009). Instead, MDA5 binds to the dsRNA backbone parallel to the helix axis and multimerizes into helical filaments along dsRNA (Berke et al., 2012, Wu et al., 2013). Less is known about LGP2, the third member of the RLRs, which was shown to bind blunt-ended dsRNA in an ATP hydrolysis-dependent manner (Bruns et al., 2013, Murali et al., 2008) and regardless the presence of 5′-ppp (Li et al., 2009b, Pippig et al., 2009). Displaying a complex phenotype, LGP2 plays negative as well as positive roles in IFN induction. In fact, it negatively regulates RIG-I (Rothenfusser et al., 2005, Yoneyama et al., 2005) while it was shown to facilitate viral dsRNA recognition (Bruns et al., 2013, Satoh et al., 2010) and to act as positive regulator of MDA5, with which it co-operates in response to polyI:C stimulation (Childs et al., 2013).
The observed non-redundant abilities of RLRs to bind to diverse RNA ligands reflect their differential capacity to recognize various viral groups. In fact, while RIG-I preferentially recognizes 5′-ppp-containing self-paired panhandle RNAs, such those exhibited by several (−)-ssRNA viruses, (+)-ssRNA viruses that produce long blunt-ended RNA duplexes are mostly detected by MDA5. Notably, these patterns partially overlap, as some viruses are recognized by both RLRs (Loo et al., 2008, Kato et al., 2006).
5. Hide, mask, hit: three viral strategies to block RLRs dsRNA detection
Obeying the ultimate goal of replicating in the host cell to generate new infectious virions, viral pathways include functionalities aimed to circumvent, downregulate or even totally suppress IFN-α/β induction, IFN-α/β signaling or ISG function (Versteeg and Garcia-Sastre, 2010). Any of the several components involved in the different phases of the innate antiviral response can be targeted by viruses. However, the inhibition of IFN-α/β production achieved by blocking the RLR pathway in its upstream portion, at the level of dsRNA recognition, is a target shared by most viral antagonists, further evidencing the potency of such PAMP in triggering a robust innate immune response (Bowie and Unterholzner, 2008). RNA viruses, of which several are pathogenic to humans, have developed a plethora of solutions to avoid dsRNA-mediated activation of RLRs. These can be ascribed to three strategies, hereby named as
-
1.
Hide, that indicates dsRNA sequestration to prevent its binding to RLRs, and is achieved either by viral proteins competing with RLRs for dsRNA binding or by shielding dsRNA through its compartmentalization within the cytoplasm (Fig. 4 A);
-
2.
Mask, that relies on the ability of viral proteins to process double-helix and 5′-ppp termini, removing the PAMP signatures recognized by RLRs (Fig. 4B);
-
3.
Hit, that involves physical interaction between viral proteins and either RLRs or their downstream adaptors to suppress their functionalities (Fig. 5 ).
Fig. 4.
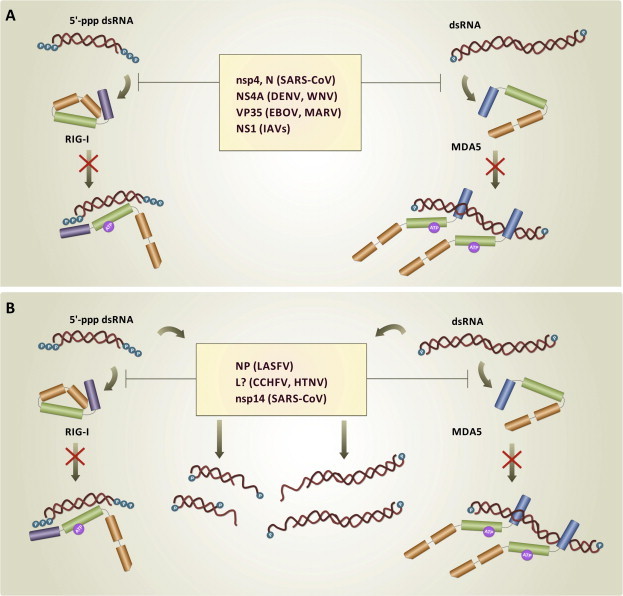
Hide and Mask strategies. Production of type I IFN-α/β is prevented by keeping RLRs from being triggered by short 5′-ppp dsRNA and long dsRNA molecules. (A) In the Hide strategy, nucleic acids are (i) bound by viral IFN-antagonists that compete with RLRs for the same binding sites on dsRNA backbone and/or terminal bases and phosphate groups; (ii) sequestered from RLRs recognition by compartmentalization into virally–assembled CMs and DMVs. (B) In the Mask strategy, viral IFN-antagonists remove PAMP signatures from dsRNA by (i) processing terminal 5′-ppp to monophosphate groups; (ii) digesting one nucleic acid strand through exoribonuclease activity.
Fig. 5.
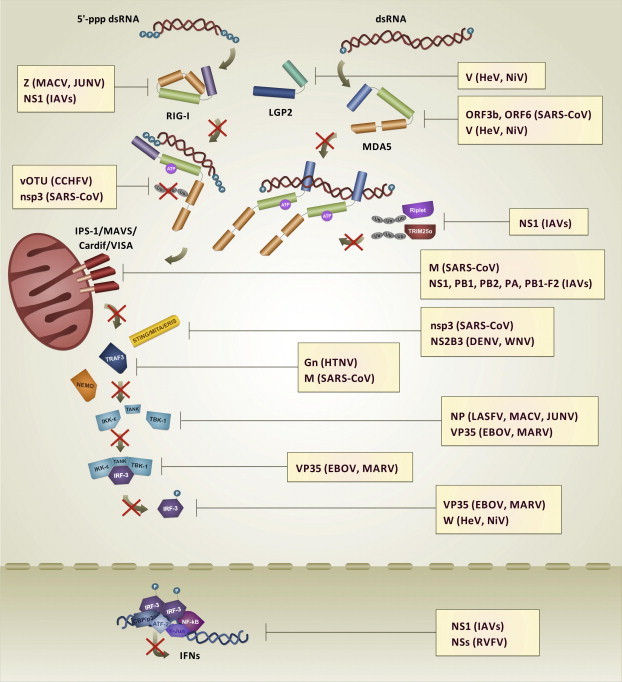
Hit strategy. In the Hit strategy, viral IFN antagonists suppress RLRs-mediated IFN-α/β induction by interacting with RLRs and their adaptor and/or effector molecules to inhibit their functionalities.
Due to the multifunctional nature of the viral proteins involved in these strategies, a certain degree of redundancy is present, with a given virus concomitantly adopting more than one strategy at a time through the action of one or more viral IFN-antagonists. Noteworthy, thanks to the recent availability of crystallographic structures of several viral proteins bound to their dsRNA ligands, some molecular details of the dsRNA Hide strategy are beginning to be unraveled. Conversely, the direct Hit towards RLRs, or their effectors, appears to be the most multifaceted strategy given the high number of cellular components involved so that, in most cases, the mechanism through which viral antagonists exert such inhibition still remain to be elucidated. According to these three strategies, we reviewed here solutions adopted by a number of RNA viruses that are highly lethal to humans and for which effective medical countermeasures are currently lacking (Table 1 ).
Table 1.
Highly pathogenic RNA viruses which encode proteins that adopt the Hide, Mask and Hit strategies to block dsRNA recognition.
| Family | Virus | Viral IFN-antagonist involved in targeting the RLRs pathway |
||
|---|---|---|---|---|
| Hide | Mask | Hit | ||
| Arenaviridae (OW) | LASFV | NP | NP, Z | |
| Arenaviridae (NW) | JUNV | NP, Z | ||
| MACV | NP, Z | |||
| Bunyaviridae | CCHFV | L (?) | vOTU | |
| HTNV | L (?) | Gn | ||
| RVFV | NSs | |||
| Coronaviridae | SARS-CoV | nsp4, N | nsp14 | ORF3b, ORF6, M, nsp3 |
| Filoviridae | EBOV | VP35 | VP35 | |
| MARV | VP35 | VP35 | ||
| Flaviviridae | DENV | NS4A | NS3B2 | |
| WNV | NS4A | NS3B2 | ||
| Henipaviridae | HeV | V, W | ||
| NiV | V, W | |||
| Orthomyxoviridae | IAVs | NS1 | NS1, PB1, PB2, PA, PB1-F2 | |
CCHFV, Crimean-Congo hemorrhagic fever virus; DENV, dengue virus; EBOV, Ebola virus; HTNV, Hantaan virus; HeV, Hendra virus; IAVs, influenza A viruses; JUNV, Junin virus; LASFV, Lassa fever virus; MACV, Machupo virus; MARV, Marburg virus; NiV, Nipah virus; RVFV, Rift Valley fever virus; SARS-CoV, severe acute respiratory syndrome coronavirus; WNV, West Nile virus.
6. Arenaviruses
Arenaviruses are mostly rodent-borne zoonotic pathogens with bi-segmented and ambisense ssRNA genomes that belong to the single genus Arenavirus of the family Arenaviridae and are the largest and worldwide distributed group of viruses causing hemorrhagic fever in humans (Charrel et al., 2011). On the basis of their antigenic properties, genetic relationships and geographical endemicity, members of this group are classified into Old World (OW) and New World (NW) arenaviruses. Among them, the OW Lassa fever virus (LASFV) and Lujo virus (LUJV), as well as the NW Chapare virus (CHPV), Guanarito virus (GTOV), Junin virus (JUNV), Machupo virus (MACV), Sabia virus (SABV) and Whitewater Arroyo virus (WWAV) have all been associated with fatal disease in humans (Briese et al., 2009, Charrel et al., 2011, Delgado et al., 2008). 5′-ppp dsRNA moieties belonging to arenaviral genomes have been shown to activate the RLR pathway (Habjan et al., 2008, Zhou et al., 2010, Huang et al., 2012). Moreover, suppression of the type I IFN response in cell culture assays by both OW and NW arenaviruses has been reported and related to the properties displayed by two of the four proteins encoded by the arenaviral genome: the nucleocapsid protein NP and (only for NW arenaviruses) the matrix protein Z (Carnec et al., 2011, Groseth et al., 2011, Huang et al., 2012, Martinez-Sobrido et al., 2006, Martinez-Sobrido et al., 2007, Müller et al., 2007).
6.1. Mask
The arenaviral NP is a multifunctional protein with essential roles in the replication and transcription of viral RNA (Hass et al., 2004, Pinschewer et al., 2003), in the encapsidation of viral genome into a ribonucleoprotein (RNP) complex (López et al., 2001) and, together with the Z protein, in virion assembly (Eichler et al., 2004). Early studies identified NP as sufficient for the inhibition of IRF-3 phosphorylation and consequent suppression of IFN induction, thereby highlighting its innate immune antagonist nature (Martinez-Sobrido et al., 2006, Martinez-Sobrido et al., 2007). Recently, structural and functional characterization of LASFV NP revealed that this property relies on the ability of the protein to process viral dsRNA with a 3′–5′ exoribonuclease activity (Fig. 6 A). In fact, while the N-terminal NP folds into a domain important for mRNA capping during transcription, a large cavity within its C-terminal domain exhibits a dsRNA 3′–5′ exoribonuclease activity with a catalytic site comprising residues D389, E391, D466, D533 and H528, and structurally resembling the active sites of the DEDDH exonuclease superfamily (Qi et al., 2010). Notably, these amino acids reside within the same NP region (residues 370–553) mapped for the anti-IFN activity of the OW lymphocytic choriomeningitis virus (LCMV), which contains critical residues along its DIEGR motif (residues 382–386) as well as at positions D459, H517 and D522 (Jiang et al., 2013, Martinez-Sobrido et al., 2009, Qi et al., 2010).
Fig. 6.
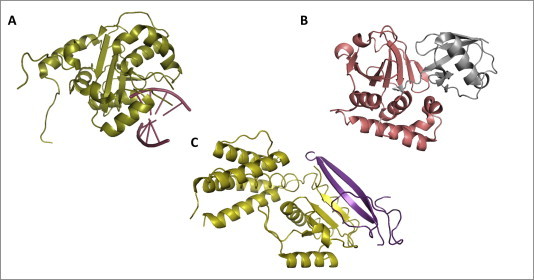
Structures of viral proteins dispaying the Mask and the Hit strategies. (A) LASFV NP masks dsRNA: LASFV may evade host cell dsRNA-induced innate immune antiviral response through the 3′–5′ exoribonuclease activity of its NP protein (highlighted in deep olive, PyMOL colors), which eliminates 5′-ppp and double-strandness PAMP signatures from viral dsRNA. Crystallographic structure by Jiang et al., 2013 (PDB: 4G9Z); (B) CCHFV vOTU hits human Ubiquitin: CCHFV may downregulate RLRs pathway-mediated type I IFN production through the activity of its vOTU protease (highlighted in deep salmon, PyMOL colors), which removes ubiquitin (highlighted in gray, PyMOL colors) from RLRs, their adaptor and/or effector molecules, impairing their proper signaling. Crystallographic structure by Capodagli et al., 2011 (PDB: 3PRP); (C) Paramyxovirus V hits MDA5: HeV and NiV inhibit RLRs pathway-mediated IFN-α/β induction by preventing MDA5 activation and signaling through its sequestration by V protein. As shown in figure for the henipavirus closely-related PIV5, V protein zinc finger domain (highlighted in violet–purple, PyMOL colors) interacts with the Hel2 domain of MDA5 (highlighted in olive, PyMOL colors), thereby inhibiting its ATPase activity. Crystallographic structure by Motz et al., 2013 (PDB: 4I1S).
Alanine point mutation at each position of the LASFV NP 3′–5′ exoribonuclease active site, as well as at the two additional residues G392 and R492, resulted in markedly reduced exoribonuclease activity, without affecting the NP cap-binding function (Hastie et al., 2011, Qi et al., 2010). Notably, two of these residues were found to be critical for IFN suppression (Martinez-Sobrido et al., 2009), as the substitution into alanine of all of them caused complete inability of LASFV NP to inhibit SeV- or polyI:C-induced activation of the IFN-α/β promoter (Hastie et al., 2011, Jiang et al., 2013, Qi et al., 2010).
Unlike other known DEDDH exonucleases, which split dsDNA to start digestion from an unpaired 3′-terminus, LASFV NP binds to dsRNA without unwinding its strands and, regardless of whether it bears overhanging or blunt-ended termini, with or without 5′-ppp, it readily digests the 3′-strand in the 5′-direction. (Hastie et al., 2011, Jiang et al., 2013, Qi et al., 2010). It is therefore possible to envision the possibility that such processing removes from the viral dsRNA any PAMP hallmark that would be otherwise recognized by RLRs. The fact that critical residues for dsRNA exoribonuclease-mediated IFN inhibition are highly conserved among the NPs of all arenaviruses strongly suggests that this form of the Mask strategy is widely shared within the family (Harmon et al., 2013, Jiang et al., 2013, Qi et al., 2010).
6.2. Hit
As a remarkable example of IFN-antagonism redundancy, the same C-terminal NP region that is responsible for dsRNA masking was also found to exert a Hit strategy in both OW and NW arenaviruses. In fact, confocal microscopy and co-immunoprecipitation (co-IP) analyses have revealed that arenaviral NPs bind to the kinase domain of IKK-ε, blocking its autocatalytic activity and inhibiting the phosphorylation of IRF-3 substrate. IKK-ε sequestration by arenaviral NPs impedes the interaction between this kinase and its mitochondrial partner MAVS, thereby shutting down signaling along the RLR pathway (Pythoud et al., 2012). Importantly, no interactions were observed between NP and other components of the pathway, such as TBK-1, MAVS, TRAF3 and even RIG-I or MDA5 (Pythoud et al., 2012), although for the latter another study reported a co-IP with the NP of LCMV (Zhou et al., 2010). Notably, the NP-IKK-ε interaction is disrupted by mutating the same NP residues that are critical for its 3′–5′ exoribonuclease activity, suggesting the existence of an overlap in the domains responsible for the two IFN-antagonist functions (Pythoud et al., 2012).
In the case of the NW arenaviruses, a Hit strategy against the RLRs pathway is also carried out, through the function exerted by the Z matrix protein, a small RING finger polypeptide that acts as a co-factor of the viral polymerase complex and also mediates both RNP incorporation into virions and their release from the plasma membrane (Fehling et al., 2012). Overexpression of the JUNV Z protein blocked IRF-3 and nuclear factor-kappa-B (NF-kB) phosphorylation and nuclear translocation, and decreased IFN-β mRNA levels in response to both SeV and 5′-ppp dsRNA stimulation (Fan et al., 2010, Martinez-Sobrido et al., 2006, Martinez-Sobrido et al., 2007). Furthermore, confocal microscopy and co-IP revealed that the Z proteins of GTOV, JUNV, MACV and SABV, but not of LCMV and LASFV, are able to interact with RIG-I, preventing its recruitment to MAVS for downstream signaling (Fan et al., 2010). The mechanism of such inhibiting interaction, as well as the molecular details in terms of involved domain/s and critical residues in the Z protein, remain to be elucidated.
7. Bunyaviruses
Viruses of the Bunyaviridae family have a tripartite RNA genome with negative or ambisense coding strategy and are classified into the five genera Orthobunyavirus, Hantavirus, Nairovirus, Phlebovirus and Tospovirus. (Walter and Barr, 2011). This group comprises zoonotic pathogens that are etiologic agents of severe disease in humans, characterized by high lethality, lack of preventive or therapeutic treatments and an emerging pattern at the human-wildlife interface (Walter and Barr, 2011). Among these, the arthropod-borne Rift Valley fever virus (RVFV) from the genus Phlebovirus and the Crimean-Congo hemorrhagic fever virus (CCHFV) from the genus Nairovirus cause fulminating hemorrhagic fever (Ikegami, 2012, Keshtkar-Jahromi et al., 2011), while the rodent-borne Hantaan virus (HTNV), Sin Nombre virus (SNV) and Andes virus (ANDV) from the genus Hantavirus cause the typical hemorrhagic fevers with renal and cardio-pulmonary syndromes, respectively (Charrel et al., 2011). A new member of the genus Phlebovirus, severe fever with thrombocytopenia syndrome virus (SFTSV), was recently discovered and identified as the causative agent of a hemorrhagic fever with high fatality rates that has emerged in several provinces of China (Yu et al., 2011, Zhang et al., 2013).
As shown by in vivo studies, IFN-α/β is poorly induced in RVFV-infected animal models, and the late onset of the type I IFN response is correlated with increased viral pathogenicity (Elliott and Weber, 2009). IFN-α/β levels are kept very low in hantavirus-infected patients during the entire course of disease, and hantaviral species that are pathogenic to humans were found to be able to suppress IFN induction and signaling in cell culture assays much more efficiently than non-pathogenic viruses (Elliott and Weber, 2009). Moreover, a biphasic pattern was observed during hantavirus infection, in which IFN production is suppressed early, but a dramatic increase in ISGs expression occurs at later stages (Matthys and Mackow, 2012). Likewise, type I IFN production and secretion is strongly delayed during CCHFV infection and virus replicating cells were found totally insensitive to IFN-α treatment (Andersson et al., 2008, Weber and Mirazimi, 2008). A similar pattern of type I IFN response dysregulation was also observed for SFTSV, showing that human monocytes are susceptible to infection, mounting a restrained antiviral response with upregulation of some IFN-inducible proteins, but with very low levels of induced type I IFN (Qu et al., 2012).
Taken together, these data indicate that the virulence and pathogenesis of bunyaviruses may be due, at least in part, to their ability to counteract cellular IFN responses (Walter and Barr, 2011). Accordingly, Mask and Hit strategies here described are adopted by members of the genera Hantavirus and Nairovirus to escape RIG-I sensing. For RVFV, as well as for other viruses of the genera Orthobunyavirus and Phlebovirus, potent inhibition of the type I IFN-based antiviral response has been found as the result of the properties displayed by their NSs proteins (Bouloy et al., 2001). Indeed, NSs proteins do not specifically target any of the above described components of the RLRs pathway; however, given that their action ultimately impedes the production of IFN-β (Elliott and Weber, 2009), the NSs strategy is de facto a Hit towards the pathway at its most basic level. By contrast, members of the genera Hantavirus and Nairovirus adopt Mask and Hit strategies aimed at directly escaping RIG-I sensing.
7.1. Mask
Digestion with a specific 5′–3′ ssRNA exoribonuclease demonstrated that HTNV and CCHFV genomic RNAs bear a monophosphate group at the 5′ terminus, which may explain why such nucleic acids are not sensed by RIG-I (Habjan et al., 2008). HTNV 5′-ppp moieties are trimmed off during the synthesis of viral genome through a “prime and realign” process that cleaves the first-incorporated nucleotide leaving a 5′ monophosphorylated terminus (Garcin et al., 1995). Furthermore, in addition to being devoid of RIG-I-stimulating 5′-ppp, the HTNV genome has complementary 5′- and 3′-strands that form perfectly paired panhandle structures that are promptly encapsidated by viral nucleocapsid proteins (Garcin et al., 1995). For CCHFV, whose genomic RNA bears a terminal pyrimidine residue at the 5′-end, in place of the purine normally used by viral polymerases to initiate RNA synthesis, a similar “prime and realign” process likely occurs during replication (Garcin et al., 1995). However, the exact mechanism and viral proteins through which CCHFV removes protruding 5′-ppp from its genome ends is currently unknown (Habjan et al., 2008).
7.2. Hit
Even though genomic HTNV and CCHFV RNAs do not significantly activate the RLRs pathway, dsRNA-like secondary structures able to stimulate RIG-I were observed in the mRNA transcripts of HTNV viral nucleoprotein (Lee et al., 2011). Moreover, while HTNV replication was efficient in RIG-I knock-down A549 alveolar epithelial cells, it was strongly inhibited by transient overexpression of RIG-I in Huh7.5 cells that do not constitutively express this helicase (Lee et al., 2011). It is not surprising that also pathogenic hantaviruses display a redundant Hit strategy to target components along the RLRs pathway. The ectopic expression of the hantaviral glycoprotein Gn, from the pathogenic New York-1 virus (NY-1V), was reported to inhibit IRF-3 phosphorylation as well as IRF-3-directed transcriptional response of IFN-β promoter (Alff et al., 2006). Subsequent studies showed that such IFN antagonism is related to a 142-residue cytoplasmic Gn tail that is able to sequester the TRAF3 adaptor by binding to its N-terminal region, and that consequently inhibits the formation of a functional TBK1-TRAF3 complex (Alff et al., 2008).
The presence of a viral homologue of the ovarian tumor protease domain (vOTU) (Fig. 6B) within the CCHFV RdRp polymerase L (Frias-Staheli et al., 2007) allows a cross-deubiquitinase proteolytic process for the removal of both poly-Ub and ISG15 from target proteins (Capodagli et al., 2011, James et al., 2011). Notably, RIG-I ubiquitination by TRIM25α stabilizes its interaction with MAVS and enhances downstream signaling (Gack et al., 2007, Gack et al., 2008). Similarly to ubiquitination, also K63-linked isopeptide-bond conjugation with ISG15 (ISGylation) of several target proteins is critical for IFN-α/β induction and antiviral activity in response to RNA virus infection, showing both positive and negative regulation of the RLRs pathway (Zhao et al., 2013). For example, IRF-3 ISGylation prevents its degradation and is required for efficient translocation and permanence into the nucleus (Lu et al., 2006), while RIG-I ISGylation lowers its cellular levels to finely tune the antiviral response strength (Kim et al., 2008). Therefore, vOTU deconjugation of Ub and ISG15 from RLRs pathway components seems to be a Hit strategy by which CCHFV evades IFN-based immune response, however further investigations are required to precisely define both mechanism of action and target proteins.
As the major virulence factor and IFN-antagonist of RVFV, NSs induces a complete shut-off of transcriptional activity in the infected cell, exerted through the sequestration of the p44 and XPD subunits of RNA polymerase II transcriptional factor TFIIH (Le May et al., 2004) and by promoting the degradation of its p62 subunit (Kalveram et al., 2011). Moreover, in addition to such generalized downregulation of host cell gene expression (Billecocq et al., 2004), the RVFV NSs was shown to directly target the IFN-β production. In fact, by recruiting the repressor protein SAP30, NSs forms with it a multiprotein complex on the IFN-β promoter, thereby inhibiting its induction (Le May et al., 2008). Recently, also the NSs of SFSTV, together with its N protein, were found to act as IFN-antagonists, as revealed by reporter gene assays that showed suppression of IFN-β and NF-kB promoters activity in response to polyI:C and IAV infection (Qu et al., 2012). However, while the mechanism by which blockage of IFN production is achieved by both of these proteins is currently unknown, it is likely that inhibition would be exerted by SFSTV NSs through a yet-to-be-determined Hit strategy, as it was found able to co-precipitate with TBK-1 (Qu et al., 2012).
8. Coronaviruses
Coronaviruses of the genus Betacoronavirus are (+)-ssRNA viruses in the family Coronaviridae, whose members cause important respiratory disease in humans (Perlman and Netland, 2009). In particular, the spillover from a zoonotic reservoir into the human population of Guandong province in southern China led in 2002 to the identification of a novel coronavirus (CoV) belonging to the genus Betacoronavirus (Ksiazek et al., 2003). Causing a “flu-like” disease characterized by severe acute respiratory syndrome (SARS) with high mortality rates, the SARS-CoV rapidly spread to many countries (Hui and Wong, 2004). More recently, a novel CoV was identified in the Middle East as the causative agent of several acute respiratory syndrome with renal failure (Zaki et al., 2012, Khan, 2013). Tentatively named Middle East respiratory syndrome (MERS)-CoV, this pathogen has been responsible for more than 100 cases of lethal infection (de Groot et al., 2013).
Patients with SARS typically show aberrant type I IFN, ISGs and cytokine responses, suggesting that the virulence and pathogenicity of the SARS-CoV are related to profound dysregulation of the innate immune antiviral response (Totura and Baric, 2012). Regarding the RLRs pathway, even though both RIG-I and MDA5 are overexpressed during SARS-CoV infection in vitro (Yoshikawa et al., 2010), no IRF-3 phosphorylation or dimerization were observed and loss of IFN induction was reported in SARS-CoV-infected fibroblasts (Thiel and Weber, 2008). A similar pattern of poor IFN production and expression of pro-inflammatory cytokine was also observed in human bronchial epithelial cells infected with the MERS-CoV, and transcriptomic analysis of host gene expression showed upregulation of RIG-I, MDA5, IRFs and other ISGs (Kindler et al., 2013, Josset et al., 2013). In addition, comparative analysis of cell tropism and innate immune evasion showed that both viruses prevent IRF-3-mediated IFN-α/β induction, with MERS-CoV showing a higher sensitivity to IFN treatment (Chan et al., 2013, de Wilde et al., 2013, Zielecki et al., 2013). Together, these data suggest that SARS-CoV and MERS-CoV employ strategies to counteract RLRs dsRNA recognition.
8.1. Hide
The observation that in SARS-CoV-infected cells no type I IFN production was found, whereas treatment with polyI:C resulted in both IRF-3 activation and IFN-α/β induction (Versteeg et al., 2007, Zhou and Perlman, 2007), strongly suggests that viral dsRNA is somehow buried and rendered inaccessible to cytoplasmic RLRs. In line with this, electron tomography studies have revealed that SARS-CoV infection alters the cytoplasmic membrane architecture by inducing the formation of a series of double-membrane vesicles (DMVs) which, interconnecting to each other without communicating with the cytosol, converge to the endoplasmic reticulum (ER) and the Golgi apparatus (Knoops et al., 2008). Membrane convolution into DMVs seems to be directed by the SARS-CoV nsp4 protein, as its mutation results in aberrant and open vesicles (Clementz et al., 2008). Furthermore, nsp3, nsp5 and nsp8 proteins, forming SARS-CoV replicase, as well as SARS-CoV genomic RNA all localize inside the DMVs, suggesting that viral RNA synthesis likely occurs at these intracellular sites (Snijder et al., 2006, Stertz et al., 2007).
Notably, such extensive membrane re-arrangment, with DMV formation and co-localization with dsRNA was also observed in Vero cells infected with MERS-CoV, (de Wilde et al., 2013). Hence, despite the substantial amount of dsRNA byproducts generated during SARS-CoV replication cycle (Weber et al., 2006), it is likely that dsRNA shielding through its segregation into DMVs microenvironment is a Hide strategy through which CoVs effectively elude dsRNA detection (Knoops et al., 2008). However, such a passive mechanism is not exclusive, since the SARS-CoV N, a 46 kDa highly basic protein with a primary function of encapsidating the viral genome, has recently been found to target RLRs activation and signaling (Lu X. et al., 2011). The SARS-CoV N protein inhibits IFN-α/β production induced by both SeV and polyI:C (Kopecky-Bromberg et al., 2007, Lu et al., 2011) whereas, by contrast, it is not able to suppress type I IFN induction upon overexpression of components such as RIG-I-CARD, MAVS, TBK1 and IKK-ε, suggesting that the N protein blocks the initial step of dsRNA recognition by RLRs (Lu X. et al., 2011). In addition, co-IP experiments have shown that the SARS-CoV N protein does not interact with either RIG-I or MDA5, thereby excluding the possibility that such a block is exerted by directly impairing RLRs function (Lu X. et al., 2011). SARS-CoV N has been reported to be an RNA-binding protein (Chang et al., 2009, Tang et al., 2005) through both its N-terminal and C-terminal domains (Chang et al., 2009, Chen et al., 2007). Therefore, it is likely that SARS-CoV N binds dsRNA to prevent RIG-I and MDA5 activation and subsequent type I IFN induction (Lu X. et al., 2011).
8.2. Mask
A 3′–5′ exoribonuclease DEDDH domain was recently identified in the SARS-CoV nsp14 protein, demonstrating its ability to digest ssRNA as well as dsRNA with one 3′-mismatched nucleotide (Bouvet et al., 2012). Primarily placed in the context of viral RNA synthesis, this nsp14 activity has been related to nucleotide misincorporation repair (i.e. proofreading) putatively exerted by the SARS-CoV polymerase complex. However, it is possible that this function is also related to the IFN-response evasion. In fact, as observed for the LASFV NP, such 3′–5′ exoribonuclease activity may serve for nsp14 degradation of dsRNA intermediates, thereby leading to inhibition of IFN induction (Bouvet et al., 2012). However, albeit plausible, the formal involvement of nsp14 in a Mask strategy to prevent dsRNA detection requires further investigation.
8.3. Hit
In addition to hiding and masking dsRNA, SARS-CoV IFN antagonism relies on directly targeting the RLR pathway. Several SARS-CoV encoded proteins have been found to display such properties, but the mechanism of action remains to be elucidated for most of them. The SARS-CoV ORF3b and ORF6 proteins are involved in suppressing IFN induction at the level of RLR signaling with their downstream adaptors. In fact, preferential localization and redistribution at the mitochondrial outer membrane of both ORF3b and ORF6 has been reported, seemingly associated with inhibition of RIG-I and MDA5 recruitment of MAVS (Freundt et al., 2009, Kopecky-Bromberg et al., 2007). Another SARS-CoV-encoded IFN antagonist protein is the structural glycosylated M protein, essential for the assembly of viral particles. SARS-CoV M exerts a double Hit strategy that results in the failure of IRF-3 phosphorylation and activation. First, SARS-CoV M interacts with RIG-I and MAVS, sequestering and re-localizing them into discrete membrane compartments associated with the Golgi apparatus (Siu et al., 2009). Second, SARS-CoV M binds TRAF3 and, stably associating with it, abolishes the formation of a functional complex with the TBK1 and IKK-ε kinases (Siu et al., 2009). Similarly, the papain-like protease (PLP) domain contained within the SARS-CoV nsp3 protein, an essential component of the viral replicase complex, interacts with the adaptor STING, impeding its dimerization and activation and disabling the STING essential function of MAVS recruiter to the TBK1-IKK-ε complex for IRF-3 phosphorylation (Devaraj et al., 2007, Sun et al., 2012). Furthermore, a redundant inhibiting effect on RLR signaling is also due to the deubiquitinating activity of SARS-CoV PLP, that has been found to remove Ub from several RLR pathway components, such as RIG-I, STING, TBK1 and IRF-3 (Clementz et al., 2010, Frieman et al., 2009).
9. Filoviruses
Filoviruses are (−)-ssRNA viruses that belong to the family Filoviridae, which includes one species in the genus Cuevavirus, five species of Ebolaviruses (EBOVs) in the genus Ebolavirus and one species of Marburgvirus (MARV) in the genus Marburgvirus, and are among the most virulent known pathogens (Kuhn et al., 2010). EBOVs and MARV are causative agents of fulminant hemorrhagic fever in humans and nonhuman primates, with up to 90% case fatality rates (Hartman et al., 2010). Even though rare and mostly confined to Sub-Saharan Africa and South-East Asia, filoviruses actually pose a worldwide concern. In fact, due to the risk of travel-imported cases, their demonstrated aerosol infectivity and transmissibility between pigs as well as their potential misuse as biological weapons in a bioterrorist attack, they are considered one of the highest priorities for global health security (Bray, 2003, MacNeil and Rollin, 2012).
The extreme lethality of EBOVs and MARV is the result of uncontrolled viral replication associated with a total impairment of the innate immune system, related, at least in part, to the properties displayed by three determinants of virulence and pathogenicity, namely the VP24, VP35 and VP40 proteins. However, only the multifunctional polymerase cofactor VP35 acts by suppressing IFN-α/β production, while the EBOV VP24 and MARV VP40 matrix proteins are involved in inhibiting the type I IFN signaling pathway (Basler and Amarasinghe, 2009, Ramanan et al., 2011). Initially identified as the only filoviral protein able to complement the growth of a mutant IAV lacking its IFN-antagonist NS1 protein (see section 12) (Basler et al., 2000), filoviral VP35 was found to suppress production of endogenous IFN-β as well as transcriptional activation of IFN-β, ISG54 and ISG56 promoters-driven reporter genes, induced by either mutant IAV infection, SeV infection or polyI:C dsRNA transfection (Basler et al., 2000, Basler et al., 2003, Hartman et al., 2004). These properties were correlated with the inhibition of IRF-3 phosphorylation, dimerization and nuclear translocation (Cárdenas et al., 2006, Hartman et al., 2004, Hartman et al., 2006), as well as inhibition of IFN-α/β promoter upon overexpression of either RIG-I, RIG-I CARD, MAVS, TBK1 and IKK-ε (Cárdenas et al., 2006). Together, these data indicate that the filoviral VP35 protein exerts its IFN-antagonism by targeting the RLR pathway.
9.1. Hide
The innate immune escape capabilities exhibited by the filoviral VP35 all reside in its uniquely folded C-terminal RNA binding domain (RBD), also called the IFN-inhibitory domain (IID), comprising a central basic patch (CBP) that is highly conserved among filoviruses and shares high sequence identity with the IAV NS1 RBD (Hartman et al., 2004, Leung et al., 2009, Leung et al., 2010a). Indeed, although the RBD/IID is sufficient by itself to display an anti-IFN phenotype, a full-length VP35 capable of homo-oligomerization is required for fully efficient IFN inhibition (Leung et al., 2009, Reid et al., 2005). EBOV VP35 was found to be able to interact with dsRNA (Cárdenas et al., 2006), binding to both blunt-ended (Feng et al., 2007, Kimberlin et al., 2010, Zinzula et al., 2009) and 5′-ppp dsRNA molecules (Leung et al., 2010b, Zinzula et al., 2012) with very high affinity and in a sequence-independent manner. Notably, residues within the CBP critical for dsRNA binding such as R305, K309, R312 and K339 were found to be important for IFN inhibition, since their substitution with alanine decreased dsRNA binding to different extents (Cárdenas et al., 2006, Zinzula et al., 2012) and led to failure in suppressing both ISG56 and IFN-β promoter activity induced by various stimuli (Cárdenas et al., 2006, Hartman et al., 2004, Hartman et al., 2006). In particular, with respect to wt VP35, R312A VP35 showed the most dramatic defects, eliciting different ISGs expression profile in infected cells, and EBOV containing R312A VP35 mutation showed delayed viral growth and attenuation of infectivity in mice (Hartman et al., 2008a, Hartman et al., 2008b). A similar defective phenotype was also generated by the double mutation K319A/R322A, causing total loss of VP35′s dsRNA binding activity, abolishing IFN inhibition and rendering the corresponding mutant EBOV avirulent in guinea pigs (Prins et al., 2010). Notably, this double mutant was immunogenic and protected animals from subsequent challenge with wt EBOV (Prins et al., 2010), strongly reinforcing the notion of a correlation between VP35 dsRNA binding, inhibition of IFN-α/β production and the pathogenicity of filoviruses.
The molecular details of the VP35 interaction with dsRNA have been solved by four crystallographic structures: two EBOV VP35 RBD/IID from the Zaire and Reston viruses bound to 8 and 18 bp blunt-ended dsRNA molecules, respectively (Kimberlin et al., 2010, Leung et al., 2010b), and two crystallographic structures of the MARV VP35 RBD/IID bound to 12 and 18 bp blunt-ended dsRNA, respectively (Bale et al., 2012, Ramanan et al., 2012). As observed in these crystals, EBOV VP35s form an asymmetric dimer at each of the dsRNA ends (Fig. 7 A). One monomer, termed the backbone binding RBD/IID, binds to the sugar-phosphate backbone of both dsRNA strands, while the second monomer, referred to as end-capping, binds to dsRNA terminal bases and the proximal phosphate backbone. As a result of this bimodal strategy, VP35 RBD/IID dimers mimic the RLRs shape and hide their recognition site at dsRNA ends, possibly accommodating the 5′-ppp hindrance into a positively-charged pocket (Leung et al., 2010b, Kimberlin et al., 2010). In contrast, MARV VP35 RBDs/IIDs have been reported to helically coat dsRNA along its entire phosphate backbone, with a footprint of four monomers every 4–5 bp (Fig. 7B) (Bale et al., 2012, Ramanan et al., 2012). Notably, even though in this case no VP35 RBDs/IIDs capping of the nucleic acid ends was observed, isothermal titration calorimetry and dot blot assays revealed that a binding event with a VP35:dsRNA stoichiometry of 1:1 almost disappears in the presence of dsRNA overhangs, thereby suggesting that an end-capping-like binding dsRNA blunt-ends modality may also take place for MARV VP35 (Bale et al., 2012, Ramanan et al., 2012).
Fig. 7.
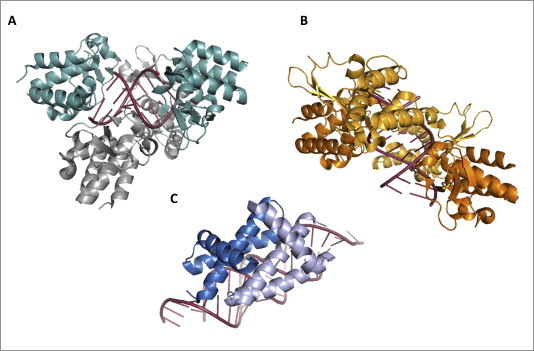
Structures of viral proteins displaying the Hide strategy. (A) EBOV VP35 hides dsRNA: EBOV suppresses IFN-α/β production through the properties of its VP35 protein, which sequesters dsRNA from RLRs detection. VP35 RBD shields RLRs recognition sites on a 8 bp dsRNA binding to its phosphate backbone (backbone-binding monomers, highlighted in gray, PyMOL colors) and to its terminal bases and phosphate groups (end-capping monomers, highlighted in light teal, PyMOL colors). Crystallographic structure by Leung et al., 2010a, Leung et al., 2010b (PDB: 3L25); (B) MARV VP35 hides dsRNA: MARV VP35 RBD monomers fully coat a 12 bp dsRNA molecule along to its phosphate backbone. Crystallographic structure by Bale et al., 2012 (PDB: 4GHA); (C) IAV NS1 hides dsRNA: IAVs suppress type I IFN-based innate immune response through the properties of the multifunctional NS1 protein. The NS1 RBD dimer (highlighted in sky blue and light blue, PyMOL colors) coats dsRNA along to its phosphate backbone, thereby preventing its recognition by RLRs and PKR. Crystallographic structure by Cheng et al., 2009 (PDB: 2ZKO).
More recently, a similar behavior was also described for the EBOV VP35 RBD/IID, and a model was proposed in which the end-capping monomer constitute the first binding event to which attachment of babckbone binding monomers follows (Bale et al., 2013). As revealed by biochemical studies, EBOV VP35 RBDs/IIDs bind with similar affinities regardless of dsRNA length (Leung et al., 2010b, Kimberlin et al., 2010, Ramanan et al., 2012, Zinzula et al., 2012), while MARV RBD/IID binding affinity decreases as dsRNA shortens (Bale et al., 2012, Ramanan et al., 2012). In addition, dsRNA overhangs presence is barely tolerated by EBOV VP35, for which a 5′-ppp is otherwise important for binding to dsRNA with high affinity (Kimberlin et al., 2010, Leung et al., 2010b, Zinzula et al., 2012), while they do not affect MARV VP35 binding function (Bale et al., 2012, Ramanan et al., 2012). Such different abilities to recognize diverse dsRNA PAMPs reflect the diverse properties of the two filoviral VP35 in antagonizing RLRs. In fact, while both EBOV and MARV VP35 were able to inhibit the dsRNA-induced ATPase activity of MDA5, only EBOV VP35 exerted the same effect on RIG-I for all tested dsRNA ligands (Leung et al., 2010b, Ramanan et al., 2012).
Overall, the architecture of the VP35-dsRNA complex in the four crystallographic structures accounts for the importance of several residues in dsRNA binding. Side chains of residues R305/294, K309/298, R312/301, K339/K328 in the Zaire/Reston EBOV VP35 CBP (Kimberlin et al., 2010, Leung et al., 2010b), as well as in the corresponding residues N261, Q263, R294, K298, S299, R301 and K328 in the MARV VP35 CBP, are all involved in direct interactions with the dsRNA phosphate backbone (Bale et al., 2012, Ramanan et al., 2012). In addition, residues R312/301, K319/308, R322/311 and K339/328 in the Zaire/Reston EBOV VP35 CBP are also involved in protein–protein interactions at the RBD/IID dimer interface, while residues I340/329 and F239/228, in the end-capping monomer, interact with the adjacent K339/328, Q274/263 and I278/267 residues, which in turn bind dsRNA terminal bases (Kimberlin et al., 2010, Leung et al., 2010b). Most importantly, the proposed model for filoviral VP35 homo-oligomers shielding dsRNA PAMP signatures has been validated by mutagenesis studies, since alanine substitutions of the same residues resulted in a decrease or loss of dsRNA binding function, as well as in diminished or abolished inhibition of SeV-induced IFN-β promoter activation and IRF-3 phosphorylation (Bale et al., 2012, Kimberlin et al., 2010, Leung et al., 2010b, Ramanan et al., 2012). Hence, given the high degree of conservation of these amino acid residues among all known EBOV and MARV species, it is plausible that dsRNA sequestration by VP35 represents a key strategy through which these pathogens circumvent host innate immunity.
9.2. Hit
In line with the other viral IFN-antagonist redundancy observed to concomitantly target multiple levels of the RLRs pathway, filoviral VP35 also employs several Hit strategies to suppress IFN-α/β induction. In fact, both EBOV and MARV VP35 were shown to block IRF-3 phosphorylation and nuclear translocation induced by TBK1 and IKK-ε overexpression, but not to inhibit IFN-β promoter activation induced by a constitutively active form of IRF-3 (Prins et al., 2009, Ramanan et al., 2012). Consistent with a target upstream in the RLRs pathway, co-IP studies showed that EBOV VP35 interacts with the N-terminal domain of TBK1 and IKK-ε, it decreases their catalytic activity and is phosphorylated by these kinases (Prins et al., 2009). In addition, overexpression of VP35 resulted in disruption of IKK-ε interactions with IRF-3, IRF-7 and MAVS (Prins et al., 2009). Therefore, filoviral VP35 targets the RLRs pathway also in a dsRNA binding-independent way by acting as alternative substrate for the TBK1-IKK-ε complex in place of IRF-3 and 7. Furthermore, these two IRFs are also directly targeted by VP35 that prevents their migration to the nucleus (Chang T.H. et al., 2009). In fact, EBOV VP35 was found to physically interact with both IRF-3 and 7 and to promote their Ub-like modification by the two members of the small Ub-like modifier cascade PIAS1 and Ubc9, thereby inhibiting the IRFs transcriptional function and subsequently suppressing the activation of IFN-β promoter (Chang T.H. et al., 2009).
Finally, a novel mechanism through which VP35 suppresses the RLRs pathway was recently described, in which the EBOV IFN-antagonist interacts with cellular PKR activator (PACT), a dsRNA-binding protein involved in the activation of RIG-I and stimulation of its ATPase activity (Fabozzi et al., 2011; Luthra et al., 2013). The formation of a complex between VP35 and PACT abolishes critical interaction of the latter with the CTD of RIG-I, thereby preventing dsRNA-induced immune signaling (Luthra et al., 2013).
10. Flaviviruses
The genus Flavivirus of the family Flaviviridae includes two mosquito-borne (+)-ssRNA viruses, dengue virus (DENV) and West Nile virus (WNV), which are emerging as global life-threatening pathogens. WNV infects millions of people, with thousands of deaths annually, while DENV has spread worldwide, with a recent increase in morbidity and outbreak frequency (Gould and Solomon, 2008, Heinz and Stiasny, 2012). DENV and WNV human infections are often asymptomatic or characterized by mild and self-limiting fever. In some cases, however, the infection develops into severe and fatal illness, characterized by hemorrhagic fever and shock syndrome for DENV, and by neurological diseases such as meningitis, encephalitis or acute flaccid paralysis in the case of WNV (Gould and Solomon, 2008, Heinz and Stiasny, 2012).
The disease severity caused by these pathogenic flaviviruses has been correlated with their ability to counteract the IFN-α/β response (Keller et al., 2006), and both DENV and WNV have been found to encode several IFN-antagonists acting on different targets (Muñoz-Jordán and Fredericksen, 2010). However, most studies have focused on characterizing DENV and WNV suppression of the TLRs-mediated IFN induction, the IFN signaling pathway and the ISGs-mediated antiviral response, whereas less is known about viral proteins and mechanisms that interfere with the RLR pathway (Morrison et al., 2012, Suthar et al., 2013). Notably, RIG-I, MDA5 and LGP2 are strongly up-regulated in several DENV- and WNV-infected cell lines (da Conceição et al., 2013, Fredericksen et al., 2008, Surasombatpattana et al., 2011, Qin et al., 2011), while viral replication is enhanced in the absence of RLR expression (Nasirudeen et al., 2011, Suthar et al., 2010). Inhibition of IRF-3 phosphorylation has been reported during productive infection by both DENV and WNV, indicating that subversion of the RLR pathway is important for their replication (Chang et al., 2006, Fredericksen and Gale, 2006, Keller et al., 2006).
10.1. Hide
DENV is able to efficiently downregulate RLR signaling (Loo et al., 2008), even though its genomic RNA can be recognized by both RIG-I and MDA5 (Rodriguez-Madoz et al., 2010a). Similarly, WNV is able to elude RIG-I recognition early post-infection (Fredericksen et al., 2004), although its RNA subgenomic fragments fold into secondary structures that have recently been shown to potently activate RIG-I (Shipley et al., 2012). Together, these data make it plausible that flavivirus counteraction of the type I IFN-response may include mechanisms that avoid dsRNA detection. Supporting this hypothesis, two studies based on 3D electron tomography have shown that DENV and WNV modify ER and Golgi apparatus morphology, inducing the formation of convoluted membranes (CMs) in the cytoplasm. Tightly associated with the viral NS4A protein, CMs invaginations wrap around other NS proteins that form the viral replication machinery which, as revealed by immuno-labeling, co-localizes into the CMs vesicles packed with dsRNA (Gillespie et al., 2010, Welsch et al., 2009). By providing a hidden compartment where RNA synthesis and virus budding take place, such membrane modifications represent a passive strategy by which DENV and WNV hide dsRNA from RLRs.
10.2. Hit
The RLRs pathway is also actively targeted by these two flaviviruses. In DENV infection, inhibition of SeV- or polyI:C-mediated transcriptional activity of the IFN-β promoter was found to be dose-dependently related to the catalytic activity of the NS2B3 protease (Rodriguez-Madoz et al., 2010b). As recently demonstrated, this correlation is due to the cleavage of the STING protein (Aguirre et al., 2012, Yu et al., 2012) by a proteolytic core, within the last 40 amino acids of NS2 and the first 180 residues of NS3, which targets the consensus sequence LRRQ96G of STING, it eliminates its ability to interact with TBK1 and activates this kinase for IRF-3 phosphorylation (Aguirre et al., 2012, Yu et al., 2012). In WNV infection, a possible Hit strategy to block RLRs signaling has been identified based on the viral protein NS2. By using a WNV subgenomic replicon, severe inhibition of IFN-β promoter-driven transcription was observed, and this phenotype was abolished by the occurrence of an adaptive alanine-to-proline substitution at position 30 of the NS2 protein (Liu et al., 2004). With respect to wt NS2 WNV, A30P NS2 mutant WNV led to faster production of higher IFN-α/β levels in infected cells and determined an attenuation of viral growth and neuro-invasiveness in mouse models (Liu et al., 2006). Furthermore, immunization of mice with this mutant WNV conferred protection against subsequent challenge with a lethal dose of a highly virulent WNV strain (Liu et al., 2006). These data reflect the importance of WNV NS2 as a determinant of virulence and pathogenesis, even though the exact mechanism by which this protein modulates IFN-α/β expression remains to be determined.
11. Henipaviruses
Within the viral family Paramyxoviridae, (+)-ssRNA viruses Hendra virus (HeV) and Nipah virus (NiV) of the genus Henipavirus have recently emerged as zoonotic bat-borne pathogens characterized by extreme lethality for humans and a broad mammalian host range (Eaton et al., 2006). Endemic in Australia and South-East Asia, respectively, HeV and NiV typically affect livestock but also cause severe neurologic and respiratory disease in humans that often progresses to encephalitis and multiorgan failure (Marsh and Wang, 2012).
As for the viruses discussed above, fatal outcome following henipavirus infection might be correlated with the ability of these pathogens to potently subvert the host innate immune system (Basler, 2012). In agreement with this concept, daily treatment with IFN-inducers prevented death in NiV-infected animals (Georges-Courbot et al., 2006). Henipaviruses counteract the type I IFN response by blocking the IFN-α/β production at multiple levels (Basler, 2012). This is the effect of the properties displayed by the four proteins encoded by the henipaviral P gene, which include the homonymous phosphoprotein P, together with viral products V, W and C (Shaw, 2009). The first three proteins are expressed by the same open reading frame (ORF), share a common N-terminal region and have a unique C-terminus. In fact, while the viral polymerase cofactor P is produced by the exact transcription of the entire gene, both V and W proteins are generated by an editing process based on the insertion of non-templated G nucleotide residues into the viral mRNA. By contrast, the C protein is expressed by an alternative ORF within the P gene, and therefore has a completely different sequence (Kulkarni et al., 2009, Lo et al., 2009). All proteins encoded by the henipaviral P gene act as determinants of virulence, inhibiting the IFN-α/β production by targeting both the TLR and RLR pathways, as well as suppressing type I IFN signaling (Basler, 2012). With regard to the counteraction of RLR pathway, the discussion is limited only to functions displayed by the V and W proteins.
11.1. Hit
The HeV and NiV V and W proteins are able to inhibit the SeV- and dsRNA-induced transcriptional activation of the ISG54 promoter by IRF-3 (Basler, 2012, Shaw, 2009). The V protein was shown to bind MDA5 (Andrejeva et al., 2004, Childs et al., 2007) and LGP2 (Parisien et al., 2009) by targeting homologous regions in the two helicases that, encompass residues 676–816 of MDA5 and residues 327–465 of LGP2, corresponding to the boundaries of their Hel2 domain (Childs et al., 2009, Parisien et al., 2009). As a consequence of this interaction with V, MDA5 dsRNA binding and homo-oligomerization were abolished (Childs et al., 2009) and ATP-hydrolysis activity of both helicases were disrupted (Parisien et al., 2009). Moreover, even though henipaviral V proteins do not directly bind RIG-I (Childs et al., 2009), their interaction with LGP2 induced the formation of a stable LGP2-RIG-I complex, rendering the latter unable to recognize 5′-ppp dsRNA ligands (Childs et al., 2012).
The V protein domain responsible for such IFN-inhibiting activity was mapped to a region comprising 49–68 amino acids at its C-terminus, with one histidine and seven cysteine residues that are highly conserved among all paramyxoviruses and fold into a zinc-finger domain to coordinate two zinc atoms (Ramachandran and Horvath, 2010). As revealed by mutagenesis studies, four residues in the zinc-coordinating domain, namely C194, C206, C208 and C211, as well as the two nearby conserved R409 and I414, are essential for the V protein’s ability to suppress MDA5 functionality (Ramachandran and Horvath, 2010). Conversely, the single amino acids R806 of MDA5, R455 of LGP2 and L714 of RIG-I are sufficient for the interaction with the viral protein, as their point mutation renders these helicases unable to co-immunoprecipitate with overexpressed V in luciferase-based cell culture assays (Rodriguez and Horvath 2013). The MDA5 crystallographic structure in complex with the V protein of the henipavirus closely-related parainfluenza virus 5 (PIV5) was recently obtained (Fig. 6C) (Motz et al., 2013). As revealed by this structure, the zinc-finger β-hairpin located at the C-terminal domain of V is folded in a way that mimics the two β-strands in the MDA5 Hel2 domain (Motz et al., 2013). As a result, upon interaction with MDA5, V displaces the β-sheet motif of the Hel2 domain and accommodates its zinc-finger β-hairpin into the MDA5 fold, thereby causing the inhibition of MDA5 ATP-hydrolysis activity, dsRNA binding ability and oligomerization properties, which ultimately inhibits the MDA5-induced production of IFN-α/β (Motz et al., 2013, Wu and Hur, 2013).
The henipaviral W protein also downregulates type I IFN production, but the molecular mechanisms by which it targets the RLR pathway are less clear. In fact, the W protein was shown to potently block transcription from promoters activated by IRF-3, such as the IFN-α/β and the ISG54 promoters (Shaw et al., 2005). Moreover, since transcription starting at these IRF-3-responsive promoters was inhibited by henipaviral W also with concomitant overexpression of both TBK1 and IKK-ε kinases, it is conceivable that the target hit by this viral protein is placed downstream along the RLRs pathway (Shaw et al., 2005). Notably, due to a frameshift-extended ORF, W is about 43 amino acids longer than V, it lacks the domain responsible for interaction with RLRs and has a unique C-terminal basic stretch that functions as a nuclear localization signals (NLS) (Lo et al., 2009). Therefore, given that W is almost exclusively nuclear and that it does not block IRF-3 phosphorylation, it is likely that this protein directly hits the transcriptional factor once in the nucleus, possibly rendering IRF-3 dimers less stable in order to prevent IFN-α/β promoter activation (Lo et al., 2009, Shaw et al., 2005). This mechanism of action would explain the ability of henipavirus W to impair both RLRs and TLR3 pathways, consistent with the hypothesis that it acts at a point in the pathways where the two signaling cascades converge (Lo et al., 2009, Shaw et al., 2005).
12. Influenza A viruses
Influenzaviruses are segmented (−)-ssRNA-viruses that belong to the three genera Influenzavirus A, B and C within the family Orthomyxoviridae. They are the most important respiratory pathogens affecting the human population. IAVs are maintained in several vertebrate host species and are subtyped according to the antigenic properties of their 16 different hemagglutinin and 9 different neuraminidase glycoproteins (Medina and Garcia-Sastre, 2011). Subtypes that circulate in humans undergo an antigenic shift due to accumulation of mutations at these two proteins, which results in overcoming pre-existing immunity to cause seasonal flu epidemics worldwide. Occasionally, profound antigenic change may derive from interspecies transmission or gene re-assortment during concomitant infection of the same host by different IAV subtypes. If such antigenic drift occurs, viruses that are new to human population can emerge and generate devastating pandemics with millions of deaths (Neumann et al., 2009, Neumann et al., 2010).
Human infections by highly pathogenic IAVs typically may result in acute respiratory distress syndrome with fatal pneumonia, due, at least in part, to the ability of IAVs to efficiently suppress the host innate immune response (Ramos and Fernandez-Sesma, 2012). Regarding interference with the dsRNA-induced production of IFN-α/β, IAVs exert their antagonism through the properties of at least three proteins, namely NS1, PB2 and PB1-F2, which target the RLR pathway at several levels and with different mechanisms (van de Sandt et al., 2012).
12.1. Hide
The notion that the multifunctional NS1 protein of IAVs is an IFN-antagonist derived from the finding that viruses deleted of its encoding gene (delNS1) showed attenuated growth and induced large amounts of IFN-α/β in IFN-competent cells, but replicated well in IFN-deficient cells (Egorov et al., 1998, García-Sastre et al., 1998). Subsequent studies showed that this phenotype was the result of a counteraction towards the RLR pathway, as abrogated virus- and dsRNA-induced activation of IRF-3, NF-kB and the ATF-2/c-Jun enhanceosome (Ludwig et al., 2002, Talon et al., 2000, Wang et al., 2000). Such inhibition was correlated with the ability of NS1 to bind dsRNA, since a binding-defective double mutant R38A/K41A NS1 led to virus attenuation in vivo and to IFN-α/β induction in cell cultures (Donelan et al., 2003, Talon et al., 2000). Notably, a third substitution at position 42 in the revertant NS1 mutant R38/K41A/S42G was reported to gain back IFN suppression capability but did not restore dsRNA binding, suggesting that, even though dsRNA binding is necessary and sufficient for efficient inhibition of IFN synthesis, it is not the sole function that mediate IFN antagonism by the IAV NS1 (Donelan et al., 2003). The impact of alternative mechanisms through which NS1 counteracts the host innate immune response (see below) was also highlighted by comparing NS1 proteins from different IAVs, which showed strain specificity in suppressing type I IFN and ISGs expression (Kochs et al., 2007). On the other hand, the importance of NS1 dsRNA-binding function for IFN antagonism was supported by the fact that the ability of RIG-I to detect IAV-derived RNAs was inhibited in the presence of NS1, but it was efficiently activated by the same PAMPs in cells infected with a virus lacking NS1 (Baum et al., 2010, Hornung et al., 2006, Pichlmair et al., 2006).
NS1 binds to blunt-ended and 5′-ppp dsRNA as well as ssRNA panhandle structures through an N-terminal RBD that encompasses residues 1–73 (Chien et al., 1997, Hatada and Fukuda, 1992, Qian et al., 1995). Moreover, NS1 proteins deleted in their C-terminal effector domains were still active in blocking activation of IFN-β promoter (Guo et al., 2007, Hayman et al., 2006). Functional proteins were obtained even when the sole NS1 N-terminal dsRNA binding domain was fused to a heterologous C-terminal domain (Wang et al., 2002), reinforcing the importance of dsRNA sequestration by preventing RLR recognition and subsequent IFN-α/β production.
Structural evidence indicates that NS1 exists as a homo-dimer and interacts with the phosphate backbone of dsRNA, so that the two RBD monomers lie on nucleic acid strands like parallel tracks on a slot (Chien et al., 2004, Liu et al., 1997, Wang et al., 1999). Along these tracks, several conserved basic and hydrophobic residues such as T5, P31, D34, R35, R38, K41, G45, R46 and T49 have been identified as critical for NS1 dsRNA-binding ability (Wang et al., 1999, Yin et al., 2007). In fact, as revealed by the crystal structures of the NS1 RBD in complex with a 21-bp dsRNA (Fig. 7C), these residues form a concave surface that recognizes the minor groove of the dsRNA A-form (Cheng et al., 2009). In particular, residues R38, S42 and T49 of each RBD, as well as arginines at positions 35 and 46 from different monomers, are involved in establishing key hydrogen bonds with the phosphate group of both dsRNA strands (Cheng et al., 2009). Furthermore, an even more evocative description of how IAVs prevent the detection of dsRNA by RLRs has come from the solved structure of a full-length NS1 of the highly pathogenic H5N1 IAV in complex with dsRNA (Bornholdt and Prasad, 2008). As shown by a combined X-ray crystallography and cryo-EM approach, NS1 dimers multimerize through alternating interactions between the RBD and the C-terminal effector domain, forming a tubular structure with a central tunnel suitable to accommodate dsRNA (Bornholdt and Prasad, 2008). Again, critical orientation is observed for key residues such as R38, which is projected to the tunnel center in order to establish direct bonds with dsRNA, while other residues like K41 contribute to enwrap the phosphate backbone all along the entire nucleic acid length (Bornholdt and Prasad, 2008).
12.2. Hit
NS1 also exerts its IFN-inhibiting properties against the RLR pathway through Hit strategies, since it was found to be able to block both RIG-I and MAVS functions by physically interacting with these components (Guo et al., 2007, Mibayashi et al., 2007, Opitz et al., 2007). Noteworthy, the presence of 5′-ppp dsRNA enhanced co-precipitation with RIG-I, and such interaction was abolished in the dsRNA binding-defective R38A/K41A mutant, suggesting either the possibility of overlapping functions in the NS1 RBD or a role of dsRNA as intermediate in the formation of the NS1-RIG-I complex (Pichlmair et al., 2006, Talon et al., 2000).
RLR activation and signaling are also blocked by NS1 through the suppression of RIG-I K63-linked ubiquitination by TRIM25α (Gack et al., 2009). Specifically, NS1 interacts with the coiled-coil domain of TRIM25α and disrupts its homo-oligomerization, which is in turn indispensable for poly-ubiquitination of RIG-I (Gack et al., 2009). Moreover, the alanine substitution of E96 and E97 residues as well as the R38A/K41A mutation responsible for loss of dsRNA binding impaired NS1 interaction with TRIM25α and caused attenuation of viral growth in vivo and increased IFN-α/β production (Gack et al., 2009). Also, the TRIM25α-orthologue Riplet was recently found to be a target, in human cells, of NS1 from human IAV strains. However, while the E96A/E97A mutant was in this case still capable of such interaction, the dsRNA binding-defective R38A/K41A NS1 could not exert it (Rajsbaum et al., 2012). Finally, the NS1 proteins of most (but not all) IAVs are able to alter innate immune responses through a global restriction of host gene expression. To achieve this, NS1 binds the 30 kDa subunit of cleavage and polyadenylation-specificity factor (CPSF30) (Nemeroff et al., 1998, Noah et al., 2003, Das et al., 2008), preventing polyadenylation of the 3′ ends of host pre-mRNA, and interacts with the poly-(A)-binding protein II (PABPII), resulting in inhibition of host pre-mRNA nuclear export (Chen et al., 1999).
In addition to NS1, the other IAV proteins PB1, PB2, PA and PB1-F2 all display IFN-antagonism against the RLR pathway by targeting the mitochondrial adaptor MAVS through a Hit strategy. The three RNA polymerase subunits PB1, PB2 and PA were all found to inhibit dsRNA-induced and RIG-I- or MDA5-mediated activation of IFN-β promoter in a gene reporter assay, and such inhibition was related to their binding to MAVS (Iwai et al., 2010). In PB2, whose inhibitory effect and binding ability were stronger, amino acids 1–37 were found to be critical for the interaction with the N-terminal 150 residues of MAVS, while an asparagine at position 9 in PB2 sequence was found to be essential for its mitochondrial localization and inhibition of the RLR pathway signaling (Graef et al., 2010, Patel et al., 2013). Regarding PB1-F2, this small protein encoded by a +1 ORF in the PB1 gene is found only in some IAVs and its presence is related to the high virulence in a strain-dependent manner (Schmolke et al., 2011, Varga and Palese, 2011). PB1-F2 strongly suppresses RIG-I-mediated IFN-α/β induction, whereas infection with delPB1-F2 viruses results in increased IRF-3 activation and production of IFN-β mRNA (Dudek et al., 2011). Initially, such activity was found to be mediated by a region spanning residues 39–87 of the PB1-F2 C-terminus, where a highly conserved sequence served as a mitochondrial targeting signal (MTS) (Gibbs et al., 2003, Varga et al., 2011, Yamada et al., 2004). More recently, the presence of a serine in place of the asparagine at position 66 within the PB1-F2 MTS was found to be related to a severe pathogenicity and a strong reduction of ISGs expression, and to be responsible for the increased virulence of the avian H5N1 and the 1918 Spanish flu IAV strains (Conenello et al., 2007, Conenello et al., 2011). In line with these data, the mechanism of action through which PB1-F2 shuts down type I IFN production has been elucidated by recent work demonstrating that PB1-F2 physically interacts with MAVS, and that the N66S mutation enhances this interaction (Varga et al., 2012). PB1-F2 also targets the transmembrane region of MAVS, dissipating the mitochondrion membrane potential that is an important factor for the recruitment of downstream components and the prosecution of the RLRs signaling cascade (Varga et al., 2012).
13. Concluding remarks and future challenges
Based on the production of type I IFN in response to viral infections, the intrinsic innate immune system is an integral part of the broader immune system of vertebrates, and both cell-mediated and humoral adaptive immune responses depend on its proper function. Prompt detection of the pathogen, which relies on the recognition of its molecular components, is a prerequisite for IFN production, and dsRNA is the hallmark of virus presence, above all PAMPs. In the cytoplasm, dsRNA recognition is assigned to the RLRs. Viruses, in turn, circumvent this detection through an array of mechanisms aimed to suppress the RLR pathway. Within this array, we have described three main viral strategies, which are respectively directed to hide dsRNA from RLRs, to mask its presence by modifying its molecular features and to directly hit RLRs and their adaptors and/or effectors to impair their functions. These strategies, even though put in place through different molecular mechanisms, represent one aspect of the overall process by which adaptation between any given virus and the organism it infects was established. However, while the result of this coevolution and interplay is a fine modulation of the maintenance host’s innate immune system, to circumvent its effects, spillover of the virus to a new host species may lead to new fortuitous interactions with diverse intracellular machineries, often causing the dramatic effects of an overwhelming infection.
In line with this consideration, for all of the highly pathogenic RNA viruses here examined, the proteins involved in countering host dsRNA-induced IFN production were also found to act as determinants of virulence and pathogenesis. Moreover, in several cases, point mutations that impair their anti-IFN functions cause a loss of virus lethality. Together with the fact that the functional domains responsible for IFN antagonism are highly conserved within each viral group, this indicates that these viral proteins are attractive and promising targets for antiviral development. Additionally, given the importance of the RLRs pathway, it is also possible to envision the strategy of developing compounds that, mimicking the triggering power of dsRNA, may be able to boost the intrinsic innate immune system to obtain a preventive/aiding activation against viral infections. To this aim, tremendous advances in our knowledge are provided by the crystallographic structures of several viral IFN-antagonists as well as by the elucidation of their mechanism of action.
Finally, it is worth noting that many questions still remain unanswered regarding the different effects of the described viral proteins on the RLR pathway in human cells, on the one side, and in the natural reservoirs of these lethal pathogens, on the other, since these animals are infected asymptomatically and do not develop disease. In fact, assuming that viral proteins have evolved to finely tune the host innate antiviral response, rather than to completely suppress it, the understanding of the subtle differences between the innate immune antiviral response in humans and in these viruses’ animal reservoirs will also be of paramount importance for the development of effective viral therapeutics, making it possible to shift the balance from a fatal outcome to the clearance of infection.
Contributor Information
Luca Zinzula, Email: lucazinzula@unica.it.
Enzo Tramontano, Email: tramon@unica.it.
References
- Aguirre S., Maestre A.M., Pagni S., Patel J.R., Savage T., Gutman D., Maringer K., Bernal-Rubio D., Shabman R.S., Simon V., Rodriguez-Madoz J.R., Mulder L.C.F., Barber G.N., Fernandez-Sesma A. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012;8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 2006;4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Alff P.J., Gavrilovskaya I.N., Gorbunova E., Endriss K., Chong Y., Geimonen E., Sen N., Reich N.C., Mackow E.R. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 2006;80:9676–9686. doi: 10.1128/JVI.00508-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alff P.J., Sen N., Gorbunova E., Gavrilovskaya I.N., Mackow E.R. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 2008;82:9115–9122. doi: 10.1128/JVI.00290-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson I., Karlberg H., Mousavi-Jazi M., Martinez-Sobrido L., Weber F., Mirazimi A. Crimean-Congo hemorrhagic fever virus delays activation of the innate immune response. J. Med. Virol. 2008;80:1397–1404. doi: 10.1002/jmv.21222. [DOI] [PubMed] [Google Scholar]
- Andrejeva J., Childs K.S., Young D.F., Carlos T.S., Stock N., Goodbourn S., Randall R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S., Julien J.-P., Bornholdt Z.A., Kimberlin C.R., Halfmann P., Zandonatti M.A., Kunert J., Kroon G.J.A., Kawaoka Y., Macrae I.J., Wilson I.A., Saphire E.O. Marburg virus VP35 can both fully coat the backbone and cap the ends of dsRNA for interferon antagonism. PLoS Pathog. 2012;8:e1002916. doi: 10.1371/journal.ppat.1002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S., Julien J.P., Bornholdt Z.A., Krois A.S., Wilson I.A., Saphire E.O. Ebolavirus VP35 coats the backbone of double-stranded RNA for interferon antagonism. J. Virol. 2013;87:10385–10388. doi: 10.1128/JVI.01452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamming D., Horvath C.M. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J. Biol. Chem. 2009;284:9700–9712. doi: 10.1074/jbc.M807365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R., Ewald S.E., Mouchess M.L., Barton G.M. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- Basler C.F., Wang X., Mühlberger E., Volchkov V., Paragas J., Klenk H.D., Garcia-Sastre A., Palese P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA. 2000;97:12289–12294. doi: 10.1073/pnas.220398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler C.F. Nipah and hendra virus interactions with the innate immune system. Curr. Top. Microbiol. Immunol. 2012;359:123–152. doi: 10.1007/82_2012_209. [DOI] [PubMed] [Google Scholar]
- Basler C.F., Amarasinghe G.K. Evasion of interferon responses by ebola and marburg viruses. J. Interferon Cytokine Res. 2009;29:511. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler C., Mikulasova A., Martinez-Sobrido L., Paragas J., Muhlberger E., Bray M., Klenk H.-D., Palese P., Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003;77:7945. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Sachidanandam R., Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. USA. 2010;107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui S.M., Paz S., Samuel S., Goulet M.L., Sun Q., Kikkert M., Iwai K., Dikic I., Hiscott J., Lin R. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS–TRAF3 complex. Cell Host Microbe. 2012;12:211–222. doi: 10.1016/j.chom.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Berke I.C., Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31:1714–1726. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke I.C., Yu X., Modis Y., Egelman E.H. MDA5 assembles into a polar helical filament on dsRNA. Proc. Natl. Acad. Sci. USA. 2012;109:18437–18441. doi: 10.1073/pnas.1212186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billecocq A., Spiegel M., Vialat P., Kohl A., Weber F., Bouloy M., Haller O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004;78:9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder M., Eberle F., Seitz S., Mücke N., Hüber C.M., Kiani N., Kaderali L., Lohmann V., Dalpke A., Bartenschlager R. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I) J. Biol. Chem. 2011;266:27278–27287. doi: 10.1074/jbc.M111.256974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt Z.A., Prasad B.V.V. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature. 2008;456:985–988. doi: 10.1038/nature07444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloy M., Janzen C., Vialat P., Khun H., Pavlovic J., Huerre M., Haller O. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J. Virol. 2001;75:1371–1377. doi: 10.1128/JVI.75.3.1371-1377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. USA. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M. Defense against filoviruses used as biological weapons. Antiviral Res. 2003;57:53–60. doi: 10.1016/s0166-3542(02)00200-0. [DOI] [PubMed] [Google Scholar]
- Bray M. Pathogenesis of viral hemorrhagic fever. Curr. Opin. Immunol. 2005;17:399–403. doi: 10.1016/j.coi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Briese T., Paweska J.T., McMullan L.K., Hutchison S.K., Street C., Palacios G., Khristova M.L., Weyer J., Swanepoel R., Egholm M., Nichol S.T., Lipkin W.I. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns A.M., Pollpeter D., Hadizadeh N., Myong S., Marko J.F., Horvath C.M. ATP hydrolysis enhances RNA recognition and antiviral signal transduction by the innate immune sensor, laboratory of genetics and physiology 2 (LGP2) J. Biol. Chem. 2013;288:938–946. doi: 10.1074/jbc.M112.424416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capodagli G.C., McKercher M.A., Baker E.A., Masters E.M., Brunzelle J.S., Pegan S.D. Structural analysis of a viral ovarian tumor domain protease from the Crimean-Congo hemorrhagic fever virus in complex with covalently bonded ubiquitin. J. Virol. 2011;85:3621–3630. doi: 10.1128/JVI.02496-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas W.B., Loo Y.M., Gale M., Jr., Hartman A.L., Kimberlin C.R., Martinez-Sobrido L., Saphire E.O., Basler C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnec X., Baize S., Reynard S., Diancourt L., Caro V., Tordo N., Bouloy M. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 2011;85:12093–12097. doi: 10.1128/JVI.00429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan R.W., Chan M.C., Agnihothram S., Chan L.L., Kuok D.I., Fong J.H., Guan Y., Poon L.L., Baric R.S., Nicholls J.M., Peiris J.S. Tropism of and innate immune responses to the novel human betacoronavirus lineage C virus in human ex vivo repsiraroy organ cultures. J. Virol. 2013;87:6604–6614. doi: 10.1128/JVI.00009-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K.-Y., Ramos A. The double-stranded RNA-binding motif, a versatile macromolecular docking platform. FEBS J. 2005;272:2109–2117. doi: 10.1111/j.1742-4658.2005.04652.x. [DOI] [PubMed] [Google Scholar]
- Chang T.-H., Liao C.-L., Lin Y.-L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 2006;8:157–171. doi: 10.1016/j.micinf.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Chang C.-K., Hsu Y.-L., Chang Y.-H., Chao F.-A., Wu M.-C., Huang Y.-S., Hu C.-K., Huang T.-H. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J. Virol. 2009;83:2255–2264. doi: 10.1128/JVI.02001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T.-H., Kubota T., Matsuoka M., Jones S., Bradfute S.B., Bray M., Ozato K. Ebola zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrel R.N., Coutard B., Baronti C., Canard B. Arenaviruses and hantaviruses: from epidemiology and genomics to antivirals. Antivir. Res. 2011;90:102–114. doi: 10.1016/j.antiviral.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Chen Z., Li Y., Krug R.M. Influenza A NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999;18:2273–2283. doi: 10.1093/emboj/18.8.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-Y., Chang C.-K., Chang Y.-W., Sue S.-C., Bai H.-I., Riang L., Hsiao C.-D., Huang T.-H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007;368:1075–1086. doi: 10.1016/j.jmb.2007.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A., Wong S.M., Yuan Y.A. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2009;19:187–195. doi: 10.1038/cr.2008.288. [DOI] [PubMed] [Google Scholar]
- Chien C.Y., Tejero R., Huang Y., Zimmerman D.E., Ríos C.B., Krug R.M., Montelione G.T. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat. Struct. Biol. 1997;4:891–895. doi: 10.1038/nsb1197-891. [DOI] [PubMed] [Google Scholar]
- Chien C.-Y., Xu Y., Xiao R., Aramini J.M., Sahasrabudhe P.V., Krug R.M., Montelione G.T. Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: evidence for a novel RNA-binding mode. Biochemistry. 2004;43:1950–1962. doi: 10.1021/bi030176o. [DOI] [PubMed] [Google Scholar]
- Childs K., Stock N., Ross C., Andrejeva J., Hilton L., Skinner M., Randall R., Goodbourn S. Mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Childs K.S., Andrejeva J., Randall R.E., Goodbourn S. Mechanism of mda-5 Inhibition by paramyxovirus V proteins. J. Virol. 2009;83:1465–1473. doi: 10.1128/JVI.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs K., Randall R., Goodbourn S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 2012;86:3411–3421. doi: 10.1128/JVI.06405-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs K., Randall R.E., Goodbourn S. LGP2 plays a critical role in sensitizing mda-5 to activation by double-stranded RNA. PLoS One. 2013;8:e64202. doi: 10.1371/journal.pone.0064202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.H. Viral polymerases. Adv. Exp. Med. Biol. 2012;726:267–304. doi: 10.1007/978-1-4614-0980-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F., Bennett M., Moldt M., Deimling T., Witte G., Schiesser S., Carell T., Hopfner K.-P. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011;12:1127–1134. doi: 10.1038/embor.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Kanjanahaluethai A., O’Brien T.E., Baker S.C. Mutation in murine coronavirus replication protein nsp4 alters assembly of double membrane vesicles. Virology. 2008;375:118–129. doi: 10.1016/j.virol.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., Li K., Mesecar A.D., Baker S.C. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conenello G.M., Zamarin D., Perrone L.A., Tumpey T., Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:1414–1421. doi: 10.1371/journal.ppat.0030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conenello G.M., Tisoncik J.R., Rosenzweig E., Varga Z.T., Palese P., Katze M.G. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 2011;85:652–662. doi: 10.1128/JVI.01987-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S., Eisenächer K., Kirchhofer A., Brzózka K., Lammens A., Lammens K., Fujita T., Conzelmann K.-K., Krug A., Hopfner K.-P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- da Conceição T.M., Rust N.M., Berbel A.C.E.R., Martins N.B., do Nascimento Santos C.A., Da Poian A.T., de Arruda L.B. Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology. 2013;435:281–292. doi: 10.1016/j.virol.2012.09.038. [DOI] [PubMed] [Google Scholar]
- Das K., Ma L.C., Xiao R., Radvansky B., Aramini J., Zhao L., Marklund J., Kuo R.L., Twu K.Y., Arnold E., Krug R.M., Montelione G.T. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. USA. 2008;105:13093–13098. doi: 10.1073/pnas.0805213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A.M., Galiano M., Gorbalenya A.E., Memish Z., Perlman S., Poon L.L.M., Snijder E.J., Stephens G.M., Woo P.C.Y., Zaki A.M., Zambon M., Ziebuhr J. Middle east respiratory syndrome coronavirus (MERS-CoV); announcement of the coronavirus study group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A.H., Raj V.S., Oudshoorn D., Bestebroer T.M., van Nieuwkoop S., Limpens R.W., Posthuma C.C., van der Meer Y., Bárcena M., Haagmans B.L., Snijder E.J., van den Hoogen B.G. MERS-coronavirus replication induces severe in vitro cytophatology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 2013;94:1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E., Ferron F., Lescar J., Canard B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011;10:51–65. doi: 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado S., Erickson B.R., Agudo R., Blair P.J., Vallejo E., Albariño C.G., Vargas J., Comer J.A., Rollin P.E., Ksiazek T.G., Olson J.G., Nichol S.T. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog. 2008;4:e1000047. doi: 10.1371/journal.ppat.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.-T.K., Baker S.C., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitte-Orr S.J., Mehta D.R., Collins S.E., Suthar M.S., Gale M., Mossman K.L. Long double-stranded RNA induces an antiviral response independent of IFN regulatory factor 3, IFN-beta promoter stimulator 1, and IFN. J. Immunol. 2009;183:6545–6553. doi: 10.4049/jimmunol.0900867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelan N.R., Basler C.F., Garcia-Sastre A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 2003;77:13257–13266. doi: 10.1128/JVI.77.24.13257-13266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek S.E., Wixler L., Nordhoff C., Nordmann A., Anhlan D., Wixler V., Ludwig S. The influenza virus PB1-F2 protein has interferon antagonistic activity. Biol. Chem. 2011;392:1135–1144. doi: 10.1515/BC.2011.174. [DOI] [PubMed] [Google Scholar]
- Eaton B.T., Broder C.C., Middleton D., Wang L.-F. Hendra and Nipah viruses: different and dangerous. Nat. Rev. Microbiol. 2006;4:23–35. doi: 10.1038/nrmicro1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov A., Brandt S., Sereinig S., Romanova J., Ferko B., Katinger D., Grassauer A., Alexandrova G., Katinger H., Muster T. Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J. Virol. 1998;72:6437–6441. doi: 10.1128/jvi.72.8.6437-6441.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler R., Strecker T., Kolesnikova L., ter Meulen J., Weissenhorn W., Becker S., Klenk H.-D., Garten W., Lenz O. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP) Virus Res. 2004;100:249–255. doi: 10.1016/j.virusres.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Elliott R.M., Weber F. Bunyaviruses and the type I interferon system. Viruses. 2009;1:1003–1021. doi: 10.3390/v1031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabozzi G., Nabel C.S., Dolan M.A., Sullivan N.J. Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J. Virol. 2011;85:2512–2523. doi: 10.1128/JVI.01160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams M.E., Guenther U.-P., Jankowsky E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L., Briese T., Lipkin W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010;84:1785–1791. doi: 10.1128/JVI.01362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehling S.K., Lennartz F., Strecker T. Multifunctional nature of the arenavirus RING finger protein Z. Viruses. 2012;4:2973–3011. doi: 10.3390/v4112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Cerveny M., Yan Z., He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007;81:182. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field A.K., Tytell A.A., Lampson G.P., Nemes M.M., Hilleman M.R. Double-stranded polynucleotides as interferon inducers. J. Gen. Physiol. 1970;56:90–96. doi: 10.1085/jgp.56.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field A.K., Lampson G.P., Tytell A.A., Hilleman M.R. Demonstration of double-stranded ribonucleic acid in concentrates of RNA viruses. Proc. Soc. Exp. Biol. Med. 1972;141:440–444. doi: 10.3181/00379727-141-36793. [DOI] [PubMed] [Google Scholar]
- Field A.K., Tytell A.A., Piperno E., Lampson G.P., Nemes M.M., Hilleman M.R. Poly I:C, an inducer of interferon and interference against virus infections. Medicine (Baltimore) 1972;51:169–174. doi: 10.1097/00005792-197205000-00002. [DOI] [PubMed] [Google Scholar]
- Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., Coyle A.J., Liao S.-M., Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Fredericksen B.L., Gale M. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 2006;80:2913–2923. doi: 10.1128/JVI.80.6.2913-2923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen B.L., Smith M., Katze M.G., Shi P.-Y., Gale M. The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen B.L., Keller B.C., Fornek J., Katze M.G., Gale M. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 2008;82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt E.C., Yu L., Park E., Lenardo M.J., Xu X.-N. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 2009;83:6631–6640. doi: 10.1128/JVI.00367-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias-Staheli N., Giannakopoulos N.V., Kikkert M., Taylor S.L., Bridgen A., Paragas J., Richt J.A., Rowland R.R., Schmaljohn C.S., Lenschow D.J., Snijder E.J., Garcia-Sastre A., Virgin H.W. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack M.U., Shin Y.C., Joo C.-H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., Jung J.U. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gack M.U., Kirchhofer A., Shin Y.C., Inn K.-S., Liang C., Cui S., Myong S., Ha T., Hopfner K.-P., Jung J.U. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc. Natl. Acad. Sci. USA. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack M.U., Albrecht R.A., Urano T., Inn K.-S., Huang I.-C., Carnero E., Farzan M., Inoue S., Jung J.U., Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre A., Egorov A., Matassov D., Brandt S., Levy D.E., Durbin J.E., Palese P., Muster T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Garcin D., Lezzi M., Dobbs M., Elliott R.M., Schmaljohn C., Kang C.Y., Kolakofsky D. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J. Virol. 1995;69:5754–5762. doi: 10.1128/jvi.69.9.5754-5762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges-Courbot M.C., Contamin H., Faure C., Loth P., Baize S., Leyssen P., Neyts J., Deubel V. Poly(I)-poly(C12U) but not ribavirin prevents death in a hamster model of Nipah virus infection. Antimicrob. Agents Chemother. 2006;50:1768–1772. doi: 10.1128/AAC.50.5.1768-1772.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlier D., Lyles D.S. Interplay etween Innate Immunity and Negative-Strand RNA Viruses: towards a Rational Model. Microbiol. Mol. Biol. Rev. 2011;75:468–490. doi: 10.1128/MMBR.00007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J.S., Malide D., Hornung F., Bennink J.R., Yewdell J.W. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 2003;77:7214–7224. doi: 10.1128/JVI.77.13.7214-7224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie L.K., Hoenen A., Morgan G., Mackenzie J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010;84:10438–10447. doi: 10.1128/JVI.00986-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E.A., Solomon T. Pathogenic flaviviruses. Lancet. 2008;371:500–509. doi: 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- Graef K.M., Vreede F.T., Lau Y.-F., McCall A.W., Carr S.M., Subbarao K., Fodor E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 2010;84:8433–8445. doi: 10.1128/JVI.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groseth A., Hoenen T., Weber M., Wolff S., Herwig A., Kaufmann A., Becker S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl. Trop. Dis. 2011;5:e1137. doi: 10.1371/journal.pntd.0001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z., Chen L.-M., Zeng H., Gomez J.A., Plowden J., Fujita T., Katz J.M., Donis R.O., Sambhara S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007;36:263–269. doi: 10.1165/rcmb.2006-0283RC. [DOI] [PubMed] [Google Scholar]
- Habjan M., Andersson I., Klingström J., Schumann M., Martin A., Zimmermann P., Wagner V., Pichlmair A., Schneider U., Mühlberger E., Mirazimi A., Weber F. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One. 2008;3:e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon B., Kozina C., Maar D., Carpenter T.S., Branda C.S., Negrete O.A., Carson B.D. Identification of critical amino acids within the nucleoprotein of Tacaribe virus important for anti-interferon activity. J. Biol. Chem. 2013;288:8702–8711. doi: 10.1074/jbc.M112.444760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A., Towner J., Nichol S. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328:177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hartman A.L., Dover J.E., Towner J.S., Nichol S.T. Reverse genetic generation of recombinant zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J. Virol. 2006;80:6430–6440. doi: 10.1128/JVI.00044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A., Bird B., Towner J., Antoniadou Z.-A., Zaki S., Nichol S. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of Ebola virus. J. Virol. 2008;82:2699. doi: 10.1128/JVI.02344-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A., Ling L., Nichol S., Hibberd M. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008;82:5348. doi: 10.1128/JVI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L., Towner J.S., Nichol S.T. Ebola and marburg hemorrhagic fever. Clin. Lab. Med. 2010;30:161–177. doi: 10.1016/j.cll.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Hass M., Gölnitz U., Müller S., Becker-Ziaja B., Günther S. Replicon system for Lassa virus. J. Virol. 2004;78:13793–13803. doi: 10.1128/JVI.78.24.13793-13803.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie K.M., Kimberlin C.R., Zandonatti M.A., Macrae I.J., Saphire E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3″ to 5″ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA. 2011;108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatada E., Fukuda R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 1992;73(Pt. 12):3325–3329. doi: 10.1099/0022-1317-73-12-3325. [DOI] [PubMed] [Google Scholar]
- Hausmann S., Marq J.-B., Tapparel C., Kolakofsky D., Garcin D. RIG-I and dsRNA-induced IFNbeta activation. PLoS One. 2008;3:e3965. doi: 10.1371/journal.pone.0003965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman A., Comely S., Lackenby A., Murphy S., McCauley J., Goodbourn S., Barclay W. Variation in the ability of human influenza A viruses to induce and inhibit the IFN-beta pathway. Virology. 2006;347:52–64. doi: 10.1016/j.virol.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Heinz F.X., Stiasny K. Flaviviruses and flavivirus vaccines. Vaccine. 2012;30:4301–4306. doi: 10.1016/j.vaccine.2011.09.114. [DOI] [PubMed] [Google Scholar]
- Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.-K., Schlee M., Endres S., Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Hou F., Sun L., Zheng H., Skaug B., Jiang Q.-X., Chen Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Kolokoltsova O.A., Yun N.E., Seregin A.V., Poussard A.L., Walker A.G., Brasier A.R., Zhao Y., Tian B., de la Torre J.C., Paessler S. Junín virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl. Trop. Dis. 2012;6:e1659. doi: 10.1371/journal.pntd.0001659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui D.S.C., Wong G.W.K. Advancements in the battle against severe acute respiratory syndrome. Expert Opin. Pharmacother. 2004;5:1687–1693. doi: 10.1517/14656566.5.8.1687. [DOI] [PubMed] [Google Scholar]
- Ikegami T. Molecular biology and genetic diversity of Rift Valley fever virus. Antiviral Res. 2012;95:293–310. doi: 10.1016/j.antiviral.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai A., Shiozaki T., Kawai T., Akira S., Kawaoka Y., Takada A., Kida H., Miyazaki T. Influenza A virus polymerase inhibits type I interferon induction by binding to interferon beta promoter stimulator 1. J. Biol. Chem. 2010;285:32064–32074. doi: 10.1074/jbc.M110.112458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James T.W., Frias-Staheli N., Bacik J.-P., Levingston Macleod J.M., Khajehpour M., Garcia-Sastre A., Mark B.L. Structural basis for the removal of ubiquitin and interferon-stimulated gene 15 by a viral ovarian tumor domain-containing protease. Proc. Natl. Acad. Sci. USA. 2011;108:2222–2227. doi: 10.1073/pnas.1013388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F., Ramanathan A., Miller M.T., Tang G.-Q., Gale M., Patel S.S., Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Kinch L.N., Brautigam C.A., Chen X., Du F., Grishin N.V., Chen Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Huang Q., Wang W., Dong H., Ly H., Liang Y., Dong C. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J. Biol. Chem. 2013;288:16949–16959. doi: 10.1074/jbc.M112.420521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josset L., Menachery V.D., Gralinski L.E., Agnihothram S., Sova P., Carter V.S., Yount B.L., Graham R.L., Baric R.S., Katze M.G. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio. 2013;4:e00165-13. doi: 10.1128/mBio.00165-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalveram B., Lihoradova O., Ikegami T. NSs proteins of rift valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J. Virol. 2011;85:6234–6243. doi: 10.1128/JVI.02255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J., Yamaguchi O., Otsu K., Tsujimura T., Koh C.-S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T.S., Fujita T., Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K.J., Takeuchi O., Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Keller B.C., Fredericksen B.L., Samuel M.A., Mock R.E., Mason P.W., Diamond M.S., Gale M. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshtkar-Jahromi M., Kuhn J.H., Christova I., Bradfute S.B., Jahrling P.B., Bavari S. Crimean-Congo hemorrhagic fever: current and future prospects of vaccines and therapies. Antiviral Res. 2011;90:85–92. doi: 10.1016/j.antiviral.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Khan G. A novel coronavirus capable of lethal human infections: an emerging picture. Virol. J. 2013;10:66. doi: 10.1186/1743-422X-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.-J., Hwang S.-Y., Imaizumi T., Yoo J.-Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008;82:1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin C.R., Bornholdt Z.A., Li S., Woods V.L., Macrae I.J., Saphire E.O. From the Cover: Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc. Natl. Acad. Sci. USA. 2010;107:314–319. doi: 10.1073/pnas.0910547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler E., Jónsdóttir H.R., Muth D., Hamming O.J., Hartmann R., Rodriguez R., Geffers R., Fouchier R.A., Drosten C., Müller M.A., Dijkman R., Thiel V. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. MBio. 2013;4:e00611-12. doi: 10.1128/mBio.00611-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H.E.V.D., Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs G., Garcia-Sastre A., Martinez-Sobrido L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007;81:7011–7021. doi: 10.1128/JVI.02581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E., Lunardi T., McCarthy A.A., Louber J., Brunel J., Grigorov B., Gerlier D., Cusack S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.-E., Humphrey C.D., Shieh W.-J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.-Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., SARS Working Group A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kuhn J.H., Becker S., Ebihara H., Geisbert T.W., Johnson K.M., Kawaoka Y., Lipkin W.I., Negredo A.I., Netesov S.V., Nichol S.T., Palacios G., Peters C.J., Tenorio A., Volchkov V.E., Jahrling P.B. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010;155:2083–2103. doi: 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S., Volchkova V., Basler C.F., Palese P., Volchkov V.E., Shaw M.L. Nipah virus edits its P gene at high frequency to express the V and W proteins. J. Virol. 2009;83:3982–3987. doi: 10.1128/JVI.02599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H., Kawai T., Akira S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Le May N., Dubaele S., Proietti De Santis L., Billecocq A., Bouloy M., Egly J.M. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell. 2004;116:541–550. doi: 10.1016/s0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- Le May N., Mansuroglu Z., Léger P., Josse T., Blot G., Billecocq A., Flick R., Jacob Y., Bonnefoy E., Bouloy M. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog. 2008;4:e13. doi: 10.1371/journal.ppat.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.-H., Lalwani P., Raftery M.J., Matthaei M., Lütteke N., Kirsanovs S., Binder M., Ulrich R.G., Giese T., Wolff T., Krüger D.H., Schönrich G. RNA helicase retinoic acid-inducible gene I as a sensor of Hantaan virus replication. J. Gen. Virol. 2011;92:2191–2200. doi: 10.1099/vir.0.032367-0. [DOI] [PubMed] [Google Scholar]
- Lehner B., Williams G., Campbell R.D., Sanderson C.M. Antisense transcripts in the human genome. Trends Genet. 2002;18:63–65. doi: 10.1016/s0168-9525(02)02598-2. [DOI] [PubMed] [Google Scholar]
- Leung D.W., Ginder N.D., Fulton D.B., Nix J., Basler C.F., Honzatko R.B., Amarasinghe G.K. Structure of the Ebola VP35 interferon inhibitory domain. Proc. Natl. Acad. Sci. USA. 2009;106:411–416. doi: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.W., Prins K.C., Borek D.M., Farahbakhsh M., Tufariello J.M., Ramanan P., Nix J.C., Helgeson L.A., Otwinowski Z., Honzatko R.B., Basler C.F., Amarasinghe G.K. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat. Struct. Mol. Biol. 2010;17:165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.W., Shabman R.S., Farahbakhsh M., Prins K.C., Borek D.M., Wang T., Mühlberger E., Basler C.F., Amarasinghe G.K. Structural and functional characterization of Reston Ebola virus VP35 interferon inhibitory domain. J. Mol. Biol. 2010;399:347–357. doi: 10.1016/j.jmb.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Lu C., Stewart M., Xu H., Strong R.K., Igumenova T., Li P. Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch. Biochem. Biophys. 2009;488:23–33. doi: 10.1016/j.abb.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Li X., Ranjith-Kumar C.T., Brooks M.T., Dharmaiah S., Herr A.B., Kao C., Li P. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J. Biol. Chem. 2009;284:13881–13891. doi: 10.1074/jbc.M900818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Lynch P.A., Chien C.Y., Montelione G.T., Krug R.M., Berman H.M. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat. Struct. Biol. 1997;4:896–899. doi: 10.1038/nsb1197-896. [DOI] [PubMed] [Google Scholar]
- Liu W.J., Chen H.B., Wang X.J., Huang H., Khromykh A.A. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 2004;78:12225–12235. doi: 10.1128/JVI.78.22.12225-12235.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.J., Wang X.J., Clark D.C., Lobigs M., Hall R.A., Khromykh A.A. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 2006;80:2396–2404. doi: 10.1128/JVI.80.5.2396-2404.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-Y., Sanchez D.J., Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol. 2011;23:57–64. doi: 10.1016/j.coi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.M., Loo Y.-M., Horner S.M., Zornetzer G.A., Katze M.G., Gale M., Jr The mitochondrial targeting chaperone 14-3-3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe. 2012;11:528–537. doi: 10.1016/j.chom.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M.K., Harcourt B.H., Mungall B.A., Tamin A., Peeples M.E., Bellini W.J., Rota P.A. Determination of the henipavirus phosphoprotein gene mRNA editing frequencies and detection of the C, V and W proteins of Nipah virus in virus-infected cells. J. Gen. Virol. 2009;90:398–404. doi: 10.1099/vir.0.007294-0. [DOI] [PubMed] [Google Scholar]
- Loo Y.-M., Fornek J., Crochet N., Bajwa G., Perwitasari O., Martinez-Sobrido L., Akira S., Gill M.A., Garcia-Sastre A., Katze M.G., Gale M. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López N., Jácamo R., Franze-Fernández M.T. Transcription and RNA replication of tacaribe virus genome and antigenome analogs require N and L proteins: Z protein is an inhibitor of these processes. J. Virol. 2001;75:12241–12251. doi: 10.1128/JVI.75.24.12241-12251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G., Reinert J.T., Pitha-Rowe I., Okumura A., Kellum M., Knobeloch K.P., Hassel B., Pitha P.M. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell. Mol. Biol. (Noisy-le-grand) 2006;52:29–41. [PubMed] [Google Scholar]
- Lu C., Xu H., Ranjith-Kumar C.T., Brooks M.T., Hou T.Y., Hu F., Herr A.B., Strong R.K., Kao C.C., Li P. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure. 2010;18:1032–1043. doi: 10.1016/j.str.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Ranjith-Kumar C.T., Hao L., Kao C.C., Li P. Crystal structure of RIG-I C-terminal domain bound to blunt-ended double-strand RNA without 5′ triphosphate. Nucleic Acids Res. 2011;39:1565–1575. doi: 10.1093/nar/gkq974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Pan J., Tao J., Guo D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes. 2011;42:37–45. doi: 10.1007/s11262-010-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig S., Wang X., Ehrhardt C., Zheng H., Donelan N., Planz O., Pleschka S., Garcia-Sastre A., Heins G., Wolff T. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J. Virol. 2002;76:11166–11171. doi: 10.1128/JVI.76.21.11166-11171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D., Ding S.C., Vela A., Kohlway A., Lindenbach B.D., Pyle A.M. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P., Ramanan P., Mire C.E., Weisend C., Tsuda Y., Yen B., Liu G., Leung D.W., Geisbert T.W., Ebihara H., Amarasinghe G.K., Basler C.F. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil A., Rollin P.E. Ebola and Marburg hemorrhagic fevers: neglected tropical diseases? PLoS Negl. Trop. Dis. 2012;6:e1546. doi: 10.1371/journal.pntd.0001546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus P.I. Interferon induction by viruses: one molecule of dsRNA as the threshold for interferon induction. Interferon. 1983;5:115–180. [PubMed] [Google Scholar]
- Marques J.T., Devosse T., Wang D., Zamanian-Daryoush M., Serbinowski P., Hartmann R., Fujita T., Behlke M.A., Williams B.R. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 2006;24:559–565. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- Marsh G.A., Wang L.-F. Hendra and Nipah viruses: why are they so deadly? Curr. Opin. Virol. 2012;2:242–247. doi: 10.1016/j.coviro.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Martinez-Sobrido L., Zúñiga E.I., Rosario D., Garcia-Sastre A., de la Torre J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006;80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sobrido L., Giannakas P., Cubitt B., Garcia-Sastre A., de la Torre J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007;81:12696–12703. doi: 10.1128/JVI.00882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sobrido L., Emonet S., Giannakas P., Cubitt B., Garcia-Sastre A., de la Torre J.C. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2009;83:11330–11340. doi: 10.1128/JVI.00763-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthys V., Mackow E.R. Hantavirus regulation of type I interferon responses. Adv. Virol. 2012 doi: 10.1155/2012/524024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R.A., Garcia-Sastre A. Influenza A viruses: new research developments. Nat. Rev. Microbiol. 2011;9:590–603. doi: 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Mibayashi M., Martinez-Sobrido L., Loo Y.-M., Cardenas W.B., Gale M., Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnier L., Sanders F.K. Replicative form of encephalomyocarditis virus ribonucleic acid. Nature. 1963;199:664–667. doi: 10.1038/199664a0. [DOI] [PubMed] [Google Scholar]
- Morin B., Kranzusch P.J., Rahmeh A.A., Whelan S.P. The polymerase of negative-stranded RNA viruses. Curr. Opin. Virol. 2013;3:103–110. doi: 10.1016/j.coviro.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J., Aguirre S., Fernandez-Sesma A. Innate immunity evasion by Dengue virus. Viruses. 2012;4:397–413. doi: 10.3390/v4030397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz C., Schuhmann K.M., Kirchhofer A., Moldt M., Witte G., Conzelmann K.-K., Hopfner K.-P. Paramyxovirus V proteins disrupt the fold of the RNA sensor MDA5 to inhibit antiviral signaling. Science. 2013;339:690–693. doi: 10.1126/science.1230949. [DOI] [PubMed] [Google Scholar]
- Müller S., Geffers R., Günther S. Analysis of gene expression in Lassa virus-infected HuH-7 cells. J. Gen. Virol. 2007;88:1568–1575. doi: 10.1099/vir.0.82529-0. [DOI] [PubMed] [Google Scholar]
- Muñoz-Jordán J.L., Fredericksen B.L. How flaviviruses activate and suppress the interferon response. Viruses. 2010;2:676–691. doi: 10.3390/v2020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali A., Li X., Ranjith-Kumar C.T., Bhardwaj K., Holzenburg A., Li P., Kao C.C. Structure and Function of LGP2, a DEX(D/H) Helicase That Regulates the Innate Immunity Response. J. Biol. Chem. 2008;283:15825–15833. doi: 10.1074/jbc.M800542200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S., Cui S., Cornish P.V., Kirchhofer A., Gack M.U., Jung J.U., Hopfner K.-P., Ha T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasirudeen A.M.A., Wong H.H., Thien P., Xu S., Lam K.-P., Liu D.X. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 2011;5:e926. doi: 10.1371/journal.pntd.0000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff M.E., Barabino S.M., Li Y., Keller W., Krug R.M. Influenza virus NS1 protein ineracts with the cellular 30 kDa subunit of CPSF inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- Nemes M.M., Tytell A.A., Lampson G.P., Field A.K., Hilleman M.R. Inducers of interferon and host resistance. VII. Antiviral efficacy of double-stranded RNA of natural origin. Proc. Soc. Exp. Biol. Med. 1969;132:784–789. doi: 10.3181/00379727-132-34309. [DOI] [PubMed] [Google Scholar]
- Neumann G., Noda T., Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature. 2009;459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G., Chen H., Gao G.F., Shu Y., Kawaoka Y. H5N1 influenza viruses: outbreaks and biological properties. Cell Res. 2010;20:51–61. doi: 10.1038/cr.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah D.L., Twu K.Y., Krug R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology. 2003;307:386–395. doi: 10.1016/s0042-6822(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Oganesyan G., Saha S.K., Guo B., He J.Q., Shahangian A., Zarnegar B., Perry A., Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2005;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- Opitz B., Rejaibi A., Dauber B., Eckhard J. IFNβ induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007;9:930–938. doi: 10.1111/j.1462-5822.2006.00841.x. [DOI] [PubMed] [Google Scholar]
- Oshiumi H., Matsumoto M., Hatakeyama S., Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009;284:807–817. doi: 10.1074/jbc.M804259200. [DOI] [PubMed] [Google Scholar]
- Oshiumi H., Miyashita M., Inoue N., Okabe M., Matsumoto M., Seya T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. 2010;8:496–509. doi: 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Oshiumi H., Miyashita M., Matsumoto M., Seya T. A distinct role of riplet-mediated K63-linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013;9:e1003533. doi: 10.1371/journal.ppat.1003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D., Maniatis T., Harrison S.C. An atomic model of the interferon-β enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien J.-P., Bamming D., Komuro A., Ramachandran A., Rodriguez J.J., Barber G., Wojahn R.D., Horvath C.M. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J. Virol. 2009;83:7252–7260. doi: 10.1128/JVI.00153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D., Schultz L.W., Umland T.C. Influenza A polymerase subunit PB2 possesses overlapping binding sites for polymerase subunit PB1 and human MAVS proteins. Virus Res. 2013;172:75–80. doi: 10.1016/j.virusres.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Patel J.R., Jain A., Chou Y.Y., Baum A., Ha T., Garcia-Sastre A. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type I interferon. EMBO Rep. 2013 doi: 10.1038/embor.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A., Lin C., Wu B., Orme-Johnson M., Liu M., Walz T., Hur S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA. 2011;108:21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 2009;7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Rehwinkel J., Kato H., Takeuchi O., Akira S., Way M., Schiavo G., Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinschewer D.D., Perez M., de la Torre J.C. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J. Virol. 2003;77:3882–3887. doi: 10.1128/JVI.77.6.3882-3887.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippig D.A., Hellmuth J.C., Cui S., Kirchhofer A., Lammens K., Lammens A., Schmidt A., Rothenfusser S., Hopfner K.-P. The regulatory domain of the RIG-I family ATPase LGP2 senses double-stranded RNA. Nucleic Acids Res. 2009;37:2014–2025. doi: 10.1093/nar/gkp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins K., Cardenas W., Basler C. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKK{varepsilon} and TBK-1. J. Virol. 2009;83:3069. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins K., Delpeut S., Leung D., Reynard O., Volchkova V., Reid S., Ramanan P., Cardenas W., Amarasinghe G., Volchkov V., Basler C. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J. Virol. 2010;84:3004. doi: 10.1128/JVI.02459-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pythoud C., Rodrigo W.W.S.I., Pasqual G., Rothenberger S., Martinez-Sobrido L., de la Torre J.C., Kunz S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 2012;86:7728–7738. doi: 10.1128/JVI.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Lan S., Wang W., Schelde L.M., Dong H., Wallat G.D., Ly H., Liang Y., Dong C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature. 2010;468:779–783. doi: 10.1038/nature09605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X.Y., Chien C.Y., Lu Y., Montelione G.T., Krug R.M. An amino-terminal polypeptide fragment of the influenza virus NS1 protein possesses specific RNA-binding activity and largely helical backbone structure. RNA. 1995;1:948–956. [PMC free article] [PubMed] [Google Scholar]
- Qin C.-F., Zhao H., Liu Z.-Y., Jiang T., Deng Y.-Q., Yu X.-D., Yu M., Qin E.-D. Retinoic acid inducible gene-I and melanoma differentiation-associated gene 5 are induced but not essential for dengue virus induced type I interferon response. Mol. Biol. Rep. 2011;38:3867–3873. doi: 10.1007/s11033-010-0502-7. [DOI] [PubMed] [Google Scholar]
- Qu B., Qi X., Wu X., Liang M., Li C., Cardona C.J., Xu W., Tang F., Li Z., Wu B., Powell K., Wegner M., Li D., Xing Z. Suppression of the interferon and NF-kB responses by severe fever with thrombocytopenia syndrome virus. J. Virol. 2012;86:8388–8401. doi: 10.1128/JVI.00612-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajsbaum R., Albrecht R.A., Wang M.K., Maharaj N.P., Versteeg G.A., Nistal-Villán E., Garcia-Sastre A., Gack M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A NS1 protein. PLoS Pathog. 2012;8:e1003059. doi: 10.1371/journal.ppat.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A., Horvath C.M. Dissociation of paramyxovirus interferon evasion activities: universal and virus-specific requirements for conserved V protein amino acids in MDA5 interference. J. Virol. 2010;84:11152–11163. doi: 10.1128/JVI.01375-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan P., Shabman R.S., Brown C.S., Amarasinghe G.K., Basler C.F., Leung D.W. Filoviral immune evasion mechanisms. Viruses. 2011;3:1634–1649. doi: 10.3390/v3091634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan P., Edwards M.R., Shabman R.S., Leung D.W., Endlich-Frazier A.C., Borek D.M., Otwinowski Z., Liu G., Huh J., Basler C.F., Amarasinghe G.K. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc. Natl. Acad. Sci. USA. 2012;109:20661–20666. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos I., Fernandez-Sesma A. Innate immunity to H5N1 influenza viruses in humans. Viruses. 2012;4:3363–3388. doi: 10.3390/v4123363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall R.E., Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J., Tan C.P., Goubau D., Schulz O., Pichlmair A., Bier K., Robb N., Vreede F., Barclay W., Fodor E., Reis e Sousa C. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. 2010;140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- Reid S., Cardenas W., Basler C. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology. 2005;341:179–189. doi: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez K.R., Horvath C.M. Amino acid requirements for MDA5 and LGP2 recognition by paramyxovirus V proteins: a single arginine distinguishes MDA5 from RIG-I. J. Virol. 2013;87:2974–2978. doi: 10.1128/JVI.02843-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Madoz J.R., Belicha-Villanueva A., Bernal-Rubio D., Ashour J., Ayllon J., Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol. 2010;84:9760–9774. doi: 10.1128/JVI.01051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Madoz J.R., Bernal-Rubio D., Kaminski D., Boyd K., Fernandez-Sesma A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J. Virol. 2010;84:4845–4850. doi: 10.1128/JVI.02514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S., Goutagny N., DiPerna G., Gong M., Monks B.G., Schoenemeyer A., Yamamoto M., Akira S., Fitzgerald K.A. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 2005;175:5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Sadler A.J., Williams B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T., Kato H., Kumagai Y., Yoneyama M., Sato S., Matsushita K., Tsujimura T., Fujita T., Akira S., Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Hartmann G. The chase for the RIG-I ligand-recent advances. Mol. Ther. 2010;16:1254–1262. doi: 10.1038/mt.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Roth A., Hornung V., Haqmann C.A., Wimmenauer V., Barchet W., Coch C., Janke M., Mihailovic A., Wardle G., Juranek S., Kato H., Kawai K., Poeck H., Fitzgerald K.A., Takeuchi O., Akira S., Tuschi T., Latz E., Ludwig J., Hartmann G. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A., Schwerd T., Hamm W., Hellmuth J.C., Cui S., Wenzel M., Hoffmann F.S., Michallet M.-C., Besch R., Hopfner K.-P., Endres S., Rothenfusser S. 5′-Triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA. 2009;106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmolke M., Manicassamy B., Pena L., Sutton T., Hai R., Varga Z.T., Hale B.G., Steel J., Pérez D.R., Garcia-Sastre A. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog. 2011;7:e1002186. doi: 10.1371/journal.ppat.1002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R.B., Sun L., Ea C.-K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Shaw M.L. Henipaviruses employ a multifaceted approach to evade the antiviral interferon response. Viruses. 2009;1:1190–1203. doi: 10.3390/v1031190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M.L., Cardenas W.B., Zamarin D., Palese P., Basler C.F. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J. Virol. 2005;79:6078–6088. doi: 10.1128/JVI.79.10.6078-6088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley J.G., Vandergaast R., Deng L., Mariuzza R.A., Fredericksen B.L. Identification of multiple RIG-I-specific pathogen associated molecular patterns within the West Nile virus genome and antigenome. Virology. 2012;432:232–238. doi: 10.1016/j.virol.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.-L., Kok K.-H., Ng M.-H.J., Poon V.K.M., Yuen K.-Y., Zheng B.-J., Jin D.-Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., van der Meer Y., Zevenhoven-Dobbe J., Onderwater J.J.M., van der Meulen J., Koerten H.K., Mommaas A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006;80:5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz S., Reichelt M., Spiegel M., Kuri T., Martinez-Sobrido L., Garcia-Sastre A., Weber F., Kochs G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology. 2007;361:304–315. doi: 10.1016/j.virol.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahle L., Garcin D., Kolakofsky D. Sendai virus defective-interfering genomes and the activation of interferon beta. Virology. 2006;351:101–111. doi: 10.1016/j.virol.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Sun W., Li Y., Chen L., Chen H., You F., Zhou X., Zhou Y., Zhai Z., Chen D., Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Clementz M.A., Banach B.S., Li K., Baker S.C., Chen Z. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surasombatpattana P., Hamel R., Patramool S., Luplertlop N., Thomas F., Desprès P., Briant L., Yssel H., Missé D. Dengue virus replication in infected human keratinocytes leads to activation of antiviral innate immune responses. Infect. Genet. Evol. 2011;11:1664–1673. doi: 10.1016/j.meegid.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Suthar M.S., Ma D.Y., Thomas S., Lund J.M., Zhang N., Daffis S., Rudensky A.Y., Bevan M.J., Clark E.A., Kaja M.-K., Diamond M.S., Gale M. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar M.S., Diamond M.S., Gale M. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013;11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- Takahasi K., Yoneyama M., Nishihori T., Hirai R., Kumeta H., Narita R., Gale M., Jr, Inagaki F., Fujita T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell. 2008;29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Takahasi K., Kumeta H., Tsuduki N., Narita R., Shigemoto T., Hirai R., Yoneyama M., Horiuchi M., Ogura K., Fujita T., Inagaki F. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 2009;284:17465–17474. doi: 10.1074/jbc.M109.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talon J., Horvath C.M., Polley R., Basler C.F., Muster T., Palese P., García-Sastre A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000;74:7989–7996. doi: 10.1128/jvi.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T.-K., Wu M.P.-J., Chen S.-T., Hou M.-H., Hong M.-H., Pan F.-M., Yu H.-M., Chen J.-H., Yao C.-W., Wang A.H.-J. Biochemical and immunological studies of nucleocapsid proteins of severe acute respiratory syndrome and 229E human coronaviruses. Proteomics. 2005;5:925–937. doi: 10.1002/pmic.200401204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Weber F. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 2008;19:121–132. doi: 10.1016/j.cytogfr.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B., Bevilacqua P.C., Diegelman-Parente A., Mathews M.B. The double-stranded-RNA-binding motif: interference and much more. Nat. Rev. Mol. Cell Biol. 2004;5:1013–1023. doi: 10.1038/nrm1528. [DOI] [PubMed] [Google Scholar]
- Totura A.L., Baric R.S. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012;2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytell A.A., Lampson G.P., Field A.K., Hilleman M.R. Inducers of interferon and host resistance. 3. Double-stranded RNA from reovirus type 3 virions (reo 3-RNA) Proc. Natl. Acad. Sci. USA. 1967;58:1719–1722. doi: 10.1073/pnas.58.4.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sandt C.E., Kreijtz J.H.C.M., Rimmelzwaan G.F. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses. 2012;4:1438–1476. doi: 10.3390/v4091438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z.T., Palese P. The influenza A virus protein PB1-F2: killing two birds with one stone? Virulence. 2011;2:542–546. doi: 10.4161/viru.2.6.17812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z.T., Ramos I., Hai R., Schmolke M., Garcia-Sastre A., Fernandez-Sesma A., Palese P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067. doi: 10.1371/journal.ppat.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z.T., Grant A., Manicassamy B., Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012;86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg G.A., Garcia-Sastre A. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 2010;13:508–516. doi: 10.1016/j.mib.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg G.A., Bredenbeek P.J., van den Worm S.H.E., Spaan W.J.M. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361:18–26. doi: 10.1016/j.virol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter C.T., Barr J.N. Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 2011;92:2467–2484. doi: 10.1099/vir.0.035105-0. [DOI] [PubMed] [Google Scholar]
- Wang W., Riedel K., Lynch P., Chien C.Y., Montelione G.T., Krug R.M. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA. 1999;5:195–205. doi: 10.1017/s1355838299981621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Li M., Zheng H., Muster T., Palese P., Beg A.A., García-Sastre A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000;74:11566–11573. doi: 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Basler C.F., Wiliams B.R., Silverman R.H., Palese P., Garcia-Sastre A. Functional replacement of the carboxy-terminal two thirds of the influenza A virus NS1 protein with short heterologous dimerization domains. J. Virol. 2002;76:12951–12962. doi: 10.1128/JVI.76.24.12951-12962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Ludwig J., Schuberth C., Goldeck M., Schlee M., Li H., Juranek S., Sheng G., Micura R., Tuschl T., Hartmann G., Patel D.J. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010;17:781–787. doi: 10.1038/nsmb.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Li S., Dorf M.E. NEMO binds ubiquitinated TANK-binding kinase 1 (TBK1) to regulate innate immune responses to RNA viruses. PLoS One. 2012;7:e43756. doi: 10.1371/journal.pone.0043756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F., Mirazimi A. Interferon and cytokine responses to Crimean Congo hemorrhagic fever virus; an emerging and neglected viral zonoosis. Cytokine Growth Factor Rev. 2008;19:395–404. doi: 10.1016/j.cytogfr.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F., Wagner W., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M., Gawanbacht A., Habjan M., Rang A., Borner C., Schmidt A.M., Veitinger S., Jacob R., Devignot S., Kochs G., Garcia-Sastre A., Weber F. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe. 2013;13:336–346. doi: 10.1016/j.chom.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K.E., Walther P., Fuller S.D., Antony C., Krijnse-Locker J., Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A. Natural antisense transcripts. RNA Biol. 2005;2:53–62. doi: 10.4161/rna.2.2.1852. [DOI] [PubMed] [Google Scholar]
- Wilkins C., Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisskirchen C., Ludersdorfer T.H., Müller D.A., Moritz E., Pavlovic J. The cellular helicase UAP56 is required for prevention of double-stranded RNA formation during influenza A virus infection. J. Virol. 2011;85:8646–8655. doi: 10.1128/JVI.02559-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Hur S. Viral counterattack against the host innate immune system. Cell Res. 2013;23:735–736. doi: 10.1038/cr.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- Xu L.-G., Wang Y.-Y., Han K.-J., Li L.-Y., Zhai Z., Shu H.-B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Yamada H., Chounan R., Higashi Y., Kurihara N., Kido H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004;578:331–336. doi: 10.1016/j.febslet.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Yin C., Khan J.A., Swapna G.V.T., Ertekin A., Krug R.M., Tong L., Montelione G.T. Conserved surface features form the double-stranded RNA binding site of non-structural protein 1 (NS1) from influenza A and B viruses. J. Biol. Chem. 2007;282:20584–20592. doi: 10.1074/jbc.M611619200. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K., Foy E., Loo Y.-M., Gale M., Akira S., Yonehara S., Kato A., Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T., Hill T.E., Yoshikawa N., Popov V.L., Galindo C.L., Garner H.R., Peters C.J., Tseng C.-T.K. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS One. 2010;5:e8729. doi: 10.1371/journal.pone.0008729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.J., Liang M.F., Zhang S.Y., Liu Y., Li J.D., Sun Y.L., Zhang L., Zhang Q.F., Popov V.L., Li C., Qu J., Li Q., Zhang Y.P., Hai R., Wu W., Wang Q., Zhan F.X., Wang X.J., Kan B., Wang S.W., Wan K.L., Jing H.Q., Lu J.X., Yin W.W., Zhou H., Guan X.H., Liu J.F., Bi Z.Q., Liu G.H., Ren J., Wang H., Zhao Z., Song J.D., He J.R., Wan T., Zhang J.S., Fu X.P., Sun L.N., Dong X.P., Feng Z.J., Yang W.Z., Hong T., Zhang Y., Walker D.H., Wang Y., Li D.X. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 2011;364:1523–1532. doi: 10.1056/NEJMoa1010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.-Y., Chang T.-H., Liang J.-J., Chiang R.-L., Lee Y.-L., Liao C.-L., Lin Y.-L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012;8:e1002780. doi: 10.1371/journal.ppat.1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zeng W., Sun L., Jiang X., Chen X., Hou F., Adhikari A., Xu M., Chen Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Liu Y., Zhao L., Li B., Yu H., Wen H., Yu X.J. An emerging hemorrhagic fever in China caused by a novel bunyavirus SFTSV. Sci. China Life Sci. 2013;56:697–700. doi: 10.1007/s11427-013-4518-9. [DOI] [PubMed] [Google Scholar]
- Zhao T., Yang L., Sun Q., Arguello M., Ballard D.W., Hiscott J., Lin R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- Zhao C., Collins M.N., Hsiang T.-Y., Krug R.M. Interferon-induced ISG15 pathway: an ongoing virus-host battle. Trends Microbiol. 2013;21:181–186. doi: 10.1016/j.tim.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Yang Y., Li S., Wang Y.-Y., Li Y., Diao F., Lei C., He X., Zhang L., Tien P., Shu H.-B. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Zhou H., Perlman S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 2007;81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Cerny A.M., Zacharia A., Fitzgerald K.A., Kurt-Jones E.A., Finberg R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010;84:9452–9462. doi: 10.1128/JVI.00155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielecki F., Weber M., Eickmann M., Spiegelberg L., Zaki A.M., Matrosovich M., Becker S., Weber F. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 2013;87:5300–5304. doi: 10.1128/JVI.03496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzula L., Esposito F., Mühlberger E., Trunschke M., Conrad D., Piano D., Tramontano E. Purification and functional characterization of the full length recombinant Ebola virus VP35 protein expressed in E. coli. Protein Expr. Purif. 2009;66:113–119. doi: 10.1016/j.pep.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Zinzula L., Esposito F., Pala D., Tramontano E. DsRNA binding characterization of full length recombinant wild type and mutants Zaire ebolavirus VP35. Antiviral Res. 2012;93:354–363. doi: 10.1016/j.antiviral.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]