

Highlights
-
•
4-Oxo- or thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-ones were synthesized.
-
•
Their anti-influenza viral activity was tested in a cell culture system.
-
•
15a, 15g, 15j, 15q, 15s, 15t, and 15x exhibited EC50 values lower than 20 μM.
-
•
Their anti-influenza viral activity was acquired by their inhibition of viral NA.
Keywords: Influenza virus, Antiviral, Dihydrofuropyridinones, Cytopathic effect reduction assay, NA inhibitor
Abstract
A target-free approach was applied to discover anti-influenza viral compounds, where influenza infected Madin–Darby canine kidney cells were treated 7500 different small organic chemicals individually and reduction of virus-induced cytopathic effect was measured. One of the hit compounds was (Z)-1-((5-fluoro-1H-indol-3-yl)methylene)-6-methyl-4-thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-one (15a) with half-maximal effective concentrations of 17.4–21.1 μM against influenza A/H1N1, A/H3N2 and B viruses without any cellular toxicity at 900 μM. To investigate the structure–activity relationships, two dozens of the hit analogs were synthesized. Among them, 15g, 15j, 15q, 15s, 15t and 15x had anti-influenza viral activity comparable or superior to that of the initial hit. The anti-influenza viral compounds efficiently suppressed not only viral protein level of the infected cells but also production of viral progeny in the culture supernatants in a dose-dependent manner. Based on a mode-of-action study, they did not affect virus entry or RNA replication. Instead, they suppressed viral neuraminidase activity. This study is the first to demonstrate that dihydrofuropyridinones could serve as lead compounds for the discovery of alternative influenza virus inhibitors.
1. Introduction
Along with seasonal influenza vaccines, antivirals have played a critical role in controlling influenza virus infection and transmission. They are most important when a viral strain which cannot be neutralized by the existing vaccines emerges, as occurred during the 2009 pandemic (Miller et al., 2013). Two classes of antivirals were approved for preventing and treating influenza. The first one comprises adamantanes, including amantadine (AMT) and rimantadine. They inhibit the ion channel function of influenza A virus matrix protein 2 (M2) that is required for endosomal acidification during the virus entry step (Das, 2012, De Clercq, 2006, Pinto et al., 1992). However, despite their high efficacy, adamantanes are no longer recommended for the treatment of influenza infections due to a very high prevalence of drug resistance among the circulating seasonal influenza viruses (Belshe et al., 1989, Saito et al., 2003). For example, in Republic of Korea during 2005–2010, the frequency of AMT resistance of seasonal influenza A was 30% for H1N1 and 76% for H3N2, and that of pandemic 2009 H1N1 virus (H1N1pdm09) was 100% (Cho et al., 2013). The second class is neuraminidase (NA) inhibitors, such as oseltamivir, zanamivir, peramivir and laninamivir (Das, 2012, De Clercq, 2006, Samson et al., 2013, von Itzstein, 2007). They suppress the NA activity required for cleavage of neuraminic acid moieties from cellular receptors and glycoproteins of newly synthesized influenza virions. These antivirals have been shown to be highly effective against currently circulating influenza viruses and thus are recommended for the coming influenza season. In fact, 99.6% and 100% of the 2009 H1N1 viruses were susceptible to oseltamivir and zanamivir, respectively, for the 2012–2013 season (CDC, 2013). However, according to earlier surveillance studies, 100% and 99.8% of A(H1N1) viruses had reduced oseltamivir susceptibility in Japan and Republic of Korea, respectively, for the 2008–2009 season (Baranovich et al., 2010, Choi et al., 2011). Moreover, Leang et al. (2013) also reported that cases of oseltamivir-resistant H1N1pdm09 infections with the H275 NA mutation increased during 2011 in Australia, compared with the first year of the pandemic. This sporadic appearance of oseltamivir resistance of seasonal influenza viruses prompts a note of caution against a potential pandemic situation caused by an NA inhibitor-resistant human influenza virus with high pathogenicity and transmissibility. Thus, development of novel antiviral agents overcoming resistance issues of the first-line antiviral drugs could be worthwhile to pursue urgently. Currently, antiviral researchers are trying to develop a second generation of antivirals by designing NA inhibitors to induce a formation of a stabilized covalent intermediate of NA or by targeting other viral proteins or cellular proteins involved in virus replication (Das, 2012, Kim et al., 2013a).
In our laboratory, a chemical library of approximately 7500 small organic compounds was screened to discover hit compounds with activity against two human influenza A viruses (H1N1 and H3N2) and one influenza B virus in a cell culture-based system. Among the hit compounds with over 50% virus growth inhibition at 20 μM, (Z)-1-((5-fluoro-1H-indol-3-yl)methylene)-6-methyl-4-thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-one (15a) was proved to be inhibitory against all three isolates (Fig. 1 A and B). This study was aimed to investigate the structure–activity relationships (SARs) of 15a analogs and their antiviral mode-of-action, and ultimately to provide insights into the design of another class of influenza inhibitors with unique chemical structures different from those of existing antivirals.
Fig. 1.
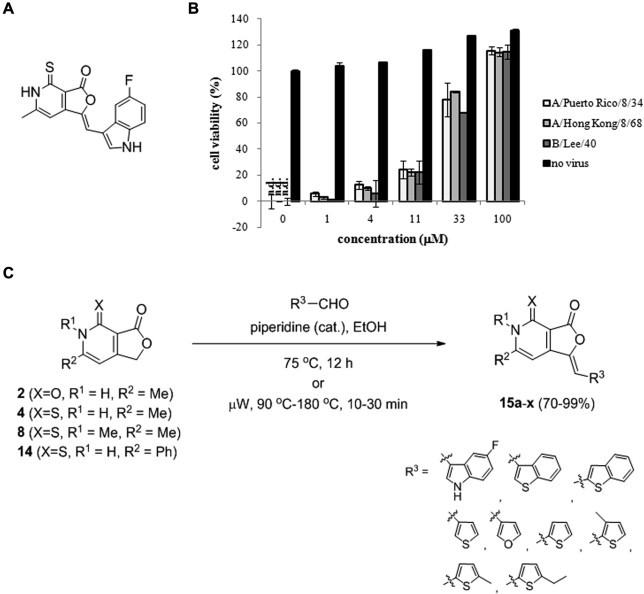
Anti-influenza virus activity of 15a and synthesis of its derivatives. (A) Chemical structure of the hit compound 15a. (B) Inhibition of influenza virus-induced cytopathic effects by 15a. MDCK cells were mock-infected (no virus) or infected with three different types of influenza viruses, such as A/Puerto Rico/8/34 (H1N1), A/Hong Kong/8/68 (H3N2), and B/Lee/40, and then treated with serially diluted 15a in cell culture media. On day 3 p.i., the cell viability was measured using the MTT assay. The viability of uninfected, DMSO-treated cells was set to 100%, while that of cells infected with each virus was only set to 0%. The data shown are the means ± standard deviations (S.D.) of two samples from three independent experiments. n.d., not detected. (C) Synthesis scheme of the chemicals 15a–x. Me, methyl; Ph, phenyl.
2. Materials and methods
2.1. Cells, viruses and antiviral control compounds
Madin–Darby canine kidney (MDCK) cells and African green monkey kidney Vero cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD). The cells were maintained in Minimum Essential Medium (MEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen) supplemented with 5% FBS, respectively, at 37 °C with 5% CO2. Human influenza viruses, A/Puerto Rico/8/34 (H1N1; PR8), A/Hong Kong/8/68 (H3N2), and B/Lee/40, were purchased from ATCC. The PR8 and A/Hong Kong/8/68 viruses were amplified by infection of 10-day-old chicken eggs, whereas the tissue culture-adapted B/Lee/40 strain was done by infection of MDCK cells in serum-free MEM containing 2 μg/ml TPCK-trypsin (Sigma, St. Louis, MO). After incubation at 37 °C for 3 days, the viruses were harvested from the egg allantoic fluid or the cell culture supernatant by centrifugation at 900 rpm for 5 min, and stored at −70 °C.
The additional cell lines and viral strains used for antiviral assays are as follows: Vero cells were infected with herpes simplex virus type 1 (HSV-1) strain F or type 2 (HSV-2) strain MS purchased from ATCC; Crandell feline kidney (CrFK) cells were infected with feline coronavirus (FECV) strain WSU 79–1683 (ATCC); HeLa cells were infected with Coxsackie B1 virus (CoxB1) strain Connecticut-5 or CoxB3 strain Nancy (ATCC); and MT-4 cells were infected with human immunodeficiency virus type 1 (HIV-1) strain IIIB or type 2 (HIV-2) ROD obtained from the National Institute for Biological Standards and Control (NIBSC, Potters Bar, United Kingdom).
AMT, ribavirin (RBV), and (−)-epigallocatechin-3-gallate (EGCG) were obtained from Sigma, and oseltamivir carboxylate (OSV-C) from US Biological (Swampscott, MA). Favipiravir (T-705; 6-flouro-3-hydroxy-2-pyrazinecarboxamide) was synthesized in-house by Dr. I.Y. Lee, according to a published method (Furuta et al., 2004).
2.2. Chemical synthesis and analysis
Four core scaffolds 2, 4, 8, and 14 were designed and synthesized to prepare 15a and its derivatives 15b–x (Fig. 1C and Supplementary data). Core compounds 2 and 4 were prepared to explore position X based on a previous report (Lena et al., 2008). Using a modification of this method, the other core compounds, 8 and 14, were synthesized. Subsequently, dihydrofuropyridinones 15a–x were prepared using the method described in the synthesis scheme (Fig. 1C). Conventional or microwave heating conditions were used, depending on the reactivity of the core scaffolds and aldehydes. The structures of the purified compounds were confirmed by 300 MHz 1H NMR (Bruker Avance 300; Bruker BioSpin, Fremont, CA) and liquid chromatography–mass spectrometry (LC/MS). LC/MS analysis was performed using a Waters 2996 Photodiode array detector and a Waters Micromass ZQ with electrospray ionization in the positive ion mode (Waters, Milford, MA) (Table S1). The purity of the prepared compounds was estimated to be >95%.
2.3. Cytopathic effect (CPE) reduction assay
MDCK cells seeded in 96-well plates were either mock-infected or infected with influenza viruses at a multiplicity of infection (MOI) of 0.001 for 1 h at 35 °C. After removing the medium, the cells were treated with test or control compounds, which were threefold serially diluted in serum-free MEM containing 2 μg/ml TPCK-trypsin. On day 3 postinfection (p.i.), the cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma), as described previously (Kao et al., 2010) with some modifications. In brief, cell monolayers were incubated with 50 μl of 3 mg/ml MTT in MEM at 37 °C for 4 h, then treated with 100 μl of MTT solvent (0.1 N HCl and 10% Triton X-100 in isopropanol). After shaking the plates for 10 min, the absorbance was read at 540 nm with a reference filter at 690 nm using a SpectraMax M3 Microplate Reader (Molecular Devices, Sunnyvale, CA). The 50% cytotoxic concentration (CC50) and 50% effective concentration (EC50) values were calculated using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Selectivity index (S.I.) was calculated as ratio of CC50 to EC50.
Antiviral assays against other viruses, such as HSV (De Logu et al., 2000), FECV (Liu et al., 2013), CoxB (Zhong et al., 2009), and HIV (Bedoya et al., 2010, Malpani et al., 2013), were performed according to the same method using MTT as mentioned above, but with a shorter incubation period with MTT of 1 h.
2.4. HA (hemagglutinin) assay
MDCK cells in 24-well plates were infected with PR8 virus at an MOI of 0.002 in the presence of dimethyl sulfoxide (DMSO), which was used as a vehicle to dissolve the test compounds, or different concentrations of chemicals. The cell culture supernatants were harvested at 12, 24, 36, and 48 h p.i. Fifty microliters of the twofold serially diluted PR8 virus-containing supernatants were mixed with the same volume of 0.5% chicken red blood cells (RBCs) for 30 min at room temperature (RT). The HA activity was determined by measuring the dilution factor of the samples required for complete HA-mediated chicken RBC agglutination (Kim et al., 2012). This experiment was performed three times in duplicate.
2.5. Plaque titration
MDCK cells were infected with PR8 virus at an MOI of 0.001 in the presence of test or control compounds for 24 h at 35 °C. To determine infectious virus titers using a plaque formation assay, culture supernatants were ten-fold serially diluted (10−1 to 10−6 in MEM) to infect fresh MDCK cells in 48-well plates for 1 h at 33 °C. After washing with phosphate-buffered saline (PBS), the cells were incubated with overlay medium [MEM containing 0.5% carboxymethylcellulose (CMC; Sigma) and 2 μg/ml of TPCK-trypsin]. The plates were incubated for 3 days at the same temperature, before the viral plaques were visualized by staining with crystal violet (Kim et al., 2012).
2.6. Quantitative RT-PCR (reverse transcription-polymerase chain reaction)
Influenza viral RNA was extracted from the culture supernatant using a QIAamp Viral RNA kit (Qiagen, Hilden, Germany), and reverse transcribed to cDNA using SuperScript III reverse transcriptase (Invitrogen) and an influenza A viral RNA-specific universal primer (5′-AGCAAAAGCAGG-3′) (Hoffmann et al., 2001). Quantitative PCR was performed using PR8 NS-specific primers (NSfwd, 5′-CATAATGGATTCAAACACTGTGTC-3′; NSrev, 5′-CCTCTTAGGGATTTCTGATCTCGG-3′) (Kim et al., 2013b) and SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA) with a CFX96 real-time PCR detection system (Bio-Rad). The viral RNA level from the virus-infected cells was defined as 100% and the relative viral RNA levels from the test samples were automatically calculated by using Bio-Rad’s CFX Manager Software. Data and standard deviations were determined from two independent experiments in triplicate.
2.7. Western blot analysis
MDCK cells in 6-well plates were inoculated with PR8 at an MOI of 0.001 in the presence of DMSO as a vehicle control, test compounds (11, 33, and 100 μM), or OSV-C (11 μM) for 24 h at 35 °C. The culture medium was removed and cells were lysed with 200 μl of Mammalian Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL). The protein lysates were separated by 10% or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and electro-transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). Viral nucleoprotein (NP) and HA protein, and cellular β-actin protein (loading control) were detected with anti-NP (Catalog No. 11675-MM03, Sino Biological, Beijing, China), -HA2 (Catalog No. 86001-RM01, Sino Biological), and -β-actin antibodies (Catalog No. A1978, Sigma), respectively. The horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-mouse IgG for anti-NP and anti-β-actin, and goat anti-rabbit IgG for anti-HA2) were purchased from Thermo Scientific (Waltham, MA). The bound antibodies were detected using the SuperSignal West Pico Chemiluminescent Substrate detection kit (Thermo Scientific, Rockford, IL).
2.8. Confocal microscopy
PR8 virus was infected into MDCK cells seeded in four-well chamber slides at an MOI of 2.5 in the absence or presence of chemicals (100 μM) and incubated for 2 h at 37 °C. The cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. After blocking with 10% normal goat serum and 1% bovine serum albumin in PBS, the slides were incubated overnight at 4 °C with an influenza A NP-specific monoclonal antibody (Catalog No. sc-80481, Santa Cruz Biotechnology, Santa Cruz, CA). Subsequently, they were labeled with Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) for 1 h at RT, followed by counterstaining with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Laser scanning confocal microscopy was performed with a Zeiss LSM 710 confocal microscope (Carl Zeiss, Thornwood, NY).
2.9. Viral RNA polymerase activity assay
Based on previous methods, DNA samples derived from four PR8 genomic RNAs, i.e., PB2, PB1, PA, and NP, were cloned individually into a vector that harbored convergent RNA polymerase I and II promoters by RT-PCR, which generated pVP-PB2, -PB1, -PA, and -NP, respectively (Hoffmann et al., 2000, Hoffmann et al., 2001, Kim et al., 2013b). The plasmid pHH21-FLuc, which expressed the influenza virus-like RNA of firefly luciferase (FLuc) flanked with 5′- and 3′-untranslated regions (UTR) of the PR8 NS gene under the control of the human RNA polymerase I promoter, was constructed as described previously (Fujii et al., 2005, Kim et al., 2013b, Pleschka et al., 1996). To determine the effects of the test compounds on the influenza viral polymerase activity, Vero cells (2 × 105 cells per well in 24-well plates) were transfected with the four viral polymerases/NP-encoding pVP DNAs and pHH21-FLuc (0.2 μg each), together with 0.05 μg of phLR-CMV (Promega, Madison, WI) expressing Renilla luciferase (RLuc), using Lipofectamine 2000 (Invitrogen). After 6 h, the supernatant was replaced with fresh medium containing 0.2% DMSO as a negative control, or increasing amounts of a compound (15a, 15q, or T-705). On day 1 post-transfection, the FLuc activity was measured by normalizing against the RLuc activity using the dual-luciferase reporter assay system (Promega).
2.10. NA assay
NA assay was performed by using a NA-XTD Influenza Neuraminidase Assay kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol with some modifications (Murtaugh et al., 2013). In brief, 25 μl of influenza A virus PR8 [1 × 105 plaque-forming units (PFU)/ml] or purified NA proteins derived from influenza H1N1 (A/California/04/2009) and its OSV-resistant H275Y mutant [NA (H275Y)] (Sino Biological) [25 μl of 40 milliunits (mU) per ml] in PBS were mixed individually with an equal volume of serially diluted test compounds in 96-well plates and incubated at 37 °C for 20 min. After adding 25 μl of 5 μM NA-XTD substrate (Applied Biosystems), the reaction mixture was incubated at RT for 20 min. The luminescent signal was maximized by the addition of 60 μl of NA-XTD accelerator and measured with a Berthold LB960 Centro microplate luminometer (Berthold Technologies, Bad Wilbad, Germany) using a 1 s/well read time. The half-maximum inhibitory concentrations (IC50) were calculated using GraphPad Prism 6.
2.11. In silico docking analysis
A reference H1N1 NA structure (A/California/04/2009) for molecular docking analysis on NA binding to 15a was obtained from the Protein Data Bank (PDB) accession No. 3TI6, which showed 3D structures of the viral NA complexed with OSV-C (Vavricka et al., 2011). The CHARMM force field parameters were first assigned for the PR8 NA protein and the binding site was defined using the advanced Define and Edit Binding Site tools. Docking calculations were carried out using ligandfit software interfaced with Accelrys Discovery Studio 3.5 (Accelrys Software Inc., San Diego, CA) (Venkatachalam et al., 2003). In detail, the ligand 15a was loaded in the protein’s binding site to have low-energy conformations using the protocol ‘Generate Conformations’ in Discovery Studio 3.5. To verify the reliability of our simulation protocol, we performed self-docking analysis using OSV-C as a reference antiviral compound. Based on the parameters used for OSV-C, 15a was in silico docked to the OSV-C binding site within the PR8 NA protein.
2.12. Statistical analysis
Statistically significant differences were evaluated by ANOVA followed by pairwise comparisons recommended for ANOVA analysis. P < 0.05 was considered significant.
3. Results
3.1. In vitro antiviral activities of 15a–x against influenza viruses
A cell culture-based screening system was established for influenza A and B viruses, i.e., PR8, A/Hong Kong/8/68, and B/Lee/40, using an MTT-based CPE reduction assay. With control antiviral compounds, including AMT, which was known to inactive against PR8 and B/Lee/40 (Kim et al., 2013b, Malpani et al., 2013), OSV-C, T-705, and RBV, the sensitivities of the different viruses were measured to confirm the reliability of the assay (Table 1 ). After screening a chemical library of about 7500 small organic compounds obtained from the Korea Chemical Bank (KRICT, Daejeon, Republic of Korea), it was observed that one of hit compounds, 15a, efficiently protected cells from influenza virus infection (Fig. 1A and B). It had the EC50 values ranging from 17.4 to 21.1 μM with no toxicity to MDCK cells at 900 μM (Table 1).
Table 1.
Structure and anti-influenza virus activity of dihydrofuropyridinones with a bicyclic substituent at R3.
| Compound | Substituent |
EC50a (μM) for influenza virus (S.I.b) |
CC50c (μM) for MDCK cells | |||||
|---|---|---|---|---|---|---|---|---|
| X | R1 | R2 | R3 | A/Puerto Rico/8/34 | A/Hong Kong/8/68 | B/Lee/40 | ||
| 15a | S | H | Me | 17.8 ± 10.6 (>50.6) | 17.4 ± 6.3 (>51.7) | 21.1 ± 9.73 (>42.7) | >900.0 | |
| 15b | O | H | Me | 22.5 ± 10.8 | 26.8 ± 16.2 | 31.1 ± 19.7 | >900.0 | |
| 15c | S | Me | Me | >100.0 | >100.0 | >100.0 | >900.00 | |
| 15d | S | H | Ph | 63.8 ± 25.2 | 32.5 ± 13.3 | 41.2 ± 7.0 | >900.0 | |
| 15e | S | H | Me | >100.0 | >100.0 | >100.0 | >900.0 | |
| 15f | S | H | Me | 26.1 ± 6.0 | 28.8 ± 6.2 | 19.8 ± 7.3 | >900.0 | |
| 15g | S | H | Me | 16.1 ± 6.0 (>55.9) | 10.5 ± 5.0 (>85.7) | 18.4 ± 12.8 (>48.9) | >900.0 | |
| 15h | S | H | Me | >100.0 | >100.0 | >100.0 | >900.0 | |
| 15i | S | Me | Me | >100.0 | >100.0 | >100.0 | >900.0 | |
| Amantadine hydrochloride | >100.0 | 2.8 ± 1.7 | >100.0 | >100.0 | ||||
| Oseltamivir carboxylate | 0.03 ± 0.01 | <0.01 | 1.3 ± 0.7 | >100.0 | ||||
| T-705d | 4.3 ± 2.4 | 6.5 ± 1.7 | 6.1 ± 0.6 | >100.0 | ||||
| Ribavirin | 18.8 ± 4.3 | 20.2 ± 2.4 | 23.7 ± 5.4 | >100.0 | ||||
Effective concentration required for reducing virus-induced cytopathicity by 50%.
Selectivity index, CC50/EC50.
Cytotoxic concentration required for reducing MDCK cell viability by 50%.
Favipiravir.
The CPE reduction assay was performed to determine the antiviral activities of the synthesized compounds 15a–i, using bicyclic R3 initially (Fig. 1C and Table 1). Based on the results, 15b had similar activities to 15a, which suggests that replacement of the sulfur at X with oxygen still maintain the antiviral activity. However, the antiviral activity was abolished at 15c, indicating that the non-substituted hydrogen at R1 or the linked position of the indole substituent play a significant role for the activities. When the methyl group at R2 was replaced with a phenyl group (15d), the EC50 values increased two- to threefold compared with those of 15a. Removal of 5-fluorine in the indole group (15e) led to the complete loss of antiviral activity. Interestingly, when the indole group at R3 was substituted with benzo[b]thiophen-3-yl or 2-yl without fluoride to yield 15f or 15g, respectively, both compounds were active. It additionally shows that the linked position, either 2 or 3, of the bicyclic substituents might not be a critical factor for their antiviral activity. However, the derivatives with 3-methylbenzo[b]thiophen-2-yl at R3 (15h and 15i), had a severe loss of inhibitory function, indicating that introduction of a methyl group close to the linked position of R3 is unfavorable. Taken together, these data suggest that sulfur and oxygen are acceptable at X, whereas substitution of the hydrogen at R1 with a methyl group or the methyl group at R2 with a phenyl group is not desirable for maintaining the antiviral activity of 15a. Furthermore, an optimal bicyclic substituent appeared to be 5-fluoroindole or unsubstituted benzo[b]thiophene.
To explore the SARs of the R3 position further, a variety of compounds were prepared with monocyclic R3 substituents 15j–x (Fig. 1C and Table 2 ). The antiviral activities of 15j, which had a thiophen-3-yl substituent at R3, were improved threefold compared with those of 15a, with EC50 values of 5.0–6.2 μM and no toxicity in uninfected MDCK cells at 900 μM. 15j was three- to fivefold more active than its bicyclic cognate 15f, which indicates that monocyclic thiophene could be a more suitable substituent than bicyclic benzo[b]thiophene at R3. To evaluate the substitution effects in the X and R2 positions, while keeping R1 and R3 of 15j intact, 15k with an oxygen at X and 15l with a phenyl at R2 were prepared, respectively. 15k exhibited significant toxicity with a CC50 value of 19.8 μM and 15l had no antiviral activities except with the A/Hong Kong/8/68 strain. Replacement of the thiophene in 15j with a furan (15m) resulted in a three- to fivefold less active compound. 15n, an oxygen version of 15m at X, was also toxic (CC50, 61.9 μM) and inactive, as found with 15k. Other compounds with imidazoles (15o and 15p) at R3 showed no anti-influenza activity, even at 100 μM. 15q, a monocyclic R3 version of 15g, was comparable to 15j, and the EC50 values ranged from 6.8–7.5 μM against all three influenza viruses tested. In contrast to 15l, 15r, a phenyl R2 version of 15q, was still active with EC50 values between 15.1 and 31.7 μM, although less potent than 15q. To investigate whether modification of the thiophene ring in 15q contributed to the influenza inhibitory effect of the monocyclic compounds, an additional series of derivatives (15s–w) was synthesized and their antiviral activity was tested. Addition of a methyl group at position 3 (15s) or position 5 (15t) of thiophene yielded comparable antiviral activities to those of 15q. Notably, 15s, a monocyclic R3 version of 15h, was far more active than 15h. Substitution with an ethyl group at position 5 (15u) was active but had a reduced inhibitor effect than that of 15t. However, the addition of an electron-withdrawing bromide at position 4 (15v) or 5 (15w) of thiophene decreased or nullified the antiviral effect. Table 1 shows that methylation at R1 (15c and 15i) resulted in the loss of the antiviral activities of bicyclic substituents. By contrast, a methyl group (15x) at R1 of monocyclic substituents maintained the antiviral activities like R1-unmethylated 15s. Taken together, these results suggest that the anti-influenza viral activities of 15a can be maintained by substituting the bicyclic R3 with a monocyclic thiophene or furan ring, but not with an imidazole ring. The alkyl and bromide substitutions of the thiophene-2-yl at R3 indicated that the addition of electron-donating groups on thiophene-2-yl appeared to be preferable to electron-withdrawing groups for improving the antiviral activities. In contrast to the bicyclic compounds, the introduction of sulfur at the X position in monocyclic derivatives was more desirable than oxygen for reducing the toxicity, whereas the substitution of hydrogen with a methyl group at R1 did not affect the antiviral activities. Particularly, the seven active compounds, including 15a, 15g, 15j, 15q, 15s, 15t, and 15x, showed S.I. values over 50.0 (Table 1, Table 2). However, calculation of their exact S.I. was not available, because of DMSO-derived cytotoxicity at a concentration higher than 1000 μM (containing 2% DMSO).
Table 2.
Structure and anti-influenza virus activity of dihydrofuropyridinones with a monocyclic substituent at R3.
| Compound | Substituent |
EC50a (μM) for influenza virus (S.I.b) |
CC50c (μM) for MDCK cells | |||||
|---|---|---|---|---|---|---|---|---|
| X | R1 | R2 | R3 | A/Puerto Rico/8/34 | A/Hong Kong/8/68 | B/Lee/40 | ||
| 15j | S | H | Me | 5.0 ± 1.0 (>180.0) | 6.2 ± 3.1 (>145.2) | 5.9 ± 2.0 (>152.5) | >900.0 | |
| 15k | O | H | Me | >19.8 | >19.8 | >19.8 | 19.8 ± 8.2 | |
| 15l | S | H | Ph | >100.0 | 27.1 ± 5.8 | >100.0 | >900.0 | |
| 15m | S | H | Me | 21.4 ± 12.6 | 34.1 ± 10.0 | 16.3 ± 8.5 | >900.0 | |
| 15n | O | H | Me | >61.9 | >61.9 | >61.9 | 61.9 ± 10.7 | |
| 15o | S | H | Me | >100.0 | >100.0 | >100.0 | >900.0 | |
| 15p | S | H | Me | >100.0 | >100.0 | >100.0 | >900.0 | |
| 15q | S | H | Me | 7.5 ± 3.2 (>120.0) | 6.8 ± 1.3 (>132.4) | 7.0 ± 3.8 (>128.6) | >900.0 | |
| 15r | S | H | Ph | 28.9 ± 12.6 | 15.1 ± 3.8 | 31.7 ± 19.4 | >900.0 | |
| 15s | S | H | Me | 17.6 ± 11.0 (>51.1) | 12.7 ± 8.2 (>70.9) | 14.0 ± 7.4 (>64.3) | >900.0 | |
| 15t | S | H | Me | 8.0 ± 3.6 (>112.5) | 12.2 ± 2.9 (>73.8) | 9.6 ± 3.5 (>93.8) | >900.0 | |
| 15u | S | H | Me | 31.7 ± 21.7 | 32.6 ± 10.2 | 46.6 ± 28.6 | >900.0 | |
| 15v | S | H | Me | 63.7 ± 36.0 | 47.1 ± 18.7 | 71.8 ± 22.2 | >900.0 | |
| 15w | S | H | Me | >100.0 | >100.0 | >100.0 | >900.0 | |
| 15x | S | Me | Me | 12.1 ± 9.5 (>74.4) | 11.7 ± 2.7 (>76.9) | 8.0 ± 3.6 (>112.5) | >900.0 | |
| Amantadine hydrochloride | >100.0 | 2.8 ± 1.7 | >100.0 | >100.0 | ||||
| Oseltamivir carboxylate | 0.03 ± 0.01 | <0.01 | 1.3 ± 0.7 | >100.0 | ||||
| T-705d | 4.3 ± 2.4 | 6.5 ± 1.7 | 6.1 ± 0.6 | >100.0 | ||||
| Ribavirin | 18.8 ± 4.3 | 20.2 ± 2.4 | 23.7 ± 5.4 | >100.0 | ||||
Effective concentration required for reducing virus-induced cytopathicity by 50%.
Selectivity index, CC50/EC50.
Cytotoxic concentration required for reducing MDCK cell viability by 50%.
Favipiravir.
In addition, using three representative active compounds, 15a, 15g and 15q, MTT-based antiviral efficacy tests were performed against additional cell lysis-inducing viruses, such as HSV, FECV, CoxB, and HIV. However, no antiviral activity was detected at a maximum concentration of 100 μM against any of the viruses (data not shown), indicating that the antiviral activity mentioned above is influenza virus-specific.
3.2. Inhibition of influenza virus replication by dihydrofuropyridinones
Based on the results of the CPE reduction assay (Table 1, Table 2), the seven compounds, 15a, 15g, 15j, 15q, 15s, 15t, and 15x, were applied to additional antiviral assays. PR8-infected MDCK cells were incubated in the presence of increasing amounts of the test compounds, as well as the control compound T-705 or OSV-C. The HA titers in the culture supernatants were first measured at 12 h intervals until 48 h p.i. (Table 3 ). In the DMSO-treated control sample, the HA titer was not detected at 12 h p.i. (data not shown). However, it reached 64 and 128 HA units (HAU) per 50 μl culture supernatant at 24 and 36 h p.i., respectively, and showed its maximum of 256 HAU/50 μl at 48 h p.i. As expected, there was considerable reduction in the HA titers of the samples treated with 11 μM T-705 or OSV-C, which resulted in 8 HAU/50 μl or no detection, even at 48 h p.i. Among the test samples, the most potent inhibitors were 15a, 15g, and 15q, where each exhibited a dose-dependent inhibitory response during the whole detection period. The others, 15j, 15s, 15t, and 15s, also exhibited dose-dependent activity at an early time point, i.e., 24 h p.i., but they were unable to suppress viral replication thereafter at a concentration of ⩽33 μM.
Table 3.
HA titers of PR8-infected MDCK cell supernatants in the presence of the selected compounds.
| Compound | Concentration (μM) | HA unita |
||
|---|---|---|---|---|
| 24 hb | 36 h | 48 h | ||
| DMSO | –c | 64 | 128 | 256 |
| 15a | 11 | n.d.d | 64 | 128 |
| 33 | n.d. | 16 | 64 | |
| 100 | n.d. | n.d. | 8 | |
| 15g | 11 | 64 | 128 | 128 |
| 33 | n.d. | 32 | 128 | |
| 100 | n.d. | 4 | 16 | |
| 15j | 11 | 32 | 128 | 256 |
| 33 | n.d. | 128 | 256 | |
| 100 | n.d. | 4 | 64 | |
| 15q | 11 | 32 | 128 | 256 |
| 33 | n.d. | 64 | 128 | |
| 100 | n.d. | 4 | 32 | |
| 15s | 11 | 64 | 128 | 256 |
| 33 | 16 | 128 | 256 | |
| 100 | n.d. | 16 | 64 | |
| 15t | 11 | 32 | 128 | 256 |
| 33 | 16 | 128 | 256 | |
| 100 | n.d. | 64 | 256 | |
| 15x | 11 | 64 | 128 | 256 |
| 33 | 32 | 128 | 256 | |
| 100 | n.d. | 64 | 128 | |
| T-705 | 11 | n.d. | 2 | 8 |
| Oseltamivir carboxylate | 11 | n.d. | n.d. | n.d. |
HA units per 50 μl supernatants.
Hours post-infection.
0.2% DMSO treated.
Not detected.
The following study was design to address whether the three most active compounds, 15a, 15g, and 15q, could have inhibitory effect on infectious virus yield. Thus, the virus was titrated by plaque assays and quantitative RT-PCR analyses. Consistent with the results of the HA assay (Table 3), they significantly inhibited the levels of infectious viral titers (Fig. 2 A) and vRNA levels (Fig. 2B) dose-dependently. Using the PR8-infected MDCK cells, the effects of the antiviral compounds, 15a, 15g, 15j, and 15q, on the expression levels of viral proteins were additionally studied using Western blot analysis (Fig. 3 ). Overall, these results suggest that the antiviral compounds among the synthesized dihydrofuropyridinones have inhibitory effects on both infectious influenza virus production and the viral protein expression.
Fig. 2.
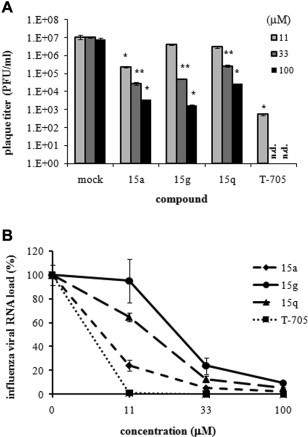
Reduction of influenza viral titers in the culture supernatants by the antiviral dihydrofuropyridinones. PR8-infected MDCK cells were incubated with different concentrations of 15a, 15g, 15q, or T-705 for 24 h. The supernatants were used for plaque titration (A) or for viral RNA quantification by quantitative RT-PCR (B). These experiments were performed twice in triplicate. P values were calculated against mock-treated, virus-infected samples. ∗P < 0.05; ∗∗P < 0.01. n.d., not detected.
Fig. 3.
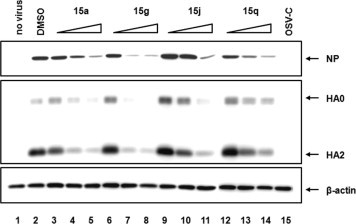
Reduction of the influenza viral protein level in cells by the active dihydrofuropyridinones. MDCK cells were mock-infected (lane 1) or infected with PR8 virus (lanes 2–15), and then incubated with either DMSO (lane 2), different concentrations (11, 33, and 100 μM) of 15a (lanes 3–5), 15g (lanes 6–8), 15j (lanes 9–11), or 15q (lanes 12–14), or 11 μM OSV-C (lane 15) for 24 h at 35 °C. The viral proteins, NP and HA, were detected using their specific primary antibodies and HRP-conjugated secondary antibodies. Cellular β-actin was used as a loading control. Each protein is indicated with an arrow at the right of the panel. The triangles at the top indicate an increase in the chemical concentration.
3.3. No effect of dihydrofuropyridinones on virus entry and RNA polymerase activity
Confocal microscopy was performed to determine whether the virus entry step is targeted by the antiviral dihydrofuropyridinones. After 2 h p.i. at 37 °C to allow viral adsorption and penetration, the intracellular localization of the viral NP protein was analyzed in the presence of 100 μM (−)-epigallocatechin-3-gallate (EGCG), an influenza virus entry inhibitor, or 15q (Fig. 4 A). This showed that EGCG reduced the overall NP signal intensity in the cells (Kim et al., 2013b). By contrast, NP distribution and intensity in cells was not affected by 15q, which was similar with that of the DMSO control. These results suggest that 15q has no effect on the virus entry. To further examine involvement of 15a and 15q in viral RNA replication, an influenza polymerase activity assay system was introduced, where an influenza virus-like RNA containing FLuc served as a reporter gene. The results clearly showed that the PR8-derived viral RNA polymerase function in Vero cells was not changed by 15a or 15q, whereas it was suppressed by the influenza viral polymerase inhibitor T-705 (Fig. 4B). All of these data suggest that the antiviral target of the dihydrofuropyridinone compounds probably could be a late step after viral RNA replication.
Fig. 4.
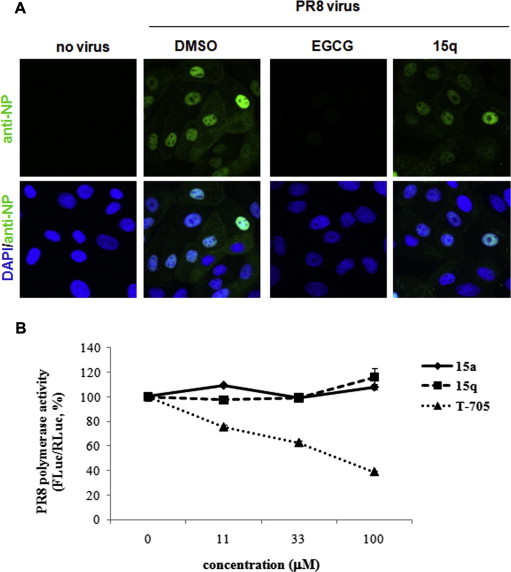
No effect of the antiviral dihydrofuropyridinones on influenza virus entry or polymerase activity. (A) Confocal microscopy. MDCK cells were infected with mock (no virus) or with PR8 at a multiplicity of infection (MOI) of 2.5 in the presence of DMSO, or 100 μM EGCG or 15q for 2 h at 37 °C. The viral NP protein was detected by confocal microscopy using an NP-specific monoclonal antibody and Alexa 488-conjugated goat anti-mouse secondary antibody (green). The nuclei were counterstained with DAPI (blue). Original magnification, 400×. (B) Vero cells were transfected with the four pVP DNAs, which express PR8 polymerase/NP proteins, pHH-FLuc, and phRL-CMV (as a transfection control). At 6 h, DMSO or increasing amounts of 15a, 15q, or T-705 (11, 33, and 100 μM) in DMEM with 5% FBS were added to each well. The FLuc expression levels in the cells were measured and normalized against that of RLuc on day 1. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Inhibition of influenza viral NA activity by dihydrofuropyridinones
To examine whether the antiviral dihydrofuropyridinones could affect the final step of virus progeny release from infected cells, the NA inhibitory activity of 15a and 15q was assessed using the whole influenza virion. Notably, they suppressed the PR8 NA activity with IC50 values of 0.6 ± 0.1 μM and 1.5 ± 0.4 μM, respectively, whereas 15i, which was inactive in the CPE reduction assay (Table 1), did not (Fig. 5 A). This finding also appeared against other viral strains, A/Hong Kong/8/68 and B/Lee/40 (data not shown). In addition, based on a previous report (Vavricka et al., 2011), in silico molecular docking analysis was performed by using 15a as a ligand for the PR8 NA protein. Conformationally, it fitted with the active site of the NA protein via interaction with amino acids R118, E222, R292 and R371 (Fig. 5B). The indole part of 15a formed cation-π interactions with R292 and R371. And fluorine of the indole in 15a was found to be involved in a weak hydrogen bond with R118, explaining the reason for the complete loss of activities of 15e (Table 1). Notably, hydrogen at the R1 position of the core skeleton made a hydrogen bond with E227. In accordance with this observation, 15g, which has a very similar structure and antiviral activity to those of 15a, was successfully loaded at the active site of NA (data not shown). However, inactive 15i, which was methylated compounds at this position, failed to be stably loaded. It was further confirmed that 15a suppressed the function of the oseltamivir-resistant NA protein [NA (H275Y)] as efficiently as that of the wild-type NA derived from influenza H1N1 (A/California/04/2009) (Fig. 5C and D). Taken together, these data suggest that the antiviral activity of dihydrofuropyridinones is expressed by their occupying the active site within NA and thereby blocking the enzymatic activity.
Fig. 5.
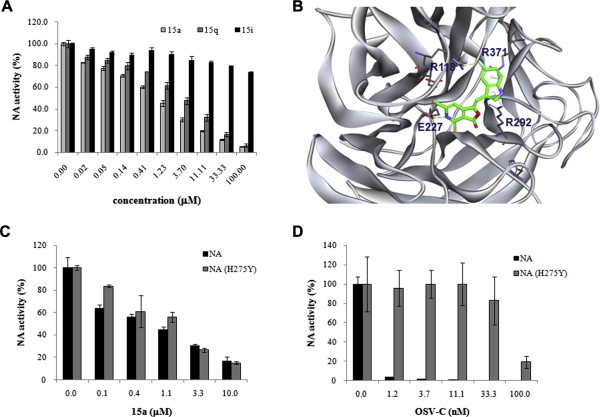
Inhibition of influenza virus NA activity by 15a and 15q. (A) NA activity assay. The enzymatic activity of the PR8 NA protein was measured in the presence of increasing amounts of 15a, 15q, or 15i using a chemiluminescent NA substrate. The data shown are the means ± S.D. for two samples from three independent experiments. (B) Docking analysis of 15a with the PR8 NA protein. The dashed lines represent interactions between the ligand and the target protein. The NA inhibitory molecule 15a is shown as a stick model, and the atom color-coding is as follows: carbon, green; nitrogen, blue; oxygen, red; sulfur, yellow; and fluorine, cyan. Inhibition assay of the purified wild-type and H275Y mutant NA proteins by 15a (C) and OSV-C (D). These proteins are derived from influenza H1N1 (A/California/04/2009) and its OSV-resistant H275Y mutant [NA (H275Y)]. Each experiment was performed independently twice in duplicate. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In the present study, the screening of a chemical library of small organic compounds deposited by KRICT chemists identified a dihydrofuropyridinone compound, 15a, which inhibited influenza A and B virus replication in cells, with EC50 values of about 20 μM and a CC50 of >900 μM (Fig. 1, and Table 1, Table 2). The chemical synthesis of its derivatives facilitated an investigation of its SARs and improved the antiviral efficacy. As a result, not only 15a, but also 15g, 15j, 15q, 15s, 15t, and 15x were found to be active. Using additional antiviral assays, which measured the titers of HA, infectious viral particles or viral RNA in the culture supernatants as well as the viral protein levels in the cell lysates, it was ensured that they could suppress production of infectious influenza viruses (Table 3, and Fig. 2, Fig. 3).
Previously, relationships between structures of dihydrofuropyridinones, such as 15e, 15f, 15g, 15j, 15m, 15q and 15t, and their inhibitory effect on cell lysis induced by the pore-forming glycoprotein perforin were investigated (Lena et al., 2008). However, there was no consensus in the chemical structures between their abilities for inhibiting the perforin-induced cytolytic effect and for suppressing influenza virus replication. For example, 15f, 15g, 15q, and 15t were active, whereas 15e was inactive in both studies. In contrast, 15j and 15m exhibited antiviral activities in the present study, but none in the previous study (Lena et al., 2008). To exclude the possibility that the anti-influenza activity of the dihydrofuropyridinones was gained from their intrinsic property improving cell viability nonspecifically, we repeated the MTT-based antiviral assay against other cell lysis-inducing viruses such as HSV, FECV, CoxB, and HIV. However, the anti-influenza viral compounds 15a, 15g, and 15q failed to show antiviral function against them (data not shown), indicating that their antiviral activity is influenza virus-specific. Furthermore, our mode-of-action study and in silico docking analysis with some active compounds suggested that their antiviral effects may occur at a late stage, particularly the NA-mediated virus release step, but not at any step of the virus entry or the viral RNA replication (Fig. 4, Fig. 5).
In conclusion, the results presented in the study suggest that the dihydrofuropyridinones, 15a, 15g, 15j, 15q, 15s, 15t, and 15x, have significant anti-influenza viral activities in cells and that their main mode-of-action is the inhibition of viral NA activity. Thus, it is anticipated that the compounds will provide initial chemical structures for the development of non-sialic acid-based NA inhibitors and facilitate the design of a novel anti-influenza drug.
Acknowledgments
We would like to thank the staff at the Korea Chemical Bank of KRICT for supplying chemicals and for characterizing their purity and identity. The authors greatly appreciate the constructive comments of Prof. Erik De Clercq. This work was supported by the Transgovernmental Enterprise for Pandemic Influenza in Korea (TEPIK) (Grant A103001).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2014.04.013.
Contributor Information
Pilho Kim, Email: pkim@krict.re.kr.
Meehyein Kim, Email: mkim@krict.re.kr.
Appendix A. Supplementary data
References
- Baranovich T., Saito R., Suzuki Y., Zaraket H., Dapat C., Caperig-Dapat I., Oguma T., Shabana I.I., Saito T., Suzuki H. Emergence of H274Y oseltamivir-resistant A(H1N1) influenza viruses in Japan during the 2008–2009 season. J. Clin. Virol. 2010;47:23–28. doi: 10.1016/j.jcv.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Bedoya L.M., Abad M.J., Calonge E., Saavedra L.A., Gutierrez C.M., Kouznetsov V.V., Alcami J., Bermejo P. Quinoline-based compounds as modulators of HIV transcription through NF-kappaB and Sp1 inhibition. Antiviral Res. 2010;87:338–344. doi: 10.1016/j.antiviral.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Belshe R.B., Burk B., Newman F., Cerruti R.L., Sim I.S. Resistance of influenza A virus to amantadine and rimantadine: results of one decade of surveillance. J. Infect. Dis. 1989;159:430–435. doi: 10.1093/infdis/159.3.430. [DOI] [PubMed] [Google Scholar]
- CDC, 2013. Influenza Antiviral Drug Resistance. Available from <http://www.cdc.gov/flu/about/qa/antiviralresistance.htm>.
- Cho H.G., Choi J.H., Kim W.H., Hong H.K., Yoon M.H., Jho E.H., Kang C., Lim Y.H. High prevalence of amantadine-resistant influenza A virus isolated in Gyeonggi Province, South Korea, during 2005–2010. Arch. Virol. 2013;158:241–245. doi: 10.1007/s00705-012-1482-9. [DOI] [PubMed] [Google Scholar]
- Choi W.Y., Yang I., Kim S., Lee N., Kwon M., Lee J.Y., Kang C. The emergence of oseltamivir-resistant seasonal influenza A (H1N1) virus in Korea during the 2008–2009 season. Osong Public Health Res. Perspect. 2011;2:178–185. doi: 10.1016/j.phrp.2011.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K. Antivirals targeting influenza A virus. J. Med. Chem. 2012;55:6263–6277. doi: 10.1021/jm300455c. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Logu A., Loy G., Pellerano M.L., Bonsignore L., Schivo M.L. Inactivation of HSV-1 and HSV-2 and prevention of cell-to-cell virus spread by Santolina insularis essential oil. Antiviral Res. 2000;48:177–185. doi: 10.1016/s0166-3542(00)00127-3. [DOI] [PubMed] [Google Scholar]
- Fujii K., Fujii Y., Noda T., Muramoto Y., Watanabe T., Takada A., Goto H., Horimoto T., Kawaoka Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005;79:3766–3774. doi: 10.1128/JVI.79.6.3766-3774.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta, Y., Egawa, H., Takahashi, K., Tsutsui, Y., Uehara, S., Murakami, M., 2004. Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue. In: Ltd., T.C.C. (Ed.), International Application No. PCT/JP2002/008250.
- Hoffmann E., Neumann G., Kawaoka Y., Hobom G., Webster R.G. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E., Stech J., Guan Y., Webster R.G., Perez D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001;146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- Kao R.Y., Yang D., Lau L.S., Tsui W.H., Hu L., Dai J., Chan M.P., Chan C.M., Wang P., Zheng B.J., Sun J., Huang J.D., Madar J., Chen G., Chen H., Guan Y., Yuen K.Y. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 2010;28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Yim J.H., Kim S.Y., Kim H.S., Lee W.G., Kim S.J., Kang P.S., Lee C.K. In vitro inhibition of influenza A virus infection by marine microalga-derived sulfated polysaccharide p-KG03. Antiviral Res. 2012;93:253–259. doi: 10.1016/j.antiviral.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Resende R., Wennekes T., Chen H.M., Bance N., Buchini S., Watts A.G., Pilling P., Streltsov V.A., Petric M., Liggins R., Barrett S., McKimm-Breschkin J.L., Niikura M., Withers S.G. Mechanism-based covalent neuraminidase inhibitors with broad-spectrum influenza antiviral activity. Science. 2013;340:71–75. doi: 10.1126/science.1232552. [DOI] [PubMed] [Google Scholar]
- Kim M., Kim S.Y., Lee H.W., Shin J.S., Kim P., Jung Y.S., Jeong H.S., Hyun J.K., Lee C.K. Inhibition of influenza virus internalization by (-)-epigallocatechin-3-gallate. Antiviral Res. 2013;100:460–472. doi: 10.1016/j.antiviral.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Leang S.K., Deng Y.M., Shaw R., Caldwell N., Iannello P., Komadina N., Buchy P., Chittaganpitch M., Dwyer D.E., Fagan P., Gourinat A.C., Hammill F., Horwood P.F., Huang Q.S., Ip P.K., Jennings L., Kesson A., Kok T., Kool J.L., Levy A., Lin C., Lindsay K., Osman O., Papadakis G., Rahnamal F., Rawlinson W., Redden C., Ridgway J., Sam I.C., Svobodova S., Tandoc A., Wickramasinghe G., Williamson J., Wilson N., Yusof M.A., Kelso A., Barr I.G., Hurt A.C. Influenza antiviral resistance in the Asia-Pacific region during 2011. Antiviral Res. 2013;97:206–210. doi: 10.1016/j.antiviral.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Lena G., Trapani J.A., Sutton V.R., Ciccone A., Browne K.A., Smyth M.J., Denny W.A., Spicer J.A. Dihydrofuro[3,4-c]pyridinones as inhibitors of the cytolytic effects of the pore-forming glycoprotein perforin. J. Med. Chem. 2008;51:7614–7624. doi: 10.1021/jm801063n. [DOI] [PubMed] [Google Scholar]
- Liu I.J., Tsai W.T., Hsieh L.E., Chueh L.L. Peptides corresponding to the predicted heptad repeat 2 domain of the feline coronavirus spike protein are potent inhibitors of viral infection. PLoS One. 2013;8:e82081. doi: 10.1371/journal.pone.0082081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpani Y., Achary R., Kim S.Y., Jeong H.C., Kim P., Han S.B., Kim M., Lee C.K., Kim J.N., Jung Y.S. Efficient synthesis of 3H,3’H-spiro[benzofuran-2,1′-isobenzofuran]-3,3′-dione as novel skeletons specifically for influenza virus type B inhibition. Eur. J. Med. Chem. 2013;62:534–544. doi: 10.1016/j.ejmech.2013.01.015. [DOI] [PubMed] [Google Scholar]
- Miller P.E., Rambachan A., Hubbard R.J., Li J., Meyer A.E., Stephens P., Mounts A.W., Rolfes M.A., Penn C.R. Supply of neuraminidase inhibitors related to reduced influenza A (H1N1) mortality during the 2009–2010 H1N1 pandemic: summary of an ecological study. Influenza Other Respir. Viruses. 2013;7(Suppl 2):82–86. doi: 10.1111/irv.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh W., Mahaman L., Healey B., Peters H., Anderson B., Tran M., Ziese M., Carlos M.P. Evaluation of three influenza neuraminidase inhibition assays for use in a public health laboratory setting during the 2011–2012 influenza season. Public Health Rep. 2013;128(Suppl. 2):75–87. doi: 10.1177/00333549131280S212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L.H., Holsinger L.J., Lamb R.A. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- Pleschka S., Jaskunas R., Engelhardt O.G., Zurcher T., Palese P., Garcia-Sastre A. A plasmid-based reverse genetics system for influenza A virus. J. Virol. 1996;70:4188–4192. doi: 10.1128/jvi.70.6.4188-4192.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R., Sakai T., Sato I., Sano Y., Oshitani H., Sato M., Suzuki H. Frequency of amantadine-resistant influenza A viruses during two seasons featuring cocirculation of H1N1 and H3N2. J. Clin. Microbiol. 2003;41:2164–2165. doi: 10.1128/JCM.41.5.2164-2165.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson M., Pizzorno A., Abed Y., Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res. 2013;98:174–185. doi: 10.1016/j.antiviral.2013.03.014. [DOI] [PubMed] [Google Scholar]
- Vavricka C.J., Li Q., Wu Y., Qi J., Wang M., Liu Y., Gao F., Liu J., Feng E., He J., Wang J., Liu H., Jiang H., Gao G.F. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog. 2011;7:e1002249. doi: 10.1371/journal.ppat.1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam C.M., Jiang X., Oldfield T., Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003;21:289–307. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 2007;6:967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- Zhong Q., Yang Z., Liu Y., Deng H., Xiao H., Shi L., He J. Antiviral activity of Arbidol against Coxsackie virus B5 in vitro and in vivo. Arch. Virol. 2009;154:601–607. doi: 10.1007/s00705-009-0346-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.