

Highlights
-
•
This article summarizes the principal invited lectures at the 28th International Conference on Antiviral Research.
-
•
Phillip Furman, Elion award recipient, described research leading to the discovery of sofosbuvir.
-
•
Dennis Liotta, Holý award recipient, described how an investigation into HIV entry inhibitors led to a new cancer therapy.
-
•
Erica Ollmann Saphire, Prusoff award recipient, explored how viral proteins remodel for different roles in replication.
-
•
Keynote talks focused on the treatment of HCV infection and on the role of Médicins Sans Frontières in the Ebola epidemic.
Keywords: Sofosbuvir, Hepatitis C virus, Ebola virus, RNA viruses, Emerging viruses, Antiviral chemistry
Abstract
The 28th International Conference on Antiviral Research (ICAR) was held in Rome, Italy from May 11 to 15, 2015. This article summarizes the principal invited lectures. Phillip Furman, the Elion award recipient, described the research leading to sofosbuvir. Dennis Liotta, who received the Holý award, described how an investigation into HIV entry inhibitors led to a new therapy for cancer patients. Erica Ollmann Saphire, winner of the Prusoff Young Investigator award, explored the world of viral proteins and how they remodel to perform different essential roles in viral replication. The keynote addresses, by Raffaele De Francesco and Michael Manns, reported on the remarkable progress made in the therapy of chronic HCV infections. A third keynote address, by Armand Sprecher, related the difficulties and successes of Médicins Sans Frontières in West Africa ravaged by the Ebola outbreak. There were three mini-symposia on RNA Viruses, Antiviral Chemistry and Emerging Viruses. There was a good collection of talks on RNA viruses (norovirus, rabies, dengue, HEV, HCV, and RSV). A highlight of the chemistry was the preparation of prodrugs for nucleotide triphosphates as this opens a door to new options. The third mini-symposium emphasized how research work in the antiviral area is continuing to expand and needs to do so with a sense of urgency. Although this meeting report covers only a few of the presentations, it aims to illustrate the great diversity of topics discussed at ICAR, bringing together knowledge and expertise from the whole spectrum of antiviral research.
1. Introduction
This article provides an overview of the invited lectures at the 28th International Conference on Antiviral Research (ICAR), sponsored by the International Society for Antiviral Research (ISAR), which was held in Rome, Italy, from May 11–15, 2015. It begins with reports of lectures by the recipients of ISAR’s three major awards, held in memory of Gertrude (Trudy) Elion, Antonín (Tony) Holý and William (Bill) Prusoff. These are followed by summaries of the three keynote addresses and the main presentations within the three mini-symposia on “RNA Viruses”, “Antiviral Chemistry” and “Emerging Viruses”. Because this review article simply provides short accounts of oral presentations, it is not generally accompanied by references to the scientific literature. Any descriptions of favorable treatment outcomes should not be taken as a recommendation for clinical use. Generally, I have added my personal comments on the meeting within the conclusion. In a few instances, I have added my own comment within the main text, indicated either by the wording or by the use of square brackets. One of my aims has been to illustrate how the great diversity of topics can stimulate thinking in other areas of antiviral research, one of the strengths of ICAR.
2. Gertrude Elion Memorial award lecture: sofosbuvir: a search for a cure. Phillip (Phil) Furman., Furman Biotech Consulting, St Augustine, FL, USA.
Having joined Burroughs Wellcome in 1975, Phil (Fig. 1 ) worked with Trudy Elion for ten years. During this time, he was involved in the development of acyclovir (Zovirax®) and its prodrug, valacyclovir (Valtrex®). In 2004, Phil joined Pharmasset. The focus of this presentation was his research at Pharmasset, leading to the identification of the activity of sofosbuvir and to an understanding of its mechanism of action against hepatitis C virus (HCV).
Fig. 1.

Bob Buckheit congratulates Phil Furman on receiving the Elion award.
Phil’s account started with the cytidine analog, PSI-6130 (Fig. 2 ). This was one of the more active compounds in development at that time, better than the Roche and Idenix nucleosides but possibly a little less active than the Merck compound. An additional important factor, PSI-6130 lacked detectable cytotoxicity (CC50 > 100 μM) in a panel of 5 cell lines (Clone A, Huh7, HepG2, CEM and PBM). Incidentally, the Merck compound showed no cytotoxicity in 3 cell lines but had a CC50 of 5 μM in CEM cells and therefore was rejected. In contrast to many reported cytotoxicity values, these are derived from assays in which the cells are replicating. It is important to compare the effect of a compound against both replicating virus and replicating cells in order to obtain valid therapeutic ratios.
Fig. 2.
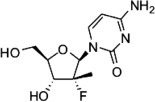
Structure of PSI-6130, 2′-α-F, 2′-C-methylcytidine.
PSI-6130 is metabolized in cells to the corresponding triphosphate (PSI-6130-TP) which was a good inhibitor of the HCV polymerase, NS5B, (Inhibition Constant, K i = 0.06 μM). However, PSI-6130 was poorly absorbed orally and there was significant conversion to the inactive uridine analog. A prodrug approach, in collaboration with Roche, resulted in RG-7128 which increased the bioavailability of PSI-6130 by 5-fold but it was not an approach which could enhance the short half-life of PSI-6130-TP (T 1/2 = 5 h). It was considered highly desirable to have a triphosphate analog with a sufficiently long half-life to enable once-a-day dosing.
While studying the metabolism of PSI-6130 in cells, it was noted that some of the monophosphate, PSI-6130-MP, was converted to the corresponding uridine-MP which was further metabolized to the uridine-TP (PSI-6206-TP). This uridine-TP analog was a less active inhibitor of the HCV polymerase than the cytidine-TP analog (K i = 0.42 μM and 0.06 μM respectively) but had a much longer half-life (T 1/2 = 38 h and 5 h respectively). The long half-life of 38 h stimulated the interest of the research team. Because the uridine analog (PSI-6206) was inactive due to lack of metabolism to PSI-6206-MP, it was decided to use the phosphoramidate prodrug strategy to deliver the PSI-6206-MP into cells. [Phosphoramidate prodrugs were first introduced by Chris McGuigan, a former President of ISAR.] Although the approach is well known, there are side chains which can influence the properties of the prodrug. Over 140 phosphoramidate prodrugs of PSI-6206 were synthesized. These were evaluated in a cascade of tests which are of particular interest.
Every successful compound has to have two basic properties, a useful efficacy and a good safety profile. The cascade started with an assay comparing the compound in an HCV replicon system vs. a ribosomal-RNA (r-RNA) cytotoxicity test. The next steps were cytotoxicity evaluations in an extensive panel of cell lines, mitochondrial toxicity and bone marrow toxicity. [In my experience, it is rare to find a compound-screening strategy giving so much emphasis to the safety profile.] Only then, the compounds were tested for triphosphate levels, pharmacokinetic studies in rats and dogs and in vivo rat toxicity tests. Just 7 prodrugs were selected for cell culture triphosphate analysis. Of these, PSI-7851 gave the best levels of PSI-6206-TP in human cells. Three prodrugs were then tested in vivo, initially in a rat pharmacokinetic test. Of these, PSI-7851 gave the highest levels of PSI-6206-TP in liver, measured either by the maximum concentration (C max) or by area-under-the-curve (AUC). Similarly, PSI-7851 was the best compound in dogs and monkeys.
The activity of PSI-7851 was evaluated in a range of HCV genotypes (1a, 1b and 2a) using replicon cells. These genotypes were all sensitive to PSI-7851, the EC90 values being within 2-fold. Also, PSI-7851 was active against genotypes 1a and 1b in an infectious virus assay. It showed no activity against a panel of other DNA and RNA viruses. Recombinant NS5B polymerases from HCV genotypes 1b, 2a, 3a and 4a were all inhibited by PSI-6206-TP (PSI-7409) (IC50 values 1.6, 2.8, 0.7 and 2.6 μM respectively). This result suggested that PSI-7851 would be active against genotypes 3 and 4 for which replicon assays were not available. In combination with interferon or RBV, there was additive activity with PSI-7851. Based on this evaluation scheme, PSI-7851 was selected as the candidate for development.
However, PSI-7851 is a mixture of two diastereomers. In anticipation of questions from the FDA, the group decided to separate the isomers. The Rp isomer (PSI-7976) was then found to be less active than the Sp isomer (PSI-7977) (EC50 = 1.1 and 0.092 μM respectively). PSI-7977 became sofosbuvir (Fig. 3 ).
Fig. 3.
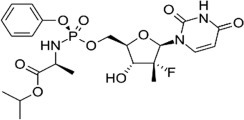
Structure of sofosbuvir (PSI-7977).
Sofosbuvir was approved by the FDA in December 2013 and by the EU in January 2014. Later in 2014, Harvoni, the first once a day fixed-dose combination therapy for HCV, was approved. This is a combination of sofosbuvir and ledipasvir, a HCV NS5A inhibitor. As of September 2014, it had been estimated that, in the USA, more than 100,000 patients had received sofosbuvir as part of their treatment and had been cured of their HCV infection. For patients with HCV, sofosbuvir has been a “game-changer”.
3. The Antonín Holý Memorial award lecture: novel therapeutics for treating viral diseases, cancers and inflammatory disorders. Dennis C Liotta, Emory University, Atlanta, GA, USA.
In memory of the late Tony Holý, a new ISAR award in medicinal chemistry was announced at the 2013 ICAR. Piet Herdewijn, the first to receive this award, gave a fascinating talk entitled “From modified nucleoside to a chemically modified genome” at the 2014 ICAR. Dennis is, therefore, only the 2nd person to receive this award (Fig. 4 ). As the title of his lecture suggests, Dennis gave us a wonderful account of how chemistry led him into a new and totally unexpected direction.
Fig. 4.

Bob Buckheit presenting the Antonín Holý award to Dennis Liotta.
Currently, there are many excellent drugs for HIV therapy but only two approved drugs target the attachment and entry of HIV into cells – maraviroc (Pfizer) and enfuvirtide (Roche). Of these, maraviroc acts as an antagonist to the cellular receptor, CCR5. However, HIV uses not only the CCR5 receptor (M-tropic virus) but also the CXCR4 receptor (T-tropic virus). The majority of patients, newly diagnosed with HIV infection, have only M-tropic HIV. In one study, just 5 of 59 (9%) had T-tropic virus, initially. In untreated patients, T-tropic virus will emerge in about half the patients, the average time being about 5 years. Could maraviroc therapy favor the selection of T-tropic HIV? This has important clinical implications because the T-tropic virus damages the immune system and thereby significantly (p < 0.001) increases the rate of progression to AIDS.
There are no approved CXCR4 antagonists but clinical trials with AMD3100 and AMD11070 did validate the concept. Development was discontinued after Phase I and Phase II trials, respectively. For a long time now, no candidates have had a good enough safety profile for progression as a therapy for HIV but there is potential as cancer chemosensitizers. On the outside of cells, CXCL12 binds to CXCR4. The chemosensitizer releases the CXCL12 from CXCR4, thus enabling immune cells to destroy the cancer cells. AMD3100 is in Phase I/II trials to enhance the effects of chemotherapy in acute myeloid leukemia (AML). To obtain similar compounds, an efficient general synthetic scheme was developed using readily available amino acid building blocks. To evaluate these compounds, several cell-based assays were established, including a 2-day attachment assay (CXCR4 HIV-1 MAGI), a 6-day infection assay (peripheral blood mononuclear cells [PBMC] and T-tropic HIV). To initiate the signaling pathway via CXCR4 receptor, CXCL12 (also known as SDF-1) binds to the receptor. To characterize the activity of any compound found to be active in the above assays, a radio-ligand displacement assay (125I-labeled SDF-1 in CEM cells) was devised. TIQ-15 was active in both the MAGI and SDF-1 assays and it had good “drug-like” properties but needs optimization.
A further aim became apparent – the potential to target both CXCR4 and CCR5. There is considerable sequence homology (65% similarity of which 33% is identical). The crystal structures have similar binding pockets. In the literature, AMD3451 had dual activity. Starting with the Aldrich database (5 million compounds) and using virtual screening for binding to both CCR5 and CXCR4, the top 300 compounds were identified. Of these, 38 compounds were purchased and tested (MAGI) to yield 13 compounds with activity at 100 μM, 3 of these active at 10 μM. One of these, “compound 2” had good activity in the MAGI (CCR5) and MAGI (CXCR4) assays (IC50 = 3.8 and 0.8 μM respectively). It was a surprise to find that compound 2 was also active as a non-nucleoside reverse transcriptase inhibitor (NNRTI) of HIV. The general synthetic route to similar compounds was shown. In three weeks, more than 50 compounds were synthesized. Having three parameters (binding to receptors CCR5, CXCR4 and NNRTI) would make structure–activity-relationships (SAR) studies challenging. It was decided to focus on the binding to the two receptors. From these, “compound 3” (Fig. 5 ) was selected (IC50 = 0.21 and 0.18 μM respectively). As an NNRTI, it was active at 10 μM. Interestingly, in the 125I-SDF-1 displacement assay, there was no effect even at 1 mM. May it be an advantage to have a compound able to inhibit the binding of HIV to the receptor but be unable to displace SDF-1?
Fig. 5.
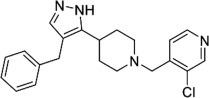
Structure of “compound 3”.
Another research approach has been to synthesize compounds containing the linking part of AMD-3100 (Table 1 ). This work led to Q-122 (MSX-122) with several major advantages. It does displace another compound (TN-14003) bound to CXCR4 but does not displace 125I-SDF-1. The crystal structure of TN-14003 bound to CXCR4 is known. Q-122 (MSX0122) showed activity in two cancer models using mice.
Table 1.
Synthesis of compounds containing the linking part of AMD-3100 led to Q-122 (MSX-122), which has several major advantages.
| AMD-3100 | Q-122 (MSX-122) |
 |
 |
| 100 nM potency⁎ 10-Step synthesis Cardiotoxicity Requires injection |
1 nM potency⁎ 1-Step synthesis No apparent toxicity Orally available |
Activity against metastatic 686 NL cancer cells (a model for head and neck cancer).
In pre-clinical testing, no toxicity was seen with doses up to 600 mg/kg in dogs or monkeys. Two Phase Ia trials have been completed – a single ascending dose study (up to 400 mg) in healthy volunteers and 56 daily doses in late stage, solid tumor patients. No toxicity was noted in these trials. In the latter trial, a woman, who had fallopian tube cancer, had a complete remission of her hot flashes during the dosing period. Therefore, a Phase Ib study was done in women with breast cancer taking tamoxifen. Such patients can be on drug for 10 years and most (up to 80%) are subject to hot flashes. The approved products for post-menopausal hot flashes are contra-indicated in cancer patients. In both dose groups (100 and 200 mg), Q-122 showed reductions in hot flashes. Encouragingly, some subjects had a complete response (going from >50 hot flashes/week to zero). Alongside the response in the primary end-point, there were improvements in various secondary end-points. The drug had a good safety profile. The mechanism for this effect is unknown.
It seems surprising that an antiviral research program can be the source of the first potential therapy to treat hot flashes in the large group of women on tamoxifen therapy. Who could have predicted that this would be the outcome?
4. The William Prusoff Young Investigator award lecture: remodel, repurpose, rearrange; how viruses leverage the few proteins they encode. Erica Ollmann Saphire, The Scripps Research Institute, La Jolla, CA, USA.
Erica started her lecture with a photograph of Bill Prusoff, noting that he has been called “Father of antiviral chemotherapy”. She felt honored to give a lecture in his name (Fig. 6 ).
Fig. 6.

Bob Buckheit presenting the Prusoff Young Investigator award to Erica Ollmann Saphire.
Introduction
Erica described how she was intrigued by viruses – they encode so few proteins yet get so much function from them. For example, filoviruses, like Ebola, encode 8 proteins and arenaviruses, such as Lassa, encode just 4 proteins. Yet these proteins perform about 60 functions within the life cycle of the virus. Another example, the filovirus matrix protein (VP40) is responsible for assembly, budding, transcriptional control and, sometimes, immunosuppression; how is this done? Erica suggests that one answer is that these viral proteins are able to remodel, repurpose and rearrange themselves to extend their functional complexity.
Remodel
Erica showed us the structures of the surface glycoprotein (GP) of Ebola and Marburg viruses, first just the protein and then as the fully glycosylated form as it exists on the viral surface. The extensive coating of sugar residues hides part of the protein from immune surveillance. This complete, fully glycosylated form does not bind to the receptor. The sugar-linked residues are cleaved off to leave a “naked” protein and the receptor binding site becomes exposed. We know that Ebola survivors have effective antibodies but what can be protective when so many protein sites are hidden?
Human antibody (KZ52) was obtained from an Ebola survivor (1995). This antibody works in the test tube, in mice and in guinea pigs but does not protect monkeys. Up to 2007, KZ52 was the best antibody against Ebola; was there still a way forward? During 2011–2012, several groups showed that combinations of antibodies are protective in monkeys. For example, MB-003 from the United States Army Medical Research Institute of Infectious Diseases (USAMRIID) is a cocktail of three antibodies, two binding to sites on the mucin domain, the third to the GP “glycan cap” which is cleaved off in the endosome. Although this cocktail does not work well in vitro, it does protect monkeys. But how is it that KZ52 works well in vitro but not in monkeys whereas MB-003 works in monkeys but not in vitro? There are many unanswered questions; how many antibodies? Which are best? What assay? Can there be synergy? To resolve such a complex set of unknowns, a large sample set is required.
The Viral Hemorrhagic Fever Immunotherapeutic Consortium (VHFIC) is a global collaboration to evaluate all available monoclonal antibodies (mAbs) to Ebola, with the eventual aim to repeat this later with other viruses. Erica credited John Dye as the major player in bringing all the interested parties together to form the VHFIC. Initially, the “tortoise” path was set up. Samples of all the antibodies were sent to Erica’s laboratory for coding and then sent to all the collaborators to test in their various assays and to determine the binding sites. Initial results suggested that antibodies binding to the “top” of the Ebola GP did not give good neutralization in vitro but were protective whereas antibodies binding to the lower part gave good neutralization and may be protective.
In parallel with the “tortoise” track, a “hare” track was initiated using 6 antibodies from two current combinations. It was envisaged that a focus on structure determination and in vivo efficacy could lead quickly to a reasonably efficacious product. As it turned out, this approach was fortuitous because the case zero of the current Ebola outbreak was in December 2013 and ZMapp, which emerged from the “hare” track, was discovered about February 2014. In August 2014, ZMapp was used on a compassionate basis to treat health care workers repatriated to the USA. ZMapp consists of three antibodies, 13C6 (binds to the top of the GP and recruits the immune system), 2G4 (binds to the base of GP) and 4G7 (also binds to base of GP). As the latter two antibodies are similar to KZ52, they enable the combination to be active in the neutralization test but also they contribute to the protection which is given by the 13C6 antibody. Are antibodies, that bind to the mucin, necessary for protection?
After the first 7 months of the “tortoise” approach (and 81 mAbs), much has been learnt. There are mAbs which bind to various parts of the Ebola GP (number of mAbs): GP1 core (15), fusion loop (2), base of GP2 (8), glycan cap (17), mucin (16) and 23 mAbs are still undefined. USAMRIID carried out the protection test in mice, ranking mAbs on % survival of 10 mice. Those mAbs binding to the base of GP2 gave the better protection (50–100%). These are similar to KZ52 and to 2 of 3 mAbs in ZMapp. There were other mAbs, giving about 50% protection in mice, which could be considered for inclusion in a combination. Those mAbs, which bound to the GP1 top, GP1 core, glycan cap or mucin, gave protection but no strong neutralization. Those mAbs, which bound to the base of GP, gave both protection and neutralization but there was only a rough correlation. The explanation is that the GP is remodeled, the mucin and glycan cap are removed and therefore the core protein is not neutralized.
Repurpose
As an example of an arenavirus nucleoprotein (NP), Erica chose Lassa virus. Erica compared the NP to a multifunction penknife – having roles in nucleocapsid assembly, immunosuppression, virion incorporation and regulation of polymerase. The N-terminal and C-terminal domains open up to reveal a new site predicted to be the RNA-binding site. But the RNA gate, on the revealed surface of the N-terminal domain, has to open before ssRNA can bind to it. In addition, the C-terminal domain has a role in immunosuppression. It is folded so that it becomes an exonuclease able to digest dsRNA. How these functions are balanced is still unknown.
Rearrange
In Ebola and Marburg viruses, the VP40 protein alone can self-assemble to form virus-like particles. It also controls transcription. In 2000, the structure of the monomer was determined. In 2003, an octameric ring structure was discovered. Eight N-terminal domains bound together to form the ring with the C-terminal domains in the center of the ring. In 2013, SEC-MALS (a light scattering device used to determine molecular weight of proteins) proved that VP40 can exist as a dimer. Using mutants, it was found that the butterfly-shaped dimer, but not the octameric ring, was essential for virus assembly. The dimers can further assemble and rearrange to form zigzag-shaped hexamers which lead to virus assembly. However, although octameric rings are not essential for virus assembly, mutants which prevent octameric ring formation are nonetheless lethal for virus replication. The ring controls transcription. In conclusion, VP40 can form dimers for trafficking about the cell, octameric rings to control transcription, and hexamers for virion assembly.
Our understanding of molecular biology is evolving
The traditional view is that a gene encodes the protein which folds so that the protein is able to perform one function. Working with Ebola virus has taught us that, although current computer programs follow the traditional view, nature uses one protein folded in different ways to perform different functions. With VP40, we see one gene encodes one protein which can fold and assemble into three distinct forms, each able to perform its own function. Erica likened VP40 to a “Transformer”, a child’s toy which can be a robot or refolded to become a truck – one form can make RNA, the other transport the virion.
Looking to the future, one can anticipate that multifunctional proteins, similar to VP40, will be found with other viruses – will some patterns emerge? Will we be able to give computer programs new rules? Looking beyond viruses, what may we find? May there be some human proteins which can fold into two forms and may this be connected with disease and/or drug therapy?
5. Keynote addresses
5.1. From the elucidation of the HCV life-cycle to the development of highly effective antivirals. Raffaele De Francesco, Istituto Nazionale di Genetica Molecolare “Romeo ed Enrica Invernizzi” (INGM), Milano, Italy.
Raffaele started his presentation with three introductory slides. The main points highlighted:
-
•
140 million people are estimated to be infected with hepatitis C virus (HCV).
-
•
3 million/year are newly infected.
-
•
1–5% will die after 20–30 years of chronic infection.
-
•
No vaccine is available.
However, there is good news – HCV is curable because there is no nuclear integration, no DNA reservoir and no latency. The objective of therapy is a “cure” defined as a sustained virological response (SVR) in which patients remain HCV-free for 12 weeks (SVR12) after the end of therapy. Virus elimination is associated with a reduced risk of liver cirrhosis and cancer. The third slide gave the milestones in the therapy of HCV (genotype 1), starting in 1991 with interferon alpha (IFNα) (7–11% cure rate), in 2001 with PegIFNα and RBV (42–46%), in 2011 with the addition of protease inhibitors (PIs, 67–75%) and in 2014 sofosbuvir usually with another antiviral (93–100%).
Raffaele gave a detailed introduction to HCV, its life-cycle and the development of antiviral compounds. The main viral targets have been the serine protease (NS3/NS4A) (inhibitors with names ending in “previr”), the protein that creates the HCV replication complex (NS5A) (names ending in “asvir”), and the RNA polymerase (NS5B) (with names ending in “buvir”). In the slide “What have we learnt so far?”, only nucleotide polymerase inhibitors have a good profile for all four key parameters: barrier to resistance, pangenotypic efficacy, antiviral potency and adverse events. [This provides my justification for focusing on nucleotide analogs as HCV polymerase inhibitors in this summary report.]
With nucleoside/nucleotide analogs, there seems to be a narrow SAR for good efficacy against HCV, virtually all active compounds having a 2′-C-methyl substituent. Of these, there appears to be a narrow SAR for safety. Development of mericitabine (Roche) and INX-089 (BMS) have been terminated. Only sofosbuvir (Fig. 3) has been approved. Three others are in development, MK-3682 (Merck), ACH-3422 (Achillion) and AL-335 (Janssen). Although these structures have not been disclosed, they are believed to have the 2′-C-methyl-2′-fluoro substituents as in SOF. SOF shows a high genetic barrier to resistance. In vitro, the S282T mutant shows cross-resistance to 2′-C-methyl nucleotides, but the resistance to SOF is modest (2 to 10-fold) and the mutant has low viral fitness. Although clinically significant resistance to SOF had not previously been reported in clinical trials, the FDA have analyzed clinical specimens by next-generation sequencing. They have highlighted three low-frequency NS5B substitutions (L159F, V321A, and S282R), which were associated with SOF failure, and one baseline polymorphism (N316) which may potentially reduce SOF efficacy in genotype 1b.
SOF has been combined with ledipasvir, a NS5A inhibitor to form the first single pill regimen (Harvoni) for HCV therapy. NS5A is an interesting target because it has no known enzymatic activity but is crucial for the formation of the HCV “membranous web” within which HCV RNA replicates. NS5A is also involved in viral particle assembly and release. Several new NS5A inhibitors are being developed which have pangenotypic activity. First generation NS5A inhibitors, including ledipasvir, have a low barrier to resistance but those in development, elbasvir (Merck), ACH-3102 and GS-5816 (Gilead) have a higher barrier to resistance and are endowed with pan-genotypic activity.
There are now various IFN-free options for potential HCV therapies. Although there are different combinations with efficacy against genotypes 1 and 4, only SOF-containing regimens are active against all genotypes (1–6), the best being SOF with daclatasvir. In clinical use, this combination (once daily for 12–24 weeks) shows activity against genotypes 1–4. Furthermore, initial data indicate that this combination will be effective against genotypes 5 and 6. In Phase III trials, the combination of SOF with Gilead’s new NS5A inhibitor (GS-5816) is being tested at once-daily dosing for 8–12 weeks against all genotypes. Other IFN-free combination are currently being tested in Phase II clinical trials aimed at developing what today is considered an aspirational regimen, once daily, single pill, 8 weeks, 100% SVR12 for all genotypes.
Looking to the future, one can envisage eradication of HCV, at least in some areas. But there remain serious hurdles, one being that there are many undiagnosed HCV infections. Currently, cost tends to limit prescription to patients with advanced disease but inclusion of patients, who may spread the disease, will become more important. Although a few patients, with chronic hepatitis B, achieve a functional cure after years of therapy, hepatitis C is the first chronic viral infection to be cured efficiently by antiviral compounds. It seems that 100% SVR in all treated patients is within reach. This has been a most exciting period for patients and physicians alike.
5.2. The MSF response to the West African Ebola outbreak. Armand Sprecher, Médicins Sans Frontières, Operational Center of Brussels, Belgium.
From 1995 to 2012, Médicins Sans Frontières (MSF) responded to 14 outbreaks of Ebola or Marburg hemorrhagic fever. In 2014, MSF’s role in the current Ebola outbreak started on the 13th March with an alert from the Ministry of Health, Guinea asking for help with an outbreak in which an infection in 15 people had led to 9 deaths. On 18th March, a MSF team arrived in Guéckédou. Just 3 days later, the infection was identified as Ebola. On 23rd March, just 10 days after the initial alert, MSF opened their Ebola treatment unit (ETU) in Guéckédou. On 25th March, the Liberian Ministry of Health notified MSF of four Ebola deaths. The next day, MSF set up an ETU in Macenta which is in Guinea but close to the border with Liberia. The next day (27th March), four Ebola cases were identified in Conakry, Guinea. On 1st April, MSF opened their ETU in Conakry. By this time, MSF had sent 60 staff out into the field. About three weeks later (26th April), MSF opened their ETU in Liberia. To my mind, the speed of this response is truly impressive.
Between November 2014 and February 2015, MSF were running 10 or more centers in Guinea, Liberia and Sierra Leone. In total, MSF opened 19 ETUs (>700 beds) in 6 countries, trained >800 people in Europe, sent out >1000 people and had >4000 local staff working in their centers. Because it is difficult to distinguish between the fever caused by malaria and Ebola, it was usual to treat all patients entering an ETU with antimalarial drugs. Of 9492 people admitted to their ETUs, 5176 were confirmed as being infected with Ebola virus. Also, mass antimalarial treatment was distributed to 350,000 people in Monrovia and 1.8 million people in the Freetown area. Another service to the community was the distribution of 70,000 “home protection kits”.
One of the notable aspects of this Ebola outbreak was the risks faced by health care workers – not just the ever present risk of becoming infected but facing hostility from the local community. In Guinea, although MSF staff were threatened on a number of occasions, forced to withdraw sometimes and had staff suffering minor injuries in attacks, no MSF staff were killed. Unfortunately, in one incident that led to the deaths of health care workers, there was an attack on a Guinean health promotion team. This incident served as a warning to illustrate the seriousness of the threat posed. Conspiracy theories spread easily. Health care workers had difficulties at home, for example, their children being isolated by their class-mates at school. Just working in protective gear was stressful – heat exhaustion limited the safe working time to about 40 min. In an 8 h shift, an individual could be expected to work about 2 h in contact with Ebola patients. The elaborate protective procedures were successful in greatly limiting the risk of contracting Ebola but could not eliminate the risk totally. Three international staff became infected but were evacuated to their home countries and survived. Among the locally recruited staff, although there were exposures that were linked to workplace events, most of the investigations into staff illness found that their chief risk was outside of their work with MSF. The staff may have had family or friends infected with Ebola or performing healthcare activities outside of the MSF center. Naturally, these people, being healthcare workers and known to have experience with Ebola, were often under significant pressure in their home communities to provide discreet services. In total, 28 staff members contracted Ebola, 14 of whom died.
During this outbreak, MSF have been carefully collecting data which has enabled them to find factors which were associated with increased survival. Factors that had little or no effect on survival rates include male/female, month of admission or time from first symptoms to admission. It was expected that giving intravenous (iv) fluid would increase survival. At the Foya ETU, up to week 29 (mid-July), a few patients were receiving iv fluid but this was suspended from weeks 30 to 33 when there was a large increase in new admissions. From week 34 onwards, an increasing proportion of patients were receiving iv fluid. Unexpectedly, the Kaplan–Meier survival estimates showed no benefit. Only two factors did increase the chance of survival, age (about 15–20 years of age was best) and viral load at baseline, the lower the viral load, the greater the chance of survival. The latter was the single most important factor.
MSF centers have participated in clinical trials. The trial with brincidofovir was terminated due to lack of available trial participants. The favipiravir trial is ongoing following publication of an encouraging sign of efficacy in those patients who initially presented with either low or moderate viral loads. A trial with convalescent plasma had recruited 93 patients by the end of April. In Guinea, frontline workers are being vaccinated with the Merck Ebola vaccine (n = 390 by the end of April, about 800 by mid-June 2015).
Of 5176 confirmed cases of Ebola infected patients being cared for in MSF centers, there were 2449 survivors. Although the epidemic has had a huge human cost, there is a great joy every time a survivor leaves to go home (Fig. 7 ). Armand Sprecher’s conclusion “Let’s not do this again”.
Fig. 7.

MSF staff member sharing and celebrating the joy of departing patients. After recovering from Ebola, the MSF patients leave their hand-print on the ELWA3 survivor wall. Image © Peter Casaer, published with permission.
5.3. Advances in HCV therapies. Michael Manns, Hanover Medical School, Hanover, Germany.
Michael started his presentation with an introduction to HCV, including data confirming that a SVR12 or SVR24 did, indeed, represent a cure. Of 366 patients who had achieved a SVR after IFN therapy, only 3 relapsed over the next 5 years, a sustained response rate of 99.4%. Over a ten-year period, a SVR virtually eliminated the risk of dying due to liver-related disease. Survival was compared to an age- and sex-matched Dutch population. Patients without SVR had significantly greater risk of dying (p = <0.001) whereas those with SVR had a normal life span (p = 0.6). Furthermore, symptoms, due to HCV infection, are not just related to liver disease but there are various other manifestations that limit professional and social life. The classification of HCV strains is continually evolving – a recent special issue in Hepatology expanded the classification into 7 genotypes and 67 subtypes.
The therapy of chronic HCV infection has developed since the first HCV PIs were approved in 2011. During the period 2011–2014, the addition of a PI to PegIFNα increased the cure rate from about 45% to about 75% but this was only for genotype 1, the duration of therapy was still long (48 weeks), side effects were common and the treatment protocol complex. With the approval of SOF in 2014, the duration of therapy with PegIFNα/SOF was only 12 weeks and the cure rate approached 90%. Also in 2014, simeprevir (SMV, PI active against genotypes 1 and 4), daclatasvir (NS5A inhibitor active against all genotypes) and SOF/ledipasvir (Harvoni, genotypes 1, 4, 5 and 6) were approved. Various antiviral combinations allowed the exploration of IFN-free therapies. In Hanover (January–June 2014), SOF/RBV (n = 207) was compared with a PI-based therapy (n = 208). The overall cure rate with SOF/RBV (52%) was much higher than with the PI therapy (21%) due to more patients being eligible for treatment (74% vs 50%), better adherence (>95% vs 49%) and better SVR (70% vs 42%), respectively. By September 2014, in a survey with 2185 patients in USA and Europe (97 from German sites), 51% of patients were on SOF/SMV, 24% on SOF/PegIFNα/RBV, 15% on SOF/SMV/RBV and 10% on SOF/RBV. SOF had become the backbone of therapies for chronic hepatitis C.
In 2015, a combination of ombitasvir/paritaprevir/ritonavir (genotypes 1 and 4) and dasabuvir (non-nucleoside inhibitor of HCV polymerase, genotype 1) were approved. For the therapy of genotype 1, therapy with ombitasvir/paritaprevir and dasabuvir gave SVR > 95%, similar to that achieved with Harvoni but the latter was a simple one pill/day treatment. Treatment guidelines are changing rapidly – the European Association for the Study of the Liver (EASL) guidelines of April 2014 have been superseded by those of April 2015. Treatment should be prioritized for patients with significant liver disease, co-infection with HIV or HBV, debilitating fatigue or at risk of transmitting HCV. The choice of therapy depends on several factors but perhaps the more important are the genotype and the stage of liver disease, moderate (compensated) or severe (decompensated) cirrhosis. Preferred treatment options usually include SOF but the hardest-to-treat patients are those with genotype 3 with cirrhosis. In such patients, other options may be better, even including PegIFNα at least for the present. Also, for patients with renal insufficiency, ombitasvir/paritaprevir/dasabuvir is a preferred option (genotypes1 and 4) with SOF/daclatasvir or pegIFNα/SOF for the other genotypes.
Looking ahead, grazoprevir/elbasvir combination is completing Phase III trials. It seems to be well tolerated in patients with renal insufficiency, including those on dialysis. HCV resistance has emerged as a problem with ledipasvir (a component of Harvoni). In patients treated with ledipasvir (without SOF), essentially all patients ended treatment with resistant virus which seems to be fully competent, the mutant virus persisting for more than 48 weeks after therapy. However, the new NS5A inhibitor (velpatasvir, GS 5816) is active against all genotypes and against HCV strains and mutants resistant to ledipasvir. Also, it appears to have a high genetic barrier to resistance. Velpatasvir will be used with SOF as a fixed-dose combination pill. Approvals for both grazoprevir/elbasvir and SOF/velpatasvir are expected in early 2016. The second nucleoside analog to receive approval (about late 2016?) for HCV therapy may be MK-3682. It seems possible that all patients with HCV infection, so long as their liver disease is not too severe, will be treatable with virtually all patients achieving a cure. Initially, the duration of therapy is likely to be 12 weeks but 8 weeks looks possible for some patient groups. So far, therapies of 4 weeks have given a substantially reduced proportion of cures.
A novel approach is to use microRNA (MiR-122) inhibitors. At the 26th International Conference on Antiviral Research (ICAR) held in San Francisco, California (2013), there was a presentation on miravirsen (Vere Hodge, 2013). Within the 5′UTR of HCV, there are two six-nucleotide regions, known as seed sites 1 and 2 (S1 & S2). These sites are highly conserved through all genotypes of HCV and bind to a host micro-RNA (miR-122) which is present in liver cells. This interaction prevents the degradation of the viral RNA and thereby enables HCV to replicate. Miravirsen is a 15-base oligonucleotide containing 8 blocked bases (5′-mCcmAttmGmTcamCamCtmCmC-3′). It binds to miR-122 and so prevents its binding to viral RNA. In a small Phase II study, the mean reduction in viral load was 3 log10 and 4/9 patients became HCV RNA undetectable.
In a similar approach, a single dose of RG-101 (2 or 4 mg/kg) was administered by s.c. injection in a placebo controlled Phase II trial (n = 28, genotypes 1, 3, 4). At 4 weeks, the mean viral load reductions were 4.1 log10 and 4.8 log10, respectively. Notably, seven patients remained HCV RNA negative for 20 weeks follow-up.
In conclusion, chronic HCV infection has become a curable disease using well-tolerated, simple once daily antiviral therapies. Presently, the main barrier to increase treatment uptake is cost but one may anticipate that costs will decline. Considering the uptake in France (with one of the higher rates of uptake, 5%), one can calculate that France will have a much reduced HCV-infected population by 2025. Within the near future, there is the prospect of being able to treat, and cure, virtually all the high-priority patients. Hopefully, the new therapies expected in 2016 will make it easier to treat all patients irrespective of genotype. When one considers how many potential HCV mutants can arise every day in untreated patients, the progress with antiviral therapies is a remarkable achievement.
6. Mini-symposium: RNA viruses
6.1. Cruising with norovirus: progress and challenges in antiviral drug discovery. Joana Rocha-Pereira, Rega Institute for Medical Research, Leuven, Belgium.
In 1968, in Norwalk, Ohio, USA, a bacteria-free filtrate was shown to cause gastroenteritis. Eventually, small virus particles (27–35 nm) were visualized in 1972 and the cloning of the Norwalk virus genome in 1990 showed that the virus belonged to the family Caliciviridae. Between 1995 and 2003, broadly reactive RT-PCR and TaqMan assays became available. Noroviruses are now known to be the major cause of epidemic gastroenteritis and food borne illness. Although norovirus outbreaks in cruise ships get a lot of publicity, in the USA (2010–2011), most outbreaks (59%) were in long-term care homes and only 4% in cruise ships. The more vulnerable populations are the elderly (increased risk of hospitalization and death), children (symptoms can last for 7 days and virus shedding can continue for 6 weeks or more) and the immunocompromised (chronic gastroenteritis may last months to years). In countries with routine rotavirus vaccination, noroviruses are the major cause of severe childhood diarrhea.
It is easy to understand why outbreaks in semi-closed environments are difficult to control and quickly become extensive. Whereas an infectious dose is about 20 virus particles, a symptomatic patient may be shedding about 109–1012 virus particles/g stool and shedding can persist for 30 days. In addition, about 30% of the individuals may be asymptomatic but shedding virus, albeit 100-fold less but still many times an infectious dose. The gut microbiota could have an important role in the infection. Infectious particles can remain on surfaces for up to 2 weeks. Virtually all individuals will be susceptible due to the great diversity of strains and the lack of long-lasting immunity.
Vaccines use recombinant proteins to form virus-like particles (rVLP, based on the self-assembly of VP1). These have shown slight efficacy and major challenges remain – cross-protection against the many strains and the duration of immunity. There are no approved drugs to treat norovirus infections.
As a possible assay to test for potential therapies, the use of murine norovirus (MNV) has been investigated. In wild-type mice, MNV gives an asymptomatic infection but in mice with innate immune deficiency, there is diarrhea, either acute or chronic. Two compounds were used to evaluate the assays, 2′-C-methylcytidine (2CMC) and favipiravir. The prodrug of 2CMC, valopicitabine, was being developed as an anti-HCV drug. Favipiravir (T-705) has been licensed in Japan for influenza treatment and is in Phase III in the USA. In a MNV cell culture assay, 2CMC was active (EC50 = 2 μM) but favipiravir was poorly active. In an acute MNV infection, 2CMC administered 1 h before infection gave 100% protection (vs no survivors in the control group) and decreasing protection when given 12, 24 or 48 h pi (90%, 50% and 40% survival, respectively). In a chronic infection, 2CMC or T-705 were administered from day 7 for 14 days and again from day 55 for 14 days. During treatment, 2CMC showed good reductions in viral loads but these returned to control levels when off treatment. T-705 was essentially inactive. In these MNV assays, 2CMC would be a good positive control.
6.2. Developments in antivirals to prevent rabies. Anthony Fooks, Animal and Plant Health Agency, Weybridge, UK.
Ever since Pasteur’s rabies vaccine, derived from rabbit spinal cord, timely post-exposure prophylaxis (PEP) has successfully protected subjects from clinical disease. The Semple and Fuenzalida vaccines use killed virus derived from sheep brain and mouse brain, respectively. These vaccines were used widely but are no longer recommended. Rabies virus grown in cell cultures, then inactivated, is used in current vaccines which promote a good antibody response and have a better safety profile than the tissue-derived vaccines. Rabies is normally transmitted from an infected mammal via a bite, often into a muscle. The incubation period is very variable, from 2 weeks to 7 years but usually about 2 months. It is this long incubation period that enables PEP to protect the subject from clinical disease. Once the virus gets to the brain, mortality is essentially 100%. Worldwide, 20 million people receive PEP each year. Nevertheless, about 60,000/year die of rabies – the majority of these not receiving any PEP. In some countries, vaccine cost remains a limiting factor.
When the exposure is severe, both vaccine and immunoglobulin are administered but this raises the cost substantially. How can the cost be reduced and availability increased? One approach is to use plants to produce monoclonal antibodies (mAbs). Potential advantages include a reduced risk of contamination with animal/human pathogens. Although the mAbs have plant-specific glycans, they neutralize rabies virus as efficiently as the original mAbs. Hamsters were inoculated with rabies virus in the leg muscle and treated with the plant-derived antibodies, 63–71–3 or E559, at 24 h post infection (pi) without co-administration of rabies vaccine. At 14 days pi, there were 8/9 and 9/9 survivors, respectively, compared to 0/4 in the control group. These two mAbs each neutralize a broad range of virus isolates. There is the potential to combine these mAbs for therapy because they bind to different epitopes.
Although there is the potential to grow transgenic plants in developing countries, it would be a great advantage to be able to use an existing oral antiviral compound. Several approaches have been tried without success. Favipiravir (T-705) is licensed in Japan for influenza therapy and it is known to be active against many RNA viruses. A cell culture assay provided the first evidence for the activity of favipiravir against rabies virus. In a preliminary test in mice, with virus inoculated directly into the brain, there was essentially no protection provided by favipiravir treatment. However, this was certainly a severe test which does not represent the human clinical situation in which the incubation period is commonly 2 months. It is hoped that further evaluation of favipiravir will continue. One can build on the experience with favipiravir used to treat patients with Ebola infection in the current outbreak in West Africa – it can be used safely at higher doses than for influenza infections and there was an indication that favipiravir therapy was beneficial to those patients whose levels of Ebola virus at the time of presentation were low or moderate. Is it possible that the same may be true for patients with rabies virus infection when the exposure has been less severe?
6.3. Dengue therapeutics: state of the art and future directions. James Whitehorn, London School of Hygiene & Tropical Medicine, London, UK and Oxford University Clinical Research Unit, Ho Chi Minh City, Viet Nam.
Dengue virus (DENV) is an emerging disease of tropical and sub-tropical regions. The transmission cycle includes species of Aedes mosquito – the spread of DENV seems to be due to the spread of these mosquitoes. There are 4 serotypes of DENV. Prior infection with one serotype is protective against the same serotype but may enhance the severity of infection with another serotype via antibody-enhanced entry of the virus into cells.
Clinical symptoms can range from mild to severe. The global burden has been estimated to be about 2 million severe cases of which 21,000 lead to death. Current treatment is supportive care. A tetravalent vaccine has been tested in young children both in Latin America and Asia. It was partially effective in preventing clinical disease but very effective in preventing serious disease. Additionally, it would be useful to have available a treatment to help patients with severe disease. Typically, the virus load is already declining by the time a patient has serious symptoms. However, clinical trials showed that the use of steroids was not beneficial.
Balapiravir is a nucleoside analog which was being developed for HCV but stopped due to safety concerns in patients on weeks/months of therapy. A trial with balapiravir showed no benefit in DENV infection. Similarly, other potential antivirals have been evaluated in clinical trials. Although none has been efficacious, these trials have given the Oxford University Clinical Research Unit good experience in organizing trials. There is great potential to collaborate with other groups, for example, with the University of Marseilles which is exploring inhibitors of viral RNA-dependent RNA polymerase.
6.4. Hepatitis E: need for new therapies. Heiner Wedemeyer, Hannover Medical School, Hannover, Germany.
Hepatitis E Virus (HEV) was first described as “water-borne hepatitis” in 1980 and designated as HEV in 1983. HEV is a spherical, positive-stranded RNA virus belonging to the family Hepeviridae. The virus codes for a protease, helicase and polymerase, potential antiviral targets. There are 5 genotypes (genotype 5 does not infect humans) but only one serotype. In Europe and USA, the most common genotype is 3 but genotype 1 is particularly severe in pregnant women. In India and Asia, genotypes 1 and 4 are common. In Germany from 2001 to 2006, there were ⩽50 cases/year but since 2007, the number of cases has been increasing to 670 in 2014. Worldwide, HEV infection causes about 70,000 deaths/year.
HEV (genotype 3) has been detected in various mammals, including pigs. There is an increased seroprevalence in individuals in contact with pigs and transmission of HEV, via pork meat, has been recorded. In the general immunocompetent population, HEV usually causes a mild to moderate, self-limiting disease. However, in immunocompromised patients such as transplant patients, the prevalence of chronic HEV is about 1–4% with progression to cirrhosis within 1–2 years and likely leading to death.
Diagnostic tests for HEV are of variable quality. For example, 5 HEV RT-PCR assays were compared using 47 samples known to contain HEV. The detection rates were 100%, 100%, 97%, 97% and 83%. This has implications for testing blood donations and blood products. The annual incidence of HEV infection in blood donors in Germany is about 0.35%. A prevalence and transmission study in southeast England noted that the prevalence of HEV (genotype 3) in the population (including blood donors) is unknown but probably widespread and that HEV had been detected in pooled plasma samples.
Ribavirin (RBV) can be used to treat HEV infected patients. In a case series in Hannover, one patient had severe acute infection (genotype 1) and 11 patients had chronic hepatitis (genotype 3). They were treated with RBV (600–800 mg/day) for 5 months. Nine patients achieved SVR, one relapsed and one developed resistant virus. Interestingly, the HEV-mutant had increased viral fitness. In a larger study (n = 59), the median duration of RBV therapy was 3 months. The majority of patients (46) achieved SVR. Unfortunately, RBV use is contra-indicated in pregnant women. Another compound with apparent activity against HEV is mycophenolate – no chronic HEV is seen in heart transplant patients receiving mycophenolate.
With only one serotype, a vaccine for HEV seems to be a realistic possibility. A recombinant HEV vaccine (rHEV), containing the capsid protein, was being developed by GlaxoSmithKline (GSK). It was evaluated in a Phase II trial. The vaccine was administered at months 0, 1 and 6. The subjects were followed to day 800. The primary end-point was the number having confirmed HEV infection from day 14 after the 3rd vaccine dose. There were 69 subjects who had HEV: 3 in the vaccine group (0.3%) and 66 in the placebo group (7.4%) (p < 0.001). The efficacy of the vaccine was 95.5%. Unfortunately, this vaccine has not been progressed to Phase III trials.
In China, an attenuated virus vaccine was evaluated in a large Phase III trial (n = 56,000 each group). The subjects were followed for 55 months after the first dose. There were 60 subjects who had HEV infection, 7 in the vaccine group, 53 in the placebo group (p < 0.001). The vaccine efficacy was 86.8%. Although this vaccine is available in China, there seem to be no plans to evaluate this vaccine in the USA or Europe. However, the World Health Organization (WHO) have recently published a statement “Hepatitis E vaccine: WHO position paper, May 2015”. In a factsheet (#280, updated July 2015), their conclusions were:
-
•
Due to the lack of sufficient information on safety, immunogenicity and efficacy in the following population subgroups, WHO does not recommend routine use of the vaccine in children aged <16 years, pregnant women, chronic liver disease patients, and patients on organ transplant waiting lists, and travellers.
-
•
There may be special situations such as outbreaks where the risk of hepatitis E or of its complications or mortality is particularly high. The current WHO position concerning routine programmes should not preclude the use of the vaccine in these specific situations.
There seems to be little prospect of a vaccine becoming widely available in the foreseeable future and ribavirin cannot be used in certain high need patients (for example, pregnant women). Therefore there is an urgent need to have safer and more efficacious drugs to treat HEV infections.
6.5. Hepatitis C virus entry inhibitors. Thomas Baumert, University of Strasbourg, Strasbourg, France.
In their keynote lectures, Raffaele De Francesco and Michael Manns have described the exciting advances in antiviral therapies for treating chronic hepatitis C. Although the current therapies give high cure rates in many cases, the cure rates are rather lower in those patients with genotype 3 and/or have very advanced liver disease. Hopefully, the new combination of SOF/ velpatasvir, (approval target date early 2016) will effectively treat patients with genotype 3. For patients with very advanced liver disease, liver transplantation will remain as an option but there is universal re-infection of the graft. After transplantation, HCV quasispecies evolve to generate HCV variants which have enhanced efficiency of viral entry into cells, have altered receptor use and are poorly neutralized by patient’s antibodies. Viral entry is regarded as a good target for prevention and therapy of liver graft infection.
There are several host proteins which are involved in the entry of HCV into a cell, but antibodies to one of these, claudin-1 (CLDN1) has shown promising efficacy with a good safety profile. CLDN1-specific antibodies (CLDN1-mAb) prevent infection in cell culture models and show strong synergy with antiviral compounds. They are effective with all genotypes, although genotypes 2a and 6 require about a ten-fold higher dose than the other genotypes. Various functional assays have not revealed any toxicities. In uPA-SCID mice grafted with human hepatocytes, CLDN1 expression and localization is similar to that in human liver. This SCID mouse model was used for several proof-of-concept studies. To test for prevention of infection, CLDN1-mAb (500 μg) was administered by intraperitoneal (ip) injection at time of infection with HCV genotype 1b and twice more in the first week. There were good circulating levels of CLDN1-mAb to about the middle of the second week but reducing to almost baseline at week 2. In the control group, each of 5 mice had rapidly increasing HCV loads during the first two weeks, then reaching a plateau to 6 weeks. All 5 of the mice treated with CLDN1-mAb had undetectable HCV levels through to 6 weeks. To test for efficacy in SCID mice chronically infected with HCV genotype 2a, CLDN1-mAb was administered weekly for 4 doses (500 μg ip/dose). In the control mice (n = 4), HCV loads remained high. In the treated group (n = 5), the viral loads became undetectable during the four-week treatment period. In 4 mice, HCV remained undetectable to week 11. In a single mouse, the mAb levels were low and this may account for the relapse after the end of therapy. However, this virus showed no resistance to CLDN1-mAb. Using this model, monotherapy with CLDN1-mAb was shown to be active against all genotypes, there was no emergence of resistance and no signs of toxicity. Longer treatment times (7 weeks) results in the elimination of HCV-infected hepatocytes in this mouse model.
This good efficacy with CLDN1-mAb appears to be due to its ability to inhibit cell–cell transmission. Over time, the HCV-infected cells are replaced by new uninfected cells. It is envisaged that CLDN1-mAb therapy offers an efficient and safe antiviral strategy, possibly in combination with antiviral compounds, for the prevention of liver graft infection. Thinking more broadly, this approach may be used to treat infections caused by other viruses, such as dengue, coxsackievirus and adenovirus.
6.6. Development of antivirals against respiratory syncytial virus. John De Vincenzo, University of Tennessee, Memphis, TN, USA.
Respiratory syncytial virus (RSV) is a highly “successful” virus, infecting virtually every child by 2 years of age, world-wide. Autumn is the peak season for RSV infections in the USA. An ongoing trial is evaluating a potential link between asthma, RSV and hospitalization. For a long time, there has been uncertainty whether the symptoms of RSV infection are caused directly by the virus or via the induced stimulation of the immune system. Recent work has shown that many respiratory cells contain RSV, all those cells with cilia becoming infected. There is no big influx of immune inflammatory cells. The virus load peaks about 3 days after the start of symptoms. Viral load is a predictor for hospitalization and virus clearance is associated with recovery. These findings have led to the conclusion that RSV directly causes the disease. Therefore, there is a potential role for antiviral therapy although the window of opportunity remains uncertain.
Palivizumab, a mAb which binds to the fusion protein of RSV, is the first drug licensed to prevent RSV infections. Motavizumab is closely related to palivizumab but has improved binding to the fusion protein. Both these mAbs are effective in preventing RSV disease but their role in reducing established disease is still uncertain.
GS-5806 (Fig. 8 ) is an inhibitor of the RSV fusion protein. It has good oral availability (ranging from 46% in rats to 100% in dogs) and a lung/plasma area-under-the-curve (AUC0–12h) ratio of 26-fold in rats. GS-5806 exhibited potent in vitro activity against 75 RSV A and B clinical isolates (mean EC50 = 0.43 nM).
Fig. 8.
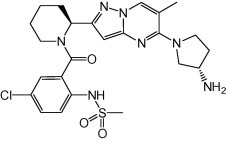
Structure of GS-5806, an inhibitor of the RSV fusion protein.
In an experimental viral challenge trial, healthy adult volunteers were inoculated with well-characterized strain of RSV on day 0. The incubation period varied from 1 to 7 days. The volunteers were monitored for the first detection of virus in upper respiratory tract, usually occurring about 2–5 days post inoculation. Therapy was started about 5–6 h after the first detection of virus in nasal wash. GS-5806 was evaluated in a dose-ranging challenge study (about 100 volunteers) comprising 7 dose groups and one placebo group. Viral loads, mucus weights and total symptom scores were measured. Oral dosing of GS-5806 once daily reduced the RSV replication and suppressed the infection-associated clinical symptoms in dose-dependent manner. Dosing of the drug for 5 days with the initial dose of 50 mg followed by 4 doses of 25 mg produced the strongest therapeutic effect with a peak viral load suppression of >4 log10 and a 10-fold reduced AUC of clinical symptoms compared to placebo treatment.
ALS-8176 is the oral prodrug of a nucleoside analog ALS-8112 (Fig. 9 ) which inhibits the RSV polymerase. In a similar volunteer challenge study (as above), treatment was started 12 h after virus was detected by PCR or 6 days after inoculation (D6), whichever was first. Although 62 volunteers were randomized to ALS-8176 or placebo, only 35 subjects met the criterion for infection (intention-to-treat population). There were three dosing regimens, each administered twice daily for 5 days (375 mg each dose, 750 mg loading dose (LD)/500 mg maintenance doses (MD), 750 mg LD/150 mg MD). In the placebo group, the viral loads were high (⩾4log10 pfu/ml) from day 2 until day 4, then reducing slowly to day 9. Similarly, the symptom scores peaked about days 2–4, reducing slowly to day 9. All doses of ALS-8176 showed good activity in all three parameters (viral load, mucus weight and symptom score). The mean time to non-detectability of RSV RNA was 1.3–2.3 days for the ALS-8176 treatment groups compared to 7.2 days in the placebo group. It was particularly noted that the loading dose of ALS-8176 reduced the viral load immediately, by about 1 log10 at 12 h. In contrast, the fusion inhibitor, GS-5806, reduced viral load but only after an initial increase at 24 h. ALS-8176 seemed to be well tolerated. [ALS-8176 has been reported to demonstrate a high barrier to the development of viral resistance (Alios BioPharma, Inc. press release, October 13, 2014)].
Fig. 9.
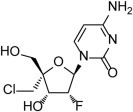
Structure of ALS-8112, an inhibitor of the RSV RNA polymerase. ALS-8176 is the oral prodrug of ALS-8112.
This volunteer challenge study seems to confirm that the symptoms caused by RSV are directly due to the virus and that a polymerase inhibitor is able to significantly reduce both viral loads and symptom scores (p < 0.001 and p < 0.05, respectively). There are ongoing Phase II studies in young children hospitalized with RSV infection. In another volunteer study, RSV and influenza were compared. Both viral loads and symptom scores increased later with RSV than influenza. This suggests that the window of opportunity for antiviral therapy will be greater for infections with RSV than influenza.
7. Mini-symposium: antiviral chemistry
7.1. Design, synthesis and biological evaluation of human DDX3 inhibitors with multiple antiviral activities. Maurizio Botta, Università degli Studi di Siena, Siena, Italy.
DDX3 is one of the human DEAD-box RNA helicases which contain the conserved motif, with amino acid sequence D–E–A–D (asp-glu-ala-asp) which gave this family of proteins the name “DEAD-box”. The focus of this presentation was to investigate the potential antiviral benefit which could be obtained by the inhibition of DDX3, a cellular cofactor. The potential benefits are easy to identify. DDX3 is involved in several viral infections but not all by the same mechanism. For HIV, the newly synthesized viral RNA is exported through the nuclear membrane via binding to DDX3. For HCV, DDX3 interacts with the viral core protein which helps build the nucleocapsid. For Japanese encephalitis virus (JEV), DDX3 binds to the viral RNA during replication. For poxvirus, DDX3 binds to the viral K7 protein which is essential to overcome the IFN-mediated cellular response. Both West Nile virus (WNV) and dengue virus (DNV) are dependent on DDX3 although the mechanism is not known.
What harmful effects may be caused by inhibition of DDX3? It has been implicated to play a role in various processes, including RNA metabolism (transcription, splicing, mRNA nuclear export and translation). However, there are two encouraging findings. DDX3 is over-expressed in several aggressive cancers and knock-down of DDX3 inhibits HIV replication but does not seem to affect cell viability.
DDX3 has two sites of particular interest, an ATPase binding site and typical helicase catalytic site. The crystal structure of DDX3 is known but only for the protein in an open form with the two sub-domains connected by a flexible linker. One domain has the ATPase binding site but the typical conserved motifs for a helicase are distributed between the two domains. To construct a model of DDX3 in the closed conformation, two approaches were tried, starting with the human protein, DDX48 (homology 37%) or the Drosophila Vasa equivalent to DDX3. The model resulting from the first approach did not have enough space for the RNA strand but the second approach led to a model in which the RNA strand fitted perfectly.
Using in silico screening of commercial databases led to several hits both for the ATPase site and the helicase site. The latter seemed to have more potential for achieving selectivity for inhibition of DDX3 – the major focus was on the optimization of compounds binding to the helicase site. Several compounds were inhibitors of DDX3 but inactive as inhibitors of DDX1. The three most active compounds inhibited at least two of the panel of 5 viruses but only one had results for all 5 viruses. This is the first demonstration of small molecules able to target an active site of DDX3 and inhibit its activity. Also, it is the first time that a single DDX3 inhibitor has shown activity against different viruses.
[Although this was a good demonstration of the concept, to assess safety, it is important to compare like with like, inhibition of viral replication with inhibition of cellular replication.] Especially for HIV and HCV, the safety profiles of current drugs set a very high standard which would have to be matched for any new therapy. Studies are still ongoing in that direction. On the other hand this approach may lead to anti-cancer treatments. Interesting data has just been received and will be published in due course.
7.2. Triphosphate prodrugs (triPPPro’s) of biologically active nucleoside analogs. Chris Meier, University of Hamburg, Hamburg, Germany.
At previous ICARs, Chris has reported on his cycloSal approach to deliver the nucleoside monophosphate (NMP) into a cell. Although there are many compounds which have poor antiviral activity due to inefficient phosphorylation to the monophosphate (MP), the limiting step may be at the phosphorylation to the di- or triphosphate (DP, TP). The focus of this presentation was the development of a strategy to synthesize prodrugs of nucleoside-triphosphates (NTPs).
If a NTP is fully masked, it becomes chemically labile in water. The chemical stability is much improved if there is a charge on the alpha (α) and beta (β) phosphorus atoms. Initially, for ease of synthesis, the terminal phosphorus was protected with two identical masking groups. In aqueous phosphate buffered saline (PBS), essentially no NTP was formed at 20 h but it was the predominant form at 432 h. When triPPPro-d4T-TP was incubated with pig liver esterase, the first masking group was removed within 5 min, the second much more slowly. By 24 h, the predominant form was the NTP with a very small amount of the NDP. Although symmetrical triPPPro-nucleotides were able to penetrate into cells, high lipophilicity was associated with desirable high transport into cells but also with high stability which led to greater amounts of NMP and NDP. When there are two different masking groups, one highly lipophilic, the other less so, the esterase cleaves the less lipophilic group first, quickly followed by chemical cleavage of the remaining part of that masking group, creating a charge on the terminal phosphorus atom. When the second ester is cleaved, the specificity for TP formation is high.
To demonstrate this approach, d4T-DP had one masking group with a methyl group, the other with C9H19 (Fig. 10 ). This prodrug was incubated with cell extracts. By 4 min, most of the methyl ester had been cleaved. Between about 15 and 60 min, d4T-DP was formed with essentially no d4T-MP. Similarly, CH3/C9H19-DiPPro-AZT-DP was prepared but in this test it was compared with the symmetrical C9H19-DiPPro-AZT-DP. The ratios of AZT-DP/AZT-MP were 5/1 and 1.5/1, respectively. Recently, comparable results were obtained in the case of the triphosphate derivatives. Again the non-symmetric derivatives gave better selectivity in the delivery of the TP versus the DP.
Fig. 10.
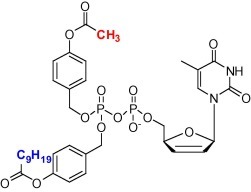
Structure of CH3/C9H19-DiPPro-d4T-DP.
To demonstrate that such masked NTPs can enter cells and regenerate the NTP inside cells, the symmetrical C8H17-TriPPPro-BCNA-TP (Fig. 11 ) was incubated with CEM cells (a lymphoblastoid CD4+ T-cell line). After incubation for 60 min, considerable amounts of BCNA-TP were detected in the cell extracts in addition to BCNA-DP. After incubation for 180 min, BCNA-TP has been dephosphorylated to give BCNA-DP as the major product as well as some amount of BCNA-MP. In contrast incubation of the parent nucleoside, BCNA, did not give any trace of phosphorylated BCNA metabolites. Here only the parent nucleoside was detected. An unsymmetrical version of BCNA-TP has been synthesized and is being evaluated.
Fig. 11.
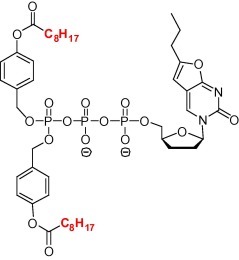
Structure of C8H17-TriPPPro-BCNA-TP.
7.3. Molecular modeling studies on the ternary complex of dengue virus polymerase. Cecilia M. Cima, Cardiff University, Cardiff, Wales, UK.
The DENV polymerase is an attractive target for antivirals as the NS5 protein, which contains the polymerase, is the most conserved protein among the four serotypes of DENV. Although the crystal structure of the free DENV polymerase is known, the polymerase, with bound template and nascent RNA strands (ternary complex), has not been crystallized. The aim of this project was to create a model of the ternary complex of DENV polymerase and then to explore the conformational changes as the polymerase initiates and then elongates the viral RNA.
A starting point was the crystal structure of the ternary complex of HCV polymerase. The template and nascent RNA strands were imported from the HCV polymerase into the DENV polymerase. Two Mg2+ ions were added to the active site and the model refined via energy minimization. This model was validated by docking known polymerase inhibitors. The model stability was confirmed by a molecular dynamics (MD) simulation. The Mg2+ ions and nucleic acid appeared to be well positioned in this model of the ternary complex.
In the free DENV polymerase crystal structure, there are two loops (loop A from residues 406 to 420 and loop B from residues 457 to 469) having unknown structures and a known priming loop. To model the initiation complex, first two Mg2+ ions were added to the active site (as in the above model), loops A and B were built according to the amino acid sequence and the priming loop (residues 782–796) was modeled in the presence of a short double-stranded (ds) RNA. Finally, an additional single-stranded (ss) RNA was introduced through a putative tunnel. As the ssRNA template enters the active site, it is encircled by three loops including A and B. The next step was to include a longer dsRNA into the model representative of the elongation phase. The priming loop was re-modeled to accommodate it and the complex submitted to a MD simulation. Specific residues in the priming loop gained contact with the nascent RNA strand.
These models indicate that the changing conformation of the loops, especially loop A and the priming loop, play an important role in enabling the polymerase to perform the different functions required for replication of viral RNA synthesis, initiation and elongation. These results are likely to be helpful in the design of DENV polymerase inhibitors.
8. Mini-symposium: emerging viruses (sponsored by Antiviral Research)
8.1. Globalization of chikungunya: 10 years to invade the world. Remi Charrel, Aix-Marseille Université Marseille, Marseille France.
Chikungunya virus (CHIKV) was first isolated in 1952 from a patient in Tanzania. Since then, there have been successive outbreaks in Africa. The first documented epidemic in Asia (Bangkok) was in 1958. By 2005, CHIKV virus had spread to India and much of tropical south-east Asia. La Réunion Island, which is east of Madagascar, is a French territory with health care comparable to that in France. In 2005, the first cases of CHIKV infections occurred in February and the outbreak peaked in May (450 cases/week). The following January, the epidemic spread rapidly, peaking at the end of January (45,000 cases/week). By June 2006, there had been 266,000 cases, about 1/3 of the island population. With such an extensive outbreak in an area with good health facilities, some clinical aspects, previously only suspected (eg myocarditis), were confirmed and other aspects were newly observed (eg neurologic forms not just in children but also in adults, fulminant hepatitis, renal failure). Elderly patients were at slightly increased risk of mortality but, generally, most patients recovered although the severe arthralgia may persist for months, even years.
There are many genotypes of CHIKV but these are related to three major lineages, (i) Eastern, Central, South African (ECSA) which includes the Indian sub-lineage, (ii) Asian lineage and (iii) the West African lineage, the latter having never produced a large outbreak but sporadic cases. The common host for CHIKV has been Aedes aegypti which is restricted to tropical regions, but Aedes albopictus survives in slightly cooler regions. As of January 2015 in Europe, Ae. aegypti is established only in the coastal regions around the east of the Black Sea and in the Portuguese island of Madeira. From 2004 to January 2015, Ae. albopictus has invaded and become established first in the north east costal region of Italy and then spreading to much of the Mediterranean coast from Spain to Greece. Within the ECSA lineage only, there have been important mutations, in particular A226V in the E1 glycoprotein, which increases viral replication in the mosquito Ae. albopictus. This expanding range of Ae. albopictus has presented CHIKV with an new opportunity.
Until 2007, CHIKV had not been known to circulate in Europe. In 2007, a single individual returning to Italy from India led to an outbreak of 197 cases. In September 2010, the first two local cases were recorded in south-eastern France, followed in 2014 with a cluster of 21 cases in Montpellier, France.
At least in recent times (see below), CHIKV had not been reported in the New World until November 2013. In the Caribbean island, St. Martin, five patients had a “dengue-like” fever but later shown to have CHIKV infection (Asian lineage). Although the Asian lineage of CHIKV was the initial strain in the New World, in 2014, the ECSA lineage was recently identified as the cause of a large outbreak in Brazil, concomitantly. Today, there have been over 1.2 million cases in the New World. Also, there is potential for the Caribbean CHIKV to spread to France – about 400 imported cases are known to have been in regions of France where Ae. albopictus is present.
This account of CHIKV ended with an unexpected twist – had this virus spread to the Caribbean and the Americas previously? There may have been a CHIKV epidemic in 1827–1828, as recorded by Henry Dickson, Professor of Medicine, South Carolina, USA. In a recent publication (Halstead, 2015), it was proposed that the epidemic in 1827–1828, then called dengue, had the characteristics of CHIKV.
8.2. Emerging viruses in the Balkans and the Mediterranean region. Anna Papa, Aristotle University of Thessaloniki, Thessaloniki, Greece
In this presentation, Anna highlighted the following viruses:
-
•
Crimean-Congo hemorrhagic fever virus (CCHFV).
-
•
Tick-borne encephalitis virus (TBEV).
-
•
West Nile virus (WNV).
-
•
Phleboviruses (7 viruses).
Crimean-Congo hemorrhagic fever virus (CCHFV)
CCHFV is a member of the family Bunyaviridae. The RNA genome in 3 segments, designated L, M and S. Transmission is by the bites of infected ticks, the main vector being Hyalomma marginatum, or by direct contact with blood or tissues of viremic patients or livestock. Various environmental factors influence tick survival and abundance. The infection is seasonal (March to November) and the group, who are at higher risk of infection, are those in contact with livestock. The course of disease – incubation period (3–7 days), fever and peak viremia (next 1–7 days), followed by a hemorrhagic phase peaking about 3 days later. The fatality rate can be up to 30%.
There are no specific treatment options. Although ribavirin is routinely used, its clinical efficacy is unproven and it is not FDA approved. Treatment with favipiravir (T-705) may be a better option as it was highly potent in cell culture and in vivo. Anna briefly mentioned a report using a mouse model (Oestereich et al., 2014). The placebo-treated mice died on days 3 and 4 post infection (p.i.) with CCHFV (100 PFU). With ribavirin, the survival time was prolonged, but the survival rate was not increased. Favipiravir was first tested at a high dose (150 mg/kg twice daily, orally) starting 1 h, 1 day or 2 days p.i. and ending on day 8. All these treatments protected mice fully with essentially no evidence of infection or disease. Only when treatment started 2 days p.i., minor changes in weight, temperature and aspartate amino transferase (AST) were detected at day 3 but not thereafter. Virus remained undetectable in blood and organs in all three groups throughout the observation period. To provide evidence for infection of the animals in the favipiravir treatment groups, the development of CCHFV-specific antibodies was investigated 21 days p.i. Only 1/10 (10%) of the animals treated 1 h p.i. developed antibodies, whereas 10/10 (100%) of the animals treated from day 1 or 2 p.i. developed antibodies. In a dose ranging study, favipiravir (15 and 3.75 mg/kg) was administered orally twice daily, starting 1 h p.i. and continuing to day 8. The higher dose gave good protection – no detectable virus in blood throughout the observation period, no virus in organs on day 3 and essentially no clinical signs of infection. However, the lower dose was only marginally active with prolonged survival time but no change in survival rate. The authors conclude that these data hold promise for clinical efficacy of favipiravir in human CCHF.
There is no widely approved vaccine but formalin-inactivated mouse-brain virus is used in Bulgaria. A novel vaccine, with the entire open reading frame (ORF) of the M-segment of CCHFV inserted into vaccinia virus, was shown to protect mice from a lethal challenge (Buttigieg et al., 2014). Studies on the low pathogenic Greek CCHFV AP92 strain may give insights into the vaccine development. . In 1975, AP92 was isolated from Rhipicephalus bursa ticks from goats in Vergina, a Greek village. This and related strains (Kosovo, Albania, and Turkey) seem to cause little or no symptoms. Seroprevalence in Greece was 6% in the area, around10% in some areas, even higher in some mountain regions in western Greece.
Tick-borne encephalitis virus (TBEV)
TBEV belongs to the family Flaviviridae. There are two types, (i) Eastern sub-type (Russian), the vector being Ixodes persulcatus, causes severe disease with about 25% mortality. (ii) Western sub-type, central Europe, the vector being Ixodes ricinus, causes a milder disease (<5% mortality). Ixodes ricinus is widely distributed in Europe. The disease course – incubation 4–14 days, fever next 2–5 days, neurological symptoms next ca. 3 weeks followed by recovery. A vaccine is available.
West Nile virus (WNV) (outbreaks in Greece)
An episode in 2010 gave a good illustration of quick collaboration between hospital and research laboratory. On 4th August, a physician in the Thessaloniki hospital noted an unusually high number of encephalitis cases. The next day, samples were sent to the reference laboratory and 10/11 cases were shown to be positive for WNV. On the night of 9–10th August, mosquito traps were set up. A positive PCR test was achieved on 12th August. There was a mutation, P249H, in the NS3 protein, previously associated with increased pathogenicity and neuroinvasiveness in humans compared to other strains. Following this observation, reported cases of West Nile neuroinvasive disease (WNND) in Greece have been collected. From 2010 to 2013, the average number of cases has been about 100 cases/year, but just 14 in 2014. Of a total 441 cases in these 5 years, 72 were fatal. What will 2015 bring?
Phleboviruses
Phleboviruses belong to the family Bunyaviridae. The disease varies from asymptomatic to serious CNS infections, depending on the virus. Several novel phleboviruses have been detected in the Mediterranean region. In Greece, 7 viruses have been reported. The disease is seasonal (in Greece, April to October). In a small study, 494 patients with fever, of whom 50% had CNS infection, were tested for a phlebovirus infection. During the period 2010–2014, there were 43 (8.7%) patients with a phlebovirus infection. Although the proportion of these patients with CNS infections varied from year to year, it was about 2/3rds.
There are many tick-borne phleboviruses found in the Mediterranean countries. As phleboviruses belong to the same family (Bunyaviridae) as CCHFV, is it possible that favipiravir may have good activity against these phleboviruses? [Favipiravir has been reported to show efficacy in Rift Valley fever rodent models.]
8.3. Middle east respiratory syndrome (MERS). Bart Haagmans, Erasmus MC, Netherlands.
Coronaviruses (CoVs) infect both humans and animals. Human CoVs normally cause symptoms typical for a common cold but, in November 2002, we saw the appearance of a severe acute respiratory disease which became known as SARS. Via air travel, SARS quickly spread around the globe. Between 1 November 2002 and 10 July 2003, there were 5910 probable cases reported. The outbreak was controlled by identification, quarantine and isolation.
In 2012, when a 60 year-old Saudi male with fever, cough and pneumonia was studied, a new CoV (MERS) was detected. In contrast to SARS, the new virus did not spread rapidly. From March 2012 to March 2015, virtually all of the 1082 confirmed cases of MERS were in the Middle East, just 15 in Europe and 10 in the rest of the world. For all cases of MERS, the Middle East is the probable source of infection. Of concern, there were 439 deaths (40%). There was a large peak of reported cases in April/May 2014 but otherwise there are normally a few (<50) cases/month. The fact that human cases were found over a relatively long period in the absence of sustained human–human transmission led to the search for a local animal reservoir in the Middle East.
MERS-CoV is related to bat CoVs and it replicates in bat cell lines, suggesting it enters cells through use of a conserved receptor. Dipeptidyl peptidase 4 (DPP4), also known as CD26, was discovered to be a functional receptor for MERS-CoV replication. When either human or bat DPP4 is expressed in non-permissive Cos7 cells, virus replicates. Notably, DPP4 is present on human epithelial cells of the lower respiratory tract but absent in upper respiratory tract epithelial cells. This could explain the poor human to human transmission of MERS compared with SARS.
To investigate which animal could be the source of zoonotic transmission of MERS-CoV, antibodies to various CoVs were sought in sheep, cows, goats, Spanish dromedaries and Omani dromedaries. Although antibodies to SARS-CoV were not detected in any of these animals, antibodies to MERS-CoV were detected in a few individual Spanish dromedaries. Moreover, virtually all the Omani dromedaries had high levels of antibodies to MERS-CoV. Using camels being presented for slaughter, DPP4 expression was detected not only in the lower respiratory tract but also the upper respiratory tract and in the nasal cavity. From nasal samples taken from camels of different ages, the presence of MERS-CoV varied with age, being more prevalent in younger animals (age <1 year). Clearly, virtually all the camels become infected in their first year. MERS-CoV antibodies were detected in a minority of people who work with camels but not in people who have no contact with camels. Those people with antibodies did not recall having any serious disease.
Would it be possible to create a vaccine effective in both camels and humans? A recombinant spike protein fragment (S1), amino acid (aa) 1–731 from the full length (aa 1–1353), previously shown to bind to bat and human cells, may be used as a candidate vaccine. Not only do human mAbs that recognize S1 also neutralize MERS-CoV in vitro, but also expression of the spike protein by a modified vaccinia Ankara virus (MVA-S) induces virus neutralizing antibodies in mice. Therefore this vaccine may be tested in camels to reduce virus excretion and subsequent camel–human transmission.
For a proof-of-concept trial of a vaccine, dromedary camels were immunized with two doses of MVA-S and challenged with MERS-CoV via a nasal spray device. After immunization, none of the control camels developed antibodies to MERS-CoV whereas all camels given the vaccine developed virus neutralizing antibodies. In addition, the camels, which were vaccinated with MVA-S, showed reduced virus excretion upon challenge with MERS-CoV.
In contrast to these encouraging results with the vaccine, treatment of patients with interferon alfa-2a and ribavirin did not greatly improve survival at 28 days (6/20 vs 4/24, p = 0.054). Although a larger trial may have shown a significant improvement, a more efficacious therapy is clearly needed.
9. Contributor presentations
9.1. Discovery and characterization of MK-8876, a novel non-nucleoside inhibitor of HCV NS5B that possesses broad genotypic potency. Steve Ludmerer, Merck & Co., Kennilworth, NJ, USA.
The polymerase of HCV (NS5B) has at least 5 regions to which known non-nucleoside inhibitors (NNI) bind. Three regions are at the base of a deep cleft, close to the catalytic site to which nucleosides bind. Starting with a benzofuran core known to bind one of the sites of this pocket, it was thought that, if this core could be extended further into the cavity, it may be possible to capture extra binding interactions. This could lead to a NNI with improved potency and a broader range of activities against different genotypes.
Starting from an in-house model of the polymerase, MK-8876 eventually emerged as a lead candidate. This compound had excellent activity against genotypes 1, 2, 3, 4 and 5 (replicon assays, EC50 ⩽ 7 nM) and lacked cytotoxicity (CC50 > 10 μM). Also, it maintained potency against known NNI resistant mutations and those identified in direct resistant selections. A mutation in the genotype 1a background, S365L, was identified but this mutation appeared to reduce viral fitness. In a Phase Ib proof-of-concept study, there were 3 patients with genotype 1a and 3 with genotype 3a. MK-8876 (800 mg) was given once daily for 7 days. However, there were two unexpected results. (i) The three patients (genotype 3a) had more modest reductions in viral loads than genotype 1a patients, a result not predicted by the in vitro potencies nor explained by the in vivo pharmacokinetics. (ii) The viral loads in two of the three patients (genotype 1a) reduced initially but rebounded during the dosing period. The mutations (S365L/S) were observed at day 5.
This study raises some interesting questions. (i) Why was the differential response between genotypes 1a and 3a not predicted from pre-clinical studies? (ii) How did the mutant virus (S365L) replicate in patients but appeared debilitated in vitro? (iii) What new pre-clinical resistance studies would accurately predict lack of clinical resistance? Such studies should be incorporated into future NNI discovery/lead optimization protocols.
9.2. Pharmacodynamic investigation of a human rhinovirus inhibitor in a hollow fiber infection model. Qin Yu, AstraZeneca R&D, Waltham, MA, USA.
By growing human cells in a hollow fiber system, it is possible to accurately mimic human pharmacokinetics – one reservoir contains active drug, the other only culture medium and a pump system controlled by computer. The cells can be infected by adding an inoculum of virus. Qin described the use of this system to investigate a protease inhibitor of human rhinovirus. What was the main factor determining efficacy, drug peak levels (C max) or trough levels (C min)?
The first step was to determine the steady-dose level which gave good protection. The lowest drug level, which gave good protection for 100 h, was 3 × EC50 concentration. Then, keeping the total daily dose equivalent to the constant 3 × EC50 concentration, once daily, twice daily and four-times daily dosing were simulated. As expected, the once daily dosing gave the highest C max and the four-times daily gave the highest C min. Once daily dosing gave poor activity, four-times daily dosing the best activity.
This hollow-fiber system is a useful tool for investigating how human pharmacokinetics can influence efficacy. For example, would a loading dose be beneficial? Qin concluded “We established the first human rhinovirus hollow fiber system”.
10. Conclusion
Our three awardees have each had remarkable careers. The Elion award was presented to Phil Furman who, in his early days, worked on acyclovir with Trudy Elion. To my mind, acyclovir is the most important “game-changing” antiviral. It may not have the biggest sales but it is the first antiviral drug to combine good clinical efficacy with a truly remarkable safety profile. After some three decades, it still stands as a benchmark for safety. Today, acyclovir, and its prodrug valacyclovir, are used worldwide to treat herpesvirus infections. In the generally healthy population, the proportion of resistant virus remains below 1%. Prior to acyclovir, I do not recall anyone predicting that an antiviral could be used clinically for three decades with less clinical problems than the best antibiotics. However, Phil hardly mentioned acyclovir but focused on the research that led to sofosbuvir, another “game-changer”, this time for patients with HCV infections.
The Holý award was presented to Dennis Liotta. Like Phil, Dennis did not describe his work that led to the invention of important antivirals for HIV therapy (it has been estimated that more than 90% of HIV-positive patients in the USA have taken a drug invented or developed by Dennis) but he chose to describe his recent research. It is a remarkable journey in which the chemistry led him into new, unexpected territory.
The Prusoff award is for a young investigator who has already made an impact in the antiviral field and is set to continue to do so. Erica Ollmann Saphire is a highly qualified recipient. In her award lecture, she described how viral proteins are able to rearrange into different forms and so fulfil different functions. I have been lucky to listen to many fine lectures at ICAR meetings over the years but Erica’s lecture sailed straight into my top-ten.
There were three excellent keynote lectures. The approvals of sofosbuvir, by the FDA in December 2013 and by the EU in January 2014, heralded a new era in the therapy of chronic HCV infections. Raffaele De Francesco set the scene, with an overview of the early antiviral agents which improved the cure rate when used with IFN but could not be used on their own due to HCV resistance. Although a few other nucleotide analogs are in development, there seems to be a narrow SAR and sofosbuvir remains the only approved nucleotide analog. Michael Manns continued the story giving a detailed account of the clinical picture – how it has changed and is continuing to change rapidly – guide-lines have been written and rewritten. Early in 2016, new drugs are expected, including sofosbuvir combined with a new NS5A inhibitor (velpatasvir, GS 5816) which is active against all genotypes and against HCV strains and mutants resistant to ledipasvir (part of Harvoni). We seem to be close to an aspirational therapy (single pill once daily for 8 or 12 weeks) giving essentially 100% cure rates in patients with any genotype. As HCV can produce 1012 virions daily in untreated patients and as it has such a great capacity for generating resistant mutants, this is a remarkable scientific achievement.
The third keynote speaker, Armand Sprecher, gave an amazing account of a remarkable organization, Médicins Sans Frontières (MSF). Just 10 days after the initial alert, MSF opened their Ebola treatment unit (ETU) in Guéckédou – an impressive achievement. The Ebola outbreak has exacted a huge human cost. However, I greatly admire all the healthcare workers who responded to the emergency, even though they were putting their own lives at risk. Armand briefly mentioned the clinical trials, including that with favipiravir. In the published results, those treated patients, who had moderate or low baseline levels of Ebola virus, generally had rapidly decreasing viral loads on days 2 and 4 after the start of therapy. With the Ebola outbreak continuing, do patients routinely have their viral loads measured? If it is unusual for viral loads to decrease at days 2 and 4 after entering an Ebola treatment center, then the viral load data gathered during the clinical trial gives the best indication that favipiravir is providing benefit to Ebola patients. Armand ended his presentation on a happier note – I follow suit in my report.
Although the Ebola outbreak is continuing, there are signs of progress. The World Health Organization (WHO), reported (Ebola Situation Report - 5 August 2015) “There were 2 confirmed cases of Ebola virus disease (EVD) reported in the week to 2 August: 1 in Guinea and 1 in Sierra Leone. This is the lowest weekly total to have been reported since March 2014, and marks a third consecutive decline in weekly case incidence.” The same report warns of the continuing difficulties facing the policy of containment. An individual was a known contact but was lost to follow-up. When Ebola infection was confirmed, intensive efforts traced over 1000 contacts. However, I am pleased to see that this WHO report mentions the trial of the Ebola ça suffit! ring vaccination trial in Guinea. Clusters of all contacts and contacts of contacts were defined and randomly allocated 1:1 to immediate vaccination or delayed (21 days later) vaccination with rVSV-ZEBOV (one dose of 2 × 107 plaque-forming units, administered intramuscularly) (Henao-Restrepo et al., 2015). Essentially, the trial’s eligibility criteria allows vaccination of all healthy adults except pregnant women. The primary end-point was the number of laboratory-confirmed Ebola virus disease with onset of symptoms at least 10 days after randomization. There were 90 clusters, with a total population of 7651 people included in the planned interim analysis. There were 16 Ebola cases in the delayed vaccination group. No new cases of Ebola virus disease were diagnosed in vaccinees from the immediate or delayed groups from 6 days post-vaccination. Following this result, all clusters are being offered immediate vaccination.
The elimination of Ebola infections seems hard to achieve but the WHO have reported (Ebola Situation Report – 9 September 2015) continuing progress. Liberia has, again, been declared free of Ebola transmission. “No new health worker infections were reported in the week to 6 September. There have been a total of 881 confirmed health worker infections reported from Guinea, Liberia, and Sierra Leone since the start of the outbreak, with 513 reported deaths.” “The Ebola ça suffit! ring vaccination Phase 3 efficacy trial of the rVSV-ZEBOV vaccine has now been extended from Guinea to Sierra Leone. Contacts and contacts of contacts associated with new confirmed cases and who meet the trial’s eligibility criteria will therefore be offered the vaccine.”
In the years to come, there will surely be new outbreaks of Ebola. The current outbreak has revealed the difficulties encountered with containment but also enabled the development of new options. Hopefully, all new outbreaks of Ebola will be quickly controlled by using the vaccine to ring-fence each index case and each patient may have a greater chance of survival if treated promptly with favipiravir. With vaccination, healthcare workers will not be putting their own lives at risk.
There were three mini-symposia, “RNA Viruses”, “Antiviral Chemistry” and “Emerging Viruses”. These mini-symposia fulfilled an important aim of ICAR, to bring together knowledge and expertise from the whole spectrum of antiviral research. There was a good collection of RNA viruses (Norovirus, rabies. dengue, HEV, HCV, and RSV). For me, the highlight of the chemistry was the preparation of prodrugs for nucleotide triphosphates as this opens a door to new options. Such an approach is able to bypass all the activation steps normally needed for a nucleoside/tide and such an approach might be beneficial for antiviral, antibiotics, antitumor or simply for chemical biology questions using non-natural nucleoside triphosphates. The third mini-symposium emphasized how the research work in the antiviral area is continuing to expand and needs to do so with a sense of urgency.
Finally, I have made a personal choice to include two further presentations. Steve Ludmerer described the work of their group with MK-8876 and ended his presentation with some important questions, to which I add another. When considering how to treat chronic HCV infection, viral resistance seems to be a more important parameter than initial potency. Of all the drugs licensed for the therapy of HCV or HIV, those showing the greatest genetic barrier to resistance are nucleoside/tide analogs. These bind to a catalytic site which is highly conserved and there are few options for mutations which remain viable. Of course, the converse is not necessarily true – some nucleoside/tide analogs may show a relatively low genetic barrier to resistance, for example, lamivudine. This presentation prompts one to consider how best to approach the therapy of other RNA viral diseases. What are the prospects for fusion inhibitors vs protease inhibitors vs nucleoside/tide polymerase inhibitors? For example, the nucleoside analog, ALS-8176, has shown excellent activity against RSV in a clinical experimental challenge trial in healthy volunteers. Also, ALS-8176 has been reported to demonstrate a high barrier to the development of viral resistance. Although infections with many RNA viruses are self-limiting in normally healthy people, such infections can be long-lasting in immunocompromised patients. It is important to have therapies which not only help those who would normally recover but also meet the requirements of those with high medical needs.
Qin Yu described a hollow fiber culture system for growing human cells which were infected with human rhinovirus. In this system, it is possible to mimic accurately the human pharmacokinetics of the drug being evaluated. Qin showed how this system can be used to determine the most efficacious dosing regimen. From Qin’s final comment, it seems that the fiber culture system is rarely used but I suggest that it is an important tool for evaluating compounds. At the 8th ICAR (1995 in Santa Fe), Hamzeh described the comparison between acyclovir (ACV) and penciclovir (PCV) in HSV-infected human cells in a hollow-fiber culture system (Hamzeh et al., 1995). In a review (Vere Hodge and Field, 2013), there is a brief summary of this work. With twice daily dosing with PCV, the trough levels of PCV-TP increased after each of the first four doses. In the Elion Lecture, Phil reported that the half-life of sofosbuvir-triphosphate was 38 h. I would expect that HCV therapy with sofosbuvir would be improved by starting with a loading dose. This fiber system could be used to prove what the optimum loading dose should be.
I hope that my summary of this ICAR will reflect some of the enthusiasm that the speakers had for their subjects and that, even if the topics are not in one’s own area of research, there is much of interest to be appreciated. Rarely, a major step forward in antiviral therapy happens within a short period of time. Within the last two years, the therapy of chronic HCV infections has been transformed. I am especially pleased that the 2015 ICAR program included outstanding talks describing this exciting progress. It takes much effort to organize so many excellent presentations and so I would like to add my thanks to the ISAR Officers and Conference Committee for another interesting and successful ICAR.
Acknowledgements
I wish to thank all those authors who have kindly provided me with copies of their presentations and for giving me valuable comments. Also, I thank the President of ISAR for asking me to prepare this meeting report.
References
- Buttigieg K.R., Dowall S.D., Findlay-Wilson S., Miloszewska A., Rayner E., Hewson R., Carroll M.W. A novel vaccine against Crimean-Congo haemorrhagic fever protects 100% of animals against lethal challenge in a mouse model. PLoS ONE. 2014;9(3):e91516. doi: 10.1371/journal.pone.0091516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead S.B. Reappearance of chikungunya, formerly called dengue, in the Americas. Emerg. Infect. Dis. 2015;21(4):557–561. doi: 10.3201/eid2104.141723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamzeh F.M., Schaad H.J., Lietman P.S. Santa Fe; New Mexico, USA: 1995. 8th International Conference on Antiviral Research. (abstract) [Google Scholar]
- Henao-Restrepo A.M., Longini I.M., Egger M., Dean N.E., Edmunds W.J. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet. 2015;386(9996):857–866. doi: 10.1016/S0140-6736(15)61117-5. [DOI] [PubMed] [Google Scholar]
- Oestereich L., Rieger T., Neumann M., Bernreuther C., Lehmann M. PLoS ONE. 2014 doi: 10.1371/journal.pntd.0002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vere Hodge R.A. Meeting report: 26th International Conference on Antiviral Research. Antiviral Res. 2013;100:276–285. doi: 10.1016/j.antiviral.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vere Hodge R.A., Field H.J. Antiviral agents for herpes simplex virus. In: De Clercq Erik., editor. vol. 67. Academic Press; Burlington: 2013. pp. 1–38. (Advances in Pharmacology). [DOI] [PubMed] [Google Scholar]