

Highlights
-
•
The 3D structure of the MERS-CoV papain-like protease (PLpro) has been determined.
-
•
The enzyme has a deficient oxyanion hole and unique S3 as well as S5 subsites.
-
•
In-vitro kinetics of the MERS-CoV PLpro are slower than for the SARS-CoV enzyme.
-
•
Restoring the oxyanion hole through mutagenesis enhances the enzyme kinetics.
-
•
The unique features of the enzyme will allow design of MERS-specific antivirals.
Keywords: MERS-CoV, Papain-like protease, Oxyanion hole, Zinc finger, Deubiquitinase, Antiviral drug design
Abstract
The Middle-East Respiratory Syndrome coronavirus (MERS-CoV) causes severe acute pneumonia and renal failure. The MERS-CoV papain-like protease (PLpro) is a potential target for the development of antiviral drugs. To facilitate these efforts, we determined the three-dimensional structure of the enzyme by X-ray crystallography. The molecule consists of a ubiquitin-like domain and a catalytic core domain. The catalytic domain displays an extended right-hand fold with a zinc ribbon and embraces a solvent-exposed substrate-binding region. The overall structure of the MERS-CoV PLpro is similar to that of the corresponding SARS-CoV enzyme, but the architecture of the oxyanion hole and of the S3 as well as the S5 specificity sites differ from the latter. These differences are the likely reason for reduced in vitro peptide hydrolysis and deubiquitinating activities of the MERS-CoV PLpro, compared to the homologous enzyme from the SARS coronavirus. Introduction of a side-chain capable of oxyanion stabilization through the Leu106Trp mutation greatly enhances the in vitro catalytic activity of the MERS-CoV PLpro. The unique features observed in the crystal structure of the MERS-CoV PLpro should allow the design of antivirals that would not interfere with host ubiquitin-specific proteases.
1. Introduction
Ten years after the outbreak of severe acute respiratory syndrome (SARS) of 2002/2003 (Hilgenfeld and Peiris, 2013), another highly pathogenic coronavirus, Middle-East Respiratory Syndrome coronavirus (MERS-CoV), has been recognized to infect humans (Zaki et al., 2012, de Groot et al., 2013). Accumulating evidence suggests camels to act as a zoonotic source of the virus (Reusken et al., 2013, Haagmans et al., 2014, Meyer et al., 2013). Limited human-to-human transmission of the virus has been described (Assiri et al., 2013). As of June 11, 2014, 683 cases of MERS have been reported, with 204 deaths (http://www.who.int). The clinical symptoms of MERS include severe pneumonia and sometimes acute renal failure (Eckerle et al., 2013). However, the majority of MERS patients had/has comorbidities, such as diabetes, lung disease, or chronic renal disease (Perlman, 2013).
SARS-CoV and MERS-CoV belong to the genus Betacoronavirus but pertain to highly distinct phylogenetic clades termed b and c, respectively (de Groot et al., 2013). In case of SARS-CoV, the best-characterized potential antiviral drug targets are the two viral proteases, the main protease (Mpro, also called 3C-like protease, 3CLpro) (Hilgenfeld and Peiris, 2013, Anand et al., 2003, Yang et al., 2003, Yang et al., 2005, Xu et al., 2005, Lu et al., 2006, Verschueren et al., 2008, Zhu et al., 2011, Kilianski et al., 2013) and the papain-like protease (PLpro) (Hilgenfeld and Peiris, 2013, Kilianski et al., 2013, Barretto et al., 2005, Ratia et al., 2006, Ratia et al., 2008, Baez-Santos et al., 2014). The latter enzyme exists in all coronaviruses (Woo et al., 2010) and has been shown to be responsible for releasing non-structural proteins (Nsp) 1, 2, and 3 from the N-terminal part of polyproteins 1a and 1ab. The three cleavage sites contain the sequence motif LXGG↓XX. In addition, the SARS-CoV PLpro has been shown to have deubiquitinating and interferon antagonism activities, thereby interfering with the host innate immune response (Barretto et al., 2005, Lindner et al., 2005, Devaraj et al., 2007, Frieman et al., 2009). Specifically, it can prevent the activation of IRF3 (interferon-regulatory factor 3) and antagonize the NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) pathway, but the detailed mechanisms involved are still unclear. Very recently, the MERS-CoV PLpro has been reported to also have proteolytic, deubiquitinating, and deISG15ylating activities in HEK293T cells (Yang et al., 2013, Mielech et al., 2014) (ISG15 = interferon-stimulated gene 15); it therefore also acts as an interferon antagonist through blocking the IRF3 pathway. Interestingly, these reports differ in their finding that the interferon-antagonizing activity of the MERS-CoV PLpro is either independent of (Yang et al., 2013) or dependent on (Mielech et al., 2014) its proteolytic activity.
In spite of the accumulating knowledge on the essential roles of the coronavirus PLpro in virus replication and evasion of the host-cell innate immune response (Devaraj et al., 2007, Frieman et al., 2009, Yang et al., 2013, Mielech et al., 2014, Lindner et al., 2007, Clementz et al., 2010), the three-dimensional structures of only two of these enzymes have been reported so far, i.e., that of the PLpro from SARS-CoV (Ratia et al., 2006) and that of the PL1pro from Transmissible Gastroenteritis Virus (TGEV) (Wojdyla et al., 2010). Here we present the crystal structure of the MERS-CoV PLpro at 2.50 Å resolution, in order to unravel the structural basis of the activities of the enzyme and facilitate structure-based drug design efforts. In addition, we report the in vitro hydrolytic activities of the enzyme towards two synthetic peptide substrates and a fluorogenic ubiquitin derivative.
2. Materials and methods
2.1. Recombinant production of MERS-CoV papain-like protease (PLpro)
The PLpro of MERS-CoV (strain 2c EMC/2012; GenBank: AFV09327.1) comprises 320 amino-acid residues, corresponding to Gln1482 – Asp1801 of pp1a, and is part of non-structural protein 3. In the interest of an easy description, we renumber Gln1482 into Gln1 here. A gene coding for the PLpro was amplified by the polymerase chain reaction (PCR) using the forward primer 5′-CTAGCTAGCcagttaacaatcgaagtcttagtg-3′ and the reverse primer 5′-CCGCTCGAGttaatcgctactgtatttttggccggg-3′. The resulting PCR product was digested with restriction enzymes NheI and XhoI for ligation into pET-28a (Novagen). Cloning was designed to include an N-terminal hexahistidine (His6) tag and a thrombin cleavage site. The recombinant plasmid was used to transform Escherichia coli strain Tuner (DE3) pLacI (Novagen). Transformed cells were grown at 37 °C overnight in LB medium, supplemented with kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL). The culture was used to inoculate LB medium the day after. When the OD600 of the culture reached 0.6–0.8, overexpression of the PLpro gene was induced for 20 h with the addition of isopropyl-D-thiogalactoside (IPTG, final concentration 0.5 mM) at 20 °C. Subsequently, the culture was harvested by centrifugation for 30 min at ∼7300×g and 4 °C. Cells were resuspended in 30 mL buffer A (20 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, pH 8.8, 10 mM β-mercaptoethanol (BME)) and lysed by sonication on ice. The lysate was centrifuged for 1 h at ∼48,000×g and 4 °C to remove the cell debris. The supernatant was applied to a HisTrap™ nickel column (GE Healthcare) and the His-tagged protein was eluted with buffer B (20 mM Tris–HCl, 500 mM NaCl, 500 mM imidazole, pH 8.8, 10 mM BME) using standard protocols. The target protein was processed overnight by thrombin (Sigma) cleavage at 4 °C to remove the His-tag. Left with six extra residues (GSHMAS) at the N-terminus after this processing step, the PLpro was further purified by gel filtration (HiLoad™ 16/60 Superdex 200 column, GE Healthcare) using buffer C (20 mM Tris–HCl, 150 mM NaCl, pH 8.8, 10 mM BME).
2.2. Crystallization and diffraction-data collection
Purified PLpro was concentrated to ∼11 mg/ml in buffer C. Crystallization was performed at 18 °C by using a Phoenix crystallization robot (Art Robbins) employing the sitting-drop vapor-diffusion method, with mixing 0.25 μl of protein and 0.25 μl of reservoir to equilibrate against 75 μl reservoir solution. The following commercially available screens were used: SaltRx™, PEG/Ion™ 1 & 2 Screen, Index™, and PEG Rx™ 1 & 2 (Hampton Research). Crystals were observed under condition 19 of Index™. Optimized crystals were subsequently obtained within one day using 0.056 M NaH2PO4, 1.344 M K2HPO4, pH 8.0, and 15% glycerol as reservoir, with mixing 2 μl of protein and 2.5 μl of reservoir to equilibrate against 500 μl reservoir solution.
Crystals were flash-cooled in a 100-K nitrogen-gas stream. A dataset to 2.50 Å resolution was collected using synchrotron radiation at wavelength 0.98 Å at beamline P11 of DESY, Hamburg. Diffraction data were processed with the program XDS (Kabsch, 2010). The space group was determined as C2, with unit-cell parameters a = 100.89 Å, b = 47.67 Å, c = 88.43 Å, β = 122.35°. Diffraction data statistics are given in Table 1 .
Table 1.
Data collection and refinement statistics.
| MERS-CoV PLpro | |
|---|---|
| Data collection statistics | |
| Space group | C2 |
| Unit-cell dimensions (Å, °) | a = 100.89, b = 47.67, c = 88.43 |
| β = 122.35 | |
| Wavelength (Å) | 0.98 |
| Vm (Å3/Da) | 2.53 |
| Solvent content (%) | 51.34 |
| Resolution range (Å) | 42.62–2.50 (2.64–2.50) |
| Number of unique reflections | 12337 |
| Rmerge | 0.059 (0.472) |
| Rpim1 | 0.025 (0.194) |
| Completeness (%) | 99.0 (98.3) |
| Mean I/σ (I) | 19.2 (3.9) |
| Multiplicity | 6.6 (6.8) |
| Refinement statistics | |
| Rcryst (%)2 | 18.7 (23.6) |
| Rfree (%)2 | 23.4 (30.3) |
| No. of atoms | |
| Protein | 2462 |
| Ligand/ion | 1 |
| Water | 94 |
| Clashscore3 | 2 |
| r.m.s.deviation in bond lengths (Å) | 0.01 |
| r.m.s.deviation in bond angles (°) | 1.13 |
| Average B-factor for all atoms (Å2) | 61 |
| Ramachandran plot | |
| Residues in favored regions (%) | 96.8 |
| Residues in additionally allowed regions (%) | 3.2 |
| Residues in outlier regions (%) | 0 |
Rpim (Weiss and Hilgenfeld, 1997).
Rcryst = ∑hkl|Fo(hkl) − Fc(hkl)|/∑hkl Fo(hkl). Rfree was calculated for a test set of reflections (4.9%) omitted from the refinement.
Clashscore is defined as the number of clashes calculated for the model per 1000 atoms (including hydrogens) of the model. Hydrogens were added by MolProbity (Chen et al., 2010).
2.3. Phase determination, model building and refinement
The structure of the MERS-CoV PLpro was solved by molecular replacement using the program BALBES (Long et al., 2008). The program selected molecule A of the SARS-CoV PLpro (PDB: 2FE8, Ratia et al., 2006) as the most suitable search model. The resulting model for the MERS-CoV PLpro was inspected and rebuilt using Coot (Emsley et al., 2010), and refined using autoBUSTER (Bricogne et al., 2011). The final refinement statistics are presented in Table 1. Atomic coordinates and structure factors have been deposited in the PDB with accession code 4P16. All figures except Fig. 3 and Supplementary Fig. 1 have been prepared using Pymol (Schrödinger; http://www.pymol.org/).
Fig. 3.

Structure-based alignment of MERS-CoV PLpro (GenBank: AFV09327.1), SARS-CoV PLpro (GenBank: AY278741.1), and TGEV PL1pro (GenBank: AJ271965.2). Secondary-structure elements of MERS-CoV PLpro (top) and SARS-CoV PLpro (bottom) are indicated. Residues of the catalytic triad are marked by black asterisks. The four cysteine residues coordinating the zinc ion are marked by black inverted triangles. The mobile β 15–β 16 loop (corresponding to BL2 of USP14) is indicated by a dashed purple box. Residues that are identical in the three CoV proteases are marked by white letters in red boxes with blue outline, while similar residues are marked by red letters and a blue outline. The figure was created by using the program ESPript (Gouet et al., 1999).
2.4. Site-directed mutagenesis
Using the pET28a-PLpro plasmid as template, site-directed mutagenesis (L106W mutation) was performed by PCR with the following primers (mutated codons shown in bold and underlined): gtacgttctctcaaatggagtgataataat and acaattattatcactccatttgagagaacg. The PCR products were digested by DpnI (Thermo Scientific) and transformed to DH5α directly. The positive clones were incubated in LB medium overnight and the bacterial culture was harvested next day. The plasmids were purified from the bacteria using the GeneJET Plasmid Miniprep Kit (Thermo Scientific). All DNA plasmids were sequenced and the correctness of the mutation was verified. The procedures for the expression of the construct carrying the L106W mutation and the purification of the corresponding protein were the same as described above for the wild-type protein.
2.5. Assays of MERS-CoV PLproin vitro activity
Before usage of the freshly prepared MERS-CoV PLpro for determination of the enzyme kinetics, the number of free (non-oxidized) cysteine residues was determined by titration with Ellman’s reagent (Riddles et al., 1983). All procedures followed the standard protocol of the Ellman’s Reagent kit (Thermo Scientific). The resulting number of 13.4 ± 0.3 free cysteine residues (out of 13 in the amino-acid sequence) showed that in the bulk of our enzyme preparation, the catalytic Cys111 was in the free state and fully reduced.
We also examined the influence of ethylene diamine tetraacetic acid (EDTA) on the enzyme’s activity, but found no effect of concentrations up to 10 mM. However, at a concentration of 20 mM, a decrease of enzymatic activity was observed, presumably due to the removal of Zn2+ from the PLpro zinc finger. Accordingly, the kinetic assays were carried out in the absence of EDTA.
All enzymatic assays were performed in 20 mM Tris–HCl, 150 mM NaCl, pH 7.9, 2 mM dithiothreitol (DTT), using a 96-well microtiter plate. Three fluorogenic substrates, Cbz-Arg-Leu-Arg-Gly-Gly-7-amino-4-methylcoumarin (Z-RLRGG-AMC) (Bachem), Z-LRGG-AMC and ubiquitin-AMC (Ub-AMC) (BostonBiochem), were used. The enzymatic cleavage reactions were monitored at 25 °C by measuring the increased fluorescence (λ ex: 360 nm; λ em: 460 nm) resulting from AMC release, using an Flx800 fluorescence spectrophotometer (BioTek). Reactions were started by addition of the substrate to the microtiter plate. The kinetic assays were run under the following conditions: 1 μM MERS-CoV PLpro with different concentrations (10–100 μM) of Z-RLRGG-AMC in a final volume of 100 μl, 1 μM MERS-CoV PLpro with different concentrations (20–160 μM) of Z-LRGG-AMC in a final volume of 100 μl, or 100 nM PLpro with different concentrations (0.2–1.2 μM) of Ub-AMC in a final volume of 50 μl. In case of the PLpro carrying the L106W mutation, the following conditions were used: 125 nM enzyme with different concentrations (10–100 μM) of Z-RLRGG-AMC in a final volume of 100 μl, 250nM enzyme with different concentrations (10–100 μM) of Z-LRGG-AMC in a final volume of 100 μl, or 125 nM PLpro with different concentrations (0.1–1.0 μM) of Ub-AMC in a final volume of 50 μl. Initial velocities were determined from the linear section of the curve. Since no saturation could be observed, the data were fitted to the equation v/[E]tot. = k app[S], where k app approximates k cat/K M, as described in Barretto et al. (2005) and Wojdyla et al. (2010). A calibration curve was generated by measuring the fluorescence of free AMC in reaction buffer at concentrations ranging from 0.005 μM to 2.5 μM.
3. Results and discussion
3.1. Overall structure of MERS-CoV PLpro
The MERS-CoV PLpro molecule is divided into two parts, the N-terminal ubiquitin-like (Ubl) domain and the catalytic domain (Fig. 1 A). The Ubl domain consists of the 62 N-terminal amino-acid residues. It comprises five β-strands, one α-helix, and one 310-helix (η) in the order β1-β2-α1-β3-β4-η1-β5. The catalytic domain forms an extended right-hand scaffold, which comprises three distinct subdomains: thumb, fingers, and palm. The thumb domain consists of six α-helices and four β-strands with the order α2-α3-β6-β7-α4-β8-α5-α6-α7-β9. The fingers domain contains β10–β13, the N-terminal 16 residues (Glu231–Leu246) of β14, η2, α8, and the C-terminal 7 residues (Phe312–Ser318) of β19. It also includes a zinc finger, in which four cysteine residues (Cys191, Cys194, Cys226, and Cys228) coordinate a zinc ion (Fig. 1A and B). The palm domain comprises six β-strands: β15–β18, the C-terminal 10 residues (Ser247–Thr256) of β14, and the N-terminal 10 residues (Asp302–Leu311) of β19. In the loop Gly271-Gly277 between β15 and β16, residues Ile272–Ala275 are not defined by electron density, indicative of high flexibility. The substrate-binding site is a solvent-exposed region between the palm and thumb domains (Fig. 1A and C). At the center of this region, the catalytic triad consisting of Cys111, His278, and Asp293 is located (Fig. 1C).
Fig. 1.

Structure of the MERS-CoV papain-like protease (PLpro). (A) Cartoon view of the enzyme’s overall structure. α-Helices (cyan) and β-strands (purple) are numbered, polypeptide segments devoid of repetitive secondary structure, including loops and turns, are brown. The ubiquitin-like (Ubl) domain is encircled by a red dashed line. The catalytic domain consists of the thumb, fingers, and palm subdomains. The structural zinc ion in the fingers domain is indicated by a gray sphere. The Cα atoms of the catalytic-site cysteine (111), histidine (278), and aspartate (293) residues are also shown (yellow, blue, and red sphere, respectively). The red arrow indicates the substrate-binding region and points to the catalytic site. (B) The four cysteine ligands (Cys191, Cys194, C226 and C228) and the structural zinc ion (gray sphere) in the zinc ribbon of the fingers domain. An Fo-Fc omit density (green; contoured at 5 σ above the mean) for the zinc is shown. Sulfur atoms are shown in yellow, oxygen in red, nitrogen in blue, and carbon in light blue. The coordinative bonds between the sulfur atoms and the zinc ion are indicated by dashed red lines. (C) The catalytic triad: Cys111, His278, and Asp293. Atom colors: carbon, yellow; oxygen, red; nitrogen, blue. A 2Fo-Fc electron density (gray; contoured at 1.0 σ above the mean) is also displayed. CME111: Cys111 covalently modified by β-mercaptoethanol.
Interestingly, the segment 283-RLKGG↓Li-289, located in the connection between the active-site residues His278 and Asp293, constitutes a potential autocleavage site for the MERS-CoV PLpro. Whereas we have some preliminary evidence for partial autoprocessing of our protease preparation at this site (data not shown), the electron density maps suggest that no cleavage has occurred in the crystallized protein at this position. Residues 283–285 are at the C-terminus of strand β16 and the two glycines are the central residues of a β-turn that leads into strand β17, which starts with Leu288. Thus, the residues concerned are part of well-defined secondary-structure elements and therefore, although largely accessible to solvent, cannot be accommodated by the substrate-binding site of the protease. However, whether or not in-trans PLpro autocleavage occurs at this position in the viral polyprotein, remains to be investigated.
3.2. Comparisons of the overall fold
3.2.1. The Ubl domain
As the Ubl domain and the catalytic domain of the MERS-CoV PLpro are two independent domains, we treated them separately in searching the Protein Data Bank (PDB) for structural similarity using the DALI server (Holm and Rosenström, 2010). However, we note that there are no variations in the relative orientation of these domains with respect to each other, as the Ubl domain is anchored to the core domain by two strong salt-bridges in MERS-CoV PLpro (Arg16...Glu64, 2.9 Å; Asp39 … His81, 3.5 Å) as well as in SARS-CoV PLpro (the TGEV PL1pro lacks the Ubl domain). The structural comparisons show that the MERS-CoV Ubl domain is similar to other proteins or domains featuring the ubiquitin fold (Table 2 , Fig. 2 A). The Ubl domain of SARS-CoV PLpro is essential for downregulating IRF3 or the NF-κB antiviral signaling pathway, but the Ubl domain alone is not sufficient to do so (Frieman et al., 2009). On the other hand, the Ubl domain is often involved in protein–protein interactions (Mueller and Feigon, 2003, Su and Lau, 2009, Hartmann-Petersen and Gordon, 2004) and it is conceivable that the Ubl domain of MERS-CoV PLpro could interfere with host signaling-pathway proteins by providing a binding scaffold, which is necessary for the catalytic domain to accomplish its function of antagonizing the host’s innate immune response.
Table 2.
Structural comparisons of MERS-CoV PLpro with other proteins.
| MERS-CoV PLpro ubiquitin-like (Ubl) domain |
||||||
|---|---|---|---|---|---|---|
| PDB/chain ID | Z score | RMSD (Å) | Cα1 | % id2 | References | |
| SARS-CoV PLpro Ubl | 2FE8/A | 11.0 | 1.1 | 61/64 | 28 | Ratia et al. (2006) |
| DSK2 | 2BWF/B | 5.5 | 2.1 | 54/77 | 17 | Lowe et al. (2006) |
| hHR23A | 1P98/A | 5.2 | 2.1 | 56/78 | 7 | Mueller and Feigon (2003) |
| MERS-CoV PLpro catalytic domain (CD) |
||||||
|---|---|---|---|---|---|---|
| PDB/chain ID | Z score | RMSD (Å) | Cα1 | % id2 | References | |
| SARS-CoV PLpro CD | 2FE8/A | 27.4 | 2.4 | 246/251 | 32 | Ratia et al. (2006) |
| TGEV PL1pro | 3MP2/A | 18.7 | 3.1 | 198/211 | 23 | Wojdyla et al. (2010) |
| USP14 | 2AYN/B | 13.0 | 3.1 | 199/337 | 14 | Hu et al. (2005) |
| USP21 | 3I3T/A | 12.8 | 3.0 | 196/303 | 15 | Ernst et al. (2013) |
| USP2 | 2HD5/A | 12.8 | 3.1 | 198/315 | 15 | Renatus et al. (2006) |
| USP7 | 1NB8/B | 10.5 | 3.5 | 198/333 | 15 | Hu et al. (2002) |
Aligned Cα atoms/total Cα atoms.
Sequence identity.
Fig. 2.

Comparison of the MERS-CoV PLpro with other coronavirus PLpros and human ubiquitin-specific protease 14 (USP14). (A) Superposition of the MERS-CoV PLpro Ubl domain (green) with the SARS-CoV PLpro Ubl domain (purple, PDB: 2FE8, chain A, Ratia et al., 2006) and yeast DSK2 (yellow, PDB: 2BWF, chain B, Lowe et al., 2006). All three adopt the globular β-grasp fold. The N and C termini of the MERS-CoV PLpro Ubl domain are marked. (B) Superposition of the catalytic domain (CD) of MERS-CoV PLpro (green) with the SARS-CoV PLpro CD (red, PDB: 2FE8, chain A, Ratia et al., 2006) and with TGEV PL1pro (blue, PDB: 3MP2, Wojdyla et al., 2010). Four regions of structural differences are apparent (encircled by dashed black lines and labeled with roman numbers). The structural zinc ion in the fingers domain of each protease is indicated by a green, red, and blue sphere, respectively. (C) Side-by-side comparison of USP14 (left, purple, PDB: 2AYN, chain B, Hu et al., 2005) and MERS-CoV PLpro CD (right, green). The Cα positions (yellow, blue, and red sphere, respectively) of the catalytic-triad cysteine, histidine, and aspartate residues are shown for each protease. The two “blocking loops”, BL1 and BL2, of USP14 are highlighted in cyan. The loop β14-β15 (salmon) of MERS-CoV, which corresponds to BL1 of USP14, is oriented towards the opposite direction in the PLpro. The dashed red line indicates the loop β15-β16 in MERS-CoV PLpro (corresponding to BL2 of USP14) that was not defined by electron density in our structure. The dashed black lines indicate disordered regions in USP14. The structural zinc ion of MERS-CoV PLpro is indicated by a gray sphere. The black arrow indicates the substrate binding region of each protease. The N and C termini of each enzyme are marked with letters in italics.
3.2.2. The catalytic domain: comparison with other coronaviral PLpros
The catalytic domain of the MERS-CoV PLpro is more similar to that of the SARS-CoV (Ratia et al., 2006) than to the PL1pro of Transmissible Gastroenteritis Virus (TGEV; Wojdyla et al., 2010) (Table 2). There are four regions of significant structural difference between the coronaviral PLpros (Fig. 2B). The first (Region I) concerns the two helices α2 and α3 in the N-terminal region of the thumb domain of the MERS-CoV and SARS-CoV enzymes, which are absent in the TGEV PL1pro (Figs. 2B and 3 ). The second region (II in Fig. 2B) of major structural differences concerns the four β-strands of the fingers domain. Two β-hairpins, which provide the cysteine residues for binding the structural zinc ion in this region, are twisted to different degrees among the three structures. The third structurally different area (III in Fig. 2B) is the connecting region between the fingers and palm domains. In the MERS-CoV PLpro, the C-terminal 10 residues of β14 extend into the palm domain and an 8-residue loop (Thr257–Val264) connects β14 and β15. In the SARS-CoV enzyme, the corresponding strand is divided into two separate β-strands, β12 and β13, the latter of which is mostly part of the palm domain, and a shorter loop formed by 5 residues (Gln256 - Leu260) connects β13 and β14. In the TGEV PL1pro, there are two loops (Ser157–Thr160 and Val166–Val170) and one 310-helix (Pro161–Phe165) connecting the fingers and palm domains. The different connections in the various PLpros might lead to different mutual orientations of the two domains, which in turn might affect the enzymatic activities of the individual PLpros. Finally, Region IV (Fig. 2B) concerns the loop between β15 and β16 (Gly271–Gly277), four residues of which are not defined by electron density in MERS-CoV PLpro. The corresponding region in TGEV PL1pro (Gly177–Gly182) and in SARS-CoV PLpro (Gly267–Gly272) is shorter by one residue compared to the MERS-CoV enzyme; it forms a loop between β8 and β9 in TGEV PL1pro and a short 310-helix between β14 and β15 in the SARS-CoV enzyme. However, this region is very flexible and adopts different positions in the three copies of the SARS-CoV PLpro in the asymmetric unit of the crystal (Ratia et al., 2006). In the recently reported crystal structure of the SARS-CoV PLpro (C112S mutant) in complex with ubiquitin (Chou et al., 2014), the β14-β15 loop shows large conformational differences compared to its position in the free enzyme. The two glycines framing this loop are absolutely conserved among the coronavirus PLpros, but the residues between them are different in each of the enzymes, suggesting that there must be differences in the interaction between the loop and the substrates.
3.2.3. Comparison with cellular ubiquitin-specific proteases
Even though the catalytic domain of the MERS-CoV PLpro only shares 12–15% sequence identity with the cellular ubiquitin-specific proteases (USPs), it features largely the same fold as the USPs with known three-dimensional structures (Table 2). A side-by-side comparison of the catalytic domain of the MERS-CoV PLpro with USP14 is shown in Fig. 2C. The most significant differences are located in the connecting region between the fingers and palm domains. The two “blocking loops”, BL1 and BL2, of USP14 regulate the deubiquitinating activity (Hu et al., 2005) (Fig. 2C). BL1 of USP14 connects the fingers and palm domains. It is a 22-residue loop between β8 and β9, exposed to the substrate-binding surface. The corresponding loop in MERS-CoV PLpro is much shorter (8 residues) and connects β14 and β15. Furthermore, this short loop does not face the substrate-binding region but rather points to the bottom of the thumb domain of MERS-CoV PLpro (Fig. 2C), and can thus not be considered a “blocking loop”. As a consequence, the substrate-binding site (indicated by the black arrow in Fig. 2C) is larger in MERS-CoV PLpro than in the USPs, probably enabling the enzyme to not only bind ubiquitin but also viral polyprotein. Connecting β10 and β11, the other blocking loop, BL2 (residues Gly427–Gly433), of USP14 is near the active site and undergoes conformational change upon substrate binding (Hu et al., 2005); this loop corresponds to β15-β16 (residues Gly271–Gly277) of MERS-CoV PLpro which unfortunately lacks electron density for four of its seven residues (see above).
3.3. The active site of MERS-CoV PLpro
The MERS-CoV PLpro possesses a catalytic triad consisting of Cys111, His278, and Asp293 (Figs. 1C and 4 A). Cys111 in the MERS-CoV PLpro is situated at the N-terminus of α4 and points into the substrate-binding cleft between the palm and thumb domains. There is clear electron density indicating that the catalytic cysteine has been modified by disulfide bond formation with β-mercaptoethanol (BME) during crystallization of the enzyme. His278 is located at the N-terminus of β16; its Nδ1 atom is 4.4 Å from the sulfur of Cys111. This large distance is likely due to the BME-modification of Cys111; in most papain-like protease structures, the distances are between 3.7 Å and 4.0 Å (Fig. 4A). Although the side-chain of His278 is somewhat displaced from its regular position in the catalytic triad, Asp293 and Cys111 align well with their counterparts in SARS-CoV PLpro and USP14 (Fig. 4A).
Fig. 4.
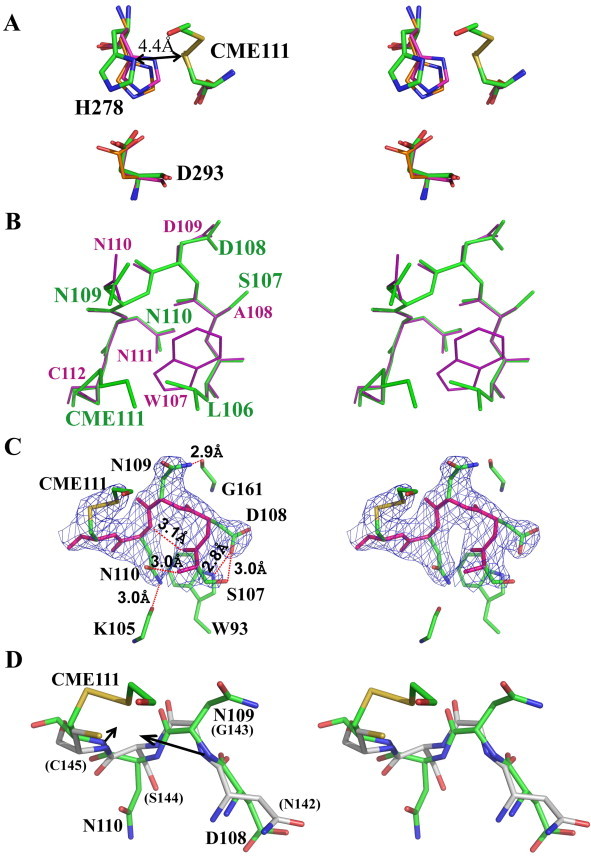
Active site of MERS-CoV PLpro. (A) Superposition (stereo view) of catalytic-triad residues of MERS-CoV PLpro (thick green sticks), SARS-CoV PLpro (thin purple sticks, PDB: 2FE8, chain A, Ratia et al., 2006), and USP14 (thin orange sticks, PDB: 2AYN, chain B, Hu et al., 2005). The distance between Nδ1 of His278 and the sulfur of Cys111 in MERS-CoV PLpro is indicated. CME111: Cys111 covalently modified by β-mercaptoethanol. (B) Superposition (stereo view) of the polypeptide segment preceding the catalytic cysteine in MERS-CoV PLpro (106-LSDNN(CME)-111, green, shown as thick sticks, with bold green labels) and SARS-CoV PLpro (107-WADNNC-112, purple, thin sticks, smaller purple labels). (C) Stereo view of the same β-turn, Ser107-Asp108-Asn109-Asn110, in MERS-CoV PLpro, with the corresponding 2Fo-Fc electron density (blue; contoured at 1.5 σ above the mean). The main chain of the β-turn and of the (modified) catalytic residue CME111 is shown in pink, while side-chains are green. Gly161, Lys105 (main chain), and Trp93 (side-chain) are displayed as well. Hydrogen bonds are indicated by dashed red lines. (D) Stereo view of a superposition of 108-DNN(CME)-111 in MERS-CoV PLpro (green) and 142-NGSC-145 in SARS-CoV Mpro (gray, PDB: 2BX3, Tan et al., 2005). Residues of MERS-CoV PLpro and SARS-CoV Mpro (in brackets) are labeled. The RMSD of all main-chain atoms between this pair of four residues is ∼0.62 Å. The two black arrows point to the presumable position of the oxyanion in the transition state of the proteolytic reaction.
The transition state of the proteolytic reaction catalyzed by papain-like cysteine proteases is stabilized in the oxyanion hole, through two hydrogen bonds usually donated by the main-chain amide of the catalytic cysteine residue and by the side-chain amide of a glutamine or asparagine residue five or six positions N-terminal to the catalytic cysteine. For example, the latter residue is Asn in USP14 and HAUSP (Hu et al., 2002, Hu et al., 2005), and Gln in the ubiquitin C-terminal hydrolases (UCH)-L3 and UCH-L1 (Johnston et al., 1997). In TGEV PL1pro, there is also a glutamine residue (Gln27) at this position (Wojdyla et al., 2010). In SARS-CoV PLpro, the corresponding residue is Trp107, the indole NH of which has been proposed to stabilize the oxyanion transition state (Fig. 4B). Accordingly, replacing Trp by Ala abrogates the protease activity (Ratia et al., 2006). Surprisingly, the corresponding residue in the MERS-CoV PLpro is Leu106, the side-chain of which is incapable of hydrogen bonding (Fig. 4B).
How then is the oxyanion transition state stabilized in MERS-CoV PLpro? Ratia et al. (2006) have discussed Asn110 as a potential additional component of the oxyanion hole in the SARS-CoV PLpro. This residue is highly conserved amongst the coronaviral PLpros, including the MERS-CoV enzyme (Asn109) and in TGEV PL1pro (Asn30). In all three coronavirus PLpros of known three-dimensional structure as well as in USP14, this Asn residue occupies position i+2 of a β-turn (Ser107-Asp108-Asn109-Asn110 in MERS-CoV) that precedes the catalytic cysteine in the polypeptide chain. However, the side-chain of Asn109 is oriented away from the oxyanion hole and is involved in a strong (2.9 Å) hydrogen-bond with the carbonyl oxygen of conserved Gly161 of the thumb subdomain (Fig. 4C); therefore, it is unlikely to undergo a conformational change that will bring it into the neighborhood of the catalytic center. The other two potential hydrogen-bonding donors in this β -turn, Ser107 and Asn110, are heavily engaged in hydrogen bonds across the turn (Fig. 4C) and therefore equally unlikely to undergo the conformational changes necessary to reorient their side-chains towards the catalytic center.
So if none of the side-chains in this β -turn is a likely component of the oxyanion hole, what about main-chain amides? We notice that the segment between residues 108 (position i+1 of the β-turn) and 111 can be superimposed (with an RMSD of ∼0.62 Å for main-chain atoms) onto the loop (142-NGSC-145) preceding the catalytic nucleophile, Cys145, in SARS-CoV main protease (Mpro; PDB: 2BX3, Tan et al., 2005) (Fig. 4D). The Mpro is a cysteine protease comprising a chymotrypsin-like fold; in these enzymes, the oxyanion hole is formed by the main-chain amides of the catalytic cysteine (or serine) and of the penultimate residue (Taranto et al., 2008, Wu et al., 2013). The penultimate residue before the catalytic nucleophile in chymotrypsin-like proteases is absolutely conserved as a glycine, whereas in MERS-CoV PLpro, the conserved Asn109 resides at this position. However, this asparagine is in a left-handed (αL) conformation, with positive ϕ and ψ angles (56° and 34°, resp.) in the Ramachandran plot, just like the conserved glycine in the catalytically competent conformation of chymotrypsin-like proteases (Verschueren et al., 2008, Tan et al., 2005). In fact, Asn and Asp are the only residues apart from Gly, for which this conformation is observed at a significant rate in protein structures (Hutchinson and Thornton, 1994). (Incidentally, in MERS-CoV PLpro, the i+1 residue of this β-turn, Asp108, is also in an αL conformation, so that we have the rare situation here of a β-turn with both i+1 and i+2 residues having positive ϕ/ψ angles). In spite of the Asn109 main-chain N atom being relatively well superimposable onto the corresponding atom of Gly143 in SARS-CoV Mpro, we note that the N-H vector of this amide does not point towards the position that would likely be assumed by the oxyanion. Whether or not the necessary (minor) rearrangement may occur, remains to be answered by elucidating the structure of a complex between the MERS-CoV PLpro and a transition-state analogue.
In any case, our mutation experiment described below clearly demonstrates that the oxyanion hole of the MERS-CoV PLpro is deficient.
3.4. The substrate-binding site of MERS-CoV PLpro
In the MERS-CoV polyproteins, the three cleavage sites for the PLpro are KLIGG↓DV (Nsp1-2), RLKGG↓AP (Nsp2-3), and KIVGG↓AP (Nsp3-4). The P1 and P2 positions are strictly conserved as glycine residues. P5 is Lys or Arg and P4 Leu or Ile, whereas P3 can be Ile, Lys, or Val. In the SARS-CoV polyprotein, cleavage by the PLpro also occurs behind LXGG, but X is either Asn or Lys (Barretto et al., 2005), never a hydrophobic residue such as Ile or Val as in MERS-CoV. The LXGG motif is also present at the C-terminus of ubiquitin. In addition to the crystal structure of the complex between the C112S mutant of SARS-CoV PLpro and ubiquitin (Chou et al., 2014), the structure of the wild-type SARS-CoV PLpro with Ubal has very recently been reported (Ratia et al., 2014; Ubal is ubiquitin with the C-terminal carboxylate reduced to an aldehyde, which forms a covalent bond with the catalytic Cys residue). However, as the PDB coordinates for the latter structure have not been released yet, we rely for our further discussion on the structures of the ubiquitin complex of the SARS-CoV PLpro (C112S mutant) and on the structure of USP14-Ubal; Hu et al., 2005). The structural homology between these two enzymes and MERS-CoV PLpro allowed us to deduce conclusions concerning the substrate-binding site of the latter. When we superimposed the substrate-binding sites of MERS-CoV PLpro, SARS-CoV PLpro(C112S), and USP14, we found a remarkable degree of structural conservation but also some important differences (Fig. 5 ).
Fig. 5.

Substrate-binding site of MERS-CoV PLpro as deduced from the homologous structures of SARS-CoV PLpro and USP14. (A) The substrate-binding site in SARS-CoV PLpro (purple, PDB: 4M0W (complex with ubiquitin), Chou et al., 2014). The five C-terminal residues of ubiquitin, RLRGG, are displayed as cyan sticks. Ubiquitin residues are labeled by cyan letters in italics, and residues of SARS-CoV PLpro involved in binding the C-terminal residues of ubiquitin are labeled with black regular letters. The salt-bridge Glu168 … Arg42 and the hydrogen bond Tyr265 … Arg74 are highlighted in insets. (B) The substrate-binding site of USP14 (red, PDB: 2AYO, complex with ubiquitin-aldehyde, Hu et al., 2005). The five C-terminal residues of ubiquitin, RLRGG-aldehyde, in the complex are displayed as blue sticks (GLZ, amino-acetaldehyde, i.e. the aldehyde of the C-terminal G76). The P1-P5 residues of ubiquitin are labeled by blue letters in italics. Residues of USP14 involved in binding the C-terminal residues of ubiquitin are labeled with regular black letters. The covalent bond between sulfur (Cys113) and carbon (GLZ76) is displayed by a green line. The salt-bridge Glu201 … Arg72 is highlighted in the inset. (C) The substrate-binding site of MERS-CoV PLpro. Residues that are different from those of SARS-CoV PLpro and USP14 are underlined. The supposed positions of the subsites S1–S5 are indicated. (D) Structure-based alignment of residues involved in defining specificity subsites in MERS-CoV PLpro, SARS-CoV PLpro, and USP14. Conserved residues are in red. Arg168 of MERS-CoV PLpro (corresponding to Glu168 and Glu201 in SARS-CoV PLpro and USP14, respectively) is in blue. Phe269 of the MERS-CoV enzyme (Tyr265 in SARS-CoV PLpro) is green. Residues involved in shaping subsites are labeled by the symbol for the corresponding subsite (S1–S5) above the alignment. Residues contributing to S1, S2, and S4, but not S3 and S5, are conserved. (E, F) Electrostatic surfaces of the substrate-binding regions of SARS-CoV PLpro (E) and MERS-CoV PLpro (F), colored according to electrostatic potential (blue, positive potential; red, negative potential). The electrostatic surfaces were calculated using the APBS plugin in PyMOL (Baker et al., 2001). The contouring level is -8 kBT/e to 8 kBT/e. Orientation is the same as in the cartoon representations in (A and C). The S3 and S5 subsites are indicated by orange labels. (E) Glu162 and Glu168 of SARS-CoV PLpro are marked. (F) Ala162 and R168 of MERS-CoV PLpro are marked.
In MERS-CoV PLpro, the substrate-binding site is lined by residues Leu106–Tyr112 and Gly161–Arg168 of the thumb subdomain, and Phe269–Tyr279, Pro250, and Thr308 of the palm subdomain (Fig. 5C and D). Asn109, Cys111, the NH of Tyr112, Gly277, and His278 (all conserved in SARS-CoV PLpro and USP14) form the spatially restricted S1 site (Fig. 5C and D), which can only accommodate glycine as the P1 residue. Pro163 (Leu163 in SARS-CoV PLpro, Gln196 in USP14), Asp164 (main chain), Gly277 (conserved), and Tyr279 (side-chain, conserved) are involved in shaping the equally restricted S2 subsite, which again is specific for glycine (Fig. 5C and D). Replacement of Tyr274 in SARS-CoV PLpro (corresponding to Tyr279 of the MERS-CoV enzyme) by Ala leads to a loss of protease activity (Barretto et al., 2005).
The S3 subsite of MERS-CoV PLpro features important differences from the one in the SARS-CoV enzyme. In the SARS-CoV PLpro(C112S)-Ub and SARS-CoV PLpro-Ubal complexes (Chou et al., 2014, Ratia et al., 2014), the main-chain amide of P3-Arg (substrate residues are indicated in italics in what follows) forms an H-bond with the side-chain OH of Tyr265 (Fig. 5A and S1A). In MERS-CoV PLpro, this residue is replaced by Phe269 (Fig. 5C and D). Replacement of Tyr265 by Phe in SARS-CoV PLpro reduces the peptidolytic activity of the enzyme by a factor of 2.4 and its deubiquitinating activity by 57% (Chou et al., 2014). Another potentially important difference between the S3 subsites of SARS-CoV PLpro and MERS-CoV PLpro is that Glu162 of the former is replaced by Ala162 in the latter (Fig. 5A and C), reducing the negative electrostatic potential of this region in the MERS-CoV PLpro (Fig. 5E and F). This might enable the MERS-CoV enzyme to accommodate hydrophobic P3 residues such as Ile or Val, as they occur at the PLpro cleavage sites of the MERS-CoV (but not the SARS-CoV) polyprotein.
The S4 subsite in MERS-CoV PLpro is a hydrophobic pocket formed by Thr308, Pro250, Phe269, and the Cβ atom of conserved Asp165. The side-chain carboxylate of the latter may interact with the amide of P4-Leu. Accordingly, replacement of this aspartate by Ala in SARS-CoV PLpro inactivates the enzyme (Chou et al., 2014). Phe269 is involved in both the S3 subsite (see above) and in making hydrophobic interactions with the P4-Leu of polyprotein cleavage sites or ubiquitin. In addition, the mobile β 15–β 16 loop probably contributes to the S4 site (as deduced from the structure of SARS-CoV PLpro in complex with ubiquitin (Chou et al., 2014)), but we are unable to provide a detailed description because of missing electron density (see above). In any case, however, the loop of the MERS-CoV enzyme is devoid of a large aromatic residue that could make interactions with Leu73 (P4) (and Leu71 (P6)) of ubiquitin, as observed for Tyr269 of SARS-CoV PLpro (Chou et al., 2014).
The S5 subsite of MERS-CoV PLpro is also different from that in USP14 and in the SARS-CoV PLpro. The side-chain of the P5-Arg forms a salt-bridge with Glu201 in the USP14-Ubal complex (Hu et al., 2005; Fig. 5B and S1B) and the same is observed for Glu168 of SARS-CoV PLpro with Ubal (Ratia et al., 2014). However, in the SARS-CoV PLpro(C112S) complex with ubiquitin (Chou et al., 2014), this same glutamate residue interacts with an arginine of the core of ubiquitin (Arg42) instead of the P5-Arg (Fig. 5A). This may be due to the position of Glu168 and hence the entire S5 site of the PLpro at the surface of the protease, near the entrance to the substrate-binding cleft. While not forming a pocket, the S5 is still important for substrate binding, because the Glu168 side-chain is at the core of a region with pronounced negative electrostatic potential (see Fig. 5E). Interestingly, this important glutamate of USP14 and SARS-CoV PLpro is replaced by Arg168 in MERS-CoV PLpro (Fig. 5D), changing the electrostatic potential of the S5 site of the latter to more positive and therefore less ideal for interacting with the P5-Arg or Arg42 of ubiquitin, or the P5 Lys/Arg of the MERS-CoV polyprotein cleavage sites (Fig. 5F). Indeed, Chou et al. (2014) have replaced Glu168 of SARS-CoV PLpro by Arg and found a 25-fold decrease in deubiquitinating activity of the enzyme.
Mesecar and colleagues have demonstrated that the SARS-CoV PLpro is a druggable target. They used their crystal structure of the enzyme (Ratia et al., 2006) in a virtual screening campaign and reported crystal structures of the complex with the optimized hit compound, GRL0617 (5-amino-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl]benzamide) and derivatives thereof (Ratia et al., 2008, Baez-Santos et al., 2014). Interestingly, the inhibitors do not bind to the catalytic center directly but near the S3 and S4 subsites as well as the mobile β 14–β 15 loop (β 15–β 16 in MERS-CoV PLpro) (Ratia et al., 2008, Baez-Santos et al., 2014). As there are important differences in the S3 subsite as well as in the mobile loop between the SARS-CoV and the MERS-CoV PLpros, it is unlikely that these compounds will inhibit the PLpro and the replication of MERS-CoV. However, virtual or real screening approaches might be expected to result in the discovery of different small-molecule compounds binding to a similar site in MERS-CoV PLpro.
3.5. In-vitro peptide-hydrolysis and deubiquitinating activities of wild-type MERS-CoV PLpro and its L106W variant
We tested the PLpro-catalyzed hydrolysis of the peptides Z-RLRGG-7-amino-4-methylcoumarin (AMC) and Z-LRGG-AMC as mimics of the cleavage sites in the viral polyproteins. For this assay (and the one described below), only freshly prepared enzyme was used, in which the catalytic cysteine residue was shown to be in a free, reduced (and hence active) state by titration with Ellman’s reagent and by the absence of an effect of adding EDTA up to a concentration of 10 mM (see Materials & Methods). We found that the peptides were hydrolyzed by MERS-CoV PLpro in vitro, and that the initial rate of hydrolysis increased with raising the substrate concentration. However, we were unable to observe saturation of the reactions. This might indicate a large Km value and/or low enzyme efficiency. We used the pseudo-first-order rate constant, kapp, to estimate an approximate kcat/Km value. The kapp rates were 1.31 ± 0.14 min− 1 mM−1 for Z-RLRGG-AMC and 1.00 ± 0.01 min−1 mM−1 for Z-LRGG-AMC, values significantly lower than those reported for the SARS-CoV PLpro (Table 3 ). This may be explained by the apparent deficiency of the oxyanion hole of the MERS-CoV PLpro and the inability of the side-chain of Phe269 to form the hydrogen bond with the main-chain amide of P3-Arg observed for the corresponding Tyr265 in SARS-CoV PLpro (Chou et al., 2014). In order to examine whether this relatively low activity is caused by the deficiency of the oxyanion hole, i.e. the lack of a side-chain available for oxyanion stabilization through hydrogen bonding, we prepared the L106W mutant of the MERS-CoV PLpro and found its peptide-hydrolyzing activities on Z-RLRGG-AMC and Z-LRGG-AMC, respectively, to be 40-fold and 60-fold higher than those of the wild-type enzyme (Table 3). This is a clear demonstration of the deficiency of the oxyanion hole in the MERS-CoV PLpro and shows that the contribution to oxyanion stabilization by the main-chain amide of residue 109, if at all existent, is far less efficient than by a tryptophan residue in position 106, as found in the SARS-CoV PLpro.
Table 3.
Kinetic parameters of MERS-CoV PLpro.
| Z-RLRGG-AMC | Z-LRGG-AMC | Ub-AMC | |
|---|---|---|---|
| MERS-CoV PLpro | |||
| WT# | 1.31 ± 0.14⁎ | 1.00 ± 0.01 | (5.06 ± 0.35) × 102 |
| L106W | 53.27 ± 5.10 | 60.16 ± 7.00 | (1.70 ± 0.58) × 103 |
| SARS-CoV PLpro | |||
| WT (Lindner et al., 2005) | ∼3.6 | ∼7.86 × 102 | |
| WT (Barretto et al., 2005) | ∼4.48 × 103 | ||
Wild-type.
kapp (min−1 mM−1).
We also tested the deubiquitinating activity of the MERS-CoV PLpro in vitro using Ub-AMC as a substrate. We determined kapp as (5.06 ± 0.35) x 102 min−1 mM−1. This value is also lower than the rate constants reported for SARS-CoV PLpro by different research groups (Table 3). Again, the less-than-perfect oxyanion hole and the replacement of Tyr265 of SARS-CoV PLpro by Phe269 in MERS-CoV PLpro may be responsible for this. In addition, the replacement of Glu168 of SARS-CoV PLpro by Arg168 in the MERS-CoV enzyme very likely weakens the interaction with ubiquitin. The deubiquitinating activity of the MERS-CoV PLpro carrying the L106W mutation was found to be 3.4-fold higher than that of the wild-type enzyme (Table 3). Our results demonstrate that the MERS-CoV PLpro displays both peptidase and deubiquitinase activities in vitro and is thus suitable for screening chemical libraries for inhibitors.
4. Conclusions
The crystal structure of the MERS-CoV PLpro provides critical information on this important potential drug target. The unique architecture of the oxyanion hole, which differs from all papain-like proteases that have been structurally characterized so far, may be of fundamental interest. Because of unique features of its S3 and S5 subsites as well as in the flexible loop (β 15–β 16) covering the substrate-binding site, we expect that the MERS-CoV PLpro will show differences in ubiquitin recognition, compared to SARS-CoV PLpro and USP14. Both peptidase and deubiquitinating activities of MERS-CoV PLpro have been demonstrated in vitro. Introduction of the L106W mutation leads to a restoration of the oxyanion hole of the PLpro and an enhancement of both catalytic activities. Furthermore, the structural differences from homologous host enzymes such as USP14 should allow the design of antivirals devoid of too many side effects.
Acknowledgements
Technical assistance by Susanne Zoske is gratefully acknowledged. We thank Linlin Zhang and Guido Hansen for discussion. We also acknowledge the staff at beamline P11 of DESY, Hamburg, Germany. This work was supported by the European Commission through its “SILVER” project (contract No. HEALTH-F3-2010-260644) and by the German Center for Infection Research (DZIF). RH acknowledges support by the DFG Cluster of Excellence “Inflammation at Interfaces” (EXC 306). This work is dedicated to Professor Wolfram Saenger on the occasion of his 75th birthday.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2014.06.011.
Appendix A. Supplementary data
Schematic representation of ubiquitin binding in the SARS-CoV PLpro(C112S)–Ub and USP14–Ubal complexes. (A) Interactions between the S1 and S5 subsites of SARS-CoV PLpro(C112S) with the C-terminal pentapetide, RLRGG, of ubiquitin. C, N, and O atoms are displayed by small black, blue, and red balls, respectively. Residues of SARS-CoV PLpro are shown as pink sticks and carry purple labels, those of Ub as cyan sticks and marked by black labels in italics. Hydrogen bonds between SARS-CoV PLpro(C112S) and Ub are marked by green dashed lines. The two salt-bridges Glu162 … Arg74 and Glu168 … Arg42 are displayed by black dashed lines. (B) Interactions between the S1 and S5 subsites of USP14 with the C-terminal tetrapeptide RLRG-GLZ (amino-acetaldehyde, i.e., the aldehyde of the C-terminal G76) of Ubal (ubiquitin with the C-terminal carboxylate reduced to an aldehyde). The C, N, O, and S atoms are displayed as small black, blue, red, and yellow balls, respectively. Residues of USP14 are shown as green sticks and indicated by black labels. Residues of RLRG-GLZ are shown as blue sticks and marked by blue labels in italics. The hydrogen bonds between USP14 and RLRG-GLZ are marked by red dashed lines. The salt-bridge between Glu201 and Arg72 is displayed by a black dashed line. The covalent bond between sulfur (Cys113) and carbon (GLZ76) is displayed by a thick black line. This figure was made using the program LigPlot+ (Laskowski & Swindells, 2011).
References
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Assiri A., McGeer A., Perl T.M., Price C.S., Al Rabeeah A.A., Cummings D.A., Alabdullatif Z.N., Assad M., Almulhim A., Makhdoom H., Madani H., Alhakeem R., Al-Tawfiq J.A., Cotton M., Watson S.J., Kellam P., Zumla A.I., Memish Z.A., KSA MERS-CoV Investigation Team Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 2013;369:407–416. doi: 10.1056/NEJMoa1306742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y.M., Barraza S.J., Wilson M.W., Agius M., Mielech A.M., Davis N.M., Baker S.C., Larsen S.D., Mesecar A.D. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014;57:2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O.S., Vonrhein C., Womack T.O. Global Phasing Ltd; Cambridge, United Kingdom: 2011. BUSTER Version 2.11.4. [Google Scholar]
- Chen V.B., Arendall W.B.I.I.I., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.Y., Lai H.Y., Chen H.Y., Cheng S.C., Cheng K.W., Chou Y.W. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr. D Biol. Crystallogr. 2014;70:572–581. doi: 10.1107/S1399004713031040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz M.A., Chen Z., Banach B.S., Wang Y., Sun L., Ratia K., Baez-Santos Y.M., Wang J., Takayama J., Ghosh A.K., Li K., Mesecar A.D., Baker S.C. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A., Galiano M., Gorbalenya A.E., Memish Z.A., Perlman S., Poon L.L., Snijder E.J., Stephens G.M., Woo P.C., Zaki A.M., Zambon M., Ziebuhr J. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the coronavirus study group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.T., Baker S.C., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle I., Müller M.A., Kallies S., Gotthardt D.N., Drosten C. In-vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East Respiratory Syndrome (MERS) coronavirus infection. Virol. J. 2013;10:359. doi: 10.1186/1743-422X-10-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst A., Avvakumov G., Tong J., Fan Y., Zhao Y., Alberts P., Persaud A., Walker J.R., Neculai A.M., Neculai D., Vorobyov A., Garg P., Beatty L., Chan P.K., Juang Y.C., Landry M.C., Yeh C., Zeqiraj E., Karamboulas K., Allali-Hassani A., Vedadi M., Tyers M., Moffat J., Sicheri F., Pelletier L., Durocher D., Raught B., Rotin D., Yang J., Moran M.F., Dhe-Paganon S., Sidhu S.S. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339:590–595. doi: 10.1126/science.1230161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P., Courcelle E., Stuart D.I., Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Haagmans B.L., Al Dhahiry S.H., Reusken C.B., Raj V.S., Galiano M., Myers R., Godeke G.J., Jonges M., Farag E., Diab A., Ghobashy H., Alhajri F., Al-Thani M., Al-Marri S.A., Al Romaihi H.E., Al Khal A., Bermingham A., Osterhaus A.D., Alhajri M.M., Koopmans M.P. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 2014;14:140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann-Petersen R., Gordon C. Integral UBL domain proteins: a family of proteasome interacting proteins. Semin. Cell Dev. Biol. 2004;15:247–259. doi: 10.1016/j.semcdb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Hilgenfeld R., Peiris M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013;100:286–295. doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M., Li P., Li M., Li W., Yao T., Wu J.W., Gu W., Cohen R.E., Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- Hu M., Li P., Song L., Jeffrey P.D., Chenova T.A., Wilkinson K.D., Cohen R.E., Shi Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–3756. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson E.G., Thornton J.M. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S.C., Larsen C.N., Cook W.J., Wilkinson K.D., Hill C.P. Crystal structure of a deubiquitinating enzyme (human UCH-L3) at 1.8 Å resolution. EMBO J. 1997;16:3787–3796. doi: 10.1093/emboj/16.13.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilianski A., Mielech A.M., Deng X., Baker S.C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013;87:11955–11962. doi: 10.1128/JVI.02105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Ménard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Lytvyn V., Qi H., Lachance P., Ziomek E., Ménard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007;466:8–14. doi: 10.1016/j.abb.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F., Vagin A.A., Young P., Murshudov G.N. BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe E.D., Hasan N., Trempe J.F., Fonso L., Noble M.E., Endicott J.A., Johnson L.N., Brown N.R. Structures of the Dsk2 UBL and UBA domains and their complex. Acta Crystallogr. D Biol. Crystallogr. 2006;62:177–188. doi: 10.1107/S0907444905037777. [DOI] [PubMed] [Google Scholar]
- Lu I.L., Mahindroo N., Liang P.H., Peng Y.H., Kuo C.J., Tsai K.C., Hsieh H.P., Chao Y.S., Wu S.Y. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006;49:5154–5161. doi: 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- Meyer B., Müller M.A., Corman V.M., Reusken C.B., Ritz D., Godeke G.J., Lattwein E., Kallies S., Siemens A., van Beek J., Drexler J.F., Muth D., Bosch B.J., Wernery U., Koopmans M.P., Wernery R., Drosten C. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg. Infect. Dis. 2013 doi: 10.3201/eid2004.131746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielech A.M., Kilianski A., Baez-Santos Y.M., Mesecar A.D., Baker S.C. MERS-CoV papain-like protease has delSGylating and deubiquitinating activities. Virology. 2014;450–451:64–70. doi: 10.1016/j.virol.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T.D., Feigon J. Structural determinants for the binding of ubiquitin-like domains to the proteasome. EMBO J. 2003;22:4634–4645. doi: 10.1093/emboj/cdg467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S. The Middle East respiratory syndrome – how worried should we be? mBio. 2013;4:e00531-13. doi: 10.1128/mBio.00531-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., Baker S.C., Ghosh A.K., Mesecar A.D. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Kilianski A., Baez-Santos Y.M., Baker S.C., Mesecar A. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10:e1004113. doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renatus M., Parrado S.G., D’Arcy A., Eidhoff U., Gerhartz B., Hassiepen U., Pierrat B., Riedl R., Vinzenz D., Worpenberg S., Kroemer M. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusken C.B., Haagmans B.L., Müller M.A., Gutierrez C., Godeke G.J., Meyer B., Muth D., Raj V.S., Smits-De Vries.L., Corman V.M., Drexler J.F., Smits S.L., El Tahir Y.E., De Sousa R., van Beek J., Nowotny N., van Maanen K., Hidalgo-Hermoso E., Bosch B.J., Rottier P., Osterhaus A., Gortázar-Schmidt C., Drosten C., Koopmans M.P. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013;13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddles P.W., Blakeley R.L., Zerner B. Reassessment of Ellman’s reagent. Methods Enzymol. 1983;91:49–60. doi: 10.1016/s0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- Su V., Lau A.F. Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell. Mol. Life Sci. 2009;66:2819–2833. doi: 10.1007/s00018-009-0048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Verschueren K.H., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R., Chen K., Shen X., Jiang H., Hilgenfeld R. PH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranto A.G., Carvalho P., Avery M.A. QM/QM studies for Michael reaction in coronavirus main protease (3CLPro) J. Mol. Graph. Model. 2008;27:275–285. doi: 10.1016/j.jmgm.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren K.H., Pumpor K., Anemüller S., Chen S., Mesters J.R., Hilgenfeld R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008;15:597–606. doi: 10.1016/j.chembiol.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M.S., Hilgenfeld R. On the use of the merging R factor as a quality indicator for X-ray data. J. Appl. Cryst. 1997;30:203–205. [Google Scholar]
- Wojdyla J.A., Manolaridis I., van Kasteren P.B., Kikkert M., Snijder E.J., Gorbalenya A.E., Tucker P.A. Papain-like protease 1 from transmissible gastroenteritis virus: crystal structure and enzymatic activity toward viral and cellular substrates. J. Virol. 2010;84:10063–10073. doi: 10.1128/JVI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C., Huang Y., Lau S.K., Yuen K.Y. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2:1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.G., Cheng S.C., Chen S.C., Li J.Y., Fang Y.H., Chen Y.H., Chou C.Y. Mechanism for controlling the monomer-dimer conversion of SARS coronavirus main protease. Acta Crystallogr. D Biol. Crystallogr. 2013;69:747–755. doi: 10.1107/S0907444913001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Ooi A., Lee H.C., Wilmouth R., Liu D.X., Lescar J. Structure of the SARS coronavirus main proteinase as an active C2 crystallographic dimer. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2005;61:964–966. doi: 10.1107/S1744309105033257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main protease. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Chen X., Bian G., Tu J., Xing Y., Wang Y., Chen Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2013;95:614–626. doi: 10.1099/vir.0.059014-0. [DOI] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhu L., George S., Schmidt M.F., Al-Gharabli S.I., Rademann J., Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 2011;92:204–212. doi: 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of ubiquitin binding in the SARS-CoV PLpro(C112S)–Ub and USP14–Ubal complexes. (A) Interactions between the S1 and S5 subsites of SARS-CoV PLpro(C112S) with the C-terminal pentapetide, RLRGG, of ubiquitin. C, N, and O atoms are displayed by small black, blue, and red balls, respectively. Residues of SARS-CoV PLpro are shown as pink sticks and carry purple labels, those of Ub as cyan sticks and marked by black labels in italics. Hydrogen bonds between SARS-CoV PLpro(C112S) and Ub are marked by green dashed lines. The two salt-bridges Glu162 … Arg74 and Glu168 … Arg42 are displayed by black dashed lines. (B) Interactions between the S1 and S5 subsites of USP14 with the C-terminal tetrapeptide RLRG-GLZ (amino-acetaldehyde, i.e., the aldehyde of the C-terminal G76) of Ubal (ubiquitin with the C-terminal carboxylate reduced to an aldehyde). The C, N, O, and S atoms are displayed as small black, blue, red, and yellow balls, respectively. Residues of USP14 are shown as green sticks and indicated by black labels. Residues of RLRG-GLZ are shown as blue sticks and marked by blue labels in italics. The hydrogen bonds between USP14 and RLRG-GLZ are marked by red dashed lines. The salt-bridge between Glu201 and Arg72 is displayed by a black dashed line. The covalent bond between sulfur (Cys113) and carbon (GLZ76) is displayed by a thick black line. This figure was made using the program LigPlot+ (Laskowski & Swindells, 2011).