

ABSTRACT
Infectious bronchitis is considered to be one of the most devastating diseases in poultry. Control of its spread is typically attempted through biosecurity measures and extensive vaccination. However, the remarkable genetic and antigenic variability of the virus, which originate from both mutations and recombination events, represents an unsolved challenge for this disease. The present study reports on the emergence and spread of recombinant clusters detected in Italy and Spain between 2012 and 2014. A total of 36 Spanish and Italian infectious bronchitis virus (IBV) field strains were investigated and genetically characterized using phylogenetic, molecular, recombination and selection pressure analyses of the complete S1 gene. Based on the partial S1 sequencing, 27 IBV strains originating from Spain and nine from Italy were initially classified as being closely related to the Guandong/Xindadi (XDN) genotype. Phylogenetic analysis of the complete S1 gene revealed that the XDN strains formed a homogeneous clade with the Spanish IBV isolates within the QX genotype, whereas there was higher variability within the Italian strains. Recombination analysis determined that these strains belonged to four groups, which originated from independent recombination events between the QX and 793B IBV genotypes. Our data support the hypothesis of two different scenarios: firstly, in Spain, the large and homogeneous clade probably originated from a single offspring of the recombinant founder, which became dominant and spread throughout the country. Secondly, the nine Italian recombinants, which are characterized by three different recombination patterns, probably represent less fitted strains, because they were less viable with respect to their recombinant parents.
KEYWORDS: Infectious bronchitis virus, recombination events, Italy, Spain, poultry
Introduction
Avian infectious bronchitis is a highly contagious viral disease that is considered to be one of the major causes of economic losses to the poultry industry worldwide. Most strains of infectious bronchitis virus (IBV) primarily only cause respiratory disease in birds; however, some strains can spread to other organs, such as the kidneys and the reproductive tract, which results in nephritis, decreased egg production and quality and significant mortality.
IBV is caused by a Coronavirus, which was first identified in 1936. The coronaviruses have a single-stranded, positive-sense, RNA genome of approximately 27 Kb. The 3′ end of the genome encodes four structural proteins (spike (S), envelope (E), membrane (M) and nucleocapsid (N)) and four non-structural accessory proteins. Genetic diversity in coronaviruses, due to adaptive evolution, is created both by recombination events and by mutations, such as substitutions, deletions and insertions, that occur when the viral genome is replicated. The high mutation rates are attributed to the minimal proofreading capabilities of the viral RNA-dependent RNA-polymerase, while recombination events are thought to result from a unique template-switching copy-choice mechanism during RNA replication (Simon-Loriere & Holmes, 2011). Similarly to other coronaviruses, the high mutation and recombination rate of IBV has led to the emergence of several new variants, particularly in Europe and North America where poultry are intensively raised. However, it is believed that only a small proportion of the total IBV genetic variability has been identified. In particular, the S1 protein has been demonstrated to have the highest variability of the whole viral genome (Cavanagh & Naqi, 2003). This protein, constituting the outermost part of the virion, is involved in host cell attachment and contains virus-neutralizing epitopes. Consequently, mutation and recombination events in the S1 gene are critically important for the emergence of new serotypes of the virus that are able to cause disease in other host species. The continuous emergence of new IBV serotypes has prompted the development of appropriate control programmes aimed to mitigate the low degree of cross-protection observed among IBV serotypes (Dolz et al., 2008).
Molecular studies performed in Italy and Spain showed the concurrent circulation of different IBV genotypes. In the last few years, the most predominant genotypes in both countries were QX and 793B (Franzo et al., 2014; Blanco et al., 2015). However, beginning in 2012, a new IBV variant related to the CK/CH/Guandong/Xindadi/0903 (XDN) variant (Ji et al., 2011) was detected in Italy and Spain. Therefore, the aim of the present study was to investigate and genetically characterize the XDN variant. We sequenced the complete S1 gene of several IBV strains isolated in Italy and Spain that were closely related to the XDN variant. In addition, we compared these sequences with the XDN reference sequence and with other IBV sequences available in GenBank to perform phylogenetic, molecular and recombination analyses.
Materials and methods
Clinical samples
Samples of the trachea, kidneys and lungs were collected from suspected IBV-infected chickens and submitted to the diagnostic laboratories in Spain and Italy during 2012–2014 for routine diagnosis. Clinical signs, geographic location, production type and the age of the birds were recorded for each sample.
Virus isolation
To isolate the virus, IBV-positive samples were inoculated into 9–11-day-old specific-pathogen-free (SPF) chicken embryonated eggs (CEEs) by the allantoic sac route as previously described elsewhere (Cavanagh & Naqi, 2003).
RNA extraction and sequencing
Viral RNA was extracted from clinical samples using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. IBV was detected using RT-PCR as previously described (Cavanagh et al., 1999). Partial sequencing of the S1 gene (464-bp) was performed using the same primers that were used for RT-PCR (Cavanagh et al., 1999). Briefly, the amplified products were separated on an agarose gel and purified using a QIAquick gel extraction kit (Qiagen, Valencia, CA, USA). Sequencing reactions were performed using a BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). The sequences were resolved using an ABI 3130 DNA automated sequencer (Applied Biosystems). The DNA sequences were combined and edited using the Lasergene sequencing analysis software package (DNASTAR, Madison, WI, USA). The samples were classified using NCBI BLAST and/or through phylogenetic analysis and comparison with reference strains. Complete S1 gene of the XDN-like strains was amplified using specific primers: IBV_S1F (TGGGTGACAGTGGAAAACTG) and IBV_S1R (TGTGTTTGTATGTACTCATC). The S1 gene sequencing was performed using a previously described protocol.
Phylogenetic and recombination analysis
To identify the Italian and Spanish recombinant strains and their probable parental genotypes, a complete collection of IBV S1 sequences was downloaded from the ViPR database (http://www.viprbrc.org/brc/home.spg?decorator=vipr). The database size was reduced by collapsing the sequences into clusters that shared 98% sequence identity using CD-HIT. The sequences obtained in the present study were aligned with one sequence representative of each cluster using the MAFFT method implemented in GUIDANCE2, and the alignment score was calculated using the same software. To account for the coding nature of the genomic region, the S1 sequences were first aligned at the amino acid level, and then, the nucleotide sequences were superimposed. Exploratory phylogenetic trees were performed on the partial and complete S1 gene using the maximum likelihood method in the IQ-TREE software (Nguyen et al., 2015). The recombination analysis was performed on this database using the RDP4 recombination analysis programme. The different method settings were adjusted according to the programme’s manual (Martin et al., 2015). To obtain a conservative estimate, a recombination event was accepted only when detected by two or more methods implemented in the programme, with a P-value lower than 0.05. To refine the recombination pattern analysis, a similar analysis was carried out on a smaller database including all recombinants and corresponding parental strains; two Massachusetts (Mass)-like strains were also added as an outgroup. Finally, a collection of recombination-free partitions was obtained for each recombination event by dividing the original alignment at the recombination breakpoints. Phylogenetic trees were reconstructed for each partition using RAXML (Silvestro & Michalak, 2012) and used to calculate per-site log likelihoods for each alignment partition. Statistical significance of the topological incongruence between segments separated by recombination breakpoints was assessed through SH, KH, ELW and AU tests implemented in CONSEL. A P-value < 0.05 was considered statistically significant in all statistical tests.
Compilation of data sets and selection pressure analysis
To better analyse the selection pressure, the IBV data set was further divided into two subsets according to their country of origin and clade (as evidenced by the phylogenetic tree). We then estimated the rates of non-synonymous (dN) and synonymous (dS) substitutions per site (ratio dN/dS) for both the Spanish and Italian data sets using the single likelihood ancestor counting and the fixed effects likelihood methods available through Datamonkey (online version of the Hy-Phy package) (Kosakovsky Pond et al., 2005). A model of best fit for nucleotide substitutions was estimated for each data set according to the Akaike information criterion. To understand the degree of variation in the S1 gene, the entropy of the aligned amino acid sequences within this gene for the different isolates was calculated using BioEdit (version 7.0.9.0). Entropy is a useful quantification of diversity at a single position of amino acid sequences. Large entropy indicates that the amino acid in the given position is prone to substitution. A higher peak indicates a larger entropy and a higher variation frequency of the amino acid site. Conserved residues will have an entropy value close to 0, but these values will increase to 1 as the variability for an amino acid position increases. An entropy value higher than 0.4 indicates the corresponding amino acid position is not conserved (Mo et al., 2013).
In addition, codon-based tests of positive selection (Z-test, MEGA5) were used to estimate the dN/dS ratio within the S1 protein to understand whether these proteins are submitted to positive selection. These analyses were conducted using the Nei–Gojobori method in MEGA5.2 (Tamura et al., 2011). The probability of rejecting the null hypothesis of strict neutrality (dN = dS) in favour of the alternative hypothesis (dN > dS) was calculated, and a P-value less than 0.05 was considered significant at the 5% level and highlighted. This method is more conservative and provides a more reliable test of the alternative hypothesis (HA: dN > dS) when the total number of substitutions between the sequences is small (Nei & Kumar, 2000).
GenBank accession numbers
GenBank accession numbers of the Spanish and Italian IBV S1 sequences used in this study are shown in Table 1.
Table 1. Origin of the isolates and cleavage recognition motifs of the IBV strains used in this study.
| Year isolated | Strain no. | Country | Province (region) | Type of chicken | Age (days) | Material sampled | Clinical signs | Cleavage recognition motifs | GenBank accession number |
|---|---|---|---|---|---|---|---|---|---|
| 2012 | 1129 | Spain | Tarragona (Catalonia) | Broilers | n.a.a | T + CTb | n.a. | HRRRR | KU934152 |
| 2012 | 1150 | Spain | Murcia (Murcia) | Broilers | n.a. | Trachea | n.a. | HRRRR | KU934153 |
| 2012 | 1254 | Spain | Lleida (Catalonia) | Broilers | n.a. | T + CT | None. Routine sampling | HRRRR | KU934154 |
| 2012 | 1384 | Spain | Barcelona (Catalonia) | Broilers | 46 | Trachea + lungs | Respiratory | HRRRR | KU934155 |
| 2012 | 1516/1 | Spain | Tarragona (Catalonia) | Layers | n.a. | Trachea + kidneys | n.a. | HRRRR | KU934156 |
| 2012 | 1516/2 | Spain | Tarragona (Catalonia) | Layers | n.a. | Trachea + kidneys | n.a. | HRRRR | KU934157 |
| 2012 | 1516/3 | Spain | Tarragona (Catalonia) | Layers | n.a. | Trachea + kidneys | n.a. | HRRRR | KU934158 |
| 2012 | 1516/4 | Spain | Tarragona (Catalonia) | Layers | n.a. | Trachea + kidneys | n.a. | HRRRR | KU934159 |
| 2012 | 2499 | Spain | Tarragona (Catalonia) | Broilers | n.a. | Trachea + kidneys | Respiratory | HRRRR | KU934160 |
| 2012 | 2828/1 | Spain | Tarragona (Catalonia) | Broiler breeders | 90 | T + CT | n.a. | HRRRR | KU934161 |
| 2012 | 5090 | Spain | Tarragona (Catalonia) | Broilers | 14 | Kidneys | Respiratory. Nephritis | HRRRR | KU934162 |
| 2012 | 8469 | Spain | Tarragona (Catalonia) | Broilers | 42 | Kidneys | Respiratory. Nephritis | HRRKR | KU934163 |
| 2012 | 9508 | Spain | Tarragona (Catalonia) | Layers | 28 | Trachea + kidneys | Respiratory | HRRKR | KU934164 |
| 2012 | 243746 | Italy | Brescia (Lombardia) | Backyard | n.a. | Trachea + lungs | Respiratory | HRRRR | KU934179 |
| 2013 | 405 | Spain | Tarragona (Catalonia) | Layers | 61 | Kidneys + caecal tonsils | Respiratory. Nephritis. | HRRKR | KU934172 |
| 2013 | 635/1 | Spain | Tarragona (Catalonia) | Layers | n.a. | Kidneys | n.a. | HRRRR | KU934166 |
| 2013 | 1410 | Spain | Tarragona (Catalonia) | Broilers | 21 | Kidneys | Respiratory. Nephritis | HRRRR | KU934167 |
| 2013 | 1585 | Spain | Tarragona (Catalonia) | Broilers | 39 | T + CT | Respiratory | HRRRR | KU934168 |
| 2013 | 1607 | Spain | Tarragona (Catalonia) | Broilers | 34 | Caecal tonsils | n.a. | HRRRR | KU934169 |
| 2013 | 1622 | Spain | Tarragona (Catalonia) | Broilers | 18 | Kidneys + caecal tonsils | Respiratory. Nephritis. | HRRRR | KU934171 |
| 2013 | 1679 | Spain | Barcelona (Catalonia) | Boilers | 33 | Kidneys + caecal tonsils | Respiratory. Nephritis. Mortality (0.3%/day). | HRRRR | KU934170 |
| 2013 | 2010 | Spain | Tarragona (Catalonia) | Broilers | 33 | Trachea + kidneys | Respiratory & lameness | HRRRR | KU934172 |
| 2013 | 4976 | Spain | Huesca (Catalonia) | Layers | 91 | Trachea | Respiratory | HRRRR | KU934173 |
| 2013 | 10861 | Spain | Tarragona (Catalonia) | Broilers | 34 | Kidneys | Respiratory. Mortality. | HRRRR | KU934174 |
| 2013 | 20588 | Italy | Verona (Veneto) | Broilers | 42 | HRRRR | KU934180 | ||
| 2013 | 80472-2 | Italy | Brescia (Lombardia) | Layers | 280 | Trachea + oviduct | Decreased egg production | HRRKR | KU934181 |
| 2014 | 445 | Spain | Sevilla (Andalucia) | Broiler breeders | n.a. | Trachea | Respiratory | HRRRQ | KU934175 |
| 2014 | 1786 | Spain | Central Spain | Layers (pullets) | 18 | Tracheal swabs | Respiratory. Nephritis | HRRRR | KU934176 |
| 2014 | 2022 | Spain | Tarragona (Catalonia) | Broilers | 36 | Trachea + kidneys | Respiratory. Nephritis | HRRRR | KU934177 |
| 2014 | 6443-1 | Spain | Tarragona (Catalonia) | Layers | 18 | Trachea | Respiratory | HRRRR | KU934178 |
| 2014 | 8318 | Italy | Brescia (Lombardia) | Broilers | 28 | Trachea + lungs | Respiratory | HRRRR | KU934182 |
| 2014 | 14810 | Italy | Verona (Veneto) | Broilers | 23 | Trachea + lungs | Respiratory | HRRRR | KU934183 |
| 2014 | 14847 | Italy | Forlì Cesena (Emilia Romagna) | Broilers | 35 | Trachea + kidneys | Respiratory | HRRRR | KU934184 |
| 2014 | 55311 | Italy | Bologna (Emilia Romagna) | Layers | 350 | Trachea + lungs | Respiratory | HRRKR | KU934185 |
| 2014 | 90334 | Italy | Forlì Cesena (Emilia Romagna) | Broilers | 40 | Trachea | Respiratory | HRRRR | KU934186 |
| 2014 | 125490 | Italy | Mantova (Lombardia) | Layers | 364 | Trachea + lungs | Respiratory | RRSRR | KU934187 |
aNo information available.
bTrachea + caecal tonsil.
Results
Viruses
A total of 36 IBV-positive samples were isolated from CEEs, identified using RT-PCR and classified as XDN variants. Of these isolates, 27 originated from Spain and nine from Italy. The first XDN variants were detected in Spain in February 2012 and in Italy in December 2012. The majority of the samples (65.2% in Spain and 55.5% in Italy) originated from 14- to 46-day-old broilers. The remaining samples came from layers, with the exception of two samples: one that was isolated from Spanish broiler breeders and the other that was isolated from an Italian backyard farm. The detailed origin and clinical record of each strain is summarized in Table 1.
Phylogenetic and molecular analyses
The exploratory phylogenetic tree based on the partial S1 gene showed a distinct clade formed by the XDN, Spanish and Italian strains with the exception of the Italy/125490/2014 strain. Within this XDN clade, two groups of sequences clustered according to the country of origin. In contrast, the Italy/125490/2014 strain was placed in the 793B clade (Supplementary Figure 1).
Phylogenetic analysis of the complete S1 gene revealed a different clade distribution. The XDN clade was placed within the QX genotype, and the strains were part of four different groups according to the country of origin. The Spanish isolates formed a homogeneous clade, whereas the Italian strains showed higher variability: five highly related strains and two distinct strains isolated in 2012 (Italy/243746/2012) and 2014 (Italy/125490/2014) (Figure 1).
Figure 1.
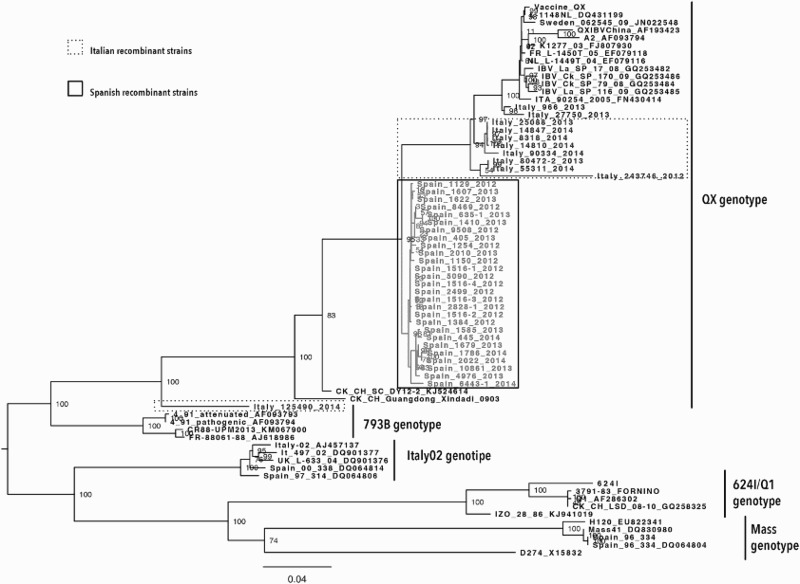
Phylogenetic relationships of Spanish and Italian IBV strains based on the sequence of the complete S1 gene. The unrooted tree was created by the maximum likelihood method implemented in the IQ software and bootstrapped with 1000 replicates. Only bootstrap values higher than 50% are shown. Spanish and Italian sequences are highlighted by boxes.
Homology analysis of the 27 Spanish IBV strains revealed the highest nucleotide and deduced amino acid sequence similarities to the QX strain, which was first isolated in Europe in 2004. The percentage of similarities for the Spanish strains to NL/L-1148/04 (DQ431199) ranged from 94.6% to 95.3% and 95.4% to 96.7% at the nucleotide and amino acid levels, respectively. Conversely, the Italian strains showed the highest nucleotide and deduced amino acid sequence similarities (88.1% to 97.5% and 87.6% to 96.7%, respectively) to the current QX strain circulating in Italy (Italy/966/2013), which indicated higher variability among the Italian strains. The two unique strains (Italy/243746/2012 and Italy/125490/2014) showed the lowest similarity.
Alignment of the S1 gene and deduced amino acid sequences of the Italian and Spanish strains showed that all strains, except the Italy/243746/2012 strain, contained 1620 nucleotides corresponding to a 540-amino-acid S1 protein. The Italy/243746/2012 strain had a deletion of six nucleotides, resulting in a S1 protein of 538 amino acids.
The S glycoprotein was translated as a precursor protein (S0) and then cleaved into two subunits, S1 and S2, by proteases during viral maturation (Jackwood et al., 2012). In this study, four different cleavage recognition motifs were identified. Of these, one motif (HRRRR) was shared by most of the Spanish (23/27) and Italian (6/9) strains. The other three motifs were: HRRKR (identified in three Spanish and two Italian strains), HRRRQ (identified in only one strain from southern Spain) and RRSRR (identified in the Italy/125490/2014 strain), which is a typical motif of the 793B genotype. The cleavage recognition motifs of the investigated strains are shown in Table 1.
Recombination analysis
The recombination analysis demonstrated that all the Italian and Spanish sequences identified in the present study originated from recombination between the QX and 793B genotypes (Supplementary Figure 2). However, the different groups demonstrated distinct recombination patterns (Figure 2). Briefly, the entire Spanish clade and all the Italian sequences, except for two (Italy/243746/2012 and Italy/125490/2014), demonstrated one recombinant region, which was located between positions 855 and 1235 in the Spanish strains and positions 84 and 1010 in the Italian strains. The Italy/243746/2012 strain had two different recombinant regions with two 793B inserts between positions 875 and 1040, and 1240 and 1315. The final strain (Italy/125490/2014) had a different recombination pattern, which was characterized by two QX inserts (one in the first part of the S1 gene between 1 and 506, and the other between 1046 and 1192).
Figure 2.
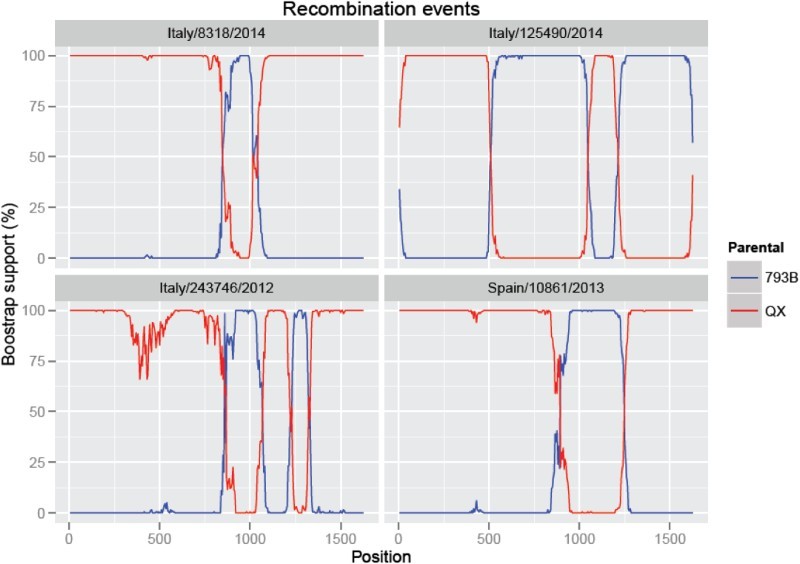
Results of Bootscan analysis performed on the strains Italy/8318/2014, Italy/125490/2014, Italy/243746/2012 and Spain/10861/2013 using a window of 200 nucleotides moved by 10 nucleotides at a time.
Selection pressure
The average dN/dS ratio in the Spanish and Italian data sets was estimated to be 0.9137 (95% CI 0.68–1.19) and 0.302 (95% CI 0.25–0.35), respectively. A codon-based Z-test for positive selection was used to analyse the dN/dS ratio in the S1 gene of both the Spanish and Italian isolates. No significant evidence for positive selection of S1 protein of the major part of both isolates was observed (P > 0.05). However, the test showed that positive selection occurred between:
the Spain/10861/2013 and Spain/1679/2013 strains (dN/dS = 2.416, P = 0.009);
the Spain/635-1/2013 strain and three isolates: Spain/8469/2012 (dN/dS = 2.614, P = 0.005), Spain/9508/2012 (dN/dS = 2.883, P = 0.002) and Spain/405/2013 (dN/dS = 2.620, P = 0.005);
the Spain/1410/2013 strain and the same three isolates above reported: Spain/8469/2012 (dN/dS = 2.614, P = 0.005), Spain/9508/2012 (dN/dS = 2.883, P = 0.002) and Spain/405/2013 (dN/dS = 2.620, P = 0.005);
the Italy/90334/2014 strain and the following strains: Italy/25088/2013 (dN/dS = 3.446, P = 0.000), Italy/8318/2014 (dN/dS = 3.446, P = 0.000), Italy/14810/2014 (dN/dS = 2.147, P = 0.017) and Italy/14847/2014 (dN/dS = 3.446, P = 0.000).
The results of the entropy analysis of the S1 gene for the amino acid sequences of the Spanish and Italian samples are shown in Supplementary Figure 3. The percentage of entropy values greater than 0.4 was 6.85% (37/540) in the Italian sequences and 1.66% (9/540) in the Spanish sequences. The majority of the positive entropy values were located within the three hypervariable regions (HVRs) located between amino acid residues 60 and 88, 115 and 140, and 275 and 292 (numbered according to the H120 reference S1 sequence) (Cavanagh et al., 1988).
Discussion
The aim of the present study was to characterize a new IBV variant that was first detected in 2012 in Spain and Italy. Italy and Spain are two European countries where poultry are intensively raised and where several field, vaccine and vaccine-derived strains are circulating. A total of 36 IBV strains, originating from both countries during 2012–2014, were sequenced in two steps. A preliminary characterization based on HVRII sequencing of the S1 gene was performed and revealed that these strains grouped together with the XDN variant. However, further phylogenetic analysis based on the whole S1 gene showed a different clade distribution, indicating that all of the XDN strains except one were placed in the QX clade.
While some studies have reported that genotyping based on sequencing the HVRs of the S1 gene is representative of the grouping based on the complete S1 gene (Lee et al., 2003), others have disagreed with these findings (Schikora et al., 2003). The results of this study showed that genotyping based on partial sequencing of the S1 gene was not representative of genotyping based on the whole gene in some of the variants. In the present study, these discrepancies were due to the presence of recombination events involving the S1 HVR. Recombination has been suggested to play an important role in viral evolution. Exchange of a long region of the genome allows viruses to rapidly explore ample areas of the sequence space, potentially leading to the emergence of variants with different features in terms of virulence, cross-protection and cell and host tropism (Simon-Loriere & Holmes, 2011). Recombination, similar to mutation, is likely to have a high fitness cost due to the destruction of optimized intra- and inter-protein interactions (Simon-Loriere & Holmes, 2011). Additionally, recombinant strains must display a fitness superior to the parental fitness to emerge and spread in the environment. In our study, convincing evidence showed four distinct recombination events affecting the S1 gene, suggesting that this gene is prone to recombination at multiple sites.
The first prerequisite for recombination is co-infection. In the last 4 years, the partial S1 gene was sequenced from samples collected in 1968 (1248 Italian and 720 Spanish samples) and the presence of different IBV genotypes was detected. In Italy the predominant genotypes were QX (37.3%), XDN (1.2%), 793B (44.1%), M41 (5%) and 624I/Q1 (10.8%) (Massi et al., 2015), whereas in Spain, the main genotypes were QX (12.2%), XDN (19.4%), 793B (64.4%) and IT02 (4%) (Blanco et al., 2015). Interestingly, the It02 serotype, which was highly distributed throughout Italy until 2007 and Spain until 2011, has recently been replaced by other serotypes, such as QX and 793B. The genotype 624I/Q1 reappeared in Italy in 2010 after 3–4 years in which it was probably circulating below the cut-off level for detection (Franzo et al., 2014; Massi et al., 2015). Considering the high prevalence of the QX and 793B genotypes in both countries, it is reasonable to speculate that these genotypes co-infect birds more frequently compared to the other genotypes. Moreover, control of IBV in the commercial poultry industry is through the use of live attenuated and inactivated vaccines. Because little or no cross-protection occurs between the different IBV serotypes or variants, different virus antigenic types must be used for vaccine development. Antigenic types, such as Mass-based or 793B-type, have been commonly used in both countries for a long time. Recently (in 2013 in Spain and 2014 in Italy), a new QX vaccine was registered, but is currently not highly distributed. The current vaccines induce a non-sterilizing immunity, which allows prolonged replication, shedding and circulation of the vaccine viruses, particularly when only part of the population is effectively vaccinated. It is likely that vaccination with live attenuated vaccines produces an environment where co-infection between field (i.e. QX genotype) and vaccine (i.e. 793B genotype) strains can enhance the likelihood of recombination. High frequencies of recombination between vaccine and field strains have frequently been reported (Cavanagh et al., 1992; McKinley et al., 2011; Jackwood et al., 2012; Feng et al., 2014). However, the shorter persistence and shedding of Mass-based vaccines (Bijlenga et al., 2004) and the low percentage of the M41 genotype in both countries (Blanco et al., 2015; Massi et al., 2015) support the absence of recombinants for this genotype. In fact, partial sequencing of the S1 gene showed that a high number of detected field strains were of the 793B vaccines, or vaccine-derived strains (Franzo et al., 2014).
Moreover, the frequency of successful inter-typic genetic exchange (i.e. those instances in which the recombinant offspring can establish itself in the host population) could be dependent on the viability of the recombinant progeny and on the “evolutionary gain” intended as the increase in fitness of the recombinant virus compared to its non-recombinant parents (Smits et al., 2003). The different recombinant groups identified in this study demonstrated different levels of fitness between the strains from the two countries, with the Spanish strains forming a large and long-lasting clade, while the Italian strains were only sporadically detected and probably represented strains not sufficiently competitive with respect to their progenitor strains. Future studies are needed to investigate if the unique recombination patterns are responsible for the virus success by either leading to deleterious modifications or producing more fit strains (more virulent viruses, immune escape mutants, etc.).
The phylogenetic and molecular analyses confirmed the results of the recombination analysis and showed a higher variation in the Italian amino acid S1 sequences compared to the Spanish ones. In addition, two different epidemiological situations were observed. In Italy, a small percentage of the QX strains were classified as XDN variants and represented 3% of the QX strains detected between 2012 and 2015 (Massi et al., 2015). In Spain, the recombinant strains showed a tendency to replace the classical QX strains and were identified as XDN in 74.5% of the QX cases identified between 2012 and 2014 (Blanco et al., 2015).
The analysis of the cleavage site motifs in the S protein revealed the presence of four different motifs, indicating that the cleavage recognition motifs underwent a continuous evolution process. Of these four, one motif was shared by the majority of the Spanish and Italian strains investigated in this study and has been reported to be one of the most common cleavage recognition sites of the S protein (Feng et al., 2014). The other three motifs were observed in a few of the isolated strains. In addition, one was present in the only strain placed in the 793B clade and was shared by the majority of the strains belonging to this genotype. We also found that individual strains with high similarities usually shared the same motifs.
Taken together, our data support the hypothesis of two different scenarios for each country. In Spain, the large and homogeneous clade probably originated from a single offspring of the recombinant founder, which became dominant and spread throughout the country. In Italy, the nine recombinants were characterized by three different recombination patterns, which probably represented less fitted strains because they were less viable with respect to their recombinant parents.
The IBV samples used in this study were grown using SPF CEEs. Although the egg passage of all of the viruses used was very low (one to two passages), the possibility of some genetic changes introduced as a result of propagation in the CEEs cannot be rule out. However, a study examining genetic changes in the highly variable S1 gene following 10 passages in embryonated eggs reported that five viruses had no changes, two viruses had only two nucleotide changes and one strain had only one change (McKinley et al., 2011).
There is a practical importance in accounting for recombination during diagnostic procedures. Firstly, a preliminary analysis using BLAST and/or phylogenetic analysis is typically performed during routine diagnostic procedures in many laboratories, which in this study led to the classification of the investigated strains into the XDN group that originated in China. This misclassification could have led us to infer new introduction events inaccurately, which would severely bias the validity and potentially the efficacy of virus control measures. Secondly, even if the cost/benefit of the extensive use of vaccination to control IBV infections is far from being understood, this study stresses the importance of improving vaccination tools in terms of both protection and application.
Supplementary Material
Acknowledgements
We would like to thank Francesca Adella, Sonia Manenti and Dr Beatrice Boniotti for their technical collaboration.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCiD
Ana Moreno http://orcid.org/0000-0002-8497-9708
References
- Bijlenga G., Cook J.K.A., Gelb J. Jr & de Wit J.J. (2004). Development and use of the H strain of avian infectious bronchitis virus from the Netherlands as a vaccine: a review. Avian Pathology, 33, 550–557. doi: 10.1080/03079450400013154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco A., Antillés N., Camprubí Q., Jové R. & Biarnes M. (2015). Bronquitis aviar: Epidemiología molecular y evolución de los diferentes genotipos predominantes en España desde 2011, LII Simposio cientifico de Avicultura, 28–30 October, Malaga (Spain).
- Cavanagh D., Davis P.J. & Cook J.K.A. (1992). Infectious bronchitis virus: evidence for recombination within the Massachusetts serotype. Avian Pathology, 21, 401–408. doi: 10.1080/03079459208418858 [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J. & Mockett A.P.A. (1988). Amino acids within hypervariable region 1 of avian coronavirus IBV (Massachusetts serotype) spike glycoprotein are associated with neutralization epitopes. Virus Research, 11, 141–150. doi: 10.1016/0168-1702(88)90039-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Mawditt K., Britton P. & Naylor C.J. (1999). Longitudinal field studies of infectious bronchitis virus and avian pneumovirus in broilers using type-specific polymerase chain reactions. Avian Pathology, 28, 593–605. doi: 10.1080/03079459994399 [DOI] [PubMed] [Google Scholar]
- Cavanagh D. & Naqi S.A. (2003). Infectious bronchitis. In Saif Y.M., Barnes H.J., Glisson J.R., Fadly A.M., McDougald L.R., & Swayne D.E. (Eds.), Diseases of Poultry, 11th edn (pp. 101–119). Ames: Iowa State Press. [Google Scholar]
- Dolz R., Pujols J., Ordonez G., Porta R. & Majo N. (2008). Molecular epidemiology and evolution of avian infectious bronchitis virus in Spain over a fourteen-year period. Virology, 374, 50–59. doi: 10.1016/j.virol.2007.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng K., Xue Y., Wang F., Chen F., Shu D. & Xie Q. (2014). Analysis of S1 gene of avian infectious bronchitis virus isolated in southern China during 2011–2012. Virus Genes, 49, 292–303. doi: 10.1007/s11262-014-1097-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzo G., Naylor C.J., Lupini C., Drigo M., Catelli E., Listorti V., Pesente P., Giovanardi D., Morandini E. & Cecchinato M. (2014). Continued use of IBV 793B vaccine needs reassessment after its withdrawal led to the genotype’s disappearance. Vaccine, 32, 6765–6767. doi: 10.1016/j.vaccine.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackwood M.W., Hall D. & Handel A. (2012). Molecular evolution and emergence of avian gammacoronaviruses. Infection, Genetics and Evolution, 12, 1305–1311. doi: 10.1016/j.meegid.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J., Xie J., Chen F., Shu D., Zuo K., Xue C., Qin J., Li H., Bi Y., Ma J. & Xie Q. (2011). Phylogenetic distribution and predominant genotype of the avian infectious bronchitis virus in China during 2008–2009. Virology Journal, 8, 184. doi: 10.1186/1743-422X-8-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., Frost D.D.W. & Muse S.V. (2005). HyPhy: hypothesis testing using phylogenies. Bioinformatics, 21, 676–679. doi: 10.1093/bioinformatics/bti079 [DOI] [PubMed] [Google Scholar]
- Lee C.-W., Hilt D.A. & Jackwood M.W. (2003). Typing of field isolates of infectious bronchitis virus based on the sequence of the hypervariable region in the S1 gene. Journal of Veterinary Diagnostic Investigation, 15, 344–348. doi: 10.1177/104063870301500407 [DOI] [PubMed] [Google Scholar]
- Martin D.P., Murrell B., Golden M., Khoosal A. & Muhire B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1, vev003. doi: 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi P., Barbieri I., Fiorentini L., Casadio M., Parigi M. & Tosi G. (2015). Analisi molecolare di ceppi del virus della bronchite infettiva aviare negli anni 2013 e 2014. Considerazioni sui genotipi circolanti in Italia e in altri paesi europei ed extra-europei. LIV Convegno Annuale Società Italiana di patologia Aviare, Forlì, 16–17 Aprile 2015.
- McKinley E.T., Jackwood M.W., Hilt D.A., Kissinger J.C., Robertson J.S., Lemke C. & Paterson A.H. (2011). Attenuated live vaccine usage affects accurate measures of virus diversity and mutation rates in avian coronavirus infectious bronchitis virus. Virus Research, 158, 225–234. doi: 10.1016/j.virusres.2011.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo M.-L., Li M., Huang B.-C., Fan W.-S., Wei P., Wei T.-C., Cheng Q.-Y., Wei Z.-J. & Lang Y.-H. (2013). Molecular characterization of major structural protein genes of avian coronavirus infectious bronchitis virus isolates in Southern China. Viruses, 5, 3007–3020. doi: 10.3390/v5123007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. & Kumar S. (2000). Molecular Evolution and Phylogenetics. Oxford: Oxford University Press. [Google Scholar]
- Nguyen L.-T., Schmidt H.A., von Haeseler A. & Minh B.Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. doi: 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikora B.M., Shih L.M. & Hietala S.K. (2003). Genetic diversity of avian infectious bronchitis virus California variants isolated between 1988 and 2001 based on the S1 subunit of the spike glycoprotein. Archives of Virology, 148, 115–136. doi: 10.1007/s00705-002-0904-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestro D. & Michalak I. (2012). RaxmlGUI. A graphical front-end for RAxML. Organisms Diversity and Evolution, 12, 335–337. doi: 10.1007/s13127-011-0056-0 [DOI] [Google Scholar]
- Simon-Loriere E. & Holmes E.C. (2011). Why do RNA viruses recombine? Nature Reviews, 9, 617–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S.L., Lavazza A., Matiz K., Horzinek M.C., Koopmans M.P. & de Groot R.J. (2003). Phylogenetic and evolutionary relationships among torovirus field variants: evidence for multiple intertypic recombination events. Journal of Virology, 77, 9567–9577. doi: 10.1128/JVI.77.17.9567-9577.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M. & Kumar S. (2011). MEGA5: molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. doi: 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.