

Abstract
As antiviral drug resistance develops and new viruses emerge there is a pressing need to develop strategies to rapidly develop antiviral therapeutics. Here we use phospho-specific flow cytometry to assess perturbations of many different cellular signaling pathways during treatment with drug combinations that are highly effective in blocking Herpes simplex virus type 1 (HSV-1) infection. We discovered two antiviral drug combinations act on distinct signaling pathways, either STAT1 or S6 phosphorylation, to block HSV-1 infection. We focused on upregulation of S6 phosphorylation by HSV-1 infection, and our subsequent finding that ribavirin antagonizes this upregulation of S6 phosphorylation. We go on to show that the S6 kinase inhibitor SL0101 blocks HSV-1 replication in vitro and in an in vivo animal model of HSV-1 infection. Overall, we have used an unbiased analysis of cellular signaling pathways during treatment by antiviral drug combinations to discover a novel antiviral drug target against HSV-1 infection. The outcomes of the approach we present highlight the importance of analyzing how antiviral drugs modulate cellular and pathogen-induced signaling as a method to discover new drug therapy targets.
Graphical abstract
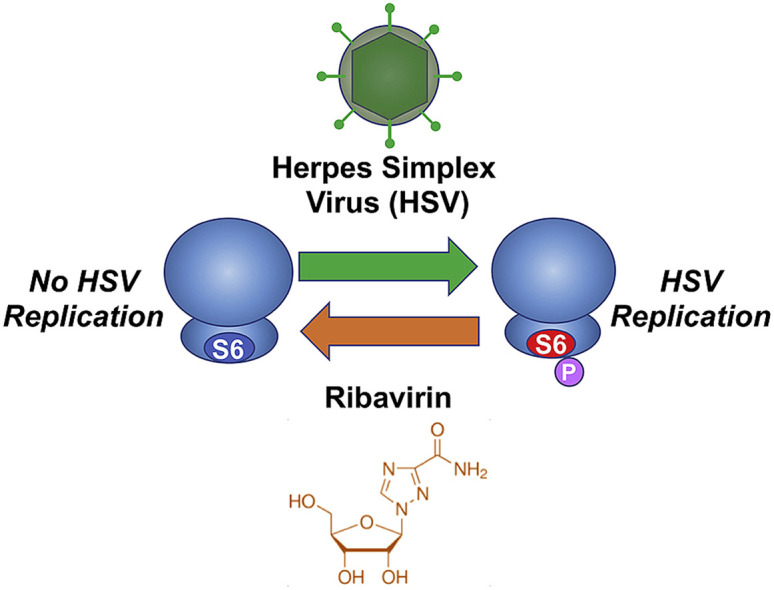
Highlights
-
•
Phospho-specific flow cytometry was used to asses anti-HSV-1 antiviral drug combinations.
-
•
HSV-1 replication induces phosphorylation of S6 ribosomal protein.
-
•
Ribavirin blocked HSV-1-induced phosphorylation of S6 ribosomal protein while blocking HSV-1 replication.
-
•
S6 kinase inhibitor blocks HSV-1 replication in vitro and an in vivo animal model of HSV-1 infection.
1. Introduction
One approach to new therapeutic drug discovery is based on the efficacy of the drug against one particular target or outcome, such as stopping infection or limiting disease pathology. However, potential side effects and off-label indications are often not found until clinical trials or post-market drug safety monitoring. Consequently, this approach to drug discovery often leads to unexpected problems or misfires that delay or block significant advancements. Techniques that can determine the best drug or combination of drugs to optimally treat a disease without using conventional drug screening approaches are needed for rapid, efficient translational medicine (Lee et al., 2017; Sun et al., 2013). Developing targeted therapies that focus on the key signaling pathways that lead to pathology and avoid bystander perturbations of the physiology of a cell are possible when looking at how a therapeutic drug combination impacts a cell using an unbiased analysis of cellular signaling (Bendall et al., 2011; Bodenmiller et al., 2012; Schweizer and Zhang, 2013).
Treatment of different disease states through combinations of multiple drugs is used to achieve better therapeutic results, reduce the impact of drug resistance, and sustain high efficacy with low toxicity often with lower doses of the individual drug components. The use of drug combinations is well documented in both cancer and infectious disease therapy (Al-Lazikani et al., 2012; Arts and Hazuda, 2012; George et al., 2015). Therapeutic drug combinations work by simultaneously controlling several cellular pathways in the complex network system. In a previous study (Ding et al., 2012), two combinations of antiviral drugs, one combination rich in interferon and one rich in Ribavirin, were determined due to their ability to inhibit Herpes simplex virus type 1 (HSV-1) infection. These combinations were identified through a Feedback System Control (FSC) technique which uses an unbiased search based on the outcome of the drug combination treatment and can identify many unexpected drug combinations offering desired therapeutic outputs (Wong et al., 2008). However, due to the focus on outputs, the FSC search methodology does not give any information on which cellular signaling pathways are stimulated by these drug combination or the mechanistic reasoning behind efficacy of the optimal drug combination.
In this study, we use phosphospecific flow cytometry, or phosphoflow, to understand the changes in cellular signaling using potent antiviral drug combinations as stimuli. The phosphorylation and dephosphorylation of proteins by kinases and phosphatases are part of the essential regulatory networks that drive the phenotype and function of cells, tissues, and organisms, especially after exposure to a drug or biological therapeutic (Sachs et al., 2005). Advances in phosphoflow have made studies at the phosphoproteomic level possible (Krutzik and Nolan, 2006). Using phosphoflow to monitor multiple indicators of cellular physiology allows us to determine how drug combinations affect the intricate cellular signaling by simultaneously interrogating distinct signaling nodes within treated cells. The insight afforded to us by phosphoflow analysis of a drug combination or disease state may allow us to better design specific therapeutics that modulate the activity of disease-related kinases or phosphatases (Krutzik et al., 2008; Bodenmiller et al., 2010).
Antivirals represent a class of drugs for which development is often focused on the singular outcome of limiting viral infection. However, side effects and potential off-label or secondary indications for the use of a drug are often only discovered after the drug is used with FDA approval. A prime example of a virus that is well studied with regards to antiviral therapy is HSV-1. HSV-1 frequently infects humans leading to diseases ranging from mild mucocutaneous lesions to life threatening infections, especially in elderly or immunocompromised patients (Whitley and Roizman, 2001). HSV-1 is characterized by its ability to develop latency in neurons of sensory ganglia and lytic outbreaks that often require drug therapy (Knickelbein et al., 2008). Although extensive research efforts have been made to unveil the mechanism of HSV-1 infection, new aspects of HSV-1 pathogenesis are frequently discovered (Tognarelli et al., 2019; Sauerbrei, 2016; Johnston and Corey, 2016).
The standard therapy for HSV management includes acyclovir (ACV), a compound developed in the 1980s. Extensive use of ACV, as is expected with antiviral therapeutics, has led to resistance to ACV being increasingly reported (Morfin and Thouvenot, 2003; Piret and Boivin, 2011). As the future clinical efficacy of ACV is being assessed, therapies with increased efficacy, reduced resistance potential, and improved pharmacokinetics will undoubtedly benefit clinical outcomes (James and Prichard, 2014). Combinatorial drugs often provide better efficacy with reduced individual drug doses compared to treatment with a single drug (Silva et al., 2016; Nowak-Sliwinska et al., 2016; Weiss et al., 2015; Ding et al., 2014). As mentioned above, two distinct drug combinations identified through the FSC technique (a mixture of ACV with either ribavirin (RIB) or interferons (IFNs)) could both effectively inhibit HSV-1 infection (Ding et al., 2012). Though both drug combinations were effective in reducing HSV-1 infection, the underlying mechanisms as to why these particular drug combinations were so potent have not been fully explored. In this study, infected cells treated with either the RIB-rich drug combination or the IFN-rich drug combination were compared side-by-side using phosphoflow technology to determine how these drug combinations affect cellular signaling networks. By not focusing on one set pathway of cellular signaling we used an unbiased approach looking across various signaling nodes in cells to better understand the antiviral mechanisms of these two drug combinations and directly identify cellular signaling pathways that are critical in the control of HSV-1 replication. This study demonstrates the power of using this approach to analyze drug combinations and how to translate the findings. Importantly, we also directly translate our findings into the signaling changes involved in the HSV-1 infection to directly target those changes with antiviral therapies.
2. Materials and methods
2.1. Cell culture and HSV-1 infections
This in vitro investigation was carried out in NIH 3T3 cells, which is a cell line derived from mouse embryo fibroblasts. NIH 3T3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum and 1% antibiotics (penicillin and streptomycin). Preparation and quantization of HSV-1 were conducted as previously described (Ding et al., 2012). Viruses were diluted in DMEM to a multiplicity of infection (MOI) of 0.1.
2.2. Drugs and solutions
IFN-α, IFN-β, and IFN-γ were purchased from PBL Interferon Source (Piscataway, NJ). RIB and ACV were purchased from Calbiochem (San Diego, CA). DMEM was purchased from CELLGRO (Manassas, VA), and penicillin and streptomycin were obtained from GIBCO (Grand Island, NY). Paraformaldehyde (PFA) was purchased from Electron Microscopy Sciences (Hatfield, PA), and phosphate buffered saline (PBS) was bought from EMD (Rockland, MA). All other plates and tubes were purchased from BD Falcon (San Jose, CA).
2.3. Drug combinations applied in the study
Based on our previous work (Ding et al., 2012), the following five drug combinations were applied and compared in the current study (Table 1 ): high dose IFNs and ACV (Optimal_A and Optimal_AA), high dose RIB and ACV (Optimal_B), low dose IFNs and ACV (Subopt_A), and low dose IFNs and RIB (Subopt_B). Each individual drug from the combinations was administrated alone or in combination at time 0 in duplicate. After treatment, samples at four time points (0.5, 4, 8, and 16 h) were collected.
Table 1.
Drug combinations tested in the current study.
| Optimal_A | Optimal_AA | Optimal_B | Subpot_A | Subpot_B |
|---|---|---|---|---|
| IFN-β | IFN-α | RIB | IFN-α | IFN-α |
| 3.1 ng/mL | 3.1 ng/mL | 25,000 ng/mL | 0.8 ng/mL | 0.8 ng/mL |
| IFN-γ | IFN-γ | IFN-β | IFN-β | IFN-β |
| 3.1 ng/mL | 3.1 ng/mL | 0.2 ng/mL | 0.8 ng/mL | 0.8 ng/mL |
| ACV | ACV | ACV | IFN-γ | IFN-γ |
| 80 ng/mL | 80 ng/mL | 80 ng/mL | 0.8 ng/mL | 0.8 ng/mL |
| ACV | RIB | |||
| 20 ng/mL | 390 ng/mL | |||
2.4. Phosphoflow
Phosphoflow was performed as previously described (Krutzik et al., 2008). Briefly, cells with or without treatment were fixed with formaldehyde (1.6% final concentration), permeabilized with 4 °C pure methanol, washed twice with staining medium (PBS containing 0.5% bovine serum albumin and 0.02% sodium azide), and finally stained with phosphospecific antibodies in staining medium. Antibodies are against Akt (also known as Protein kinase B), ERK (extracellular signal-related kinase), JNK (c-Jun N-terminal kinases), cJun, MAPKAPK2 (Mitogen-activated protein kinase-activated protein kinase 2), NFĸB (Nuclear Factor ĸB), p38 (Mitogen-activated protein kinase), S6 (ribosomal protein S6), STAT1 (signal transducer and activator of transcription), STAT3, STAT5, and STAT6. Flow cytometry was performed on a BD FACSCanto II flow cytometer. After acquisition, data were analyzed using FlowJo software to generate median fluorescent intensity (MFI) values for each phosphoprotein in each treatment group. The median values were exported to Microsoft Excel for further analysis. All experiments were done with isotype controls to determine background staining. In addition, all experiments were done in triplicates. Data presented as single flow cytometry plot is representative of all three experiments. Data from the triplicates was further converted to bar graphs by averaging data and plotting with error bars representing the standard error.
2.5. Assessing pS6 kinase inhibitor on in vitro HSV-1 infection
To validate the cause-effect relationships between pS6 signaling and HSV-1 infection, we used a pS6 kinase inhibitor, SL0101, to evaluate the effect of anti-HSV-1 drug combinations on down-regulating the S6 signaling pathway. SL0101 at three doses (25, 50, and 100 μM) was compared with RIB at 100 μM and ACV at 100 μM. Cells were infected with HSV-1 at the same time that drugs were administered. We also examined the effects of these treatments on pAKT, pERK, pMAPKAPK2, p-p38, p-p70, p-p90RSK, pS6, pSTAT1, and pSTAT3 using phosphoflow.
2.6. Animal model of HSV-1 infection
In vivo compound treatment and stimulations were performed on female KOS strain mice. All animal handling was performed in accordance with University of California, Los Angeles (UCLA) Animal Research Committee guidelines and animals were monitored for changes to health regularly. Each mouse received either 25 μL of 2.9 × 107 PFU/mL HSV-1 or 25 μL PBS vehicle via vaginal administration on day 0. HSV-1 infection was allowed to occur for 24 h post virus administration. The first vaginal lavage was collected at 24 h post infection (day 1) followed immediately by a 50 μL drug treatment via vaginal administration. The second vaginal lavage was collected at 48 h post-infection (this was 24 h post-first-round drug treatment) (day 2) followed immediately by a 50 μL drug treatment via vaginal administration. The third vaginal lavage was collected at 72 h post-infection (24 h post-second-round drug treatment) (day 3). HSV-1 viral copy number in vaginal lavage was quantified by plaque assay as described below. To assess Optimal_B efficacy, twenty-seven mice were randomly distributed into three groups each containing nine mice. One group was untreated, one was treated with ACV at 80 ng/mL, and one was treated with drug combination Optimal_B. To evaluate SL0101, an RSK p90 inhibitor, eighteen mice were divided into two groups (an untreated control group and a SL0101 treated group). SL0101 at dose of 200 μM was applied to the treated group.
2.7. Plaque assay to quantify HSV-1 titer
Plaque assays were performed on Vero cell monolayers. Each lavage was serially diluted eight times with DMEM at a ratio of 1:10. During this process, 0.4 mL of lavage dilution was added to the Vero cell monolayer, and virus absorption was allowed to proceed for 1 h at 37 °C with gentle shaking every 20 min. After absorption, excess virus was removed by three washes with DMEM. 1 mL of agar overlay medium was added on top of the Vero cell monolayer. After plaques started to form the agar overlay was removed, and the cell monolayer was stained with 0.1% crystal violet. Plaques were counted under a microscope and bar graphs were produced based on averages of three experimental data points. Statistical differences were determined using a t-Test between viral levels at day 2 or day 3, SL0101 or untreated samples, or fold change in virus levels at day 2 versus day 1 and day 3 versus day 1.
3. Results
3.1. Cellular signaling analysis of cells stimulated with antiviral drug combinations
To better understand how the therapeutic drug combinations were able to inhibit HSV-1 replication, we used phosphoflow to simultaneously monitor the phosphorylated states of 12 cellular signaling nodes (Akt, ERK, JNK, cJun, MAPKAPK2, NFĸB, p38, S6, STAT1, STAT3, STAT5, and STAT6) in individual cells upon drug stimulation. We chose these different signaling nodes to highlight key signal transduction cascades that if modulated would lead to direct changes in the state of the cells. This system is robust enough to track the changes in cellular physiology, as indicated by changes in cellular signaling, induced by the different drug combinations to provide definitive insight into how the cell is affected by these drugs individually and in their optimal combinations by looking at the panel of phosphoproteins. Each individual drug from the combinations was administrated alone and in combination in duplicate at time 0. Samples were collected at four time points post simulation (0.5, 4, 8, and 16 h).
Using this approach, we were able to discern the exact changes in treated cells that were signatures of the optimal drug combinations. An overview of the phosphoprotein panel shown in Supplementary Fig. 1 indicates that most proteins phosphorylation states were unaffected in cells treated with drug combinations. However, analysis of these panels of signaling nodes revealed that levels of phosphorylated STAT1 (pSTAT1) and phosphorylated S6 (pS6) were noticeably regulated by treatment with different drug combinations (Fig. 1 A). Sixteen hours after drug treatment, we found that stimulation with individual revealed that, as expected, Interferons including IFN-β and IFN-γ induced upregulation of pSTAT1 in a dose-dependent manner (Fig. 1B, left panel, yellow histograms). In addition, we see that most of the drugs individually cause minimal to no change to the high levels of pS6 proteins, except the higher dose of Ribavirin (Fig. 1B, right panel, black histogram). The optimal combinations followed these key individual drugs with the IFN-rich treatments (Optimal_A) inducing increased levels of pSTAT1 but induced no obvious changes in pS6 (Fig. 1C). Conversely, RIB-rich treatments (Optimal_B) decreased levels of pS6 but induced no obvious changes in pSTAT1 (Fig. 1C). While both of these drug combinations were discovered for their effectiveness at blocking HSV-1 replication (Ding et al., 2012), these two drug combinations seems to change cellular signaling in distinct ways.
Fig. 1.

Cellular Signaling Analysis of Drug Stimulated Cells. A. Host cells were treated with the five drug combinations listed in Table 1 as well as each individual drugs. As a reminder: high dose IFNs and ACV (Optimal_A and Optimal_AA), high dose RIB and ACV (Optimal_B), low dose IFNs and ACV (Subopt_A), and low dose IFNs and RIB (Subopt_B). Drug treatments are indicated at the bottom of the panel. Samples were collected 0.5, 4, 8, and 16 h after drug stimulations. Although 12 phosphoproteins were examined for each sample (pAkt, pERK, pJNK, p-cJun, pMAPKAPK2, pNFĸB, p-p38, pS6, pSTAT1, pSTAT3, pSTAT5, and pSTAT6), here we show only pS6 and pSTAT1. The complete panel is shown in Supplementary Fig. 1. B. Comparison of pSTAT1 and pS6 signaling after 16 h of individual drug treatments indicates IFN up-regulated pSTAT1 signaling and RIB down-regulated pS6 signaling. C. Comparison of two optimal drug combination treatments shows that the IFN-rich treatment up-regulated pSTAT1 signaling and RIB-rich treatment down-regulated pS6 signaling. All experimental data shown is representative data from three independent experiments. Flow cytometry histograms are color-coded as in a heatmap with black being unchanged and shades of yellow denoting increases. Quantitation of plots from B and C are shown in Supplemental Fig. 2.
3.2. Monitoring STAT1 and S6 as distinct signals of antiviral drug efficacy
Based on the results described above, we next focused on the phosphorylated states of STAT1 and S6 after drug treatment of cells. Monitoring pSTAT1 over time showed that pSTAT1 was upregulated, in a dose-dependent manner, as early as 0.5 h and this was extended up to 16 h after drug treatments (Fig. 2 A and B). In contrast, adding drug combinations containing RIB lowered pS6 in a dose-dependent manner. pS6 signaling was not an instant marker for RIB, as pS6 decreased only after 8 h of drug treatments (Fig. 2C and D). When plotting pSTAT1 and pS6 signaling side-by-side along the time course, pSTAT1 was an instant and long-lasting marker for IFN, whereas pS6 was a later period marker for RIB (Fig. 2E).
Fig. 2.

STAT1 and S6 Phosphorylation States After Antiviral Drug Stimulation. A. pSTAT1 signaling was identified as a marker for IFN rich treatment, but not for RIB rich treatment. B. pSTAT1 is an instant, long-lasting marker for IFN treatment. Activation of pSTAT1 was available as early as 0.5 h after drug stimulations and continued for up to 16 h after IFN drug treatments. C. pS6 signaling was identified as a marker for RIB rich treatment, but not for IFN rich treatment. D. pS6 signaling was not an instant marker but a later-period marker for RIB treatment, as pS6 level did not change until 8 h after drug treatments. E. Quantification of pSTAT1 and pS6 signaling in samples treated with Optimal_A and Optimal_B indicates pSTAT1 signaling is Optimal_A specific and pS6 is optimal_B specific. Unit for drugs is ng/ml. As a reminder: high dose IFNs and ACV (Optimal_A and Optimal_AA), high dose RIB and ACV (Optimal_B), low dose IFNs and ACV (Subopt_A), and low dose IFNs and RIB (Subopt_B). For Part E, data plotted is mean ± standard deviation. All experimental data shown is representative data from three independent experiments.
3.3. Monitoring cellular signaling and HSV-1 infection after post-infection treatment
To better understand how these drugs induced changes to the cellular signaling network would lead to limiting HSV-1 replication, we next systemically evaluated changes in the phosphoprotein panel during viral infections treated with drug combinations. Compared to uninfected cells treated with drug combinations alone, cells pre-infected with HSV-1 followed by drug treatments provide a more realistic model for studying the changes in cellular physiology under optimal drug combination treatments, as drug treatments would almost always take place after infection is detected (Fig. 3 A). We used HSV-1 that was engineered to express GFP (Green Fluorescent Protein) as a way to track HSV-1 replication within infected cells. This allowed us to distinguish changes in cellular signaling induced by infection on the single cell level.
Fig. 3.

Cellular Signaling During Drug-Treated Viral Infection. A. Host cells were infected with HSV-1, and 8 h post-infection they were treated with drugs. B. 0.5 h after drug treatments, all infected samples had approximately equivalent viral infection levels/percentages, which indicates that the drugs would not instantly lead to viral reduction. C. 16 h post-treatment (24 h post-infection), pAKT, pMAPKAPK2, pNFĸB, pS6 and pSTAT1 were found to be involved during the viral infection and inhibition process. As a reminder: high dose IFNs and ACV (Optimal_A and Optimal_AA), high dose RIB and ACV (Optimal_B), low dose IFNs and ACV (Subopt_A), and low dose IFNs and RIB (Subopt_B). All experimental data shown is representative data from three independent experiments. Flow cytometry histograms are color-coded as in a heatmap with black being unchanged and shades of yellow denoting increases. Quantitation of pSTAT1 from B are shown in Supplemental Fig. 3. Quantitation of plots from C are shown in Supplemental Fig. 4 with GFP levels in 4A, pAKT levels in 4B, pMAPKAPK2 levels in 4C, pNFĸB levels in 4D, pS6 levels in 4E and pSTAT1 levels in 4F. Red arrows in Supplemental Fig. 4 A, E and F indicate Optimal_B and Ribavirin treatments across the analysis.
All samples infected with HSV-1 were done so at a multiplicity of infection (MOI) of 0.1 (+HSV) for 8 h after which the cells were treated with drugs. All conditions had equivalent GFP levels/percentages 0.5 h after drug treatment (Fig. 3B). The GFP level in +HSV samples was low across all treatments, indicating that viral infection was still in progress and that the majority of cells remained uninfected. Neither IFN-rich nor RIB-rich treatments lowered levels of viral infection in an instantaneous manner (Fig. 3C). 16 h after drug treatments, GFP levels in +HSV samples increased extensively throughout the cell population indicating that the viral infection was able to infect the whole cell population during this time frame. In this model with virus infection prior to drug treatment, drug combination Optimal_B (RIB-rich combination) outperformed Optimal_A (IFN-rich combination) in viral inhibition at 16 h post treatment. Although Optimal_A and Optimal_B combinations were identified in a drug-virus co-infection model (Ding et al., 2012), drug combination Optimal_A barely reduced the viral infection level in cells infected with virus prior to treatment (Fig. 3C). This may be due to the previous work using a viral co-infection/treatment model for determining the optimal drug combinations.
3.4. Changes in distinct signaling nodes after post-infection treatment of viral infection
The results in Fig. 3 suggest that IFN does not directly eradicate virus in already infected cells; instead IFN inhibits viral infection by targeting uninfected cells and preparing them to avoid further viral invasion, which is in line with the known biological mechanisms of IFN. Once viral infection had begun in the cell population, IFN could not reduce the level of viral infection. In contrast, Optimal_B (RIB-rich combination) was effective in this study, in which cells were infected prior to drug treatment. These results indicate that RIB can directly target viral replication inside the cells.
Differences in phosphoprotein signaling became more complex 16 h after treatment (Fig. 3C). pAKT, pMAPKAPK2, pNFĸB, pS6, and pSTAT1 were regulated to various extents by the different drug combinations. In particular, RIB treatments led to both a decrease in pS6 levels and a reduction in HSV-1 viral burden (Fig. 4 A). pSTAT1 levels were diminished in HSV-1 infected cells, but pSTAT1 levels were recovered by IFN treatments (Fig. 4B). HSV-1 infection decreased pMAPKAPK2 levels, whereas blocking HSV-1 infection with ACV containing treatments increased pMAPKAPK2 signaling (Fig. 4C). pNFĸB was induced in HSV-1 infected cells, but ACV treatments reduced pNFĸB levels to baseline (Fig. 4D). ACV containing treatments that reduced HSV-1 levels appeared to induce pAKT levels in the HSV-1 positive population (Fig. 4E). Overall, when HSV-1 viral infection was present, phosphoprotein changes became more complex. However, the most significant changes in phosphorylation status for drug combination optimal_A was still pSTAT1, and the most significant changes in phosphorylation status for drug combination optimal_B was still pS6. The correlation between IFN, pSTAT1 status, and HSV-1 infection has been widely reported and part of the known biology of IFN. However, the lack of agreement about the mechanisms of RIB and the high efficacy of the RIB-rich drug combination in blocking HSV-1 encouraged us to focus our further investigation into the correlations among RIB, pS6 levels, and HSV-1 infection.
Fig. 4.

Drug Treatment of Viral Infection Affecting Distinct Signaling Nodes. Host cells were pre-infected with HSV-1 for 8h and then received drug treatments for 16 h. The signaling alterations became more complex compared to host cells treated only with drugs but without virus. A. RIB reduced both pS6 level and viral burden. B. pSTAT1 signaling was blocked in HSV-1 infected cells, but it was recovered by IFN treatments. C. pMAPKAPK2 level was decreased by HSV-1 treatment, whereas blocking HSV-1 infection increased pMAPKAPK2 signaling. D. pNFĸB was induced in HSV-1 infected cells, whereas ACV containing treatments reduced pNFĸB level to baseline. E. ACV treatments that reduced HSV-1 levels appeared to induce pAKT levels in the HSV-1 positive population. As a reminder: high dose IFNs and ACV (Optimal_A). All experimental data shown is representative data from three independent experiments.
3.5. Monitoring cellular signaling and HSV-1 infection with simultaneous infection and treatment
To confirm the correlations among RIB, pS6 signaling, and HSV-1 infection obtained in the initial screening, we investigated the state of cellular signaling in cells simultaneously treated with the drug combinations and infected with HSV-1 (Fig. 5 A). In order to allow viral infection to propagate adequately, we focused on the time point 16 h after cells were treated with drug combinations and infected with HSV-1. In this model, Optimal_A and Optimal_B both profoundly reduced HSV-1 infections compared to unstimulated controls (Fig. 5B). This result is in line with conclusions in the previously published findings (Ding et al., 2012). When we further compared HSV-1 infection levels (Fig. 5C) and pS6 levels (Fig. 5D), HSV-1 infection induced an increase in pS6 levels by 5–10 fold, and RIB or the RIB-containing drug combination reduced percent infection and pS6 levels following a similar pattern. Percent infection plotted against pS6 levels revealed that viral infection followed a log-linear relationship with pS6 level (Fig. 5E). Thus, using an unbiased screen to monitor changes in cellular phosphorylation states allowed us to identify the key changes in cellular physiology that connect the efficacy of a given antiviral drug combination to its antiviral effect.
Fig. 5.

Signaling In Simultaneous Infection and Treatment. A. Host cells stimulated with drug combinations and HSV-1 infection simultaneously for 16 h. B. Both drug combinations Optimal_A and Optimal_B profoundly reduced HSV-1 infection compared to unstimulated controls in the co-treated model. Data points are average of data from three experimental ± standard deviation. Flow cytometry histograms are color-coded as in a heatmap with black being unchanged and shades of yellow denoting increases. C. Drug treatments versus percent HSV-1 infection. Data points are average of data from three experimental ± standard deviation. D. Drug treatments versus pS6 level showed similar patterns as viral infection. E. HSV-1 infection levels and S6 phosphorylation levels followed a log-linear correlation. Data points are average of data from three experimental ± standard deviation. As a reminder: high dose IFNs and ACV (Optimal_A and Optimal_AA), high dose RIB and ACV (Optimal_B), low dose IFNs and ACV (Subopt_A), and low dose IFNs and RIB (Subopt_B). All experimental data shown is representative data from three independent experiments. For Parts C, D and E, data plotted is mean ± standard deviation. In Part E, the correlation coefficient, R2, is displayed.
3.6. Translating cellular analysis of RIB signaling to discovery of a small molecule inhibitor of HSV-1 inhibition in vitro
Now that we understood that modulation of the S6 phosphorylation state is critical target in the control of HSV-1 replication we sought to validate the cause-effect relationships between pS6 signaling and HSV-1 infection using the pS6 kinase inhibitor, SL0101. To evaluate the effect of down-regulating the S6 pathway on HSV-1 replication we looked at viral replication 16 h after treatment with SL0101. At 50 μM of SL0101 there is a slightly decreased GFP level in +HSV-1 samples, while SL0101 at 100 μM or RIB at 100 μM profoundly decrease the viral infection level (Fig. 6 A). We also examined the effects of these treatments on pAKT, pERK, pMAPKAPK2, p-p38, p-p70, p-p90RSK, pS6, pSTAT1, and pSTAT3. When HSV-1 was not present, RIB and SL0101 both showed general signaling regulation across multiple pathways. With viral stimulation, however, SL0101 at 25 and 50 μM no longer resulted in dramatic signaling regulation. This result shows that HSV-1 infection could overcome changes induced by low dose SL0101. However, at 100 μM, SL0101 treatment resulted in a general decrease in signaling across multiple pathways. RIB at 100 μM appeared to be much more specific in signaling regulation, as only pS6 was significantly down-regulated (Fig. 6B).
Fig. 6.

A Small Molecule That Mimics Antiviral Drug Signaling Modulation. A. pS6 kinase inhibitor in HSV-1 inhibition. SL0101 displayed a RIB-like anti-HSV-1 behavior. B. SL0101 showed a general decrease in signaling across multiple pathways, particularly at 100 μM. C. SL0101 appeared to decrease all signaling pathways that have basal activity. SL0101 inhibited pS6 at 50 μM when no virus was present, but it was not effective at that dose when virus was present. When HSV-1 was present, SL0101 required as high as 100 μM to show inhibition on pS6. Flow cytometry histograms are color-coded as in a heatmap according to the scale in B. All experimental data shown is representative data from three independent experiments. In part C, *, p value < 0.05 **, p value < 0.01. Quantitation of plots from B are shown in Supplemental Fig. 6 with pAKT levels in 6A, pMAPKAPK2 levels in 6B, p-p70S6K levels in 6C and pS6 levels in 6D.
It is important to note that broad spectrum modulation of signaling pathways by a drug may lead to drug-induced toxicity. Therefore, it is possible that SL0101 could be more toxic than RIB even though such toxicity was not obvious based on observation of cellular morphology in our in vitro study (Supplementary Fig. 5A) and SL0101 only induced toxicity in an MTT assay at concentrations above what we used in this study (Supplementary Fig. 5B). SL0101 inhibited pS6 at 50 μM when virus was absent, but it was not effective at that dose when virus was present (Fig. 6C). When virus was present, SL0101 only down-regulated pS6 at 100 μM. Furthermore, with HSV infection present, SL0101 did not obviously reduce viral infection levels at 50 μM, but infection levels were diminished at 100 μM. Therefore, SL0101 regulates pS6 signaling in a dose-dependent manner in uninfected cells, but SL0101 treatment inhibits HSV infection at a high dose in concordance with changes in pS6 during viral infection.
3.7. Translating cellular signaling analysis: small molecule inhibition of HSV-1 infection in an animal model
The ultimate goal of our system using phosphoflow is to better understand how drug therapies affect cellular physiology so that we can quickly develop more targeted therapeutic modalities for treating disease. In this study we used a mouse model of vaginal HSV-1 infection as a way to assess the in vivo efficacy of the drug combinations we previously discovered. In addition, this acts as a proof of principle that our platform can determine key signaling nodes that should be manipulated as therapy of disease. Fig. 7 A depicts our setup of a model of HSV-1 vaginal infection in mice. To ensure that the animal model could be used to evaluate HSV-1 inhibition efficacy in vivo, we first tested ACV alone and drug combination Optimal_B (RIB-rich treatments). Mice treated with ACV showed decreased HSV-1 viral load, especially on day 3. Remarkably, mice treated with drug combination Optimal_B had HSV-1 levels that were 70-fold lower than those of untreated mice on day 3 (Fig. 7B). This result indicated that this animal model could be applied as an effective in vivo model to examine HSV-1 inhibition efficacy of other drug treatments.
Fig. 7.

Translating Cellular Signaling Analysis to a Small Molecule Treatment of HSV Infection. SL0101 (RSK p90 inhibitor) inhibited HSV-1 infection in vivo. A. Demonstration of the animal model used in the current study. B. Drug combination Optimal_B (high dose RIB and ACV) effectively prevented HSV-1 infection in the animal model, indicating that the animal model was valid for further evaluations on the anti-HSV-1 potency of SL0101. C. SL0101 significantly reduced viral burden in vivo. D. Unlike drug combination Optimal_B, which started to show strong an anti-HSV-1 effect on day 2, SL0101 took longer (day 3) to induce viral inhibition. Each experiment used nine animals for each data point. *, p value < 0.05 **, p value < 0.01.
We then tested SL0101 for its in vivo anti-HSV-1 effect using this animal model (Fig. 7A). Mice treated with SL0101 did not show significant viral reduction compared to the untreated group on day 2. However, the treated group showed more than 12 fold viral reduction compared to untreated group on day 3 (Fig. 7C). When we compared the fold change in HSV-1 levels (e.g. levels on day 2 divided by day 1 levels (day2/day1) and levels on day 3 divided by day 1 levels (day3/day1)), the benefit of SL0101 treatment in limiting HSV-1 replication became much clearer, especially for day3/day1. Unlike drug combination Optimal_B, which started to show strong an anti-HSV-1 effect on day 2, the antiviral effect of SL0101 was delayed by about a day (Fig. 7D).
4. Discussion
Analysis of alternations in cellular signaling induced by any one drug, combination of drugs, or the disease state can rapidly elucidate the intricate changes in cellular signaling which is crucial for determining how to therapeutically apply drugs for a proper balance between therapeutic outcomes and potential side effects. Phosphoflow technology is a robust tool that can monitor many cellular signaling pathways that can be modulated during drug treatment (George et al., 2015; Hildebrand and Kubatzky, 2017; Coppin et al., 2017). Using this technology one can rapidly interrogates the state of key signaling nodes in diverse cellular pathways. We focused our work on changes in phosphorylation states in a panel of key signaling nodes representative of the major cellular processes that could be altered by the drug treatments being used.
Here we present a study focusing on HSV-1 infection. While HSV-1 infection can be well controlled by currently available antivirals, we must develop alternative therapeutic strategies due to several reasons. These reasons include the appearance of anti-viral resistant strains, the possibility of HSV-1 inducing enhanced disease in immunocompromised individuals, and the lack of an effective HSV-1 vaccine. Here we analyze cellular signaling on a system level to determine how antiviral drug combinations modulate many different pathways and discovered that they have different molecular signatures when assayed by phosphoflow. Our results reveal that HSV-1 infection can be targeted using antivirals that modulate one of two pathways: STAT1 or S6. Combinations containing RIB cause changes in S6 phosphorylation, whereas IFN-containing combinations lead to changes in STAT1 phosphorylation. STAT1 is a well-known cellular inducer of the antiviral state (Horvath and Darnell, 1996; Liu et al., 2012). This antiviral state efficiently blocks viral growth and thus it makes sense as a marker of antiviral activity.
The impact that modulation of S6 phosphorylation would have on viral replication is not a well-understood idea. While HSV-1 replication either induced or required upregulated levels of S6 phosphorylation, blocking these heightened levels of S6 phosphorylation by RIB was sufficient to block HSV-1 replicaiton. We used these findings to focus on the phosphorylation of S6 as a new target when developing therapeutics against HSV-1 infection. Using this focus, we used a small molecule (SL0101), an inhibitor of an S6 kinase that can reduce S6 phosphorulation and thereby mimic the efficacy of one of the drug combination by lowering S6 phosphorylation. We went on to show that SL0101 could effectively block the replication of HSV-1 in an animal model of vaginal HSV infection.
HSV-1 is one of the most well studied viruses known so it is intriguing that our analysis of cellular signaling modulation in infected cells, using an unbiased approach to discovered that S6 phosphorylation is a new target for antivirals targeting HSV-1. Given the importance of the S6 protein control of ribosomal translation, further studies of how HSV-1 replication benefits from this change in S6 and how common this method of manipulation of cellular translation is among different viruses is needed (Ruvinsky and Meyuhas, 2006). Could it be that the efficacy of RIB is conveyed via changes in S6 induced by viruses? If the theme of S6 manipulation by viruses is common in other viral disease, S6 phosphorylation modulators may be a new class of antivirals.
The efficacy of the S6 phosphorylation inhibition in reducing HSV-1 replication in an animal model of vaginal HSV-1 infection shows how important it is to understand the changes in cellular signaling that are effective in blocking viral infection. However, results of this study revealed that even without HSV-1 infection, RIB directly lowers the level of phosphorylated S6. This finding is important because it underscores that regulation of S6 phosphorylation occurs in cells with and without viral infection. Consequently, the consequences of changing the phosphorylation state of a key ribosomal protein by any drug may be immense. We did not see any obvious changes in cellular morphology nor changes in cell viability as assessed by MTT assay when cells were treated with SL0101, suggesting that minimal cellular toxicity was induced by the SL0101. However, further investigations into how SL0101 can impact cellular physiology are warranted in order to develop effective counter measures against side effects that may be induced by this type of antiviral.
RIB has become a go-to drug in antiviral therapy, especially when looking to target the replication of viruses with few options for treatment. At one time, RIB was the major treatment for Hepatitis C Virus infection but now RIB-independent therapies have become commonplace in the clinic (Kish et al., 2017). RIB remains the first line of antiviral therapy against other viruses on the world stage, including MERS-CoV (Middle East Respiratory Syndrome-Corona Virus), Lassa Fever Virus, and Zika virus (Kamiyama et al., 2017; Carrillo-Bustamante et al., 2017; Chong et al., 2015). However, a different mechanism of action may be at work for every virus that RIB effectively treats, including nucleoside base level changes and catastrophic error induction in cells (Te et al., 2007; Crotty et al., 2002).
5. Conclusions
In conclusion, we have used phosphoflow technology to evaluate possible mechanisms of therapeutic drug combination and elucidate how these drug combinations modulate cellular signaling so as to design better antiviral therapeutics. Specifically, we were able to identify STAT1 and S6 phosphorylation as a key signaling nodes involved in HSV-1 replication and demonstrate that therapeutic drug combinations that effectively block HSV-1 replication were modulating these signaling nodes to block HSV-1 replication. We extended our findings to develop a proof of principle in a mouse model of vaginal HSV-1 infection by targeting S6 phosphorylation directly with the S6 kinase inhibitor SL0101. The efficacy of targeting this phosphorylation step shows that using analysis of cellular signaling modulation can determine new, efficacious antivirals and drug targets.
Author contributions
XD, DMJr, CMH, GPN, and DJS wrote the manuscript. XD, POK, AAG, YZ and DJS conducted experiments. XD, YZ, DMJr and DJS analyzed data. XD, POK, AAG, GC, CMH, GPN and DJS designed experiments.
Acknowledgements
The work was supported by National Institutes of Health Nanomedicine Development Center, grant number PN2EY018228 as well as NIH 1R15AI138847 for D.J.S. and Science and Technology Innovation Zone, grant number 17-163-15-XJ-002-002-09 for X.D..
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.antiviral.2019.104598.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Fig. S1.

Panel of Phosphoprotein States in Stimulated Cells. The phosphoproteins are listed on the top of the figure, and the time points are listed to the right of the figure. Drug treatments run across each block in the figure. The median fluorescent intensity (MFI) is listed to the left of the figure. MFI staining correlates to the phosphorylation level of the stained protein. NIH 3T3 cells were treated with drug combinations for 0.5, 4, 8, or 16 h, respectively. The cells were then fixed, permeabilized, and fluorescently labeled with phosphospecific antibodies. The phosphoproteins were stained with either a 488 fluorescent tag or a 647 fluorescent tag according to established protocols [13]. All experimental data shown is representative data from three independent experiments.
Fig. S2.

Quantification of Flow Cytometry Plots fromFig. 1. Mean Fluorescent Intensity (MFI) for the histogram plots represented in Fig. 1 (N = 3) were quantitated relative to unstimulated and plotted in bar graphs along with standard error shown in errors bars across experiments. The plots from Fig. 1B are shown in A and the plots from Fig. 1C are shown in B.
Fig. S3.

Quantification of Flow Cytometry Plots fromFig. 3B. The average of Mean Fluorescent Intensity (MFI) for the histogram plots represented in Fig. 3B (N = 3) were quantitated relative to unstimulated and plotted in bar graphs along with standard error shown in errors bars across experiments. Green bars represent HSV infected cells while black bars represented mock infected cells.
Fig. S4.

Quantification of Flow Cytometry Plots from Fig. 3C. The average of Mean Fluorescent Intensity (MFI) for the histogram plots represented in Fig. 3C (N = 3) were quantitated relative to unstimulated and plotted in bar graphs along with standard error shown in errors bars across experiments. Green bars represent HSV infected cells while black bars represented mock infected cells. A shows GFP levels, B shows pAKT levels, C shows pMAPKAPK2 levels, D shows pNFĸB levels, E shows pS6 levels and F shows pSTAT1 levels. Red arrows in A, E and F indicate Optimal_B and Ribavirin treatments across the analysis.
Fig. S5.

SL0101 Does Not Induce Cellular Toxicity. A. NIH3T3 cells used in the previous in vitro experiments were subjected to treatment with mock conditions or 100 mm of SL0101 for 48 h. After which they were observed for changes in morphology that would be a sign of SL0101 induced cytotoxicity. No changes were seen. Images shown are representative of several (N = 5) images taken within each well. B. SL0101 was added to cells in increasing amounts and a standard MTT assay was used to determine toxicity. Cell viability from the MTT assay was plotted against SL0101 concentrations. Data points were done in triplicates and averaged of the values for each point were graphed with error bars representing standard error of triplicates shown.
Fig. S6.

Quantification of Flow Cytometry Plots from Fig. 6C. The average of Mean Fluorescent Intensity (MFI) for the histogram plots represented in Fig. 6C (N = 3) were quantitated relative to unstimulated and plotted in bar graphs along with standard error shown in errors bars across experiments. Green bars represent HSV infected cells while black bars represented mock infected cells. A shows pAKT levels, B shows pMAPKAPK2 levels, C shows p-p70S6K levels, and D shows pS6 levels.
References
- Al-Lazikani B., Banerji U., Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012;30(7):679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- Arts E.J., Hazuda D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. 2012;2(4) doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall S.C. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 2010;3(153):rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 2012;30(9):858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Bustamante P. Determining Ribavirin's mechanism of action against Lassa virus infection. Sci. Rep. 2017;7(1):11693. doi: 10.1038/s41598-017-10198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Y.P. Antiviral treatment guidelines for Middle East respiratory Syndrome. Infect Chemother. 2015;47(3):212–222. doi: 10.3947/ic.2015.47.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppin E. Flow cytometric analysis of intracellular phosphoproteins in human monocytes. Cytometry B Clin Cytom. 2017;92(3):207–210. doi: 10.1002/cyto.b.21207. [DOI] [PubMed] [Google Scholar]
- Crotty S., Cameron C., Andino R. Ribavirin's antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. (Berl.) 2002;80(2):86–95. doi: 10.1007/s00109-001-0308-0. [DOI] [PubMed] [Google Scholar]
- Ding X. Cascade search for HSV-1 combinatorial drugs with high antiviral efficacy and low toxicity. Int. J. Nanomed. 2012;7:2281–2292. doi: 10.2147/IJN.S27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X. Discovery of a low order drug-cell response surface for applications in personalized medicine. Phys. Biol. 2014;11(6) doi: 10.1088/1478-3975/11/6/065003. [DOI] [PubMed] [Google Scholar]
- George A.A. Phosphoflow-based evaluation of mek inhibitors as small-molecule therapeutics for B-cell precursor acute lymphoblastic leukemia. PLoS One. 2015;10(9) doi: 10.1371/journal.pone.0137917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand D., Kubatzky K.F. Phospho-flow analysis of primary mouse cells after HDAC inhibitor treatment. Methods Mol. Biol. 2017;1510:233–243. doi: 10.1007/978-1-4939-6527-4_17. [DOI] [PubMed] [Google Scholar]
- Horvath C.M., Darnell J.E., Jr. The antiviral state induced by alpha interferon and gamma interferon requires transcriptionally active Stat1 protein. J. Virol. 1996;70(1):647–650. doi: 10.1128/jvi.70.1.647-650.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S.H., Prichard M.N. Current and future therapies for herpes simplex virus infections: mechanism of action and drug resistance. Curr Opin Virol. 2014;8:54–61. doi: 10.1016/j.coviro.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Johnston C., Corey L. Current concepts for genital herpes simplex virus infection: diagnostics and pathogenesis of genital tract shedding. Clin. Microbiol. Rev. 2016;29(1):149–161. doi: 10.1128/CMR.00043-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama N. Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremia in ZIKV-infected STAT1-deficient mice. Antivir. Res. 2017;146:1–11. doi: 10.1016/j.antiviral.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish T., Aziz A., Sorio M. Hepatitis C in a new era: a review of current therapies. PT. 2017;42(5):316–329. [PMC free article] [PubMed] [Google Scholar]
- Knickelbein J.E. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322(5899):268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik P.O., Nolan G.P. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat. Methods. 2006;3(5):361–368. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- Krutzik P.O. High-content single-cell drug screening with phosphospecific flow cytometry. Nat. Chem. Biol. 2008;4(2):132–142. doi: 10.1038/nchembio.2007.59. [DOI] [PubMed] [Google Scholar]
- Lee B.Y. Drug regimens identified and optimized by output-driven platform markedly reduce tuberculosis treatment time. Nat. Commun. 2017;8:14183. doi: 10.1038/ncomms14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.Y. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. U. S. A. 2012;109(11):4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfin F., Thouvenot D. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 2003;26(1):29–37. doi: 10.1016/s1386-6532(02)00263-9. [DOI] [PubMed] [Google Scholar]
- Nowak-Sliwinska P. Optimization of drug combinations using feedback system control. Nat. Protoc. 2016;11(2):302–315. doi: 10.1038/nprot.2016.017. [DOI] [PubMed] [Google Scholar]
- Piret J., Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 2011;55(2):459–472. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I., Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 2006;31(6):342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Sachs K. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308(5721):523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- Sauerbrei A. Optimal management of genital herpes: current perspectives. Infect. Drug Resist. 2016;9:129–141. doi: 10.2147/IDR.S96164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer L., Zhang L. Enhancing cancer drug discovery through novel cell signaling pathway panel strategy. Cancer Growth Metastasis. 2013;6:53–59. doi: 10.4137/CGM.S11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A. Output-driven feedback system control platform optimizes combinatorial therapy of tuberculosis using a macrophage cell culture model. Proc. Natl. Acad. Sci. U. S. A. 2016 doi: 10.1073/pnas.1600812113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Vilar S., Tatonetti N.P. High-throughput methods for combinatorial drug discovery. Sci. Transl. Med. 2013;5(205):205rv1. doi: 10.1126/scitranslmed.3006667. [DOI] [PubMed] [Google Scholar]
- Te H.S., Randall G., Jensen D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol Hepatol (N Y) 2007;3(3):218–225. [PMC free article] [PubMed] [Google Scholar]
- Tognarelli E.I. Herpes simplex virus evasion of early host antiviral responses. Front Cell Infect Microbiol. 2019;9:127. doi: 10.3389/fcimb.2019.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A. A streamlined search technology for identification of synergistic drug combinations. Sci. Rep. 2015;5:14508. doi: 10.1038/srep14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley R.J., Roizman B. Herpes simplex virus infections. Lancet. 2001;357(9267):1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- Wong P.K. Closed-loop control of cellular functions using combinatory drugs guided by a stochastic search algorithm. Proc. Natl. Acad. Sci. U. S. A. 2008;105(13):5105–5110. doi: 10.1073/pnas.0800823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.